DE102004018548A1 - Durch Strahlung und Feuchtigkeit härtende Zusammensetzungen auf Basis Silan-terminierter Polymere, deren Herstellung und Verwendung - Google Patents
Durch Strahlung und Feuchtigkeit härtende Zusammensetzungen auf Basis Silan-terminierter Polymere, deren Herstellung und Verwendung Download PDFInfo
- Publication number
- DE102004018548A1 DE102004018548A1 DE200410018548 DE102004018548A DE102004018548A1 DE 102004018548 A1 DE102004018548 A1 DE 102004018548A1 DE 200410018548 DE200410018548 DE 200410018548 DE 102004018548 A DE102004018548 A DE 102004018548A DE 102004018548 A1 DE102004018548 A1 DE 102004018548A1
- Authority
- DE
- Germany
- Prior art keywords
- polymer
- base
- photolatent
- silyl
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Die
Erfindung betrifft härtbare
Zusammensetzungen, die mindestens ein silylterminiertes Polymer
und mindestens eine photolatente Base enthalten, wobei das silylterminierte
Polymer aus einem linearen oder verzweigten, silangruppenfreien
Basispolymer zusammengesetzt ist, welches endständige Silangruppen trägt. Ferner
wird ein Verfahren zur Herstellung solcher Zusammensetzungen sowie
deren Verwendung beschrieben.
Description
- Die vorliegende Erfindung betrifft Zusammensetzungen, die durch Bestrahlung und Feuchtigkeit aushärten, sowie deren Herstellung und Verwendung.
- Feuchtigkeitshärtende Klebstoffzusammensetzungen werden im zunehmenden Maße als Beschichtungs-, Dichtungs- und Klebemassen im Bauwesen und in der Automobilindustrie verwendet. An solche Massen werden hohe Anforderungen, insbesondere hinsichtlich Verarbeitbarkeit und Aushärtgeschwindigkeit gestellt.
- Aus dem Stand der Technik sind eine Vielzahl verschiedener Polymere bekannt, die als vernetzende Bestandteile in Dichtungs-, Beschichtungs- und Klebemassen eingesetzt werden. Solche Massen sollen üblicherweise rasch aushärten, damit die verklebten, beschichteten, abgedichteten oder verfugten Substrate möglichst bald ihrer eigentlichen Bestimmung zugeführt werden können. Andererseits ist es wünschenswert, daß die Massen eine gute Verarbeitbarkeit aufweisen, die es dem Fachmann auf dem Gebiet der Klebe- und Dichtungstechnik ermöglicht größere Bereiche gleichmäßig mit den Massen zu behandeln, ohne daß Teile bereits aushärten. Der Anwendungstechniker befindet sich somit in einem Dilemma zwischen optimaler Aushärtgeschwindigkeit einerseits und Verarbeitbarkeit andererseits. Idealerweise sollten derartige Massen oder Zusammensetzungen solange gleichmäßig verarbeitbar sein, wie das Aufbringen der Zusammensetzungen auf das Substrat dauert. Anschließend sollten die Zusammensetzungen möglichst rasch aushärten.
- Häufig werden in Dichtungs-, Beschichtungs- und Klebemassen silylgruppenterminierte organische Polymere (STOP) als vernetzende Bestandteile eingesetzt. Erhältlich sind STOP praktisch aus nahezu sämtlichen Basispolymeren, indem organofunktionelle alkoxylierte Silane mit reaktiven Enden des Basispolymers zur Reaktion gebracht werden. Beispielsweise können Polyurethane mit endständigen Isocyanatgruppen mit Aminosilanen zur Reaktion gebracht werden. Es ist jedoch auch möglich hydroxyterminierte Polyether, Polyurethane oder Polyester mit Isocyanatosilanen zu STOP umzusetzen. Die physikalischen Eigenschaften der resultierenden STOP werden dabei maßgeblich durch das Basispolymer vorgegeben, wodurch mannigfaltige Variationsmöglichkeiten eröffnet werden.
- Häufig werden in Dichtungs-, Beschichtungs- und Klebemassen auch über Alkoxygruppen vernetzende RTV-1-Silikonkautschuke (Alkoxy-Silikone) als vernetzende Bestandteile eingesetzt. Erhältlich sind diese im allgemeinen durch Umsetzung eines Polydimethylsiloxans mit endständigen OH-Gruppen mit Trialkoxyorganosilanen (Vernetzen) in Gegenwart von Katalysatoren. Die physikalischen Eigenschaften der resultierenden Alkoxy-Silikone werden dabei maßgeblich durch das Basissilikon und den gewählten Vernetzen vorgegeben, wodurch auch hier mannigfaltige Variationsmöglichkeiten eröffnet werden.
- Eine Vernetzung beziehungsweise Härtung der STOP und Alkoxy-Silikone erfolgt üblicherweise basenkatalysiert in Gegenwart von Feuchtigkeit, insbesondere in Gegenwart von Luftfeuchtigkeit, durch hydrolytische Abspaltung der silanständigen Alkoxygruppen unter Freisetzung von Alkanolen und einer damit einhergehenden Quervernetzung der Polymere. Da bereits Luftfeuchtigkeit ausreicht die katalysatorhaltigen Zusammensetzungen auszuhärten, ist zwar üblicherweise eine schnelle Aushärtung gewährleistet, diese geht jedoch zu Lasten einer begrenzten Verarbeitungsdauer. Es werden kurze Hautbildungszeiten erreicht, was jedoch beispielsweise bei Verfugungsarbeiten dazu führt, daß nur kleine Bereiche gleichmäßig verfugt werden können, da ein Glätten bereits in der Verfestigung befindlicher Fugen ein Aufreißen der bereits gebildeten Haut mit sich bringt und somit ungleichmäßige Ergebnisse erhalten werden.
- Um ein zu schnelles Aushärten zu vermeiden muß daher auf weniger reaktive Systeme ausgewichen werden. So besteht zum Beispiel die Möglichkeit die Basenkonzentration herabzusetzen, das heißt die Katalysatormenge zu reduzieren. Auch ist es möglich die Polymere selbst weniger reaktiv zu gestalten, indem schwächer reaktive organofunktionelle Silane als Endgruppen bzw. Vernetzer angebracht werden.
- Zu den schwächer reaktiven Silanen gehören beispielsweise die sogenannten γ-Silane, bei welchen die organofunktionellen Gruppen durch einen Propylenspacer vom Siliciumatom getrennt sind. Um jedoch eine zufriedenstellende Reaktivität der γ-Silane zu gewährleisten, sind fast ausschließlich deren Trialkoxy-Vertreter einsetzbar, wobei als Alkoxygruppe zudem alleine die Methoxygruppe eine vertretbare Reaktivität bei der feuchtigkeitsinduzierten Härtung besitzt.
- Der Einsatz trialkoxylierter γ-Silane ist unter dem Gesichtspunkt des Umweltschutzes nachteilig, da bei der hydrolytischen Quervernetzung eine um 50 % höhere Menge Alkanol freigesetzt wird. Die Tendenz geht daher in Richtung einer Begrenzung der Freisetzung flüchtiger organischer Substanzen. Somit geht die Verwendung weniger reaktiver trialkoxylierter γ-Silane zur Verlängerung der Verarbeitbarkeit mit der Inkaufnahme einer höheren Umweltbelastung einher. Sollte die Umweltbelastung verringert werden, so wäre der Einsatz von beispielsweise dialkoxylierten α-Silanen, die anstelle des Propylenspacers einen Methylenspacer aufweisen, dem von trialkoxylierten γ-Silanen vorzuziehen, wodurch sich jedoch wiederum die Verarbeitbarkeitszeit verkürzt.
- Ferner sind ethoxylierte γ-Silane häufig zu reaktionsträge, weshalb methoxylierte γ-Silane eingesetzt werden, wobei bei der Vernetzung Methanol freigesetzt wird. Aufgrund der Toxizität von Methanol ist der Einsatz von Klebstoffen, Dichtstoffen und ähnlichen Zusammensetzungen, welche trimethoxylierte STOP enthalten, in schlecht belüftbaren Räumen nachteilig.
- Üblicherweise erfordert der Einsatz wenig reaktiver γ-Silane als Endcapper bzw. Vernetzer zusätzlich die Verwendung metallorganischer Verbindungen beim Härtungsprozeß. Besonders zinnorganische Katalysatoren sind aber in jüngerer Zeit aufgrund ihrer toxischen Eigenschaften in Verruf geraten. Darüber hinaus schließt der Einsatz derartiger Katalysatoren in der Regel das Vorliegen von Esterbindungen in diesen Systemen aus, da die Katalysatoren eine Esterspaltung beschleunigen und damit gegebenenfalls Eigenschaftsveränderungen des Systems hervorrufen. Damit ist der Hersteller derartiger Systeme, beispielsweise der Hersteller von Kleb- oder Dichtstoffsystemen für den Heimwerkerbereich, in der Formulierungsfreiheit eingeschränkt.
- Eine der Aufgaben der vorliegenden Erfindung bestand daher darin, die obengeschilderten Nachteile zu überwinden und das Verarbeitungs-Aushärtungs-Dilemma zu lösen. Insbesondere sollte auch der Einsatz reaktiver silyl-terminierter Polymere, wie z.B. α-silyl-terminierter Polymere in Kleb- und Dichtstoffsystemen bei langer Verarbeitbarkeit ermöglicht werden, indem der Zeitpunkt der Härtung in das Ermessen des Anwenders gestellt wird. Günstigerweise sollten die Zusammensetzungen auch eine hervorragende Lagerstabilität besitzen.
- Diese Aufgaben konnten überraschenderweise durch Bereitstellung härtbarer Zusammensetzungen gelöst werden, die mindestens ein silyl-terminiertes Polymer und mindestens eine photolatente Base enthalten, wobei das silyl-terminierte Polymer aus einem linearen oder verzweigten, silangruppenfreien Basispolymer-Rest besteht, welcher endständige Silangruppen trägt. Bevorzugt sind insbesondere silylterminierte organische Polymere und/oder silyl-terminierte Silikonpolymere.
- Die entsprechenden organischen Polymere oder Silikonpolymere enthalten dabei innerhalb ihrer Hauptkette oder Hauptketten keine Silangruppen, sondern nur an den jeweiligen Kettenenden der Polymerketten. So sind in einem linearen Polymer nur die beiden Enden des Polymers silyl-terminiert, ein solches Polymer wird im folgenden als divalent oder zweiwertig bezeichnet. Bei der Verwendung eines beispielsweise Glycerol gestarteten Polymers können sich ausgehend von den drei Hydroxygruppen des Glycerols drei unabhängige silanfreie Polymerstränge bilden, wobei wiederum erst am Strangende eine Silangruppe als Terminus angebracht sein kann. Ein derart verzweigtes Polymer mit drei Enden wird im folgenden als trivalent oder dreiwertig bezeichnet. Analog werden Polymere mit vier Enden als vierwertig bezeichnet.
- Das Fehlen weiterer Silangruppen innerhalb der Basispolymerkette, z.B. als Seitengruppen ermöglicht eine Kontrolle der Vernetzungsdichte, sowie den Erhalt der basispolymerspezifischen Eigenschaften. Bei einer Vernetzung über Seitengruppen könnte beispielsweise die in vielen Fällen gewünschte Elastizität der gehärteten Zusammensetzungen verloren gehen.
- Die in den erfindungsgemäßen, härtbaren Zusammensetzungen enthaltenen silylterminierten organischen Polymere besitzen vorzugsweise die allgemeine Formel (I)
- In Formel (I) stellt A den zwei-, drei- oder vierwertigen Rest eines beliebigen organischen Polymers dar (z = 2 bis 4). Bei den in Frage kommenden organischen Polymeren kann es sich um Polymerisate oder Polykondensate handeln.
- Vorzugsweise handelt es sich bei den organischen Polymeren um Polyurethane, Polyether, Polyester, Polyacrylate, Polyvinylester, Polyolefine oder statistische Copolymere und Blockcopolymere dieser Polymere. Besonders bevorzugt sind Polyurethane, Polyester und Polyether, sowie deren Copolymerisate, wie beispielsweise Polyether-Polyester-Copolymere. Typische gewichtsmittlere Molmassen derartiger Reste A liegen im Bereich von etwa 100 bis 50000 g/mol, vorzugsweise 1000 bis 30000 g/mol. Gängige Viskositäten der Basispolymere liegen beispielsweise im Bereich von 1000 mPa·s bis 700000 mPa·s, vorzugsweise 5000 mPa·s bis 300000 mPa·s.
- Die in Formel (I) dargestellten Reste X können innerhalb eines Moleküls gleich oder unterschiedlich sein und bilden die Brückenglieder zwischen dem zwei-, drei- oder vierwertigen Rest des Basispolymers und der Methylengruppe des endständigen Silylrests. Die Art der Reste X unterliegt keinen bestimmten Beschränkungen. Somit sind sämtliche üblichen aus dem Polymerbereich bekannten Verknüpfungsreaktionen denkbar, um die Reste X zu erzeugen.
- So können endständige am Basispolymer befindliche Gruppen wie beispielweise Hydroxylgruppen, Aminogruppen oder Isocyanatogruppen direkt mit der an der Alkylengruppe des Silans befindlichen organofunktionellen Gruppe zur Reaktion gebracht werden. Die Silane weisen hierbei beispielweise primäre oder sekundäre Aminogruppen, Epoxygruppen oder Isocyanatogruppen auf. Somit können geeignete Reaktionspartner beispielsweise Aminogruppe und Isocyanatogruppe, Hydroxygruppe und Isocyanatogruppe, Hydroxygruppe und Epoxygruppe, Aminogruppe und Epoxygruppe, Thiolgruppe und Isocyanatogruppe, Epoxygruppe und Thiolgruppe, Carbamatgruppe und Aminogruppe oder Acrylatgruppe und Aminogruppe sein.
- Es ist jedoch auch möglich die Reste X indirekt zu erzeugen, indem zum Beispiel ein hydroxyterminiertes Basispolymer, beispielsweise ein hydroxyterminierter Polyether, zunächst mit einer difunktionellen Verbindung, bei welcher wenigstens eine Funktion hydroxyreaktiv ist, zur Reaktion gebracht wird. Eine solche difunktionelle Verbindung kann beispielsweise ein Diisocyanat, wie Hexamethylendiisocyanat, sein. Das Reaktionsprodukt aus hydroxyterminiertem Polyether und Hexamethylendiisocyanat kann dann wiederum mit einem isocyanatreaktiven Silan umgesetzt werden.
- Typische Reste X können beispielsweise -O(CO)NH-, -NH(CO)O-, -NH(CO)NH-, -(CO)(NH)L(NH)(CO)NR3-, -NR3(CO)(NH)L(NH)(CO)-, worin R3 ein aliphatischer, cycloaliphatischer oder aromatischer Rest mit 1 bis 12 Kohlenstoffatomen ist und L ein aliphatischer, cycloaliphatischer oder aromatischer Rest mit 1 bis 12 Kohlenstoffatomen ist, -O(CO)-, -(CO)O-, -SH(CO)NH-, oder -NH(CO)SH-, sein.
- Die Reste R1 und R2 können gleich oder unterschiedlich sein und stellen einen geradkettigen oder verzweigten Alkyl- oder Alkenylrest mit 1 bis 6 Kohlenstoffatomen, zum Beispiel Methyl, Ethyl, n-Propyl, iso-Propyl, iso-Propenyl-, n-Butyl, iso-Butyl, tert.-Butyl, Pentyl oder Hexyl dar. Bevorzugt sind hierbei Methyl und Ethyl aufgrund der höheren Reaktivität der Reste OR2 bei der Hydrolyse. Unter diesen Resten ist aufgrund der Toxizität von Methanol der Ethyl-Rest bevorzugt.
- Die Werte für n können in der obigen Formel (I) für jeden der z Reste unabhängig voneinander 0 oder 1 sein. Im Fall, daß n = 0 ist, handelt es sich um eine endständige trialkoxylierte Silylgruppe. Wenn n hingegen 1 beträgt, liegt eine endständige dialkoxylierte Silylgruppe vor. Je nachdem, ob n = 0 oder 1 ist und z = 2, 3 oder 4 beträgt, stehen 4 bis 12 Alkoxy- oder Alkenyloxygruppen für die hydrolytische Vernetzungsreaktion zur Verfügung.
- Die Werte für u betragen vorzugsweise 1 bis 3, wobei im Fall von u = 1 von α-Silylgruppen die Rede ist und es sich bei u = 3 um γ-Silylgruppen handelt.
- Alternativ zu den zwei-, drei- oder vierwertigen Resten der organischen Polymere können auch zwei bis vierwertige Silikone eingesetzt werden.
- Da die härtbaren Zusammensetzungen vorzugsweise in gehärtetem Zustand noch elastische Eigenschaften besitzen sollen, sind die Silangruppen nur terminal an den Enden der Polymerstränge angebracht. Hierdurch wird ermöglicht die durch die Art der Polymerstränge eingebrachte Charakteristik zu erhalten und die Vernetzungsdichte über die endständigen Silangruppen zu regeln.
- Neben den STOP oder Alkoxy-Silikonen enthalten die erfindungsgemäßen härtbaren Zusammensetzungen sogenannte photolatente Basen. Unter photolatenten Basen sind vorzugsweise organische Basen mit einem oder mehreren basischen Stickstoffatomen zu verstehen, die zunächst in einer blockierten Form vorliegen und erst nach Bestrahlung mit UV-Licht, sichtbarem Licht oder IR-Strahlung durch Spaltung des Moleküls die basische Form freisetzen.
- Die Wahl der photolatenten Base hängt einerseits von den zu vernetzenden silanterminierten Polymeren ab, andererseits von den weiteren fakultativen Bestandteilen der Zusammensetzung. Bedarf die Vernetzungsreaktion einer starken Base, so muß eine photolatente bzw. photolabile Base gewählt werden, die bei Bestrahlung eine starke Base freisetzt. Enthält die Zusammensetzung weitere UV-absorbierende Bestandteile, so wird vorzugsweise eine photolatente Base gewählt, deren Aktivierungswellenlänge, d.h. Wellenlänge der Abspaltung der freien Base, nicht mit den anderen UV-absorbierenden Bestandteilen interferiert.
- Da der Zeitpunkt der Bestrahlung frei gewählt werden kann und somit der Kontakt der härtbaren Polymere mit den freien Basen durch den Anwender bestimmbar ist, kann der Härtungsbeginn ins Belieben und Ermessen des Anwenders gestellt werden.
- Prinzipiell können alle photolatenten Basen als geschützte Katalysatoren eingesetzt werden. So kommen beispielsweise auch o-Nitrobenzyloxycarbonylamine, Benzoincarbamate, α,α-Dimethylbenzoyloxycarbonylamine, Formanilid-Derivate oder O-Acyloxime in Frage. Derartige Verbindungen wurden beispielsweise von Cameron et al. im J. Am. Chem. Soc. 118 (1996) 12925, J. Chem. Soc. Perkin Trans. I (1997) 2429 und J. Org. Chem. 55 (1990) 5919, von Nishikubo et al. im Polym. J. 29 (1997) 450 und Polym. J. 25 (1993) 365, sowie Ito et al. im J. Poly. Sci. Part A: Polym. Chem. 32 (1994) 2177 beschrieben.
- Bevorzugt werden jedoch die erst kürzlich beschriebenen photolatenten tertiären Amine und Amidine. Diese sind beispielsweise in der publizierten internationalen Patentanmeldung WO 03/014226 A1 oder in der Veröffentlichung „New latent amines for the coatings industry" von T. Jung, K. Dietlinker und J. Benkhoff (Farbe & Lack 109 (10/2003) 34) beschrieben.
- Beispiele für geeignete Basen sind unter anderem tertiäre Amine und Amidine, wie Diazabicyclooctan, N-Alkylmorpholine, Tetramethylguanidin (TMG), Diazabicyclononen (DBN), Diazabicycloundecen (DBU) und Imidazol.
- Besonders geeignete Amidine sind photolabile Diazabicyclononane, insbesondere 5-Benzyl-1,5-diazabicyclo[4.3.0]nonan, wobei der 5-Benzyl-Rest auch ein- oder mehrfach substituiert sein kann. Geeignete Substituenten am 5-Benzyl-Rest sind beispielsweise Halogenreste, wie Chlor oder Brom, Alkylreste, wie Methyl, Ethyl, oder Propyl, Nitrilreste, Nitrogruppen, Alkoxygruppen, wie Methoxy oder Ethoxy oder an den 5-Benzylrest ankondensierte aromatische Reste, wobei zum Beispiel aus einem 5-(Benzyl)rest ein 5-(Naphth-2-ylmethyl)rest oder ein 5-(Anthracen-9-yl-methyl)rest ableitbar ist. Auch kann anstelle des 5-Benzylrests beispielsweise ein 5-(Anthrachinon-2-yl-methyl)rest treten. Neben den möglichen Substitutionen am 5-Benzyl-Rest kann auch der Diazacyclononanrest weiter substituiert sein, wie zum Beispiel in 5-Benzyl-2-methyl-1,5-diazabicyclo[4.3.0]nonan.
- Neben den photolabilen Diazabicyclononanen, besteht auch die Möglichkeit photolabile Diazabicycloundecane, wie beispielsweise 8-Benzyl-1,8-diazabicyclo[5.4.0]undecane und dessen Derivate einzusetzen. Der 8-Benzyl-Rest kann analog dem 5-Benzyl-Rest des 5-Benzyl-1,5-diazabicyclo[4.3.0]nonans weiter substituiert sein oder ersetzt werden. Auch hier besteht die Möglichkeit einer weiteren Substitution am Diazabicyclononane-Rest.
- Es können auch photolatente Basen eingesetzt werden, die zwei abspaltbare Basen in einem Molekül enthalten. Ein Vertreter dieser Art ist beispielsweise das 1,4-Bis(1,5-Diazabicyclo[4.3.0]nonanylmethyl)benzol.
- Die Synthese der oben genannten photolatenten Basen ist unter anderem in der WO 03/033500 A1 beschrieben.
- Die erfindungsgemäßen Zusammensetzungen können neben den härtbaren Bestandteilen, d.h. vorzugsweise den STOP und/oder Alkoxy-Silikonen, sowie den photolatenten Basen, noch weitere Additive enthalten. Diese Additive werden in der Regel in wasserfreiem Zustand zugegeben.
- Zu diesen Additiven zählen beispielsweise Photosensibilisatoren, Füllstoffe, wie Calciumcarbonat, Kalkmehl, Glasfasern, Silikate, gefällte Kieselsäuren, pyrogene Kieselsäuren, Zeolithe, Bentonite, gemahlene Mineralstoffe, Talk, Kaolin, Glimmer, Bariumsulfat, Metalloxide und Metallhydroxide, Ruß, Graphit, synthetische Fasern oder Glaskugeln, Glashohlkugeln, Glasfasern, Kunststoffkugeln, PVC-Pulver, Pigmente, Weichmacher, wie beispielsweise Ester der Abietinsäure, Adipinsäure, Azalainsäure, Benzoesäure, Buttersäure, Essigsäure, höhere Fettsäuren, Glykolsäure, Phosphorsäure, Phthalsäure, Propionsäure, Sebacinsäure, Sulfonsäure, Trimellithsäure, Zitronensäure oder reine sowie gemischte Ether monofunktionaler, linearer oder verzweigter C4-C16-Alkohole, Feuchtigkeitsstabilisatoren, wie Isocyanate, Vinylsilane, Carbamatosilane, Flammschutzmittel, Aufheller, rheologiesteuernde Additive, Antistatika, Antimykotika, Verlaufmittel, Emulgatoren, UV-Schutz-Additive, Alterungsschutz-Additive, Tackifier und Reaktivverdünner.
- Unter den angegebenen Additiven sind insbesondere Photosensibilisatoren von Bedeutung, da sich diese positiv auf die Quantenausbeute bei der Photoaktivierung der photolatenten Basen auswirken.
- Beispiele für Photosensibilisatoren sind aromatische Ketone, wie zum Beispiel substituierte und unsubstituierte Benzophenone, Thioxanthone, Anthrachinone oder Farbstoff, wie Oxazine, Acridine, Phenazine und Rhodamine.
- Besonders geeignet sind substituierte Benzophenone und Thioxanthone. Beispiele für geeignete Vertreter der Benzophenone und Thioxanthone sind neben Benzophenon und Thioxanthon selbst, beispielsweise auch 4,4'-Bis(dimethylamino)benzophenon, 4,4'-Bis-(diethylamino)benzophenon, 4,4'-Bis(ethylmethylamino)benzophenon, 4,4'-Diphenylbenzophenon, 4,4'-Diphenoxybenzophenon, 4,4'-Bis(p-isopropylphenoxy)benzophenon, 4-Methylbenzophenon, 2,4,6-Trimethylbenzophenon, 4-Phenylbenzophenon, 2-Methoxycarbonylbenzophenon, 4-Benzoyl-4'-methyldiphenylsulfid, 4-Methoxy-3,3'-methylbenzophenon, Isopropylthioxanthon, Chlorthioxanthon, 1-Chlor-4-propoxythioxanthon, 2,4-Dimethylthioxanthon, 2,4-Diethylthioxanthon und 1,3-Dimethyl-2-(2-ethylhexyloxy)thioxanthon. Auch Mischungen dieser Substanzen sind als Photosensibilisatoren einsetzbar.
- Weitere Photosensibilisatoren sind beispielsweise 3-Acylcoumarine, wie 3-Benzoylcoumarin, 3-Benzoyl-7-methoxycoumarin, 3-Benzoyl-5,7-di(propoxy)coumarin, 3-Benzoyl-6,8-dichlorcoumarin, 3-Benzoyl-6-chlorcoumarin, 3, 3'-carbonylbis[5,7-di(propoxy)coumarin], 3,3'-Carbonylbis(7-methoxycoumarin), 3,3'-carbonylbis(7-diethylaminocoumarin), 3-Isobutyroylcoumarin, 3-Benzoyl-5,7-dimethoxycoumarin, 3-Benzoyl-5,7-diethoxycoumarin, 3-Benzoyl-5,7-dibutoxycoumarin, 3-Benzoyl-5,7-di(methoxyethoxy)coumarin, 3-Benzoyl-5,7-di (allyloxy)coumarin, 3-Benzoyl-7-dimethylaminocoumarin, 3-Benzoyl-7-diethylaminocoumarin, 3-Isobutyroyl-7-dimethylaminocoumarin, 5,7-Dimethoxy-3-(1-naphthoyl)coumarin, 5,7-Dimethoxy-3-(1-naphthoyl)coumarin, 3-Benzoylbenzo[f]coumarin, 7-Diethylamino-3-thienoylcoumarin, 3-(4-Cyanobenzoyl)-5,7-dimethoxycoumarin oder 3-(Aroylmethylen)thiazoline, wie 3-Methyl-2-benzoylmethylennaphthothiazolin, 3-Methyl-2-benzoylmethylenbenzothiazolin, 3-Methyl-2-propionylmethylen-p-naphthothiazolin oder andere Carbonylverbindungen, wie beispielsweise Acetophenon, 3-Methoxyacetophenon, 4-Phenylacetophenon, 2-Acetylnaphthalin, 2-Naphthaldehyd, 9,10-Anthrachinon, 9-Fluorenon, Dibenzosuberon, Xanthon, 2,5-Bis(4-diethylaminobenzyliden)cyclopentanon, 2-(4-Dimethylaminobenzyliden)indan-1-on oder 3-(4-Dimethylaminophenyl)-1-indan-S-yl-propenon, 3-Phenylthiophthalimid oder N-Methyl-3,5-di(ethylthio)phthalimid.
- Die Photosensibilisatoren werden vorzugsweise in Mengen von 0,01 Gew.-% bis 5 Gew.-% bezogen auf das Gesamtgewicht der erfindungsgemäßen Zusammensetzung eingesetzt.
- Die erfindungsgemäße härtbare Zusammensetzung enthält vorzugsweise bis 99 Gew.-%, bevorzugter 5 bis 95 Gew.-%, noch bevorzugter mehr als 10 Gew.-% des silyl-terminierten Polymers bezogen auf die Gesamtzusammensetzung, sowie vorzugsweise 0,01 bis 20 Gew.-%, bevorzugter 1 bis 10 Gew.-% noch bevorzugter 2 bis 5 Gew.-% der photolatenten Base bezogen auf die Gesamtzusammensetzung und gegebenenfalls bis vorzugsweise 10 Gew.-%, bevorzugter bis 5 Gew.-%, noch bevorzugter bis 3 Gew.-% eines Photosensibilisators bezogen auf die Gesamtzusammensetzung. Des weiteren können die oben genannten Additive enthalten sein, worunter vorzugsweise bis 80 Gew.-% Füllstoffe fallen.
- Die vorliegende Erfindung stellt auch ein Verfahren zur Herstellung der erfindungsgemäßen Zusammensetzungen zur Verfügung. Dieses erfindungsgemäße Verfahren umfaßt in einem ersten Schritt die Umsetzung eines Basispolymers mit 2 bis 4 endständigen reaktiven Gruppen mit einem oder mehreren Silanen der allgemeinen Formel:
- Beschränkungen hinsichtlich der verwendbaren Silane gibt es nicht, solange diese mit den endständigen Gruppen des Basispolymers auf eine Weise verbunden werden können, die den Erhalt von mindestens zwei R2O-Gruppen in der Silanendgruppe gewährleistet. Somit sind prinzipiell auch Verbindungen der obigen Formel mit n = 0, unter Verlust einer R2O Gruppe, mit dem Basispolymer zum Beispiel durch Kondensation und Freisetzung von R2OH bei Erhalt der Gruppe W verbindbar. In einem solchen Fall ist sogar die Reaktivität der Gruppe W gegenüber den endständigen reaktiven Gruppen des Basispolymers entbehrlich.
- Zu den im erfindungsgemäßen Verfahren bevorzugt einsetzbaren α-Silanen gehören unter anderem (N-Cyclohexylaminomethyl)methyl-diethoxysilan, (N-Cyclohexylamino-methyl)triethoxysilan, (N-Phenylaminomethyl)methyldimethoxysilan, (N-Phenylamino-methyl)trimethoxysilan, (Methacryloxymethyl)methyldimethoxysilan, (Methacryloxymethyl)trimethoxysilan, (Methacryloxymethyl)methyldiethoxysilan, (Methacryloxymethyl)triethoxysilan, (Isocyanatomethyl)methyldimethoxysilan, (Isocyanatomethyl)trimethoxysilan, oder N-(Trimethoxysilylmethyl)-O-methylcarbamat. Diese Produkte sind beispielsweise von Wacker, Burghausen, Deutschland unter der Handelsbezeichnung GENIOSIL® XL erhältlich.
- Es können jedoch auch reaktionsträgere Silane, wie beispielsweise γ-Silane eingesetzt werden. Beispiele für entsprechende γ-Silane sind N-(2-Aminoethyl)(3-aminopropyl)trimethoxysilan, (3-Aminopropyl)triethoxysilan, (3-Methacryloxypropyl)trimethoxysilan, (3-Isocyanatopropyl)trimethoxysilan, (3-Glycidoxypropyl)trimethoxysilan, (3-Glycidoxypropyl)triethoxysilan oder (3-Triethoxysilyl)propylbernsteinsäureanhydrid. Diese Produkte sind beispielsweise von Wacker, Burghausen, Deutschland unter der Handelsbezeichnung GENIOSIL® GF erhältlich.
- Bevorzugt werden jedoch die α-Silane aufgrund ihrer höheren Reaktivität bei der Basen katalysierten Vernetzung eingesetzt. Bei der Verwendung von γ-Silanen kann es unter Umständen nötig sein weitere vernetzungsübliche Katalysatoren zuzugeben. Solche konventionellen Katalysatoren können dann jedoch üblicherweise in geringerer Menge eingesetzt werden, da eine Ergänzung durch die aus den photolabilen Basen freigesetzten Basen erfolgt.
- Die in den erfindungsgemäßen Zusammensetzungen enthaltenen photolatenten Basen können je nach Substitutionsmuster über einen weiten Wellenlängenbereich hinweg, der vom Ultraviolett-Bereich über sichtbares Licht in den Infrarot-Bereich reicht, gespalten werden. Die gewählte Wellenlänge hängt hierbei von der Aktivierungswellenlänge der verwendeten photolatenten Base ab. Bevorzugte Wellenlängen liegen im Bereich von etwa 200 bis 700 nm. Sowohl Sonnenlicht als auch Kunstlicht sind zur Bestrahlungshärtung, beziehungsweise Freisetzung der aktiven Basenform geeignet. Es können sowohl punktförmige Lichtquellen als auch flächige Lichtquellen eingesetzt werden. Beispielhaft seien als Lichtquellen Kohlebogenlampen, Xenonbogenlampen, Quecksilberlampen niedrigen, mittleren und hohen Drucks, Metallhalogenlampen, mikrowellenstimulierte Metalldampflampen, Excimerenlampen, superactinische Fluoreszenzröhren, Fluoreszenzlampen, Glühlampen, Elektronenblitzröhren, Xenonblitzröhren, Scheinwerfer, Elektronen- und Röntgenstrahlen emittierende Röhren, Leuchtdioden und Laserlichtquellen genannt. Der Bestrahlungsabstand mit Kunstlichtquellen kann von 1 cm bis zu 2 m oder mehr betragen, je nach Stärke der Lichtquelle und Empfindlichkeit der photolatenten Basen.
- Des weiteren betrifft die vorliegende Erfindung die Verwendung der erfindungsgemäßen Zusammensetzungen als lagerstabile Klebstoffe, Dichtungsmassen, Spachtelmassen und Montageschaumstoffe.
- Die Erfindung betrifft darüber hinaus die Verwendung der erfindungsgemäßen Zusammensetzungen in einem Verfahren, umfassend die Schritte:
- (A) Aufbringen der Zusammensetzung auf ein Substrat,
- (B) Einwirkenlassen von Feuchtigkeit auf die Zusammensetzung und
- (C) Bestrahlen der Zusammensetzung mit Strahlung einer Wellenlänge, bei welcher die photolatente Base in ihre freie Form überführt wird.
- Hierbei kann Feuchtigkeit bereits während des Aufbringens der Zusammensetzung auf das Substrat einwirken, d.h. Schritt (B) zugleich mit Schritt (A) erfolgen. Es ist jedoch auch denkbar Schritt (A) unter Ausschluß von Feuchtigkeit durchzuführen und dann mit Schritt (B) und Schritt (C) in beliebiger Reihenfolge fortzufahren. Es kann jedoch auch, während noch Feuchtigkeit auf das aufgebrachte Substrat einwirkt, mit der Bestrahlung begonnen werden, so daß (A), (B) und (C) quasi gleichzeitig ablaufen oder in beliebiger Weise miteinander verschachtelt sind.
- Als Substrate eignen sich insbesondere Metalle, beispielsweise Aluminium, Stahl, Kupfer oder Legierungen und dergleichen, Polymere, wie beispielsweise Polyacrylate, Polymethacrylate, Polyvinylchloride, Polycarbonate und dergleichen, keramische und mineralische Materialien, Glas, Marmor, Beton, Zement, Granit, Sandstein, Spiegel, Holz, Leder, Textilien, Papier, Pappe, Gummi und dergleichen.
- Im folgenden wird die Erfindung durch Beispiele veranschaulicht. Diese sollen lediglich der Erläuterung dienen und nicht als beschränkend erachtet werden.
- Polymer 1: Herstellung eines α-silanterminierten Polymeren (Zinn-Katalyse)
- 282 g (15 mmol) Polypropylenglykol 18000 (Hydroxyzahl = 6,0) wurden in einem 500 ml Dreihalskolben bei 100°C im Vakuum getrocknet. Unter Stickstoffatmosphäre wurde bei 80°C 0,06 g (0,02%) Dibutylzinnlaurat hinzugegeben und anschließend mit 5,5 g (34 mmol) Isocyanatomethyldimethoxymethylsilan (%NCO = 25,7) versetzt. Nach einstündigem Rühren bei 80°C wurde das entstandene Polymer abgekühlt und mit 6 g Carbamatomethyltrimethoxysilan versetzt. Das klare, farblose Produkt wurde feuchtigkeitsdicht unter Stickstoffatmosphäre in einem Glasgefäß gelagert.
- Polymer 2: Herstellung eines α-silanterminierten Polymeren (Titan-Katalyse)
- 282 g (15 mmol) Polypropylenglykol 18000 (Hydroxyzahl = 6,0) wurden in einem 500 ml Dreihalskolben bei 100°C im Vakuum getrocknet. Unter Stickstoffatmosphäre wurde bei 80°C 0,14g (0,05%) Titantetraisopropylat hinzugegeben und anschließend mit 5,5 g (34 mmol) Isocyanatomethyldimethoxymethylsilan (%NCO = 25,7) versetzt. Nach einstündigem Rühren bei 80°C wurde das entstandene Polymer abgekühlt und mit 6 g Carbamatomethyltrimethoxysilan versetzt. Das klare, orangegelbe Produkt wurde feuchtigkeitsdicht unter Stickstoffatmosphäre in einem Glasgefäß gelagert.
- PL-DBN: Herstellung der photolatenten Base 5-Benzyl-1,5-diazabicyclo[4.3.0]nonan
- 5-Benzyl-1,5-diazabicyclo[4.3.0]nonan (PL-DBN) wurde entsprechend WO 03/033500 A1 aus Benzylchlorid und 1,5-diazabicyclo[4.3.0]nonan hergestellt:
Hierzu wurden 63 g (0,5 mol) 1,5-diazabicyclo[4.3.0]non-5-en (DBN) in 1000 ml tert. Butylmethylether gelöst und anschließend portionsweise mit 9,5 g (0,25 mol) Lithiumaluminiumhydrid bei Raumtemperatur versetzt. Anschließend wird drei Stunden unter Rückfluß erhitzt. Bei 0°C wurde vorsichtig mit 10 ml Wasser und anschließend 10 ml 10%iger NaOH-Lösung versetzt. Es wurden weitere 25 ml Wasser zugegeben und anschließend der Niederschlag abfiltriert. Die organische Phase wurde abgetrennt, getrocknet und das Lösungsmittel abdestilliert. Das Rohprodukt wurde im Vakuum destilliert. - 42 g (1 mol) Natriumhydroxid und 11,6 g (0,07 mol) Kaliumjodid wurden in 700 ml Methylenchlorid suspendiert. Es wurden 88,6 g (0,7 mol) Benzylchlorid und 89,4 g (0,7 mol) 1,5-diazabicyclo[4.3.0]nonan zugegeben und 48 h bei Raumtemperatur gerührt. Es wurden 400 ml Wasser zugegeben und die organische Phase abgetrennt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt und zum resultierenden Öl wurden 1000 ml Hexan gegeben. Die ausgefallenen Salze wurden abfiltriert und das organische Lösungsmittel entfernt. Das Rohprodukt wurde im Vakuum destilliert. Das Endprodukt kristallisierte beim Abkühlen aus.
- Herstellung von Zusammensetzungen und Katalyseversuche:
- Die Polymere 1 und 2 wurden mit verschiedenen Katalysatoren versetzt und anschließend in einem 1 mm dicken Film auf eine Glasplatte aufgetragen. Ein Teil der Proben wurde im Dunkeln belassen und ein anderer Teil nach 15 Minuten mit einer UV Lampe (Typ: Loctite 97034, 200W Quecksilber-Höchstdrucklampe, 220–500 nm) für 90 Sekunden bestrahlt. Es wurden die Hautbildungszeit (Skin over time/SOT) sowie die Zeit zur Bildung einer klebfreien Schicht (Tack free time/TFT) bestimmt.
- Zur Bestimmung der Lagerstabilität wurden die Proben in einem geschlossenen Behälter für 7 Tage gelagert und die Viskositätserhöhung gemessen. Tabelle 1Silan GF96 = Aminopropyltrimethoxysilan (Haftvermittler und Cokatalysator)
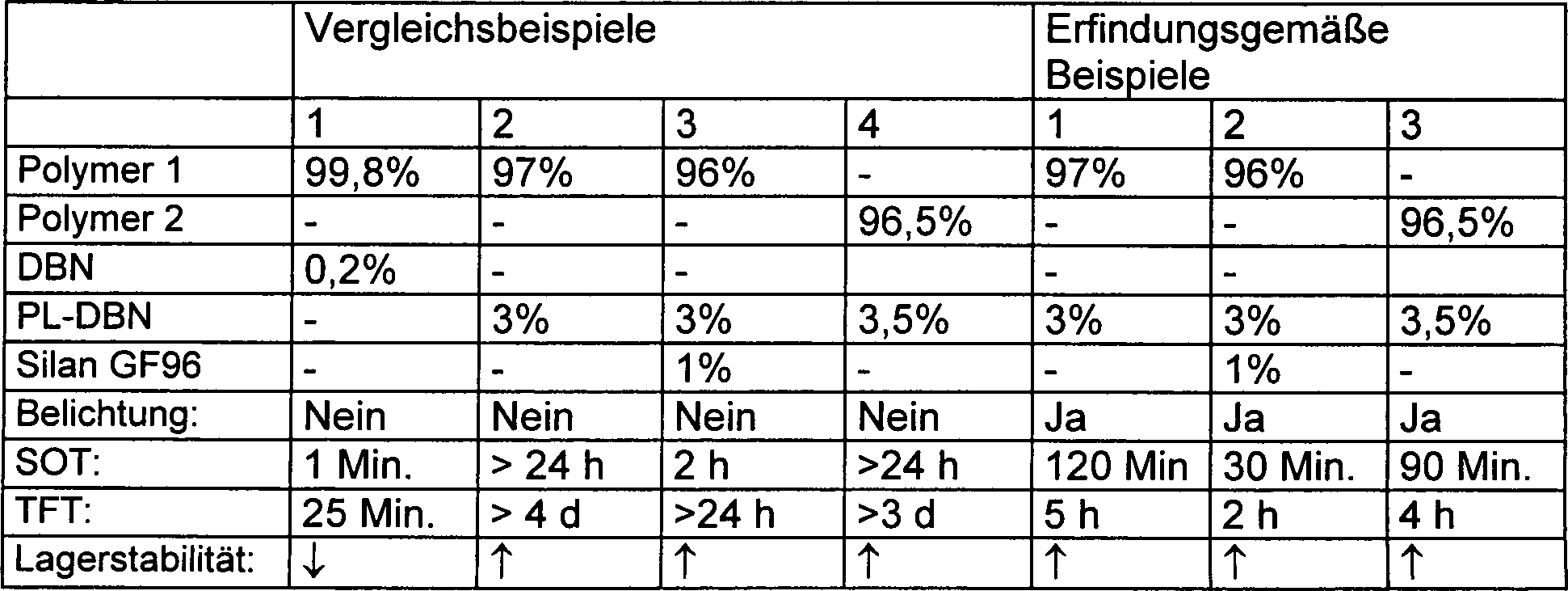
- Vergleichsbeispiel 1 enthält neben Polymer 1 auch 0,2 Gew.-% freies 1,5-diazabicyclo[4.3.0]nonan (DBN) als Base. Hierbei kommt es bereits nach 1 min zur unerwünscht schnellen Hautbildung, wodurch der Verarbeitbarkeitszeitraum für praktische Anwendungen viel zu kurz ist.
- Hinsichtlich ihrer Zusammensetzung direkt vergleichbar sind Vergleichsbeispiele 2, 3 und 4 mit den erfindungsgemäßen Beispielen 1, 2 und 3. Einziger Unterschied ist, daß die erfindungsgemäßen Zusammensetzungen einer Belichtung unterzogen wurden, wodurch die photolatente Base (PL-DBN) in die freie Basenform überführt wurde. Diese Beispiele belegen klar die erhöhte Lagerstabilität der Zusammensetzungen gegenüber Vergleichsbeispiel 1, sowie die Wirkung der Bestrahlung bei der Freisetzung der freien Base als Härtungskatalysator.
- Polymer 3: Herstellung eines α- und γ-silanterminierten Polymeren
- 155,1 g (19 mmol) Polypropylenglykol 8000 (Hydroxyzahl = 14,0) wurden in einem 500 ml Dreihalskolben bei 100°C im Vakuum getrocknet. Unter Stickstoffatmosphäre wurde bei 80°C 0,06 g Dibutylzinnlaurat hinzugegeben und anschließend mit 15,3 g (87 mmol) Toluendiisocyanat (TDI) (%NCO = 47,8) versetzt. Nach einstündigem Rühren bei 80°C wurde das entstandene Polymer mit 103,4 g (105 mmol) Polytetrahydrofuran (PolyTHF) 1000 (Hydroxyzahl = 114) versetzt und eine weitere Stunde bei 80°C gerührt. Es wurde mit einer Mischung aus 10,2 g (45 mmol) Isocyanatopropyltrimethoxysilan (%NCO = 18,3) und 5,5 g (34 mmol) Isocyanatomethyldimethoxymethylsilan (%NCO = 25,7) versetzt und eine weitere Stunde bei 80°C gerührt. Das Polymer wurde abgekühlt und mit 6 g Vinyltrimethoxysilan versetzt. Das Produkt wurde feuchtigkeitsdicht unter Stickstoffatmosphäre in einem Glasgefäß gelagert.
- Herstellung von Zusammensetzungen und Katalyseversuche
- Das Polymer 3 wurde mit verschiedenen Katalysatoren/Photosensibilatoren versetzt und anschließend in einem 1 mm dicken Film auf eine Glasplatte aufgetragen. Ein Teil der Proben wurde im Dunkeln belassen und ein anderer Teil nach 15 Minuten mit einer LED Lampe für 5 Minuten bestrahlt. Es wurden die Hautbildungszeit (Skin over time/SOT) sowie die Zeit zur Bildung einer klebfreien Schicht (Tack free time/TFT) wie oben bestimmt. Tabelle 2Genocure ITX: Thioxanthone (Photo-Sensibilisator)
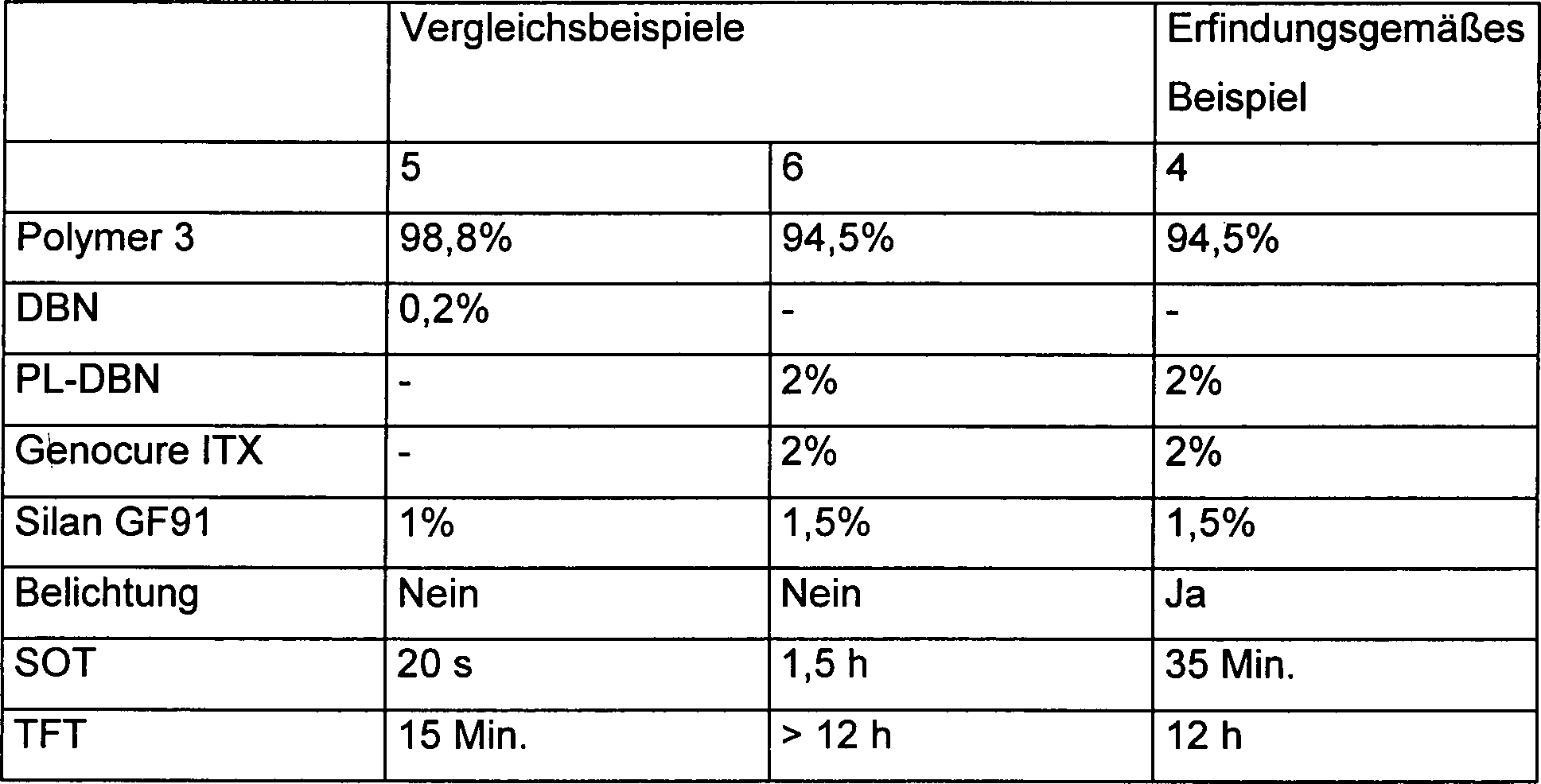
Silan GF91 = N-Aminoethyl-3-aminopropyltrimethoxysilan (Haftvermittler und Cokatalysator) - Wie Vergleichsbeispiel 5 zeigt bewirkt die direkte Zugabe einer freien Base (DBN) die quasi sofortige Hautbildung (20 s). In Gegenwart einer photolatenten Base (PL-DBN) findet ohne Belichtung selbst in einer Zusammensetzung, welche weitere Haftvermittler und Cokatalysatoren enthält nur eine langsame Hautbildung statt, die mit einer langen TFT einhergeht (Vergleichsbeispiel 6). Das erfindungsgemäße Beispiel 4 belegt wiederum die vorteilhafte Wirkung der Bestrahlung auf die erfindungsgemäßen Zusammensetzungen, was sich in einer verkürzten SOT und TFT zeigt.
- Alkoxy-Silikon 1: Herstellung eines Alkoxy-Silikons (Aceton-System):
- 75 g α,ω-Bishydroxy-Polydimethylsiloxan (Viskosität 20000 mPa·s/23°C) wurden bei 35°C mit 3,9 g Vinyltriisopropenoxysilan und 0,05 g Tetramethylguanidine versetzt und für 30 Minuten gerührt. Bei Raumtemperatur wurden 20 g einer hydrophoben pyrogenen Kieselsäure eingemischt und anschließend mit 3 g 5-Benzyl-1,5-diazabicyclo[4.3.0]nonan (PL-DBN) versetzt. Mittels Vakuum wurde entstandenes Aceton entfernt und die Masse entgast. Die Masse war in einem geschlossenen Behälter auch nach mehrmaligem Öffnen über Monate stabil.
- Alkoxy-Silikon 2: Herstellung eines Alkoxy-Silikons (Methoxy-System):
- 60 g α,ω-Bishydroxy-Polydimethylsiloxan (Viskosität 20000 mPa·s/23°C) wurden bei 35°C mit 4 g Vinyltrimethoxysilan versetzt und für 30 Minuten gerührt. Es wurde 0,01 g Butyllithium zugegeben und 60 Min. bei 50°C unter Stickstoffatmosphäre und Vakuum gerührt. Bei Raumtemperatur wurden 35 g vorgetrocknetes CaCO3 eingemischt und anschließend mit 3 g 5-Benzyl-1,5-diazabicyclo[4.3.0]nonan (PL-DBN) versetzt. Mittels Vakuum wurde die Masse entgast. Die Masse war in einem geschlossenen Behälter auch nach mehrmaligem Öffnen über Monate stabil.
- Aushärtung der Alkoxy-Silikone
- Die Alkoxy-Silikone 1 und 2 wurden in 1 mm dicken Filmen auf eine Glasplatte aufgetragen. Ein Teil der Proben wurde im Dunkeln belassen und ein anderer Teil nach 15 Minuten mit einer UV Lampe (Typ: Loctite 97034, 200W Quecksilber-Höchstdrucklampe, 220–500 nm) für 60 Sekunden bestrahlt. Es wurden die Hautbildungszeit (Skin over time/SOT) sowie die Zeit zur Bildung einer klebfreien Schicht (Tack free time/TFT) wie oben bestimmt. Tabelle 3DBU: 1,8-diazabicyclo[5.4.0]undecen

Claims (13)
- Härtbare Zusammensetzung enthaltend (i) mindestens ein silyl-terminiertes Polymer, und (ii) mindestens eine photolatente Base, wobei das silyl-terminierte Polymer aus einem linearen oder verzweigten, silangruppenfreien Basispolymer-Rest besteht, welcher endständige Silangruppen trägt.
- Härtbare Zusammensetzung gemäß Anspruch 1, dadurch gekennzeichnet, daß das silyl terminierte Polymer ein silyl terminiertes organisches Polymer der allgemeinen Formel (I)
- Härtbare Zusammensetzung gemäß Anspruch 1, dadurch gekennzeichnet, daß das silyl terminierte Polymer ein Alkoxy-Silikon ist.
- Härtbare Zusammensetzung gemäß Anspruch 1 oder 2, dadurch gekennzeichnet, daß A ein zwei-, drei- oder vierwertige Rest eines organischen Polymers gewählt aus der Gruppe bestehend aus „Polyurethan, Polyether, Polyester, Polyacrylat, Polyvinylester und Polyolefine oder einem Copolymer aus zwei oder mehreren dieser Polymere" ist.
- Härtbare Zusammensetzung gemäß einem der Ansprüche 1 bis 3, dadurch gekennzeichnet, daß die photolatente Base ein photolatentes an 5-Position substituiertes 1,5-Diazabicyclo[4.3.0]nonan oder eine photolatentes an 8-Position substituiertes 1,8-Diazabicyclo[5.4.0]undecan ist.
- Härtbare Zusammensetzung gemäß Anspruch 5, wobei die 5-Position des substituierten 1,5-Diazabicyclo[4.3.0]nonans oder die 8-Position des substituierten 1,8-Diazabicyclo[5.4.0]undecans einen Rest, gewählt aus der Gruppe bestehend aus „Benzyl, gegebenenfalls ein- oder mehrfach substituiert mit Halogen, Alkyl, Nitrit, Nitro, Alkoxy; Naphth-2-ylmethyl; Anthracen-9-yl-methyl; und Anthrachinon-2-yl-methyl" trägt.
- Härtbare Zusammensetzung gemäß einem der vorangegangenen Ansprüche, wobei die photolatente Base durch Bestrahlung mit einer Strahlung der Wellenlänge von 200 nm bis 700 nm die freie Base abspaltet.
- Härtbare Zusammensetzung gemäß einem der vorangegangenen Ansprüche, wobei 5 bis 98 Gew.-% mindestens eines silyl terminierten Polymers, und 0,01 bis 20 Gew.-% mindestens einer photolatenten Base, sowie bis 5 Gew.-% eines oder mehrerer Photosensibilisatoren, vorzugsweise gewählt aus der Gruppe bestehend aus „Benzophenonen, Thioxanthonen und Anthrachinonen", und bis 80 Gew.-% eines oder mehrerer Füllstoffe, enthalten sind.
- Härtbare Zusammensetzung gemäß einem der Ansprüche 1, 2 oder 4 bis 8, wobei u = 1 ist.
- Härtbare Zusammensetzung gemäß einem der Ansprüche 1, 2 oder 4 bis 9, wobei das gewichtsmittlere Molekulargewicht des Rests A zwischen 100 und 50000 g/mol beträgt.
- Verfahren zur Herstellung der erfindungsgemäßen Zusammensetzungen, welches (a) einen ersten Schritt, in welchem ein organisches Basispolymer mit endständigen reaktiven Gruppen mit einem oder mehreren Silanen der allgemeinen Formel:
- Verwendung der härtbaren Zusammensetzung aus einem der Ansprüche 1 bis 8 oder erhältlich nach dem Verfahren gemäß Anspruch 9 als Klebstoff, Dichtmasse, Spachtelmasse oder Montagemasse.
- Verwendung der härtbaren Zusammensetzung aus einem der Ansprüche 1 bis 8 oder nach dem Verfahren gemäß Anspruch 9 in einem Verfahren, umfassend die Schritte: (A) Aufbringen der Zusammensetzung auf ein Substrat, (B) Einwirkenlassen von Feuchtigkeit auf die Zusammensetzung und (C) Bestrahlen der Zusammensetzung mit Strahlung einer Wellenlänge, bei welcher die photolatente Base in ihre freie Form überführt wird, wobei die Reihenfolge der Schritte (B) und (C), sowie deren Startzeitpunkte frei wählbar sind.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE200410018548 DE102004018548A1 (de) | 2004-04-14 | 2004-04-14 | Durch Strahlung und Feuchtigkeit härtende Zusammensetzungen auf Basis Silan-terminierter Polymere, deren Herstellung und Verwendung |
| PCT/EP2005/003706 WO2005100482A1 (de) | 2004-04-14 | 2005-04-08 | Durch strahlung und feuchtigkeit härtende zusammensetzungen auf basis silan-terminierter polymere, deren herstellung und verwendung |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE200410018548 DE102004018548A1 (de) | 2004-04-14 | 2004-04-14 | Durch Strahlung und Feuchtigkeit härtende Zusammensetzungen auf Basis Silan-terminierter Polymere, deren Herstellung und Verwendung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| DE102004018548A1 true DE102004018548A1 (de) | 2005-11-10 |
Family
ID=34963803
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE200410018548 Ceased DE102004018548A1 (de) | 2004-04-14 | 2004-04-14 | Durch Strahlung und Feuchtigkeit härtende Zusammensetzungen auf Basis Silan-terminierter Polymere, deren Herstellung und Verwendung |
Country Status (2)
| Country | Link |
|---|---|
| DE (1) | DE102004018548A1 (de) |
| WO (1) | WO2005100482A1 (de) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102007038030A1 (de) * | 2007-08-10 | 2009-02-19 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzungen aus Dimethoxysilanen |
| DE102008020980A1 (de) * | 2008-04-25 | 2009-10-29 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzungen enthaltend silylierte Polyurethane auf Basis von Polyetherblockpolymeren |
| DE102008043218A1 (de) | 2008-09-24 | 2010-04-01 | Evonik Goldschmidt Gmbh | Polymere Werkstoffe sowie daraus bestehende Kleber- und Beschichtungsmittel auf Basis multialkoxysilylfunktioneller Präpolymerer |
| EP2194086A1 (de) | 2008-12-05 | 2010-06-09 | Evonik Goldschmidt GmbH | Verfahren zur Modifizierung von Oberflächen |
| DE102009028640A1 (de) | 2009-08-19 | 2011-02-24 | Evonik Goldschmidt Gmbh | Härtbare Masse enthaltend Urethangruppen aufweisende silylierte Polymere und deren Verwendung in Dicht- und Klebstoffen, Binde- und/oder Oberflächenmodifizierungsmitteln |
| WO2018069530A1 (en) | 2016-10-14 | 2018-04-19 | Basf Se | Stabilizer composition |
| WO2018069526A1 (en) | 2016-10-14 | 2018-04-19 | Basf Se | Hardenable polymer composition |
| WO2020148211A1 (en) | 2019-01-16 | 2020-07-23 | Basf Se | Stabilizer composition for sealants and adhesives |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050234208A1 (en) | 2004-04-14 | 2005-10-20 | Matthias Koch | Fast curing polydiorganosiloxanes |
| DE102007000833A1 (de) | 2007-10-08 | 2009-04-09 | Wacker Chemie Ag | Silan-substituierte RAFT-Reagenzien und Silan-vernetzbare Polymere |
| DE102009022631A1 (de) | 2009-05-25 | 2010-12-16 | Evonik Goldschmidt Gmbh | Härtbare Silylgruppen enthaltende Zusammensetzungen und deren Verwendung |
| DE102009022630A1 (de) | 2009-05-25 | 2010-12-02 | Evonik Goldschmidt Gmbh | Emulsionen auf Basis Silylgruppen tragender Hydroxylverbindungen |
| DE102009034607A1 (de) | 2009-07-24 | 2011-01-27 | Evonik Goldschmidt Gmbh | Neuartige Siliconpolyethercopolymere und Verfahren zu deren Herstellung |
| DE102009028636A1 (de) | 2009-08-19 | 2011-02-24 | Evonik Goldschmidt Gmbh | Neuartige Urethangruppen enthaltende silylierte Präpolymere und Verfahren zu deren Herstellung |
| DE102010001528A1 (de) | 2010-02-03 | 2011-08-04 | Evonik Goldschmidt GmbH, 45127 | Neue Partikel und Kompositpartikel, deren Verwendungen und ein neues Verfahren zu deren Herstellung aus Alkoxysilylgruppen tragenden Alkoxylierungsprodukten |
| DE102010038774A1 (de) | 2010-08-02 | 2012-02-02 | Evonik Goldschmidt Gmbh | Modifizierte Alkoxylierungsprodukte, die zumindest eine nicht-terminale Alkoxysilylgruppe aufweisen, mit erhöhter Lagerstabilität und erhöhter Dehnbarkeit der unter deren Verwendung hergestellten Polymere |
| DE102010038768A1 (de) | 2010-08-02 | 2012-02-02 | Evonik Goldschmidt Gmbh | Modifizierte Alkoxylierungsprodukte mit mindestens einer nicht-terminalen Alkoxysilylgruppe mit erhöhter Lagerstabilität und erhöhter Dehnbarkeit der unter deren Verwendung hergestellten Polymere |
| DE102010041978A1 (de) | 2010-10-05 | 2012-04-05 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzung mit speziellem Katalysator/Weichmacher-System |
| US9616460B2 (en) | 2011-12-02 | 2017-04-11 | Toyota Motor Engineering & Manufacturing North America, Inc. | Terminate-on-demand cationic polymerization method for forming a two-dimensional coating |
| DE102012203737A1 (de) | 2012-03-09 | 2013-09-12 | Evonik Goldschmidt Gmbh | Modifizierte Alkoxylierungsprodukte, die zumindest eine nicht-terminale Alkoxysilylgruppe aufweisen und mehrere Urethangruppen enthalten und deren Verwendung |
| DE102013213655A1 (de) | 2013-07-12 | 2015-01-15 | Evonik Industries Ag | Härtbare Silylgruppen enthaltende Zusammensetzungen mit verbesserter Lagerstabilität |
| DE102013216777A1 (de) | 2013-08-23 | 2015-02-26 | Evonik Industries Ag | Bei Raumtemperatur härtbare Silikonharz-Zusammensetzungen |
| DE102013216751A1 (de) | 2013-08-23 | 2015-02-26 | Evonik Industries Ag | Modifizierte Alkoxylierungsprodukte, die Alkoxysilylgruppen aufweisen und Urethangruppen enthalten und deren Verwendung |
| DE102013216787A1 (de) | 2013-08-23 | 2015-02-26 | Evonik Degussa Gmbh | Guanidingruppen aufweisende semi-organische Siliciumgruppen enthaltende Verbindungen |
| DE102013216781A1 (de) | 2013-08-23 | 2015-02-26 | Evonik Industries Ag | Beschichtungsmassen |
| DE102013218981A1 (de) | 2013-09-20 | 2015-03-26 | Evonik Industries Ag | Raumtemperaturhärtendes Silikon-Polyester-Bindemittel |
| DE102013218976A1 (de) | 2013-09-20 | 2015-04-16 | Evonik Industries Ag | Hydroxylgruppenhaltiges Silikon-Polyester-Acrylat-Bindemittel |
| EP3380542A1 (de) | 2015-11-26 | 2018-10-03 | Evonik Degussa GmbH | Bindemittelsysteme enthaltend alkoxysilylgruppen tragende präpolymere und epoxidverbindungen sowie deren verwendung |
| DE102016111590A1 (de) | 2016-06-24 | 2017-12-28 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Einkomponentenmasse auf Basis von Alkoxysilanen und Verfahren zum Fügen oder Vergießen von Bauteilen unter Verwendung der Masse |
| EP3461864B1 (de) | 2017-09-28 | 2025-10-29 | Evonik Operations GmbH | Härtbare zusammensetzung auf basis von polysiloxanen |
| EP3470475B1 (de) | 2017-10-13 | 2021-01-27 | Evonik Operations GmbH | Härtbare zusammensetzung für beschichtungen mit anti-adhäsiver eigenschaft |
| JP7698427B2 (ja) | 2020-02-18 | 2025-06-25 | エボニック オペレーションズ ゲーエムベーハー | 抗菌性を有するコーティングを作製するための組成物 |
| EP3929253A1 (de) | 2020-06-26 | 2021-12-29 | Evonik Operations GmbH | Zusammensetzung zur herstellung von beschichtungen mit antimikrobieller eigenschaft |
| EP4074784A1 (de) | 2021-04-13 | 2022-10-19 | Evonik Operations GmbH | Zusammensetzung zur herstellung von beschichtungen mit verbesserten leuchtstoffen |
| JP2022183014A (ja) | 2021-05-27 | 2022-12-08 | エボニック オペレーションズ ゲーエムベーハー | 有機官能性修飾ポリブタジエンに基づくユニバーサル接着促進剤 |
| JP2022183009A (ja) | 2021-05-27 | 2022-12-08 | エボニック オペレーションズ ゲーエムベーハー | ポリエステル-ポリエーテル修飾ポリブタジエンおよびその製造方法 |
| JP2023138394A (ja) | 2022-03-17 | 2023-10-02 | エボニック オペレーションズ ゲーエムベーハー | フラックス処理アップコンバージョン蛍リン光体の調製方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0966503B1 (de) * | 1997-03-14 | 2003-05-28 | Minnesota Mining And Manufacturing Company | Auf-anfrage-härtung von feuchtigkeithärtbaren zusammensetzungen mit reaktiven funktionellen silangruppen |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9210653D0 (en) * | 1992-05-19 | 1992-07-01 | Ici Plc | Silane functional oligomer |
| DE10139132A1 (de) * | 2001-08-09 | 2003-02-27 | Consortium Elektrochem Ind | Alkoxyvernetzende einkomponentige feuchtigkeitshärtende Massen |
| DE10317881A1 (de) * | 2003-04-17 | 2004-11-11 | Consortium für elektrochemische Industrie GmbH | Isocyanatfreie schäumbare Mischungen mit verbessertem Brandverhalten |
-
2004
- 2004-04-14 DE DE200410018548 patent/DE102004018548A1/de not_active Ceased
-
2005
- 2005-04-08 WO PCT/EP2005/003706 patent/WO2005100482A1/de not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0966503B1 (de) * | 1997-03-14 | 2003-05-28 | Minnesota Mining And Manufacturing Company | Auf-anfrage-härtung von feuchtigkeithärtbaren zusammensetzungen mit reaktiven funktionellen silangruppen |
Non-Patent Citations (1)
| Title |
|---|
| Encyclopedia of Polymer Science and Engineering, 2. Ed, Vol. 15, S. 252 * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102007038030B4 (de) * | 2007-08-10 | 2009-07-09 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzungen aus Dimethoxysilanen |
| DE102007038030A1 (de) * | 2007-08-10 | 2009-02-19 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzungen aus Dimethoxysilanen |
| WO2009130297A3 (de) * | 2008-04-25 | 2011-01-06 | Henkel Ag & Co. Kgaa | Härtbare zusammensetzungen enthaltend silylierte polyurethane auf basis von polyetherblockpolymeren |
| DE102008020980A1 (de) * | 2008-04-25 | 2009-10-29 | Henkel Ag & Co. Kgaa | Härtbare Zusammensetzungen enthaltend silylierte Polyurethane auf Basis von Polyetherblockpolymeren |
| DE102008043218A1 (de) | 2008-09-24 | 2010-04-01 | Evonik Goldschmidt Gmbh | Polymere Werkstoffe sowie daraus bestehende Kleber- und Beschichtungsmittel auf Basis multialkoxysilylfunktioneller Präpolymerer |
| DE102009022628A1 (de) | 2008-12-05 | 2010-06-10 | Evonik Goldschmidt Gmbh | Verfahren zur Modifizierung von Oberflächen |
| EP2194086A1 (de) | 2008-12-05 | 2010-06-09 | Evonik Goldschmidt GmbH | Verfahren zur Modifizierung von Oberflächen |
| DE102009028640A1 (de) | 2009-08-19 | 2011-02-24 | Evonik Goldschmidt Gmbh | Härtbare Masse enthaltend Urethangruppen aufweisende silylierte Polymere und deren Verwendung in Dicht- und Klebstoffen, Binde- und/oder Oberflächenmodifizierungsmitteln |
| EP2289972A1 (de) | 2009-08-19 | 2011-03-02 | Evonik Goldschmidt GmbH | Härtbare Masse enthaltend Urethangruppen aufweisende silylierte Polymere und deren Verwendung in Dicht- und Klebstoffen, Binde- und/oder Oberflächenmodifizierungsmitteln |
| EP3184576A1 (de) | 2009-08-19 | 2017-06-28 | Evonik Degussa GmbH | Härtbare masse enthaltend urethangruppen aufweisende silylierte polymere und deren verwendung in dicht- und klebstoffen, binde- und/oder oberflächenmodifizierungsmitteln |
| WO2018069530A1 (en) | 2016-10-14 | 2018-04-19 | Basf Se | Stabilizer composition |
| WO2018069526A1 (en) | 2016-10-14 | 2018-04-19 | Basf Se | Hardenable polymer composition |
| US11001698B2 (en) | 2016-10-14 | 2021-05-11 | Basf Se | Stabilizer composition |
| US11098151B2 (en) | 2016-10-14 | 2021-08-24 | Basf Se | Hardenable polymer composition |
| WO2020148211A1 (en) | 2019-01-16 | 2020-07-23 | Basf Se | Stabilizer composition for sealants and adhesives |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005100482A1 (de) | 2005-10-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE102004018548A1 (de) | Durch Strahlung und Feuchtigkeit härtende Zusammensetzungen auf Basis Silan-terminierter Polymere, deren Herstellung und Verwendung | |
| DE102007038030B4 (de) | Härtbare Zusammensetzungen aus Dimethoxysilanen | |
| EP1529071B1 (de) | Polymermassen auf basis alkoxysilanterminierter polymere mit regulierbarer härtungsgeschwindigkeit | |
| EP1678254B1 (de) | Festigkeitsoptimierte polymere mit gemischten oxyalkyleneiheiten | |
| EP2076568B1 (de) | Zusammensetzungen aus teilweise silylterminierten polymeren | |
| BRPI0617946A2 (pt) | polìmero modificado por a-etoxissilano, processo para a preparação e uso do mesmo, composição endurecìvel, bem como uso da mesma | |
| EP2222751B1 (de) | Härtbare zusammensetzungen aus silanen mit drei hydrolisierbaren gruppen | |
| DE10139132A1 (de) | Alkoxyvernetzende einkomponentige feuchtigkeitshärtende Massen | |
| JP2013510200A (ja) | 2−プロピルヘプタノールを基礎とするエステルを含有する接着剤およびシーラント | |
| DE60028117T2 (de) | Mit Feuchtigkeit härtbare Ein-Komponenten-Zusammensetzung | |
| EP2468759A1 (de) | Sekundäre Aminosilane | |
| EP3475374B1 (de) | Einkomponentenmasse auf basis von alkoxysilanen und verfahren zum fugen oder vergiessen von bauteilen unter verwendung der masse | |
| KR20020013437A (ko) | 프라이머용 접착 증진제 및 프라이머 조성물 | |
| DE10134634A1 (de) | Über Alkoxygruppen vernetzende RTV-1-Siliconkautschuk-Mischungen | |
| DE10348555A1 (de) | Lagerstabiles, Silylgruppen tragendes Polyurethan | |
| EP3613786A1 (de) | Trocknungsmittel für feuchtigkeitshärtende zusammensetzungen | |
| JP4988083B2 (ja) | 室温硬化性組成物 | |
| US10072113B2 (en) | Composition, curable composition, production method therefor, and cured product | |
| JP4125413B2 (ja) | ウレタンプレポリマー組成物および1液型ウレタンシーラント | |
| JPH0513999B2 (de) | ||
| WO2020239593A1 (en) | Silyl terminated prepolymer and composition comprising the same | |
| WO2015012259A1 (ja) | 反応促進剤、およびこれを用いたウレタン化合物、チオウレタン化合物、アミド化合物またはウレア化合物の製造方法 | |
| JPH0873594A (ja) | オキサゾリジン環を有するオルガノポリシロキサンおよびその組成物 | |
| DE102006035846A1 (de) | λ5Si-Silicat-funktionelle Prepolymere | |
| JP2004123901A (ja) | 硬化性組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| OP8 | Request for examination as to paragraph 44 patent law | ||
| 8131 | Rejection |