EP1682135B1 - Salz eines Tetrahydropyranylcyclopentyltetrahydropyridopyridin-Derivats als CCR-2 Antagonisten - Google Patents
Salz eines Tetrahydropyranylcyclopentyltetrahydropyridopyridin-Derivats als CCR-2 Antagonisten Download PDFInfo
- Publication number
- EP1682135B1 EP1682135B1 EP04796120A EP04796120A EP1682135B1 EP 1682135 B1 EP1682135 B1 EP 1682135B1 EP 04796120 A EP04796120 A EP 04796120A EP 04796120 A EP04796120 A EP 04796120A EP 1682135 B1 EP1682135 B1 EP 1682135B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- added
- trifluoromethyl
- solution
- mol
- pyran
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 CC(C)[C@@](C1)(C=C[C@]1N*)C(O)=O Chemical compound CC(C)[C@@](C1)(C=C[C@]1N*)C(O)=O 0.000 description 1
- ANVYHALMQXHQSG-NTSWFWBYSA-N COC([C@H](C1)C=C[C@H]1N)=O Chemical compound COC([C@H](C1)C=C[C@H]1N)=O ANVYHALMQXHQSG-NTSWFWBYSA-N 0.000 description 1
- UBNQRXPWESAWSF-NWDGAFQWSA-N Cc1ccc(C)[n]1[C@@H](C1)C=C[C@@H]1C(OC)=O Chemical compound Cc1ccc(C)[n]1[C@@H](C1)C=C[C@@H]1C(OC)=O UBNQRXPWESAWSF-NWDGAFQWSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- WO-A-0213824 discloses piperidine compounds bearing a cyclopentyl group as CCR-2 modulators.
- WO-A-0076512 discloses piperidine compounds substituted by a cyclopentylmethyl group as CCR-5 and/or CCR-3 modulators.
- the present invention provides the succinate salt of ((1R,3S)-3-isopropyl-3- ⁇ [3-(trifluoromcthyl)-7,8-dihydro-1,6-naphthridin-6(5H)-yl]carbonyl)cyclopentyl)[(3S,4S)-3-methoxytetrahydro-2H-pyran-4-yl]amine, its use in therapy and describes an efficient synthesis of it.
- the present invention provides the succinate salt of ((1R,3S)-3-isopropyl-3- ⁇ [3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl]carbonyl ⁇ cyclopentyl) [(3S, 4S)-3-methoxytetrahydro-2H-pyran-4-yl]amine and its use in therapy for treating, ameliorating or reducing the risk of an inflammatory and immunoregulatory disorder or disease or of rheumatoid arthritis.
- the present invention is also dedicated to a pharmaceutical composition which comprises this compound and an inert carrier.
- the hydrogenation step is accomplished prior to the formation of compound 2.
- N-((1R,3S)-3-isopropyl-3- ⁇ [3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl]carbonyl ⁇ cyclopentyl)-N-[(cis-3S,4S)-3-methoxytetrahydro-2H-pyran-4-yl]amine, 1 is synthesized by sequentially coupling three building block compounds 4, 5 and 6 (Schemes 3, 4 and 5, below).
- compound 1 may be purified employing crystallization, either as a succinate salt or other salt such as a benzenesulfonate salt, followed by a salt break. Purification in this manner is described in the examples which follow.
- (1R,4S)-4-aminocyclopent-2-ene-1-carboxylic acid is converted into the corresponding methyl ester by treatment with thionyl chloride in methanol.
- Methyl (1 R ,4 S )-4-(2,5-dimethyl-1 H -pyrrol-1-yl)cyclopent-2-ene-1-carboxylate is then synthesized by reacting the (1R,4S)-4-aminocyclopent-2-ene-1-carboxylate methyl ester with 2,4-pentanedione in the presence of DIEA.
- intermediates may be synthesized having protecting groups other than 2,5-dimethyl-1H-pyrrol-1-yl.
- protecting group "PG 1" includes but is not limited to tert-butoxycarbonyl, benzyloxycarbonyl, alkyloxycarbonyl, allyloxycarbonyl, benzoyl, formyl, acetyl, trifluoroacetyl, 2-nitrobenzenesulfonyl, 4-nitrobenzenesulfonyl, 2,4-dinitrobenzenesulfonyl, benzyl, triphenylmethyl, imines (such as diphenylmethylene) and other protecting groups known in the art, as exemplified in Greene, T; Wuts, P. G. M. Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, Inc., New York, NY 1999 .
- Phosphorous oxychloride (POCl 3 ) is added to chilled DMF at a rate such that the temperature remained below 10 °C (approx. 1 h).
- the reaction mixture is warmed to RT and then 3,3,3-trifluoropropanoic acid is added (exotherm to approx. 45 °C).
- the reaction mixture is warmed to 50°C and held at this temperature for approx 4 h.
- Upon completion of the vinamidinium formation the reaction mixture is allowed to cool to RT.
- the reaction mixture is then added concurrently with 5 N NaOH to a solution of NaPF 6 in water cooled to 0°C.
- the rates of addition of the two solutions are controlled such that the temperature of the aqueous slurry remains below 10 °C and the pH is between 3 and 4 (addition time takes about 2 h).
- the resulting yellow slurry is aged for one hour at 0 °C then filtered to collect solids.
- the filtercake is slurry washed with ice cold water (2 times) and the cake dried with nitrogen/vacuum. Typical yield 85 %.
- a solution of the protected piperidone, for instance the BOC protected piperidone, in THF is then added to a cooled (-20 °C) solution of lithium hexamethyldisilylamide (LiN(TMS) 2 ) in THF, keeping the temperature below 10°C, to form the lithium enolate (approx 45 min).
- the enolate solution is transferred to a chilled (-20 °C) slurry of the CF 3 DT in THF at a rate such that the internal temperature remains below -10 °C (approx 45 min). This mixture is aged for 2 h at -20°C then acetic acid is added and the mixture warmed to room temperature over 30 minutes.
- the organic layer from the naphthyridine formation is concentrated and solvent switched to methanol.
- HCl in IPA is then added and the mixture heated at 60 °C until the deprotection is complete (about 1 h).
- water is added and the pH adjusted to appox 10.5.
- IPAc is added and the layers separated.
- the aqueous layer is back extracted with IPAc two additional times.
- the combined organic layers are concentrated and solvent switched to IPA.
- This IPA solution is filtered to remove inorganic salts, the filtercake washed with IPA and the combined filtrate and washes reconcentrated to a total volume of about 5 mL/g. After the free base/IPA solution to 60°C HCl in IPA is added over 30 minutes.
- piperidones having protecting groups (“PG 2 ”) other than tert- butoxycarbonyl may also be used.
- Other protecting groups useful in the synthesis of the naphthiridine include, but are not limited to benzyloxycarbonyl, alkyloxycarbonyl, allyloxycarbonyl, benzoyl, acetyl, formyl, trifluoroacetyl, 2-nitrobenzenesulfonyl, 4-nitrobenzenesulfonyl, 2,4-dinitrobenzenesulfonyl, N-benzyl, triphenylmethyl, and other protecting groups known in the art, as exemplified in Greene, T; Wuts, P. G. M. Protective Groups in Organic Synthesis, 3rd Ed, John Wiley & Sons, Inc., New York, NY 1999 .
- the starting pyranone is converted to its dipropyl ketal by treatment with tripropylorthoformate in the presence of an acid catalyst.
- the crude ketal is then heated in the presence of chlorobenzene. Under these conditions, elimination of propanol provides the propyl enol ether.
- the reaction is driven by the removal of propanol by distillation during the reaction sequence.
- the crude enol ether is then oxidized under modified Sharpless asymmetric dihydroxylation conditions. In this reaction N-methylmorpholine oxide is used as the stoichiometric oxidant.
- the reaction typically gives the product ⁇ -hydroxyketone in about 80 to 85 % ee.
- the ⁇ -hydroxyketone is not isolated directly but an aqueous solution of Na 2 S 2 O 5 is added to form the bisulfite adduct of the ketone.
- racemic bisulfite adduct crystallizes. The racemate is removed by filtration and the resulting mother liquor is typically 95 to 99 % enantiomeric excess (ee).
- the acetone is removed in vacuo and isopropanol is added to give the crystalline bisulfite adduct with high enantiomeric excess.
- This is treated with HCl and methanol with trimethylorthoformate as a water scavenger, to provide the dimethylketal.
- the hydroxyl group is then methylated using NaOt-Bu and Me 2 SO 4 . Adding water and HCl to the reaction mixture provides the target ⁇ -methoxypyranone in about 96% ee.
- Step 1 6- ⁇ [(1S,4S)-4-(2,5-dimethyl-1H-pyrrol-1-yl)-1-isopropylcyclopent-2-en-1-yl]carbonyl ⁇ -3-(trifluoromethyl)-5,6,7,8-tetrahydro-l,6-naphthyridine
- the reaction solution was cooled to ⁇ 15°C and tetrahydronaphthyridine-HCl salt (670 g, 73.2wt% as free base equivalent, 2.42 mol) was added.
- the temperature was increased to 23 °C and another portion of diisopropylethylamine (0.93 L, 5.34 mol) was added with cooling at ⁇ 25 °C over 15 min.
- the mixture was aged for >1 h and was then diluted with 5% NaHCO 3 (16 L).
- the product was extracted with ethyl acetate (EtOAc) (16 L).
- EtOAc ethyl acetate
- the organic phase was washed with water (10 L) and concentrated under vacuum with displacement of the EtOAc with methanol to a volume of 10 L.
- the assay yield of the amide was quantitative (1.055 kg, 2.45 mol).
- Step 2 (1S,4S)-4-isopropyl-4- ⁇ [3-(trifluorornetlzyl)-7,8-dilzydro-1,6-naphthyridin-6(5H)-yl]carboyzyl ⁇ cyclopent-2-en-1-amine
- the amide (1.055 kg, 2.45 mol) in methanol (10 L) was added to hydroxylamine-HCl (1 kg, 14.4 mol), 50% hydroxylamine in water (1 L, 16.3 mol) and water (5 L).
- the resulting slurry was heated to reflux (71 °C) and maintained at this temperature for 6 h.
- the reaction solution was cooled to room temperature and the pH was adjusted to 11.0 with 10 N NaOH.
- the reaction solution was diluted with water (12 L) and the product was extracted with chlorobenzene three times (14 L, 13L, and 13L). Each organic layer was washed once with the same water (10 L).
- the organic layers containing product (0.79 assay kg, 2.24 mol, 92% yield) were combined and concentrated.
- Step 3 (3S,4S)-N-((1S,4S)-4-isopropyl-4- ⁇ [3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl]carbonyl ⁇ cyclopent-2-en-1-yl)-3-methoxytetrahydro-2H-pyran-4-amine
- the amine dihydrochloride salt (777 g, 552 g as free base, 1.56 mol) was slurried in IPAc (3 L). The mixture was cooled in an ice bath and n -Bu 3 N (860 mL, 3.61 mol) was added followed by isopropanol (260 mL, 3.40 mol). Sodium triacetoxyborohydride (724 g, 3.42 mol) was added at 5°C. After 1 h, a solution of the methoxypyranone in IPAc (1.76 L of a 160 g/L solution, 2.17 mol) was added to the batch at 1°C. After 6 h, the mixture was partitioned between saturated aq.
- the acetonitrile phase contained 654 g (1.4 mol, 90%) (35,45)-N-((1S,4S)-4-isopropyl-4- ⁇ [3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)carbonyl ⁇ cyclopent-2-en-1-yl)-3-methoxytetrahydro-2H-pyran-4-amine.
- the solvent was removed in vacuo to provide an oil that was used in the final hydrogenation step.
- Step 4 ((1R,3S)-3-Isopropyl-3- ⁇ [3-(trifluoronzethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl]carbonyl ⁇ cyclopentyl)[(3S,4S)-3-methoxytetrahydro-2H-pyran-4-yl]amine
- the crude coupled cyclopentene of Step 3 (640 g) was diluted with methanol (3.2 L) and the solution was concentrated to an oil. Dilution with methanol (3.2 L) and concentration were repeated two additional times. After the final concentration the oil was diluted with methanol (6.4 L) and charged to an autoclave. The catalyst 5% Pd/C (256 g) was charged to the autoclave as a slurry in methanol (1.4 L). The hydrogenation (40 psi) was run overnight (18 h) at 25°C. The batch was filtered through solka floc ( ⁇ 1.5 in depth) in an 8 L sintered-glass funnel. The autoclave was rinsed with methanol (5.0 L) and this rinse was used to wash the filter cake.
- Benzenesulfonate Salt The resulting oil from the Example 1 was dissolved in IPA (1.54 L) and transferred to crystallization flask. An IPA (2 X 385 mL) rinse was added to the batch. The solution was warmed to 56°C at which point benzenesulfonic acid (283 g, 1.79 mol) was added which resulted in an increase in temperature to 71°C. The solution was cooled to 60 °C and benzenesulfonate salt seed (1 g) was added. The thin slurry was aged for 30 min to develop a thick seed bed after which heptane (4.62 L) was added over 50 min.
- the slurry was aged for 2 h at 65°C, then allowed to cool to room temperature overnight (-9 h).
- the solids were filtered, washed with 2:1 heptane/ethanol (2 X 2.3 L, 2 mL/g free base), and dried under vacuum with a N 2 stream for 2 h.
- the filtercake was broken up and further dried with N 2 /vacuum for ⁇ 48 h to afford 1.328 kg of the succinate salt (91.6 %).
- the filtercake was dried by passing N 2 through it with vacuum for 3 h then the filtercake was broken up and further dried with N 2 /vacuum for approximately 48 h. After completion of drying the bulk drug was stored in a doubled polybag within a fiber drum to await milling. A total of 3.018 kg of the succinate salt (93.9 % yield) was obtained.
- the solid aminocyclopentene methyl ester salt (1.076 kg, 6.059 mol) was dissolved in MeOH (3 L, 2M) at 20 °C under nitrogen.
- Diisopropylethylamine (DIEA, 0.78 kg, 6.059 mol) was added followed by acetonyl acetone (0.711 kg, 6.241 mol).
- the batch had an exotherm increasing the temperature to 32-35 °C.
- the reaction mixture was then aged at 25 °C for 16 h.
- the batch was diluted with IPAc (9-10 L) and washed with 10% NH 4 Cl (2 x 3 L) and 5% brine (2 x 3 L).
- the IPA batch was dried over sodium sulfate, filtered, and concentrated to an oil.
- LHMDS lithium hexamethyldisilazide
- the organic layer was washed with 6% aq NH 4 Cl (10 L), 5% brine (2 x 10 L), and concentrated to an oil.
- the air-sensitive alkylated pyrrole methyl ester (1419 g, 98% yield) was stored at 5-7 °C under nitrogen until saponified.
- Step 4 (1S,4S)-4-(2,5-dimethyl-1H-pyrrol-1-yl)-1-isopropylcyclopent-2-ene-1-carboxylic acid
- the alkylated pyrrole methyl ester (1.38 kg, 5.197 mol) was dissolved in MeOH (7.7 L). DI water (2.5 L) was added followed by 10N NaOH (2.08 L, 20.786 mol). The batch was then heated to 65 °C for 16 h. The batch was cooled to 10 °C. The product was crystallized by adjusting the pH to 4.5 with concd HCl. The slurry was aged for 1 h and DI water (15 L) was charged to the batch. The slurry was aged 18 h at 20-25°C.
- Hexafluorophosphoric acid (980 mL, 60 wt% aqueous) was added to water (7.1 L) with cooling at 4 °C.
- Sodium hydroxide (5 N, 2.0 L) was added slowly keeping the internal temperature below 15 °C.
- the solution was then cooled to 0 °C.
- Sodium hydroxide (5 N) was charged to a 2 L addition funnel and added concurrently with the TFPA/POCl 3 /DMF reaction mixture at a rate such that the internal temperature remained below 5 °C and the pH varied from 3.05 to 3.6 (approx. 3.2 during most of the addition).
- the resulting yellow slurry was then aged for 60 min at ⁇ 0 °C.
- Step 2 N-(tert-Butoxycarbonyl)-3-(trifluoromethyl)-5,6,7,8-tetrahydro-1,6-naphthyridine
- N-BOC-4-piperidone (672 g) in THF (2.72 L) was charged to a solution of lithium bis(trimethylsilyl) amide (LHMDS) (3.55 L, 1.0 M solution in THF) in THF (3.7 L) at -12°C over 45 min.
- LHMDS lithium bis(trimethylsilyl) amide
- the reaction mixture was allowed to warm to room temperature. This mixture was added over 30 min to a slurry of CF 3 DT (1.17 kg) in THF (5.45 L) cooled to -24 °C. The reaction mixture was then aged for 2 h at ⁇ -20 °C. Acetic acid (295 mL) was added over 3 min.
- the reaction mixture was warmed to 20°C over 1 h 15 min and ammonium acetate (741 g) was added in one portion.
- the reaction mixture was warmed to 64 °C and aged for 2 h at this temperature.
- the reaction mixture was cooled to room temperature then diluted with water (15.4 L) and methylcyclohexane (15.4 L).
- the mixture was agitated, the stirring stopped, and the layers were allowed to settle.
- Step 3 3-(trifluoromethyl)-5,6,7,8-tetrahydro-1,6-naphthyridine
- the organic layer from the naphthyridine formation containing N-(tert-butoxycarbonyl)-3-(trifluoromethyl)-5,6,7,8-tetrahydro-1,6-naphthyridine (34.2 kg total weight containing 989 g of BOC naphthyridine) was concentrated to an oil.
- the residue was diluted with methanol (6 L) and the solution was concentrated to an oil.
- Methanol (6 L) was added to the residue and this solution was concentrated to 2.3 L.
- the solution was diluted with methanol to a volume of 7.3 L and 4.58 M HCl in IPA (3.6 L) was added.
- the solution was heated to 55 °C and aged for 1 h. After cooling to room temperature water (5 L) was added.
- Step 1- tetrahyro-4H-pyran-4-one: A 22 L round-bottomed-flask equipped with a mechanical stirrer, thermocouple, and nitrogen inlet was charged with tetrahydro-4H-pyran-4-one (3.00 kg, 30.0 mol) and tripropylorthoformate (5.70 kg, 30.0 mol). The pump used for the transfer was flushed with chlorobenzene (300 mL) and the flush was added to the batch. The resulting solution was cooled in an ice bath to 5 °C. Amberlyst-15 (60 g), previously washed with DI water then propanol and dried, was added in one portion. The mixture was stirred at ambient temperature for 16 h.
- the batch temperature was maintained between 122 and 125 °C during the distillation. After 16 h, GC assay indicated > 9:1 conversion.
- the vacuum was increased to 50 mm Hg and the reaction was distilled to provide a 53 wt% solution of 3 (3.13 kg, 73 % yield) and chlorobenzene.
- Step 2- 3,4-dihydroxy-tetrahydro-2-H-pyran-4-sulfonic acid, sodium salt A 50 L round-bottomed-flask equipped with a mechanical stirrer, nitrogen inlet and thermocouple was charged with acetone (12.7 L) and water (1.28 L). Hydroquinidine-1,4-phthalazinediyl diether (DHQD 2 PHAL, 54.8 g, 0.070 mol), potassium osmate dihydrate (12.95 g, 0.035 mol), and 4-methylmorpholine N-oxide monohydrate (NMO, 1.078 kg, 7.74 mol) were added sequentially to the solvent and the resulting solution was cooled to 0 °C.
- DHQD 2 PHAL Hydroquinidine-1,4-phthalazinediyl diether
- NMO 4-methylmorpholine N-oxide monohydrate
- the propylenol ether (1.85 kg of a 54 wt % solution, 7.03 mol) was added over 7 h while maintaining a reaction temperature of about 0 °C.
- the solids were removed by filtration and the filtrate was concentrated in vacuo to remove the acetone.
- Isopropanol (28 L) was added over 3.5 h to provide a colorless slurry.
- the solids were collected on a frit, rinsed with isopropanol (6 L) and dried in a vacuum oven to provide 958 g of 92 wt% (57 % isolated yield) product that was 97.2 % ee.
- Step 3 4,4-dimethoxytetrahydro-2H pyran-3-ol: A 22 L round bottomed flask equipped with a mechanical stirrer, 5 L dropping funnel, nitrogen inlet and thermocouple was charged with bisulfite adduct (893 g of a 92 wt% solid, 4.06 mol)MeOH (8.1 L) and trimethylorthoformate (948 g, 8.93 mol). The resulting slurry was warmed to 50 °C and a 1.89 M solution of HCl in MeOH (2.48 L, 4.69 mol) was added via dropping funnel over 40 min.
- the slurry was cooled to 7 °C and 50 wt% NaOH (340 mL) was added as a slow stream.
- the solids were collected on a frit and the filtrate was solvent switched to toluene using a total of 12 L toluene.
- the batch was concentrated to about 3 L then the solids were removed by filtration and the cake was rinsed with THF (2 L).
- Step 4 (3R)-3-Methoxytetrahydro-4H-pyran-4-one:
- NaOt-Bu (494 g, 5.14 mol) was added in one portion to provide a clear yellow solution.
- the flask was immersed in an ice bath and Me 2 SO 4 (737 g, 5.84 mol) was added over 20 min maintaining an internal temp of below 36 °C. The cold bath was removed and the reaction mixture was aged for 4 h to provide a crude solution.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Claims (6)
- Die Verbindung der Formel (I):

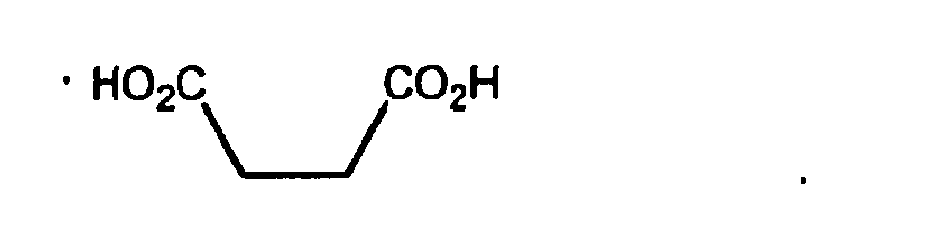
- Verwendung der Verbindung der Formel (I) nach Anspruch 1 zur Herstellung eines Medikaments zur Behandlung, Verbesserung oder Verringerung des Risikos einer entzündlichen und immunregulatorischen Störung oder Erkrankung.
- Verwendung der Verbindung der Formel (I) nach Anspruch 1 zur Herstellung eines Medikaments zur Behandlung, Verbesserung oder Verringerung des Risikos von rheumatoider Arthritis.
- Eine pharmazeutische Zusammensetzung, die einen inerten Träger und eine Verbindung der Formel (I) nach Anspruch 1 enthält.
- Eine Verbindung der Formel (I) nach Anspruch 1 zur Verwendung in der Therapie.
- Eine Verbindung der Formel (I) nach Anspruch 1 zur Verwendung bei der Behandlung, Verbesserung oder Verringerung des Risikos einer entzündlichen und immunregulatorischen Störung oder Erkrankung oder von rheumatoider Arthritis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US51473503P | 2003-10-27 | 2003-10-27 | |
| PCT/US2004/035069 WO2005044264A1 (en) | 2003-10-27 | 2004-10-25 | Ccr-2 antagonist salt |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP1682135A1 EP1682135A1 (de) | 2006-07-26 |
| EP1682135A4 EP1682135A4 (de) | 2009-11-25 |
| EP1682135B1 true EP1682135B1 (de) | 2011-07-27 |
Family
ID=34572774
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP04796120A Expired - Lifetime EP1682135B1 (de) | 2003-10-27 | 2004-10-25 | Salz eines Tetrahydropyranylcyclopentyltetrahydropyridopyridin-Derivats als CCR-2 Antagonisten |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US7473696B2 (de) |
| EP (1) | EP1682135B1 (de) |
| JP (1) | JP4542550B2 (de) |
| KR (1) | KR20060101476A (de) |
| CN (1) | CN1870998B (de) |
| AT (1) | ATE517622T1 (de) |
| AU (1) | AU2004287416C1 (de) |
| BR (1) | BRPI0415836A (de) |
| CA (1) | CA2543201C (de) |
| CO (1) | CO5690606A2 (de) |
| EC (1) | ECSP066524A (de) |
| IL (1) | IL175009A0 (de) |
| IS (1) | IS8400A (de) |
| MA (1) | MA28104A1 (de) |
| MX (1) | MXPA06004647A (de) |
| NO (1) | NO20062377L (de) |
| NZ (1) | NZ546447A (de) |
| RU (1) | RU2317295C1 (de) |
| UA (1) | UA81365C2 (de) |
| WO (1) | WO2005044264A1 (de) |
| ZA (1) | ZA200602752B (de) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY129850A (en) | 2002-04-29 | 2007-05-31 | Merck Sharp & Dohme | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity |
| US20060030582A1 (en) * | 2002-04-29 | 2006-02-09 | Demartino Julie | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity |
| MXPA05005660A (es) | 2002-11-27 | 2005-10-18 | Incyte Corp | Derivados de 3-aminopirrolidina como moduladores de receptores de quimocina. |
| US7393844B2 (en) * | 2003-03-18 | 2008-07-01 | Merck & Co., Inc. | Tetrahydropyranyl cyclopentyl heterocyclic amide modulators of chemokine receptor activity |
| AR045875A1 (es) * | 2003-10-27 | 2005-11-16 | Merck & Co Inc | Procedimiento para la preparacion del antagonista ccr-2 |
| BRPI0417605B8 (pt) | 2003-12-18 | 2021-05-25 | Incyte Corp | n-[2-((3s)-3-{[4-hidróxi-4-(5-pirimidin-2- ilpiridin-2-il)ciclohexil]amino}-pirrolidin-1-il)-2- oxoetil]-3-(trifluor-metil)benzamida, ou um sal farmaceuticamente aceitável do mesmo e composição que o compreende. |
| JP2007537264A (ja) | 2004-05-11 | 2007-12-20 | インサイト コーポレイション | ケモカインレセプタのモジュレータとしての3−(4−ヘテロアリールシクロヘキシルアミノ)シクロペンタンカルボキサミド |
| GB0413605D0 (en) * | 2004-06-17 | 2004-07-21 | Addex Pharmaceuticals Sa | Novel compounds |
| WO2006004741A2 (en) | 2004-06-28 | 2006-01-12 | Incyte Corporation | 3-aminocyclopentanecarboxamides as modulators of chemokine receptors |
| JP4116670B2 (ja) | 2004-06-28 | 2008-07-09 | インサイト コーポレイション | ケモカイン受容体の調節剤としての3−アミノシクロペンタンカルボキサミド |
| CA2669917A1 (en) | 2006-11-17 | 2008-05-22 | Dawn M. George | Aminopyrrolidines as chemokine receptor antagonists |
| US20090076065A1 (en) * | 2007-09-17 | 2009-03-19 | Protia, Llc | Deuterium-enriched mk-0812 |
| US7741327B2 (en) | 2008-04-16 | 2010-06-22 | Hoffmann-La Roche Inc. | Pyrrolidinone glucokinase activators |
| US8178689B2 (en) | 2010-06-17 | 2012-05-15 | Hoffman-La Roche Inc. | Tricyclic compounds |
| WO2012125663A2 (en) * | 2011-03-17 | 2012-09-20 | Merck Sharp & Dohme Corp. | Cyclohexane substituted amino cyclopentane derivatives as useful ccr2 antagonists |
| WO2013149376A1 (en) | 2012-04-02 | 2013-10-10 | Abbott Laboratories | Chemokine receptor antagonists |
| EP2962691A4 (de) * | 2013-02-28 | 2016-08-31 | Santen Pharmaceutical Co Ltd | Mittel zur prävention oder behandlung von erkrankungen der hinteren teile des auges mit einem tetrahydropyranyl-amino-cyclopentylcarbonyl-tetrahydropyridopyridin-derivat als wirkstoff |
| ES2904252T3 (es) | 2015-05-21 | 2022-04-04 | Chemocentryx Inc | Moduladores de CCR2 |
| CN118267479A (zh) | 2017-09-25 | 2024-07-02 | 凯莫森特里克斯股份有限公司 | 使用趋化因子受体2(ccr2)拮抗剂和pd-1/pd-l1抑制剂的联合治疗 |
| IL275898B2 (en) | 2018-01-08 | 2025-05-01 | Chemocentryx Inc | Methods for treating solid tumors with CCR2 antagonists |
| US20190269664A1 (en) | 2018-01-08 | 2019-09-05 | Chemocentryx, Inc. | Methods of treating solid tumors with ccr2 antagonists |
| CN112341350A (zh) * | 2020-10-19 | 2021-02-09 | 江苏威奇达药业有限公司 | 酶法拆分γ-内酰胺后副产物的处理方法 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RO108347B1 (ro) * | 1989-10-30 | 1994-04-28 | Bellon Labor Sa Roger | DERIVATI DE BENZO-(b)-NAFTIRIDIN-1,8 SI PROCEDEU DE PREPARARE A ACESTORA |
| GB9417249D0 (en) * | 1994-08-26 | 1994-10-19 | Wellcome Found | A novel salt |
| JPH09291034A (ja) * | 1996-02-27 | 1997-11-11 | Yoshitomi Pharmaceut Ind Ltd | 縮合ピリジン化合物およびその医薬としての用途 |
| RU2174123C2 (ru) * | 1996-10-28 | 2001-09-27 | Новартис Аг | Производные 8-арил-1,7-нафтиридина и фармацевтическая композиция, обладающая противовоспалительной активностью |
| US6312689B1 (en) * | 1998-07-23 | 2001-11-06 | Millennium Pharmaceuticals, Inc. | Anti-CCR2 antibodies and methods of use therefor |
| FR2787790A3 (fr) * | 1998-12-23 | 2000-06-30 | Sanofi Sa | Procede de preparation du (r)-(+)-3-{1°2-(4-benzoyl-2-(3,4- difluorophenyl)morpholin-2-yl)ethyl!-4-phenylpiperidin-4-yl} -1,1-dimethyluree, de ses sels, solvats et/ou hydrates |
| AU5473400A (en) | 1999-06-11 | 2001-01-02 | Merck & Co., Inc. | Cyclopentyl modulators of chemokine receptor activity |
| EP1318811B1 (de) * | 2000-08-17 | 2006-08-30 | Merck & Co., Inc. | Cyclopentylmodulatoren der chemokin-rezeptor-aktivität |
| CZ20031315A3 (cs) * | 2000-11-28 | 2004-04-14 | Pfizer Products Inc. | Soli isothiazolkarboxamidu a jejich použití jako prostředků proti hyperproliferaci |
| EP1461040B1 (de) * | 2001-11-29 | 2006-03-15 | Pfizer Products Inc. | Succinate des 5,8,14-triazatetracyclo[10.3.1.0 2,11 .0 4,9 ]-hexadeca-2(11),3,5,7,9,-pentaens und pharmazeutische zusammensetzungen |
| GEP20063872B (en) * | 2001-12-12 | 2006-07-10 | Pfizer Prod Inc | Salt forms of e-2-methoxy-n-(3-{4-[3-methyl-4-(6-methyl-pyridin-3-yloxy)-phenylamino]-quina-zolin-6-yl}-allyl)-acetamide and method of production |
| JP4459807B2 (ja) * | 2002-04-29 | 2010-04-28 | メルク エンド カムパニー インコーポレーテッド | テトラヒドロピラニルシクロペンチルテトラヒドロイソキノリン系のケモカイン受容体活性調節剤 |
| EP1501507B1 (de) * | 2002-04-29 | 2008-05-28 | Merck & Co., Inc. | Tetrahydropyranyl-cyclopentyl-tetrahydropyridopyridin-modulatoren der chemokin-rezeptor-aktivität |
| MY129850A (en) | 2002-04-29 | 2007-05-31 | Merck Sharp & Dohme | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity |
| AR045875A1 (es) * | 2003-10-27 | 2005-11-16 | Merck & Co Inc | Procedimiento para la preparacion del antagonista ccr-2 |
-
2004
- 2004-10-25 AT AT04796120T patent/ATE517622T1/de not_active IP Right Cessation
- 2004-10-25 CA CA2543201A patent/CA2543201C/en not_active Expired - Fee Related
- 2004-10-25 AU AU2004287416A patent/AU2004287416C1/en not_active Ceased
- 2004-10-25 NZ NZ546447A patent/NZ546447A/en unknown
- 2004-10-25 BR BRPI0415836-9A patent/BRPI0415836A/pt not_active IP Right Cessation
- 2004-10-25 WO PCT/US2004/035069 patent/WO2005044264A1/en not_active Ceased
- 2004-10-25 RU RU2006118352/04A patent/RU2317295C1/ru not_active IP Right Cessation
- 2004-10-25 MX MXPA06004647A patent/MXPA06004647A/es active IP Right Grant
- 2004-10-25 US US10/577,584 patent/US7473696B2/en not_active Expired - Fee Related
- 2004-10-25 KR KR1020067008066A patent/KR20060101476A/ko not_active Ceased
- 2004-10-25 UA UAA200605772A patent/UA81365C2/uk unknown
- 2004-10-25 CN CN2004800315947A patent/CN1870998B/zh not_active Expired - Fee Related
- 2004-10-25 EP EP04796120A patent/EP1682135B1/de not_active Expired - Lifetime
- 2004-10-25 JP JP2006538125A patent/JP4542550B2/ja not_active Expired - Fee Related
-
2006
- 2006-04-04 ZA ZA200602752A patent/ZA200602752B/xx unknown
- 2006-04-06 IS IS8400A patent/IS8400A/is unknown
- 2006-04-20 IL IL175009A patent/IL175009A0/en unknown
- 2006-04-24 MA MA28957A patent/MA28104A1/fr unknown
- 2006-04-25 CO CO06039063A patent/CO5690606A2/es unknown
- 2006-04-26 EC EC2006006524A patent/ECSP066524A/es unknown
- 2006-05-24 NO NO20062377A patent/NO20062377L/no not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| ZA200602752B (en) | 2007-08-29 |
| US7473696B2 (en) | 2009-01-06 |
| JP4542550B2 (ja) | 2010-09-15 |
| EP1682135A1 (de) | 2006-07-26 |
| MA28104A1 (fr) | 2006-08-01 |
| JP2007509940A (ja) | 2007-04-19 |
| CN1870998A (zh) | 2006-11-29 |
| KR20060101476A (ko) | 2006-09-25 |
| BRPI0415836A (pt) | 2007-01-02 |
| AU2004287416A1 (en) | 2005-05-19 |
| ATE517622T1 (de) | 2011-08-15 |
| AU2004287416C1 (en) | 2010-09-09 |
| NO20062377L (no) | 2006-05-24 |
| IL175009A0 (en) | 2006-08-20 |
| CA2543201A1 (en) | 2005-05-19 |
| ECSP066524A (es) | 2006-10-10 |
| RU2317295C1 (ru) | 2008-02-20 |
| CA2543201C (en) | 2010-12-21 |
| UA81365C2 (en) | 2007-12-25 |
| CN1870998B (zh) | 2010-10-20 |
| WO2005044264A1 (en) | 2005-05-19 |
| MXPA06004647A (es) | 2006-06-27 |
| NZ546447A (en) | 2009-02-28 |
| EP1682135A4 (de) | 2009-11-25 |
| US20070135474A1 (en) | 2007-06-14 |
| IS8400A (is) | 2006-04-06 |
| CO5690606A2 (es) | 2006-10-31 |
| AU2004287416B2 (en) | 2009-11-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1682135B1 (de) | Salz eines Tetrahydropyranylcyclopentyltetrahydropyridopyridin-Derivats als CCR-2 Antagonisten | |
| US9126906B2 (en) | Asymmetric synthetic processes for the preparation of aminosulfone compounds | |
| US11370739B2 (en) | Sacubitril intermediate and preparation method thereof | |
| CN100545145C (zh) | 用于制备1,2-二氨基化合物的不使用叠氮化物的方法 | |
| AU2004287810B2 (en) | Process for the preparation of CCR-2 antagonist | |
| JP2019210273A (ja) | エドキサバンの製造方法 | |
| AU8680598A (en) | Alpha 1a adrenergic receptor antagonist | |
| US5962724A (en) | High enantio-selective process for producing pure enantiomeric cyclopentane and cyclopentene-(β)-amino acids | |
| US7232907B2 (en) | Process for production of naphthyridine-3-carboxylic acid derivatives | |
| US20130197229A1 (en) | Method of making azaindazole derivatives | |
| KR20040079324A (ko) | 4-아미노메틸-3-알콕시이미노피롤리딘 메탄설폰산염의신규한 제조 방법 | |
| EP2448916B1 (de) | Herstellung von trans-4-aminocyclopentan-2-en-1-carboxylsäurederivaten | |
| EP4276101B1 (de) | Verfahren zur herstellung eines pyrrolopyridinderivats | |
| US20040236118A1 (en) | Pyrrolidine derivatives and method of synthesizing these | |
| JP2019526554A (ja) | Nep阻害剤合成のための新規な方法および中間体 | |
| USH1737H (en) | 7-oxabicycloheptane carboxylic acid prostaglandin analog intermediates useful in the preparation of anti-thrombotic and anti-vasospastic compounds and method for preparing same | |
| HK1099208A (zh) | Ccr-2拮抗劑鹽 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20060529 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LI LU MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: HR LT LV |
|
| RAX | Requested extension states of the european patent have changed |
Extension state: LT Payment date: 20060529 Extension state: LV Payment date: 20060529 Extension state: HR Payment date: 20060529 |
|
| A4 | Supplementary search report drawn up and despatched |
Effective date: 20091027 |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: C07D 471/04 20060101ALI20091016BHEP Ipc: A61P 37/02 20060101ALI20091016BHEP Ipc: A61P 29/00 20060101ALI20091016BHEP Ipc: A61K 31/4375 20060101AFI20050526BHEP Ipc: A61P 43/00 20060101ALI20091016BHEP Ipc: A61P 37/00 20060101ALI20091016BHEP |
|
| 17Q | First examination report despatched |
Effective date: 20091222 |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: MERCK SHARP & DOHME CORP. |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| RTI1 | Title (correction) |
Free format text: SALT OF A TETRAHYDROPYRANYL CYCLOPENTYL TETRAHYDROPYRIDOPYRIDINE DERIVATIVE AS CCR2-ANTAGONIST |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LI LU MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: HR LT LV |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: NV Representative=s name: SERVOPATENT GMBH |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: T3 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602004033695 Country of ref document: DE Effective date: 20110915 |
|
| LTIE | Lt: invalidation of european patent or patent extension |
Effective date: 20110727 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 517622 Country of ref document: AT Kind code of ref document: T Effective date: 20110727 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20111128 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: BE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20111028 Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: SI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 Ref country code: MC Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20111031 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 |
|
| 26N | No opposition filed |
Effective date: 20120502 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PFUS Owner name: SCHERING CORPORATION, US Free format text: FORMER OWNER: MERCK SHARP + DOHME CORP., US Ref country code: CH Ref legal event code: PFA Owner name: MERCK SHARP & DOHME CORP. Free format text: SCHERING CORPORATION#2000 GALLOPING HILL ROAD#KENILWORTH, NJ 07033 (US) -TRANSFER TO- MERCK SHARP & DOHME CORP.#126 EAST LINCOLN AVENUE#RAHWAY, NEW JERSEY 07065 (US) |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MM4A |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602004033695 Country of ref document: DE Effective date: 20120502 |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: 732E Free format text: REGISTERED BETWEEN 20120802 AND 20120808 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: SD Effective date: 20120828 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: TD Effective date: 20120924 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20111025 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: MERCK SHARP & DOHME CORP., US Free format text: FORMER OWNER: MERCK SHARP & DOHME CORP., RAHWAY, US Effective date: 20121213 Ref country code: DE Ref legal event code: R082 Ref document number: 602004033695 Country of ref document: DE Representative=s name: ABITZ & PARTNER, DE Effective date: 20121213 Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: MERCK SHARP & DOHME CORP., US Free format text: FORMER OWNER: MERCK & CO., INC., WHITEHOUSE STATION, US Effective date: 20110726 Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: SCHERING CORPORATION, US Free format text: FORMER OWNER: MERCK SHARP & DOHME CORP., RAHWAY, US Effective date: 20121213 Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: SCHERING CORPORATION, US Free format text: FORMER OWNER: MERCK & CO., INC., WHITEHOUSE STATION, US Effective date: 20110726 Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: MERCK SHARP & DOHME CORP., RAHWAY, US Free format text: FORMER OWNER: MERCK & CO., INC., WHITEHOUSE STATION, N.J., US Effective date: 20110726 Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: MERCK SHARP & DOHME CORP., RAHWAY, US Free format text: FORMER OWNER: MERCK SHARP & DOHME CORP., RAHWAY, N.J., US Effective date: 20121213 Ref country code: DE Ref legal event code: R082 Ref document number: 602004033695 Country of ref document: DE Representative=s name: ABITZ & PARTNER PATENTANWAELTE MBB, DE Effective date: 20121213 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: TP Owner name: SCHERING CORPORATION, US Effective date: 20130318 Ref country code: FR Ref legal event code: CD Owner name: SCHERING CORPORATION, US Effective date: 20130319 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20111107 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R082 Ref document number: 602004033695 Country of ref document: DE Representative=s name: ABITZ & PARTNER, DE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20111025 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20111027 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R082 Ref document number: 602004033695 Country of ref document: DE Representative=s name: ABITZ & PARTNER, DE Effective date: 20130514 Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: MERCK SHARP & DOHME CORP., US Free format text: FORMER OWNER: SCHERING CORPORATION, RAHWAY, US Effective date: 20130514 Ref country code: DE Ref legal event code: R081 Ref document number: 602004033695 Country of ref document: DE Owner name: MERCK SHARP & DOHME CORP., RAHWAY, US Free format text: FORMER OWNER: SCHERING CORPORATION, RAHWAY, US Effective date: 20130514 Ref country code: DE Ref legal event code: R082 Ref document number: 602004033695 Country of ref document: DE Representative=s name: ABITZ & PARTNER PATENTANWAELTE MBB, DE Effective date: 20130514 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20110727 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20131029 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 20131010 Year of fee payment: 10 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: V1 Effective date: 20150501 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141031 Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141031 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20150501 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20150924 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20150924 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20151030 Year of fee payment: 12 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R119 Ref document number: 602004033695 Country of ref document: DE |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20161025 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20170630 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20161025 Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20161102 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20170503 |