CN1289331A - 具有哌啶结构的法尼转移酶抑制剂及其制备方法 - Google Patents
具有哌啶结构的法尼转移酶抑制剂及其制备方法 Download PDFInfo
- Publication number
- CN1289331A CN1289331A CN99802581A CN99802581A CN1289331A CN 1289331 A CN1289331 A CN 1289331A CN 99802581 A CN99802581 A CN 99802581A CN 99802581 A CN99802581 A CN 99802581A CN 1289331 A CN1289331 A CN 1289331A
- Authority
- CN
- China
- Prior art keywords
- methyl
- piperidin
- naphthalen
- pyrrole
- imidazol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Communicable Diseases (AREA)
- Vascular Medicine (AREA)
- Virology (AREA)
- Urology & Nephrology (AREA)
- Oncology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
Abstract
本发明涉及一种新的由化学式(1)表示的对法尼转移酶表现出抑制活性的哌啶衍生物或其药物可接受的盐,其中的A、E和G如说明书所定义;本发明也涉及化学式(1)的化合物的制备方法;本发明也涉及用于化学式(1)的化合物的制备中的中间体;本发明还涉及含有化学式(1)的化合物作为一种活性组分的药物组合物。
Description
技术领域
本发明涉及一种新的由下列化学式(1)表示的对法尼转移酶表现出抑制活性的哌啶衍生物或其药物可接受的盐, 其中的A、E和G如后面的描述所定义。
其中的A、E和G如后面的描述所定义。
本发明也涉及化学式(1)的化合物的制备方法;涉及用于化学式(1)的化合物的制备中的中间体;还涉及含有化学式(1)的化合物作为一种活性组分的药物组合物。
背景技术
哺乳动物Ras蛋白在与细胞生长和变异相关的信号传输进程中作为分子开关起作用。Ras原致癌基因家族由三个成员组成:N-,K-,和H-Ras,它们是高度均匀的四类蛋白质的代号;即,189个残基的H、N-Ras蛋白和二个同形的分别为188和189个残基的K-Ras-4B和K-Ras-4A蛋白。开关机理的化学基础包括不活泼的(关)鸟苷二磷酸(GDP)键合状态和活泼的(开)鸟苷三磷酸(GTP)键合状态之间的蛋白质的循环(Bourne,H.R.;Sanders,D.A.;McCormick,F.;Nature,1991,349,117)。生物化学和结构研究已经表明,位于GTP的磷酰基的邻位的、导致鸟苷三磷酸酶活性降低的残基12、13和61的点突变与许多人类癌症,特别是胰腺癌,膀胱癌,结肠癌等有关(Bos,J.L.,Cancer Res.,1989,49,4682)。
Ras蛋白作为细胞溶质的前体被合成,在一系列转录后的改性后最后集中于质膜的细胞质面(Gibbs,J.B.,Cell 1991,65,1)。这些通过改变电荷状态或特殊结构以增加疏水性的一系列生物化学改性使得Ras蛋白更容易附着在细胞膜上。在该一系列改性中第一和必须的步骤是在由法尼蛋白转移酶(FTase)催化的反应中在C-端的CAAX主体(motif)(C,半胱氨酸;A,通常为脂肪族残基;X,任何其它的氨基酸)的半胱氨酸残基中加入法尼基团部分。缺乏C-端半胱氨酸的Ras突变体不能被法尼基化,不能集中到原生质和不能在培养中转化哺乳动物细胞(Hancock,J.F.,Magee,A.I.,Childs,J.E.,Marshall,C.J.,Cell 1989,57,1167),证明这一改性对Ras功能是必须的。后来的转录后的改性,AAX残基的分裂,法尼化半胱氨酸的羧甲基化,以及位于H-和N-Ras蛋白中的CAAX主体的上游的半胱氨酸的十六酰化,对于Ras膜缔合作用或细胞的转化活性都不是必须的。有趣的是,不同于H-和N-Ras,K-Ras-4B具有一个被称为多元区域的多赖氨酸富集区域,而不是具有十六酰化所需要的半胱氨酸,从而促进法尼化的Ras蛋白与细胞膜的阴离子类脂层键合。催化强制改性的FTase抑制剂从而已被建议用作肿瘤的抗癌试剂,其中Ras致癌基因有助于转化(Buses,J.E.等,Chemistry&Biology,1995,2,787)。近来已充分证明,许多FTase抑制剂对于体外和动物模型中的转化的细胞和肿瘤细胞系中的阻滞Ras法尼化,信号传输和转化作用是有效的并具有特殊的能力(Kohl.N.E.等,Proc.Natl.Acad.Sci.USA.1994,91,9141;Kohl.N.E.等,NatureMedicine,1995,1,792)。
但是,大多数抑制剂涉及实际上作为Ras底物摹拟和肽的CAAX主体,或者含有巯基(USP5,141,851;Kohl.N.E.等,Science,1993,260,1934;Graham等,PCT/US95/12224;Sebti,S.M.等,J.Biol.Chem.,1995,270,26802;James,G.L.等,Science,1993,260,1937;Bishop,W.R.等,J.Biol.Chem.,1995,270,30611)。最近报道了一种模仿FTase催化步骤的新型肽模拟抑制剂(Poulter,C.D.等,J.Am.Chem.Soc.,1996,118,8761)。抑制剂设计的化学基础涉及到反应机理。这就是说,通过酶转移异戊烯基是亲电性的取代,该反应在跃迁状态需要正电荷。
但是上面描述的这些抑制剂对Ras蛋白特别是K-Ras-4B的致瘤功能的抑制具有有限的活性和选择性,K-Ras-4B被发现是人类癌症中最普遍的。因此需要具有有效地抑制K-Ras活性的能力的新的抑制剂。
关于再狭窄和血管增生疾病,已经发现细胞Ras的抑制作用防止了活体内血管损伤后的平滑肌增生(Indolfi C.等,Nature Med.,1995,1(6),541-545)。这一报道确定地支持了法尼转移酶抑制剂在该疾病中的作用,表现出对血管平滑肌的累积和增生的抑制作用。
本发明的公开
本发明人致力于开发一种具有模拟FTase的催化反应的中间状态的结构特性的化合物,结果发现按照本发明的哌啶衍生物能够有效地抑制该酶。
因此,本发明的目的是提供一种能抑制FTase的活性的化学式(1)的哌啶衍生物,该衍生物的制备方法,以及一种可有效地用于化学式(1)化合物的制备方法中的新的中间体。
本发明的另一目的是提供一种含有化学式(1)化合物作为活性组分的药物组合物。实施本发明的最好方式
本发明的第一个目的是提供一种能抑制法尼转移酶活性的由下列化学式(1)表示的哌啶衍生物及其药物可接受的盐:其中
A代表氢、低碳烷基或
 其中
其中
B代表CH2、C=O或SO2,
D代表一个选自下列组中的基团: 在取代基D的定义中,m表示0-3的整数,n表示1-3的整数,X代表氢、苯基、苯氧基、低碳烷基、低碳烷氧基、卤素、硝基、或者被苄基或低碳烷基非强制取代的氨基,
在取代基D的定义中,m表示0-3的整数,n表示1-3的整数,X代表氢、苯基、苯氧基、低碳烷基、低碳烷氧基、卤素、硝基、或者被苄基或低碳烷基非强制取代的氨基,
R1和R2各自分别代表氢、低碳烷基、C3-C6环烷基、被C3-C6环烷基取代的低碳烷基、芳基、或杂芳基,
E代表氢、苯基、萘基或
 其中
其中
R3和R4各自分别代表氢、低碳烷基、芳基或
(其中Y代表O或S,n′表示2-4的整数,R5代表低碳烷基),
G代表选自下列组中的基团: 其中
其中
Z代表O、S、SO2或N-R6(其中R6代表氢或低碳烷基),
R7和R8各自分别代表氢、低碳烷基、低碳烷氧基、卤素、氰基、羟基羰基、氨基羰基、氨基硫代羰基、羟基、苯基或苯氧基。
特别是,按照本发明的化合物具有与已知的法尼转移酶抑制剂完全不同的结构,并且从不包括硫醇基团部分。
在对化学式(1)化合物的取代基的定义中,术语“低碳烷基”是指具有1-4个碳原子的直链或支链烷基,包括甲基,乙基,异丙基,异丁基和叔丁基;术语“环烷基”是指环状烷基包括环己基;术语“芳基”是指6-14元的单环、双环或三环芳基;术语“杂芳基”是指含有选自由氧,氮和硫所组成的组中的杂原子的6-14元的单环、双环或三环芳基。
而且,本发明的化学式(1)的化合物可以形成药物可接受的盐。这种盐包括含有药物可接受阴离子的无毒的酸加成盐,例如与无机酸如盐酸、硫酸、硝酸、磷酸、氢溴酸、氢碘酸等所形成的盐,与有机羧酸如酒石酸、甲酸、柠檬酸、乙酸、三氯乙酸、三氟乙酸、葡糖酸、苯甲酸、乳酸、富马酸、马来酸、天冬氨酸等所形成的盐,或与磺酸如甲磺酸、苯磺酸、对甲苯磺酸、萘磺酸等所形成的盐;碱加成盐,例如与吡啶或氨所形成的盐;以及金属加成盐,例如与碱金属或碱土金属所形成的盐如锂盐。另外,本发明还包括化学式(1)化合物的溶剂化物如其醇盐或水合物。这些化合物可以用常规的转化方法制备。
在按照本发明的化学式(1)的化合物中,优选的化合物包括下列化合物,其中
A代表氢、低碳烷基,或
 其中
其中
B代表CH2、C=O或SO2,
D代表一个选自下列组中的基团:
在取代基D的定义中,
m表示0-1的整数,
n表示1-2的整数,
X代表氢,
R1和R2各自分别代表氢或低碳烷基,
E代表氢、苯基、萘基或
 其中
其中
R3和R4各自分别代表氢、低碳烷基、或2-甲氧基乙基,
G代表选自下列组中的基团: 其中
其中
Z代表O或N-R6(其中R6代表甲基),
R7和R8各自分别代表氢。
按照本发明的化学式(1)化合物的典型例子列于下列表1中。表1-1表1-2 表1-3表1-4
表1-3表1-4 表1-5表1-6
表1-5表1-6 表1-7
表1-7 表1-8
表1-8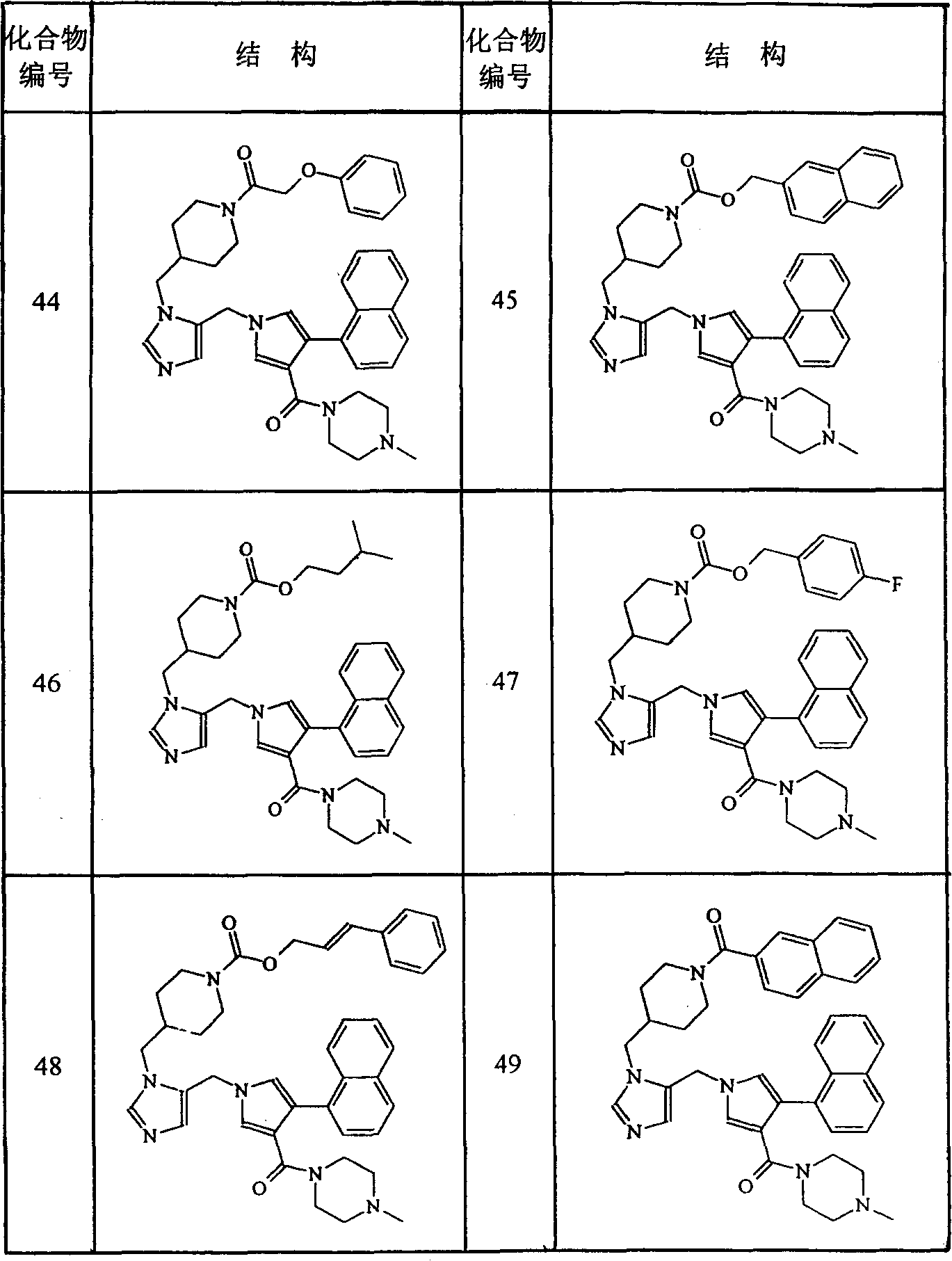 表1-9表1-10
表1-9表1-10
本发明的另一个目的是提供上述定义的化学式(1)的哌啶衍生物的制备方法。
按照本发明,化学式(1)的哌啶衍生物可以通过一种方法制备,其特征在于
(a)将下列化学式(2a)表示的化合物: 其中Cbz代表苄氧基羰基,并且在整个本说明书中具有相同的意义,在一种溶剂中在一种碱存在下与下列化学式(3)表示的化合物反应:其中E和G如前面的描述所定义;然后将保护基Cbz脱除,得到由下列化学式(1a)表示的化合物:
其中Cbz代表苄氧基羰基,并且在整个本说明书中具有相同的意义,在一种溶剂中在一种碱存在下与下列化学式(3)表示的化合物反应:其中E和G如前面的描述所定义;然后将保护基Cbz脱除,得到由下列化学式(1a)表示的化合物: 其中E和G如前面的描述所定义;
其中E和G如前面的描述所定义;
(b)将化学式(1a)的化合物在一种溶剂中与由下列化学式(4)表示的化合物反应:
A′-W (4)其中A′与A相同,所不同的是A′不为氢,W代表氢、羟基或活泼的离去基团,优选卤素,得到由下列化学式(1b)表示的化合物:其中A′,E和G如前面的描述所定义;
(c)将化学式(1a)的化合物在一种溶剂中与下列化学式(5)表示的化合物反应:
A″-N=C=O (5)其中A″代表低碳烷基、苄基或C3-C6环烷基,得到由下列化学式(1c)表示的化合物: 其中A″,E和G如前面的描述所定义;
其中A″,E和G如前面的描述所定义;
(d)将化学式(1a)的化合物在一种溶剂中在一种还原剂存在下与下列化学式(6)表示的化合物反应:
D-CHO (6)其中D如前面的描述所定义,得到由下列化学式(1d)表示的化合物:其中D,E和G如前面的描述所定义;或者
(e)将化学式(1a)的化合物在一种溶剂中与碳酰氯和下列化学式(7)表示的化合物反应:
D′H (7)其中D′代表选自下列组中的基团: 其中m,n,X,R1和R2如前面的描述所定义,得到由下列化学式(1e)表示的化合物:
其中m,n,X,R1和R2如前面的描述所定义,得到由下列化学式(1e)表示的化合物: 其中D′,E和G如前面的描述所定义。
其中D′,E和G如前面的描述所定义。
但是,本发明的化合物也可以用通过本领域现有技术熟知的各种合成路线的结合所设计的任何方法进行常规的制备,这种结合对于本领域普通技术人员是容易进行的。该方法的变量(a)-(e)将在下面进行更具体的解释。
在用于制备本发明化合物的方法变量(a)-(e)中,可以使用对反应没有不利影响的任何惰性溶剂,优选的是选自由二甲基甲酰胺,二氯甲烷,四氢呋喃,氯仿和二甲基乙酰胺所组成的组中的一种或多种。作为方法变量(a)中的碱,可以使用选自由氢化钠,叔丁醇钾,二(三甲基硅基)酰胺钠和二(三甲基硅基)酰胺钾所组成的组中的一种或多种。而且,方法(a)中从哌啶环的1-位脱除苄氧基羰基的脱保护反应可以使用常规的反应条件进行,优选的是在一种醇溶剂中使用Pd(OH)2/C或Pd/C在氢气气氛中进行。
在方法变量(b)中,将方法变量(a)中得到的化学式(1a)的化合物在前面所述溶剂中并且非必须地在叔胺碱存在下与化学式(4)的化合物偶联,得到化学式(1b)的化合物。当化学式(4)的化合物中的W为羟基时,该反应优选的是在一种偶联试剂存在下进行。作为偶联试剂,可以使用由1-羟基苯并三唑(HOBT)和由碳二亚胺类如二环己基碳二亚胺(DCC),1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(EDC),1,1′-二羰基二咪唑(CDI)等所组成的组中的一种或多种物质所形成的混合物。
化学式(1)的化合物,其中B为C=O,D为低碳烷基、苄基或被C3-C6环烷基取代的氨基[=化学式(1c)的化合物],可以通过将方法变量(a)中得到的化学式(1a)的化合物与化学式(5)的异氰酸酯衍生物反应而制备。
在方法变量(d)中,在一种还原剂存在下进行还原氨化反应。作为该反应中可以使用的还原剂,可以使用一般认为是弱还原剂的那些还原剂,例如氢气气氛下的Pd/C,三乙酸基硼氢化钠或氰基硼氢化钠。
另一方面,下列化学式(2)表示的化合物,包括在方法变量(a)中用作起始原料的化学式(2a)的化合物,是一种新化合物。因此本发明的另一个目的是提供化学式(2)的化合物:其中Y代表羟基或氯。
化学式(2)的新的中间体可以通过下列方法制备,其特征在于:
(f)将下列化学式(8)表示的化合物: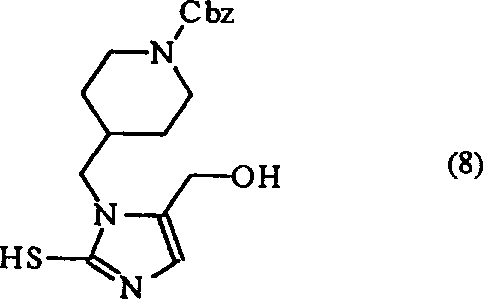 在一种有机溶剂中在硝酸存在下进行脱硫化反应,得到下列化学式(2b)表示的化合物:;或者
在一种有机溶剂中在硝酸存在下进行脱硫化反应,得到下列化学式(2b)表示的化合物:;或者
(g)将化学式(2b)的化合物与亚硫酰氯反应,得到化学式(2a)的化合物:
为了使化学式(8)的化合物脱硫化,在本发明中使用10%的硝酸。此时由于庞大的苄氧基羰基哌啶基的溶解度问题,必须在反应溶液中加入少量的有机溶剂。乙酸乙酯或四氢呋喃可以用作该有机溶剂。但是,也可以使用其它已知的方法作为脱硫条件从化学式(8)的化合物制备化学式(2b)的化合物。另外,这样获得的化学式(2b)的化合物可以与亚硫酰氯反应,有效地制备化学式(2a)的化合物。
如下面的反应路径1所描述的那样,在化学式(2)的化合物的制备中用作起始原料的化学式(8)的化合物可以通过一种方法从4-(氨基甲基)哌啶进行合成,在该方法中包括保护,苄氧羰基化和脱保护成胺化合物,然后在KSCN存在下与二羟基丙酮反应。J.Med.Chem.,33,1312-1329,1990,其中详细描述了一种类似的反应,可以作为该特定反应条件的参考。
反应路径1
在上述反应路径1中
CbzCl代表氯甲酸苄基酯,并且在整个本说明书中具有相同的意义。
用作化学式(1)化合物的制备中的反应试剂的化学式(3)的化合物如下面描述的反应路径2所述可以从1-萘甲醛或1-萘甲酸进行合成。反应路径2
在上述反应路径2中
TosMIC代表甲苯磺酰基甲基异腈,并且在整个本说明书中具有相同的意义。
可以用于上述反应路径2中第一步和第二步中的溶剂包括四氢呋喃,乙腈和二甲基甲酰胺。作为碱,可以使用选自由叔丁醇钾,1,8-二氮杂二环[5,4,0]十一-7-烯(DBU),氢氧化钾和氢氧化钠所组成的组中的一种或多种。
本发明方法中的反应条件,包括反应物的量,反应温度,反应时间等,可以由本领域普通技术人员根据特定反应物很容易地确定。
另外,上述方法中制备的以自由碱形式存在的化学式(1)的化合物可以按照本领域熟知的常规方法很容易地转化为前面所述盐的形式。
当反应完成后,所得产物可以通过常规处理方法如色谱法和重结晶法等进一步分离和/或纯化。
按照上述方法制备的化学式(1)的化合物表现出对法尼转移酶的抑制活性,因此可以有效地用作抗癌试剂。因此,本发明也提供一种药物组合物,该组合物含有与药物可接受的载体结合的、作为活性组分的、如前面所定义的化学式(1)的新化合物或者其药物可接受的盐。特别是,化学式(1)的化合物可以非常有效地用于治疗癌症,再狭窄症,动脉硬化症,和由δ肝炎和相应病毒引起的传染病。
当本发明的活性化合物用于临床的目的时,优选的给药量范围为每天每千克体重5-200mg。总的日剂量可以是一次或多次给药。但是病人的特定给药剂量可以根据所使用的特定化合物,目标病人的体重,性别,卫生条件,饮食,给药时间或方法,排泄速度,试剂的混合比例,所治疗的疾病的严重程度等来变化。
本发明的化合物可以以注射剂或口服剂的形式给药。注射剂,如消毒水溶液或油性悬浮注射液,可以按照已知的方法用合适的分散试剂,润湿剂或悬浮剂进行制备。可用于制备注射剂的溶剂包括水,林格氏流体和等渗NaCl溶液,消毒固定油也可以方便地用作溶剂或悬浮介质。任何非刺激性的固定油包括单-、二-甘油酯都可以用于该目的。脂肪酸如油酸也可以用于注射剂。
作为口服给药的固体制剂,可以使用胶囊,片剂,丸剂,粉剂和颗粒剂等,优选的是胶囊和片剂。将片剂和丸剂配进包有肠溶衣的制剂中也是所希望的。固体制剂可以通过将本发明的化学式(1)的活性化合物与至少一种载体混合而制备,所述载体选自由惰性稀释剂如蔗糖、乳糖、淀粉等,润滑剂如硬脂酸镁,崩解剂和粘合剂所组成的组中。
下面的实施例将对本发明进行更具体的说明。但是应该理解,下列实施例只是用来说明本发明,而不是对本发明的保护范围进行任何形式的限定。用于获得化学式(1)的化合物的起始原料的制备方法也在下面的制备例中进行了详细的说明。制备例1:4-(5-氯甲基-1H-咪唑-1-基甲基)-哌啶-1-羧酸苄基酯的合成
1-1) 4-氨基甲基-哌啶-1-羧酸苄基酯
将22.2克(0.2摩尔)4-氨基甲基哌啶溶解在250毫升甲苯中,然后向其中加入21.2克(0.2摩尔)苯甲醛。将该混合物用迪安-斯塔克设备回流3小时,冷却至0℃,然后在搅拌下向其中滴加入34.2克(0.2摩尔)氯甲酸苄基酯。将该混合物搅拌3小时后,在室温下向其中加入1N的硫酸氢钾水溶液(220毫升)。将混合物用200毫升乙醚萃取3次,然后用氢氧化钠将水层碱化。将水溶液用氯化钠饱和并用100毫升二氯甲烷萃取3次。将有机溶液用硫酸镁干燥并进行减压蒸馏,得到38克(产率91%,分子量248)标题化合物。
1H NMR(CDCl3)δ1.11(s,2H),1.49(s,3H),1.70(d,2H),2.57(d,2H),2.78(s,2H),4.20(s,2H),5.12(s,2H),7.34-7.35(m,5H)
FAB(M+H):249
1-2)4-(5-羟基甲基-2-巯基-1H-咪唑-1-基甲基)-哌啶-1-羧酸苄基酯
将24.8克(0.1摩尔)制备例1-1)中制备的化合物和6.0克(0.1摩尔)乙酸溶解在50毫升异丙醇中,然后将所得溶液加入到由12.6克(0.13摩尔)硫氰酸钾、9.0克(0.05摩尔)1,3-二羟基丙酮二聚物和10.0克(0.17摩尔)乙酸溶解在50毫升正丁醇中所形成的溶液中。将所得整个混合物搅拌48小时。减压蒸馏除去溶剂,向所得残余物中加入200毫升乙酸乙酯,将所得混合物用100毫升水洗涤3次。将有机层用硫酸镁干燥,减压蒸馏除去溶剂,得到27克(75毫摩尔,产率75%,分子量361)标题化合物。
1H NMR(CDCl3)δ1.22(d,2H),1.57(d,2H),2.30(s,1H),2.72(s,2H),3.96(s,2H),4.15(d,2H),4.46(s,2H),5.10(s,2H),6.62(s,1H),7.26-7.37(m,5H)
FAB(M+H):362
1-3)4-(5-羟基甲基-1H-咪唑-1-基甲基)-哌啶-1-羧酸苄基酯
将18.05克(50毫摩尔)制备例1-2)中制备的化合物加入到100毫升硝酸(10%)和10毫升乙酸乙酯的混合物中。将所得整个混合物用冰水冷却,并在室温下搅拌3小时。将所得混合物用4N氢氧化钠水溶液碱化,然后用100毫升乙酸乙酯萃取二次。将所得有机萃取物用硫酸镁干燥,并在减压下蒸馏,得到12.3克(38毫摩尔,产率75%,分子量329)标题化合物。
1H NMR(CDCl3)δ1.16(d,2H),1.56(d,2H),1.98(s,1H),2.70(s,2H),3.88(d,2H),4.18(s,2H),4.49(s,1H),4.52(br,1H),4.58(s,2H),5.10(s,2H),6.82(s,1H),7.27-7.40(m,5H)
FAB(M+H):330
1-4)4-(5-氯甲基-1H-咪唑-1-基甲基)-哌啶-1-羧酸苄基酯
将9.9克(30毫摩尔)制备例1-3)中制备的化合物溶解在50毫升氯仿中,在0℃下向其中缓慢滴加入7.1克(60毫摩尔)亚硫酰氯。将所得混合物搅拌2小时,减压蒸馏除去溶剂,并在真空中除去残留的盐酸,得到9.9克(产率95%,分子量347.5)标题化合物的盐酸盐。
1H NMR(CDCl3)δ1.12(d,2H),1.53(d,2H),2.65(s,2H),3.82(d,2H),4.22(s,2H),4.42(s,1H),4.49(s,3H),5.12(s,2H),6.60(s,1H),7.30-7.41(m,5H)
FAB(M+H):349制备例2:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯的合成
2-1)3-(萘-1-基)-丙烯酸乙酯
将22.4克(0.10摩尔)三乙基膦酰基乙酸酯溶解在500毫四氢呋喃中,向其中缓慢加入12.4克(1.1摩尔)叔丁醇钾。向所得溶液中缓慢加入溶解在20毫升四氢呋喃中的15.6克(0.10摩尔)1-萘甲醛,然后将所得溶液搅拌8小时。减压蒸馏除去有机溶剂,将所得残余物溶解在乙酸乙酯中,用水洗涤二次,用硫酸镁干燥并浓缩。将所得浓缩物进行硅胶柱色谱提纯(洗脱液:正己烷/乙酸乙酯=95/5,v/v),得到20.3克(0.090摩尔,产率90%)标题化合物。
1H NMR(CDCl3)δ1.33(t,3H),4.10(q,2H),6.75(q,1H),7.50(m,3H),7.73(d,1H),7.85(m,2H),8.10(d,1H),8.21(d,1H)
FAB 227(M+H)
2-2)3-(乙氧基羰基)-4-(萘-1-基)-1H-吡咯
将5克(18.9毫摩尔)制备例2-1)中制备的3-(萘-1-基)-丙烯酸乙酯和3.68克(18.9毫摩尔)甲苯磺酰基甲基异腈溶解在100毫升四氢呋喃中。向所得溶液中缓慢加入溶解在100毫升四氢呋喃中的2.55克(22.7毫摩尔)叔丁醇钾,然后将所得溶液回流30分钟。向该反应溶液中加入100毫升水以终止反应,减压下除去溶剂。将残余物用乙醚萃取,用氯化钠水溶液洗涤,并用硫酸镁干燥。减压下除去溶剂,所得残余物进行硅胶柱色谱提纯(洗脱液:乙酸乙酯/正己烷=1/3,v/v),得到3.85克(14.5毫摩尔,产率77%)标题化合物。
1H NMR(CDCl3)δ1.27(t,3H),4.07(q,2H),6.76(s,1 H),7.28-7.47(m,5H),7.59(s,1H),7.82(m,2H),9.99(s,1H)
FAB 266(M+H)
2-3)3-羟基羰基-4-(萘-1-基)-1H-吡咯
将2.64克(10毫摩尔)制备例2-2)中制备的化合物溶解在50毫升50%的乙醇中,向其中加入2.24克(40毫摩尔)氢氧化钾。将反应溶液回流7小时,冷却至室温,将pH调节至4-5,用乙酸乙酯萃取并用硫酸镁干燥。减压下除去溶剂,得到1.62克(8.1毫摩尔,产率81%)标题化合物。该化合物不经纯化直接用于下一步反应。
1H NMR(CDCl3)δ6.60(s,1H),7.32-7.49(m,5H),7.54(s,1H),7.84(m,2H),9.92(s,1H)
FAB 238(M+H)
2-4)3-(吗啉-4-基)羰基-4-(萘-1-基)-1H-吡咯
将234毫克(1毫摩尔)制备例2-3)中制备的化合物溶解在2毫升二甲基甲酰胺中,向其中加入230毫克(1.2毫摩尔)EDC和162毫克(1.7毫摩尔)HOBT,将所得混合物在0℃下搅拌5分钟。向该反应溶液中加入87毫克(1毫摩尔)吗啉,然后在室温下搅拌5小时。减压下除去溶剂,向所得残余物中加入10毫升饱和碳酸钾水溶液。将该溶液用乙酸乙酯萃取,用10毫升1N的盐酸水溶液洗涤,用氯化钠水溶液和水洗涤,用硫酸镁干燥,然后浓缩,得到243毫克(0.8毫摩尔,产率80%)标题化合物。
1H NMR(CDCl3)δ2.13-3.52(br,8H),6.54(s,1H),7.31-7.51(m,5H),7.53(s,1H),7.81(m,2H),9.93(s,1H)
FAB 307(M+H)
2-5)3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯
将234毫克(1毫摩尔)制备例2-3)中制备的化合物溶解在2毫升二甲基甲酰胺中,向其中加入230毫克(1.2毫摩尔)EDC,101毫克(1毫摩尔)三乙胺和162毫克(1.7毫摩尔)HOBT,将所得混合物在0℃下搅拌5分钟。向该反应溶液中加入124毫克(1毫摩尔)N-(2-甲氧基乙基)-N-甲基胺盐酸化物,然后在室温下搅拌5小时。减压下除去溶剂,向所得残余物中加入10毫升饱和碳酸钾水溶液。将该溶液用20毫升乙酸乙酯萃取,用10毫升1N的盐酸水溶液洗涤,用氯化钠水溶液和水洗涤,用硫酸镁干燥,然后浓缩,得到246毫克(0.8毫摩尔,产率80%)标题化合物。
1H NMR(CDCl3)δ2.46(s,2H),2.80-3.40(m,8H),3.40(s,1H),6.80(s,1H),7.00(s,1H),7.42(m,4H),7.73(d,1H),7.81(d,1H),8.17(d,1H),10.66(s,1H)
FAB 309(M+H)制备例3:4-羟甲基-2-(2-丙基)噻唑的合成
3-1)2-甲基硫代丙酰胺
将3.0克(43毫摩尔)异丁腈溶解在由30毫升用硫化氢气体饱和的吡啶和9毫升三乙胺组成的溶剂混合物中,将所得溶液在室温下搅拌12小时。在减压下除去溶剂,然后将残余物溶解在200毫升乙酸乙酯中,用0.5N HCl和水洗涤。有机层用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:乙酸乙酯/正己烷=1/1,v/v),得到3.1克(30毫摩尔,产率70%)标题化合物。
1H NMR(CDCl3)δ1.26(d,3H),1.28(d,3H),2.92(m,1H),7.38(br,1H),8.32(br,1H)
3-2)4-乙氧基羰基-2-(2-丙基)噻唑
将3.0克(29毫摩尔)制备例3-1)中制备的化合物和5.6克(29毫摩尔)溴丙酮酸乙酯溶解在50毫升乙醇中,将所得混合物回流3小时。在减压下除去溶剂,将残余物进行硅胶柱色谱提纯(洗脱液:乙酸乙酯/正己烷=1/4,v/v),得到4.5克(23毫摩尔,产率79%)标题化合物。
1H NMR(CDCl3)δ1.31-1.35(m,9H),3.34(m,1H),4.35(q,2H),8.11(s,1H)
3-3)4-羟甲基-2-(2-丙基)噻唑
在0℃下将95毫克(2.5毫摩尔)氢化锂铝加入到3毫升四氢呋喃中,向其中缓慢加入500毫克(2.51毫摩尔)制备例3-2)中制备的化合物。将所得混合物在室温下搅拌10分钟,向其中小心地加入10毫升水。将所得溶液用乙酸乙酯萃取。将有机层用无水硫酸镁干燥并浓缩,然后将浓缩物进行硅胶柱色谱提纯(洗脱液:乙酸乙酯/正己烷=7/3,v/v),得到220毫克(1.40毫摩尔,产率56%)标题化合物。
1H NMR(CDCl3)δ1.34(d,3H),1.36(d,3H),3.27(m,1H),4.71(s,2H),5.22(s,1H),7.03(s,1H)制备例4:2-(2-丙基)噻唑-4-羧酸的合成
将200毫克(1.00毫摩尔)制备例3-2)中制备的化合物溶解在由四氢呋喃,甲醇和水(1.0毫升/0.6毫升/0.3毫升)所组成的溶剂混合物中,向其中加入63毫克(1.5毫摩尔)氢氧化锂。将该反应混合物在室温下搅拌1小时,在减压下除去溶剂。向残余物中加入水,然后用稀盐酸水溶液将pH调节至6,并用乙酸乙酯萃取。所得有机层用无水硫酸镁干燥并浓缩,得到130毫克(1.40毫摩尔,产率76%)标题化合物。该化合物不经纯化直接用于下一步反应。
1H NMR(CDCl3+CD3OD)δ1.25(m,6H),3.30(m,1H),8.05(s,1H)制备例5:3-(萘-1-基)羰基-1H-吡咯的合成
5-1) N-甲基-1-萘异羟肟酸甲酯
将3.44克(20毫摩尔)1-萘甲酸溶解在20毫升二甲基甲酰胺中。向该溶液中加入4.6克(24毫摩尔)EDC,2.02克(20毫摩尔)三乙胺和3.24克(24毫摩尔)HOBT,将所得混合物在0℃下搅拌5分钟。向其中加入1.85毫克(20毫摩尔)N,O-二甲基羟胺盐酸化物,然后将该混合物在室温下搅拌5小时。减压下除去溶剂,向所得残余物中加入100毫升饱和碳酸钾水溶液,然后用乙酸乙酯萃取。有机层先后用1N的盐酸水溶液、氯化钠水溶液和水洗涤,用无水硫酸镁干燥,然后浓缩,得到3.04克(1.05毫摩尔)标题化合物。
1H NMR(CDCl3)δ2.42(s,3H),3.24(s,3H),7.47(m,4H),7.67(d,1H),7.74(m,2H)
FAB 216(M+H)
5-2)1-(萘-1-基)-丙-2-烯-1-酮
将2.03克(9.4毫摩尔)制备例5-1)中制备的化合物溶解在20毫升干燥的四氢呋喃中,在0℃下向其中缓慢加入20毫升1N的乙烯基镁溴化物-四氢呋喃溶液。将该混合物在室温下搅拌30分钟,向其中加入20毫升1N的盐酸,并将所得混合物用50毫升乙酸乙酯萃取。所得有机层用无水硫酸镁干燥,减压下除去其中的溶剂,得到1.63克(9毫摩尔,产率96%)标题化合物。
1H NMR(CDCl3)δ6.92(m,1H),7.51(m,4H),7.74(d,1H),7.85(m,2H),7.98(d,1H),8.31(d,1H)
5-3)3-(萘-1-基)羰基-1H-吡咯
将901毫克(5毫摩尔)制备例5-2)中制备的化合物和1.01克(5.5毫摩尔)甲苯磺酰基甲基异腈溶解在10毫升四氢呋喃中,然后向其中缓慢加入溶解在10毫升四氢呋喃中的555毫克(5.5毫摩尔)叔丁醇钾。将该反应溶液搅拌30分钟,向该溶液中加入10毫升水以终止反应。减压下除去溶剂。向残余物中加入20毫升水,然后用乙酸乙酯萃取,用氯化钠水溶液洗涤,然后用无水硫酸镁干燥。减压下除去溶剂,所得残余物进行硅胶柱色谱提纯(洗脱液:乙酸乙酯/正己烷=1/3,v/v),得到884毫克(4毫摩尔,产率80%)标题化合物。
1H NMR(CDCl3)δ 6.57(s,1H),6.66(s,1H),6.79(s,1H),7.36(m,3H),7.48(d,1H),7.77(d,1H),7.82(d,1H),8.04(d,1H),9.91(s,1H)实施例1:1-[1-(1-苄氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-(吗啉-4-基)羰基-4-(萘-1-基)-1H-吡咯(1)的合成
将612毫克(2.0毫摩尔)制备例2-4)中制备的化合物溶解在10毫升二甲基甲酰胺中,在0℃下向其中加入264毫克(6.6毫摩尔)氢化钠(60%),然后将所得混合物搅拌5分钟。向该混合物中加入765毫克(2.2毫摩尔)制备例1-4)中制备的化合物,将整个混合物在室温下搅拌5小时。减压蒸馏除去溶剂,向残余物中加入10毫升水。将所得混合物用20毫升乙酸乙酯萃取二次,用硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=90/10,v/v),得到930毫克(产率75%)标题化合物。
1H NMR(CDCl3)δ1.11(d,2H),1.51(m,3H),2.30(br,1H),2.54-3.41(br,9H),3.75(d,2H),4.18(s,2H),5.10(s,2H),5.18(s,2H),6.75(s,1H),7.18(s,1H),7.20-7.53(m,10H),7.71(s,1H),7.82(d,1H),7.88(d,1H),8.07(d,1H)
FAB(M+H)618实施例2:3-(吗啉-4-基)羰基-4-(萘-1-基)-1-[1-(哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(2)的合成
将227毫克(0.36毫摩尔)实施例1中制备的化合物溶解在5毫升甲醇中,向其中加入20毫克氢氧化钯炭(Pd(OH)2/C),将该混合物在1atm的氢气压力下反应2小时。将反应物过滤并除去溶剂。所得残余物进行硅胶柱色谱提纯(洗脱液:氨水/甲醇=15/85,v/v),得到120毫克(0.26毫摩尔,产率74%)标题化合物。
1H NMR(CD3OD)δ1.07(m,2H),1.25-1.48(m,3H),2.25(br,3H),2.40(m,2H),2.60-3.40(m,8H),3.78(d,2H),5.22(s,2H),6.88(s,1H),7.12(d,2H),7.26(m,1H),7.35(m,3H),7.63(s,1H),7.75(d,1H),7.80(d,1H),7.93(d,1H)
FAB(M+H)484实施例3:1-[1-(1-乙酰基哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-(吗啉-4-基)羰基-4-(萘-1-基)-1H-吡咯(3)的合成
将30毫克(62微摩尔)实施例2中制备的化合物加入到2毫升二氯甲烷中,然后用注射器向其中加入5.4毫克(6.9微摩尔)乙酰氯。将该混合物反应2小时。减压下除去溶剂,所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=80/20,v/v),得到26毫克(5.3微摩尔,产率85%)标题化合物。
1H NMR(CDCl3+CD3OD)δ1.09-1.35(m,3H),1.45-1.75(m,4H),2.08(s,3H),2.10(br,1H),2.30(br,1H),2.44(t,1H),2.96(t,2H),3.08(br,2H),3.30(br,1H),3.79(d,1H),3.89(d,2H),4.55(d,1H),5.25(s,2H),6.80(s,1H),7.18(s,1H),7.28-7.52(m,5H),7.83(d,1H),7.99(d,1H),8.01(d,1H),8.06(s,1H)
FAB(M+H)526实施例4:1-[1-(1-甲磺酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-(吗啉-4-基)羰基-4-(萘-1-基)-1H-吡咯(4)的合成
将30毫克(62微摩尔)实施例2中制备的化合物加入到2毫升二氯甲烷中,然后用注射器向其中加入7.8毫克(6.9微摩尔)甲磺酰氯。将该混合物反应2小时。减压下除去溶剂,所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=90/10,v/v),得到25毫克(4.6微摩尔,产率85%)标题化合物。
1H NMR(CDCl3+CD3OD)δ1.08(m,2H),1.35-1.65(m,3H),2.25(br,2H),2.45(t,2H),2.65(s,3H),2.75-3.40(br,6H),3.54(d,2H),3.82(d,2H),5.23(s,2H),6.91(s,1H),7.14(m,2H),7.26(d,1H),7.32-7.50(m,3H),7.68(s,1H),7.76(d,1H),7.82(d,1H),7.93(d,1H)
FAB (M+H) 562实施例5:1-[1-(1-苄氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(5)的合成
将616毫克(2.0毫摩尔)制备例2-5)中制备的化合物溶解在10毫升二甲基甲酰胺中,在0℃下向其中加入264毫克(6.6毫摩尔)氢化钠(60%),然后将所得混合物搅拌5分钟。向该混合物中加入765毫克(2.2毫摩尔)制备例1-4)中制备的化合物,将整个混合物在室温下搅拌5小时。减压蒸馏除去溶剂,向残余物中加入10毫升水。将所得混合物用20毫升乙酸乙酯萃取二次,用硫酸镁干燥,浓缩,然后进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=90/10,v/v),得到930毫克(产率75%)标题化合物。
1H NMR(CDCl3)δ1.11(s,2H),1.35-1.65(m,3H),2.39(s,2H),2.70(br,4H),2.90-3.20(m,5H),3.32(s,1H),3.78(d,2H),4.16(br,2H),5.08(s,2H),5.16(s,2H),6.74(s,1H),7.10(s,1H),7.21-7.50(m,10H),7.76(d,1H),7.84(d,1H),7.91(s,1H),8.07(d,1H)
FAB(M+H)620实施例6:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-[1-(哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(6)的合成
将227毫克(0.36毫摩尔)实施例5中制备的化合物溶解在5毫升甲醇中,向其中加入20毫克氢氧化钯炭,然后将所得混合物在1atm的氢气压力下反应2小时。将反应物过滤并除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:氨水/甲醇=15/85,v/v),得到128毫克(0.26毫摩尔,产率74%)标题化合物。
1H NMR(CDCl3)δ1.10-1.30(br,3H),1.47(d,2H),2.30-2.60(m,4H),2.68(br,1H),2.90-3.18(m,6H),3.29(s,1H),3.63(m,2H),4.04(br,2H),5.06(s,2H),6.71(s,1H),7.04(s,1H),7.12(s,1H),7.26-7.57(m,5H),7.71(d,1H),7.79(d,1H),8.05(d,1H)
FAB(M+H)486实施例7:1-[1-(1-乙酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(7)的合成
将30毫克(62微摩尔)实施例6中制备的化合物加入到2毫升二氯甲烷中,然后用注射器向其中加入5.4毫克(6.9微摩尔)乙酰氯。将所得混合物反应2小时。减压下除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=80/20,v/v),得到27.8毫克(5.3微摩尔,产率85%)标题化合物。
1H NMR(CDCl3)δ1.05-1.35(br,3H),1.51(m,3H),2.04(s,3H),2.41(br,3H),2.72(br,1H),2.88-3.22(m,6H),3.33(br,1H),3.75(d,1H),4.01(d,2H),4.58(d,1H),5.28(s,2H),6.79(s,1H),7.18(s,1H),7.25-7.55(m,5H),7.78(d,1H),7.86(d,1H),8.09(d,1H),8.78(s,1H)
FAB(M+H)528实施例8:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-1-[1-(1-甲磺酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-4-(萘-1-基)-1H-吡咯(8)的合成
将30毫克(62微摩尔)实施例6中制备的化合物加入到2毫升二氯甲烷中,然后用注射器向其中加入7.8毫克(6.9微摩尔)甲磺酰氯。将所得混合物反应2小时。减压下除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=90/10,v/v),得到26毫克(4.6微摩尔,产率85%)标题化合物。
1H NMR(CDCl3)δ1.25(br,4H),1.55(br,2H),2.43(br,4H),2.70(s,3H),2.81-3.24(m,6H),3.34(br,1H),3.68(d,2H),4.04(d,2H),5.27(s,2H),6.80(s,1H),7.15(s,1H),7.25-7.55(m,5H),7.78(d,1H),7.87(d,1H),8.05(d,1H),8.64(s,1H)
FAB(M+H)564实施例9:1-{1-[1-(N-苄基氨基甲酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(9)的合成
将62毫克(128微摩尔)实施例6中制备的化合物加入到2毫升二氯甲烷中,向其中加入20.5毫克(153微摩尔)异氰酸苄基酯。将所得混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到62毫克(产率78%)标题化合物。
1H NMR(CDCl3)δ1.01(br,2H),1.30-1.51(m,3H),2.42(s,1H),2.50-2.72(m,6H),2.90-3.10(m,6H),3.30(s,1H),3.42(s,3H),3.30(d,1H),4.92(d,2H),5.25(br,1H),6.72(s,1H),7.01(s,1H),7.15-7.30(m,7H),7.55(m,3H),7.60(s,1H),7.75(d,1H),7.85(d,1H),8.07(d,1H)
FAB(M+H)619实施例10:1-{1-[1-(N-丁基氨基甲酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(10)的合成
将62毫克(128微摩尔)实施例6中制备的化合物加入到2毫升二氯甲烷中,向其中加入15毫克(153微摩尔)异氰酸丁基酯。将所得混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到61毫克(产率85%)标题化合物。
1H NMR(CDCl3)δ0.91(t,3H),1.07(s,2H),1.57(m,4H),2.55(br,2H),2.61(br,2H),2.71-2 90(m,1H),3.0-3.25(m,8H),3.31(br,1H),3.40(m,3H),3.73(br,2H),3.95(m,2H),4.85(br,1H),5.15(s,2H),6.71(s,1H),7.11(s,1H),7.35(m,5H),7.74(d,1H),7.85(d,1H),7.91(s,1H),8.07(s,1H)
FAB(M+H)585实施例11:1-{1-[1-(N-环己基氨基甲酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(11)的合成
将62毫克(128微摩尔)实施例6中制备的化合物加入到2毫升二氯甲烷中,向其中加入19毫克(153微摩尔)异氰酸环己基酯。将所得混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到49毫克(产率65%)标题化合物。
1H NMR(CDCl3)δ1.09(m,5H),1.35(m,3H),1.45(dd,2H),1.60(dd,1H),1.67(dd,2H),1.82(dd,2H),2.42(t,2H),2.50-2.80(m,3H),2.90-3.20(m,3H),3.37(br,1H),3.50(s,3H),3.61(m,1H),3.72(d,2H),3.90(dd,2H),4.51(br,1H),5.18(s,2H),6.72(s,1H),7.08(s,1H),7.22(s,1H),7.31(t,1H),7.55(br,3H),7.61(s,1H),7.75(d,1H),7.86(d,1H),8.08(d,1H)
FAB(M+H)611实施例12:1-[1-(1-庚酰基-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(12)的合成
将62毫克(128微摩尔)实施例6中制备的化合物溶解在2毫升二氯甲烷中,向其中加入19毫克(128微摩尔)庚酰氯。将所得混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到31毫克(产率40%)标题化合物。
1H NMR(CDCl3)δ 0.90(t,5H),1.04(m,2H),1.20-1.40(br,7H),1.50-1.70(br,4H),2.19(t,2H),2.30(t,2H),2.41(br,1H),2.71(br,1H),2.75-3.10(m,4H),3.12(s,1H),3.35(s,1H),3.70(s,2H),3.81(d,1H),4.60(d,1H),5.12(s,2H),6.75(s,1H),7.10(s,1H),7.21(s,1H),7.31(t,1H),7.40-7.50(m,3H),7.61(s,1H),7.75(d,1H),7.85(d,1H),8.07(d,1H)
FAB(M+H)598实施例13:1-{1-[1-(4-甲氧基苄基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(13)的合成
将62毫克(128微摩尔)实施例6中制备的化合物溶解在2毫升二氯甲烷中,向其中加入23毫克(128微摩尔)4-甲氧基苯基乙酰基氯。将所得混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到50毫克(产率62%)标题化合物。
1H NMR(CDCl3)δ0.78(m,1H),1.07(m,1H),1.30(br,1H1.40(dd,1H),1.51(dd,1H),2.40(br,3H),2.72(br,1H),2.85(br,1H2.85-3.10(m,3H),3.17(br,1H),3,31(br,1H),3.40-3.75(m,5H3.75-3.90(m,5H),4.48(d,1H),5.09(s,2H),6.71(s,1H),6.81(m,2H7.02-7.15(m,3H),7.21(br,1H),7.31(br,1H),7 35-7.45(m,3H),7.56(1H),7.75(d,1H),7.85(d,1H),8.08(d,1H)
FAB(M+H)634实施例14:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-[1-(1-苯氧基乙酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(14)的合成
将62毫克(128微摩尔)实施例6中制备的化合物溶解在2毫升二氯甲烷中,向其中加入23毫克(128微摩尔)苯氧基乙酰氯。将所得混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到50毫克(产率63%)标题化合物。
1H NMR(CDCl3)δ1.10(m,2H),1.40(br,1H),1.57(m,2H),2.40(br,3H),2.70(br,1H),2.90-3.20(m,7H),3.31(br,1H),3.85(br,2H),3.98(d,1H),4.50(d,1H),4.61(m,2H),5.21(s,2H),6.70(s,1H),6.87(d,2H),6.98(t,1H),7.17(s,1H),7.20-7.40(m,4H),7.40-7.50(rm,3H),7.70(d,1H),7.73(d,1H),8.10(d,1H),8.14(s,1H)
FAB(M+H)620实施例15:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(2-苯基乙基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(15)的合成
将62毫克(128微摩尔)实施例6中制备的化合物溶解在2毫升二氯甲烷中,向其中加入22毫克(128微摩尔)3-苯基丙酰氯。将所得混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到49毫克(产率63%)标题化合物。
1H NMR(CDCl3)δ0.80(m,1H),1.01(m,1H),1.31(br,1H),1.42(d,1H),1.52(d,1H),2.35(m,3H),2.60(m,2H),2.51(t,1H),2.78(br,1H),2.80(m,1H),2.90-3.01(m,4H),3.01(s,2H),3.11(s,1H),3.31(br,1H),3.60-3.81(m,3H),4.61(d,1H),5.15(s,2H),6.75(s,1H),7.12(s,1H),7.15-7.35(m,7H),7.45-7.50(m,3H),7.71(s,1H),7.79(d,1H),7.81(d,1H),8.08(d,1H)
FAB(M+H)618实施例16:1-{1-[1-(4-联苯基乙酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(16)的合成
将62毫克(128微摩尔)实施例6中制备的化合物溶解在2毫升二甲基甲酰胺中,向其中加入27毫克(128微摩尔)4-联苯基乙酸,14毫克(128毫摩尔)1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸化物和17毫克(128毫摩尔)N-羟基苯并三唑。将所得混合物反应3小时,除去溶剂,然后向残余物中加入10毫升乙酸乙酯。将所得溶液用10毫升水洗涤二次,再用10毫升6N的碳酸氢钠水溶液进一步洗涤。减压下除去溶剂,将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到49毫克(产率62%)标题化合物。
1H NMR(CDCl3)δ0.80(m,1H),1.10(m,1H),1.30(m,1H),1.42(d,1H),1.55(d,1H),2.35(m,3H),2.75(br,1H),2.85-3.24(m,7H),3.32(br,1H),3.60-3.80(m,4H),3.90(d,1H),4.61(d,1H),5.12(s,2H),6.72(s,1H),7.01(s,1H),7.20(s,1H),7.30(d,2H),7.32(t,1H),7.40-7.50(m,5H),7.50-7.60(m,5H),7.70(s,1H),7.72(d,1H),7.78(d,1H),8.09(d,1H)
FAB(M+H)680实施例17:1-[1-(1-甲氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(17)的合成
按照实施例7中的相同步骤制得产率为85%的标题化合物,所不同的是用甲氧基羰基氯代替乙酰氯。
1H NMR(CDCl3)δ1.05(br,2H),1.32(br,1H),1.53(br,2H),2.31-2.72(m,5H),3.03~3.33(m,7H),3.62(s,3H),3.66(m,2H),4.13(br,2H),5.12(s,2H),6.71(s,1H),7.03(s,1H),7.14(s,1H),7.24-7.43(m,5H),7.74(d,1H),7.82(d,1H),8.10(d,1H)
FAB(M+H)544实施例18:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-[1-(1-丙酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(18)的合成
按照实施例7中的相同步骤制得产率为82%的标题化合物,所不同的是用丙酰氯代替乙酰氯。
1H NMR(CDCl3)δ1.12(m,5H),1.40(m,1H),1.61(m,2H),2.35(q,2H),2.41(m,3H),2.70(br,1H),2.85(m,1H),3.02(m,5H),3.17(br,1H),3.31(br,1H),3.75(m,3H),4.55(m,1H),5.17(s,2H),6.69(s,1H),7.09(s,1H),7.41(m,5H),7.74(d,1H),7.83(d,1H),7.89(s,1H),8.05(d,1H)
FAB(M+H)542实施例19:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(萘-1-基甲基氧羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(19)的合成
将1.19克(7.52毫摩尔)1-萘甲醇溶解在15毫升甲苯中,向其中加入1.04克(7.53毫摩尔)碳酸钾。在0℃下向该溶液中加入3.89毫升(1.93M的甲苯溶液)的碳酰氯溶液,在室温下将整个溶液搅拌2小时。将反应物过滤除去固体物质,然后向其中加入0.108克(0.222毫摩尔)实施例6中制备的化合物和0.046毫升(0.33毫摩尔)三乙胺。将所得混合物在室温下搅拌1小时,减压下蒸馏除去溶剂。向残余物中加入10毫升饱和碳酸氢钠水溶液,然后用乙酸乙酯萃取,用无水硫酸镁干燥,浓缩。将浓缩物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到65毫克(0.097毫摩尔,产率44%)标题化合物。
1H-NMR(CDCl3)δ1.10(br,2H),1.50(br,2H),2.36(s,2H),2.58-2.85(br,4H),2.90-3.23(br,6H),3.31(s,1H),3.70(s,2H),4.08(br,1H),4.25(br,1H),5.12(s,2H),5.51(s,2H),6.70(s,1H),7.04(s,1H),7.18(s,1H),7.29(m,1H),7.38-7.65(m,7H),7.70(s,1H),7.75(d,1H),7.79-7.95(m,3H),8.00(d,1H),8.07(d,1H)
FAB(M+1)670
实施例20-26
下列表2中的物理化学数据所代表的化合物根据实施例19的相同步骤获得。表2-1
表2-2
实施例27:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(萘-2-基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(27)的合成
| 化合物编号 | 1H-NMR(CDCl3) | FAB(M+1) |
| 20 | 1.08(br,2H),1.61(br,2H),2.41(s,2H),2.71(br,4H),2.95-3.25(br,6H),3.30(br,1H),3.69(d,2H),4.15(br,2H),5.07(s,2H),5.24(s,2H),6.68(s,1H),7.08(s,1H),7.20(s,1H),7.31(m,1H),7.40-7.60(m,6H),7.66(s,1H),7.73-7.97(m,6H),8.12(d,1H) | 670 |
| 21 | 1.15(m,2H),1.62(m,10H),2.10(m,5H),2.42(s,2H)2.71(br,4H),3.04(s,4H),3.14(s,2H),3.35(s,1H),3.76(d,2H),4.17(br,2H),4.59(s,2H),5.10(m,1H),5.19(s,2H),5.32(m,1H),7.10(s,1H),7.23(s,1H),7.36(m,1H),7.42(m,3H),7.75(s,1H),7.80(m,2H),7.86(d,1H),8.10(d,1H) | 666 |
| 22 | 1.04(m,2H),1.30-1.60(m,4H),1.64(s,3H),1.70(s,3H),2.38(br,2H),2.58(br,2H),2.67(br,1H),2.99(s,3H),3.08(br,2H),3.29(br,1H),3.66(d,2H),4.07(br,2H),4.50(d,2H),5.08(s,2H),5.20-5.30(m,1H),6.69(s,1H),7.03(s,1H),7.15(s,1H),7.29-7.55(m,5H),7.52(d,1H),7.80(d,1H),8.04(d,1H) | 598 |
| 化合物编号 | 1H-NMR(CDCl3) | FAB(M+1) |
| 23 | 0.89(d,6H),1.06(d,2H),1.35-1.80(m,6H),2.39(br,2H),2.50-2.89(br,3H),2.90-3.20(br,5H),3.31(br,1H),3.71(d,2H),3.90(br,1H),4.00-4.25(m,4H),5.12(s,2H),6.71(s,1H),7.08(s,1H),7.20(s,1H),7.38(d,1H),7.40(m,3H),7.73(s,1H),7.75(d,1H),7.80(d,1H),8.05(d,1H) | 600 |
| 24 | 1.38(br,6H),2.64(br,9H),3.68(br,2H),3.74(d,2H),4.12(br,2H),5.02(s,2H),5.13(s,2H),6.72(s,1H),7.01(m,4H),7.39(m,6H),7.74(m,2H),7.83(d,1H),8.05(d,1H) | 638 |
| 25 | 1.11(br,3H),1.51(d,2H),3.01(br,12H),3.74(d,2H),4.14(s,2H),4.68(s,2H),5.13(s,2H),6.25(m,1H),6.56(d,1H),6.73(s,1H),7.08(s,1H),7.15-7.65(m,10H),7.73(d,1H),7.77(s,1H)7.82(d,1H),8.06(d,1H) | 646 |
| 26 | 1.24(m,2H),1.33(m,6H),1.38(m,2H),1.53(m,2H),2.41(m,2H),2.76(m,2H),3.01-3.32(m,8H),3.43(m,2H),3.69(m,2H),4.15(m,2H),5.11(s,2H),6.73(s,1H),7.06(s,1H),7.19(s,1H),7.25-7.45(m,5H),7.65(s,1H),7.77(d,1H),7.83(d,1H),8.09(d,1H) | 669 |
将100毫克(0.206毫摩尔)实施例6中制备的化合物和39毫克(0.22毫摩尔)2-萘甲酸溶解在1毫升二甲基甲酰胺中,向其中加入59毫克(0.31毫摩尔)EDC和42毫克(0.31毫摩尔)HOBT,然后将所得混合物在室温下搅拌2小时。减压蒸馏除去溶剂。将残余物溶解在乙酸乙酯中,并用饱和碳酸氢钠水溶液洗涤。有机层用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到83毫克(0.13毫摩尔,产率63%)标题化合物。
1H-NMR(CDCl3)δ1.16(br,2H),1.60(br,2H),2.40(br,2H),2.60-3.23(m,9H),3.31(br,1H),3.71(s,3H),4.45(br,1H),4.70(br,1H),5.10(s,2H),6.72(d,1H),7.04(s,1H),7.17(s,1H),7.31(d,1H),7.35-7.55(m,6H),7.58(s,1H),7.73(d,1H),7.75-7.95(m,5H),8.04(d,1H)
FAB(M+1)640
实施例28-29
下列表3中的物理化学数据所代表的化合物根据实施例27的相同步骤获得,所不同的是分别用反-肉桂酸和2-(2-丙基)噻唑-4-羧酸代替2-萘甲酸。
表3
实施例30:1-{1-[1-(N-苄基-N-甲基氨基甲酰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(30)的合成
| 化合物编号 | 1H-NMR(CDCl3) | FAB(M+1) |
| 28 | 1.06(s,2H),1.37(s,1H),1.55(d,2H),2.25-2.60(br,4H),2.70(br,1H),2.90-3.2(br,6H),3.30(br,1H),3.68(s,2H),4.06(m,1H),4.65(s,1H),5.10(s,2H),6.73(s,1H),6.83(d,1H),7.04(s,1H),7.17(s,1H),7.25-7.52(m,9H),7.56(s,2H),7.71(d,1H),7.80(d,1H),8.07(d,1H) | 616 |
| 29 | 1.22(m,2H),1.34(d,3H),1.36(d,3H),1.38(m,2H),1.47(m,2H),2.38(m,2H),2.52(m,2H),3.00-3.14(m,6H),3.25(m,2H),3.68(m,2H),4.43(m,1H),4.62(m,1H),5.09(s,2H),6.72(s,1H),7.04(s,1H),7.16(s,1H),7.37-7.44(m,5H),7.65(s,1H),7.72(d,1H),7.81(d,1H),8.07(d,1H) | 639 |
将100毫克(0.206毫摩尔)实施例6中制备的化合物溶解在1毫升四氢呋喃中,向其中加入27毫克(0.23毫摩尔)N-苄基-N-甲基胺。然后在0℃下向其中滴加入0.16毫升(1.93M的甲苯溶液)的碳酰氯溶液。将所得混合物在室温下搅拌1小时,向其中加入1毫升水,然后将该溶液用乙酸乙酯萃取。有机层用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=93/7,v/v),得到81毫克(0.128毫摩尔,产率62%)标题化合物。
1H-NMR(CDCl3)δ1.20(m,2H),1.49(d,2H),2.40(br,2H),2.56-2.85(m,8H),2.93-3.20(m,5H),3.29(br,1H),3.52(m,4H),3.65(d,2H),3.73(m,2H),4.31(s,2H),5.14(s,2H),6.71(d,1H),7.07(s,1H),7.29(m,10H),7.73(d,1H),7.75(s,1H),7.82(d,1H),8.05(d,1H)
FAB(M+1)633实施例31:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢喹啉-1-基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(31)的合成
按照实施例30中的相同步骤制得标题化合物,所不同的是用1,2,3,4-四氢喹啉代替N-苄基-N-甲基胺。实施例32:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢异喹啉-2-基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(32)的合成
按照实施例30中的相同步骤制得标题化合物,所不同的是用1,2,3,4-四氢异喹啉代替N-苄基-N-甲基胺。实施例33:1-{1-[1-(4-联苯基甲基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(33)的合成
将100毫克(0.206毫摩尔)实施例6中制备的化合物溶解在3毫升四氢呋喃中,向其中加入45毫克(0.24毫摩尔)4-苯基苯甲醛和52毫克(0.24毫摩尔)三乙酰氧基硼氢化钠,将所得混合物在室温下搅拌10小时。向该反应溶液中加入1毫升1N的HCl-甲醇溶液,然后搅拌30分钟,碱化,并用乙酸乙酯萃取。有机层用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=93/7,v/v),得到100毫克(0.154毫摩尔,产率75%)标题化合物。实施例34:3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(4-苯氧基苄基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(34)的合成
按照实施例33中的相同步骤制得标题化合物,所不同的是用4-苯氧基苯甲醛代替4-苯基苯甲醛。
实施例31-34中制得的化合物的物理化学数据列于下列表4中。
表4
实施例35:1-[1-(1-异丁氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(35)的合成
| 化合物编号 | 1H-NMR(CDCl3) | FABMS(M+1) |
| 31 | 1.12(m,2H),1.43(d,2H),1.89(m,2H),2.39(s,2H),2.57(t,2H),2.70(m,3H),2.90-3.20(br,7H),3.30(s,1H),3.52(m,2H),3.69(d,2H),3.75(d,2H),5.10(s,2H),6.69(d,1H),6.84(m,2H),7.04(m,3H),7.16(s,1H),7.31(m,1H),7.37(m,3H),7.61(s,1H),7.74(d,1H),7.81(d,1H),8.04(d,1H) | 645 |
| 32 | 1.12(m,2H),1.43(d,2H),1.89(m,2H),2.39(s,2H),2.75(t,2H),2.85-3.15(br,5H),3.41(br,2H),3.52(br,4H),3.69(d,2H),3.75(d,2H),4.37(s,2H),5.11(s,2H),6.74(s,1H),7.00-7.70(m,11H),7.79(d,1H),7.84(d,1H),8.06(d,1H) | 645 |
| 33 | 1.31(m,3H),1.50(m,2H),1.70(m,4H),1.91(m,1H),2.05(s,1H),2.41(s,2H),2.75(m,1H),2.90(m,1H),3.01(br,2H),3.09(m,1H),3.31(m,1H),3.51(s,2H),3.71(s,2H),5.12(s,2H),6.72(s,1H),7.10(s,1H),7.17(s,1H),7.20-7.70(m,14H),7.79(d,1H),7.84(d,1H),8.07(d,1H) | 652 |
| 34 | 1.31(m,2H),1.51(m,2H),1.90(m,4H),1.91(m,1H),2.05(s,1H),2.42(s,2H),2.75(m,1H),2.87(m,2H),3.01(br,2H),3.09(m,1H),3.31(m,1H),3.51(s,2H),3.71(s,2H),5.13(s,2H),6.72(s,1H),6.95-7.70(m,16H),7.79(d,1H),7.84(d,1H),8.07(d,1H) | 668 |
按照实施例7中的相同步骤制得产率为80%的标题化合物,所不同的是用氯甲酸异丁基酯代替乙酰氯。
1H-NMR(CDCl3)δ0.86(d,6H),1.04(m,2H),1.31(br,1H),1.47(m,2H),1.86(m,1H),2.38(br,2H),2.61(m,3H),2.99(br,3H),3.07(br,2H),3.29(br,1H),3.42(br,1H),3.66(d,2H),3.77(d,2H),4.08(br,2H),5.08(s,2H),6.69(s,1H),7.03(s,1H),7.14(s,1H),7.32(m,1H),7.37(m,3H),7.52(s,1H),7.72(d,1H),7.80(d,1H),8.03(d,1H)
FAB(M+1)586实施例36:1-{1-[1-(苄氧基羰基)-哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(萘-1-基)羰基-1H-吡咯(36)的合成
将62.6毫克(0.28毫摩尔)制备例5-3)中制备的化合物溶解在1毫升二甲基甲酰胺中,向其中加入60毫克(1.5毫摩尔)氢化钠。将所得混合物在室温下搅拌30分钟,向其中加入115毫克(0.30毫摩尔)制备例1-4)中制备的化合物,然后将所得混合物搅拌1小时。减压下除去溶剂,向残余物中加入5毫升饱和碳酸氢钠水溶液。将该溶液用20毫升乙酸乙酯萃取。有机层用氯化钠水溶液洗涤,用无水硫酸镁干燥,减压下浓缩。将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到110毫克(0.21毫摩尔,产率75%)标题化合物。
1H NMR(CDCl3)δ0.93-1.49(br,5H),2.50(s,2H),3.58(d,2H),4.18(br,2H),5.05(s,2H),5.12(s,2H),6.63(s,1H),6.70(s,1H),7.09(s,1H),7.12(s,1H),7.28-7.60(m,10H),7.89(d,1H),7.95(d,1H),8.10(d,1H)
FAB:533(M+H)实施例37:1-[1-(1-乙酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(萘-1-基)羰基-1H-吡咯(37)的合成
将110毫克(0.211毫摩尔)实施例36中制备的化合物溶解在10毫升甲醇中,向其中加入20毫克Pd(OH)2/C。然后将该混合物在氢气气氛下搅拌3小时以除去苄氧基羰基。将反应物用绿土过滤以除去催化剂,并在减压下除去溶剂。将该未纯化的残余物溶解在5毫升二甲基甲酰胺中,向其中加入16.5毫升(0.232毫摩尔)乙酰氯。将该反应溶液在室温下搅拌30分钟,并在减压下除去溶剂。向所得残余物中加入5毫升饱和碳酸氢钠水溶液,然后用20毫升乙酸乙酯萃取。有机层用氯化钠水溶液洗涤,用无水硫酸镁干燥,然后在减压下浓缩。将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=9/1,v/v),得到20.3毫克(0.046毫摩尔,产率22%)标题化合物。
1H NMR(CDCl3)δ1.11(m,3H),1.41(s,3H),2.06(s,3H),2.27(m,1H),2.78(m,1H),3.68(m,2H),4.58(d,1H),5.11(s,2H),6.69(s,1H),6.70(s,1H),7.11(s,1H),7.20(s,1H),7.50(m,3H),7.60(m,1H),7.90(m,2H),7.98(d,1H),8.12(d,1H)
FAB:441(M+1)制备例6:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯的合成
将234毫克(1毫摩尔)制备例2-3)中制备的化合物溶解在2毫升二甲基甲酰胺中,向其中加入230毫克(1.2毫摩尔)EDC和162毫克(1.7毫摩尔)HOBT,将所得混合物在0℃下搅拌5分钟。向该反应溶液中加入88毫克(1毫摩尔)N-甲基哌嗪,然后在室温下搅拌5小时。减压下除去溶剂。向残余物中加入10毫升饱和碳酸钾水溶液。将所得混合物用乙酸乙酯萃取,并用饱和氯化钠水溶液和水洗涤,然后浓缩。浓缩物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=85/15,v/v),得到240毫克(0.75毫摩尔)标题化合物。
1H NMR(CDCl3)δ1.13(br,2H),1.88(s,3H),1.75-2.08(br,2H),2.98(br,2H),3.41(br,2H),6.85(s,1H),7.12(s,1H),7.35-7.58(m,4H),7.76(d,1H),7.82(d,1H),8.11(d,1H),10.20(br,1H)
FAB 320(M+H)制备例7:3-{N-[2-(N,N-二甲基氨基)乙基]-N-甲基}氨基甲酰基-4-(萘-1-基)-1H-吡咯的合成
将234毫克(1毫摩尔)制备例2-3)中制备的化合物溶解在2毫升二甲基甲酰胺中,向其中加入230毫克(1.2毫摩尔)EDC,101毫克(1毫摩尔)三乙胺和162毫克(1.7毫摩尔)HOBT,将所得混合物在0℃下搅拌5分钟。向该反应溶液中加入102毫克(1毫摩尔)N,N,N′-三甲基乙二胺,然后在室温下搅拌5小时。减压下除去溶剂。向残余物中加入10毫升饱和碳酸钾水溶液。将所得混合物用乙酸乙酯萃取,并用饱和氯化钠水溶液和水洗涤,浓缩,然后将浓缩物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=85/15,v/v),得到257毫克(0.8毫摩尔)标题化合物。
1H NMR(CDCl3)δ1.89(br,3H),2.15(br,4H),2.44(br,2H),2.75(br,1H),3.0(br,1H),3.36(br,2H),6.84(s,1H),7.07(s,1H),7.38-7.43(m,4H),7.78(d,1H),7.83(d,1H),8.1(br,1H),10.05(br,1H)
FAB 322(M+H)实施例38:1-[1-(1-苄氧基羰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(38)的合成
将612毫克(2.0毫摩尔)制备例6中制备的化合物溶解在10毫升二甲基甲酰胺中,在0℃下向其中加入264毫克(6.6毫摩尔)氢化钠(60%),然后将所得混合物搅拌5分钟。向其中加入765毫克(2.2毫摩尔)制备例1-4)中制备的化合物,然后将所得混合物在室温下搅拌5小时。减压蒸馏除去溶剂,向残余物中加入10毫升水。将所得溶液用20毫升乙酸乙酯萃取二次,用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=90/10,v/v),得到930毫克(产率74%)标题化合物。
1H NMR(CDCl3)δ0.86(m,2H),1.07(m,2H),1.24(m,2H),1.38(m,1H),1.52(m,2H),2.65(br,2H),3.00-3.50(br,4H),3.69(d,2H),4.16(br,2H),5.09(s,2H),5.11(s,2H),6.73(d,1H),7.12(s,1H),7.21(s,1H),7.25-7.32(m,6H),7.35-7.41(m,4H),7.78(d,1H),7.83(d,1H),8.01(d,1H)
FAB(M+H)631,C38H42N6O3实施例39:1-[1-(1-苄氧基羰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-{N-[2-(N,N-二甲基氨基)乙基]-N-甲基}氨基甲酰基-4-(萘-1-基)-1H-吡咯(39)的合成
将612毫克(2.0毫摩尔)制备例7中制备的化合物溶解在10毫升二甲基甲酰胺中,在0℃下向其中加入264毫克(6.6毫摩尔)氢化钠(60%),然后将所得混合物搅拌5分钟。向其中加入765毫克(2.2毫摩尔)制备例1-4)中制备的化合物,然后将所得混合物在室温下搅拌5小时。减压蒸馏除去溶剂,向残余物中加入10毫升水。将所得溶液用20毫升乙酸乙酯萃取二次,用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=90/10,v/v),得到870毫克(产率69%)标题化合物。
1H NMR(CDCl3+CD3OD)δ1.00(m,2H),1.31-1.40(m,3H),2.54-2.70(m,9H),3.50-3.80(m,6H),4.01(br,2H),4.50(s,1H),4.96(s,2H),5.07(s,2H),6.65(s,1H),7.01(s,1H),7.03(s,1H),7.13(s,1H),7.18-7.30(m,7H),7.45(s,1H),7.52(s,1H),7.64(d,1H),7.72(d,1H),7.80(d,1H)
FAB(M+H)633:C38H44N6O3实施例40:1-[1-(1-甲氧基羰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(40)的合成
40-1)3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-[(哌啶-4-基)甲基-1H-咪唑-5-基]甲基-1H-吡咯
将227毫克(0.36毫摩尔)实施例38中制备的化合物溶解在5毫升甲醇中,向其中加入2克氢氧化钾,将所得混合物在回流下反应8小时。将该反应溶液冷却,用10毫升乙酸乙酯萃取二次,用无水硫酸钠干燥,然后在减压下蒸发,得到80%产率的标题化合物。
1H NMR(CDCl3)δ1.15(br,2H),1.48(d,2H),1.75-1.98(m,6H),2.45(t,2H),2.91(br,1H),3.02(d,2H),3.31(br,1H),3.50-3.85(m,7H),5.10(s,2H),6.70(s,1H),7.09(m,1H),7.13(s,1H),7.30(t,1H),7.35-7.50(m,4H),7.74(d,1H),7.80(d,1H),8.01(d,1H)
FAB(M+H):497,C30H36N6O
40-2)1-[1-(1-甲氧基羰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯
将30毫克(62微摩尔)实施例40-1)中制备的化合物加入到2毫升二氯甲烷中,用注射器向其中加入5.4毫克(6.9微摩尔)氯甲酸甲酯。将该混合物反应2小时。减压下除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=85/15,v/v),得到27.8毫克(50微摩尔,产率80%)标题化合物。
1H NMR(CDCl3)δ1.06(m,4H),1.40(m,1H),1.51(d,2H),1.93(s,3H),2.02(br,1H),2.60(br,3H),2.98-3.60(br,4H),3.64(s,3H),3.69(d,2H),4.10(br,2H),5.14(s,2H),6.73(d,1H),7.12(s,1H),7.18(s,1H),7.30(t,1H),7.35-7.55(m,4H),7.77(d,1H),7.82(d,1H),8.02(d,1H)
FAB(M+H):555,C32H38N6O3实施例41:3-(4-甲基哌嗪-1-基)羰基-1-[1-(1-甲磺酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-4-(萘-1-基)-1H-吡咯(41)的合成
将30毫克(62微摩尔)实施例40-1)中制备的化合物加入到2毫升二氯甲烷中,用注射器向其中加入7.8毫克(6.9微摩尔)甲磺酰氯。将该混合物反应2小时。减压下除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=90/10,v/v),得到25毫克(4.5微摩尔,产率87%)标题化合物。
1H NMR(CDCl3)δ1.03(m,4H),1.43(m,1H),1.52(d,2H),1.98(s,3H),2.03(br,1H),2.62(br,3H),2.04-3.65(br,4H),3.64(s,3H),3.69(d,2H),4.10(br,2H),5.13(s,2H),6.72(d,1H),7.11(s,1H),7.19(s,1H),7.31(t,1H),7.32-7.53(m,4H),7.78(d,1H),7.83(d,1H),8.01(d,1H)
FAB(M+H):575,C32H38N6O3
实施例42:1-[1-(1-乙酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(4-甲
基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(42)的合成
将30毫克(62微摩尔)实施例40-1)中制备的化合物加入到2毫升二氯甲烷中,用注射器向其中加入5.4毫克(6.9微摩尔)乙酰氯。将该
混合物反应2小时。减压下除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=80/20,v/v),得到26毫克(4.8微摩尔,产率78%)标题化合物。
1H NMR(CDCl3)δ1.00-1.12(m,2H),1.32-1.45(m,2H),1.52-1.58(m,2H),1.90-2.10(m,8H),2.35(m,1H),2.93(t,1H),3.07(m,1H),3.10-3.70(br,4H),3.69(d,1H),7.75(d,1H),4.57(d,1H),5.12(s,2H),6.74(d,1H),7.12(d,1H),7.20(s,1H),7.34(d,1H),7.39-7.47(m,4H),7.78(d,1H),7.85(d,1H),8.01(d,1H)
FAB(M+H):539,C32H38N6O2实施例43:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(2-苯基乙基羰基)-哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(43)的合成
将62毫克(125微摩尔)实施例40-1)中制备的化合物溶解在2毫升二氯甲烷中,向其中加入22毫克(128微摩尔)3-苯基丙酰氯。将该混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到49毫克(产率62%)标题化合物。
1H NMR(CDCl3)δ0.77(m,1H),0.90-1.20(m,2H),1.35(m,1H),1.43(d,1H),1.51(d,1H),1.91(s,3H),1.80-2.00(br,2H),2.34(t,1H),2.55(m,3H),2.75(t,1H),2.85(br,5H),3.63(m,3H),3.72(d,1H),4.60(d,1H),5.11(s,2H),6.71(d,1H),7.09(d,1H).7.14-7.35(m,7H),7.38(m,4H),7.75(d,1H),7.81(d,1H),8.01(d,1H)
FAB(M+H):629,C39H44N6O2实施例44:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-[1-(1-苯氧基乙酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-1H-吡咯(44)的合成
将62毫克(125微摩尔)实施例40-1)中制备的化合物溶解在2毫升二氯甲烷中,向其中加入23毫克(128微摩尔)苯氧基乙酰氯。将该混合物反应3小时,除去溶剂,然后将所得残余物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到50毫克(产率63%)标题化合物。
1H NMR(CDCl3)δ0.90-1.12(m,4H),1.28(m,1H),1.46(d,2H),1.70-2.00(br,1H),1.84(s,3H),2.38(t,1H),2.86(t,1H),2.80-3.50(br,5H),3.59(m,2H),3.89(d,1H),4.48(d,1H),4.58(q,2H),5.05(s,2H),6.69(d,1H),6.86(d,2H),6.91(t,1H),7.04(d,1H),7.13(d,1H),7.20-7.30(m,3H),7.30-7.60(m,4H),7.71(d,1H),7.77(d,1H),7.98(d,1H)
FAB(M+H):631,C38H42N6O3实施例45:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(萘-2-基甲氧基)羰基哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(45)的合成
将1.19克(7.52毫摩尔)(萘-2-基)甲醇溶解在15毫升甲苯中,向其中加入1.04克(7.53毫摩尔)碳酸氢钾。在0℃下向该溶液中加入3.89毫升(1.93M的甲苯溶液)的碳酰氯溶液,在室温下将整个溶液搅拌2小时。将反应物过滤除去固体物质,然后向其中加入0.108克(0.222毫摩尔)实施例40-1)中制备的化合物和0.046毫升(0.33毫摩尔)三乙胺。将所得混合物在室温下搅拌1小时,减压下蒸馏除去溶剂。向残余物中加入10毫升饱和碳酸氢钠水溶液,然后用乙酸乙酯萃取,用无水硫酸镁干燥,浓缩。将浓缩物进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到65毫克(0.097毫摩尔,产率43%)标题化合物。
1H NMR(CDCl3)δ0.94(m,3H),1.45(br,3H),2.61(m,3H),2.96(s,3H),3.15(m,2H),3.75(m,4H),4.06(d,2H),4.71(br,2H),5.06(s,2H),5.18(s,1H),6.77(s,1H),7.15-8.00(m,17H)
FAB(M+H):681,C42H44O3N6
实施例46-48
下列表5中所列的化合物根据实施例45的相同步骤获得。
表5
实施例49:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(萘-2-基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(49)的合成
| 化合物编号 | NMR(CDCl3) | FAB(M+1) |
| 46 | 0.89(d,6H),1.07(m,2H),1.37(m,1H),1.47-1.50(m,5H),1.65(m,1H),1.80-2.10(br,4H),2.59(br,2H),3.01-3.60(br,5H),3.90-4.20(m,5H),5.11(s,2H),6.73(d,1H),7.12(d,1H),7.18(s,1H),7.31(t,1H),7.31-7.54(m,4H),7.77(d,1H),7.79(d,1H),8.01(d,1H),C36H46N6O3 | 611 |
| 47 | 1.00-1.12(m,3H),1.38(m,1H),1.51(d,3H),1.95(s,3H),2.63(br,3H),3.00-3.60(br,4H),3.68(d,2H),4.12(br,3H),5.05(s,2H),5.11(s,2H),6.72(d,1H),7.00-7.07(m,2H),7.12(s,1H),7.20(s,1H),7.25-7.35(m,3H),7.36-7.52(m,4H),7.78(d,1H),7.83(d,1H),8.03(d,1H),C38H41FN6O3 | 649 |
| 48 | 1.08(br,2H),1.20(d,1H),1.45-1.60(m,4H),3.25(br,2H),3.72(m,2H),4.01-4.21(br,4H),4.71(d,1H),5.15(s,1H),6.26-6.29(m,1H),6.60(d,1H),6.76(d,1H),7.14-7.45(m,8H),7.45-6.62(m,3H),7.63(s,1H),7.82(d,1H),7.83(d,1H)7.98(d,1H),C40H44N6O3 | 657 |
将100毫克(0.206毫摩尔)实施例40-1)中制备的化合物和39毫克(0.22毫摩尔)2-萘甲酸溶解在1毫升二甲基甲酰胺中,向其中加入59毫克(0.31毫摩尔)EDC和42毫克(0.31毫摩尔)HOBT,然后将所得混合物在室温下搅拌2小时。减压蒸馏除去溶剂。将残余物溶解在乙酸乙酯中,并用饱和碳酸氢钠水溶液洗涤。有机层用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=95/5,v/v),得到88毫克(0.14毫摩尔,产率68%)标题化合物。
1H NMR(CDCl3)δ1.05(br,3H),1.38(m,1H),1.56(d,2H),1.70-1.90(br,6H),2.36(br,1H),2.47(t,1H),2.82-3.07(br,3H),3.32(br,2H),3.63(t,2H),4.07(br,1H),4.67(d,1H),5.09(s,2H),6.73(d,1H),6.84(d,1H),7.08(d,1H),7.31(d,1H),7.25-7.55(m,10H),7.58(d,1H),7.73(d,1H),7.80(d,1H),8.02(d,1H)
FAB(M+1):627,C39H42N6O2
实施例50-51
下列表6中所列化合物根据实施例49的相同步骤获得,所不同的是分别用反-肉桂酸和2-(2-丙基)噻唑-4-羧酸代替2-萘甲酸。表6
实施例52:1-{1-[1-(N-苄基-N-甲基氨基甲酰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(52)的合成
| 化合物编号 | NMR(CDCl3) | FAB(M+1) |
| 50 | 1.08(br,3H),1.33-1.45(m,1H),1.58(d,2H),1.75-1.95(br,2H),1.83(s,3H),2.36(br,1H),2.47(br,1H),2.85-3.10(br,3H),3.15-3.50(br,2H),3.62-3.80(m,2H),4.02-4.15(br,1H),4.62-4.78(br,1H),5.10(s,2H),6.74(d,1H),6.82(d,1H),7.09(d,1H),7.18(s,1H),7.30-7.55(m,10H),7.60(d,1H),7.76(d,1H),7.80(d,1H),8.03(d,1H),C41H42N6O2 | 627 |
| 51 | 1.01(br,1H),1.24(m,2H),1.37(d,7H),1.40-1.65(m,3H),1,70-2.02(m,7H),2.59(br,1H),2.92(br,2H),3.28(br,2H),3.71(d,2H),4.49(br,1H),4.65(br,1H),5.13(s,2H),6.73(d,1H),7.09(d,1H),7.18(s,1H),7.32(d,1H),7.35-7.50(m,4H),7.68(s,1H),7.70(d,1H), 7.84(d,1H),8.03(d,1H),C37H43N7O2S | 650 |
将100毫克(0.206毫摩尔)实施例40-1)中制备的化合物溶解在1毫升四氢呋喃中,在0℃下向其中加入27毫克(0.23毫摩尔)N-苄基-N-甲基胺。向其中滴加入0.16毫升(1.93M的甲苯溶液)的碳酰氯溶液,将所得溶液在室温下搅拌1小时。向该溶液中加入1毫升水,然后用乙酸乙酯萃取。有机层用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=93/7,v/v),得到86毫克(0.133毫摩尔,产率64%)标题化合物。
1H NMR(CDCl3+CF3COOH)δ1.24(m,3H),1.52(m,4H),2.44(s,3H),2.65-3.00(m,8H),3.04(s,2H),3.63(d,2H),4.00(br,1H),4.17(d,2H),4.32(s,2H),5.52(s,2H),7.21-7.63(m,12H),7.94(d,1H),7.96(d,1H),8.01(d,1H),9.06(s,1H)
FAB(M+1):644,C39H45N7O2实施例53:1-{1-[1-(N,N-二甲基氨基甲酰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(53)的合成
按照实施例52中的相同步骤制得表7所列标题化合物,所不同的是用N,N-二甲基胺代替N-苄基-N-甲基胺。实施例54:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢喹啉-1-基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(54)的合成
按照实施例52中的相同步骤制得表7所列标题化合物,所不同的是用1,2,3,4-四氢喹啉代替N-苄基-N-甲基胺。实施例55:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢异喹啉-2-基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(55)的合成
按照实施例52中的相同步骤制得表7所列标题化合物,所不同的是用1,2,3,4-四氢异喹啉代替N-苄基-N-甲基胺。
表7
实施例56:1-{1-[1-(4-联苯基甲基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(56)的合成
| 化合物编号 | NMR(CDCl3) | FABMS(M+1) |
| 53 | 1.00-1.30(m,3H),1.31-1.67(m,3H),1.70-2.05(m,6H),2.59(m,2H),2.74(s,6H),2.89(m,2H),3.20-3.50(m,2H),3.68(m,4H),5.10(s,2H),6.74(d,1H),7.12(d,1H),7.20(s,1H),7.34(d,1H),7.39-7.47(m,4H),7.78(d,1H),7.85(d,1H),8.01(d,1H),C33H41N7O2 | 568 |
| 54 | 1.03-1.30(m,4H),2.31-2.51(m,3H),1.70-2.20(m,10H),2.57(t,2H),2.72(t,1H),2.90(br,2H),3.31(br,2H),3.54(t,1H),3.66(m,2H),3.81(d,1H),5.11(s,2H),6.68(d,1H),6.83(t,1H),6.92(d,1H),7.1(m,2H),7.12(d,1H),7.18(s,1H),7.31(d,1H),7.44(m,4H),7.76(d,1H),7.82(d,1H),8.02(d,1H);C40H45N7O2 | 656 |
| 55 | 0.9-2.1(m,12H),2.72(t,2H),2.80-3.95(m,12H),4.37(s,2H),5.12(s,2H),6.72(s,1H),7.00-7.70(m,11H),7.78(d,1H),7.82(d,1H),8.05(d,1H);C40H45N7O2 | 656 |
将100毫克(0.206毫摩尔)实施例40-1)中制备的化合物溶解在3毫升四氢呋喃中,向其中加入45毫克(0.24毫摩尔)4-苯基苯甲醛和52毫克(0.24毫摩尔)三乙酰氧基硼氢化钠,将所得溶液在室温下搅拌10小时。向该反应溶液中加入1毫升1N的HCl-甲醇溶液,然后搅拌30分钟,碱化,并用乙酸乙酯萃取。有机层用无水硫酸镁干燥,浓缩,并进行硅胶柱色谱提纯(洗脱液:二氯甲烷/甲醇=93/7,v/v),得到100毫克(0.151毫摩尔,产率75%)标题化合物。
FAB MS(M+1):663实施例57:3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(4-苯氧基苄基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(57)的合成
按照实施例56中的相同步骤制得标题化合物,所不同的是用4-苯氧基苯甲醛代替4-苯基苯甲醛。
FAB MS(M+1):679试验例1Ras法尼转移酶体外抑制活性的分析
在本试验例中,使用按照改进的Pompliano方法(Pompliano等,Biochemistry,1992,31,3800)的基因重组技术制备的法尼转移酶,并使用按照已知方法(见Chung等,Biochimica et Biophysica Acta,1992,278,1129)纯化后的、韩国专利申请号第97-14409中描述的Ras底物(Ras-CVLS)蛋白。
酶反应在50微升50mM的HEPES钠缓冲溶液中进行,该缓冲溶液含有25mM的氯化钾,25mM的氯化镁,10mM的DTT和50μM的氯化锌。使用1.5μM的Ras底物蛋白,0.15μM的法尼焦磷酸氚和4.5nM的法尼转移酶。
更具体地说,在第一步中,将法尼转移酶加入到上述缓冲溶液中,在37℃保持反应30分钟,然后通过加入1毫升含有1M盐酸的乙醇溶液终止反应。将形成的沉淀用Hopper采集机(Hopper#FH 225V)吸附到GF/B滤纸上进行滤纸汇集,用乙醇洗涤,然后用LKB β计数器测定干滤纸的放射性。在底物的不饱和状态测定酶滴定度,底物中Ras底物蛋白和法尼转移酶的浓度具有定量关系。将溶解在二甲亚砜(DMSO)中的本发明化合物以小于总反应溶液5%的量加入到反应溶液中,然后测定其酶抑制活性。酶抑制活性以有试验化合物存在时结合到Ras底物蛋白中的法尼基的量与没有试验化合物存在时的量相比的百分数表示。试验化合物的IC50定义为50%的酶活性被抑制时的浓度。
为了评价本发明化合物的选择性酶抑制活性,测定了对香叶基香叶基转移酶的抑制活性。香叶基香叶基转移酶是按照从Schaber方法(Schaber等,J.Biol.Chem.1990,265,14701)改进的方法从牛脑中提纯而来,并对焦磷酸香叶基香叶基酯和Ras-CVIL底物蛋白进行与法尼转移酶基本上相同的试验步骤。
试验结果列于下面的表8中。试验例2Ras法尼转移酶体内抑制活性的分析
在本试验例中,使用了表示为C-Harvey-Ras蛋白的具有转化活性的Rat2细胞系和用H-Ras的熔凝蛋白转化的Rat2细胞系(韩国专利申请号97-14409),该H-Ras用多元赖氨酸区域在K-Ras的C-端取代。该试验通过改进的Declue方法(Declue.J.E.等,Cancer Research,1991,51,712)进行。下面将对该试验方法进行详细描述。
将转化的Rat2成纤维细胞系的3×105个细胞喷在60mm的细胞培养盘上,并在细胞培养器中于37℃培养48小时,在达到50%或更多的致密度后,将其用试验化合物处理。使用的是溶解在二甲亚砜(DMSO)中的本发明化合物。在对比组和试验组中都使用1%浓度的二甲亚砜。在用化合物处理4小时后,将用每1毫升介质中含150μCi的[35S]放射性同位素标记的蛋氨酸加入其中,培养20小时后,用生理盐水洗涤该细胞。用1毫升的冷细胞溶解缓冲溶液(50mM的HEPES钠缓冲溶液,含有5mM的氯化镁,1mM的DTT,1%的NP 40,1mM的EDTA,1mM的PMSF,2μM的亮抑蛋白酶肽,2μM的胃蛋白酶抑制剂A,和2μM的抗蛋白酶)溶解该细胞,并用12000g×5分钟的高速离心获得上层清液,该上层清液中细胞被溶解。测定上层清液中放射性同位素的量并作为标准,得到免疫沉淀反应中的定量结果,然后加入Y13-259,一种特定结合在Ras蛋白上的单克隆抗体(Furth,M.E.等,J.Virol,1982,43,294),并在4℃反应15小时。将蛋白A(与山羊抗鼠科免疫球蛋白抗体结合)-琼脂糖悬浮液加入到该溶液中,在4℃反应1小时,然后,为了除去非特定结合产物,用一种缓冲溶液(50mM的三氯化物(Tris chloride)缓冲溶液,含有50mM的氯化钠,0.5%的双羟基胆酸钠,0.5%的NP 40,和0.1%的SDS)洗涤免疫沉淀物。将该沉淀物加入到一种电泳用缓冲溶液中并煮沸,然后用13.5%的SDS聚丙烯酰胺凝胶进行电泳。电泳以后,将该凝胶固定并干燥。然后将该凝胶暴露于X射线胶片,显影并印相。从试验结果测定结合有或没有结合Ras蛋白的法尼基的蛋白的谱带的强度,抑制50%的法尼基结合的试验化合物的浓度定义为CIC50,即体内Ras法尼转移酶抑制活性。试验结果列于下面的表8中。表8-1
表8-2
表8-3
| 化合物编号 | H-RasIC50(μM) | H-RasCIC50(μM) | K-RasIC50(μM) | K-RasCIC50(μM) |
| 1 | 0.0085 | 0.1 | 2.4 | 1-10 |
| 2 | 0.009 | 0.1 | 6 | 10-100 |
| 3 | 0.001 | 0.01-0.1 | 0.016 | 10-50 |
| 4 | 0.0036 | 0.01-0.1 | 0.026 | 10-50 |
| 5 | 0.0025 | 0.01-0.1 | 0.01-0.1 | 1-10 |
| 6 | 0.008 | 0.01-0.1 | 0.01-1 | 1-10 |
| 7 | 0.0018 | 0.01-0.1 | 0.01-0.1 | 10-100 |
| 8 | 0.0012 | 0.01-0.1 | 0.01-0.1 | 10-50 |
| 9 | 0.001-0.01 | 0.01-0.1 | 0.01-1 | 1-50 |
| 10 | 0.001-0.01 | 0.01-0.1 | 0.01-1 | 1-50 |
| 11 | 0.001-0.01 | 0.01-0.1 | 0.01-1 | 1-50 |
| 12 | 0.001-0.01 | 0.01-0.1 | 0.01-1 | 1-50 |
| 13 | 0.001-0.01 | 0.01-0.1 | 0.01-1 | 1-50 |
| 14 | 0.0021 | 0.01-0.1 | 0.01-1 | 1-50 |
| 15 | 0.001 | 0.01-0.1 | 0.01-1 | 1-50 |
| 16 | 0.001-0.01 | 0.01-0.1 | 0.01-1 | 10-100 |
| 17 | 0.001-0.01 | 0.01-0.1 | 0.01-1 | 10-100 |
| 18 | 0.001 | 0.01-0.1 | 0.01-1 | 1-50 |
| 19 | 0.007 | 0.01 | 0.02 | 1-10 |
| 20 | 0.006 | 0.01 | 0.01 | 1-10 |
| 21 | 0.01-0.1 | 0.01-0.1 | 0.05 | 1-10 |
| 22 | 0.009 | 0.01-0.1 | 0.02 | 1-10 |
| 23 | 0.008 | 0.01-0.1 | 0.02 | 1-10 |
| 24 | 0.006 | 0.01-0.1 | 0.015 | 1-10 |
| 化合物编号 | H-RasIC50(μM) | H-RasCIC50(μM) | K-RasIC50(μM) | K-RasCIC50(μM) |
| 25 | 0.006 | 0.01-0.1 | 0.027 | 1-10 |
| 26 | 0.004 | 0.01-0.1 | 0.01-0.1 | 10-50 |
| 27 | 0.009 | 0.01-0.1 | 0.015 | 1-10 |
| 28 | 0.012 | 0.01-0.1 | 0.008 | 1-10 |
| 29 | 0.0025 | 0.01-0.1 | 0.01-0.1 | 10-50 |
| 30 | 0.0025 | 0.01-0.1 | 0.006 | 1-10 |
| 31 | 0.004 | 0.01-0.1 | 0.02 | 10-50 |
| 32 | 0.002 | 0.01-0.1 | 0.012 | 1-10 |
| 33 | 0.005 | 0.01-0.1 | 0.01-0.1 | 1-10 |
| 34 | 0.011 | 0.01-0.1 | 0.01-0.1 | 1-10 |
| 35 | 0.006 | 0.01-0.1 | 0.01-0.1 | 1-10 |
| 36 | 0.2 | 10 | >100 | 50 |
| 37 | 0.35 | 1-10 | 10-100 | 10-50 |
| 38 | 0.0038 | 0.0125 | 0.015 | 2.5 |
| 39 | 0.3 | 1 | 1.5 | 30-100 |
| 40 | 0.0016 | 0.03 | 0.0042 | 10-50 |
| 41 | 0.003 | 0.05 | 0.01 | 10-50 |
| 42 | 0.0012 | 0.025 | 0.006 | 10-50 |
| 43 | 0.002 | 0.05 | 0.01 | 10-50 |
| 44 | 0.002 | 0.05 | 0.011 | 10-50 |
| 45 | 0.0018 | 0.035 | 0.012 | 10 |
| 46 | 0.0022 | 0.025 | 0.016 | 10-50 |
| 47 | 0.0033 | 0.0125 | 0.0065 | 4 |
| 48 | 0.0033 | 0.0125 | 0.007 | 1 |
| 化合物编号 | H-RasIC50(μM) | H-RasCIC50(μM) | K-RasIC50(μM) | K-RasCIC50(μM) |
| 49 | 0.0018 | 0.35 | 0.012 | 10 |
| 50 | 0.0017 | 0.03 | 0.008 | 10-50 |
| 51 | 0.003 | 0.005 | 0.01 | 10-50 |
| 52 | 0.0023 | 0.05 | 0.01 | 10-50 |
| 53 | 0.003 | 0.05 | 0.0085 | 10-50 |
| 54 | 0.011 | 0.025 | 0.04 | 10-50 |
| 55 | 0.002 | 0.025 | 0.04 | 10-50 |
| 56 | 0.005 | 0.05 | 0.02 | 5 |
| 57 | 0.011 | 0.025 | 0.01 | 10 |
Claims (11)
1.一种由下列化学式(1)表示的哌啶衍生物或者其药物可接受的盐:其中
A代表氢、低碳烷基或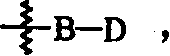 其中
其中
B代表CH2、C=O或SO2,
D代表一个选自下列组中的基团: 在取代基D的定义中,
在取代基D的定义中,
m表示0-3的整数,
n表示1-3的整数,
X代表氢、苯基、苯氧基、低碳烷基、低碳烷氧基、卤素、硝基、或者被苄基或低碳烷基非强制取代的氨基,
R1和R2各自分别代表氢、低碳烷基、C3-C6环烷基、被C3-C6环烷基取代的低碳烷基、芳基、或杂芳基,
E代表氢、苯基、萘基或
其中
R3和R4各自分别代表氢、低碳烷基、芳基或
(CH2)n′-Y-R5 (其中Y代表O或S,n′表示2-4的整数,R5代表低碳烷基),
G代表选自下列组中的基团: 其中
其中
Z代表O、S、SO2或N-R6(其中R6代表氢或低碳烷基),
R7和R8各自分别代表氢、低碳烷基、低碳烷氧基、卤素、氰基、羟基羰基、氨基羰基、氨基硫代羰基、羟基、苯基或苯氧基。
2.权利要求1的化合物,其中A代表氢、低碳烷基或
其中
B代表CH2、C=O或SO2,
D代表一个选自下列组中的基团:在取代基D的定义中,
m表示0-1的整数,
n表示1-2的整数,
X代表氢,
R1和R2各自分别代表氢或低碳烷基,
E代表氢、苯基、萘基或
 其中
其中
R3和R4各自分别代表氢、低碳烷基、或2-甲氧基乙基,
G代表选自下列组中的基团: 其中
其中
Z代表O或N-R6(其中R6代表甲基),
R7和R8各自分别代表氢。
3.权利要求1的化合物,该化合物选自下列化合物所组成的组中:1-[1-(1-苄氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-(吗啉-4-基)羰基-4-(萘-1-基)-1H-吡咯(1)、3-(吗啉-4-基)羰基-4-(萘-1-基)-1-[1-(哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(2)、1-[1-(1-乙酰基哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-(吗啉-4-基)羰基-4-(萘-1-基)-1H-吡咯(3)、1-[1-(1-甲磺酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-(吗啉-4-基)羰基-4-(萘-1-基)-1H-吡咯(4)、1-[1-(1-苄氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(5)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-[1-(哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(6)、1-[1-(1-乙酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(7)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-1-[1-(1-甲磺酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-4-(萘-1-基)-1H-吡咯(8)、1-{1-[1-(N-苄基氨基甲酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(9)、1-{1-[1-(N-丁基氨基甲酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(10)、1-{1-[1-(N-环己基氨基甲酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(11)、1-[1-(1-庚酰基-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(12)、1-{1-[1-(4-甲氧基苄基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(13)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-[1-(1-苯氧基乙酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(14)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(2-苯基乙基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(15)、1-{1-[1-(4-联苯基乙酰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(16)、1-[1-(1-甲氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(17)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-[1-(1-丙酰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-1H-吡咯(18)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(萘-1-基甲氧基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(19)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(萘-2-基甲氧基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(20)、1-{1-[1-(3,7-二甲基八-2,6-二烯-1-基氧羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(21)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-1-{1-[1-(3-甲基-2-丁烯-1-基氧羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-4-(萘-1-基)-1H-吡咯(22)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-1-{1-[1-(3-甲基-丁烷-1-基氧羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-4-(萘-1-基)-1H-吡咯(23)、1-{1-[1-(4-氟苄氧基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(24)、1-{1-[1-(肉桂氧基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(25)、1-{1-[1-(2-异丙基噻唑-4-基甲氧基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(26)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(萘-2-基羰基)-哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(27)、1-[1-(1-肉桂酰基哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(28)、1-{1-[1-(2-异丙基噻唑-4-基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(29)、1-{1-[1-(N-苄基-N-甲基氨基甲酰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(30)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢喹啉-1-基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(31)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢异喹啉-2-基羰基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(32)、1-{1-[1-(4-联苯基甲基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(33)、3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1-{1-[1-(4-苯氧基苄基)哌啶-4-基甲基]-1H-咪唑-5-基甲基}-1H-吡咯(34)、1-[1-(1-异丁氧基羰基-哌啶-4-基甲基)-1H-咪唑-5-基甲基]-3-[N-(2-甲氧基乙基)-N-甲基]氨基甲酰基-4-(萘-1-基)-1H-吡咯(35)、1-{1-[1-(苄氧基羰基)-哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(萘-1-基)羰基-1H-吡咯(36)、1-[1-(1-乙酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(萘-1-基)羰基-1H-吡咯(37)、1-[1-(1-苄氧基羰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(38)、1-[1-(1-苄氧基羰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-{N-[2-(N,N-二甲基氨基)乙基]-N-甲基}氨基甲酰基-4-(萘-1-基)-1H-吡咯(39)、1-[1-(1-甲氧基羰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(40)、3-(4-甲基哌嗪-1-基)羰基-1-[1-(1-甲磺酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-4-(萘-1-基)-1H-吡咯(41)、1-[1-(1-乙酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(42)、3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(2-苯基乙基羰基)-哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(43)、3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-[1-(1-苯氧基乙酰基哌啶-4-基)甲基-1H-咪唑-5-基]甲基-1H-吡咯(44)、3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(萘-2-基甲氧基)羰基哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(45)、1-{1-[1-(3-甲基丁氧基)羰基哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(46)、1-{1-[1-(4-氟苄氧基)羰基哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(47)、1-{1-[1-(肉桂氧基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(48)、3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(萘-2-基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(49)、1-{1-[1-(肉桂酰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(50)、1-{1-[1-(2-异丙基噻唑-4-基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(51)、1-{1-[1-(N-苄基-N-甲基氨基甲酰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(52)、1-{1-[1-(N,N-二甲基氨基甲酰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(53)、3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢喹啉-1-基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(54)、3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(1,2,3,4-四氢异喹啉-2-基羰基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(55)、1-{1-[1-(4-联苯基甲基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1H-吡咯(56)、和3-(4-甲基哌嗪-1-基)羰基-4-(萘-1-基)-1-{1-[1-(4-苯氧基苄基)哌啶-4-基]甲基-1H-咪唑-5-基}甲基-1H-吡咯(57)。
4.权利要求1所定义的化学式(1)的哌啶衍生物的制备方法,其特征在于:
(a)将下列化学式(2a)表示的化合物:其中Cbz代表苄氧基羰基,在一种溶剂中在一种碱存在下与下列化学式(3)表示的化合物反应:其中E和G如权利要求1所定义,然后将保护基Cbz脱除,得到由下列化学式(1a)表示的化合物:其中E和G如权利要求1所定义;
(b)将化学式(1a)的化合物在一种溶剂中与由下列化学式(4)表示的化合物反应:
A′-W (4)其中A′与权利要求1中定义的A相同,所不同的是A′不为氢,W代表氢,羟基或活泼的离去基团,得到由下列化学式(1b)表示的化合物: 其中A′如前面的描述所定义,E和G如权利要求1所定义;
其中A′如前面的描述所定义,E和G如权利要求1所定义;
(c)将化学式(1a)的化合物在一种溶剂中与下列化学式(5)表示的化合物反应:
A″-N=C=O (5)其中A″代表低碳烷基,苄基或C3-C6环烷基,得到由下列化学式(1c)表示的化合物: 其中A″如前面的描述所定义,E和G如权利要求1所定义;
其中A″如前面的描述所定义,E和G如权利要求1所定义;
(d)将化学式(1a)的化合物在一种溶剂中在一种还原剂存在下与下列化学式(6)表示的化合物反应:
D-CHO (6)其中D如权利要求1所定义,得到由下列化学式(1d)表示的化合物: 其中D,E和G如权利要求1所定义;或者
其中D,E和G如权利要求1所定义;或者
(e)将化学式(1a)的化合物在一种溶剂中与碳酰氯和下列化学式(7)表示的化合物反应:
D′H (7)其中D′代表选自下列组中的基团:其中m,n,X,R1和R2如权利要求1所定义,得到由下列化学式(1e)表示的化合物:其中D′如前面的描述所定义,E和G如权利要求1所定义。
5.一种由下列化学式(2)表示的化合物: 其中Y代表羟基或氯。
其中Y代表羟基或氯。
6、权利要求5所定义的化学式(2)的化合物的制备方法,其特征在于:
(f)将下列化学式(8)表示的化合物:在一种有机溶剂中在硝酸存在下进行脱硫化反应,得到下列化学式(2b)表示的化合物:
;或者
(g)将化学式(2b)的化合物与亚硫酰氯(SOCl2)反应,得到下列化学式(2a)表示的化合物: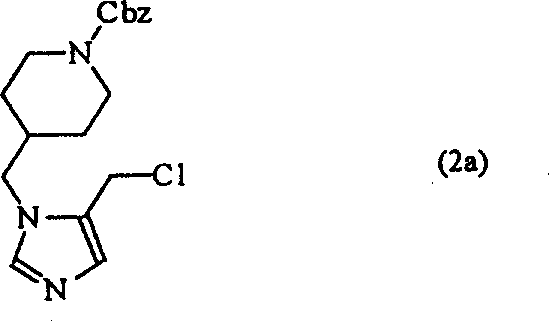
7.一种药物组合物,该组合物含有一种药物可接受的载体和作为活性组分的治疗有效量的如权利要求1所定义的化学式(1)的化合物或者其药物可接受的盐。
8.用于治疗或预防癌症的权利要求7的药物组合物。
9.用于治疗或预防再狭窄症的权利要求7的药物组合物。
10.用于治疗或预防动脉硬化症的权利要求7的药物组合物。
11.用于治疗或预防由δ肝炎和相关病毒引起的传染病的权利要求7的药物组合物。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR19980002776 | 1998-02-02 | ||
| KR1019980002777A KR100388789B1 (ko) | 1998-02-02 | 1998-02-02 | 피롤구조를갖는파네실전이효소억제제및그의제조방법 |
| KR1019980028340A KR100388792B1 (ko) | 1998-02-02 | 1998-07-14 | 피페리딘구조를갖는파네실전이효소억제제및그의제조방법 |
| KR1998/28340 | 1998-08-07 | ||
| KR10-1998-0032150A KR100388794B1 (ko) | 1998-08-07 | 1998-08-07 | 피페리딘구조를갖는파네실전이효소억제제및그의제조방법 |
| KR1998/2776 | 1998-08-07 | ||
| KR1998/2777 | 1998-08-07 | ||
| KR1998/32150 | 1998-08-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1289331A true CN1289331A (zh) | 2001-03-28 |
| CN1158277C CN1158277C (zh) | 2004-07-21 |
Family
ID=27483262
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB99802581XA Expired - Fee Related CN1158277C (zh) | 1998-02-02 | 1999-02-01 | 具有哌啶结构的法尼转移酶抑制剂及其制备方法 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US6436960B1 (zh) |
| EP (1) | EP1058683B1 (zh) |
| JP (1) | JP3283032B2 (zh) |
| CN (1) | CN1158277C (zh) |
| AT (1) | ATE229017T1 (zh) |
| AU (1) | AU745855B2 (zh) |
| BR (1) | BR9908545A (zh) |
| CA (1) | CA2320233C (zh) |
| DE (1) | DE69904302T2 (zh) |
| ES (1) | ES2185307T3 (zh) |
| PT (1) | PT1058683E (zh) |
| WO (1) | WO1999038862A1 (zh) |
Families Citing this family (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100388794B1 (ko) * | 1998-08-07 | 2003-10-10 | 주식회사 엘지생명과학 | 피페리딘구조를갖는파네실전이효소억제제및그의제조방법 |
| KR100388792B1 (ko) * | 1998-02-02 | 2003-09-22 | 주식회사 엘지생명과학 | 피페리딘구조를갖는파네실전이효소억제제및그의제조방법 |
| ATE277039T1 (de) * | 1999-04-13 | 2004-10-15 | Lg Chem Investment Ltd | Farnesyltransferase-inhibitoren die eine pyrrolstruktur haben und verfahren zu ihrer herstellung |
| KR20010063274A (ko) * | 1999-12-22 | 2001-07-09 | 성재갑 | 〔1-{〔1-(1,3-벤조디옥솔-5-일메틸-1h-이미다졸-5-일〕메틸}-4-(1-나프틸)-1h-피롤-3-일〕(4-메틸-1-피페라지닐)메타논의 약학적 조성물 |
| US7035932B1 (en) * | 2000-10-27 | 2006-04-25 | Eric Morgan Dowling | Federated multiprotocol communication |
| US7132100B2 (en) | 2002-06-14 | 2006-11-07 | Medimmune, Inc. | Stabilized liquid anti-RSV antibody formulations |
| US7425618B2 (en) | 2002-06-14 | 2008-09-16 | Medimmune, Inc. | Stabilized anti-respiratory syncytial virus (RSV) antibody formulations |
| US7563810B2 (en) | 2002-11-06 | 2009-07-21 | Celgene Corporation | Methods of using 3-(4-amino-1-oxo-1,3-dihydroisoindol-2-yl)-piperidine-2,6-dione for the treatment and management of myeloproliferative diseases |
| US8034831B2 (en) | 2002-11-06 | 2011-10-11 | Celgene Corporation | Methods for the treatment and management of myeloproliferative diseases using 4-(amino)-2-(2,6-Dioxo(3-piperidyl)-isoindoline-1,3-dione in combination with other therapies |
| EP2133340B1 (en) | 2002-12-20 | 2013-01-16 | Glaxo Group Limited | Novel benzazepine derivatives |
| JP4764818B2 (ja) | 2003-04-11 | 2011-09-07 | メディミューン,エルエルシー | 組換えil−9抗体およびその使用 |
| EP1660186B1 (en) | 2003-08-18 | 2013-12-25 | MedImmune, LLC | Humanization of antibodies |
| AU2004282189B2 (en) | 2003-10-17 | 2011-11-17 | Incyte Holdings Corporation | Substituted cyclic hydroxamates as inhibitors of matrix metalloproteinases |
| US20060121042A1 (en) | 2004-10-27 | 2006-06-08 | Medimmune, Inc. | Modulation of antibody specificity by tailoring the affinity to cognate antigens |
| JP5153613B2 (ja) | 2005-03-18 | 2013-02-27 | メディミューン,エルエルシー | 抗体のフレームワーク・シャッフル |
| EP1893647A2 (en) | 2005-06-23 | 2008-03-05 | MedImmune, Inc. | Antibody formulations having optimized aggregation and fragmentation profiles |
| ES2439994T3 (es) | 2006-08-28 | 2014-01-27 | Kyowa Hakko Kirin Co., Ltd. | Anticuerpos antagonistas monoclonales humanos específicos de LIGHT humano |
| CA2682292A1 (en) | 2007-03-30 | 2008-10-09 | Medimmune, Llc | Aqueous formulation comprising an anti-human interferon alpha antibody |
| US9540443B2 (en) | 2011-01-26 | 2017-01-10 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies |
| GB201107985D0 (en) | 2011-05-13 | 2011-06-29 | Astrazeneca Ab | Process |
| CN116574185A (zh) | 2012-07-25 | 2023-08-11 | 塞尔德克斯医疗公司 | 抗kit抗体及其用途 |
| WO2014059028A1 (en) | 2012-10-09 | 2014-04-17 | Igenica, Inc. | Anti-c16orf54 antibodies and methods of use thereof |
| SG11201509982UA (zh) | 2013-06-06 | 2016-04-28 | Igenica Biotherapeutics Inc | |
| KR102313341B1 (ko) | 2013-08-26 | 2021-10-18 | 바이오엔테크 리서치 앤드 디벨롭먼트 인코포레이티드 | 시알릴-루이스 a에 대한 사람 항체 코드화 핵산 |
| GB201403775D0 (en) | 2014-03-04 | 2014-04-16 | Kymab Ltd | Antibodies, uses & methods |
| DK3154583T3 (da) | 2014-06-04 | 2021-03-22 | Biontech Res And Development Inc | Humane monoklonale antistoffer mod gangliosid gd2 |
| PL3333191T3 (pl) | 2014-12-11 | 2021-05-04 | Pierre Fabre Médicament | Przeciwciała przeciwko c10orf54 i ich zastosowania |
| MX2017011194A (es) | 2015-03-03 | 2018-04-10 | Kymab Ltd | Anticuerpos, usos y métodos. |
| KR102815803B1 (ko) | 2015-12-02 | 2025-06-05 | 주식회사 에스티사이언스 | 당화된 btla(b- 및 t-림프구 약화인자)에 특이적인 항체 |
| CN114470194A (zh) | 2015-12-02 | 2022-05-13 | 斯特库伯株式会社 | 与btn1a1免疫特异性结合的抗体和分子及其治疗用途 |
| EP3534947A1 (en) | 2016-11-03 | 2019-09-11 | Kymab Limited | Antibodies, combinations comprising antibodies, biomarkers, uses & methods |
| US12215151B2 (en) | 2017-05-31 | 2025-02-04 | Stcube & Co., Inc. | Methods of treating cancer using antibodies and molecules that immunospecifically bind to BTN1A1 |
| KR20200015602A (ko) | 2017-05-31 | 2020-02-12 | 주식회사 에스티큐브앤컴퍼니 | Btn1a1에 면역특이적으로 결합하는 항체 및 분자 및 이의 치료적 용도 |
| CN110997724A (zh) | 2017-06-06 | 2020-04-10 | 斯特库伯株式会社 | 使用结合btn1a1或btn1a1-配体的抗体和分子治疗癌症的方法 |
| US11707522B2 (en) | 2017-10-13 | 2023-07-25 | Boehringer Ingelheim International Gmbh | Human antibodies to Tn antigen |
| KR102767692B1 (ko) | 2018-07-20 | 2025-02-17 | 피에르 파브르 메디카먼트 | Vista의 수용체 |
| AU2024265078A1 (en) | 2023-05-04 | 2025-12-11 | Revolution Medicines, Inc. | Combination therapy for a ras related disease or disorder |
| IL326136A (en) | 2023-08-07 | 2026-03-01 | Revolution Medicines Inc | RMC-6291 for use in the treatment of a disease or disorder associated with the RAS protein |
| US20250154171A1 (en) | 2023-10-12 | 2025-05-15 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025240847A1 (en) | 2024-05-17 | 2025-11-20 | Revolution Medicines, Inc. | Ras inhibitors |
| US20250375445A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IE903957A1 (en) * | 1989-11-06 | 1991-05-08 | Sanofi Sa | Aromatic amine compounds, their method of preparation and¹pharmaceutical compositions in which they are present |
| AU3748393A (en) * | 1992-03-25 | 1993-10-21 | Pfizer Inc. | Antiviral peptides |
| GB9312806D0 (en) * | 1993-06-22 | 1993-08-04 | Boots Co Plc | Therapeutic agents |
| JP2001519766A (ja) * | 1996-04-03 | 2001-10-23 | メルク エンド カンパニー インコーポレーテッド | ファルネシルタンパク質トランスフェラーゼの阻害剤 |
| CA2250232A1 (en) * | 1996-04-03 | 1997-10-09 | Allen I. Oliff | A method of treating cancer |
| CA2250353A1 (en) * | 1996-04-03 | 1997-10-09 | Christopher J. Dinsmore | Inhibitors of farnesyl-protein transferase |
| AU704792B2 (en) * | 1996-04-03 | 1999-05-06 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| EP0891353A4 (en) * | 1996-04-03 | 2001-08-08 | Merck & Co Inc | FARNESYL PROTEIN TRANSFERASE INHIBITORS |
| EP1045846B1 (en) * | 1997-11-28 | 2003-05-02 | Lg Chemical Limited | Imidazole derivatives having an inhibitory activity for farnesyl transferase and process for preparation thereof |
-
1999
- 1999-02-01 CN CNB99802581XA patent/CN1158277C/zh not_active Expired - Fee Related
- 1999-02-01 ES ES99901979T patent/ES2185307T3/es not_active Expired - Lifetime
- 1999-02-01 PT PT99901979T patent/PT1058683E/pt unknown
- 1999-02-01 WO PCT/KR1999/000051 patent/WO1999038862A1/en not_active Ceased
- 1999-02-01 EP EP99901979A patent/EP1058683B1/en not_active Expired - Lifetime
- 1999-02-01 BR BR9908545-3A patent/BR9908545A/pt not_active Application Discontinuation
- 1999-02-01 AU AU21886/99A patent/AU745855B2/en not_active Ceased
- 1999-02-01 AT AT99901979T patent/ATE229017T1/de not_active IP Right Cessation
- 1999-02-01 US US09/601,426 patent/US6436960B1/en not_active Expired - Fee Related
- 1999-02-01 DE DE69904302T patent/DE69904302T2/de not_active Expired - Fee Related
- 1999-02-01 JP JP2000529330A patent/JP3283032B2/ja not_active Expired - Fee Related
- 1999-02-01 CA CA002320233A patent/CA2320233C/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| BR9908545A (pt) | 2001-10-02 |
| ES2185307T3 (es) | 2003-04-16 |
| DE69904302T2 (de) | 2003-08-14 |
| DE69904302D1 (de) | 2003-01-16 |
| CA2320233C (en) | 2004-07-27 |
| EP1058683A1 (en) | 2000-12-13 |
| WO1999038862A1 (en) | 1999-08-05 |
| JP3283032B2 (ja) | 2002-05-20 |
| CA2320233A1 (en) | 1999-08-05 |
| ATE229017T1 (de) | 2002-12-15 |
| AU745855B2 (en) | 2002-04-11 |
| US6436960B1 (en) | 2002-08-20 |
| CN1158277C (zh) | 2004-07-21 |
| EP1058683B1 (en) | 2002-12-04 |
| PT1058683E (pt) | 2003-04-30 |
| JP2002501918A (ja) | 2002-01-22 |
| AU2188699A (en) | 1999-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1158277C (zh) | 具有哌啶结构的法尼转移酶抑制剂及其制备方法 | |
| CN1049651C (zh) | 金属蛋白酶抑制剂 | |
| CN1061977C (zh) | 芳基取代的杂环 | |
| CN1280287C (zh) | 晶体的制备方法 | |
| CN1176651C (zh) | N-(3-苯并呋喃基)脲衍生物 | |
| CN1279680A (zh) | 具有法尼基转移酶抑制活性的咪唑衍生物及其制备方法 | |
| CN1255162A (zh) | 蛋白酶抑制剂 | |
| CN1198804C (zh) | 酰胺化合物及其用途 | |
| CN87107371A (zh) | 螺-取代的戊二酰胺利尿剂 | |
| CN1134148A (zh) | 抗血栓形成的脒基苯丙氨酸和脒基吡啶丙氨酸的衍生物 | |
| CN1330637A (zh) | 用于治疗炎性疾病的化合物 | |
| CN1227540A (zh) | 具有mmp和tnf抑制活性的异羟肟酸与羧酸的衍生物 | |
| CN1281433A (zh) | 作为基质金属蛋白酶(mmp)抑制剂的异羟肟酸衍生物 | |
| CN1282243A (zh) | 用于拮抗ccr5的含n-酰苯胺衍生物的药物组合物 | |
| CN1073166A (zh) | 羟肟酸衍生物 | |
| CN1036064C (zh) | 新的具有ngf生成促进活性的苯衍生物的制备方法 | |
| CN1291993C (zh) | N-取代苯并噻唑基-1-取代苯基-O,O-二烷基-α-氨基膦酸酯类衍生物及制备方法和用途 | |
| CN1058019C (zh) | 环状氨基酸衍生物 | |
| CN1180358A (zh) | 作为基质金属蛋白酶抑制剂的桥式吲哚 | |
| CN1349542A (zh) | 杂环化合物、其中间体和弹性蛋白酶抑制剂 | |
| CN1756740A (zh) | 具有2,6-二取代苯乙烯基的含氮杂环衍生物 | |
| CN1291993A (zh) | 用作半胱氨酸活性依赖性酶抑制剂的噻二唑化合物 | |
| CN1187360C (zh) | 有羧基肽酶b抑制活性的膦酸衍生物 | |
| CN1101349A (zh) | 新的磷酸化的芳基乙醇胺衍生物 | |
| CN1311771A (zh) | 苯甲脒衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |