CN101096366A - 二硫代二噻唑酮的合成方法 - Google Patents
二硫代二噻唑酮的合成方法 Download PDFInfo
- Publication number
- CN101096366A CN101096366A CNA2006100905790A CN200610090579A CN101096366A CN 101096366 A CN101096366 A CN 101096366A CN A2006100905790 A CNA2006100905790 A CN A2006100905790A CN 200610090579 A CN200610090579 A CN 200610090579A CN 101096366 A CN101096366 A CN 101096366A
- Authority
- CN
- China
- Prior art keywords
- minutes
- accordance
- reaction
- add
- dimercapto
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000010189 synthetic method Methods 0.000 title claims abstract description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 23
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims abstract description 21
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 claims abstract description 20
- 238000006243 chemical reaction Methods 0.000 claims abstract description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 28
- 238000000034 method Methods 0.000 claims description 13
- 238000001035 drying Methods 0.000 claims description 11
- 239000012046 mixed solvent Substances 0.000 claims description 3
- 238000001953 recrystallisation Methods 0.000 claims description 3
- 239000007864 aqueous solution Substances 0.000 claims description 2
- 230000035484 reaction time Effects 0.000 claims description 2
- 239000002904 solvent Substances 0.000 claims description 2
- -1 disulfo-dithiazole ketone Chemical class 0.000 claims 2
- 238000005406 washing Methods 0.000 claims 1
- 239000000047 product Substances 0.000 abstract description 20
- BIGYLAKFCGVRAN-UHFFFAOYSA-N 1,3,4-thiadiazolidine-2,5-dithione Chemical compound S=C1NNC(=S)S1 BIGYLAKFCGVRAN-UHFFFAOYSA-N 0.000 abstract description 15
- 239000007795 chemical reaction product Substances 0.000 abstract description 2
- 231100000956 nontoxicity Toxicity 0.000 abstract description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 238000001291 vacuum drying Methods 0.000 description 20
- 239000002244 precipitate Substances 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- 239000012467 final product Substances 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 239000012153 distilled water Substances 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 239000004519 grease Substances 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- 239000005069 Extreme pressure additive Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 230000001050 lubricating effect Effects 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000003064 anti-oxidating effect Effects 0.000 description 1
- 239000007866 anti-wear additive Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- 239000010687 lubricating oil Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 238000002161 passivation Methods 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 150000004867 thiadiazoles Chemical class 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
Landscapes
- Lubricants (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
Abstract
一种二硫代二噻唑酮的合成方法,是将2,5-二巯基-1,3,4-噻二唑(DMTD)与过氧化氢或过氧乙酸混合,在35℃~55℃的温度下进行反应。反应完成后,将反应粗产物水洗,重结晶,干燥,得到目的产物。本发明与现有技术相比,具有操作简单,安全无毒,后续处理简单的特点,同时具有很高的收率。
Description
技术领域
本发明是关于一种合成二硫代二噻唑酮的方法。
背景技术
极压、抗磨添加剂是润滑油、润滑脂中非常重要的一类添加剂。近年来,噻二唑衍生物的极压抗磨作用得到了进一步的认识,作为润滑脂高性能极压抗磨剂开始崭露头角。这类极压抗磨剂不含磷和重金属元素,为无灰添加剂,而且还具有抗氧、金属钝化等功能,是环境友好、性能优异的多功能极压抗磨剂,在润滑油、润滑脂中将得到越来越广泛的应用。
二硫代二噻唑酮是2,5-二巯基-1,3,4-噻二唑的二聚物,是优秀的润滑脂极压添加剂,其商品牌号有Vanlube829,其结构式如下:

Eiichi Shouji,Yasuyuki Yokoyama,John M.Pope,Noboru Oyama(J.Phys.Chem.B 1997,101,2861-2866)公开了一种典型的合成二硫代二噻唑酮的方法。先将2,5-二巯基-1,3,4-噻二唑与甲醇混合,在氮气保护下加入碘,通过氧化反应得二硫代二噻唑酮,产率达88%。实验中采用氮气保护,使之在无氧的条件下进行,反应条件较为苛刻。实验中用到甲醇,甲醇易挥发,且具有一定毒性,对人的眼睛危害较大。
发明内容
本发明提出了一种二硫代二噻唑酮的合成方法,操作方法简单,易于控制,安全无毒,且具有很高的产率。
本发明提供的合成方法包括:
将2,5-二巯基-1,3,4-噻二唑(DMTD)与过氧化氢或过氧乙酸混合,在35℃~55℃的温度下进行反应。
过氧化氢水溶液的质量浓度为30%~70%,优选30%~50%。DMTD与过氧化氢按照2∶1摩尔比进行反应,投料时过氧化氢可适当过量,以确保DMTD反应完全。
过氧乙酸水溶液的质量浓度为10%~40%。DMTD与过氧乙酸按照2∶1摩尔比进行反应,投料时过氧乙酸可适当过量,以确保DMTD反应完全。
反应温度为35℃~55℃,优选40℃~50℃。
反应时间为10分钟~90分钟,优选30分钟~60分钟。
为了便于反应,DMTD最好先在水或醇中溶解,其中的醇为C2~C4的直链醇。反应完成后,将反应粗产物水洗,重结晶,干燥,得到目的产物。
重结晶用溶剂为C2~C4的直链醇或醇/水混合溶剂,优选乙醇。
抽真空干燥温度为70℃~120℃,优选70℃~90℃。
抽真空干燥的时间为30分钟~90分钟。
以过氧化氢作为氧化剂,本发明合成方法的反应方程式如下:
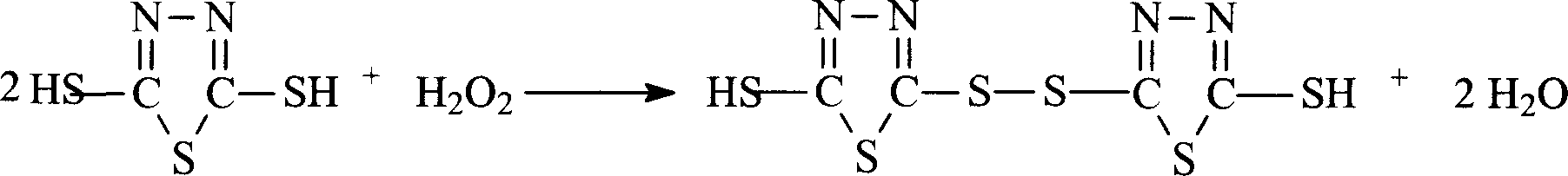
以过氧乙酸作为氧化剂,本发明合成方法的反应方程式如下:

本发明与现有技术相比,具有操作简单,安全无毒,后续处理简单的特点,同时具有很高的收率。
具体实施方式
下面将通过实例说明本发明。本发明包含但不限于以下实例。实例中所用试剂,除特别说明外,均为分析纯试剂。
实例1
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入蒸馏水,加热至45℃恒温15min使其溶解。加入30%的过氧化氢溶液10.2ml。反应一个小时后将产物倒入水中。将沉淀用乙醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度80℃,真空干燥时间60min。得最终产物。产率90%。
实例2
在三口烧瓶中加入30%的过氧化氢溶液10.2ml,加热到40℃。称取2,5-二巯基-1,3,4-噻二唑30g,加入到过氧化氢溶液中。控制反应温度不超过50℃,一个小时后将产物倒入水中。将沉淀用乙醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度90℃,真空干燥时间90min。得最终产物。产率92%。
实例3
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入丙醇,55℃恒温15min使其溶解。加入30%的过氧化氢溶液10.2ml。滴加完毕后,30分钟后将产物倒入水中。将沉淀用丙醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度90℃,真空干燥时间60min。得最终产物。产率88%。
实例4
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入正丁醇,55℃恒温15min使其溶解。加入30%的过氧化氢溶液10.2ml。滴加完毕后,30分钟后将产物倒入水中。将沉淀用正丁醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度90℃,真空干燥时间60min。得最终产物。产率88%。
实例5
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入乙醇/水(体积比为7/3)混合溶剂,45℃恒温15min使其溶解。加入50%的过氧化氢溶液5.7ml。反应90分钟后将产物倒入水中。将沉淀用乙醇/水的混合溶液重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度70℃,真空干燥时间60min。得最终产物。产率90%。
实例6
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入蒸馏水,35℃恒温15min使其溶解。加入70%的过氧化氢溶液3.8ml。滴加完毕后,一个小时后将产物倒入水中。将沉淀用乙醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度120℃,真空干燥时间30min。得最终产物。产率85%。
实例7
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入蒸馏水,加热至45℃恒温15min使其溶解。加入10%的过氧乙酸溶液71.3ml。反应一个小时后将产物倒入水中。将沉淀用乙醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度80℃,真空干燥时间60min。得最终产物。产率88%。
实例8
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入蒸馏水,加热至45℃恒温15min使其溶解。加入20%的过氧乙酸溶液34.0ml。反应一个小时后将产物倒入水中。将沉淀用乙醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度90℃,真空干燥时间60min。得最终产物。产率90%。
实例9
在三口烧瓶中加入2,5-二巯基-1,3,4-噻二唑30g,加入蒸馏水,加热至45℃恒温15min使其溶解。加入40%的过氧乙酸溶液15.5ml。反应一个小时后将产物倒入水中。将沉淀用乙醇重结晶,得一浅黄色固体。将产物放入真空干燥箱,干燥温度90℃,真空干燥时间60min。得最终产物。产率90%。
对比例1
取Eiichi Shouji,Yasuyuki Yokoyama等人公开的一种典型的合成二硫代二噻唑酮的方法为参照。取甲醇50ml,2,5-二巯基-1,3,4-噻二唑2.0g加入到200ml原底烧瓶中。取碘1.69g加入到20ml的甲醇中。在氮气保护下,强烈搅拌15min后,将碘的甲醇溶液加入到三口烧瓶中,与2,5-二巯基-1,3,4-噻二唑的甲醇溶液混合。1小时后,将反应混合物倒入水中。沉淀用甲醇重结晶。得浅黄色固体。产率为88%。
对比表明,本发明的方法,操作条件上明显简单于典型的合成方法且安全、无毒。产率普遍高于典型的合成方法的产率。适合于工业上的应用。
Claims (9)
1.一种二硫代二噻唑酮的合成方法,包括:将2,5-二巯基-1,3,4-噻二唑与过氧化氢或过氧乙酸混合,在35℃~55℃的温度下进行反应。
2.按照权利要求1所述的方法,其特征在于,过氧化氢水溶液的质量浓度为30%~70%。
3.按照权利要求1所述的方法,其特征在于,过氧乙酸水溶液的质量浓度为10%~40%。
4.按照权利要求1所述的方法,其特征在于,反应温度为40℃~50℃。
5.按照权利要求1所述的方法,其特征在于,反应时间为10分钟~90分钟。
6.按照权利要求1所述的方法,其特征在于,2,5-二巯基-1,3,4-噻二唑先在水或醇中溶解,其中的醇为C2~C4的直链醇。
7.按照权利要求1所述的方法,其特征在于,反应完成后,将反应粗产物水洗,重结晶,抽真空干燥,得到目的产物。
8.按照权利要求7所述的方法,其特征在于,重结晶用溶剂为C2~C4的直链醇或醇/水混合溶剂。
9.按照权利要求7所述的方法,其特征在于,干燥温度为70℃~120℃,干燥的时间为30分钟~90分钟。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB2006100905790A CN100569762C (zh) | 2006-06-29 | 2006-06-29 | 二硫代二噻唑酮的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB2006100905790A CN100569762C (zh) | 2006-06-29 | 2006-06-29 | 二硫代二噻唑酮的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101096366A true CN101096366A (zh) | 2008-01-02 |
| CN100569762C CN100569762C (zh) | 2009-12-16 |
Family
ID=39010471
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2006100905790A Active CN100569762C (zh) | 2006-06-29 | 2006-06-29 | 二硫代二噻唑酮的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN100569762C (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2947549A1 (fr) * | 2009-07-06 | 2011-01-07 | Mlpc Internat | Procede de preparation de bis-dmtd |
| WO2012168642A1 (fr) | 2011-06-06 | 2012-12-13 | Mlpc International | Composition de vulcanisation pour polymères insaturés |
| CN110194751A (zh) * | 2019-07-01 | 2019-09-03 | 新乡市瑞丰新材料股份有限公司 | 一种噻二唑衍生物的制备方法 |
| CN111732598A (zh) * | 2020-07-09 | 2020-10-02 | 吉首大学 | 一类含二噻唑并噻二唑双杂环的化合物及制备方法 |
| CN117756745A (zh) * | 2022-09-16 | 2024-03-26 | 中国石油天然气股份有限公司 | 高纯度双噻二唑衍生物及其制备方法 |
-
2006
- 2006-06-29 CN CNB2006100905790A patent/CN100569762C/zh active Active
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2947549A1 (fr) * | 2009-07-06 | 2011-01-07 | Mlpc Internat | Procede de preparation de bis-dmtd |
| EP2272836A1 (fr) | 2009-07-06 | 2011-01-12 | MLPC International | Procédé de préparation de bis-DMTD |
| CN101941951A (zh) * | 2009-07-06 | 2011-01-12 | Mlpc国际公司 | 双-dmtd的制备方法 |
| JP2011012064A (ja) * | 2009-07-06 | 2011-01-20 | Mlpc Internatl | ビス−dmtdの調製方法 |
| US10316005B2 (en) | 2009-07-06 | 2019-06-11 | Mlpc International | Process for the preparation of bis-DMTD |
| CN101941951B (zh) * | 2009-07-06 | 2014-09-03 | Mlpc国际公司 | 双-dmtd的制备方法 |
| US10087308B2 (en) | 2011-06-06 | 2018-10-02 | Mlpc International | Vulcanization composition for unsaturated polymers |
| WO2012168642A1 (fr) | 2011-06-06 | 2012-12-13 | Mlpc International | Composition de vulcanisation pour polymères insaturés |
| CN110194751A (zh) * | 2019-07-01 | 2019-09-03 | 新乡市瑞丰新材料股份有限公司 | 一种噻二唑衍生物的制备方法 |
| CN110194751B (zh) * | 2019-07-01 | 2021-02-26 | 新乡市瑞丰新材料股份有限公司 | 一种噻二唑衍生物的制备方法 |
| CN111732598A (zh) * | 2020-07-09 | 2020-10-02 | 吉首大学 | 一类含二噻唑并噻二唑双杂环的化合物及制备方法 |
| CN117756745A (zh) * | 2022-09-16 | 2024-03-26 | 中国石油天然气股份有限公司 | 高纯度双噻二唑衍生物及其制备方法 |
| CN117756745B (zh) * | 2022-09-16 | 2025-10-17 | 中国石油天然气股份有限公司 | 高纯度双噻二唑衍生物及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN100569762C (zh) | 2009-12-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Chen et al. | Direct thiocyanation of ketene dithioacetals under transition-metal-free conditions | |
| Ramazani et al. | The reaction of (N‐isocyanimino) triphenylphosphorane with an electron‐poor α‐haloketone in the presence of aromatic carboxylic acids: A novel three‐component reaction for the synthesis of disubstituted 1, 3, 4‐oxadiazole derivatives | |
| JP4232986B2 (ja) | 有機アンモニウム・タングステン酸塩およびモリブデン酸塩化合物、ならびにこのような化合物を調製する方法 | |
| Jiang et al. | KI-catalyzed C–S bond formation via an oxidation relay strategy: efficient access to various α-thio-β-dicarbonyl compounds | |
| CN117384080B (zh) | 一种n-甲基-2-苯基吲哚的合成方法 | |
| Hu et al. | Copper-catalyzed direct oxidative C (sp2)-H α-sulfenylation of enaminones with disulfides or thiophenols: Synthesis of polyfunctionalized aminothioalkenes | |
| RU2003136765A (ru) | Синтез 4-фенилмасляной кислоты | |
| CN101096366A (zh) | 二硫代二噻唑酮的合成方法 | |
| CN108290834B (zh) | 硫代羧酸酯的制造方法 | |
| CN102516000B (zh) | 一种合成芳基三氟甲巯基化合物的方法 | |
| Hassan et al. | New access to pyrazole, oxa (thia) diazole and oxadiazine derivatives | |
| US3948803A (en) | Catalyst | |
| Zhao et al. | Green synthesis of 1, 2, 4-thiadizoles from thioamides in water using molecular oxygen as an oxidant | |
| Mukhopadhyay et al. | Nanopowder zinc titanate in aqueous medium: An expeditious catalyst for the synthesis of propargylamines via C–H bond activation | |
| Thompson et al. | Synthesis of 5-aminothiazoles as building blocks for library synthesis | |
| Murai | Synthesis of thioamides | |
| WO2013035650A1 (en) | Method for producing alpha - hydroxy ketone compound | |
| Antonow et al. | Facile oxidation of electron-poor benzo [b] thiophenes to the corresponding sulfones with an aqueous solution of H 2 O 2 and P 2 O 5 | |
| Lv et al. | Dioxygen-triggered oxidative cleavage of the C–S bond towards C–N bond formation | |
| CN101456806A (zh) | 一种羧酸铋的制备方法 | |
| CN105541757B (zh) | [5‑(邻羟基苯基亚甲基亚氨基)‑1,3,4‑噻二唑‑2‑基]硫乙酸酯、制备方法及其应用 | |
| Vahabi et al. | Palladium-catalyzed, unsymmetrical homocoupling of thiophenes via carbon–sulfur bond activation: a new avenue to homocoupling reactions | |
| US8697886B2 (en) | Di(aminoguanidium) 4,4′,5,5′-tetranitro-2,2′-biimidazole, and preparation method thereof | |
| US7495107B2 (en) | Method for manufacturing isoxazole derivative or dihydroisoxazole derivative | |
| Metwally et al. | Utility of cyclohexanethiols in organic synthesis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |