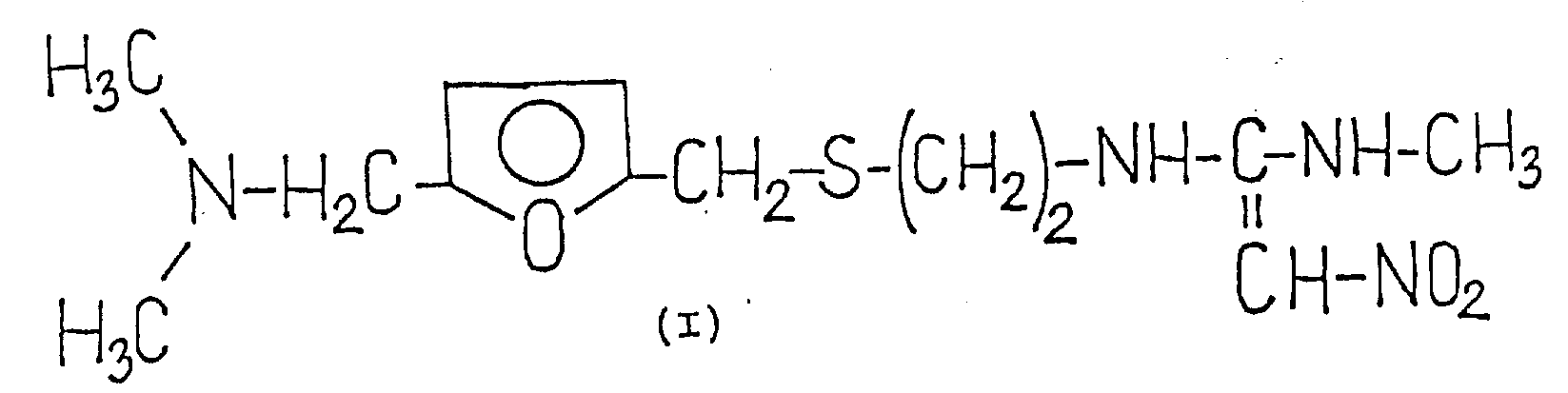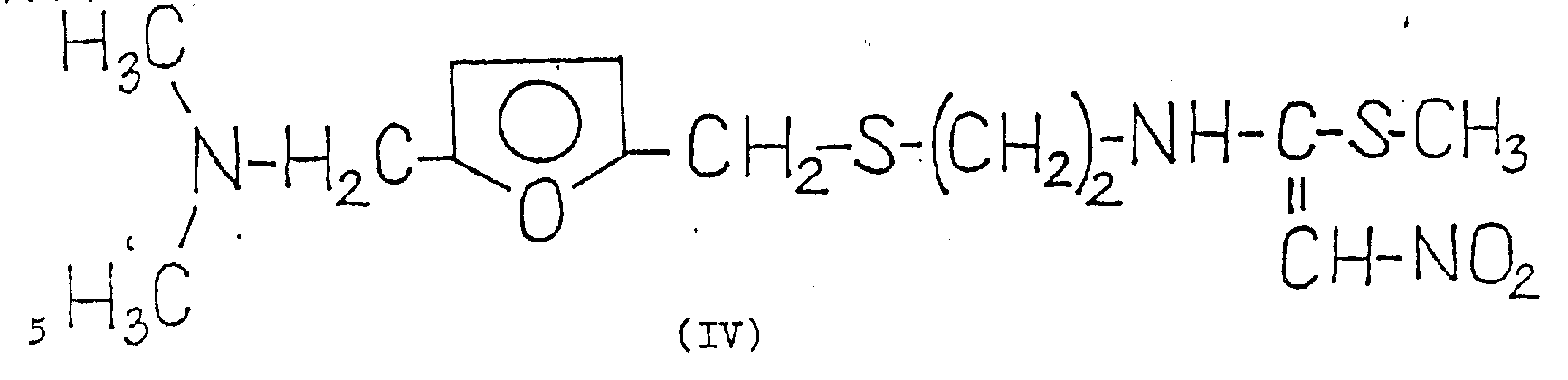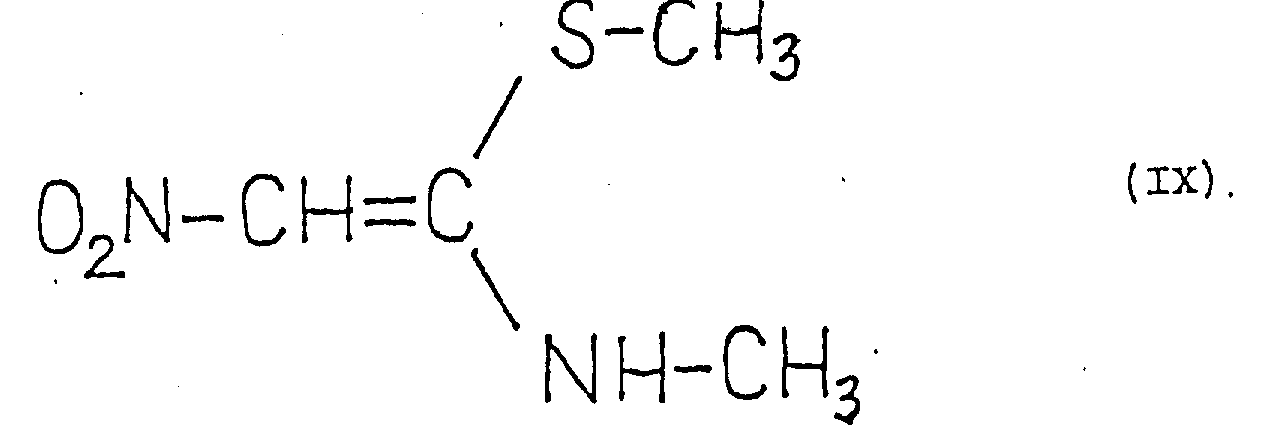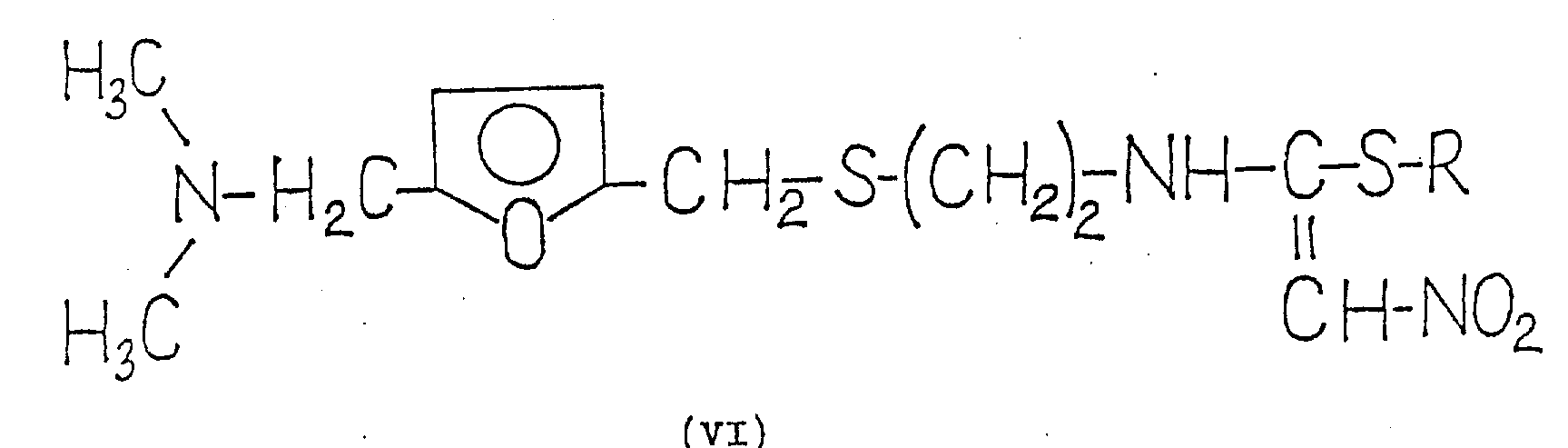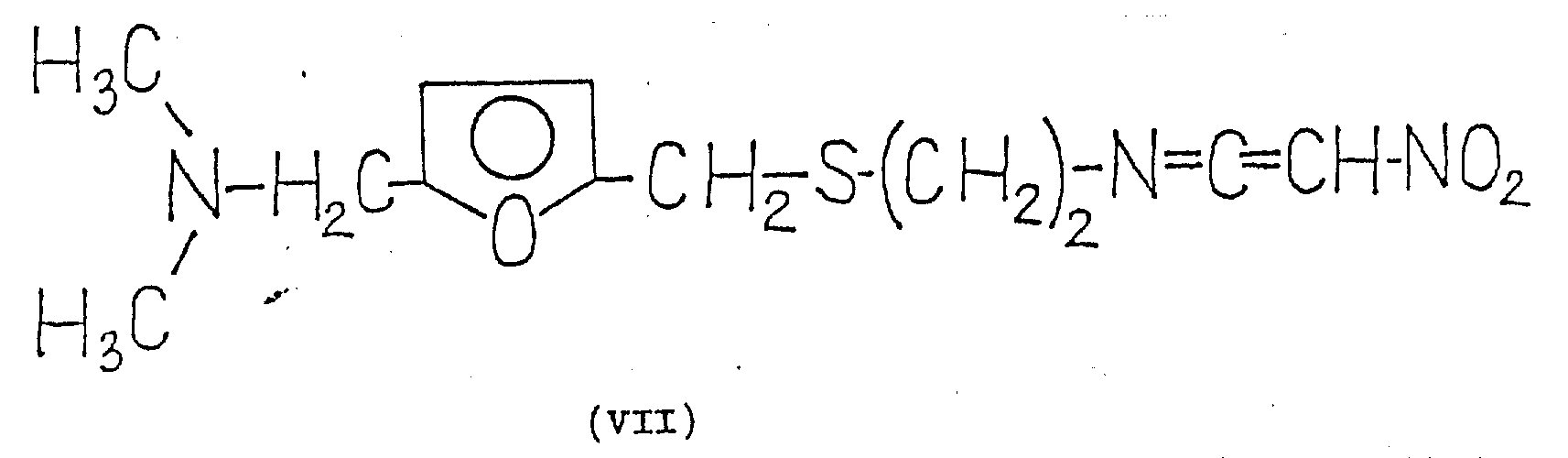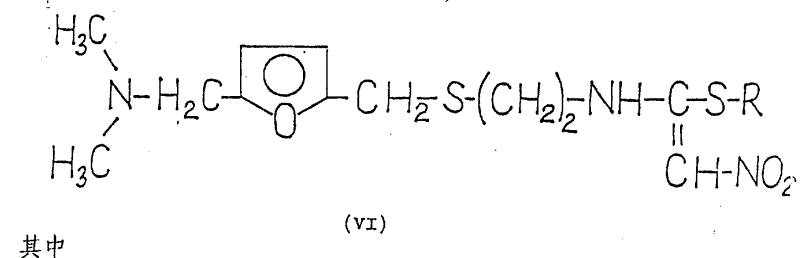Process for the preparation of basic thioethers and salts thereof
The present invention relates to a novel process for the preparation of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylamino-2-nitroethylene and its hydrochloride of formula (I).
The compounds of formula (i) [ trade name raney tincture (ranitidine) ] are attractive anti-gastric and duodenal ulcer agents as selective histamine H-2 receptor antagonists. The hydrochloride salt of the compound of formula (i) is useful for therapeutic purposes.
The following processes have been described in the literature for the preparation of the hydrochloride salt of the compound of formula (I).
a) According to example 32 of published German patent application No. 2, 734, 070 (page 75), the hydrochloride of the compound of formula (I) is prepared by dissolving the base of formula (I) in ethanol and precipitating the hydrochloride salt with ethyl acetate in a yield of 89.6% based on the starting material.
b) The hydrochloride of the compound of formula (i) is prepared according to example 1 of belgian patent specifications 890, 574 (page 8) by adding concentrated aqueous hydrochloric acid to the solution and then precipitating the hydrochloride formed with isopropanol. The yield was 93.9% calculated from compound (I).
The starting material for both processes is a base of formula (I). Thus, based on the prior art processes, in order to form hydrochloric acid, it is necessary first to prepare a base of the formula (I) of satisfactory quality. The base is prepared according to example 24 of the above-cited published German patent application No. 2, 734, 070 (page 68) by first reacting 2- [ (2-aminoethyl) -thiomethyl ] -5-dimethylaminomethylfuran of the formula (II) with 1, 1-bis (methylthio) -2-nitroethylene of the formula (III) in acetonitrile at reflux temperature for 14 hours, then removing the solvent and boiling the crude distillation residue at reflux with a solution of methanol [ hereinafter, this process is referred to as literature process "A"). The yield was 79.9% according to the specification. A significant disadvantage of this process is that the first step of this two-step reaction is characteristic. Another disadvantage is associated with the end product, and example 15 of the above-cited published German patent application (page 61), although it shows its significance in terms of yield and quality of the base of formula (I), does not give the extraction conditions for recrystallization.
The process described in example 24 of this German patent application has been found to form, in the first step, a very impure product of formula (IV) in the form of an oil, from which the base of formula (I) is not obtained in a quality suitable for the preparation of the hydrochloride of the base of formula (I).
[ according to our experiments, the crude product of formula (IV) obtained using the process described in the above example had to be purified by a laborious and cumbersome process (e.g.column chromatographic separation) and the product losses were high ]. We have further found that even with the pure compounds of the formula (IV), it is not possible to prepare bases of the formula (I) in a quality which meets the above requirements by the process described in example 24.
Another process for the preparation of bases of formula (I) is reported in Hungarian patent application No. 1827/83 (publication No. T/31115), according to which a compound of formula (IX) is converted with a heavy metal salt, such as silver nitrate or cuprous chloride, in the presence of a proton-binding agent into 1-methyl-3-nitroketene imine (keteneimine) of formula (V), which is subjected to an addition reaction with a base of formula (II) to give a base of formula (I).
O2N-CH=C=N-CH3(Ⅴ)
There are two examples of this patent application, however, none of the examples address yield. [ A method for purifying a crude base of the formula (I) has not been reported either ]. Hereinafter, this method is generally referred to as literature method "B". Spectroscopic experiments were cited for identification and quality issues of the resulting products, but are not specifically described. On the other hand, the base obtained is said to be identical to the raney tincture base obtained in example 15 of the published german patent application No. 2, 734, 070, which, however, according to our own studies, is not suitable for the preparation of raney tincture hydrochloride of satisfactory purity.
The object of the present invention is to provide a process for preparing bases of the formula (I) and their hydrochlorides which enables these compounds to be produced in high yields and to be put into industrial production in a simple manner.
It has been found that this object is fully achieved. 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1- (substituted thio) -2-nitroethylene of the general formula (VI) is converted in situ in a simple manner in high yield into 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -3-nitroketiminate of the formula (VII), and the ketene imine of the formula (VII) is then reacted with methylamine.
Wherein
R represents optionally substituted alkyl or oxoalkyl and aryl, aralkyl or oxoaralkyl.
This recognition is unexpected for several reasons. Hitherto, only the recently published literature method "B" has been described in the literature for the structurally simple nitroketene imines of the formula (V). It was not expected that the structurally more complex nitroketene imines of the formula (VII) could be formed in an unexpectedly short time (within a few minutes) from the compounds of the formula (VI), but that the starting materials of the formula (VI) were also good, and that the products of the formula (VII) formed were also good without any change in the part which did not participate in the reaction. It has likewise not been expected that the addition of the structurally complex nitroketene imines of the formula (VII) to methylamine gives the bases of the formula (I) in very high purity in a very short time, and that, according to our observations, there is hardly any side reaction.
Accordingly, the present invention relates to a novel process for the preparation of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylamino-2-nitroethylene and its hydrochloride which comprises 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1- (substituted thio) -2-nitroethylene of the general formula (VI) wherein
R represents optionally substituted alkyl or oxoalkyl, aryl, aralkyl or oxoaralkyl having 1 to 4 carbon atoms, with a reagent capable of cleaving thiols in an organic solvent in the presence of (or without) an acid binder, then reacting the resulting 1- { [2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -3-nitroketene imine of the formula (VII) in situ with methylamine, if desired, and finally separating off the resulting base of the formula (I) and purifying and/or, if desired, converting it into the hydrochloride.
The compounds of formula (VII) formed as intermediates in the process of the invention are novel compounds. The formation of this compound is supported by the fact that 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) ethyl ] } -amino-1-methylthio-2-nitroethylene [ compound of formula (IV) ] reacts with silver nitrate to obtain silver methanethiolate. The reaction was in almost quantitative yield as described below.
For the preparation of compounds of the formula (VII), one of the compounds of the formula (VI), i.e.compounds of the formula (IV), can be regarded as a known substance on the basis of comparison with the already published German patent application 2, 734, 070, which describes the modification b) of claim 37 (page 7) and the general formula reported in example 24 (page 68), but the properties of this substance, including the chemical name, are not given in this patent application. No other compounds of the formula (VI) are mentioned in this patent application. According to example 1 of published European patent application No. 0,002,930, the melting point of the compound of formula (IV) is 71 ℃. To the best of our knowledge, other compounds of formula (VI) have not been shown in the literature. Thus, in addition to compounds of formula (IV), compounds of formula (VI) are novel. These compounds can be prepared in a known manner by reacting a base of the formula (II) with a compound of the formula (VIII),
wherein R has the meaning as defined above.
Some of the compounds of the general formula (VIII) are known [ Acta chem. Scand.21, 2797 (1967); chem. Ber.100, 591 (1967) ]. The novel compounds of the formula (VIII) can be prepared in a manner known per se.
As a compound of the general formula (VI), compounds of the formula (IV) are useful, for example, for preparing nitroketene imines of the formula (VII). Metal salts, metal oxides or finely divided metals may be used as reagents capable of cleaving thiols. It is expedient to use, for example, silver nitrate or chlorous-ylidene chloride as metal salt. Organic bases are suitable acid binders. The formation of the compound of formula (VII) and the reaction with methylamine is preferably carried out at room temperature or at a temperature slightly below room temperature. Organic solvents, such as lower aliphatic alcohols like ethanol, are preferred.
In a preferred embodiment of the invention, an absolute ethanolic solution or suspension of the compound of formula (IV) is treated with silver nitrate at 5-30 ℃ to form a silver mercaptide precipitate, methylamine is added immediately, the reaction mixture is filtered, the solvent is removed from the filtrate, and the residue is treated in the usual way, consisting of the base of formula (I) except for the corresponding salt of methylamine. The raney tincture obtained is recrystallized if necessary and, if necessary, further converted into its hydrochloride salt.
In another preferred embodiment of the invention, an ethanol solution of methylamine containing silver nitrate is added dropwise to an ethanol suspension of the compound of formula (IV) and then treated as described above.
The advantages of the method of the invention can be summarized as follows:
each step of the process of the invention, i.e.the formation of the compound of formula (VII) and the reaction with methylamine, is carried out rapidly at a temperature at or slightly below room temperature. This leads to a stabilization of the compound of the formula (VII) and thus no contamination of the base of the formula (I) formed by the decomposition products. Another result is energy savings. Because the reaction time is short (production cycle is short), the equipment can be effectively utilized.
The reaction proceeds rapidly, in large part because normally gaseous methylamine can be used in excess as desired and excess methylamine can be readily removed after the reaction without any heat. In contrast, when the base of formula (I) is prepared by process "B" of the above-cited reference, the compound of formula (IX) is first converted into nitroketene imine of formula (V), which is then subjected to an addition reaction with the base of formula (II), only in stoichiometric amounts. Therefore, the addition reaction cannot be accelerated by using an excess of the base of formula (II). Since this base is unstable and heat sensitive, it can only be removed under high vacuum at higher temperatures, and this base inevitably partly decomposes during the distillation. In contrast to the fact that the base of formula (I) obtained is heavily contaminated with residual base (II) or with decomposition products of the base (II) which are present in excess during distillation, in the process "A", which in the literature gives off one mole of methyl mercaptan per step, the toxic gaseous methyl mercaptan is incorporated in the harmless form, i.e. as a non-volatile metal mercaptide, in the process of the invention and therefore does not pollute the environment.
The yield of the base of formula (I) obtained by applying the process of the invention is almost quantitative. The crude product contains only very small amounts of impurities (salts of the acid binder used). The base of formula (I) is obtained in very high yields (about 75%) in very pure form by simple purification procedures. This base can be used to form the hydrochloride salt. The hydrochloride salt thus formed does not require further purification.
Based on these facts, the process of the present invention can also easily prepare the base of formula (I) and its hydrochloride salt industrially in high yield.
The process of the invention is illustrated in detail by the following non-limiting examples.
Example 1
Preparation of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylamino-2-nitroethylene [ base of formula (I) ]:
51.0g (0.30 mole) of silver nitrate was dissolved in 4000ml of absolute ethanol at 10 ℃ and added to a solution of 99.5g (0.30 mole) of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylthio-2-nitroethylene [ compound of the formula (IV) ] in 2500ml of absolute ethanol over 1 minute to yield immediately silver methanethiolate. 557ml of a 29.2% solution of methylamine in ethanol were then added and the mixture was stirred at room temperature for 2 hours. The resulting precipitate (silver methanethiolate in a yield of 98.1% based on its weight and composition) was filtered off, and the filtrate was evaporated to dryness at room temperature. The evaporation residue is dissolved in 600ml of water and extracted 8 times with 1200ml of ethyl acetate, if necessary with adjustment of the pH to 10. The organic phases are combined, dried and evaporated under reduced pressure. The residue was evaporated to give crude Raney tincture in 79g (84%).
To prepare the hydrochloride, crude Raney tincture base is dissolved in 380ml of absolute ethanol and the pH is adjusted to 5.5 at 0-5 ℃ with concentrated aqueous hydrochloric acid. The mixture was stirred at 0 ℃ for a further 40 minutes.
The crystalline precipitate was filtered off, washed with ethanol and dried to give Raney tincture hydrochloride in a yield of 57.45g (54.6%) m.p.143 ℃ and a second crop of hydrochloride in a yield of 23.15g (22.0%) m.p.143 ℃.
1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylthio) -2-nitroethylene [ compound of the formula (IV) ] used as starting material can be prepared, for example, according to example 1 of published European patent application No. 0, 002,930.
Example 2
Preparation of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylamino-2-nitroethylene [ base of formula (I) ] and its hydrochloride:
a solution of 2.55g (0.015 mole) of silver nitrate in 200ml of absolute ethanol is added to a solution of 5g (0.015 mole) of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylthio-2-nitroethylene [ compound of formula (IV) ] in 125ml of absolute ethanol at 6 ℃ with stirring over 1 minute, immediately forming silver methanethiolate. The mixture was stirred at 5 ℃ for a further 3 minutes, after which 30ml of an 27% solution of methylamine in ethanol were added. The mixture was stirred at room temperature for 2 hours and then filtered. The filtrate was left overnight at 5 ℃ and filtered again, and the filtrate was evaporated at room temperature under reduced pressure. After addition of 50ml of water to the residue, the pH was adjusted to 5.5 with 1N aqueous hydrochloric acid. The mixture was extracted twice with 70ml of dichloromethane and the aqueous phase was adjusted to pH 10 with 1N sodium hydroxide solution and extracted four times with 70ml of dichloromethane. Finally the extract was combined with the organic phase, dried, evaporated and four times the volume of ethyl acetate added to the evaporation residue. The precipitated crystals were filtered and dried to give 3.02g (64.0%) of the desired base, m.p.68-70. After repeated evaporation of the mother liquor, the residue recrystallizes giving a further 0.55g (11.2%) of the desired base.
The base obtained in example 2 can be converted into its hydrochloride, for example, in the following manner.
3.5g of crude Raney tincture was dissolved in 17.5ml of anhydrous ethanol, and the pH of the solution was adjusted to 5-5.5 by adding concentrated aqueous hydrochloric acid solution at 0 ℃ with stirring. The mixture was stirred at 0 ℃ for a further 40 minutes and left overnight at 0-5 ℃. The precipitated crystals were filtered, washed with ethanol and dried to give 3.72g (84.4%) of the desired hydrochloride salt, m.p.143 ℃.
Example 3
Preparation of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylamino-2-nitroethylene [ base of formula (I) ]:
a solution prepared from 0.85g (0.005 mole) of silver nitrate in 10ml of methylamine ethanol was added to 1.66g (0.005 mole) of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylthio-2-nitroethylene [ compound of formula (IV) ] suspended in 10ml of anhydrous ethanol at room temperature with stirring. The mixture was stirred at room temperature for a further 30 minutes, filtered and evaporated under reduced pressure. Then, the procedure described in example 2 was followed to give 1.21g (77.1%) of the desired base, m.p.68-70 ℃.
Example 4
Preparation of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylamino-2-nitroethylene [ base of formula (I) ]:
adding a solution containing 0.85g (0.005 mole) of silver nitrate in 60ml of allyl alcohol to a solution containing 1.66g (0.005 mole) of 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylthio-2-nitroethylene [ a compound having a molecular formula (IV) ] in 20ml of allyl alcohol in portions at 8-10 ℃ for 2-3 minutes while stirring. At the same temperature, 8ml of methylamine ethanol solution is added within 1-2 minutes. The mixture was stirred at room temperature for two hours. Then, the procedure described in example 2 was followed to give 0.95g (60.5%) of the desired base, m.p.70-72 ℃.
Example 5
Comparative experiment
To confirm that in the process of the present invention, 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -amino-1-methylthio-2-nitroetheno [ compound of formula (IV) ] was first reacted with silver nitrate and then, while precipitating silver methanethiol, the resulting 1- {2- [ 5-dimethylaminomethyl-2- (furanmethylthio) -ethyl ] } -3-nitroethenone imine [ compound of formula (VII) ] was reacted with methylamine, the following comparative experiment was carried out.
The procedure described in example 1 was followed, except that after addition of silver nitrate, the precipitate formed in the solution was filtered off, washed, dried and analyzed. The weight of the precipitate was 2.28g (98.1%).
Elemental analysis: CHSAg (molecular weight 154.98)
Calculated values: 7.75 percent of C, 1.95 percent of H and 69.6 percent of AgC
Experimental values: c7.69%, H1.36%, Ag69.1%