ES2699256T3 - Piridina y derivados de pirazina - Google Patents
Piridina y derivados de pirazina Download PDFInfo
- Publication number
- ES2699256T3 ES2699256T3 ES12715279T ES12715279T ES2699256T3 ES 2699256 T3 ES2699256 T3 ES 2699256T3 ES 12715279 T ES12715279 T ES 12715279T ES 12715279 T ES12715279 T ES 12715279T ES 2699256 T3 ES2699256 T3 ES 2699256T3
- Authority
- ES
- Spain
- Prior art keywords
- amino
- pyridin
- atoms
- formula
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 title description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 title description 3
- 150000003216 pyrazines Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 217
- -1 furo [3,2-b] pyridyl Chemical group 0.000 claims abstract description 109
- 150000003839 salts Chemical class 0.000 claims abstract description 67
- 239000000203 mixture Substances 0.000 claims abstract description 64
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 28
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 26
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 24
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 21
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 15
- 125000004076 pyridyl group Chemical group 0.000 claims abstract description 14
- 229910052740 iodine Inorganic materials 0.000 claims abstract description 12
- 229910052794 bromium Inorganic materials 0.000 claims abstract description 11
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 11
- 125000002098 pyridazinyl group Chemical group 0.000 claims abstract description 9
- 125000000714 pyrimidinyl group Chemical group 0.000 claims abstract description 9
- 125000002757 morpholinyl group Chemical group 0.000 claims abstract description 8
- 125000004193 piperazinyl group Chemical group 0.000 claims abstract description 8
- 125000003386 piperidinyl group Chemical group 0.000 claims abstract description 8
- 125000003226 pyrazolyl group Chemical group 0.000 claims abstract description 8
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims abstract description 8
- 125000001544 thienyl group Chemical group 0.000 claims abstract description 8
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 6
- 125000005871 1,3-benzodioxolyl group Chemical group 0.000 claims abstract description 3
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims abstract description 3
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims abstract description 3
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims abstract description 3
- 125000005052 dihydropyrazolyl group Chemical group N1(NCC=C1)* 0.000 claims abstract description 3
- 125000004925 dihydropyridyl group Chemical group N1(CC=CC=C1)* 0.000 claims abstract description 3
- 125000005054 dihydropyrrolyl group Chemical group [H]C1=C([H])C([H])([H])C([H])([H])N1* 0.000 claims abstract description 3
- 125000002541 furyl group Chemical group 0.000 claims abstract description 3
- 125000002883 imidazolyl group Chemical group 0.000 claims abstract description 3
- 125000005945 imidazopyridyl group Chemical group 0.000 claims abstract description 3
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 claims abstract description 3
- 125000001041 indolyl group Chemical group 0.000 claims abstract description 3
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 claims abstract description 3
- 125000000842 isoxazolyl group Chemical group 0.000 claims abstract description 3
- 125000002971 oxazolyl group Chemical group 0.000 claims abstract description 3
- 125000003373 pyrazinyl group Chemical group 0.000 claims abstract description 3
- 125000000168 pyrrolyl group Chemical group 0.000 claims abstract description 3
- 125000005493 quinolyl group Chemical group 0.000 claims abstract description 3
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 3
- 125000005942 tetrahydropyridyl group Chemical group 0.000 claims abstract description 3
- 125000003831 tetrazolyl group Chemical group 0.000 claims abstract description 3
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 claims abstract description 3
- 125000000335 thiazolyl group Chemical group 0.000 claims abstract description 3
- 125000001425 triazolyl group Chemical group 0.000 claims abstract description 3
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims abstract 3
- 238000000034 method Methods 0.000 claims description 96
- 238000011282 treatment Methods 0.000 claims description 45
- 206010028980 Neoplasm Diseases 0.000 claims description 38
- 239000002253 acid Substances 0.000 claims description 22
- 201000011510 cancer Diseases 0.000 claims description 22
- 239000003814 drug Substances 0.000 claims description 22
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 13
- 238000002360 preparation method Methods 0.000 claims description 12
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- 230000008569 process Effects 0.000 claims description 9
- 206010030348 Open-Angle Glaucoma Diseases 0.000 claims description 8
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 7
- 201000006366 primary open angle glaucoma Diseases 0.000 claims description 7
- 201000001320 Atherosclerosis Diseases 0.000 claims description 6
- 230000001028 anti-proliverative effect Effects 0.000 claims description 6
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 6
- 229940127089 cytotoxic agent Drugs 0.000 claims description 5
- 239000002254 cytotoxic agent Substances 0.000 claims description 5
- 231100000599 cytotoxic agent Toxicity 0.000 claims description 5
- 239000002834 estrogen receptor modulator Substances 0.000 claims description 5
- 201000008482 osteoarthritis Diseases 0.000 claims description 5
- 102000027483 retinoid hormone receptors Human genes 0.000 claims description 5
- 108091008679 retinoid hormone receptors Proteins 0.000 claims description 5
- 239000000849 selective androgen receptor modulator Substances 0.000 claims description 5
- 201000009273 Endometriosis Diseases 0.000 claims description 4
- 201000004681 Psoriasis Diseases 0.000 claims description 4
- 208000017442 Retinal disease Diseases 0.000 claims description 4
- 206010038923 Retinopathy Diseases 0.000 claims description 4
- 206010040070 Septic Shock Diseases 0.000 claims description 4
- 239000004037 angiogenesis inhibitor Substances 0.000 claims description 4
- 229940121369 angiogenesis inhibitor Drugs 0.000 claims description 4
- 208000037976 chronic inflammation Diseases 0.000 claims description 4
- 230000006020 chronic inflammation Effects 0.000 claims description 4
- 206010020718 hyperplasia Diseases 0.000 claims description 4
- 230000004770 neurodegeneration Effects 0.000 claims description 4
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 4
- 238000001959 radiotherapy Methods 0.000 claims description 4
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 4
- 230000036303 septic shock Effects 0.000 claims description 4
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims description 3
- 239000002671 adjuvant Substances 0.000 claims description 3
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 claims description 3
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims description 3
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 claims description 3
- 102000005454 Dimethylallyltranstransferase Human genes 0.000 claims description 2
- 108010006731 Dimethylallyltranstransferase Proteins 0.000 claims description 2
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 2
- 229940122440 HIV protease inhibitor Drugs 0.000 claims 2
- 239000004030 hiv protease inhibitor Substances 0.000 claims 2
- 229940121649 protein inhibitor Drugs 0.000 claims 2
- 239000012268 protein inhibitor Substances 0.000 claims 2
- 229940075993 receptor modulator Drugs 0.000 claims 2
- 102000004357 Transferases Human genes 0.000 claims 1
- 108090000992 Transferases Proteins 0.000 claims 1
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 claims 1
- 125000004429 atom Chemical group 0.000 abstract description 8
- PBGKNXWGYQPUJK-UHFFFAOYSA-N 4-chloro-2-nitroaniline Chemical compound NC1=CC=C(Cl)C=C1[N+]([O-])=O PBGKNXWGYQPUJK-UHFFFAOYSA-N 0.000 abstract 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 119
- 238000005481 NMR spectroscopy Methods 0.000 description 104
- 238000006243 chemical reaction Methods 0.000 description 101
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 66
- 235000002639 sodium chloride Nutrition 0.000 description 51
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 49
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 45
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 42
- 210000004027 cell Anatomy 0.000 description 42
- 201000010099 disease Diseases 0.000 description 38
- DTQVDTLACAAQTR-DYCDLGHISA-N trifluoroacetic acid-d1 Chemical compound [2H]OC(=O)C(F)(F)F DTQVDTLACAAQTR-DYCDLGHISA-N 0.000 description 38
- DGGFKPSPROBKQP-UHFFFAOYSA-N 2-amino-5-bromo-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(Br)C=C1C(=O)NC1=CC=NC=C1 DGGFKPSPROBKQP-UHFFFAOYSA-N 0.000 description 37
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 30
- 238000012360 testing method Methods 0.000 description 28
- 239000000243 solution Substances 0.000 description 27
- 239000004480 active ingredient Substances 0.000 description 25
- 229910052739 hydrogen Inorganic materials 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 108091000080 Phosphotransferase Proteins 0.000 description 22
- 102000020233 phosphotransferase Human genes 0.000 description 22
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 20
- BMIBJCFFZPYJHF-UHFFFAOYSA-N 2-methoxy-5-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound COC1=NC=C(C)C=C1B1OC(C)(C)C(C)(C)O1 BMIBJCFFZPYJHF-UHFFFAOYSA-N 0.000 description 18
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 16
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 16
- 239000007787 solid Substances 0.000 description 16
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- 238000009472 formulation Methods 0.000 description 15
- 239000008194 pharmaceutical composition Substances 0.000 description 15
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 15
- FEWKHURGVUMFHP-UHFFFAOYSA-N 4-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoic acid Chemical compound NC1=NC=C(C=2C=CC(=CC=2)C(O)=O)C=C1C(=O)NC1=CC=NC=C1 FEWKHURGVUMFHP-UHFFFAOYSA-N 0.000 description 14
- 239000002585 base Substances 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 14
- 239000000843 powder Substances 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 102000001253 Protein Kinase Human genes 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 239000003112 inhibitor Substances 0.000 description 12
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 12
- 108060006633 protein kinase Proteins 0.000 description 12
- 239000003826 tablet Substances 0.000 description 12
- DLIPMSJZHRSUMD-UHFFFAOYSA-N [5-(morpholin-4-ylmethyl)thiophen-2-yl]boronic acid Chemical compound S1C(B(O)O)=CC=C1CN1CCOCC1 DLIPMSJZHRSUMD-UHFFFAOYSA-N 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- 230000001225 therapeutic effect Effects 0.000 description 11
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 10
- 206010006187 Breast cancer Diseases 0.000 description 10
- 208000026310 Breast neoplasm Diseases 0.000 description 10
- 230000000875 corresponding effect Effects 0.000 description 10
- 208000035475 disorder Diseases 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 125000006239 protecting group Chemical group 0.000 description 10
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 10
- NUKYPUAOHBNCPY-UHFFFAOYSA-N 4-aminopyridine Chemical compound NC1=CC=NC=C1 NUKYPUAOHBNCPY-UHFFFAOYSA-N 0.000 description 9
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 239000000460 chlorine Substances 0.000 description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 9
- 239000001257 hydrogen Substances 0.000 description 9
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 9
- 239000011541 reaction mixture Substances 0.000 description 9
- 230000019491 signal transduction Effects 0.000 description 9
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 8
- 201000009030 Carcinoma Diseases 0.000 description 8
- 239000002775 capsule Substances 0.000 description 8
- 238000004113 cell culture Methods 0.000 description 8
- 230000004663 cell proliferation Effects 0.000 description 8
- 238000004587 chromatography analysis Methods 0.000 description 8
- 235000019253 formic acid Nutrition 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical group [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 108010014186 ras Proteins Proteins 0.000 description 7
- 102000016914 ras Proteins Human genes 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 239000012453 solvate Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- 241000282412 Homo Species 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 241000124008 Mammalia Species 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 6
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 230000004783 oxidative metabolism Effects 0.000 description 6
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 239000006228 supernatant Substances 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 5
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 5
- NZMICYAXDXTDJV-UHFFFAOYSA-N 1-(2-methoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound C1=NN(CCOC)C=C1B1OC(C)(C)C(C)(C)O1 NZMICYAXDXTDJV-UHFFFAOYSA-N 0.000 description 5
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical class CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 5
- DUSRIWXDTCMIII-UHFFFAOYSA-N 2-amino-5-bromo-n-(2-ethoxypyridin-4-yl)pyridine-3-carboxamide Chemical compound C1=NC(OCC)=CC(NC(=O)C=2C(=NC=C(Br)C=2)N)=C1 DUSRIWXDTCMIII-UHFFFAOYSA-N 0.000 description 5
- IEPDTLRHISNBLB-UHFFFAOYSA-N 2-amino-5-bromopyridine-3-carboxylic acid Chemical compound NC1=NC=C(Br)C=C1C(O)=O IEPDTLRHISNBLB-UHFFFAOYSA-N 0.000 description 5
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 125000002252 acyl group Chemical group 0.000 description 5
- 150000001298 alcohols Chemical class 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000011230 binding agent Substances 0.000 description 5
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 5
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 230000014509 gene expression Effects 0.000 description 5
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 5
- 238000007327 hydrogenolysis reaction Methods 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 208000008443 pancreatic carcinoma Diseases 0.000 description 5
- 230000026731 phosphorylation Effects 0.000 description 5
- 238000006366 phosphorylation reaction Methods 0.000 description 5
- 229940115272 polyinosinic:polycytidylic acid Drugs 0.000 description 5
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 235000015424 sodium Nutrition 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 5
- KFYIPIUHUMDOFZ-UHFFFAOYSA-N 3-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoic acid Chemical compound NC1=NC=C(C=2C=C(C=CC=2)C(O)=O)C=C1C(=O)NC1=CC=NC=C1 KFYIPIUHUMDOFZ-UHFFFAOYSA-N 0.000 description 4
- MTNAQEKMSVDTAQ-UHFFFAOYSA-N 3-amino-6-bromopyrazine-2-carboxylic acid Chemical compound NC1=NC=C(Br)N=C1C(O)=O MTNAQEKMSVDTAQ-UHFFFAOYSA-N 0.000 description 4
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 4
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 206010009944 Colon cancer Diseases 0.000 description 4
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 4
- 101001011382 Homo sapiens Interferon regulatory factor 3 Proteins 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- 102100029843 Interferon regulatory factor 3 Human genes 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 4
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 4
- 238000010171 animal model Methods 0.000 description 4
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 229910052791 calcium Inorganic materials 0.000 description 4
- 239000011575 calcium Substances 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 4
- 239000007884 disintegrant Substances 0.000 description 4
- 229960004979 fampridine Drugs 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 239000012442 inert solvent Substances 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 210000004072 lung Anatomy 0.000 description 4
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 239000002674 ointment Substances 0.000 description 4
- 201000002528 pancreatic cancer Diseases 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 238000003825 pressing Methods 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 238000003797 solvolysis reaction Methods 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 239000000454 talc Substances 0.000 description 4
- 229910052623 talc Inorganic materials 0.000 description 4
- 235000012222 talc Nutrition 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- NOYDTSIGOXANBU-UHFFFAOYSA-N 2-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoic acid Chemical compound NC1=NC=C(C=2C(=CC=CC=2)C(O)=O)C=C1C(=O)NC1=CC=NC=C1 NOYDTSIGOXANBU-UHFFFAOYSA-N 0.000 description 3
- VQFCBWIBSRXIOR-UHFFFAOYSA-N 2-amino-5-(4-formylphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=CC(C=O)=CC=2)C=C1C(=O)NC1=CC=NC=C1 VQFCBWIBSRXIOR-UHFFFAOYSA-N 0.000 description 3
- LUAMPNIJKVRSAF-UHFFFAOYSA-N 2-amino-5-[1-(2-hydroxyethyl)pyrazol-4-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C2=CN(CCO)N=C2)C=C1C(=O)NC1=CC=NC=C1 LUAMPNIJKVRSAF-UHFFFAOYSA-N 0.000 description 3
- QMBYYYMPAXVILK-UHFFFAOYSA-N 2-amino-5-[1-[2-(oxan-2-yloxy)ethyl]pyrazol-4-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C2=CN(CCOC3OCCCC3)N=C2)C=C1C(=O)NC1=CC=NC=C1 QMBYYYMPAXVILK-UHFFFAOYSA-N 0.000 description 3
- NZLKKUAFPHRJPL-UHFFFAOYSA-N 2-amino-5-[1-[2-(oxan-2-yloxy)ethyl]pyrazol-4-yl]pyridine-3-carboxylic acid Chemical compound C1=C(C(O)=O)C(N)=NC=C1C1=CN(CCOC2OCCCC2)N=C1 NZLKKUAFPHRJPL-UHFFFAOYSA-N 0.000 description 3
- GQPLDGSOAKXPSV-UHFFFAOYSA-N 2-amino-5-bromo-n-(2-methylpyridin-4-yl)pyridine-3-carboxamide Chemical compound C1=NC(C)=CC(NC(=O)C=2C(=NC=C(Br)C=2)N)=C1 GQPLDGSOAKXPSV-UHFFFAOYSA-N 0.000 description 3
- BRPXFVUIIPGOQD-UHFFFAOYSA-N 2-amino-5-bromo-n-furo[3,2-b]pyridin-7-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(Br)C=C1C(=O)NC1=CC=NC2=C1OC=C2 BRPXFVUIIPGOQD-UHFFFAOYSA-N 0.000 description 3
- ZCWSTRWTRMGFFQ-UHFFFAOYSA-N 4-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]-2-fluorobenzoic acid Chemical compound NC1=NC=C(C=2C=C(F)C(C(O)=O)=CC=2)C=C1C(=O)NC1=CC=NC=C1 ZCWSTRWTRMGFFQ-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 description 3
- 208000024827 Alzheimer disease Diseases 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 0 C*1=CC(NC(*C(C(N)=NC=*)=CCC(C=*C(N2*CN*C2)=C)=CN)=*)=CCC1 Chemical compound C*1=CC(NC(*C(C(N)=NC=*)=CCC(C=*C(N2*CN*C2)=C)=CN)=*)=CCC1 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 3
- 101100452374 Mus musculus Ikbke gene Proteins 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 229910002651 NO3 Inorganic materials 0.000 description 3
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 3
- 108700020796 Oncogene Proteins 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 150000001342 alkaline earth metals Chemical class 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 239000003708 ampul Substances 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 3
- 201000008275 breast carcinoma Diseases 0.000 description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 229910052805 deuterium Inorganic materials 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 230000002349 favourable effect Effects 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 206010017758 gastric cancer Diseases 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 208000014829 head and neck neoplasm Diseases 0.000 description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 208000013403 hyperactivity Diseases 0.000 description 3
- 230000003463 hyperproliferative effect Effects 0.000 description 3
- 208000027866 inflammatory disease Diseases 0.000 description 3
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 3
- 201000005249 lung adenocarcinoma Diseases 0.000 description 3
- 210000004324 lymphatic system Anatomy 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000003550 marker Substances 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 239000002207 metabolite Substances 0.000 description 3
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 231100000590 oncogenic Toxicity 0.000 description 3
- 230000002246 oncogenic effect Effects 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 239000006072 paste Substances 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 208000000587 small cell lung carcinoma Diseases 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 235000017550 sodium carbonate Nutrition 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 210000002784 stomach Anatomy 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- VXWBQOJISHAKKM-UHFFFAOYSA-N (4-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=C(C=O)C=C1 VXWBQOJISHAKKM-UHFFFAOYSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- 125000005919 1,2,2-trimethylpropyl group Chemical group 0.000 description 2
- 125000006218 1-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000006219 1-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical group COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- DVJYLGFGQGGRCW-UHFFFAOYSA-N 2-amino-5-(2-aminophenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=CC=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 DVJYLGFGQGGRCW-UHFFFAOYSA-N 0.000 description 2
- GJVSHKMQEGTVTJ-UHFFFAOYSA-N 2-amino-5-(furan-3-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C2=COC=C2)C=C1C(=O)NC1=CC=NC=C1 GJVSHKMQEGTVTJ-UHFFFAOYSA-N 0.000 description 2
- MOWFLFQCPWZPGY-UHFFFAOYSA-N 2-amino-5-[4-[2-methoxyethyl(methyl)carbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)N(C)CCOC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 MOWFLFQCPWZPGY-UHFFFAOYSA-N 0.000 description 2
- KEZNARMJBJOFQB-UHFFFAOYSA-N 2-amino-5-[5-(piperidin-1-ylmethyl)thiophen-2-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCCCC3)=CC=2)C=C1C(=O)NC1=CC=NC=C1 KEZNARMJBJOFQB-UHFFFAOYSA-N 0.000 description 2
- NWMWANWILIXYPE-UHFFFAOYSA-N 2-amino-5-bromo-n-(3-chloropyridin-4-yl)pyridine-3-carboxamide Chemical compound NC1=NC=C(Br)C=C1C(=O)NC1=CC=NC=C1Cl NWMWANWILIXYPE-UHFFFAOYSA-N 0.000 description 2
- QMEXPCISXNXZPH-UHFFFAOYSA-N 2-amino-5-bromo-n-(3-methylpyridin-4-yl)pyridine-3-carboxamide Chemical compound CC1=CN=CC=C1NC(=O)C1=CC(Br)=CN=C1N QMEXPCISXNXZPH-UHFFFAOYSA-N 0.000 description 2
- AQBJMFPFHDWBEY-UHFFFAOYSA-N 2-amino-5-bromo-n-pyridazin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(Br)C=C1C(=O)NC1=CC=NN=C1 AQBJMFPFHDWBEY-UHFFFAOYSA-N 0.000 description 2
- WAGKKVIMOUZTOC-UHFFFAOYSA-N 2-amino-5-bromo-n-pyrimidin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(Br)C=C1C(=O)NC1=CC=NC=N1 WAGKKVIMOUZTOC-UHFFFAOYSA-N 0.000 description 2
- FXOQQOTXASSILN-UHFFFAOYSA-N 2-amino-n-pyridin-4-yl-5-(1h-pyrrol-3-yl)pyridine-3-carboxamide Chemical compound NC1=NC=C(C2=CNC=C2)C=C1C(=O)NC1=CC=NC=C1 FXOQQOTXASSILN-UHFFFAOYSA-N 0.000 description 2
- DYCXKPUDVYQTMX-UHFFFAOYSA-N 2-amino-n-pyridin-4-yl-5-(2-sulfamoylphenyl)pyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C(=CC=CC=2)S(N)(=O)=O)C=C1C(=O)NC1=CC=NC=C1 DYCXKPUDVYQTMX-UHFFFAOYSA-N 0.000 description 2
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 2
- KOHBEDRJXKOYHL-UHFFFAOYSA-N 2-methoxy-n-methylethanamine Chemical compound CNCCOC KOHBEDRJXKOYHL-UHFFFAOYSA-N 0.000 description 2
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 2
- 125000005916 2-methylpentyl group Chemical group 0.000 description 2
- LFGSHASYSHJIGU-UHFFFAOYSA-N 3-amino-6-[5-(morpholin-4-ylmethyl)thiophen-2-yl]-n-pyridin-4-ylpyrazine-2-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCOCC3)=CC=2)N=C1C(=O)NC1=CC=NC=C1 LFGSHASYSHJIGU-UHFFFAOYSA-N 0.000 description 2
- 125000005917 3-methylpentyl group Chemical group 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- 241000699800 Cricetinae Species 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical compound [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102100030708 GTPase KRas Human genes 0.000 description 2
- 101000584612 Homo sapiens GTPase KRas Proteins 0.000 description 2
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 2
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- 206010024305 Leukaemia monocytic Diseases 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- 239000007993 MOPS buffer Substances 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- 102000008230 Toll-like receptor 3 Human genes 0.000 description 2
- 108010060885 Toll-like receptor 3 Proteins 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 2
- 239000000853 adhesive Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 229940072056 alginate Drugs 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- 229940050390 benzoate Drugs 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 238000001574 biopsy Methods 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 230000000711 cancerogenic effect Effects 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 231100000315 carcinogenic Toxicity 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 2
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 2
- 229960002023 chloroprocaine Drugs 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 125000004431 deuterium atom Chemical group 0.000 description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 2
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 2
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 229940012356 eye drops Drugs 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 238000002875 fluorescence polarization Methods 0.000 description 2
- 208000010749 gastric carcinoma Diseases 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 230000009368 gene silencing by RNA Effects 0.000 description 2
- 208000005017 glioblastoma Diseases 0.000 description 2
- 229940050410 gluconate Drugs 0.000 description 2
- 229960002989 glutamic acid Drugs 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 210000003128 head Anatomy 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000015788 innate immune response Effects 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 230000005445 isotope effect Effects 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 201000005264 laryngeal carcinoma Diseases 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 201000005296 lung carcinoma Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 208000026037 malignant tumor of neck Diseases 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 2
- 229960003194 meglumine Drugs 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- CMPWXMFWOPOFHN-UHFFFAOYSA-N methyl 4-[[2-amino-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carbonyl]amino]pyridine-3-carboxylate Chemical compound COC(=O)C1=CN=CC=C1NC(=O)C1=CC(C=2SC(CN3CCOCC3)=CC=2)=CN=C1N CMPWXMFWOPOFHN-UHFFFAOYSA-N 0.000 description 2
- 229940095102 methyl benzoate Drugs 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 201000006894 monocytic leukemia Diseases 0.000 description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 150000002828 nitro derivatives Chemical class 0.000 description 2
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 2
- 229910000510 noble metal Inorganic materials 0.000 description 2
- 229940049964 oleate Drugs 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 230000036470 plasma concentration Effects 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 2
- 229960004919 procaine Drugs 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 230000009822 protein phosphorylation Effects 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 150000003222 pyridines Chemical class 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000003462 sulfoxides Chemical class 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- ZLDLXTVBAOZFLF-UHFFFAOYSA-N tert-butyl n-[2-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]phenyl]carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 ZLDLXTVBAOZFLF-UHFFFAOYSA-N 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000011287 therapeutic dose Methods 0.000 description 2
- 201000002510 thyroid cancer Diseases 0.000 description 2
- 210000001685 thyroid gland Anatomy 0.000 description 2
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229960000281 trometamol Drugs 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 210000003932 urinary bladder Anatomy 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- BMKDZUISNHGIBY-ZETCQYMHSA-N (+)-dexrazoxane Chemical compound C([C@H](C)N1CC(=O)NC(=O)C1)N1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-ZETCQYMHSA-N 0.000 description 1
- JKFZMIQMKFWJAY-RQJQXFIZSA-N (1r,3s,5z)-5-[(2e)-2-[(3as,7as)-1-[(2r)-6-hydroxy-6-methylhept-4-yn-2-yl]-7a-methyl-3a,5,6,7-tetrahydro-3h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C1(/[C@@H]2CC=C([C@]2(CCC1)C)[C@@H](CC#CC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C JKFZMIQMKFWJAY-RQJQXFIZSA-N 0.000 description 1
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 1
- DGUWACLYDSWXRZ-UHFFFAOYSA-N (2-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC=C1C=O DGUWACLYDSWXRZ-UHFFFAOYSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- ZUQBAQVRAURMCL-DOMZBBRYSA-N (2s)-2-[[4-[2-[(6r)-2-amino-4-oxo-5,6,7,8-tetrahydro-1h-pyrido[2,3-d]pyrimidin-6-yl]ethyl]benzoyl]amino]pentanedioic acid Chemical compound C([C@@H]1CC=2C(=O)N=C(NC=2NC1)N)CC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 ZUQBAQVRAURMCL-DOMZBBRYSA-N 0.000 description 1
- PSVUJBVBCOISSP-SPFKKGSWSA-N (2s,3r,4s,5s,6r)-2-bis(2-chloroethylamino)phosphoryloxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OC[C@H]1O[C@@H](OP(=O)(NCCCl)NCCCl)[C@H](O)[C@@H](O)[C@@H]1O PSVUJBVBCOISSP-SPFKKGSWSA-N 0.000 description 1
- REHVCPNQQBDOJJ-UHFFFAOYSA-N (3-ethoxycarbonylphenyl)boronic acid Chemical compound CCOC(=O)C1=CC=CC(B(O)O)=C1 REHVCPNQQBDOJJ-UHFFFAOYSA-N 0.000 description 1
- YZYGXFXSMDUXJT-UHFFFAOYSA-N (3-fluoro-4-methoxycarbonylphenyl)boronic acid Chemical compound COC(=O)C1=CC=C(B(O)O)C=C1F YZYGXFXSMDUXJT-UHFFFAOYSA-N 0.000 description 1
- HJBGZJMKTOMQRR-UHFFFAOYSA-N (3-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC(C=O)=C1 HJBGZJMKTOMQRR-UHFFFAOYSA-N 0.000 description 1
- WFWQWTPAPNEOFE-UHFFFAOYSA-N (3-hydroxyphenyl)boronic acid Chemical compound OB(O)C1=CC=CC(O)=C1 WFWQWTPAPNEOFE-UHFFFAOYSA-N 0.000 description 1
- ALTLCJHSJMGSLT-UHFFFAOYSA-N (3-methoxycarbonylphenyl)boronic acid Chemical compound COC(=O)C1=CC=CC(B(O)O)=C1 ALTLCJHSJMGSLT-UHFFFAOYSA-N 0.000 description 1
- QBYGJJSFMOVYOA-UHFFFAOYSA-N (4-boronophenyl)azanium;chloride Chemical compound Cl.NC1=CC=C(B(O)O)C=C1 QBYGJJSFMOVYOA-UHFFFAOYSA-N 0.000 description 1
- ZLNFACCFYUFTLD-UHFFFAOYSA-N (4-ethoxycarbonylphenyl)boronic acid Chemical compound CCOC(=O)C1=CC=C(B(O)O)C=C1 ZLNFACCFYUFTLD-UHFFFAOYSA-N 0.000 description 1
- COIQUVGFTILYGA-UHFFFAOYSA-N (4-hydroxyphenyl)boronic acid Chemical compound OB(O)C1=CC=C(O)C=C1 COIQUVGFTILYGA-UHFFFAOYSA-N 0.000 description 1
- XTVMZKCCFLOJBL-UHFFFAOYSA-N (4-pyrazol-1-ylphenyl)boronic acid Chemical compound C1=CC(B(O)O)=CC=C1N1N=CC=C1 XTVMZKCCFLOJBL-UHFFFAOYSA-N 0.000 description 1
- NRIYPIBRPGAWDD-UHFFFAOYSA-N (5-methylthiophen-2-yl)boronic acid Chemical compound CC1=CC=C(B(O)O)S1 NRIYPIBRPGAWDD-UHFFFAOYSA-N 0.000 description 1
- ZKSNZYLCOXUJIR-VOKUKXJJSA-N (5s,5ar,8ar,9r)-5-[[(2r,4ar,6r,7r,8r,8as)-7-(dimethylamino)-8-hydroxy-2-methyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxin-6-yl]oxy]-9-(4-hydroxy-3,5-dimethoxyphenyl)-5a,6,8a,9-tetrahydro-5h-[2]benzofuro[6,5-f][1,3]benzodioxol-8-one Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)N(C)C)[C@@H]3[C@@H]2C(OC3)=O)=C1 ZKSNZYLCOXUJIR-VOKUKXJJSA-N 0.000 description 1
- DLROLUIVVKTFPW-LVEBQJTPSA-N (5s,5as,8ar,9r)-9-(4-hydroxy-3,5-dimethoxyphenyl)-5-(4-nitroanilino)-5a,6,8a,9-tetrahydro-5h-[2]benzofuro[5,6-f][1,3]benzodioxol-8-one Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](NC=3C=CC(=CC=3)[N+]([O-])=O)[C@@H]3[C@@H]2C(OC3)=O)=C1 DLROLUIVVKTFPW-LVEBQJTPSA-N 0.000 description 1
- WTSKMKRYHATLLL-UHFFFAOYSA-N (6-benzoyloxy-3-cyanopyridin-2-yl) 3-[3-(ethoxymethyl)-5-fluoro-2,6-dioxopyrimidine-1-carbonyl]benzoate Chemical compound O=C1N(COCC)C=C(F)C(=O)N1C(=O)C1=CC=CC(C(=O)OC=2C(=CC=C(OC(=O)C=3C=CC=CC=3)N=2)C#N)=C1 WTSKMKRYHATLLL-UHFFFAOYSA-N 0.000 description 1
- OPBPMGYBSDKJBT-DQHLZUIQSA-N (7s,9r,10r)-9-ethyl-4,6,9,10,11-pentahydroxy-7-[(2r,4s,5s,6s)-5-hydroxy-6-methyl-4-morpholin-4-yloxan-2-yl]oxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@]([C@@H](C3=C(O)C=4C(=O)C5=CC=CC(O)=C5C(=O)C=4C(O)=C32)O)(O)CC)CCOCC1 OPBPMGYBSDKJBT-DQHLZUIQSA-N 0.000 description 1
- BSRQHWFOFMAZRL-BODGVHBXSA-N (7s,9s)-7-[(2r,4s,5s,6s)-5-[(2s,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-4-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-8,10-dihydro-7h-tetracene-5,12-dione;hydron;chloride Chemical compound Cl.C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@@H](O)C[C@H](O[C@@H]2C3=C(O)C=4C(=O)C5=CC=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)O[C@H]1C BSRQHWFOFMAZRL-BODGVHBXSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- ZGNLFUXWZJGETL-YUSKDDKASA-N (Z)-[(2S)-2-amino-2-carboxyethyl]-hydroxyimino-oxidoazanium Chemical compound N[C@@H](C\[N+]([O-])=N\O)C(O)=O ZGNLFUXWZJGETL-YUSKDDKASA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- KPZGRMZPZLOPBS-UHFFFAOYSA-N 1,3-dichloro-2,2-bis(chloromethyl)propane Chemical compound ClCC(CCl)(CCl)CCl KPZGRMZPZLOPBS-UHFFFAOYSA-N 0.000 description 1
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 1
- IWEGDQUCWQFKHS-UHFFFAOYSA-N 1-(1,3-dioxolan-2-ylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(CC2OCCO2)N=C1 IWEGDQUCWQFKHS-UHFFFAOYSA-N 0.000 description 1
- MZNMZWZGUGFQJP-UHFFFAOYSA-N 1-[11-(dodecylamino)-10-hydroxyundecyl]-3,7-dimethylpurine-2,6-dione Chemical compound O=C1N(CCCCCCCCCC(O)CNCCCCCCCCCCCC)C(=O)N(C)C2=C1N(C)C=N2 MZNMZWZGUGFQJP-UHFFFAOYSA-N 0.000 description 1
- YWRBLSYOLXMZGA-UHFFFAOYSA-N 1-[2-(oxan-2-yloxy)ethyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(CCOC2OCCCC2)N=C1 YWRBLSYOLXMZGA-UHFFFAOYSA-N 0.000 description 1
- GVSCNAOZDQCWJJ-UHFFFAOYSA-N 1-[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC(N2N=CC=C2)=C1 GVSCNAOZDQCWJJ-UHFFFAOYSA-N 0.000 description 1
- YRVXXFZZQUNLSQ-UHFFFAOYSA-N 1-[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfonylpiperidin-4-ol Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC(S(=O)(=O)N2CCC(O)CC2)=C1 YRVXXFZZQUNLSQ-UHFFFAOYSA-N 0.000 description 1
- KDPVLCCGXRCQCV-UHFFFAOYSA-N 1-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl]methyl]piperidine Chemical compound O1C(C)(C)C(C)(C)OB1C(S1)=CC=C1CN1CCCCC1 KDPVLCCGXRCQCV-UHFFFAOYSA-N 0.000 description 1
- VWOUUXTUYSWOCT-UHFFFAOYSA-N 1-[[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl]methyl]pyrrolidine Chemical compound O1C(C)(C)C(C)(C)OB1C(S1)=CC=C1CN1CCCC1 VWOUUXTUYSWOCT-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical compound CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- ROZCIVXTLACYNY-UHFFFAOYSA-N 2,3,4,5,6-pentafluoro-n-(3-fluoro-4-methoxyphenyl)benzenesulfonamide Chemical compound C1=C(F)C(OC)=CC=C1NS(=O)(=O)C1=C(F)C(F)=C(F)C(F)=C1F ROZCIVXTLACYNY-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- 125000003870 2-(1-piperidinyl)ethoxy group Chemical group [*]OC([H])([H])C([H])([H])N1C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- VLROJECCXBBKPZ-UHFFFAOYSA-N 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC=C1O VLROJECCXBBKPZ-UHFFFAOYSA-N 0.000 description 1
- XWNJMSJGJFSGRY-UHFFFAOYSA-N 2-(benzylamino)-3,7-dihydropurin-6-one Chemical compound N1C=2N=CNC=2C(=O)N=C1NCC1=CC=CC=C1 XWNJMSJGJFSGRY-UHFFFAOYSA-N 0.000 description 1
- QUNOQBDEVTWCTA-UHFFFAOYSA-N 2-[2-[3-[2-(1,3-dioxobenzo[de]isoquinolin-2-yl)ethylamino]propylamino]ethyl]benzo[de]isoquinoline-1,3-dione Chemical compound C1=CC(C(=O)N(CCNCCCNCCN2C(C=3C=CC=C4C=CC=C(C=34)C2=O)=O)C2=O)=C3C2=CC=CC3=C1 QUNOQBDEVTWCTA-UHFFFAOYSA-N 0.000 description 1
- XFYVLZVRMQJRCB-UHFFFAOYSA-N 2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazol-1-yl]acetamide Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(CC(N)=O)N=C1 XFYVLZVRMQJRCB-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- QDJHWYVWXZCCGP-UHFFFAOYSA-N 2-amino-5-(1-benzofuran-3-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C3=CC=CC=C3OC=2)C=C1C(=O)NC1=CC=NC=C1 QDJHWYVWXZCCGP-UHFFFAOYSA-N 0.000 description 1
- IGGCQUOVHYNLKC-UHFFFAOYSA-N 2-amino-5-(1-benzothiophen-2-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2SC3=CC=CC=C3C=2)C=C1C(=O)NC1=CC=NC=C1 IGGCQUOVHYNLKC-UHFFFAOYSA-N 0.000 description 1
- ZIRAJQJDABKSFH-UHFFFAOYSA-N 2-amino-5-(1-benzylpyrazol-4-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C2=CN(CC=3C=CC=CC=3)N=C2)C=C1C(=O)NC1=CC=NC=C1 ZIRAJQJDABKSFH-UHFFFAOYSA-N 0.000 description 1
- DWBFATQGRAYQFC-UHFFFAOYSA-N 2-amino-5-(1-methylpyrazol-3-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound CN1C=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=N1 DWBFATQGRAYQFC-UHFFFAOYSA-N 0.000 description 1
- VSGIYMAWHNSRDN-UHFFFAOYSA-N 2-amino-5-(1-methylpyrazol-4-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=NN(C)C=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 VSGIYMAWHNSRDN-UHFFFAOYSA-N 0.000 description 1
- HQNUSOQVFGXSKJ-UHFFFAOYSA-N 2-amino-5-(1-methylpyrrol-2-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound CN1C=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 HQNUSOQVFGXSKJ-UHFFFAOYSA-N 0.000 description 1
- PJBRSXFQFUBGPW-UHFFFAOYSA-N 2-amino-5-(1h-indol-2-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2NC3=CC=CC=C3C=2)C=C1C(=O)NC1=CC=NC=C1 PJBRSXFQFUBGPW-UHFFFAOYSA-N 0.000 description 1
- BUOPARGTGBRMBL-UHFFFAOYSA-N 2-amino-5-(1h-pyrazol-4-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C2=CNN=C2)C=C1C(=O)NC1=CC=NC=C1 BUOPARGTGBRMBL-UHFFFAOYSA-N 0.000 description 1
- PIRUNCQFLKAMMO-UHFFFAOYSA-N 2-amino-5-(2-formylphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C(=CC=CC=2)C=O)C=C1C(=O)NC1=CC=NC=C1 PIRUNCQFLKAMMO-UHFFFAOYSA-N 0.000 description 1
- HZMZVFPIFFKUGZ-UHFFFAOYSA-N 2-amino-5-(2-hydroxyphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C(=CC=CC=2)O)C=C1C(=O)NC1=CC=NC=C1 HZMZVFPIFFKUGZ-UHFFFAOYSA-N 0.000 description 1
- AWUPQCCQFJACKG-UHFFFAOYSA-N 2-amino-5-(3,5-dimethyl-1h-pyrazol-4-yl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound CC1=NNC(C)=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 AWUPQCCQFJACKG-UHFFFAOYSA-N 0.000 description 1
- DYSJFRZBYRYUCO-UHFFFAOYSA-N 2-amino-5-(3-aminophenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 DYSJFRZBYRYUCO-UHFFFAOYSA-N 0.000 description 1
- ARMYQZBDNOLOQT-UHFFFAOYSA-N 2-amino-5-(3-carbamoylphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC(=O)C1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 ARMYQZBDNOLOQT-UHFFFAOYSA-N 0.000 description 1
- JNEFOEIYOOSMPT-UHFFFAOYSA-N 2-amino-5-(3-formylphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=C(C=O)C=CC=2)C=C1C(=O)NC1=CC=NC=C1 JNEFOEIYOOSMPT-UHFFFAOYSA-N 0.000 description 1
- GJRMXVZWYBJIEK-UHFFFAOYSA-N 2-amino-5-(3-hydroxyphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=C(O)C=CC=2)C=C1C(=O)NC1=CC=NC=C1 GJRMXVZWYBJIEK-UHFFFAOYSA-N 0.000 description 1
- ABSILEKDPLDCAC-UHFFFAOYSA-N 2-amino-5-(3-pyrazol-1-ylphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=C(C=CC=2)N2N=CC=C2)C=C1C(=O)NC1=CC=NC=C1 ABSILEKDPLDCAC-UHFFFAOYSA-N 0.000 description 1
- ZYABARSOZHAPRS-UHFFFAOYSA-N 2-amino-5-(4-aminophenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(N)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 ZYABARSOZHAPRS-UHFFFAOYSA-N 0.000 description 1
- UCTCDQXMIPNKLE-UHFFFAOYSA-N 2-amino-5-(4-carbamoylphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)N)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 UCTCDQXMIPNKLE-UHFFFAOYSA-N 0.000 description 1
- NVARDTPVYGIUAK-UHFFFAOYSA-N 2-amino-5-(4-hydroxyphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=CC(O)=CC=2)C=C1C(=O)NC1=CC=NC=C1 NVARDTPVYGIUAK-UHFFFAOYSA-N 0.000 description 1
- RRQPRTMYJUAVMC-UHFFFAOYSA-N 2-amino-5-(4-pyrazol-1-ylphenyl)-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=CC(=CC=2)N2N=CC=C2)C=C1C(=O)NC1=CC=NC=C1 RRQPRTMYJUAVMC-UHFFFAOYSA-N 0.000 description 1
- ZTLALBZROUPCEA-UHFFFAOYSA-N 2-amino-5-[1-(2-amino-2-oxoethyl)pyrazol-4-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=NN(CC(=O)N)C=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 ZTLALBZROUPCEA-UHFFFAOYSA-N 0.000 description 1
- JNYKPUGUQRHMIP-UHFFFAOYSA-N 2-amino-5-[1-(2-methoxyethyl)pyrazol-4-yl]-n-(2-methylpyridin-4-yl)pyridine-3-carboxamide Chemical compound C1=NN(CCOC)C=C1C1=CN=C(N)C(C(=O)NC=2C=C(C)N=CC=2)=C1 JNYKPUGUQRHMIP-UHFFFAOYSA-N 0.000 description 1
- XUWPXRANFZZJDO-UHFFFAOYSA-N 2-amino-5-[1-(2-methoxyethyl)pyrazol-4-yl]-n-(3-methylpyridin-4-yl)pyridine-3-carboxamide Chemical compound C1=NN(CCOC)C=C1C1=CN=C(N)C(C(=O)NC=2C(=CN=CC=2)C)=C1 XUWPXRANFZZJDO-UHFFFAOYSA-N 0.000 description 1
- TXFDIBNKJINUPM-UHFFFAOYSA-N 2-amino-5-[1-(2-methoxyethyl)pyrazol-4-yl]-n-pyridazin-4-ylpyridine-3-carboxamide Chemical compound C1=NN(CCOC)C=C1C1=CN=C(N)C(C(=O)NC=2C=NN=CC=2)=C1 TXFDIBNKJINUPM-UHFFFAOYSA-N 0.000 description 1
- QKKSJPYVDJXBQN-UHFFFAOYSA-N 2-amino-5-[1-(2-methoxyethyl)pyrazol-4-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=NN(CCOC)C=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 QKKSJPYVDJXBQN-UHFFFAOYSA-N 0.000 description 1
- KZXWFUQZOSWUCY-UHFFFAOYSA-N 2-amino-5-[1-(2-morpholin-4-ylethyl)pyrazol-4-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C2=CN(CCN3CCOCC3)N=C2)C=C1C(=O)NC1=CC=NC=C1 KZXWFUQZOSWUCY-UHFFFAOYSA-N 0.000 description 1
- OSNTWBYUHHEIEN-UHFFFAOYSA-N 2-amino-5-[2-(diethylcarbamoyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound CCN(CC)C(=O)C1=CC=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 OSNTWBYUHHEIEN-UHFFFAOYSA-N 0.000 description 1
- PSLOFOVVSFJXNI-UHFFFAOYSA-N 2-amino-5-[2-(ethylsulfamoyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound CCNS(=O)(=O)C1=CC=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 PSLOFOVVSFJXNI-UHFFFAOYSA-N 0.000 description 1
- IMNMNIUTDUKZDE-UHFFFAOYSA-N 2-amino-5-[3-(4-hydroxypiperidin-1-yl)sulfonylphenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=C(C=CC=2)S(=O)(=O)N2CCC(O)CC2)C=C1C(=O)NC1=CC=NC=C1 IMNMNIUTDUKZDE-UHFFFAOYSA-N 0.000 description 1
- OIGQPJIJVQOWKU-UHFFFAOYSA-N 2-amino-5-[3-(4-methylphenyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C)=CC=C1C1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 OIGQPJIJVQOWKU-UHFFFAOYSA-N 0.000 description 1
- CLRDJFABEAWJEA-UHFFFAOYSA-N 2-amino-5-[3-(aminomethyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NCC1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 CLRDJFABEAWJEA-UHFFFAOYSA-N 0.000 description 1
- LNVIMMSSULYKOT-UHFFFAOYSA-N 2-amino-5-[3-(diethylcarbamoyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound CCN(CC)C(=O)C1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 LNVIMMSSULYKOT-UHFFFAOYSA-N 0.000 description 1
- AUSVSBMPJIHXPC-UHFFFAOYSA-N 2-amino-5-[3-(hydroxymethyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=C(CO)C=CC=2)C=C1C(=O)NC1=CC=NC=C1 AUSVSBMPJIHXPC-UHFFFAOYSA-N 0.000 description 1
- JXJRZMHXHPJSQC-UHFFFAOYSA-N 2-amino-5-[3-[2-methoxyethyl(methyl)carbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound COCCN(C)C(=O)C1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 JXJRZMHXHPJSQC-UHFFFAOYSA-N 0.000 description 1
- BXVBTVBEBIPUNG-UHFFFAOYSA-N 2-amino-5-[3-fluoro-4-[2-methoxyethyl(methyl)carbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=C(F)C(C(=O)N(C)CCOC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 BXVBTVBEBIPUNG-UHFFFAOYSA-N 0.000 description 1
- RRGSYJMAFSLHGH-UHFFFAOYSA-N 2-amino-5-[4-(2-aminoethylcarbamoyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)NCCN)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 RRGSYJMAFSLHGH-UHFFFAOYSA-N 0.000 description 1
- ADKMAFGYKZIFHH-UHFFFAOYSA-N 2-amino-5-[4-(2-methoxyethylcarbamoyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)NCCOC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 ADKMAFGYKZIFHH-UHFFFAOYSA-N 0.000 description 1
- PEOMWWQSESFHCQ-UHFFFAOYSA-N 2-amino-5-[4-(3-methoxypropylcarbamoyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)NCCCOC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 PEOMWWQSESFHCQ-UHFFFAOYSA-N 0.000 description 1
- KEWBKCZSLZPQRY-UHFFFAOYSA-N 2-amino-5-[4-(aminomethyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(CN)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 KEWBKCZSLZPQRY-UHFFFAOYSA-N 0.000 description 1
- RIXOODYIPKBCHT-UHFFFAOYSA-N 2-amino-5-[4-(diethylcarbamoyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)N(CC)CC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 RIXOODYIPKBCHT-UHFFFAOYSA-N 0.000 description 1
- MEDODHFQPOHQCU-UHFFFAOYSA-N 2-amino-5-[4-(hydroxymethyl)phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=CC(CO)=CC=2)C=C1C(=O)NC1=CC=NC=C1 MEDODHFQPOHQCU-UHFFFAOYSA-N 0.000 description 1
- PJMOMAQCHBHIED-UHFFFAOYSA-N 2-amino-5-[4-[2-(dimethylamino)ethyl-ethylcarbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)N(CCN(C)C)CC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 PJMOMAQCHBHIED-UHFFFAOYSA-N 0.000 description 1
- MQXGUYZAOFLZEX-UHFFFAOYSA-N 2-amino-5-[4-[2-(dimethylamino)ethyl-methylcarbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)N(C)CCN(C)C)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 MQXGUYZAOFLZEX-UHFFFAOYSA-N 0.000 description 1
- NHNOHNJHTZRPJD-UHFFFAOYSA-N 2-amino-5-[4-[bis(2-hydroxyethyl)carbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=CC(=CC=2)C(=O)N(CCO)CCO)C=C1C(=O)NC1=CC=NC=C1 NHNOHNJHTZRPJD-UHFFFAOYSA-N 0.000 description 1
- DATWKHYWCCNMCU-UHFFFAOYSA-N 2-amino-5-[4-[bis(2-methoxyethyl)carbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C1=CC(C(=O)N(CCOC)CCOC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 DATWKHYWCCNMCU-UHFFFAOYSA-N 0.000 description 1
- PBIDDCFAQOZAMU-UHFFFAOYSA-N 2-amino-5-[4-[methyl-[(3-methylimidazol-4-yl)methyl]carbamoyl]phenyl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound C=1C=C(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)C=CC=1C(=O)N(C)CC1=CN=CN1C PBIDDCFAQOZAMU-UHFFFAOYSA-N 0.000 description 1
- ISADMOXHYKRVEW-UHFFFAOYSA-N 2-amino-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]-n-pyridazin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCOCC3)=CC=2)C=C1C(=O)NC1=CC=NN=C1 ISADMOXHYKRVEW-UHFFFAOYSA-N 0.000 description 1
- BXMGOFSMDOETKZ-UHFFFAOYSA-N 2-amino-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCOCC3)=CC=2)C=C1C(=O)NC1=CC=NC=C1 BXMGOFSMDOETKZ-UHFFFAOYSA-N 0.000 description 1
- BSTMMSHLHCVWOV-UHFFFAOYSA-N 2-amino-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]-n-pyrimidin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCOCC3)=CC=2)C=C1C(=O)NC1=CC=NC=N1 BSTMMSHLHCVWOV-UHFFFAOYSA-N 0.000 description 1
- YXNUVQPEHUKSLS-UHFFFAOYSA-N 2-amino-5-[5-(morpholin-4-ylmethyl)thiophen-3-yl]-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=C(CN3CCOCC3)SC=2)C=C1C(=O)NC1=CC=NC=C1 YXNUVQPEHUKSLS-UHFFFAOYSA-N 0.000 description 1
- CUZMBULNHNVCJE-UHFFFAOYSA-N 2-amino-5-bromo-n-(2,6-dimethylpyridin-4-yl)pyridine-3-carboxamide Chemical compound CC1=NC(C)=CC(NC(=O)C=2C(=NC=C(Br)C=2)N)=C1 CUZMBULNHNVCJE-UHFFFAOYSA-N 0.000 description 1
- NTZSYPNVZSSBBV-UHFFFAOYSA-N 2-amino-5-bromo-n-(4-methoxyphenyl)pyridine-3-carboxamide Chemical compound C1=CC(OC)=CC=C1NC(=O)C1=CC(Br)=CN=C1N NTZSYPNVZSSBBV-UHFFFAOYSA-N 0.000 description 1
- XHTZECPADAGUPT-UHFFFAOYSA-N 2-amino-5-bromo-n-(6-methoxypyridin-3-yl)pyridine-3-carboxamide Chemical compound C1=NC(OC)=CC=C1NC(=O)C1=CC(Br)=CN=C1N XHTZECPADAGUPT-UHFFFAOYSA-N 0.000 description 1
- YQJHBHLODKPXHK-UHFFFAOYSA-N 2-amino-n-(2,6-dimethylpyridin-4-yl)-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound CC1=NC(C)=CC(NC(=O)C=2C(=NC=C(C=2)C=2SC(CN3CCOCC3)=CC=2)N)=C1 YQJHBHLODKPXHK-UHFFFAOYSA-N 0.000 description 1
- CMRSPQFHPQBQIK-UHFFFAOYSA-N 2-amino-n-(2-ethoxypyridin-4-yl)-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound C1=NC(OCC)=CC(NC(=O)C=2C(=NC=C(C=2)C=2SC(CN3CCOCC3)=CC=2)N)=C1 CMRSPQFHPQBQIK-UHFFFAOYSA-N 0.000 description 1
- KGMZRZBOGQECSK-UHFFFAOYSA-N 2-amino-n-(2-methylpyridin-4-yl)-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound C1=NC(C)=CC(NC(=O)C=2C(=NC=C(C=2)C=2SC(CN3CCOCC3)=CC=2)N)=C1 KGMZRZBOGQECSK-UHFFFAOYSA-N 0.000 description 1
- RQMDUJAJDRLURS-UHFFFAOYSA-N 2-amino-n-(2-methylpyridin-4-yl)-5-[5-(morpholin-4-ylmethyl)thiophen-3-yl]pyridine-3-carboxamide Chemical compound C1=NC(C)=CC(NC(=O)C=2C(=NC=C(C=2)C=2C=C(CN3CCOCC3)SC=2)N)=C1 RQMDUJAJDRLURS-UHFFFAOYSA-N 0.000 description 1
- IGJNKGGEJHEBLW-UHFFFAOYSA-N 2-amino-n-(3-chloropyridin-4-yl)-5-[1-(2-methoxyethyl)pyrazol-4-yl]pyridine-3-carboxamide Chemical compound C1=NN(CCOC)C=C1C1=CN=C(N)C(C(=O)NC=2C(=CN=CC=2)Cl)=C1 IGJNKGGEJHEBLW-UHFFFAOYSA-N 0.000 description 1
- BHMSKCZYCFPYKC-UHFFFAOYSA-N 2-amino-n-(3-chloropyridin-4-yl)-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCOCC3)=CC=2)C=C1C(=O)NC1=CC=NC=C1Cl BHMSKCZYCFPYKC-UHFFFAOYSA-N 0.000 description 1
- XNLQEOMWHKGKJO-UHFFFAOYSA-N 2-amino-n-(3-methylpyridin-4-yl)-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound CC1=CN=CC=C1NC(=O)C1=CC(C=2SC(CN3CCOCC3)=CC=2)=CN=C1N XNLQEOMWHKGKJO-UHFFFAOYSA-N 0.000 description 1
- UANVBHMEXMGHSB-UHFFFAOYSA-N 2-amino-n-(4-methoxyphenyl)-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound C1=CC(OC)=CC=C1NC(=O)C1=CC(C=2SC(CN3CCOCC3)=CC=2)=CN=C1N UANVBHMEXMGHSB-UHFFFAOYSA-N 0.000 description 1
- NUVZIKCAHXIPAU-UHFFFAOYSA-N 2-amino-n-(6-methoxypyridin-3-yl)-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound C1=NC(OC)=CC=C1NC(=O)C1=CC(C=2SC(CN3CCOCC3)=CC=2)=CN=C1N NUVZIKCAHXIPAU-UHFFFAOYSA-N 0.000 description 1
- DPNJNXJSLFVKOS-UHFFFAOYSA-N 2-amino-n-[3-(methylcarbamoyl)pyridin-4-yl]-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound CNC(=O)C1=CN=CC=C1NC(=O)C1=CC(C=2SC(CN3CCOCC3)=CC=2)=CN=C1N DPNJNXJSLFVKOS-UHFFFAOYSA-N 0.000 description 1
- FIRCCCGMXJDUAZ-UHFFFAOYSA-N 2-amino-n-furo[3,2-b]pyridin-7-yl-5-[1-(2-methoxyethyl)pyrazol-4-yl]pyridine-3-carboxamide Chemical compound C1=NN(CCOC)C=C1C1=CN=C(N)C(C(=O)NC=2C=3OC=CC=3N=CC=2)=C1 FIRCCCGMXJDUAZ-UHFFFAOYSA-N 0.000 description 1
- SVSGSVOKSPDTIL-UHFFFAOYSA-N 2-amino-n-furo[3,2-b]pyridin-7-yl-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound C1=C(C(=O)NC=2C=3OC=CC=3N=CC=2)C(N)=NC=C1C(S1)=CC=C1CN1CCOCC1 SVSGSVOKSPDTIL-UHFFFAOYSA-N 0.000 description 1
- DPUDZPAMJPBTGY-UHFFFAOYSA-N 2-amino-n-pyridin-4-yl-5-(1h-pyrrol-2-yl)pyridine-3-carboxamide Chemical compound NC1=NC=C(C=2NC=CC=2)C=C1C(=O)NC1=CC=NC=C1 DPUDZPAMJPBTGY-UHFFFAOYSA-N 0.000 description 1
- XIXUGUKTZKBHOE-UHFFFAOYSA-N 2-amino-n-pyridin-4-yl-5-(3-sulfamoylphenyl)pyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=C(C=CC=2)S(N)(=O)=O)C=C1C(=O)NC1=CC=NC=C1 XIXUGUKTZKBHOE-UHFFFAOYSA-N 0.000 description 1
- MRXIXCQHUIFLME-UHFFFAOYSA-N 2-amino-n-pyridin-4-yl-5-(4-sulfamoylphenyl)pyridine-3-carboxamide Chemical compound NC1=NC=C(C=2C=CC(=CC=2)S(N)(=O)=O)C=C1C(=O)NC1=CC=NC=C1 MRXIXCQHUIFLME-UHFFFAOYSA-N 0.000 description 1
- SPYTVKZEORYOEZ-UHFFFAOYSA-N 2-amino-n-pyridin-4-yl-5-[5-(pyrrolidin-1-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCCC3)=CC=2)C=C1C(=O)NC1=CC=NC=C1 SPYTVKZEORYOEZ-UHFFFAOYSA-N 0.000 description 1
- BBJKFMYLMLVUDU-UHFFFAOYSA-N 2-amino-n-pyridin-4-yl-5-thiophen-3-ylpyridine-3-carboxamide Chemical compound NC1=NC=C(C2=CSC=C2)C=C1C(=O)NC1=CC=NC=C1 BBJKFMYLMLVUDU-UHFFFAOYSA-N 0.000 description 1
- PAMGXYQVJKXRMX-UHFFFAOYSA-N 2-bromo-n-ethylbenzenesulfonamide Chemical compound CCNS(=O)(=O)C1=CC=CC=C1Br PAMGXYQVJKXRMX-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- NJRXVEJTAYWCQJ-UHFFFAOYSA-L 2-mercaptosuccinate Chemical compound OC(=O)CC([S-])C([O-])=O NJRXVEJTAYWCQJ-UHFFFAOYSA-L 0.000 description 1
- IBZKBSXREAQDTO-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)ethanamine Chemical compound COCCNCCOC IBZKBSXREAQDTO-UHFFFAOYSA-N 0.000 description 1
- ASUDFOJKTJLAIK-UHFFFAOYSA-N 2-methoxyethanamine Chemical compound COCCN ASUDFOJKTJLAIK-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- IWBFMNLPFLCMBG-UHFFFAOYSA-N 2-methylpyridine;platinum Chemical compound [Pt].CC1=CC=CC=N1 IWBFMNLPFLCMBG-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- FFRFGVHNKJYNOV-DOVUUNBWSA-N 3',4'-Anhydrovinblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C=C(C2)CC)N2CCC2=C1NC1=CC=CC=C21 FFRFGVHNKJYNOV-DOVUUNBWSA-N 0.000 description 1
- VPSARXNVXCRDIV-UHFFFAOYSA-N 3-(4-boronophenyl)propanoic acid Chemical compound OB(O)C1=CC=C(CCC(O)=O)C=C1 VPSARXNVXCRDIV-UHFFFAOYSA-N 0.000 description 1
- XXDNMKSTCJGGQP-UHFFFAOYSA-N 3-[4-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]phenyl]propanoic acid Chemical compound NC1=NC=C(C=2C=CC(CCC(O)=O)=CC=2)C=C1C(=O)NC1=CC=NC=C1 XXDNMKSTCJGGQP-UHFFFAOYSA-N 0.000 description 1
- OFVJCTMBIKLRNL-UHFFFAOYSA-N 3-amino-6-[1-(1,3-dioxolan-2-ylmethyl)pyrazol-4-yl]-n-pyridin-4-ylpyrazine-2-carboxamide Chemical compound NC1=NC=C(C2=CN(CC3OCCO3)N=C2)N=C1C(=O)NC1=CC=NC=C1 OFVJCTMBIKLRNL-UHFFFAOYSA-N 0.000 description 1
- VWYUAZMGQGZOBN-UHFFFAOYSA-N 3-amino-6-[5-(piperidin-1-ylmethyl)thiophen-2-yl]-n-pyridin-4-ylpyrazine-2-carboxamide Chemical compound NC1=NC=C(C=2SC(CN3CCCCC3)=CC=2)N=C1C(=O)NC1=CC=NC=C1 VWYUAZMGQGZOBN-UHFFFAOYSA-N 0.000 description 1
- SHFRXFJUEROSKH-UHFFFAOYSA-N 3-amino-6-bromo-n-pyridin-4-ylpyrazine-2-carboxamide Chemical compound NC1=NC=C(Br)N=C1C(=O)NC1=CC=NC=C1 SHFRXFJUEROSKH-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- FAXDZWQIWUSWJH-UHFFFAOYSA-N 3-methoxypropan-1-amine Chemical compound COCCCN FAXDZWQIWUSWJH-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- TVOJIBGZFYMWDT-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CNN=C1 TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- OZBUFFXESDBEHG-FXILSDISSA-N 4-[[(2e,4e,6e,8e)-3,7-dimethyl-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetraenoyl]amino]benzoic acid Chemical compound C=1C=C(C(O)=O)C=CC=1NC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OZBUFFXESDBEHG-FXILSDISSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- AMUMGAXTCHBNPU-UHFFFAOYSA-N 4-[[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl]methyl]morpholine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CSC(CN2CCOCC2)=C1 AMUMGAXTCHBNPU-UHFFFAOYSA-N 0.000 description 1
- GFFXZLZWLOBBLO-BWVDBABLSA-N 4-amino-1-[(2r,4s,5r)-3-(fluoromethylidene)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@H]1C(=CF)[C@H](O)[C@@H](CO)O1 GFFXZLZWLOBBLO-BWVDBABLSA-N 0.000 description 1
- PULHLIOPJXPGJN-BWVDBABLSA-N 4-amino-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)-3-methylideneoxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@H]1C(=C)[C@H](O)[C@@H](CO)O1 PULHLIOPJXPGJN-BWVDBABLSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- JWZUXKYELFWKNJ-UHFFFAOYSA-N 4h-acridin-3-one Chemical compound C1=CC=C2C=C(C=CC(=O)C3)C3=NC2=C1 JWZUXKYELFWKNJ-UHFFFAOYSA-N 0.000 description 1
- DCRFLNHKMKYMQP-UHFFFAOYSA-N 5-(4-acetyl-1h-pyrrol-2-yl)-2-amino-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound CC(=O)C1=CNC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 DCRFLNHKMKYMQP-UHFFFAOYSA-N 0.000 description 1
- RWJIWEQYLOKEBO-UHFFFAOYSA-N 5-(5-acetylthiophen-2-yl)-2-amino-n-pyridin-4-ylpyridine-3-carboxamide Chemical compound S1C(C(=O)C)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 RWJIWEQYLOKEBO-UHFFFAOYSA-N 0.000 description 1
- LCNUUUROLYPHHR-UHFFFAOYSA-N 5-[2-(dimethylamino)ethylamino]-4h-inden-1-ol Chemical compound C1C(NCCN(C)C)=CC=C2C(O)=CC=C21 LCNUUUROLYPHHR-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- GBOQUHPYCRYKGV-UHFFFAOYSA-N 5-nitro-2-(2-pyrrolidin-1-ylethyl)benzo[de]isoquinoline-1,3-dione Chemical compound O=C1C(C=23)=CC=CC3=CC([N+](=O)[O-])=CC=2C(=O)N1CCN1CCCC1 GBOQUHPYCRYKGV-UHFFFAOYSA-N 0.000 description 1
- KAEVHZSIYLATMK-UHFFFAOYSA-N 6-n-[bis(aziridin-1-yl)phosphoryl]-2-n,2-n,7-trimethylpurine-2,6-diamine Chemical compound C=12N(C)C=NC2=NC(N(C)C)=NC=1NP(=O)(N1CC1)N1CC1 KAEVHZSIYLATMK-UHFFFAOYSA-N 0.000 description 1
- RGVRUQHYQSORBY-UHFFFAOYSA-N 7-(4-amino-5-hydroxy-6-methyloxan-2-yl)oxy-6,9,11-trihydroxy-9-(2-hydroxyethyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(CCO)CC1OC1CC(N)C(O)C(C)O1 RGVRUQHYQSORBY-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- KABRXLINDSPGDF-UHFFFAOYSA-N 7-bromoisoquinoline Chemical compound C1=CN=CC2=CC(Br)=CC=C21 KABRXLINDSPGDF-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 208000003200 Adenoma Diseases 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- 239000012099 Alexa Fluor family Substances 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 108020004491 Antisense DNA Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 101710127675 Antiviral innate immune response receptor RIG-I Proteins 0.000 description 1
- 102100037435 Antiviral innate immune response receptor RIG-I Human genes 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-N Betaine Natural products C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- CUWBAOQFLODAEN-UHFFFAOYSA-N C1=C2C=CC(=O)CC2=CC2=C1OCO2 Chemical compound C1=C2C=CC(=O)CC2=CC2=C1OCO2 CUWBAOQFLODAEN-UHFFFAOYSA-N 0.000 description 1
- FOQJHZPURACERJ-UHFFFAOYSA-N CB1OC(C)(C)C(C)(C)O1 Chemical compound CB1OC(C)(C)C(C)(C)O1 FOQJHZPURACERJ-UHFFFAOYSA-N 0.000 description 1
- OVRKATYHWPCGPZ-UHFFFAOYSA-N CC1CCOCC1 Chemical compound CC1CCOCC1 OVRKATYHWPCGPZ-UHFFFAOYSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 206010048832 Colon adenoma Diseases 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229930188224 Cryptophycin Natural products 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 101710113436 GTPase KRas Proteins 0.000 description 1
- DSLZVSRJTYRBFB-UHFFFAOYSA-N Galactaric acid Natural products OC(=O)C(O)C(O)C(O)C(O)C(O)=O DSLZVSRJTYRBFB-UHFFFAOYSA-N 0.000 description 1
- 239000001828 Gelatine Substances 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 108010010369 HIV Protease Proteins 0.000 description 1
- 108010033040 Histones Proteins 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101001032342 Homo sapiens Interferon regulatory factor 7 Proteins 0.000 description 1
- 101000595548 Homo sapiens TIR domain-containing adapter molecule 1 Proteins 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 239000003458 I kappa b kinase inhibitor Substances 0.000 description 1
- 108060006678 I-kappa-B kinase Proteins 0.000 description 1
- 102000001284 I-kappa-B kinase Human genes 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 102100038070 Interferon regulatory factor 7 Human genes 0.000 description 1
- 101710085994 Interferon-induced helicase C domain-containing protein 1 Proteins 0.000 description 1
- 102100027353 Interferon-induced helicase C domain-containing protein 1 Human genes 0.000 description 1
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-NUEINMDLSA-N Isotretinoin Chemical compound OC(=O)C=C(C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-NUEINMDLSA-N 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- MLFKVJCWGUZWNV-UHFFFAOYSA-N L-alanosine Natural products OC(=O)C(N)CN(O)N=O MLFKVJCWGUZWNV-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 108010043135 L-methionine gamma-lyase Proteins 0.000 description 1
- 125000002842 L-seryl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])O[H] 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 208000030289 Lymphoproliferative disease Diseases 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102100024299 Maternal embryonic leucine zipper kinase Human genes 0.000 description 1
- 101710154611 Maternal embryonic leucine zipper kinase Proteins 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 108090000744 Mitogen-Activated Protein Kinase Kinases Proteins 0.000 description 1
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- 102000006386 Myelin Proteins Human genes 0.000 description 1
- 108010083674 Myelin Proteins Proteins 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- ONXPDKGXOOORHB-BYPYZUCNSA-N N(5)-methyl-L-glutamine Chemical compound CNC(=O)CC[C@H](N)C(O)=O ONXPDKGXOOORHB-BYPYZUCNSA-N 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 1
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- JJSOUZFXFLSQSI-UHFFFAOYSA-N NC1=NC=C(C=2C(=CC=CC=2)NC(O)=O)C=C1C(=O)NC1=CC=NC=C1 Chemical compound NC1=NC=C(C=2C(=CC=CC=2)NC(O)=O)C=C1C(=O)NC1=CC=NC=C1 JJSOUZFXFLSQSI-UHFFFAOYSA-N 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 1
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- HRHKSTOGXBBQCB-UHFFFAOYSA-N Porfiromycine Chemical compound O=C1C(N)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)C3N(C)C3CN12 HRHKSTOGXBBQCB-UHFFFAOYSA-N 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 101150093978 RALB gene Proteins 0.000 description 1
- 108090000944 RNA Helicases Proteins 0.000 description 1
- 102000004409 RNA Helicases Human genes 0.000 description 1
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 102000004278 Receptor Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000873 Receptor Protein-Tyrosine Kinases Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- OWPCHSCAPHNHAV-UHFFFAOYSA-N Rhizoxin Natural products C1C(O)C2(C)OC2C=CC(C)C(OC(=O)C2)CC2CC2OC2C(=O)OC1C(C)C(OC)C(C)=CC=CC(C)=CC1=COC(C)=N1 OWPCHSCAPHNHAV-UHFFFAOYSA-N 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- OCOKWVBYZHBHLU-UHFFFAOYSA-N Sobuzoxane Chemical compound C1C(=O)N(COC(=O)OCC(C)C)C(=O)CN1CCN1CC(=O)N(COC(=O)OCC(C)C)C(=O)C1 OCOKWVBYZHBHLU-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 239000012317 TBTU Substances 0.000 description 1
- 102100036073 TIR domain-containing adapter molecule 1 Human genes 0.000 description 1
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 241001582429 Tetracis Species 0.000 description 1
- 241000906446 Theraps Species 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- XMYKNCNAZKMVQN-NYYWCZLTSA-N [(e)-(3-aminopyridin-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=NC=CC=C1N XMYKNCNAZKMVQN-NYYWCZLTSA-N 0.000 description 1
- FIOBFNCJMPVZAV-UHFFFAOYSA-N [2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]carbamic acid Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC=C1NC(O)=O FIOBFNCJMPVZAV-UHFFFAOYSA-N 0.000 description 1
- XSMVECZRZBFTIZ-UHFFFAOYSA-M [2-(aminomethyl)cyclobutyl]methanamine;2-oxidopropanoate;platinum(4+) Chemical compound [Pt+4].CC([O-])C([O-])=O.NCC1CCC1CN XSMVECZRZBFTIZ-UHFFFAOYSA-M 0.000 description 1
- AFHOBSCDNXGFMO-UHFFFAOYSA-N [2-(hydroxymethyl)phenyl]boronic acid Chemical compound OCC1=CC=CC=C1B(O)O AFHOBSCDNXGFMO-UHFFFAOYSA-N 0.000 description 1
- CKXIPXAIFMTQCS-LRDUUELOSA-N [2-[(2s,4s)-4-[(2r,3r,4r,5s,6s)-3-fluoro-4,5-dihydroxy-6-methyloxan-2-yl]oxy-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-3,4-dihydro-1h-tetracen-2-yl]-2-oxoethyl] 3-aminopropanoate Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)COC(=O)CCN)[C@@H]1O[C@@H](C)[C@@H](O)[C@@H](O)[C@H]1F CKXIPXAIFMTQCS-LRDUUELOSA-N 0.000 description 1
- KPJCFIVGCAFAHO-UHFFFAOYSA-N [3-(4-methylphenyl)phenyl]boronic acid Chemical compound C1=CC(C)=CC=C1C1=CC=CC(B(O)O)=C1 KPJCFIVGCAFAHO-UHFFFAOYSA-N 0.000 description 1
- NPTBTFRGCBFYPZ-UHFFFAOYSA-N [3-(aminomethyl)phenyl]boronic acid;hydrochloride Chemical compound [Cl-].[NH3+]CC1=CC=CC(B(O)O)=C1 NPTBTFRGCBFYPZ-UHFFFAOYSA-N 0.000 description 1
- CWLNHPXWZRALFS-UHFFFAOYSA-N [3-[(2-methylpropan-2-yl)oxycarbonylamino]phenyl]boronic acid Chemical compound CC(C)(C)OC(=O)NC1=CC=CC(B(O)O)=C1 CWLNHPXWZRALFS-UHFFFAOYSA-N 0.000 description 1
- XYDGTCUDXZTZHW-UHFFFAOYSA-N [3-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]phenyl]carbamic acid Chemical compound NC1=NC=C(C=2C=C(NC(O)=O)C=CC=2)C=C1C(=O)NC1=CC=NC=C1 XYDGTCUDXZTZHW-UHFFFAOYSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- HUZNRXFJHYNUMV-UHFFFAOYSA-N [4-(aminomethyl)phenyl]boronic acid;hydron;chloride Chemical compound Cl.NCC1=CC=C(B(O)O)C=C1 HUZNRXFJHYNUMV-UHFFFAOYSA-N 0.000 description 1
- PZRPBPMLSSNFOM-UHFFFAOYSA-N [4-(hydroxymethyl)phenyl]boronic acid Chemical compound OCC1=CC=C(B(O)O)C=C1 PZRPBPMLSSNFOM-UHFFFAOYSA-N 0.000 description 1
- LIQODXNTTZAGID-UHFFFAOYSA-N [4-[5-[(7,8-dihydroxy-2-methyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxin-6-yl)oxy]-8-oxo-5a,6,8a,9-tetrahydro-5h-[2]benzofuro[5,6-f][1,3]benzodioxol-9-yl]-2,6-dimethoxyphenyl] dihydrogen phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(OC3C(C(O)C4OC(C)OCC4O3)O)C3C2C(OC3)=O)=C1 LIQODXNTTZAGID-UHFFFAOYSA-N 0.000 description 1
- NGGKDFAWSYIDOG-UHFFFAOYSA-N [5-(piperidin-1-ylmethyl)thiophen-2-yl]boronic acid Chemical compound S1C(B(O)O)=CC=C1CN1CCCCC1 NGGKDFAWSYIDOG-UHFFFAOYSA-N 0.000 description 1
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- UVIQSJCZCSLXRZ-UBUQANBQSA-N abiraterone acetate Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CC[C@@H](CC4=CC[C@H]31)OC(=O)C)C=C2C1=CC=CN=C1 UVIQSJCZCSLXRZ-UBUQANBQSA-N 0.000 description 1
- 229960004103 abiraterone acetate Drugs 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 229950005033 alanosine Drugs 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000005278 alkyl sulfonyloxy group Chemical group 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- VJZITPJGSQKZMX-XDPRQOKASA-N amrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC=C4C(=O)C=3C(O)=C21)(N)C(=O)C)[C@H]1C[C@H](O)[C@H](O)CO1 VJZITPJGSQKZMX-XDPRQOKASA-N 0.000 description 1
- 229960002550 amrubicin Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 229950001104 anhydrovinblastine Drugs 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003816 antisense DNA Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 1
- 239000008122 artificial sweetener Substances 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 125000005279 aryl sulfonyloxy group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- TWHSQQYCDVSBRK-UHFFFAOYSA-N asulacrine Chemical compound C12=CC=CC(C)=C2N=C2C(C(=O)NC)=CC=CC2=C1NC1=CC=C(NS(C)(=O)=O)C=C1OC TWHSQQYCDVSBRK-UHFFFAOYSA-N 0.000 description 1
- 229950011088 asulacrine Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 108010044540 auristatin Proteins 0.000 description 1
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 230000008238 biochemical pathway Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 229950008548 bisantrene Drugs 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- UBJAHGAUPNGZFF-XOVTVWCYSA-N bms-184476 Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC(C)=O)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)C=3C=CC=CC=3)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OCSC)C(=O)C1=CC=CC=C1 UBJAHGAUPNGZFF-XOVTVWCYSA-N 0.000 description 1
- GMJWGJSDPOAZTP-MIDYMNAOSA-N bms-188797 Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](OC(C)=O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)C=4C=CC=CC=4)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)OC)C(=O)C1=CC=CC=C1 GMJWGJSDPOAZTP-MIDYMNAOSA-N 0.000 description 1
- LHHCSNFAOIFYRV-DOVBMPENSA-N boceprevir Chemical compound O=C([C@@H]1[C@@H]2[C@@H](C2(C)C)CN1C(=O)[C@@H](NC(=O)NC(C)(C)C)C(C)(C)C)NC(C(=O)C(N)=O)CC1CCC1 LHHCSNFAOIFYRV-DOVBMPENSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000009134 cell regulation Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- YOQPCWIXYUNEET-UHFFFAOYSA-N chembl307697 Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(O)=CC=1)=NNC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O YOQPCWIXYUNEET-UHFFFAOYSA-N 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 208000024207 chronic leukemia Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 238000005352 clarification Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- POADTFBBIXOWFJ-VWLOTQADSA-N cositecan Chemical compound C1=CC=C2C(CC[Si](C)(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 POADTFBBIXOWFJ-VWLOTQADSA-N 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 108010006226 cryptophycin Proteins 0.000 description 1
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 description 1
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960000605 dexrazoxane Drugs 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 229950009278 dimesna Drugs 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- KQYGMURBTJPBPQ-UHFFFAOYSA-L disodium;2-(2-sulfonatoethyldisulfanyl)ethanesulfonate Chemical compound [Na+].[Na+].[O-]S(=O)(=O)CCSSCCS([O-])(=O)=O KQYGMURBTJPBPQ-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 1
- 229930188854 dolastatin Natural products 0.000 description 1
- 239000012154 double-distilled water Substances 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 241001493065 dsRNA viruses Species 0.000 description 1
- 238000004043 dyeing Methods 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229950004438 elinafide Drugs 0.000 description 1
- 229950005450 emitefur Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- RFBZWPFBCXBBJS-UHFFFAOYSA-N ethyl 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound CCOC(=O)C1=CC=CC=C1B1OC(C)(C)C(C)(C)O1 RFBZWPFBCXBBJS-UHFFFAOYSA-N 0.000 description 1
- FDWHSRIVEQFTPV-UHFFFAOYSA-N ethyl 2-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoate Chemical compound CCOC(=O)C1=CC=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 FDWHSRIVEQFTPV-UHFFFAOYSA-N 0.000 description 1
- MHOWHQYJXCQHLN-UHFFFAOYSA-N ethyl 3-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoate Chemical compound CCOC(=O)C1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 MHOWHQYJXCQHLN-UHFFFAOYSA-N 0.000 description 1
- NGQDJMZWCKAKRN-UHFFFAOYSA-N ethyl 4-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 NGQDJMZWCKAKRN-UHFFFAOYSA-N 0.000 description 1
- JEFPWOBULVSOTM-PPHPATTJSA-N ethyl n-[(2s)-5-amino-2-methyl-3-phenyl-1,2-dihydropyrido[3,4-b]pyrazin-7-yl]carbamate;2-hydroxyethanesulfonic acid Chemical compound OCCS(O)(=O)=O.C=1([C@H](C)NC=2C=C(N=C(N)C=2N=1)NC(=O)OCC)C1=CC=CC=C1 JEFPWOBULVSOTM-PPHPATTJSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- DBEPLOCGEIEOCV-WSBQPABSSA-N finasteride Chemical compound N([C@@H]1CC2)C(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)NC(C)(C)C)[C@@]2(C)CC1 DBEPLOCGEIEOCV-WSBQPABSSA-N 0.000 description 1
- 229960004039 finasteride Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000005454 flavour additive Substances 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 229960002258 fulvestrant Drugs 0.000 description 1
- PUTFLLAMOPARNC-UHFFFAOYSA-N furo[3,2-b]pyridin-7-amine Chemical compound NC1=CC=NC2=C1OC=C2 PUTFLLAMOPARNC-UHFFFAOYSA-N 0.000 description 1
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 1
- 229950011325 galarubicin Drugs 0.000 description 1
- 229950004410 galocitabine Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229950011595 glufosfamide Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 235000015220 hamburgers Nutrition 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- NAQMVNRVTILPCV-UHFFFAOYSA-N hexane-1,6-diamine Chemical compound NCCCCCCN NAQMVNRVTILPCV-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 229910000043 hydrogen iodide Inorganic materials 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- JBFYUZGYRGXSFL-UHFFFAOYSA-N imidazolide Chemical compound C1=C[N-]C=N1 JBFYUZGYRGXSFL-UHFFFAOYSA-N 0.000 description 1
- 230000003832 immune regulation Effects 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 210000005007 innate immune system Anatomy 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- NICJCIQSJJKZAH-AWEZNQCLSA-N irofulven Chemical compound O=C([C@@]1(O)C)C2=CC(C)=C(CO)C2=C(C)C21CC2 NICJCIQSJJKZAH-AWEZNQCLSA-N 0.000 description 1
- 229950005254 irofulven Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 229960005280 isotretinoin Drugs 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 210000000867 larynx Anatomy 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- UGFHIPBXIWJXNA-UHFFFAOYSA-N liarozole Chemical compound ClC1=CC=CC(C(C=2C=C3NC=NC3=CC=2)N2C=NC=C2)=C1 UGFHIPBXIWJXNA-UHFFFAOYSA-N 0.000 description 1
- 229950007056 liarozole Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229920006008 lipopolysaccharide Polymers 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229950008991 lobaplatin Drugs 0.000 description 1
- 229950000909 lometrexol Drugs 0.000 description 1
- 229960003538 lonidamine Drugs 0.000 description 1
- WDRYRZXSPDWGEB-UHFFFAOYSA-N lonidamine Chemical compound C12=CC=CC=C2C(C(=O)O)=NN1CC1=CC=C(Cl)C=C1Cl WDRYRZXSPDWGEB-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- RVFGKBWWUQOIOU-NDEPHWFRSA-N lurtotecan Chemical compound O=C([C@]1(O)CC)OCC(C(N2CC3=4)=O)=C1C=C2C3=NC1=CC=2OCCOC=2C=C1C=4CN1CCN(C)CC1 RVFGKBWWUQOIOU-NDEPHWFRSA-N 0.000 description 1
- 229950002654 lurtotecan Drugs 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 208000002780 macular degeneration Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- MMIPFLVOWGHZQD-UHFFFAOYSA-N manganese(3+) Chemical compound [Mn+3] MMIPFLVOWGHZQD-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- OETHQSJEHLVLGH-UHFFFAOYSA-N metformin hydrochloride Chemical compound Cl.CN(C)C(=N)N=C(N)N OETHQSJEHLVLGH-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- MHHYKAZYCHIGCZ-UHFFFAOYSA-N methyl 3-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoate Chemical compound COC(=O)C1=CC=CC(C=2C=C(C(N)=NC=2)C(=O)NC=2C=CN=CC=2)=C1 MHHYKAZYCHIGCZ-UHFFFAOYSA-N 0.000 description 1
- ZCXAETZQYHEVJV-UHFFFAOYSA-N methyl 4-[(2-amino-5-bromopyridine-3-carbonyl)amino]pyridine-3-carboxylate Chemical compound COC(=O)C1=CN=CC=C1NC(=O)C1=CC(Br)=CN=C1N ZCXAETZQYHEVJV-UHFFFAOYSA-N 0.000 description 1
- QMZHLDPYLWPXNH-UHFFFAOYSA-N methyl 4-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]-2-fluorobenzoate Chemical compound C1=C(F)C(C(=O)OC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 QMZHLDPYLWPXNH-UHFFFAOYSA-N 0.000 description 1
- FWKICXPIAZMWKY-UHFFFAOYSA-N methyl 4-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 FWKICXPIAZMWKY-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 230000003228 microsomal effect Effects 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- HVOYZOQVDYHUPF-UHFFFAOYSA-N n,n',n'-trimethylethane-1,2-diamine Chemical compound CNCCN(C)C HVOYZOQVDYHUPF-UHFFFAOYSA-N 0.000 description 1
- CYSDRDWNFDQHMX-UHFFFAOYSA-N n-(2-acetamidopyridin-4-yl)-2-amino-5-[5-(morpholin-4-ylmethyl)thiophen-2-yl]pyridine-3-carboxamide Chemical compound C1=NC(NC(=O)C)=CC(NC(=O)C=2C(=NC=C(C=2)C=2SC(CN3CCOCC3)=CC=2)N)=C1 CYSDRDWNFDQHMX-UHFFFAOYSA-N 0.000 description 1
- DJUHRLKLKTTYSF-UHFFFAOYSA-N n-(2-acetamidopyridin-4-yl)-2-amino-5-bromopyridine-3-carboxamide Chemical compound C1=NC(NC(=O)C)=CC(NC(=O)C=2C(=NC=C(Br)C=2)N)=C1 DJUHRLKLKTTYSF-UHFFFAOYSA-N 0.000 description 1
- RLRHPCKWSXWKBG-UHFFFAOYSA-N n-(2-azaniumylethyl)carbamate Chemical compound NCCNC(O)=O RLRHPCKWSXWKBG-UHFFFAOYSA-N 0.000 description 1
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 description 1
- TVYPSLDUBVTDIS-FUOMVGGVSA-N n-[1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-methyloxolan-2-yl]-5-fluoro-2-oxopyrimidin-4-yl]-3,4,5-trimethoxybenzamide Chemical compound COC1=C(OC)C(OC)=CC(C(=O)NC=2C(=CN(C(=O)N=2)[C@H]2[C@@H]([C@H](O)[C@@H](C)O2)O)F)=C1 TVYPSLDUBVTDIS-FUOMVGGVSA-N 0.000 description 1
- ZDUZYDDAHVZGCI-UHFFFAOYSA-N n-[2-(dimethylamino)ethyl]-9-hydroxy-5,6-dimethylpyrido[4,3-b]carbazole-1-carboxamide Chemical compound CN1C2=CC=C(O)C=C2C2=C1C(C)=C1C=CN=C(C(=O)NCCN(C)C)C1=C2 ZDUZYDDAHVZGCI-UHFFFAOYSA-N 0.000 description 1
- XBGNERSKEKDZDS-UHFFFAOYSA-N n-[2-(dimethylamino)ethyl]acridine-4-carboxamide Chemical compound C1=CC=C2N=C3C(C(=O)NCCN(C)C)=CC=CC3=CC2=C1 XBGNERSKEKDZDS-UHFFFAOYSA-N 0.000 description 1
- GWLFIMOOGVXSMZ-UHFFFAOYSA-N n-[[1-[2-(diethylamino)ethylamino]-7-methoxy-9-oxothioxanthen-4-yl]methyl]formamide Chemical compound S1C2=CC=C(OC)C=C2C(=O)C2=C1C(CNC=O)=CC=C2NCCN(CC)CC GWLFIMOOGVXSMZ-UHFFFAOYSA-N 0.000 description 1
- WLNSKTSWPYTNLY-UHFFFAOYSA-N n-ethyl-n',n'-dimethylethane-1,2-diamine Chemical compound CCNCCN(C)C WLNSKTSWPYTNLY-UHFFFAOYSA-N 0.000 description 1
- YZCXLAHTHBVYGB-UHFFFAOYSA-N n-methyl-1-(3-methylimidazol-4-yl)methanamine Chemical compound CNCC1=CN=CN1C YZCXLAHTHBVYGB-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 229950007221 nedaplatin Drugs 0.000 description 1
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 1
- 229960000801 nelarabine Drugs 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 1
- 229960002653 nilutamide Drugs 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- XHWRWCSCBDLOLM-UHFFFAOYSA-N nolatrexed Chemical compound CC1=CC=C2NC(N)=NC(=O)C2=C1SC1=CC=NC=C1 XHWRWCSCBDLOLM-UHFFFAOYSA-N 0.000 description 1
- 229950000891 nolatrexed Drugs 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 238000012758 nuclear staining Methods 0.000 description 1
- 230000005937 nuclear translocation Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 230000006548 oncogenic transformation Effects 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000003232 p-nitrobenzoyl group Chemical group [N+](=O)([O-])C1=CC=C(C(=O)*)C=C1 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 208000003154 papilloma Diseases 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- WBXPDJSOTKVWSJ-ZDUSSCGKSA-N pemetrexed Chemical compound C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 WBXPDJSOTKVWSJ-ZDUSSCGKSA-N 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229950004317 pinafide Drugs 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 239000011505 plaster Substances 0.000 description 1
- HRGDZIGMBDGFTC-UHFFFAOYSA-N platinum(2+) Chemical compound [Pt+2] HRGDZIGMBDGFTC-UHFFFAOYSA-N 0.000 description 1
- 229950008499 plitidepsin Drugs 0.000 description 1
- UUSZLLQJYRSZIS-LXNNNBEUSA-N plitidepsin Chemical compound CN([C@H](CC(C)C)C(=O)N[C@@H]1C(=O)N[C@@H]([C@H](CC(=O)O[C@H](C(=O)[C@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(OC)=CC=2)C(=O)O[C@@H]1C)C(C)C)O)[C@@H](C)CC)C(=O)[C@@H]1CCCN1C(=O)C(C)=O UUSZLLQJYRSZIS-LXNNNBEUSA-N 0.000 description 1
- 108010049948 plitidepsin Proteins 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 230000001023 pro-angiogenic effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 239000011241 protective layer Substances 0.000 description 1
- 229950007401 pumitepa Drugs 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- ABMYEXAYWZJVOV-UHFFFAOYSA-N pyridin-3-ylboronic acid Chemical compound OB(O)C1=CC=CN=C1 ABMYEXAYWZJVOV-UHFFFAOYSA-N 0.000 description 1
- OYRRZWATULMEPF-UHFFFAOYSA-N pyrimidin-4-amine Chemical compound NC1=CC=NC=N1 OYRRZWATULMEPF-UHFFFAOYSA-N 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- NGXSWUFDCSEIOO-UHFFFAOYSA-N pyrrolidin-3-amine Chemical compound NC1CCNC1 NGXSWUFDCSEIOO-UHFFFAOYSA-N 0.000 description 1
- 150000004944 pyrrolopyrimidines Chemical class 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 229940100552 retinamide Drugs 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- OWPCHSCAPHNHAV-LMONGJCWSA-N rhizoxin Chemical compound C/C([C@H](OC)[C@@H](C)[C@@H]1C[C@H](O)[C@]2(C)O[C@@H]2/C=C/[C@@H](C)[C@]2([H])OC(=O)C[C@@](C2)(C[C@@H]2O[C@H]2C(=O)O1)[H])=C\C=C\C(\C)=C\C1=COC(C)=N1 OWPCHSCAPHNHAV-LMONGJCWSA-N 0.000 description 1
- 208000007442 rickets Diseases 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229950010372 sobuzoxane Drugs 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- RCOSUMRTSQULBK-UHFFFAOYSA-N sodium;propan-1-olate Chemical compound [Na+].CCC[O-] RCOSUMRTSQULBK-UHFFFAOYSA-N 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 201000000498 stomach carcinoma Diseases 0.000 description 1
- 238000013337 sub-cultivation Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- 239000005061 synthetic rubber Substances 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229960003102 tasonermin Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- IQFQQHFUYZETBN-UHFFFAOYSA-N tert-butyl n-[2-[[4-[6-amino-5-(pyridin-4-ylcarbamoyl)pyridin-3-yl]benzoyl]amino]ethyl]carbamate Chemical compound C1=CC(C(=O)NCCNC(=O)OC(C)(C)C)=CC=C1C1=CN=C(N)C(C(=O)NC=2C=CN=CC=2)=C1 IQFQQHFUYZETBN-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229940100611 topical cream Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 1
- 229960000977 trabectedin Drugs 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 239000003558 transferase inhibitor Substances 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 229960005526 triapine Drugs 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- WTVXIBRMWGUIMI-UHFFFAOYSA-N trifluoro($l^{1}-oxidanylsulfonyl)methane Chemical group [O]S(=O)(=O)C(F)(F)F WTVXIBRMWGUIMI-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229960000875 trofosfamide Drugs 0.000 description 1
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 1
- 229950010147 troxacitabine Drugs 0.000 description 1
- RXRGZNYSEHTMHC-BQBZGAKWSA-N troxacitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)OC1 RXRGZNYSEHTMHC-BQBZGAKWSA-N 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 230000010472 type I IFN response Effects 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- 229940099259 vaseline Drugs 0.000 description 1
- 229960005212 vindesine sulfate Drugs 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 1
- 229960000641 zorubicin Drugs 0.000 description 1
- VLCYCQAOQCDTCN-ZCFIWIBFSA-N α-difluoromethylornithine Chemical compound NCCC[C@@](N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-ZCFIWIBFSA-N 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Ophthalmology & Optometry (AREA)
- Dermatology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Vascular Medicine (AREA)
- Immunology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Molecular Biology (AREA)
- Hospice & Palliative Care (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
Abstract
Compuestos de la fórmula I:**Fórmula** en la que X representa CH, R representa Het, R1 representa piridilo, pirimidilo, piridazinilo, o furo[3,2-b]piridilo, cada uno de los cuales está sin sustituir o monoo disustituido por Hal, A, OR5, CN, COOA, COOH, CON(R5)2 y/o NR5COA'; Het representa furilo, tienilo, pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, piridilo, pirimidinilo, triazolilo, tetrazolilo, tiadiazol, piridazinilo, pirazinilo, indolilo, isoindolilo, bencimidazolilo, indazolilo, quinolilo, 1,3- benzodioxolilo, benzotiofenilo, benzofuranilo o imidazopiridilo, cada uno de los cuales está sin sustituir o mono-, di- o trisustituido por A, COA, (CH2)pHet2, OH, OA, Hal, (CH2)pN(R5)2, NO2, CN, (CH2)pCOOR5, (CH2)pCON(R5)2, NR5COA, (CH2)pCOHet2 y/o (CH2)pfenilo, Het2 representa dihidropirrolilo, pirrolidinilo, tetrahidroimidazolilo, dihidropirazolilo, tetrahidropirazolilo, dihidropiridilo, tetrahidropiridilo, piperidinilo, morfolinilo, hexahidropiridazinilo, hexahidropirimidinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o mono sustituido por OH y/o A, A' representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en el que de 1-7 átomos de H pueden reemplazarse por F, A representa alquilo no ramificado o ramificado que tiene 1-10 átomos de C, en el que uno o dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N, O, S y/o por grupos -CH=CH- y/o además de 1-7 átomos de H pueden reemplazarse por F, R5 representa H o alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en el que de 1-7 átomos de H pueden reemplazarse por F, Hal representa F, Cl, Br o I, n representa 0, 1, 2, 3 o 4, p representa 0, 1 o 2, q representa 1, 2, 3 o 4, y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones.
Description
DESCRIPCIÓN
Piridina y derivados de pirazina
Campo de la invención
La invención tuvo el objetivo de encontrar nuevos compuestos que tienen propiedades valiosas, en particular, aquellos que pueden usarse para la preparación de medicamentos. La presente invención se relaciona con compuestos de piridina que son capaces de inhibir una o más cinasas. Los compuestos tienen aplicaciones en el tratamiento de una variedad de trastornos, que incluyen cáncer, choque séptico, glaucoma primario de ángulo abierto (POAG, por sus siglas en inglés), hiperplasia, artritis reumatoide, psoriasis, arterosclerosis, retinopatía, osteoartritis, endometriosis, inflamación crónica y/o enfermedades neurodegenerativas, tales como enfermedad de Alzheimer.
La presente invención se refiere a compuestos y con el uso de los compuestos, en los que la inhibición, regulación y/o modulación de la transducción de señal por las cinasas, en particular, los receptores de tirosina cinasas, además de las composiciones farmacéuticas que comprenden estos compuestos, y con el uso de los compuestos para el tratamiento de enfermedades inducidas por cinasas.
Debido a que las proteínas cinasas regulan casi cada uno de los procesos celulares, incluyendo el metabolismo, la proliferación celular, la diferenciación celular y la supervivencia celular, éstas son blancos atractivos para la intervención terapéutica para diferentes estados de enfermedades. Por ejemplo, el ciclo celular y la angiogénesis, en los que las proteínas cinasas juegan un papel primordial, son los procesos celulares asociados con numerosas afecciones de enfermedades, tales como, pero no se limitan a: cáncer, enfermedades inflamatorias, angiogénesis anormales y enfermedades relacionadas con las mismas, aterosclerosis, degeneración macular, diabetes, obesidad y dolor.
En particular, la presente invención se refiere a compuestos y al uso de los compuestos, en los que juega un papel la inhibición regulación y/o modulación de la transducción de señal por TBK1 y IKKs.
Uno de los principales mecanismos mediante los cuales es afectada la regulación celular, es a través de la transducción de señales extracelulares a través de la membrana que, en cambio, modulan las rutas bioquímicas dentro de la célula. La fosforilación proteínica representa un curso mediante el cual se propagan las señales intracelulares de molécula a molécula, resultando por último en una respuesta celular. Estas cascadas de transducción de señal se regulan altamente y a menudo se traslapan, como es evidente a partir de la existencia de muchas proteínas cinasas, así como fosfatasas. La fosforilación de proteínas se presenta predominantemente en los residuos de serina, treonina o tirosina, y, por lo tanto, las proteínas cinasas se han clasificado por su especificidad del sitio de fosforilación, es decir, serina/treonina cinasas y tirosina cinasas. Dado que la fosforilación es un proceso ubicuo dentro de las células, y dado que los fenotipos celulares están influencias ampliamente por la actividad de estas rutas, actualmente se cree que un número de estos estados de enfermedades y/o enfermedades se atribuyen a una activación aberrante o mutaciones funcionales en los componentes moleculares de las cascadas de la cinasa. En consecuencia, se ha dedicado a la caracterización de estas proteínas y compuestos que son capaces de modular su actividad (para una revisión ver: Weinstein-Oppenheimer et al. Pharma. &. Therap., 2000, 88, 229-279).
IKKs y TBK1 son serina/treonina cinasas que son altamente homólogas entre sí y a otras cinasas IkB. Las dos cinasas juegan un papel integral en el sistema inmune innato. Los virus de ARN de doble hebra se reconocen por los receptores 3 y 4 similares a Toll y las helicasas de ARN, RIG-I y MDA-5, y resultan en la activación de la cascada de señalización de TRIF-TBK1/IKKe-IRF3, que resulta en una respuesta de interferón de tipo I.
En 2007, Boehm et al. describió IKKs como un nuevo oncogen de cáncer de mama [J.S. Boehm et al., Cell 129, 1065 1079, 2007]. Se investigaron 354 cinasas con respecto a su capacidad para recapitular el fenotipo de transformación de Ras, junto con una forma activada de la cinasa Mek MAPK. IKKs se identificó en la presente como un oncogen cooperativo. Además, los autores fueron capaces de mostrar que IKKs se amplifica y sobreexpresa en numerosas líneas celulares de cáncer de mama y muestras de tumores. La reducción en la expresión génica por medio de la interferencia de ARN en las células de cáncer de mama induce la apoptosis y daña la proliferación de la misma. Eddy et al., obtuvieron descubrimientos similares en 2005, lo cual fundamenta la importancia de IKKs en las enfermedades de cáncer de mama [S.F. Eddy et al., Cancer Res. 2005; 65 (24), 11375-11383].
Un efecto protumorigénico de TBK1 se indicó por primera vez en 2006. En una selección de una biblioteca de genes que comprende 251.000 ADNc, Korherr et al., identificaron precisamente tres genes, TRIF, TBK1 y IRF3, los cuales están involucrados típicamente en la defensa inmune innata como los factores proangiogénicos [C. Korherr et al., PNAS, 103, 4240-4245, 2006]. En 2006, Chien et al. [Y. Chien et al., Cell 127, 157-170, 2006] publicaron que las células de TBK1 -/- solo pueden transformarse a un grado limitado usando Ras oncogénicos, lo que sugiere una participación de TBK1 en la transformación mediada por Ras. Además, fueron capaces de mostrar que una disminución mediada por ARNi de TBK1 desencadena la apoptosis en las células MCF-7 y Panc-1. Barbie et al.,
publicaron recientemente que TBK1 es de importancia esencial en numerosas líneas celulares de cáncer con K-Ras mutado, lo que sugiere que la intervención de TBK1 podría ser de importancia terapéutica en los tumores correspondientes [D.A. Barbie et al., Nature Letters 1-5, 2009].
Las enfermedades causadas por las proteínas cinasas se caracterizan por una actividad o hiperactividad anómala de tales proteínas cinasas. La actividad anómala se relaciona, ya sea con: (1) la expresión en las células que no expresan usualmente estas proteínas cinasas; (2) aumento en la expresión de cinasas, lo que resulta en la proliferación celular indeseada, tal como cáncer; (3) aumento de la actividad de cinasa, lo que resulta en la proliferación celular indeseada, tal como cáncer, y/o en la hiperactividad de las proteínas cinasas correspondientes. La hiperactividad se refiere a la amplificación del gen que codifica una cierta proteína cinasa o la generación de un nivel de actividad que puede correlacionarse con una enfermedad de proliferación celular (es decir, la gravedad de uno o más síntomas de la enfermedad de proliferación celular aumenta con el aumento del nivel de cinasa). La biodisponibilidad de una proteína cinasa también puede ser influenciada por la presencia o ausencia de un grupo de proteínas de unión de esta cinasa.
IKKs y TBK1 son cinasas de Ser/Thr altamente homólogas implicadas críticamente en la respuesta inmune innata por medio de la inducción de los interferones de tipo 1 y otras citosinas. Estas cinasas se estimulan en respuesta a la infección viral/bacteriana. La respuesta inmune a la infección viral y bacteriana involucra la unión de antígenos, tal como lipopolisacáridos (LSP) bacterianos, ARN de doble hebra viral (ARNds) a receptores similares a Toll, después la activación subsecuente de la ruta TBK1. TBK1 y IKKs activados fosforilan IRF3 y IRF7, lo que desencadena la dimerización de la translocación nuclear de estos factores de transcripción reguladores de interferones, induciendo finalmente una cascada de señalización que conduce a la producción de IFN.
Últimamente, IKKs y TBK1 también han sido implicados en el cáncer. Se ha mostrado que IKKs coopera con MEK activado para transformar las células humanas. Además, IKKs frecuentemente se amplifica/sobreexpresa en las líneas celulares de cáncer de mama y los tumores derivados de pacientes. TBK1 se induce en condiciones hipóxicas y se expresa a niveles significativos en muchos tumores sólidos.
Además, TBK1 se requiere para ayudar a la transformación Ras oncogénica, y la actividad de cinasa TBK1 se eleva en las células transformadas y requiere su supervivencia en cultivo. De manera similar, se encontró que la señalización de TBK1 y NF-kB son esenciales en los tumores KRAS mutantes. Han identificado TBK1 como una pareja de síntesis letal de KRAS oncogénico. Lit.: Y.-H. Ou et al., Molecular Cell 41,458-470, 2011; D.A. Barbie et al., nature, 1-5, 2009.
Por lo tanto, los compuestos de acuerdo con la invención o una sal farmacéuticamente aceptable de los mismos, se administran para el tratamiento de cáncer, incluyendo carcinomas sólidos, tales como, por ejemplo, carcinomas (por ejemplo, de los pulmones, páncreas, tiroides, vejiga o colon), enfermedades mieloides (por ejemplo, leucemia mieloide) o adenomas (por ejemplo, adenoma de colon velloso).
Además, los tumores incluyen leucemia monocítica, cerebro, urogenital, sistema linfático, estómago, carcinoma de laringe y pulmón, que incluye adenocarcinoma pulmonar y carcinoma de pulmón de células pequeñas, carcinoma pancreático y/ mama.
Los compuestos además son apropiados para el tratamiento de deficiencia inmune inducida por VIH-1 (Virus de inmunodeficiencia humana de tipo 1).
Serán consideradas las enfermedades hiperproliferativas como el como cáncer, como el cáncer de cerebro, cáncer de pulmón, cáncer epitelial escamoso, cáncer de vejiga, cáncer de estómago, cáncer pancreático, cáncer de hígado, cáncer renal, cáncer colorrectal, cáncer de mama, cáncer de cabeza, cáncer de cuello, cáncer de esófago, cáncer ginecológico, cáncer de tiroides, linfomas, leucemia crónica y leucemia aguda. En particular, el crecimiento de células similares a cáncer es una enfermedad que representa un objetivo de la presente invención. Por lo tanto, la presente invención se refiere a compuestos de acuerdo con la invención, como medicamentos y/o principios activos de medicamentos, en el tratamiento y/o profilaxis de tales enfermedades, y con el uso de los compuestos de acuerdo con la invención para la preparación de un farmacéutico para el tratamiento y/o profilaxis de tales enfermedades y con un proceso para el tratamiento de las enfermedades que comprenden la administración de uno o más compuestos de acuerdo con la invención, a un paciente en necesidad de tal administración.
Puede mostrarse que los compuestos de acuerdo con la invención, tienen una acción antiproliferativa. Los compuestos de acuerdo con la invención se administran a un paciente que tiene una enfermedad hiperproliferativa, por ejemplo, para inhibir el crecimiento tumoral, para reducir la inflamación asociada con una enfermedad linfoproliferativa, para inhibir el rechazo de transplantes o el daño neurológico debido a la reparación de tejido, etc. Los presentes compuestos son apropiados para los propósitos profilácticos o terapéuticos. Como se usa en el presente documento, el término “tratamiento” se usa para referirse a la prevención de enfermedades y el tratamiento de las afecciones preexistentes. La prevención de la proliferación/vitalidad se logra mediante la administración de los compuestos de acuerdo con la invención, antes del desarrollo de la enfermedad manifiesta, por ejemplo, para prevenir el crecimiento tumoral. De manera alternativa, los compuestos se usan para el tratamiento de las enfermedades en curso, estabilizando o
mejorando los síntomas clínicos del paciente.
El huésped o paciente pueden pertenecer a cualquier especie de mamífero, por ejemplo, una especie de primate, particularmente los humanos; roedores, incluyendo ratones, ratas y hámsteres; conejos; caballos, vacas, perros, gatos, etc. Los modelos animales son de interés para las investigaciones experimentales, proporcionando un modelo para el tratamiento de una enfermedad humana.
La susceptibilidad de una célula particular al tratamiento con los compuestos de acuerdo con la invención, puede determinarse mediante una prueba in vitro. Típicamente, un cultivo de la célula se incuba con un compuesto de acuerdo con la invención a diferentes concentraciones durante un periodo de tiempo que es suficiente para permitir que los principios activos induzcan la muerte celular o inhiban la proliferación celular, la viabilidad o la migración celular, a menudo entre aproximadamente una hora y una semana. La prueba in vitro puede llevarse a cabo usando células cultivadas de una muestra de biopsia. Después se determina la cantidad de las células que permanecen después del tratamiento. La dosis varía dependiendo del compuesto específico usado, la enfermedad específica, el estado del paciente, etc. Una dosis terapéutica es típicamente suficiente, para reducir considerablemente la población celular indeseada en el tejido blanco, mientras que se mantiene la viabilidad del paciente. En general, el tratamiento se continúa hasta que se ha presentado una reducción considerable, por ejemplo, en por lo menos aproximadamente 50 % de reducción en la carga celular, y puede continuarse hasta que esencialmente no se detectan más células en el cuerpo.
Hay muchas enfermedades asociadas con la degradación de la proliferación celular y la muerte celular (apoptosis). Las condiciones de interés incluyen, pero no se limitan a, las siguientes. Los compuestos de acuerdo con la invención, son apropiados para el tratamiento de varias condiciones, en donde hay una proliferación y/o migración de células de músculo liso y/o células inflamatorias en la capa íntima de un vaso, resultando en un flujo de sangre restringido a través de tal vaso, por ejemplo, en el caso de las lesiones oclusivas neoíntimas. Las enfermedades vasculares de injerto oclusivo de interés incluyen aterosclerosis, enfermedad vascular coronaria después de injerto, estenosis de injerto venosos, restenosis prostética perianastómica, restenosis después de angioplastia o colocación de stent y similares.
Además, los compuestos de acuerdo con la invención, pueden usarse para obtener efectos aditivos o sinérgicos en algunas quimioterapias y radioterapias de cáncer existentes y/o restaurar la eficacia de algunas quimioterapias y radioterapias de cáncer existentes.
El término “método” se refiere a las maneras, medios, técnicas y procedimientos para realizar una tarea dada, que incluye, pero no se limita a, las maneras, medios, técnicas y procedimientos conocidos por, o fácilmente desarrollados de las maneras, medios, técnicas y procedimientos conocidos por los practicantes de la química, farmacología, biología, bioquímica y las artes médicas.
El término “administración” como se usa en el presente documento, se refiere a un método para unir un compuesto de la presente invención y una cinasa diana entre sí, de tal manera que el compuesto puede afectar la actividad enzimática de la cinasa ya sea directamente; es decir, interactuando con la cinasa misma o indirectamente; es decir, interactuando con otra molécula en la que es dependiente la actividad catalítica de la cinasa. Como se usa en el presente documento, la administración puede realizarse ya sea in vitro, es decir, en un tubo de prueba, o in vivo, en células o tejidos de un organismo viviente.
En el presente documento, el término “tratamiento” incluye abrogar, inhibir sustancialmente, disminuir o revertir la progresión de una enfermedad o trastorno, mejorar sustancialmente los síntomas clínicos de una enfermedad o trastorno o prevenir sustancialmente la aparición de los síntomas clínicos de una enfermedad o trastorno.
En el presente documento, el término “prevenir” se refiere a un método para evitar que un organismo adquiera un trastorno o enfermedad en primer lugar.
Para cualquier compuesto usado en esta invención, una cantidad terapéuticamente eficaz, también se refiere en el presente documento, como una dosis terapéuticamente eficaz, puede estimarse inicialmente de las pruebas de cultivo celular. Por ejemplo, una dosis puede formularse en modelos animales para lograr un intervalo de concentración circulante que incluye la CI50 o la CI100, como se determina en el cultivo celular. Esta información puede usarse para determinar más exactamente las dosis útiles en los humanos. Las dosificaciones iniciales también pueden estimarse a partir de los datos in vivo. Usando estas guías iniciales, un experimentado en la técnica podría determinar una dosificación eficaz en los humanos.
Además, puede determinarse la toxicidad y la eficacia terapéutica de los compuestos descritos en el presente documento, mediante los procedimientos farmacéuticos estándares en los cultivos celulares o los animales experimentales, por ejemplo, determinando el LD50 o ED50. La relación de dosis entre el efecto tóxico y terapéutico es el índice terapéutico, y puede expresarse como la relación entre LD50 y ED50. Se prefieren los compuestos que
exhiben altos índices terapéuticos. Los datos obtenidos de estas pruebas de cultivos celulares y estudios en animales pueden usarse para formular un intervalo de dosificación que no es tóxico para el uso en el humano. La dosificación de tales compuestos se encuentra preferentemente dentro de un intervalo de concentraciones circulantes que excluyen el ED50 con poca o sin toxicidad. La dosificación puede variar dentro de este intervalo, dependiendo de la forma de dosificación empleada y de la ruta de administración usada. La formulación exacta, la ruta de administración y la dosificación pueden elegirse por el médico individual, desde el punto de vista de la afección del paciente (ver, por ejemplo, Fingl et al., 1975, En: The Pharmacological Basis of Therapeutics, capítulo 1, página 1).
La cantidad y el intervalo de dosificación pueden ajustarse individualmente, para proporcionar niveles en plasma del compuesto activo, los cuales son suficientes para mantener el efecto terapéutico. Las dosificaciones usuales en el paciente para el intervalo de administración oral de aproximadamente 50-2000 mg/kg/día, comúnmente de aproximadamente 100-1000 mg/kg/día, preferentemente de aproximadamente 150-700 mg/kg/día y más preferentemente de aproximadamente 250-500 mg/kg/día.
Preferentemente, los niveles en suero terapéuticamente efectivos serán logrados administrando dosis múltiples cada día. En los casos de la administración local o la captación selectiva, la concentración local eficaz del fármaco no puede relacionarse con la concentración en plasma. Un experto en la materia será capaz de optimizar las dosificaciones locales terapéuticamente efectivas sin la experimentación indebida.
Las enfermedades o trastornos preferidos, que los compuestos descritos en el presente documento pueden ser útiles para prevenir, tratar y/o estudiar, son los trastornos proliferativos celulares, especialmente el cáncer, tales como, pero no se limitan a, papiloma, blastoglioma, sarcoma de Kaposi, melanoma, cáncer de pulmón, cáncer de ovario, cáncer de próstata, carcinoma de células escamosas, astrocitoma, cáncer de cabeza, cáncer de cuello, cáncer de piel, cáncer de hígado, cáncer de vejiga, cáncer de mama, cáncer de pulmón, cáncer de útero, cáncer de próstata, carcinoma testicular, cáncer colorrectal, cáncer de tiroides, cáncer pancreático, cáncer gástrico, carcinoma hepatocelular, leucemia, linfoma, enfermedad de Hodgkin y enfermedad de Burkitt.
Técnica anterior
Otros derivados heterocíclicos y su uso como agentes anti-tumorales se han descrito en WO 2007/129044.
Otros derivados de piridina y pirazina se han descrito en el uso para el tratamiento de cáncer en WO 2009/053737 y para el tratamiento de otras enfermedades en WO 2004/055005.
Otros derivados heterocíclicos se han descrito como inhibidores de IKKs en WO 2009/122180.
Las pirrolopirimidinas se han descrito como inhibidores de IKKs y TBK1 en WO 2010/100431.
Los derivados de pirimidina se han descrito como inhibidores de IKKs y TBK1 en WO 2009/030890.
En el documento WO2010100431 se describen otros inhibidores de IKKs y TBK1 para uso en el tratamiento de trastornos hiperproliferativos, neurogenerativos, inflamatorios o autoinmunes.
Sumario de la invención
La invención se relaciona con compuestos de la fórmula I:

en la que:
X representa CH,
R representa Het,
R1 representa piridilo, pirimidilo, piridazinilo, o furo[3,2-b]piridilo, cada uno de los cuales está sin sustituir o monoo disustituido por Hal, A, OR5, CN, COOA, COOH, CON(R5)2 y/o NR5COA';
Het representa furilo, tienilo, pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, piridilo, pirimidinilo, triazolilo, tetrazolilo, tiadiazol, piridazinilo, pirazinilo, indolilo, isoindolilo, bencimidazolilo, indazolilo, quinolilo, 1,3-benzodioxolilo, benzotiofenilo, benzofuranilo o imidazopiridilo, cada uno de los cuales está sin sustituir o mono-, di- o trisustituido por A, COA, (CH2)pHet2, OH, OA, Hal, (CH2)pN(R5)2 , NO2 , CN, (CH2)pCOOR5, (CH2)pCON(R5)2 , NR5COA, (CH2)pCOHet2 y/o (CH2)pfenilo,
Het2 representa dihidropirrolilo, pirrolidinilo, tetrahidroimidazolilo, dihidropirazolilo, tetrahidropirazolilo, dihidropiridilo, tetrahidropiridilo, piperidinilo, morfolinilo, hexahidropiridazinilo, hexahidropirimidinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o mono sustituido por OH y/o A,
A' representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en el que de 1-7 átomos de H pueden reemplazarse por F,
A representa alquilo no ramificado o ramificado que tiene de 1-10 átomos de C, en el que uno o dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N, O, S y/o por grupos -CH=CH- y/o además de 1-7 átomos de H pueden reemplazarse por F,
R5 representa H o alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en el que de 1-7 átomos de H pueden reemplazarse por F,
Hal representa F, Cl, Br o I,
n representa 0, 1,2, 3 o 4,
p representa 0, 1 o 2,
q representa 1,2, 3 o 4,
y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones.
La invención también se relaciona con formas ópticamente activas (estereoisómeros), sales, enantiómeros, racematos, diastereómeros y los hidratos y solvatos de estos compuestos. El término solvatos de los compuestos significa aducciones de moléculas de disolventes inertes en los compuestos, las cuales se forman debido a su fuerza atractiva mutua. Los solvatos son, por ejemplo, mono- o dihidratos o alcóxidos. Por supuesto, la invención también se relaciona con los solvatos de las sales.
El término derivados usados farmacéuticamente significa, por ejemplo, las sales de los compuestos de acuerdo con la invención y también los llamados compuestos de profármacos.
La expresión “cantidad eficaz” representa la cantidad de un medicamento o de un principio activo farmacéutico que causa en un tejido, sistema, animal o humano, una respuesta biológica o médica que se busca o se desea, por ejemplo, por un investigador o médico.
Además, la expresión “cantidad terapéuticamente eficaz” representa una cantidad que, comparada con un sujeto correspondiente que no ha recibido esta cantidad, tiene la siguiente consecuencia:
tratamiento mejorado, curación, prevención o eliminación de una enfermedad, síndrome, afección, complicación, trastorno o efectos secundarios o incluso la reducción en el avance de una enfermedad, afección o trastorno.
La expresión “cantidad terapéuticamente eficaz” también abarca las cantidades que son efectivas para aumentar la función fisiológica normal.
La invención también se refiere al uso de mezclas de los compuestos de la fórmula I, por ejemplo, mezclas de dos diastereómeros, por ejemplo, en la relación 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 o 1:1000.
Preferentemente, éstas son mezclas de compuestos estereoisoméricos.
La invención se refiere a compuestos de la fórmula I y con un proceso para la preparación de los compuestos de la fórmula I, de acuerdo con las reivindicaciones 1-6 y con sales usadas farmacéuticamente, tautómeros y estereoisómeros de las mismas, caracterizados porque:
a) un compuesto de la fórmula II

en la que Y representa un Br o I,
X y R1 tienen los significados indicados en la reivindicación 1,
se hace reaccionar con un compuesto de la fórmula III:
R-L III
en la que R tiene el significado indicado en la reivindicación 1 y
L representa un ácido borónico o un grupo éster del ácido borónico,
o
b) un compuesto de la fórmula IV:

en la que R y X tienen los significados indicados en la reivindicación 1 y
L1 representa Cl, Br, I o un grupo OH libre o modificado funcionalmente de forma reactiva,
se hace reaccionar con un compuesto de la fórmula V:
R1-NH2 V
en la que R1 tiene el significado indicado en reivindicación 1,
y/o una base o ácido de la fórmula I se convierte en una de sus sales.
Anteriormente y a continuación, los radicales R1, R y X tienen los significados indicados para la fórmula I, a menos que se indique expresamente lo contrario.
A representa alquilo, es no ramificado (lineal) o ramificado, y tiene 1,2, 3, 4, 5, 6, 7, 8, 9 o 10 átomos de C. A representa preferentemente metilo, además etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo o tere-butilo, además también pentilo, 1-, 2- o 3-metilbutilo, 1,1-, 1,2- o 2,2-dimetilpropilo, 1 -etilpropilo, hexilo, 1- , 2-, 3- o 4-metilpentilo, 1,1-, 1,2-, 1,3-, 2,2-, 2,3- o 3,3-dimetilbutilo, 1 - o 2-etilbutilo, 1 -etil-1 -metilpropilo, 1 -etil-2-metilpropilo, 1,1,2- o 1,2,2-trimetilpropilo, además, preferentemente, por ejemplo, trifluorometilo.
Preferentemente, A representa particularmente alquilo que tiene 1, 2, 3, 4, 5 o 6 átomos de C, preferentemente metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, tere-butilo, pentilo, hexilo, trifluorometilo, pentafluoroetilo o 1,1,1-trifluoroetilo.
Uno o dos grupos CH y/o CH2 en A también pueden reemplazarse por átomos de N, O o S y/o por grupos -CH=CH-. De esta manera, A también representa, por ejemplo, 2-metoxietilo.
Más preferentemente, A representa alquilo no ramificado o ramificado, que tiene de 1-6 átomos de C, en el que uno o
dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N y/o O y/o además de 1-7 átomos de H pueden reemplazarse por F.
A’ representa alquilo, es no ramificado (lineal) o ramificado, y tiene de 1,2, 3, 4, 5 o 6 átomos de C. Preferentemente, A representa metilo, además etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo o ferc-butilo, además también pentilo, 1-, 2- o 3-metilbutilo, 1,1-, 1,2- o 2,2-dimetilpropilo, 1 -etilpropilo, hexilo, 1 -, 2-, 3- o 4-metilpentilo, 1, 1-, 1,2-, 1,3-, 2,2 , 2,3- o 3,3-dimetilbutilo, 1- o 2-etilbutilo, 1 -etil-1 -metilpropilo, 1 -etil-2-metilpropilo, 1,1,2- o 1,2,2-trimetilpropilo, además preferentemente, por ejemplo, trifluorometilo.
A’ representa preferentemente alquilo que tiene de 1, 2, 3 o 4 átomos de C, preferentemente metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, ferc-butilo o trifluorometilo.
Het representa preferentemente tienilo, pirazolilo, piridilo, cada uno de los cuales está sin sustituir o mono- o disustituido por A, (CH2)pHet2, (CH2)pCON(R5)2 y/o (CH2)pfenilo.
Het2 representa preferentemente pirrolidinilo, piperidinilo, morfolinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o monosustituido por OH y/o A.
R1 representa preferentemente piridilo, pirimidilo, piridazinilo o furo[3,2-b]piridilo, cada uno de los cuales está sin sustituir o monosustituido por Hal, A, OR5, COOA, COOH, CON(R5)2 y/o nR5COA’.
R5 representa preferentemente H, alquilo que tiene de 1,2, 3 o 4 átomos de C, más preferentemente H, metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, ferc-butilo o trifluorometilo.
Hal representa preferentemente F, Cl o Br, pero también I, preferentemente particularmente F o Cl.
A través de toda la invención, todos los radicales que se presenten más de una vez pueden ser idénticos o diferentes, es decir, son independientes entre sí.
Los compuestos de la fórmula I también puede tener uno o más centros quirales y, por lo tanto, se pueden presentar en varias formas estereoisoméricas. La fórmula I abarca todas estas formas.
Por consiguiente, la invención se refiere, en particular, a los compuestos de la fórmula I, en la que por lo menos uno de estos radicales tiene uno de los significados preferidos indicados anteriormente. Algunos grupos preferidos de los compuestos pueden expresarse por las siguientes sub-fórmulas Ia a Ig, las cuales conforman a la fórmula I y en la que los radicales no designados en mayor detalle, tienen el significado indicado para la fórmula I, pero en la que en Ic, Het representa tienilo, pirazolilo, piridilo, cada uno de los cuales está sin sustituir o mono- o disustituido por A, (CH2)pHet2, (CH2)pCON(R5)2 y/o (CH2)pfenilo;
en Ie, Het2 representa pirrolidinilo, piperidinilo, morfolinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o monosustituido por OH y/o A;
en If, A representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que uno o dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N y/o O y/o además de 1 -7 átomos de H pueden reemplazarse por F;
en Ig, X representa CH,
R representa Ar o Het,
R1 representa piridilo, pirimidilo, piridazinilo o furo[3,2-b]piridilo, cada uno de los cuales está sin sustituir o monosustituido por Hal, A, OR5, COOA, COOH, cON(R5)2 y/o NR5COA’,
Het representa tienilo, pirazolilo, piridilo, cada uno de los cuales está sin sustituir o mono- o disustituido por A, (CH2)pHet2, (CH2)pCON(R5)2 y/o (CH2)pfenilo,
Het2 representa pirrolidinilo, piperidinilo, morfolinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o monosustituido por OH y/o A,
A’ representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que 1-7 átomos de H pueden reemplazarse por F,
A representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que uno o dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N y/o O y/o además de 1-7 átomos de H pueden reemplazarse por F,
R5 representa H o alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que 1-7 átomos de H pueden reemplazarse por F,
Hal representa F, Cl, Br o I,
n representa 0, 1,2, 3 o 4,
p representa 0, 1 o 2,
q representa 1,2, 3 o 4,
y sales usadas farmacéuticamente, tautómeros y estereoisómeros de las mismas, incluyendo sus mezclas en todas las relaciones.
Los compuestos de la fórmula I y también los materiales de partida para su preparación, además, se preparan por los métodos conocidos per se, como se describe en la literatura (por ejemplo, en los trabajos estándares, tales como Houben-Weyl, Methoden der organischen Chemie [Methods of Organic Chemistry], Georg-Thieme-Verlag, Stuttgart), para ser preciso en condiciones de reacción que son conocidas y apropiadas para tales reacciones. También puede hacerse uso en la presente de variantes conocidas per se, las cuales no se mencionan en la presente en mayor detalle. Los compuestos de la fórmula I pueden obtenerse preferentemente haciendo reaccionar los compuestos de la fórmula II con un compuesto de la fórmula III.
En general, se conocen los compuestos de la fórmula II y de la fórmula III. Si éstos son nuevos, pueden prepararse por los métodos conocidos per se.
La reacción se lleva a cabo en condiciones estándares conocidas, como la reacción de Suzuki para la persona experta en la materia.
En los compuestos de la fórmula III, L representa preferentemente:

Dependiendo de las condiciones usadas, el tiempo de reacción es entre unos pocos minutos y 14 días, la temperatura de reacción es entre aproximadamente -30° y 140°, normalmente entre 0° y 110°, en particular, entre aproximadamente 60° y 110°.
Ejemplos de los disolventes inertes apropiados son los hidrocarburos, tales como hexano, éter de petróleo, benceno, tolueno o xileno; hidrocarburos clorados, tales como tricloroetileno, 1,2-dicloroetano, tetracloruro de carbono, cloroformo o diclorometano; alcoholes, tales como metanol, etanol, isopropanol, n-propanol, n-butanol o íerc-butanol; éteres, tales como éter dietílico, éter diisopropílico, tetrahidrofurano (THF) o dioxano; éteres de glicol, tales como etilenglicol monometil o monoetil éter, etilenglicol dimetil éter (diglimo); cetonas, tales como acetona o butanona; amidas, tales como acetamida, dimetilacetamida o dimetilformamida (DMF); nitrilos, tales como acetonitrilo; sulfóxidos, tales como sulfóxido de dimetilo (DMSO); disulfuro de carbono; ácidos carboxílicos, tales como ácido fórmico o ácido acético; compuestos nitro, tales como nitrometano o nitrobenceno; ésteres, tales como acetato de etilo, o mezclas de tales disolventes.
Se da preferencia particular al etanol, tolueno, dimetoxietano, acetonitrilo, diclorometano, DMF y/o agua.
Además, preferentemente, los compuestos de la fórmula I pueden obtenerse haciendo reaccionar un compuesto de la fórmula IV con un compuesto de la fórmula V.
En general, se conocen los compuestos de la fórmula IV y de la fórmula V. Sin embargo, si éstos son nuevos, pueden prepararse por los métodos conocidos per se.
En los compuestos de la fórmula IV, L1 representa preferentemente Cl, Br, I o un grupo OH libre o modificado reactivamente, tal como, por ejemplo, un éster activado, una imidazolida o alquilsulfoniloxi que tiene de 1 -6 átomos de C (preferentemente, metilsulfoniloxi o trifluorometilsulfoniloxi) o arilsulfoniloxi que tiene de 6-10 átomos de C (preferentemente fenil- o p-tolilsulfoniloxi).
En general, la reacción se lleva a cabo en presencia de un agente de enlace ácido, preferentemente una base orgánica, tal como DIPEA, trietilamina, dimetilanilina, piridina o quinolina.
También puede ser favorable la adición de un hidróxido de metal alcalino o alcalinotérreo, carbonato o bicarbonato u otra sal de un ácido débil de los metales alcalinos o alcalinotérreos, preferentemente de potasio, sodio, calcio o cesio.
Dependiendo de las condiciones usadas, el tiempo de reacción está entre unos cuantos minutos y 14 días, la temperatura de reacción es entre aproximadamente -30° y 140°, normalmente entre -10° y 90°, en particular entre aproximadamente 0° y aproximadamente 70°.
Ejemplos de los disolventes inertes apropiados son hidrocarburos, tales como hexano, éter de petróleo, benceno, tolueno o xileno; hidrocarburos clorados, tales como tricloroetileno, 1,2-dicloroetano, tetracloruro de carbono, cloroformo o diclorometano; alcoholes, tales como metanol, etanol, isopropanol, n-propanol, n-butanol o ferc-butanol; ésteres, tales como éter dietílico, éter diisopropílico, tetrahidrofurano (THF) o dioxano; éteres de glicol, tales como etilenglicol monometil o monoetil éter, etilenglicol dimetil éter (diglimo); cetonas, tales como acetona o butanona; amidas, tales como acetamida, dimetilacetamida o dimetilformamida (DMF); nitrilos, tales como acetonitrilo; sulfóxidos, tales como dimetilsulfóxido (DMSO); disulfuro de carbono; ácidos carboxílicos, tales como ácido fórmico o ácido acético; compuestos nitro, tales como nitrometano o nitrobenceno; ésteres, tales como acetato de etilo o mezclas de tales disolventes.
Se da preferencia particular a acetonitrilo, diclorometano y/o DMF.
La escisión de un éter se lleva a cabo por los métodos, como se conocen para la persona experta en la materia.
Un método estándar de la escisión de éter, por ejemplo, de un éter de metilo, es el uso de tribromuro de boro.
Los grupos retirados hidrogenocatalíticamente, por ejemplo, la escisión de un éter de bencilo, puede romperse, por ejemplo, mediante el tratamiento con hidrógeno en presencia de un catalizador (por ejemplo, un catalizador de metal noble, tal como paladio, de manera ventajosa sobre un soporte, tal como carbono). Los disolventes apropiados en la presente son los indicados anteriormente, en particular, por ejemplo, alcoholes, tal como metanol o etanol o amidas, tal como DMF. En general, la hidrogenólisis se lleva a cabo a temperaturas entre aproximadamente 0 y 100° y presiones entre aproximadamente 0,1 y 200 MPa (1 y 200 bar), preferentemente a 20-30° y 0,1-1 MPa (1-10 bar).
Los ésteres pueden saponificarse, por ejemplo, usando ácido acético o usando NaOH o KOH en agua, agua/THF o agua/dioxano, a temperaturas entre 0 y 100°.
Las alquilaciones sobre el nitrógeno se llevan a cabo en condiciones estándares, como se conocen para la persona experta en la materia.
Los compuestos de la fórmula I además pueden obtenerse liberándolos de sus derivados funcionales por solvólisis, en particular hidrólisis o por hidrogenólisis.
Los materiales de partida apropiados para la solvólisis o hidrogenólisis son los que contienen los grupos amino y/o hidroxilo protegidos correspondientes, en lugar de uno o más grupos amino e/o hidroxilo, preferentemente los que portan un grupo protector amino en lugar de un átomo de H enlazado a un átomo de N, por ejemplo, los que conforman a la fórmula I, pero contienen un grupo NHR’ (en el que R’ representa un grupo protector amino, por ejemplo, BOC o CBZ) en lugar de un grupo NH2.
Además, se da preferencia a los materiales de partida que portan un grupo protector hidroxilo en lugar del átomo de H de un grupo hidroxilo, por ejemplo, los que conforman a la fórmula I, pero contienen un grupo R”O-fenilo (en el que R” representa un grupo protector hidroxilo), en lugar de un grupo hidroxifenilo.
También es posible para una pluralidad de - idénticos o diferentes - grupos amino e/o hidroxilo protegidos que estén presentes en la molécula del material de partida. Si los grupos protectores presentes son diferentes entre sí, éstos pueden, en muchos casos, escindirse selectivamente.
La expresión “grupo protector amino” es conocida en términos generales y se refiere a grupos que son apropiados para proteger (bloquear) un grupo amino contra las reacciones químicas, pero son fáciles de retirar después de que
la reacción química deseada se ha llevado a cabo en cualquier parte de la molécula. Típicos de estos grupos son, en particular, los grupos acilo, arilo, aralcoximetilo o aralquilo insustituidos o sustituidos. Dado que los grupos protectores amino se retiran después de la reacción deseada (o secuencia de reacción), además, su tipo y tamaño no es crucial; sin embargo, se da preferencia a los que tienen 1-20, en particular, 1-8 átomos de C. La expresión “grupo acilo” se entenderá en el sentido más amplio, en conjunto con el presente proceso. Éste incluye los grupos acilo derivados de ácidos alifáticos, aromáticos o carboxílicos heterocíclicos o ácidos sulfónicos, y, en particular, los grupos alcoxicarbonilo, ariloxicarbonilo y especialmente aralcoxicarbonilo. Ejemplos de tales grupos acilo son alcanoilo, tales como acetilo, propionilo, butirilo; aralcanoilo, tal como fenilacetilo; aroilo, tal como benzoilo, tolilo; ariloxialcanoilo, tal como POA; alcoxicarbonilo, tal como metoxicarbonilo, etoxicarbonilo, 2,2,2-tri-cloroetoxicarbonilo, BOC, 2-yodoetoxicarbonilo; aralcoxicarbonilo, tal como CBZ (“carbobenzoxi”), 4-metoxibenciloxicarbonilo, FMOC; arilsulfonilo, tal como Mtr, Pbf, Pmc. Los grupos protectores amino preferidos son BOC y Mtr, además CBZ, Fmoc, bencilo y acetilo.
Asimismo, la expresión “grupo protector hidroxilo” es conocida en términos generales y se relaciona con los grupos que son apropiados para proteger un grupo hidroxilo contra las reacciones químicas, pero son fáciles de remover después de que se ha llevado a cabo la reacción química deseada en cualquier parte de la molécula. Los grupos típicos son los grupos arilo, aralquilo o acilo insustituidos o sustituidos mencionados anteriormente, además también los grupos alquilo. La naturaleza y el tamaño de los grupos protectores hidroxilo no es crucial, dado que se retiran nuevamente después de la reacción química o la secuencia de reacción deseada; se da preferencia a los grupos que tienen 1-20, en particular 1-10, átomos de C. Ejemplos de los grupos protectores hidroxilo son, inter alia, terc-butoxicarbonilo, bencilo, p-nitrobenzoilo, p-toluensulfonilo, terc-butilo y acetilo, en donde se prefieren particularmente bencilo y terc-butilo. Los grupos COOH en ácido aspártico y ácido glutámico se protegen preferentemente en la forma de sus ésteres de terc-butilo (por ejemplo, Asp(OBut)).
Los compuestos de la fórmula I se liberan de sus derivados funcionales - dependiendo del grupo protector usado -por ejemplo, usando ácidos fuertes, usando ventajosamente TFA o ácido perclórico, pero también usando otros ácidos inorgánicos fuertes, tales como ácido clorhídrico o ácido sulfúrico, ácidos carboxílicos orgánicos fuertes, tales como ácido tricloroacético o ácido sulfónico, tales como ácido bencen- o p-toluensulfónico. Es posible la presencia de un disolvente inerte adicional, pero no siempre es necesaria. Los disolventes inertes apropiados son preferentemente orgánicos, por ejemplo, ácidos carboxílicos, tales como ácido acético, éteres, tales como tetrahidrofurano o dioxano, amidas, tal como DMF, hidrocarburos halogenados, tal como diclorometano, además también alcoholes, tales como metanol, etanol o isopropanol, y agua. Además, son apropiadas las mezclas de los disolventes mencionados anteriormente. TFA se usa preferentemente en exceso sin la adición de un disolvente adicional, se usa preferentemente ácido perclórico en la forma de una mezcla de ácido acético y ácido perclórico al 70 % en la relación 9:1. Las temperaturas de reacción para la escisión son ventajosamente entre aproximadamente 0 y aproximadamente 50°, preferentemente entre 15 y 30° (temperatura ambiente).
Los grupos BOC, OBut, Pbf, Pmc y Mtr, por ejemplo, pueden escindirse preferentemente usando TFA en diclorometano o usando aproximadamente HCl de 3 a 5 N en dioxano a 15-30°, el grupo FMOC puede escindirse usando una solución de aproximadamente 5 a 50 % de dimetilamina, dietilamina o piperidina en DMF a 15-30°.
Los grupos protectores removidos hidrogenocatalíticamente (por ejemplo, CBZ o bencilo) pueden escindirse, por ejemplo, por el tratamiento con hidrógeno en presencia de un catalizador (por ejemplo, un catalizador de metal noble, tal como paladio, ventajosamente sobre un soporte, tal como carbono). Los disolventes apropiados son los indicados anteriormente, en particular, por ejemplo, alcoholes, tales como metanol o etanol, o amidas, tal como DMF. En general, la hidrogenólisis se lleva a cabo a temperaturas entre aproximadamente 0 y 100° y presiones entre aproximadamente 0,1 y 20 Mpa (1 y 200 bar), preferentemente a 20-30° y 0,1-1 MPa (1-10 bar). La hidrogenólisis del grupo CBZ tiene éxito, por ejemplo, en 5 a 10 % de Pd/C en metanol o usando formato de amonio (en lugar de hidrógeno) sobre Pd/C en metanol/DMF a 20-30°.
Sales farmacéuticas y otras formas
Los compuestos de acuerdo con la invención pueden usarse en su forma no salina final. Por otro lado, la presente invención también abarca el uso de estos compuestos en la forma de sus sales farmacéuticamente aceptables, que pueden derivarse de diferentes ácidos y bases orgánicos e inorgánicos por lo procedimientos conocidos en la técnica. Las formas salinas farmacéuticamente aceptables de los compuestos de la fórmula I se preparan, en su mayor parte, por los métodos convencionales. Si el compuesto de la fórmula I contiene un grupo carboxilo, puede formarse una de sus sales apropiadas haciendo reaccionar el compuesto con una base apropiada, para dar la sal de adición de base correspondiente. Estas bases son, por ejemplo, hidróxidos de metales alcalinos, que incluyen hidróxido de potasio, hidróxido de sodio e hidróxido de litio; hidróxidos de metales alcalinotérreos, tales como hidróxido de bario e hidróxido de calcio, alcóxidos de metales alcalinos, por ejemplo, etóxido de potasio y propóxido de sodio; y varias bases orgánicas, tales como piperidina, dietanolamina y N-metilglutamina. Asimismo, se incluyen las sales de aluminio de los compuestos de la fórmula I. En el caso de algunos compuestos de la fórmula I, las sales de adición ácida pueden formarse mediante el tratamiento de estos compuestos con ácidos orgánicos e inorgánicos farmacéuticamente aceptables, por ejemplo, haluros de hidrógeno, tales como cloruro de hidrógeno, bromuro de hidrógeno o yoduro de hidrógeno, otros ácidos minerales y las sales correspondientes de los mismos, tales como sulfato, nitrato o fosfato y
similares, y alquil- y monoarilsulfonatos, tales como etansulfonato, toluensulfonato y bencensulfonato, y otros ácidos orgánicos y las sales correspondientes de los mismos, tales como acetato, trifluoroacetato, tartrato, maleato, succinato, citrato, benzoato, salicilato, ascorbato y similares. Por lo tanto, las sales de adición ácida farmacéuticamente aceptables de los compuestos de la fórmula I incluyen las siguientes: acetato, adipato, alginato, arginato, aspartato, benzoato, bencensulfonato (besilato), bisulfato, bisulfito, bromuro, butirato, canforato, canforsulfonato, caprilato, cloruro, clorobenzoato, citrato, ciclopentanpropionato, digluconato, dihidrógenofosfato, dinitrobenzoato, dodecilsulfato, etansulfonato, fumarato, galacterato (de ácido múcico), galacturonato, glucoheptanoato, gluconato, glutamato, glicerofosfato, hemi-succinato, hemisulfato, heptanoato, hexanoato, hipurato, clorhidrato, bromhidrato, yodhidrato, 2-hidroxietansulfonato, yoduro, isetionato, isobutirato, lactato, lactobionato, malato, maleato, malonato, mandelato, metafosfato, metansulfonato, metilbenzoato, monohidrógenofosfato, 2-naftalen-sulfonato, nicotinato, nitrato, oxalato, oleato, palmoato, pectinato, persulfato, fenilacetato, 3-fenilpropionato, fosfato, fosfonato, ftalato, pero esto no representa una restricción.
Además, las sales de bases de los compuestos de acuerdo con la invención incluyen sales de aluminio, amonio, calcio, cobre, hierro (III), hierro (II), litio, magnesio, manganeso (III), manganeso (II), potasio, sodio y zinc, pero esto no se pretende que represente una restricción. De las sales mencionados anteriormente, se da preferencia a amonio; las sales de metales alcalinos, sodio y potasio, y las sales de metales alcalinotérreos, calcio y magnesio. Las sales de los compuestos de la fórmula I, las cuales se derivan de las bases orgánicas no tóxicas farmacéuticamente aceptables incluyen las sales de aminas primarias, secundarias y terciarias, aminas sustituidas, que también incluyen las aminas sustituidas que se presentan naturalmente, aminas cíclicas, y resinas de intercambio iónico básicas, por ejemplo, arginina, betaína, cafeína, cloroprocaína, colina, N,N’-dibenciletilendiamina (benzatina), diciclohexilamina, dietanolamina, dietilamina, 2-dietil-aminoetanol, 2-dimetilaminoetanol, etanolamina, etilendiamina, N-etilmorfolina, N-etilpiperidina, glucamina, glucosamina, histidina, hidrabamina, isopropilamina, lidocaína, lisina, meglumina, N-metil-D-glucamina, morfolina, piperazina, piperidina, resinas de poliamina, procaína, purinas, teobromina, trietanolamina, trietilamina, trimetilamina, tripropilamina y tris(hidroximetil)-metilamina (trometamina), pero esto no se pretende que represente una restricción.
Los compuestos de la presente invención, la cuales contienen grupos que contienen nitrógeno básico, pueden cuaternizarse usando agentes, tales como haluros de alquilo (C1-C4), por ejemplo, metilo, etilo, isopropilo y cloruro, bromuro y yoduro de ferc-butilo; sulfatos de alquilo di(C1-C4), por ejemplo, sulfato de dimetilo, dietilo y diamilo; haluros de alquilo (C10-C18), por ejemplo, cloruro, bromuro y yoduro de decilo, dodecilo, laurilo, miristilo y estearilo; y haluros de arilalquilo (C1-C4), por ejemplo, cloruro de bencilo y bromuro de fenetilo. Los compuestos solubles en agua y aceite de acuerdo con la invención, pueden prepararse usando estas sales.
Las sales farmacéuticas mencionadas anteriormente, las cuales se prefieren, incluyen acetato, trifluoroacetato, besilato, citrato, fumarato, gluconato, hemisuccinato, hipurato, clorhidrato, bromhidrato, isetionato, mandelato, meglumina, nitrato, oleato, fosfonato, pivalato, fosfato de sodio, estearato, sulfato, subsalicilato, tartrato, tiomalato, tosilato y trometamina, pero esto no se pretende que represente una restricción.
Las sales de adición ácida de los compuestos básicos de la fórmula I, se preparan poniendo la base libre en contacto con una cantidad suficiente del ácido deseado, causando la formación de la sal de una manera convencional. La base libre puede regenerarse poniendo en contacto la forma salina con una base y aislando la base libre de una manera convencional. Las formas de la base libre difieren en ciertos aspectos de las formas salinas correspondientes de las mismas, con respecto a algunas propiedades físicas, tales como solubilidad en disolventes polares; para los propósitos de la invención, sin embargo, las sales de otra manera corresponden a las formas de bases libres respectivas de las mismas.
Como se menciona, las sales de adición de bases farmacéuticamente aceptables de los compuestos de la fórmula I, se forman con metales o aminas, tales como metales alcalinos y metales alcalinotérreos o aminas orgánicas. Los metales preferidos son sodio, potasio, magnesio y calcio. Las aminas orgánicas preferidas son N,N’-dibenciletilendiamina, cloroprocaína, colina, dietanolamina, etilendiamina, N-metil-D-glucamina y procaína.
Las sales de adición de bases de los compuestos ácidos, de acuerdo con la invención, se preparan poniendo en contacto la forma de ácido libre con una cantidad suficiente de la base deseada, causando la formación de la sal de una manera convencional. El ácido libre puede regenerarse poniendo en contacto la forma salina con un ácido y aislando el ácido libre de una manera convencional. Las formas del ácido libre difieren en cierto aspecto de las formas salinas correspondientes de las mismas, con respecto a ciertas propiedades físicas, tales como solubilidad en disolventes polares; para los propósitos de la invención, sin embargo, las sales de otra manera corresponden a las formas de ácido libre respectivas de las mismas.
Si un compuesto, de acuerdo con la invención, contiene más de un grupo que es capaz de formar sales farmacéuticamente aceptables de este tipo, la invención también abarca sales múltiples. Las formas salinas múltiples típicas incluyen, por ejemplo, bitartrato, diacetato, difumarato, dimeglumina, difosfato, disodio y triclorhidrato, pero esto no se pretende que represente una restricción.
Con respecto a lo establecido anteriormente, puede observarse que la expresión “sal farmacéuticamente aceptable” en el presente aspecto, se toma para significar un principio activo que comprende un compuesto de la fórmula I, en la forma de una de sus sales, en particular si esta forma salina imparte propiedades farmacocinéticas mejoradas sobre el principio activo, comparado con la forma libre del principio activo o cualquier otra forma salina del principio activo usado previamente. La forma salina farmacéuticamente aceptable del principio activo también puede proporcionar este principio activo por primera vez con una propiedad farmacocinética deseada, la cual no tuvo una influencia positiva previa e incluso puede tener una influencia positiva sobre la farmacodinámica de este principio activo con respecto a su eficacia terapéutica en el cuerpo.
Isótopos
Además, se pretende que un compuesto de la fórmula I incluya las formas marcadas con isótopos del mismo. Una forma marcada con un isótopo de un compuesto de la fórmula I es idéntica a este compuesto, además del hecho de que uno o más átomos del compuesto se han reemplazado por un átomo o átomos que tienen una masa atómica o número de masa que difiere de la masa atómica o número de masa del átomo que usualmente se presenta de forma natural. Ejemplos de los isótopos que ya están comercialmente disponibles y que pueden incorporarse en un compuesto de la fórmula I por los métodos bien conocidos, incluyen: isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, por ejemplo, 2H, 3H, 13C, 1C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36Cl, respectivamente. Un compuesto de la fórmula I, un profármaco, del mismo o una sal farmacéuticamente aceptable de cualquiera, que contiene uno o más de los isótopos mencionados anteriormente y/u otros isótopos de otros átomos, se pretende que sean parte de la presente invención. Un compuesto marcado con un isótopo de la fórmula I, puede usarse en un número de maneras benéficas. Por ejemplo, un compuesto marcado con un isótopo de la fórmula I en la que, por ejemplo, se ha incorporado un radioisótopo, tal como 3H o 14C, es apropiado para las pruebas de distribución de medicamentos y/o tejidos de sustrato. Estos radioisótopos, es decir, tritio (3H) y carbono 14 (14C), se prefieren particularmente debido a su preparación simple y excelente capacidad de detección. La incorporación de isótopos más pesados, por ejemplo deuterio (2H), en un compuesto de la fórmula I tiene ventajas terapéuticas debido a su mayor estabilidad metabólica de este compuesto marcado con el isótopo. La mayor estabilidad metabólica se traduce directamente en un aumento en la vida media in vivo o menores dosificaciones, lo que bajo la mayoría de las circunstancias, representaría una modalidad preferida de la presente invención. Un compuesto marcado con un isótopo de la fórmula I a menudo puede prepararse llevando a cabo los procedimientos descritos en los esquemas de síntesis y la descripción relacionada, en la parte de los ejemplos y en la parte de preparación en el presente texto, reemplazando un reactivo no marcado con un isótopo mediante un reactivo marcado con un isótopo fácilmente disponible.
El deuterio (2H) también puede incorporarse en un compuesto de la fórmula I, con el propósito de manipular el metabolismo oxidativo del compuesto por medio del efecto de isótopo cinético primario. El efecto del isótopo cinético primario es un cambio de la velocidad para una reacción química, que resulta del intercambio de los núcleos isotópicos, que, en cambio, es causado por el cambio en las energías de estado terrestre necesarias para la formación del enlace covalente después del intercambio isotópico. El intercambio de un isótopo más pesado a menudo resulta en una disminución de la energía de estado terrestre para un enlace químico y, de esta manera, causa una reducción en la velocidad en la ruptura del enlace que limita la velocidad. Si se presenta la ruptura del enlace en, o en la vecindad de una región puntual de silla a lo largo del coordinado de una reacción de multi-producto, las relaciones de distribución del producto pueden alterarse sustancialmente. Para una explicación: si el deuterio se enlaza a un átomo de carbono en una posición no intercambiable, son típicas las diferencias de la velocidad de kM/kD = 2-7. Si esta diferencia de velocidad se aplica exitosamente a un compuesto de la fórmula I que es susceptible a la oxidación, el perfil de este compuesto puede modificarse drásticamente in vivo y resultar en propiedades farmacocinéticas mejoradas.
Cuando se descubren y desarrollan los agentes terapéuticos, la persona experimentada en la técnica intente optimizar los parámetros farmacocinéticos, mientras que se mantienen las propiedades in vitro deseables. Es razonable asumir que muchos compuestos con perfiles farmacocinéticos pobres son proclives del metabolismo oxidativo. Las pruebas microsómicas hepáticas in vitro actualmente disponibles, proporcionan una información valiosa sobre el curso del metabolismo oxidativo de este tipo, que en cambio, permite el diseño racional de los compuestos deuterados de la fórmula I con una estabilidad mejorada por medio de la resistencia a tal metabolismo oxidativo. De esta manera, se obtienen mejoramientos significativos en los perfiles farmacocinéticos de los compuestos de la fórmula I, y pueden expresarse cuantitativamente en términos de los aumentos en la vida media in vivo (t/2), la concentración al efecto terapéutico máximo (Cmáx), el área bajo la curva de respuesta a la dosis (AUC), y F; y en términos de la clarificación reducida, la dosis y los costos de materiales.
A continuación se pretende ilustrar lo anterior: un compuesto de la fórmula I, el cual tiene múltiples sitios potenciales de adherencia para el metabolismo oxidativo, por ejemplo, átomos de hidrógeno bencílicos y átomos de hidrógeno enlazados a un átomo de nitrógeno, se prepara como una serie de análogos en los que se reemplazan varias combinaciones de átomos de hidrógeno por átomos de deuterio, de modo que algunos, la mayoría o todos estos átomos de hidrógeno se han reemplazado por átomos de deuterio. Las determinaciones de la vida media permiten una determinación favorable y precisa del grado al que se ha mejorado el mejoramiento en la resistencia al metabolismo oxidativo. De esta manera, se determina que la vida media del compuesto padre puede prolongarse
hasta 100 %, como resultado del intercambio de deuterio-hidrógeno de este tipo.
El intercambio de deuterio-hidrógeno es un compuesto de la fórmula I también puede usarse para obtener una modificación favorable del espectro del metabolito del compuesto iniciador para disminuir o eliminar los metabolitos tóxicos indeseados. Por ejemplo, si surge un metabolito tóxico a través de la escisión del enlace oxidativo de carbonohidrógeno (C-H), puede asumirse razonablemente que el análogo deuterado disminuirá o eliminará ampliamente la producción del metabolito indeseado, aun si la oxidación particular no es una etapa de determinación de la velocidad. La información adicional sobre el estado de la técnica con respecto al intercambio de deuterio-hidrógeno puede encontrarse, por ejemplo, en Hanzlik et al., J. Org. Chem. 55, 3992-3997, 1990, Reider et al., J. Org. Chem. 52, 3326 3334, 1987, Foster, Adv. Drug Res. 14, 1-40, 1985, Gillette et al, Biochemistry 33(10) 2927-2937, 1994 y Jarman et al. Carcinogenesis 16(4), 683-688, 1993.
Además, la invención se refiere a medicamentos que comprenden por lo menos un compuesto de la fórmula I y/o las sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones, y opcionalmente, excipientes y/o adyuvantes.
Las formulaciones farmacéuticas pueden administrarse en la forma de unidades de dosificación que comprenden una cantidad predeterminada de principio activo por unidad de dosificación. Tal unidad puede comprender, por ejemplo, 0,5 mg a 1 g, preferentemente 1 mg a 700 mg, particularmente preferentemente de 5 mg a 100 mg, de un compuesto de acuerdo con la invención, dependiendo de la afección tratada, el método de administración y la edad, el peso y la afección del paciente, o las formulaciones farmacéuticas pueden administrarse en la forma de unidades de dosificación que comprenden una cantidad predeterminada de principio activo por unidad de dosificación. Las formulaciones de la unidad de dosificación preferidas son las que comprenden una dosis diaria o una parte de la dosis, como se indicó anteriormente, o una fracción correspondiente de la misma de un principio activo. Además, las formulaciones farmacéuticas de este tipo pueden prepararse usando un proceso que, en general, se conoce en la técnica farmacéutica.
Las formulaciones farmacéuticas pueden adaptarse para la administración por la vía de cualquier método deseado apropiado, por ejemplo, por los métodos oral (incluyendo bucal o sublingual), rectal, nasal, tópica (incluyendo bucal, sublingual o transdérmica), vaginal o parenteral (incluyendo subcutánea, intramuscular, intravenosa o intradérmica). Tales formulaciones pueden prepararse usando todos los procesos conocidos en la técnica farmacéutica mediante, por ejemplo, la combinación del principio activo con el o los excipientes o adyuvantes.
Las formulaciones farmacéuticas adaptadas para la administración oral pueden administrarse como unidades separadas, tales como, por ejemplo, cápsulas o comprimidos; polvos o gránulos; soluciones o suspensiones en líquidos acuosos o no acuosos; espumas comestibles o alimentos de espuma; o emulsiones líquidas de aceite en agua o emulsiones líquidas de agua en aceite.
De esta manera, por ejemplo, en el caso de la administración oral en la forma de un comprimido o cápsula, el componente del principio activo puede combinarse con un excipiente inerte farmacéuticamente aceptable oral, no tóxico, tal como, por ejemplo, etanol, glicerol, agua y similares. Los polvos se preparan triturando el compuesto hasta un tamaño fino apropiado y mezclándolo con un excipiente farmacéutico triturado de una manera similar, tal como, por ejemplo, un carbohidrato comestible, tal como, por ejemplo, almidón o manitol. Asimismo, puede estar presente un saborizante, conservador, dispersante y un colorante.
Las cápsulas se producen preparando una mezcla en polvo, como se describió anteriormente, y llenando las capas de gelatina formadas con la misma. Los deslizantes y lubricantes, tales como, por ejemplo, ácido silícico altamente disperso, talco, estearato de magnesio, estearato de calcio o polietilenglicol en la forma sólida, pueden adicionarse a la mezcla en polvo antes de la operación de llenado. Asimismo, un desintegrante o solubilizante, tal como, por ejemplo, agar-agar, carbonato de calcio o carbonato de sodio, pueden adicionarse para mejorar la disponibilidad del medicamento después que se ha tomado la cápsula.
Además, si se desea o es necesario, asimismo pueden incorporarse en la mezcla aglomerantes, lubricantes y desintegrantes, así como colorantes. Los aglomerantes apropiados incluyen almidón, gelatina, azúcares naturales, tales como, por ejemplo, glucosa o beta-lactosa, edulcorantes elaborados de maíz, caucho natural y sintético, tal como, por ejemplo, acacia, tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol, ceras y similares. Los lubricantes usados en estas formas de dosificación incluyen oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro de sodio y similares. Los desintegrantes incluyen, sin restringirse a los mismos, almidón, metilcelulosa, agar, bentonita, goma xantana y similares. Los comprimidos se formulan, por ejemplo, preparando una mezcla en polvo, granulando o prensando en seco la mezcla, adicionando un lubricante y un desintegrante y prensando la mezcla completa para dar los comprimidos. Una mezcla en polvo se prepara mezclando el compuesto triturado de una manera apropiada con un diluyente o una base, como se describió anteriormente, y opcionalmente con un aglomerante, tal como, por ejemplo, carboximetilcelulosa, un alginato, gelatina o polivinilpirrolidona, un retardante de disolución, tal como, por ejemplo, parafina, un acelerador de absorción, tal como, por ejemplo, una sal cuaternaria y/o un absorbente, por ejemplo, bentonita, caolín o fosfato de dicalcio. La mezcla en
polvo puede granularse humectándola con un aglomerante, tal como, por ejemplo, jarabe, pasta de almidón, mucílago de acadia o soluciones de celulosa o materiales poliméricos, y prensándola a través de un tamiz. Como una alternativa a la granulación, la mezcla en polvo puede correrse a través de una máquina de tableteo, para dar grumos de forma no uniforme, los cuales se rompen para formar gránulos. Los gránulos pueden lubricarse mediante la adición de ácido esteárico, una sal de estearato, talco o aceite mineral, para evitar la adherencia a los moldes de vaciado del comprimido. La mezcla lubricada después se presiona para dar los comprimidos. Los compuestos de acuerdo con la invención también pueden combinarse con un excipiente inerte de flujo libre y después prensarse directamente para dar los comprimidos sin llevar a cabo las etapas de granulación o prensado en seco. Podría estar presente una capa protectora transparente u opaca que consiste de una capa de sellado de laca, una capa de azúcar o un material polimérico y una capa brillante de cera. Pueden adicionarse colorantes a estos recubrimientos para diferenciar entre las diferentes unidades de dosificación.
Los líquidos orales, tales como, por ejemplo, solución, jarabes y elíxires, pueden prepararse en la forma de unidades de dosificación, de modo que una cantidad dada comprende una cantidad preespecificada del compuesto. Los jarabes pueden prepararse disolviendo el compuesto en una solución acuosa con un saborizante apropiado, mientras que los elíxires se preparan usando un vehículo alcohólico no tóxico. Las suspensiones pueden formularse por dispersión del compuesto en un vehículo no tóxico. Asimismo, pueden adicionarse solubilizantes y emulsificantes, tales como, por ejemplo, alcoholes de isostearilo etoxilados y éteres de polioxietilen sorbitol, conservadores, aditivos saborizantes, tales como, por ejemplo, aceite de hierbabuena o edulcorantes naturales o sacarina, u otros edulcorantes artificiales y similares.
Las formulaciones de la unidad de dosificación para la administración oral, si se desea, pueden encapsularse en microcápsulas. La formulación también puede prepararse de tal manera que la liberación se prolongue o se retarde, tal como, por ejemplo, por recubrimiento o acoplando el material particulado en polímeros, cera y similares.
Los compuestos de la fórmula I y las sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, también pueden administrarse en la forma de sistemas de liberación de liposomas, tales como, por ejemplo, vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas pueden formarse de diferentes fosfolípidos, tales como, por ejemplo, colesterol, estearilamina o fosfatidilcolinas.
Los compuestos de la fórmula I y las sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos también pueden suministrarse usando anticuerpos monoclonales como vehículos individuales a los que se acoplan las moléculas del compuesto. Los compuestos también pueden acoplarse a polímeros solubles como vehículos de medicamentos dirigidos. Tales polímeros pueden abarcar polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamidofenol, polihidroxietilaspartamidofenol o polilisina de óxido de polietileno, sustituidos por radicales de palmitoilo. Los compuestos además pueden acoplarse a una clase de polímeros biodegradables que son apropiados para obtener una liberación controlada de un medicamento, por ejemplo, ácido poliláctico, poli-epsiloncaprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidroxipiranos, policianoacrilatos y copolímeros de bloque reticulados o anfipáticos de hidrogeles.
Las formulaciones farmacéuticas adaptadas para la administración transdérmica pueden administrarse como emplastos independientes para el contacto prolongado, cercano con la epidermis del destinatario. De esta manera, por ejemplo, el principio activo puede liberarse del emplasto por iontoforesis, como se describe en términos generales en Pharmaceutical Research, 3(6), 31 8 (1986).
Los compuestos farmacéuticos adaptados para la administración tópica pueden formularse como ungüentos, cremas, suspensiones, lociones, polvos, soluciones, pastas, geles, atomizaciones, aerosoles o aceites.
Para el tratamiento del ojo u otro tejido externo, por ejemplo la boca y la piel, las formulaciones se aplican preferentemente como un ungüento o crema tópica. En el caso de la formulación proporcione un ungüento, el principio activo puede emplearse ya sea con una base de crema parafínica y miscible en agua. De manera alternativa, el principio activo puede formularse para dar una crema con una base de crema de aceite en agua, o una base de agua en aceite.
Las formulaciones farmacéuticas adaptadas para la aplicación tópica al ojo incluyen gotas para los ojos, en las que el principio activo se disuelve o se suspende en un vehículo apropiado, en particular, un disolvente acuoso.
Las formulaciones farmacéuticas adaptadas para la aplicación tópica en la boca abarcan grageas, pastillas y lavados bucales.
Las formulaciones farmacéuticas adaptadas para la administración rectal, pueden administrarse en la forma de supositorios o enemas.
Las formulaciones farmacéuticas adaptadas para la administración nasal, en las que la sustancia de vehículo en un
sólido, comprende un polvo grueso que tiene un tamaño de partícula, por ejemplo, en el intervalo de 20-500 micrómetros, que se administra de la manera en que toma la inhalación, es decir, por inhalación rápida por medio de los pasajes nasales desde un recipiente que contiene el polvo mantenido cerca de la nariz. Las formulaciones apropiadas para la administración como una atomización nasal o gotas para la nariz, con un líquido como una sustancia de vehículo, abarcan soluciones del principio activo en agua o aceite.
Las formulaciones farmacéuticas adaptadas para la administración por inhalación abarcan polvos o rocíos finamente particulados, los cuales pueden generarse por varios tipos de distribuidores presurizados con aerosoles, nebulizadores o insufladores.
Las formulaciones farmacéuticas adaptadas para la administración vaginal, pueden administrarse como pesarios, tampones, cremas, geles, pastas, espumas o formulaciones en atomización.
Las formulaciones farmacéuticas adaptadas para la administración parenteral incluyen soluciones en inyección estériles acuosas y no acuosas, que comprenden antioxidantes, amortiguadores, bacteriostáticos y solutos, por medio de los cuales la formulación se hace isotónica con la sangre del destinatario a ser tratado; y suspensiones estériles acuosas y no acuosas, las cuales pueden comprender un medio de suspensión y espesadores. Las formulaciones pueden administrarse en recipientes de dosis simple o multidosis, por ejemplo, ámpollas y viales, y almacenarse en el estado secado por congelado (liofilizado), de modo que solo es necesaria la adición del líquido del vehículo estéril, por ejemplo, agua para propósitos de inyección, inmediatamente antes del uso. Las soluciones y las suspensiones de inyección preparadas de acuerdo con la receta pueden prepararse a partir de polvos, gránulos y comprimidos estériles.
Por supuesto, además de los constituyentes mencionados particularmente antes, las formulaciones también pueden comprender otros agentes útiles en la técnica con respecto al tipo particular de formulación; de esta manera, por ejemplo, las formulaciones que son apropiadas para la administración oral pueden comprender saborizantes.
Una cantidad terapéuticamente eficaz de un compuesto de la fórmula I depende de un número de factores, que incluyen, por ejemplo, la edad y el peso del animal, la afección precisa que requiere el tratamiento, y su gravedad, la naturaleza de la formulación y el método de administración, y finalmente se determina por el doctor o veterinario tratante. Sin embargo, una cantidad eficaz de un compuesto de acuerdo con la invención para el tratamiento de crecimiento neoplásico, por ejemplo, carcinoma de colon o mama, en general, está en un intervalo de 0,1 a 100 mg/kg de peso corporal por día. De esta manera, la cantidad real por día para un mamífero adulto que pesa 70 kg usualmente está entre 70 y 700 mg, en donde esta cantidad puede administrarse como una sola dosis por día o usualmente en una serie de dosis en partes (tales como, por ejemplo, dos, tres, cuatro, cinco o seis) por día, de modo que la dosis diaria total es la misma. Una cantidad eficaz de una sal o solvato o de un derivado fisiológicamente funcional del mismo, puede determinarse como la fracción de la cantidad eficaz del compuesto de acuerdo con la invención per se. Puede asumirse que las dosis similares son apropiadas para el tratamiento de otras afecciones mencionadas anteriormente.
Además, la invención se refiere a medicamentos que comprenden por lo menos un compuesto de la fórmula I y/o las sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo los compuestos de los mismos en todas las relaciones, y por lo menos un principio activo de un medicamento adicional.
La invención también se relaciona con un kit que consiste de empaques separados de:
(a) una cantidad eficaz de un compuesto de la fórmula I y/o las sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, que incluyen mezclas de los mismos en todas las relaciones,
y
(b) una cantidad eficaz de un principio activo de medicamento adicional.
El grupo comprende recipientes apropiados, tales como cajas, botellas individuales, bolsas o ámpollas. El grupo, por ejemplo, puede comprender ámpollas separadas, que contiene cada una, una cantidad eficaz de un compuesto de la fórmula I y/o las sales usadas farmacéuticamente y estereoisómeros de las mismas en todas las relaciones, y una cantidad eficaz de un principio activo de medicamento adicional en la forma disuelta o liofilizada.
USO
La invención se refiere a los compuestos de la fórmula I para el uso para el tratamiento de cáncer, choque séptico, glaucoma primario de ángulo abierto (POAG, por sus siglas en inglés), hiperplasia, artritis reumatoide, psoriasis, arterosclerosis, retinopatía, osteoartritis, endometriosis, inflamación crónica y/o enfermedades neurodegenerativas, tales como enfermedad de Alzheimer.
La invención se refiere a compuestos de la fórmula I, para la preparación de un medicamento para el tratamiento de cáncer, choque séptico, glaucoma primario de ángulo abierto (POAG), hiperplasia, artritis reumatoide, psoriasis,
arterosclerosis, retinopatía, osteoartritis, endometriosis, inflamación crónica y/o enfermedades neurodegenerativas, tales como enfermedad de Alzheimer.
Los presentes compuestos son apropiados como principios activos farmacéuticos para los mamíferos, especialmente para los humanos, en el tratamiento y control de enfermedades cancerígenas y enfermedades inflamatorias.
El huésped o paciente puede pertenecer a cualquier especie de mamífero, por ejemplo, una especie de primate, particularmente humanos; roedores, incluyendo ratones, ratas y hámsteres; conejos; caballos, vacas, perros, gatos, etc. Los modelos animales son de interés para las investigaciones experimentales, proporcionando un modelo para el tratamiento de una enfermedad humana.
La susceptibilidad de una célula particular al tratamiento con los compuestos de acuerdo con la invención, puede determinarse por las pruebas in vitro. Típicamente, un cultivo de una célula se combina con un compuesto de acuerdo con la invención, a diferentes concentraciones durante un periodo de tiempo que es suficiente para permitir que los agentes activos, tal como anti IgM, induzcan una respuesta celular, tal como expresión de un marcador superficial, usualmente entre aproximadamente una hora y una semana. La prueba in vitro puede llevarse a cabo usando células cultivadas de sangre o de una muestra de biopsia. La cantidad del marcador superficial expresada, se valora por citometría de flujo, usando los anticuerpos específicos que reconocen el marcador.
La dosis varía dependiendo del compuesto específico usado, la enfermedad específica, el estado del paciente, etc. Una dosis terapéutica es típicamente suficiente para reducir considerablemente la población celular indeseada en el tejido blanco, mientras que se mantiene la viabilidad del paciente. En general, el tratamiento se continúa hasta que se ha presentado una reducción considerable, por ejemplo, en por lo menos una reducción de aproximadamente 50 % en la carga celular, y puede continuarse hasta que esencialmente no se detectan células en el cuerpo.
Para la identificación de una ruta de transducción de señal y para la detección de las interacciones entre las diferentes rutas de transducción de señal, los diferentes científicos han desarrollado modelos o sistemas de modelos apropiados, por ejemplo, modelos de cultivos celulares (por ejemplo, (por ejemplo Khwaja et al., EMBO, 1997, 16, 2783-93) y modelos de animales transgénicos (por ejemplo, White et al., Oncogene, 2001,20, 7064-7072). Para la determinación de algunas etapas en la cascada de transducción de señal, la interacción de compuestos puede usarse para modular la señal (por ejemplo, Stephens et al., Biochemical J., 2000, 351, 95-105). Los compuestos de acuerdo con la invención, también pueden usarse como reactivos para probar las rutas de transducción de señal dependientes de cinasa en animales y/o modelos de cultivo celular o en las enfermedades clínicas mencionadas en esta solicitud.
La medición de la actividad de cinasa es una técnica que es bien conocida para la persona experta en la materia en la técnica. Los sistemas de prueba genéricos para la determinación de la actividad de cinasa usando sustratos, por ejemplo, histona (por ejemplo, Alessi et al., FEBS Lett. 1996, 399, 3, páginas 333-338) o la proteína de mielina básica, se describen en la literatura (por ejemplo, Campos-González, R. and Glenney, Jr., J.R. 1992, J. Biol. Chem. 267, página 14535).
Para la identificación de los inhibidores de cinasa, están disponibles diferentes sistemas de prueba. En la prueba de proximidad de centelleo (Sorg et al., J. of. Biomolecular Screening, 2002, 7, 11-19) y la prueba de placa instantánea, se mide la fosforilación radioactiva de una proteína o péptido como sustrato con yATP. En presencia de un compuesto inhibidor, es detectable una señal radioactiva disminuida, o ninguna. Además, las tecnologías de transferencia de energía de resonancia de fluorescencia de tiempo resuelto, homogénea (HTR-FRET, por sus siglas en inglés) y la polarización de fluorescencia (FP) son apropiadas como los métodos de prueba (Sills et al., J. of Biomolecular Screening, 2002, 191 -214).
Otros métodos de prueba de ELISA no radioactivos usan fosfoanticuerpos específicos (fosfo-ABs). Fosfo-AB une solamente el sustrato fosforilado. Esta unión se puede detectar por quimioluminiscencia usando un segundo anticuerpo anti-oveja conjugado de peroxidasa (Ross et al., 2002, Biochem. J.).
La presente invención abarca el uso de los compuestos de la fórmula I y/o las sales fisiológicamente aceptables, tautómeros y solvatos de los mismos para la preparación de un medicamento para el tratamiento o prevención de cáncer. Los carcinomas preferidos para el tratamiento originan del grupo carcinoma cerebral, carcinoma de tracto urogenital, carcinoma del sistema linfático, carcinoma de estómago, carcinoma de laringe y carcinoma de pulmón, cáncer de intestino. Un grupo adicional de las formas preferidas de cáncer son leucemia monocítica, adenocarcinoma de pulmón, carcinomas de pulmón de células pequeñas, cáncer pancreático, glioblastomas y carcinoma de mama.
También se abarca el uso de los compuestos de la fórmula I y/o sales fisiológicamente aceptables, tautómeros y solvatos de los mismos, para la preparación de un medicamento para el tratamiento y/o control de una enfermedad inducida por un tumor en un mamífero, en el que, para este método, una cantidad terapéuticamente eficaz de un compuesto de acuerdo con la invención, se administra a un mamífero enfermo en necesidad de tal tratamiento. La cantidad terapéutica varía de acuerdo con la enfermedad particular y puede determinarse para la persona experta en
la materia en la técnica sin un esfuerzo indebido.
Se da preferencia particular al uso para el tratamiento de una enfermedad, en donde la enfermedad cancerígena es un tumor sólido.
El tumor sólido se selecciona preferentemente del grupo de tumores de epitelio escamoso, vejiga, estómago, riñones, de cabeza y cuello, esófago, cerviz, tiroides, intestino, hígado, cerebro, próstata, tracto urogenital, el sistema linfático, el estómago laringe y/o pulmón.
Además, el tumor sólido se selecciona preferentemente del grupo de adenocarcinoma de pulmón, carcinoma de pulmón de células pequeñas, cáncer pancreático, glioblastomas, carcinoma de colon y carcinoma de mama.
Además, se da preferencia al uso para el tratamiento de un tumor de la sangre y el sistema inmune, preferentemente para el tratamiento de un tumor seleccionado del grupo de leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfática aguda y/o leucemia linfática crónica.
Además, la invención se refiere al uso de compuestos de acuerdo con la invención para el tratamiento de patologías óseas, en donde la patología ósea se origina del grupo de osteosarcoma, osteoartritis y raquitismo.
Los compuestos de la fórmula I también pueden administrarse al mismo tiempo que otros agentes terapéuticos bien conocidos que se seleccionan para su utilidad particular contra la afección que es tratada.
Los presentes compuestos también son apropiados para la combinación con los agentes anti-cáncer conocidos. Estos agentes anti-cáncer conocidos incluyen los siguientes: moduladores del receptor de estrógeno, moduladores del receptor de andrógeno, moduladores del receptor retinoide, agentes citotóxicos, agentes antiproliferativos, inhibidores de transferasa de la proteína prenil transferasa, inhibidores de HMG-CoA reductasa, inhibidores de VIH proteasa, inhibidores de transcriptasa inversa e inhibidores de angiogénesis adicionales. Los presentes compuestos son particularmente apropiados para la administración al mismo tiempo que la radioterapia.
“Moduladores del receptor de estrógeno” se refiere a los compuestos que interfieren con, o inhiben la unión del estrógeno al receptor, sin considerar el mecanismo. Ejemplos de los moduladores del receptor de estrógeno incluyen, pero no se limitan a, tamoxifeno, raloxifeno, idoxifeno, LY353381, LY117081, toremifeno, fulvestrant, 2,2-dimetilpropanoato de 4-[7-(2,2-dimetil-1 -oxopropoxi-4-metil-2-[4-[2-(1 -piperidinil)etoxi]fenil]-2H-1 -benzopiran-3-il]fenilo, 4,4’-dihidroxibenzofenon-2,4-dinitrofenilhidrazona y SH646.
“Moduladores del receptor de andrógeno” se refiere a los compuestos que interfieren con o inhiben la unión de andrógenos al receptor, sin considerar el mecanismo. Ejemplos de los moduladores del receptor de andrógeno incluyen finasterida y otros inhibidores de 5a-reductasa, nilutamida, flutamida, bicalutamida, liarozol y acetato de abiraterono.
“Moduladores del receptor retinoide” se refiere a compuestos que interfieren con o inhiben la unión de retinoides al receptor, sin considerar el mecanismo. Ejemplos de los moduladores del receptor retinoide incluyen bexaroteno, tretinoina, ácido 13-cis-retinoico, ácido 9-cis-retinoico, a-difluorometilornitina, ILX23-7553, trans-N-(4’-hidroxifenil)retinamida y N-4-carboxifenilretinamida.
“Agentes citotóxicos” se refiere a los compuestos que resultan en la muerte celular principalmente a través de la acción directa sobre la función celular o inhiben o interfieren con la miosis celular, que incluyen agentes de alquilación, factores de necrosis tumoral, intercaladores, inhibidores de microtubulina e inhibidores de topoisomerasa.
Ejemplos de los agentes citotóxicos incluyen, pero no se limitan a, tirapazimina, sertenef, caquectina, ifosfamida, tasonermina, lonidamina, carboplatina, altretamina, prednimustina, dibromodulcitol, ranimustina, fotemustina, nedaplatina, oxaliplatina, temozolomida, heptaplatina, estramustina, tosilato de improsulfan, trofosfamida, nimustina, cloruro de dibrospidio, pumitepa, lobaplatina, satraplatina, profiromicina, cisplatina, irofulven, dexifosfamida, cisamindicloro(2-metilpiridin)platino, bencilguanina, glufosfamida, GPX100, tetracloruro de (trans,trans,trans)bis-mu-(hexan-1,6-diamin)-mu-[diamin-platino (II)] bis[diamin(cloro) platino (II)], diansidinilspermina, trióxido arsénico, 1-(11-dodecilamino-10-hidroxiundecil)-3,7-dimetilxantina, zorubicina, idarubicina, daunorubicina, bisantreno, mitoxantrono, pirarubicina, pinafida, valrubicina, amrubicina, antineoplastona, 3’-deamino-3’-morfolino-13-desoxo-10-hidroxicarminomicina, anamicina, galarubicina, elinafida, MEN10755 y 4-demetoxi-3-deamino-3-aziridinil-4-metilsulfonildaunorubicina (ver WO 00/50032).
Ejemplos de los inhibidores de microtubulina incluyen paclitaxel, sulfato de vindesina, 3’,4’-dideshidro-4’-desoxi-8’-norvincaleucoblastina, docetaxol, rizoxina, dolastatina, isetionato de mivobulina, auristatina, cemadotina, RPR109881, BMS184476, vinflunina, criptoficina, 2,3,4,5,6-pentafluoro-N-(3-fluoro-4-metoxifenil)bencensulfonamida, anhidrovinblastina, N,N-dimetil-L-valil-L-valil-N-metil-L-valil-L-prolil-L-prolina-t-butilamida, TDX258 y BMS188797.
Los inhibidores de topoisomerasa son, por ejemplo, topotecano, hicaptamina, irinotecano, rubitecano, 6-etoxipropionil-3’,4’-O-exobencilidencartreusina, 9-metoxi-N,N-dimetil-5-nitropirazolo[3,4,5-il]acridin-2-(6H)propanamina, 1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1 H,12H-benzo[de]pirano[3’,4’:b,7]indolizino[1 ,2b]quinolin-10,13(9H,15H)-diona, lurtotecano, 7-[2-(N-isopropilamino)etil]-(20S)camptotecina, BNP1350, BNPI1100, Bn80915, BN80942, fosfato de etopósido, tenipósido, sobuzoxano, 2’-dimetilamino-2’-desoxietopósido, GL331, N-[2-(dimetilamino)etil]-9-hidroxi-5,6-dimetil-6H-pirido[4,3-b]carbazol-1-carboxamida, asulacrina, (5a,5aB,8aa,9b)-9-[2-[N-[2-(di-metilamino)etil]-N-metilamino]etil]-5-[4-hidroxi-3,5-dimetoxifenil]-5,5a,6,8,8a,9-hexohidrofuro(3’,4’:6,7)nafto(2,3-d)-1,3-dioxol-6-ona, 2,3-(metilen-dioxi)-5-metil-7-hidroxi-8-metoxibenzo[c]fenantridinio, 6,9-bis[(2-amino-etil)amino]benzo[g]isoquinolin-5,10-diona, 5-(3-aminopropilamino)-7,10-dihidroxi-2-(2-hidroxietilaminometil)-6H-pirazolo[4,5,1-de]acridin-6-ona, N-[1 -[2(dietilamino)etilamino]-7-metoxi-9-oxo-9H-tioxanten-4-ilmetil]formamida, N-(2-(dimetilamino)etil)acridin-4-carboxamida, 6-[[2-(dimetilamino)etil]amino]-3-hidroxi-7H-indeno[2,1-c]quinolin-7-ona y dimesna.
Los “agentes antiproliferativos” incluyen oligonucleótidos de ARN y ADN antisentido, tales como G3139, ODN698, RVASKRAS, GEM231 y INX3001 y antimetabolitos, tales como enocitabina, carmofur, tegafur, pentostatina, doxifluridina, trimetrexato, fludarabina, capecitabina, galocitabina, ocfosfato de citarabina, fosteabina sódica hidratada, raltitrexed, paltitrexid, emitefur, tiazofurina, decitabina, nolatrexed, pemetrexed, nelzarabina, 2’-desoxi-2’-metilidencitidina, 2’-fluorometilen-2’-desoxicitidina, N-[5-(2,3-dihidrobenzofuril)sulfonil]-N’-(3,4-dicriloroprienil)urea, N6-[4-desoxi-4-[N2-[2(E),4(E)-tetradecadienoil]glicilamino]-L-glicero-B-L-manohepto-piranosil]adenina, aplidina, ecteinascidina, troxacitabina, ácido 4-[2-amino-4-oxo-4,6,7,8-tetrahidro-3H-pirimidino[5,4-b]-1,4-tiazin-6-il-(S)-etil]-2,5-tienoil-L-glutámico, minopterina, 5-fluorouracilo, alanosina, éster del ácido 11-acetil-8-(carbamoiloximetil)-4-formil-6-metoxi-14-oxa-1,11-diazatetraciclo(7,4.1,0.0)tetradeca-2,4,6-trien-9-ilacético, eswainsonina, lometrexol, dexrazoxana, metioninasa, 2’-ciano-2’-desoxi-N4-palmitoil-1-B-D-arabinofuranosil citosina y 3-aminopiridin-2-carboxaldehído tiosemicarbazona. Los “agentes antiproliferativos” también incluyen anticuerpos monoclonales para factores de crecimiento diferentes de los listados bajo “inhibidores de angiogénesis”, tales como trastuzumab, y genes supresores de tumores, tal como p53, que pueden liberarse por medio de la transferencia génica mediada por virus recombinantes (ver, por ejemplo, la patente US No. 6,069,134).
Prueba para la inhibición de IKKe
IKKe - Prueba de cinasa (IKKepsilon)
Sumario
La prueba de cinasa se realiza ya sea como una prueba de placa instantánea de 384 pozos (para por ejemplo, medición de Topcount).
IKKs 1 nM, péptido kBa(19-42) biotinilado 800 nM (Biotina-C6-C6-GLKKERLLDDRHDSGLDSMKDEE) y ATP 10 pM (picado con 33P-ATP/pozo 0,3 pCi) se incuban en un volumen total de 50 pl (MOPS 10 mM, acetato de Mg 10 mM, EGTA 0,1 mM, ditiotreitol 1 mM, 0,02 % de Brij35, BSA al 0,1 %, BioStab al 0,1 %, pH 7,5) con o sin el compuesto de prueba durante 2 horas a 30°C. La reacción se detiene con 25 pl de EDTA 200 mM. Después de 30 minutos a temperatura ambiente, el líquido se retira y cada pozo se lava tres veces con 100 pl de solución de cloruro de sodio al 0,9 %. La reacción no específica se determina en presencia de MSC2119074 3 pM (BX-795). La radioactividad se mide con Topcount (PerkinElmer). Los resultados (por ejemplo, los valores CI50) se calculan con las herramientas del programa proporcionadas por el departamento IT (por ejemplo, AssayExplorer, Symyx).
Prueba para la inhibición de TBK1
Prueba enzimática
Sumario
La prueba de cinasa se realiza como una prueba de placa instantánea de 384 pozos (por ejemplo, medición Topcount.
La cinasa de unión TANK 0,6 nM (TBK1), el péptido derivado de MELK biotinilado 800 (Biotina-Ah-Ah-AKPKGNKDYHLQTCCGSLAYRRR) y aTp 10 pM (picado con 33P-ATP/pozo 0,25 pCi) se incuban en un volumen total de 50 pl (MOPS 10 mM, acetato de Mg 10 mM, EGTA 0,1 mM, DTT 1 mM, Brij35 al 0,02 %, BSA al 0,1 %, pH 7,5) con o sin el compuesto de prueba durante 120 minutos a 30°C. La reacción se detiene con 25 pl de EDTA 200 mM. Después de 30 minutos a temperatura ambiente, el líquido se retira y cada pozo se lava tres veces con 100 pl de solución de cloruro de sodio al 0,9 %. La reacción no específica se determina en presencia de estaurosporina 100 nM. La radioactividad se mide en un Topcount (PerkinElmer). Los resultados (por ejemplo, los valores CI50) se calculan con las herramientas del programa proporcionadas por el departamento IT (por ejemplo, AssayExplorer, Symyx).
Prueba celular
Inhibición de la respuesta a la dosis de Fosfo-IRF3 @ Ser 386
célula/MDAMB468/INH/PHOS/IMAG/plRF3
1. Alcance
Aunque TBK1 y IKKs se conocen mejor como jugadores clave en la respuesta inmune innata, los recientes descubrimientos se han enfocado hacia un papel para TBK1 y IKKs en la transformación oncogénica inducida por Ras. TBK1 se identificó como un efector de RalB en la ruta del factor de intercambio nucleotídico de (Ral)-guanina similar a Ras (GEF, por sus siglas en inglés) que se requiere para la transformación inducida por Ras. TBK1 activa directamente IRF3 que, con la fosforilación, homodimeriza y desplaza al núcleo en donde activa los procesos involucrados con la inflamación, regulación inmune, supervivencia y proliferación celular.
Esta prueba se ha concebido para valorar la eficacia/potencia de los compuestos inhibidores de TBK1/IKKs con base en la detección inmunocitoquímica de fosfo-IRF3 localizado nuclear, un blanco directamente en la dirección 3’ de TBK1.
El tratamiento con poliinosina-ácido policitidílico (poly(I:C), un análogo sintético de ARN de doble hebra (ARNds), un patrón molecular asociado con la infección viral que se reconoce por el receptor 3 similar a Toll (TLR3) se usa para inducir la actividad de TBK1/IKKe y la fosforilación de IRF3 en Ser386.
2. PANORAMA DE LA PRUEBA
Día 1: Las células MDA-MB-468 se separan con HyQ-Tase, contada, y sembrada en una placa de superficie TC de fondo claro de 384 pozos a una densidad de 10,000 células por pozo en un volumen total de 35 ul de medio completo. Alternativamente, las células se siembran directamente de las ampolletas congeladas.
Día 2: Las células se pre-tratan con los compuestos inhibidores durante 1 h antes de la estimulación de Poly(I:C). Después de 2 h de incubación con Poly(I:C), las células se fijan en (para)formaldehído (PFA) y se permeabilizan con metanol (MeOH). Las células después se bloquean y se incuban con un anticuerpo anti-pIRF3 a 4°C durante la noche. Día 3: El anticuerpo primario se lava, se añade AlexaFluor488-conjugado secundario, las células se tiñen y se cuentan con yoduro de propidio seguido de la adquisición de la imagen en un lector de alto contenido IMX Ultra.
3. Reactivos, Materiales
células: ATCC HTB 132, Burger lab (MP-CB 2010-327 o MDA-MB-468/10)
medio de placas= medio de cultivo:
RPMI 1640, Invitrogen n.231870
10 % de FCS, Invitrogen n.° 10270-106
Glutamax 2 mM, Invitrogen n.° 35050-038
Natrium-Pyruvat 1 mM, Invitrogen n.° 11360
1 % de Pen/estrep
37°C, 5 % de CO2
placas: placas de cultivo celular de fondo de 384 pozos de fondo oscuro/claro, Falcon n.° 353962 o Greiner n.° 781090
subcultivación: HyQ-Tase, Thermo Scientific (HyClone) n.° SV30030,01
Otros reactivos:
Poli(I:C) (LMW), Invivogen n.° tlrl-picw (preparar 20 mg/ml de patrón en PBS estéril, desnaturalizar 30 min, 55°C en un baño de agua, enfriar lentamente a RT (temperatura ambiente), almacenar a -20°C en alícuotas) Inhibidor de referencia: MSC2119074A-4 = BX-795 (CI50: 200-800 nM)
Control de inhibidor: MSC2119074A-4 10 pM = BX-795
control neutro : DMSO al 0,5 %
una curva de respuesta de la dosis de 10 puntos con MSC2119074 A-4 = BX-795 se incluye en cada experimento
Hepes, Merck n.° 1,10110
PBS 1 x DPBS, Invitrogen n.° 14190
Formaldehído (libre de metanol, 16 %, Grado EM ultrapuro), Polysciences n.° 18814 (almacenar a RT), concentración final: 4 %
Metanol, Merck n.° 1,06009,101 1 (pre-enfriado a -202C)
Suero de cabra, PAA n.° B15-035 (almacenar a 4°C, tiempo largo -20°C), concentración final: 10 %
BSA (IgG y libre de proteasa, 30 %), US-Biological n.° A1317 (almacenar a 4°C, tiempo largo -20°C), concentración final: 2 %
Tween 20 Detergente, Calbiochem n.° 655204 (almacenar a RT), (preparar un patrón al 10 % en agua; concentración final: 0,1 %)
Conejo anti-pIRF-3 mAb, Epitomics n.° 2526-B (almacenar a -20°C), concentración final: 1:2000 en PBS/2 % de BSA
Cabra-anti-Conejo-488 Alexa Fluor, Invitrogen n.° A11034 o n.° A11008 (almacenar a 4°C, en la oscuridad), concentración final: 1:2000 en PBS/2 % de BSA/0,1 % de Tween
Yoduro de propidio (PI), Fluka n.° 81845, 1 mg/ml en H2O (almacenar a 4°C, oscuridad), concentración final: 0,2 pg/ml.
4. Procedimiento
Sembrar 10.000 células/pozo/35 ul de RPMI completo 10 % de FCS en placas de cultivo de fondo de 384 pozos de fondo oscuro/claro
I
Incubar durante 2 h a temperatura ambiente en la banca seguido de la incubación adicional durante 22 h a 37°C,
5 % de CO2 y 90 % de rH (humedad relativa)
I
Tratamiento del compuesto: Añadir 5 gl de los compuestos prediluidos, reactivos estándares o de control (concentración de 8 veces)
dilución del compuesto de los patrones de DMSO en Hepes 20 mM, pH 7,2; concentración final de DMSO: 0,5 % de dilución en serie de los compuestos de los patrones 10 mM (Remp) 10 etapas, 3,16 veces en DMSO 30 gM, 9,49 gM, 3 gM, 0,95 gM, 0,3 gM, 0,095 gM, 0,03 gM, 0,0095 gM, 0,003 gM, 0,00095 gM Incubar durante 60 minutos a 37°C, 5 % de CO2 y 90 % de humedad relativa
I
Tratamiento de estimulación: Añadir 10 gl de Poli(I:C) a todos los pozos excepto para los controles no estimulados, de modo que se obtenga una concentración final de 100 ug/ml (patrón 20 mg/ml ^ 1:40 en PBS)
(concentración de 5 veces)
Incubar durante 120 minutos a 37°C, 5 % de CO2 y 90 % de rH
I
aspirar completamente el sobrenadante
I
Fijar las células: Añadir 100 gl de 4 % de paraformaldehído en PBS
Incubar durante 15 minutos a RT
I
Lavar 3x con 80 |jl de PBS (poder lavador de Tecan), aspirar completamente el sobrenadante colocado en la placa sobre hielo
I
Permeabilizar las células: Añadir rápidamente 100 j l de MeOH frío a -202C (depósito pre-enfriado) Incubar durante 10 minutos a RT o 4°C
I
Lavar una vez con 80 j l de PBS (poder lavador de Tecan), aspirar completamente el sobrenadante
I
Unión no específica de bloque: Añadir 30 j1 de suero de cabra al 10 % en PBS/2 % de BSA Agitar en Multidrop Combi (17 segundos)
Incubar durante 60 minutos a 37°C
I
Aspirar completamente el sobrenadante
I
Teñido primario: Añadir 25 j l de anticuerpo primario diluido 1:2000 en PBS/2 % de BSA Agitar en Multidrop Combi (17 segundos)
Incubar O/N a 4°C
I
Lavar 3x con 80 j l de PBS (poder lavador de Tecan), aspirar completamente el sobrenadante Teñido secundario y teñido nuclear: Añadir 25 j l de anticuerpo secundario (1:2000) y
0,2 jg/m l de yoduro de propidio en PBS/2 % de BSA/0,1 % de Tween
Agitar en Multidrop Combi (17 segundos)
Incubar durante 75 minutos a 37°C
I
Lavar 3x con 80 j l de PBS (poder lavador de Tecan), aspirar completamente el sobrenadante
I
Repartir 80 j l de PBS en todos los pozos
I
Sellar las placas con sellos adhesivos transparentes
I
Adquisición de la imagen a IMX Ultra (ajustes de exploración Metaexpress 3,1. TBK 10x_pin8)
I
Análisis de la imagen (Metaexpress 3,1. <registro celular>, TBK1-Cellscoring)
I
Análisis de dados y reporte con el explorador de prueba
Condiciones de HPLC/EM:
columna: Chromolith SpeedROD RP-18e, 50 x 4,6 mm2
gradiente: A:B = 96:4 a 0:100
Caudal: 2,4 ml/min
eluyente A: agua ácido fórmico al 0,05 %
eluyente B: acetonitrilo ácido fórmico al 0,04 %
longitud de onda: 220 nm
Espectroscopia de masa: modo positivo
RMN 1H: constante de acoplamiento J [Hz].
Esquema de síntesis general preferido para manufacturar los compuestos de la fórmula I


Procedimiento experimental:
A una solución agitada del ácido 5-bromo-2-amino-nicotínico (500 mg, 2,3 mol, 1 eq.) y 4-aminopiridina (260 mg, 2,7 mol, 1,2 eq.) en DMF seca (5 ml) se le añaden HATU (1,31 g, 3,4 mol, 1,5 eq.) y N-metilmorfolina (690 mg, 6,9 mol, 3 eq.) y se deja agitar durante 3 h. Después de completar la reacción, la mezcla de reacción se concentra; se añade agua, el sólido se precipita y se filtra, se lava con NaHCO3 y agua para dar el producto.
RMN 1H (400 MHz, DMSO-d6) ó 10,51 (s, 1H), 8,47-8,46 (d, J = 5,4 Hz, 2H), 8,25-8,22 (dd, J = 2,4, 7,4 Hz, 2H), 7,69 7,67 (d, J = 6,2 Hz, 2H).

A una solución en agitación de 2-amino-5-bromo-N-piridin-4-il-nicotinamida (1 eq.) en tolueno:etanol (4:1), se le añaden ácido borónico sustituido (1,2 eq.), Na2CO3 2 M (1,5 eq.) y se desgasifica durante 15 minutos bajo una
atmósfera de N2. A esta mezcla de reacción, se añade Pd(PPh3)4 (0,012 eq.) y se calienta a 100°C durante 18 horas. Después de completar la reacción, la mezcla se pasa a través de una cama de Celite para remover las impurezas inorgánicas. El filtrado se concentra al vacío. La cromatografía de columna proporciona el compuesto puro.
Ejemplo 1
La preparación de 2-amino-5-(5-piperidin-1 -ilmetil-tiofen-2-il)-N-piridin-4-il-nicotinamida (“A1”) se lleva a cabo de manera análoga al siguiente esquema de reacción:

1.1.2-Amino-5-bromo-N-piridin-4-il-nicotinamida:
2,0 g de ácido 2-amino-5-bromonicotínico y 1,06 g de 4-amino-piridina se disuelven en 20 ml de DMF. Se añaden a la solución 5,26 g de HATU (hexafluorofosfato de (2-(7-aza-1H-benzotriazol-1-il)-1,1,3,3-tetrametil-uronio) y 3,04 ml de N-metilmorfolina.
La mezcla se agita durante 5 h a temperatura ambiente. La DMF se evapora y el residuo se tritura con agua. El sólido se filtra y se lava con solución de NaHCO3 y agua. Se obtienen 2,5 g de un sólido de color pardo claro;
HPLC/EM: 1,13 min, [M+H] = 293;
RMN 1H (400 MHz, DMSO-do) 5 [ppm] 10,51 (s, 1H, NH), 8,48 (d, J = 6,0, 2H), 8,25 (dd, J = 10,8, 2,3, 2H), 7,69 (d, J = 6,3, 2H), 7,18 (s, 2H, NH2).
1.2. 2-Amino-5-(5-piperidin-1 -ilmetil-tiofen-2-il)-N-piridin-4-il-nicotinamida (“A1 ”)
200 mg de 2-amino-5-bromo-N-piridin-4-il-nicotinamida y 218 mg de pinacol éster del ácido 5-(1 -piperidinilmetil)-tiofen-2-borónico se disuelven en 8 ml de DMF. Se añaden 0,84 ml de una solución 2 molar de Na2CO3 en nitrógeno. Se añaden 7,81 mg de tetracis(trifenilfosfin)-paladio (0). La mezcla se agita durante 6 h a 100°C.
La mezcla de reacción se enfría a temperatura ambiente y se evapora la DMF. Se añade agua y el precipitado resultante se filtra, se lava y se seca. El sólido se tritura con acetato de etilo y se filtra. Se obtienen 47 mg del producto deseado “A1 ” como un sólido de color pardo claro;
HPLC/EM: 1,08 min, [M+H] = 394;
RMN 1H (500 MHz, DMSO-do) 5 [ppm] 10,58 (s, 1H, NH), 8,48 (d, J=6,2, 2H), 8,44 (d, J=2,3, 1H), 8,22 (d, J=2,3, 1H), 7,71 (dd, J=4,8, 1,5, 2H), 7,25 (d, J=3,5, 1H), 7,13 (s, 2H, NH2), 6,93 (d, J=3,5, 1H), 3,61 (s, 2H), 2,37 (a, 4H), 1,51 (m, 4H), 1,39 (m, 2H).
Los siguientes compuestos se obtienen de manera análoga:
2-Amino-N-piridin-4-il-5-(5-pirrolidin-1-ilmetil-tiofen-2-il)-nicotinamida (“A2”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con pinacol éster del ácido 5-(1-pirrolidinil-metil)tiofen-2-borónico da el compuesto “A2”;
HPLC/EM: 1,16 min, [M+H] = 380;
RMN 1H (400 MHz, DMSO-de) S [ppm] 10,51 (s, 1H, NH), 8,51-8,46 (m, 3H), 8,27 (d, J=2,4, 1H), 8,24 (d, J=2,4, 1H), 7,69 (m, 3H), 7,18 (s, 2H), 4,70 (s, 2H), 3,54 (m, 2H), 3,23 (m, 2H), 2,12 (m, 2H), 1,98 (m, 2H);
2-Amino-5-5-metil-tiofen-2-il)-N-piridin-4-il-nicotinamida (“A3”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con pinacol éster del ácido 5-metiltiofen-2-borónico da el compuesto “A3”;
HPLC/EM: 1,45 min, [M+H] = 311;
RMN 1H (400 MHz, DMSO-d6) S [ppm] 10,74 (s, 1H, NH), 8,53 (d, J=4,9, 2H), 8,40 (d, J=2,3, 1H), 8,22 (d, J=2,1, 1H), 7,79 (d, J=11,8, 2H), 7,22 (d, J=3,5, 1H), 7,12 (s, 2H, NH2), 6,86-6,74 (m, 1H), 2,47 (s, 3H);
2-Amino-5-(3-pirazol-1 -il-fenil)-N-piridin-4-il-nicotinamida (“A4”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con 1-[3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-fenil]-1 H-pirazol da el compuesto “A4”;
HPLC/EM: 1,44 min, [M+H] = 357;
RMN 1H (400 MHz, DMSO-de) S [ppm] 11,40 (s, 1H, NH), 8,74 (t, J=7,2, 2H), 8,72 (s, 1H), 8,62 (d, J=2,3, 1H), 8,53 (d, J=2,4, 1H), 8,19 (d, J=7,2, 2H), 8,16 (t, J=1,8, 1H), 7,83 (ddd, J=7,9, 2,1, 1,0, 1H), 7,78 (d, J=1,6, 1H), 7,67 (dd, J=6,6, 1,4, 1H), 7,60 (t, J=7,9, 1H), 7,38 (s, 2H), 6,58 (dd, J=2,4, 1,8, 1H);
2-Amino-5-(4-pirazol-1 -il-fenil)-N-piridin-4-il-nicotinamida (“A5”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido [4-(1 H-pirazol-1 -il)fenil]borónico da el compuesto “A5”;
HPLC/EM: 1,38 min, [M+H] = 357;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 10,60 (s, 1H, NH), 8,59 (d, J=2,4, 1H), 8,54 (d, J=2,4, 1H), 8,52-8,45 (m, 2H), 8,40 (d, J=2,4, 1H), 7,98-7,92 (m, 2H), 7,88-7,84 (m, 2H), 7,78-7,72 (m, 3H), 7,16 (d, J=15,2, 2H), 6,60-6,56 (m, 1H);
2-Amino-5-[1-(2-metoxi-etil)-1H-pirazol-4-il]-N-piridin-4-il-nicotinamida (“A6”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con 1-(2-metoxi-etil)-4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1 H-pirazol da el compuesto “A6” ;
HPLC/EM: 1,05 min, [M+H] = 339;
RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 11,37 (s, 1H, NH), 8,75 (d, J=7,2, 2H), 8,51 (d, J=2,2, 1H), 8,34 (d, J=2,3, 1H), 8,18 (d, J=7,2, 2H), 8,12 (s, 1H), 7,89 (s, 1H), 4,29 (t, J=5,3, 3H), 3,72 (t, J=5,3, 3H), 3,23 (s, 3H);
2-Amino-N-piridin-4-il-5-(4-sulfamoil-fenil)-nicotinamida (“A7”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con pinacol éster del ácido bencen-sulfonamida-4-borónico, da el compuesto “A7” ;
HPLC/EM: 1,12 min, [M+H] = 370;
RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 10,57 (s, 1H, NH), 8,61 (d, J = 2,3, 1H), 8,55-8,46 (m, 2H), 8,42 (d, J = 2,4, 1H), 7,92 (m, 4H), 7,80-7,65 (m, 2H), 7,35 (s, 2H), 7,24 (s, 2H);
2-Amino-N-(2-metil-piridin-4-il)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A8”)

La reacción de 2-amino-5-bromo-N-(2-metil-piridin-4-il)-nicotinamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico, da el compuesto “A8”;
HPLC/EM: 1,02 min, [M+H] = 410;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 10,49 (s, 1H, NH), 8,44 (d, J=2,3, 1H), 8,35 (d, J=5,6, 1H), 8,23 (d, J=2,4, 1H), 7,59 (d, J=1,7, 1H), 7,53 (dd, J=5,6, 1,9, 1H), 7,32-7,27 (d, J=3,5, 1H), 7,12 (s, 2H), 6,96 (d, J=3,5, 1H), 3,71 (s, 2H), 3,61 -3,53 (m, 4H), 2,45 (s, 3H), 2,43 (m, 4H);
2-Amino-N-(2-etoxi-piridin-4-il)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A9”)
La reacción de 2-amino-5-bromo-N-(2-etoxi-piridin-4-il)-nicotinamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico, da el compuesto “A9”;
HPLC/EM: 1,37 min, [M+H] = 440;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 10,51 (s, 1H, NH), 8,44 (d, J=2,3, 1H), 8,19 (d, J=2,3, 1H), 8,06 (d, J=5,7, 1H), 7,26 (m, 2H), 7,22 (d, J=1,6, 1H), 7,12 (s, 2H), 6,96 (d, J=3,5, 1H), 4,30 (q, J=7,0, 2H), 3,66 (s, 2H), 3,58 (m, 4H), 2,42 (a, 4H), 1,31 (t, J= 7,0, 3H);
Ejemplo comparativo 2
Piridin-4-ilamida del ácido 3-amino-6-(5-morfolin-4-ilmetil-tiofen-2-il)-pirazin-2-carboxílico (“A10”)
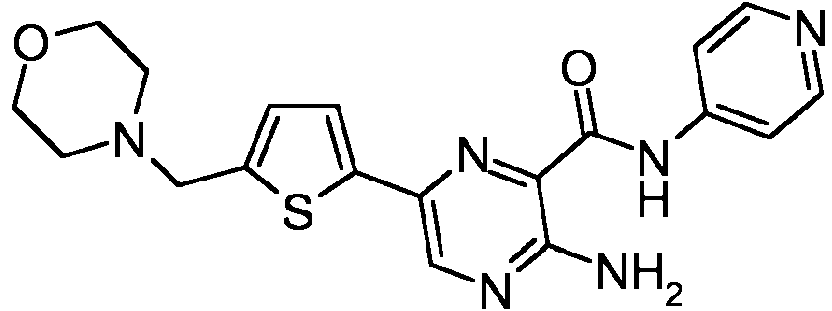
2.1. Piridin-4-ilamida del ácido 3-amino-6-bromo-pirazin-2-carboxílico

Se obtiene el compuesto del título del ácido 3-amino-6-bromo-pirazin-2-carboxílico y 4-aminopiridina, usando el mismo método como se describe en la etapa 1 para “A1” ;
HPLC/EM: 1,18 min, [M+H] = 294.
2.2. Piridin-4-ilamida del ácido 3-amino-6-(5-morfolin-4-ilmetil-tiofen-2-il)-pirazin-2-carboxílico (“A10”)
El compuesto del título se prepara de manera análoga a la etapa 2 para “A1”; HPLC/EM: 1,08 min, [M+H] = 397. Ejemplo 3
Piridin-4-ilamida del ácido 6-amino-6’-piperazin-1-il-[3,3’]bipiridinil-5-carboxílico (“A11”)

3.1. ferc-butil éster del ácido 4-[6’-amino-5’-(piridin-4-ilcarbamoil)-[3,3’]bipiridinil-6-il]-piperazin-1-carboxílico

El compuesto del título se obtiene a partir de 2-amino-5-bromo-N-(2-etoxi-piridin-4-il)-nicotinamida y pinacol éster del ácido 2-(4-ferc-butoxicarbonilpiperazin-1-il)piridin-5-borónico, de manera análoga a la etapa 2 para “A1”; HPLC/EM: 1,43 min, [M+H] = 476.
3.2. 1,1 g de ferc-butil éster del ácido 4-[6’-amino-5’-(piridin-4-ilcarbamoil)-[3,3’]bipiridinil-6-il]-piperazin-1-carboxílico se disuelven en 25 ml de dioxano. Se añaden 11 ml de HCl en dioxano (4 molar). La mezcla se agita 3 h a temperatura ambiente. La mezcla se filtra y el sólido se lava con dioxano. El producto se purifica por cromatografía; HPLC/EM: 1,02 min, [M+H] = 376;
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 9,08 (d, J=2,2, 1H), 8,84 (d, J=7,3, 2H), 8,77 (d, J=2,1, 1H), 8,67 (d, J=2,3, 1H), 8,48 (dd, J=9,5, 2,4, 1H), 8,38 (d, J=7,3, 2H), 7,54 (d, J=9,5, 1H), 4,09-3,96 (m, 4H), 3,47-3,35 (m, 4H).
Ejemplo 4
Los siguientes compuestos se obtienen de manera análoga al ejemplo 2.
Piridin-4-ilamida del ácido 3-amino-6-(1-[1,3]dioxolan-2-ilmetil-1H-pirazol-4-il)-pirazin-2-carboxílico (“A12”) (ejemplo comparativo)

La reacción de piridin-4-ilamida del ácido 3-amino-6-bromo-pirazin-2-carboxílico (síntesis descrita para “A10”) con 1-[1,3]dioxolan-2-ilmetil-4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1H-pirazol, da el compuesto “A12" HPLC/EM: 1,02 min, [M+H] = 376;
RMN 1H (500 MHz, DMSO-do) 5 [ppm] 10,55 (s, 1H, NH), 8,71 (s, 1H), 8,55-8,41 (m, 3H), 8,27 (s, 1H), 7,91-7,86 (m, 2H), 7,51 (s, 2H), 5,22 (t, J=4,3, 1H), 4,30 (d, J=4,3, 2H), 3,96-3,70 (m, 4H);
Piridin-4-ilamida del ácido 3-amino-6-(5-piperidin-1-ilmetil-tiofen-2-il)-pirazin-2-carboxílico (“A13”) (ejemplo comparativo)

La reacción de piridin-4-ilamida del ácido 3-amino-6-bromo-pirazin-2-carboxílico (síntesis descrita para “A10”) con 1-[5-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-tiofen-2-ilmetil]-piperidina, da el compuesto “A13”;
HPLC/EM: 1,13 min, [M+H] = 395;
RMN 1H (400 MHz, DMSO-de) 5 [ppm] 10,41 (s, 1H, NH), 8,80 (s, 1H), 8,51 (dd, J=4,8, 1,6, 2H), 7,82 (dd, J=4,8, 1,6, 2H), 7,67 (d, J=3,6, 1H), 7,63 (s, 2H), 6,98 (d, J=3,6, 1H), 3,64 (s, 2H), 2,40 (s, 4H), 1,59-1,32 (m, 6H);
Piridin-4-ilamida del ácido 3-amino-6-(5-pirrolidin-1 -ilmetM-tiofen-2-M)-pirazin-2-carboxílico (“A14”) (ejemplo comparativo)

La reacción de piridin-4-ilamida del ácido 3-amino-6-bromo-pirazin-2-carboxílico (síntesis descrita para “A10”) con 1-[5-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-tiofen-2-ilmetil]-pirrolidina, da el compuesto “A14”;
HPLC/EM: 1,08 min, [M+H] = 381;
RMN 1H (500 MHz, DMSO-de) 5 [ppm] 10,40 (s, 1H, NH), 8,83 (s, 1H), 8,51 (dd, J=4,8, 1,5, 2H), 7,88-7,75 (m, 2H), 7,67 (d, J=3,6, 1H), 7,63 (s, 2H), 6,96 (d, J= 3,6, 1H), 3,78 (s, 2H), 2,51 (m, 4H), 1,78-1,62 (m, 4H);
2-Amino-N-(2-metil-piridin-4-il)-5-(5-morfolin-4-ilmetil-tiofen-3-il)-nicotinamida (“A15”)

La reacción de 2-amino-5-bromo-N-(2-metil-piridin-4-il)-nicotinamida con 4-[4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-tiofen-2-ilmetil]-morfolina, da el compuesto “A15”;
HPLC/EM: 0,99 min, [M+H] = 410;
RMN 1H (500 MHz, DMSO-de) 5 [ppm] 10,45 (s, 1 H, NH), 8,53 (d, J=2,3, 1 H), 8,35 (d, J=5,6, 1H), 8,30 (d, J=2,3, 1H), 7,69 (d, J=1,4, 1 H), 7,60 (d, J=1,7, 1H), 7,56 -7,50 (m, 1H), 7,44 (s, 1 H), 7,04 (s, 2H), 3,71 (s, 2H), 3,61 -3,58 (m, 4H), 2,45 (m, 7H).
Ejemplo 5
2-Amino-5-(5-morfolin-4-ilmetil-tiofen-2-il)-N-piridazin-4-il-nicotinamida (“A16”)
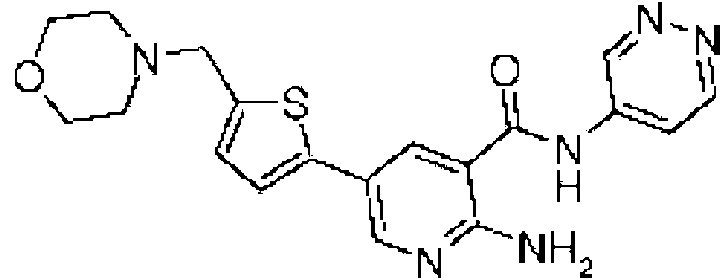
5.1.2-Amino-5-bromo-N-piridazin-4-il-nicotinamida
El compuesto del título se obtiene a partir del ácido 2-am¡no-5-bromon¡cotín¡co y 4-amlnoplrldazlna de manera análoga a “A1” en la etapa 1;
HPLC/EM: 1,40 mln, [M+H] = 294.
5.2. 2-Am¡no-5-(5-morfol¡n-4-¡lmet¡l-tlofen-2-¡l)-N-p¡r¡daz¡n-4-¡l-n¡cot¡nam¡da se obtiene a partir de 2-amlno-5-bromo-N-plrldazln-4-ll-nlcotlnamlda y plnacol éster del ácido 5-(4-morfollnllmet¡l)t¡ofen-2-borón¡co de manera análoga a “A1 ”, en la etapa 2; HPLC/EM: 1,15 mln, [M+H] = 397;
RMN 1H (500 MHz, DMSO-de) 5 [ppm] 10,78 (s, 1H, NH), 9,47 (d, J=1,9, 1H), 9,08 (d, J=5,9, 1H), 8,48 (d, J=2,3, 1H), 8,27 (d, J=2,3, 1H), 8,03 (dd, J=5,9, 2,7, 1H), 7,27 (d, J=3,1, 1H), 7,20 (s, 2H), 6,98 (s, 1H), 3,68 (s, 2H), 3,59 (s, 4H), 2,48-2,35 (m, 4H).
Los s¡gu¡entes compuestos se obt¡enen de manera análoga.
2-Amlno-5-(1 -bencll-1 H-p¡razol-4-ll)-N-(2-met¡l-p¡r¡d¡n-4-¡l)-n¡cot¡nam¡da (“A17”)

La reacción de 2-amlno-5-bromo-N-(2-met¡l-p¡r¡d¡n-4-¡l)-n¡cot¡nam¡da con 4-[4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-tiofen-2-ilmetil]-morfolina, da el compuesto “A17”);
HPLC/EM: 1,41 mln, [M+H] = 385;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 10,45 (s, 1H, NH), 8,53 (d, J=2,3, 1H), 8,35 (d, J=5,6, 1H), 8,30 (d, J=2,3, 1H), 7,69 (d, J=1,4, 1H), 7,60 (d, J=1,7, 1H), 7,55 (dd, J=5,6, 1,9, 1H), 7,44 (s, 1H), 7,04 (s, 2H), 3,71 (s, 2H), 3,63-3,57 (m, 4H), 2,45 (s m, 7H);
2-Amino-5-(3-pirazol-1 -¡l-fen¡l)-N-p¡r¡dazin-4-¡l-n¡cot¡nam¡da (“A18”)

La reacción de 2-am¡no-5-bromo-N-p¡r¡dazin-4-¡l-n¡cot¡nam¡da (ver la etapa 1 en la síntesis de “A16”) y 1-[3-(4,4,5,5-tetrametil-[1,3,2]d¡oxaborolan-2-¡l)-fen¡l]-1H-p¡razol, da el compuesto “A18”;
HPLC/EM: 1,15 mln, [M+H] = 397;
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 11,06 (s, 1H, NH), 9,54 (d, J=2,1, 1H), 9,14 (d, J=5,9, 1H), 8,67 (dd, J=4,1,2,4, 2H), 8,58 (d, J=2,3, 1H), 8,19 (t, J=1,8, 1H), 8,14 (dd, J=6,0, 2,7, 1H), 7,89-7,80 (m, 1H), 7,77 (d, J=1,7, 1H), 7,68 (d, J=7,9, 1H), 7,59 (t, J=7,9, 1H), 7,37 (s, 2H), 6,60-6,56 (m, 1H);
2-Am¡no-N-(3-met¡l-p¡ridin-4-il)-5-(4-pirazol-1 -il-fen¡l)-n¡cot¡nam¡da (“A19”)

La reacción de 2-am¡no-5-bromo-N-(3-met¡l-p¡r¡din-4-¡l)-n¡cot¡nam¡da con el ácido [4-(1 H-plrazol-1 -il)fen¡l]borónico, da el compuesto “A19” ;
HPLC/EM: 1,41 mln, [M+H] = 371;
RMN 1H (500 MHz, DMSO-de) 5 [ppm] 10,07 (s, 1H, NH), 8,59 (d, J=2,3, 1H), 8,55 (d, J=2,4, 1H), 8,45 (s, 1H), 8,44 (d, J=2,3, 1H), 8,40 (d, J=5,3, 1H), 7,96-7,91 (m, 2H), 7,90-7,83 (m, 2H), 7,76 (d, J=1,6, 1H), 7,53 (d, J=5,3, 1H), 7,17 (s, 2H), 6,60-6,54 (m, 1H), 2,28 (s, 3H);
2-Amino-5-[1-(2-metoxi-etil)-1H-pirazol-4-il]-N-(2-metil-piridin-4-il)-nicotinamida (“A20”)

La reacción de 2-amino-5-bromo-N-(2-metil-piridin-4-il)-nicotinamida con 1-(2-metoxi-etil)-4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1 H-pirazol, da el compuesto “A20”;
HPLC/EM: 1,11 min, [M+H] = 353;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 10,44 (s, 1H, NH), 8,43 (d, J=2,2, 1H), 8,36 (d, J=5,6, 1H), 8,20 (d, J=2,2, 1H), 8,09 (s, 1H), 7,86 (d, J=0,5, 1H), 7,62 (s, 1H), 7,55 (d, J=5,6, 1H), 6,94 (s, 2H), 4,33-4,22 (m, 2H), 3,76-3,66 (m, 2H), 3,25 (s, 3H), 2,46 (s, 3H);
2-Amino-N-(3-metil-piridin-4-il)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A21”)

La reacción de 2-amino-5-bromo-N-(3-metil-piridin-4-il)-nicotinamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico, da el compuesto “A21” ; HPLC/EM: 0,97 min, [M+H] = 410;
RMN 1H (500 MHz, DMSO-ds) 5 [ppm] 10,8 (a, 1H, NH), 8,72 (d, J=2,2, 1H), 8,70 (s, 1H), 8,67 (d, J=6,7, 1H), 8,52 8,47 (m, 2H), 7,54 (d, J=3,7, 1H), 7,34 (d, J=3,7, 1H), 4,61 (s, 2H), 4,01-3,87 (m, 2H), 3,67 (m, 2H), 3,33 (m, 2H), 3,14 (m, 2H);
2-Amino-5-[1-(2-metoxi-etil)-1H-pirazol-4-il]-N-(3-metil-piridin-4-il)-nicotinamida (“A22”)

La reacción de 2-amino-5-bromo-N-(3-metil-piridin-4-il)-nicotinamida con 1-(2-metoxi-etil)-4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1 H-pirazol, da el compuesto “A22”; HPLC/EM: 1,07 min, [M+H] = 353;
2-Amino-5-(4’-metil-bifenil-3-il)-N-piridin-4-il-nicotinamida (“A23”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido [3-(p-tolil)fenil]-borónico, da el compuesto “A23”;
HPLC/EM: 1,90 min, [M+H] = 381;
RMN 1H (500 MHz, DMSO-ds) 5 [ppm] 11,4 (s, 1H, NH), 9,03 (d, J=2,2, 1H), 8,75 (d, J=6,6, 2H), 8,71 (d, J=2,1, 1H), 8.25 (d, J=7,3, 2H), 8,00 (s, 1H), 7,69 (dd, J=10,1,8,7, 2H), 7,62 (d, J=8,1, 2H), 7,56 (t, J=7,7, 1H), 7,37 (a, 2H, NH2), 7.25 (d, J=8,1 ,2H), 2,31 (s, 3H);
2-Amino-5-[1-(2-metoxi-etil)-1 H-pirazol-4-il]-N-piridazin-4-il-nicotinamida (“A24")

La reacción de 2-amino-5-bromo-N-piridazin-4-il-nicotinamida (ver la etapa 1 en la síntesis de “A16") con 1-(2-metoxietil)-4-(4,4,5,5-tetrametil-[1,3,2]dioxa-borolan-2-il)-1H-pirazol, da el compuesto “A24";
HPLC/EM: 1,21 min, [M+H] = 340;
RMN 1H (500 MHz, DMSO-ds) 5 [ppm] 10,98 (a, 1H, NH), 9,51 -9,48 (m, 1H), 9,17 (d, J=6,1, 1H), 8,48 (d, J=2,2, 1H), 8,42 (d, J=1,6, 1H), 8,14 (m, 2H), 7,91 (s, 1H), 7,36 (a, 2H), 4,29 (t, J=5,3, 2H), 3,72 (t, J=5,3, 3H), 3,25 (s, 3H);
2-Amino-5-(1 -carbamoilmetil-1 H-pirazol-4-il)-N-piridin-4-il-nicotinamida (“A25")

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con 2-[4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-pirazol-1-il]-acetamida, da el compuesto “A25"; HPLC/EM: 0,94 min, [M+H] = 338;
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 10,51 (s, 1H, NH), 8,48 (dd, J=4,8, 1,5, 2H), 8,44 (d, J=2,3, 1H), 8,22 (d, J=2,3, 1H), 8,08 (d, J=0,5, 1H), 7,88 (d, J=0,6, 1H), 7,72 (dd, J=4,8, 1,6, 2H), 7,49 (s, 1H), 7,24 (s, 1H), 6,94 (s, 2H), 4,78 (s, 2H);
2-Amino-N-(2,6-dimetil-piridin-4-il)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A26")

La reacción de 2-amino-5-bromo-N-(2,6-dimetil-piridin-4-il)-nicotinamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico, da el compuesto “A26"; HPLC/EM: 1,09 min, [M+H] = 424;
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 10,43 (s, 1H, NH), 8,43 (d, J=2,3, 1H), 8,21 (d, J=2,3, 1H), 7,43 (s, 2H), 7,26 (d, J=3,5, 1H), 7,14 (s, 2H), 6,96 (d, J=3,5, 1H), 3,68 (s, 2H), 3,64-3,54 (m, 4H), 2,53-2,47 (m, 4H), 2,43 (s, 6H).
Ejemplo 6
2-Amino-5-(5-morfolin-4-ilmetil-tiofen-2-il)-N-pirimidin-4-il-nicotinamida (“A27”)

6.1. La reacción del ácido 2-amino-5-bromo-piridin-3-carboxílico con pirimidin-4-amina de acuerdo con la síntesis descrita para “A1 ”, etapa 1, da 2-amino-5-bromo-N-pirimidin-4-il-piridin-3-carboxamida.
6.2. “A27” se prepara de 2-amino-5-bromo-N-pirimidin-4-il-piridin-3-carboxamida y pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico de acuerdo con la etapa 2 de la síntesis de “A1” ;
HPLC/EM: 1,27 min, [M+H] = 397;
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 11,34 (s, 1H, NH), 8,97 (d, J=0,9, 1H), 8,72 (d, J=5,8, 1H), 8,46 (d, J=2,3, 1H), 8,40 (d, J=2,3, 1H), 8,14 (dd, J=5,8, 1,2, 1H), 7,33 (d, J=3,5, 1H), 7,25 (s, 2H), 6,97 (d, J=3,4, 1H), 3,69 (s, 2H), 3,65 3,56 (m, 4H), 2,45 (s, 4H).
Ejemplo 7
2-Amino-N-furo[3,2-b]piridin-7-il-5-[1-(2-metoxi-etil)-1H-pirazol-4-il]-nicotinamida (“A28”)

7.1. La reacción del ácido 2-amino-5-bromo-piridin-3-carboxílico con furo[3,2-b]piridin-7-amina de acuerdo con la síntesis descrita para “A1”, etapa 1, da 2-amino-5-bromo-N-furo[3,2-b]piridin-7-il-piridin-3-carboxamida.
7.2. “A28” se obtiene a partir de 2-amino-5-bromo-N-furo[3,2-b]piridin-7-il-piridin-3-carboxamida y 1-(2-metoxi-etil)-4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1H-pirazol de acuerdo con el procedimiento descrito para “A1”, etapa 2; HPLC/EM: 1,22 min, [M+H] = 379;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 10,70 (s, 1H, NH), 8,48-8,44 (m, 2H), 8,34 (d, J=2,0, 1H), 8,32 (d, J=2,2, 1H), 8,10 (s, 1H), 7,88 (s, 1H), 7,60 (d, J=5,3, 1H), 7,15 (d, J=2,2, 1H), 7,02 (s, 2H), 4,28 (t, J=5,3, 2H), 3,71 (t, J=5,3, 2H), 3,25 (s, 3H).
Los siguientes compuestos se obtienen de manera análoga:
2-Amino-N-furo[3,2-b]piridin-7-il-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A29”)

La reacción de 2-amino-5-bromo-N-furo[3,2-b]piridin-7-il-piridin-3-carboxamida (síntesis descrita anteriormente) con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico, da el compuesto “A29”;
HPLC/EM: 1,17 min, [M+H] = 436;
RMN 1H (500 MHz, DMSO-ds) 5 [ppm] 10,85 (s, 1H, NH), 8,48 (t, J=3,5, 2H), 8,36 (d, J=2,3, 1H), 8,33 (d, J=2,2, 1H), 7,62 (d, J=5,3, 1H), 7,30 (d, J=3,5, 1H), 7,23 (s, 2H), 7,16 (d, J=2,2, 1H), 6,98 (d, J=3,5, 1H), 3,68 (s, 2H), 3,62-3,56 (m, 4H), 2,44 (m, 4H);
Metil éster del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A30”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 4-metoxicarbonil)-benzoico, da el compuesto “A30”; HPLC/EM: 1,48 min, [M+H] = 349;
Ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A31 ”)

190 mg de metil éster del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico se disuelven en 20 ml de MeOH. Se añaden 175 mg de Na2CO3 en 6 ml de agua. La mezcla de reacción se agita durante la noche a 50°C. El material deseado “A31” se purifica por cromatografía;
HPLC/EM: 1,28 min, [M+H] = 335;
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 13,24-12,61 (m, 1H, OH), 11,36 (s, 1H, NH), 8,74 (d, J=6,7, 2H), 8,69 (d, J=2,4, 1H), 8,51 (d, J=2,4, 1H), 8,18 (d, J=7,2, 2H), 8,06-7,98 (m, 2H), 7,90-7,81 (m, 2H), 7,40 (s, 2H);
Ácido 3-{4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-fenil}-propiónico (“A32”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 3-(4-boronofenil)-propanoico, da el compuesto “A32”; HPLC/EM: 1,28 min, [M+H] = 363;
RMN 1H (500 MHz, DMSO-ds) 5 [ppm] 12,09 (s, 1H, OH), 10,52 (s, 1H, NH), 8,48 (dd, J=6,7, 4,2, 3H), 8,31 (d, J=2,2, 1H), 7,72 (d, J=6,1, 2H), 7,62 (d, J=8,1, 2H), 7,32 (d, J=8,1, 2H), 7,09 (s, 2H), 2,86 (t, J=7,5, 2H), 2,56 (t, J=7,5, 2H);
2-Amino-N-(3-cloro-piridin-4-il)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A33”)

La reacción de 2-amino-5-bromo-N-(3-cloro-4-piridil)piridin-3-carboxamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico, da el compuesto “A33”; HPLC/EM: 1,25 min, [M+H] = 343;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 10,30 (s, 1H, NH), 8,70 (s, 1H), 8,53 (d, J=5,3, 1H), 8,49 (d, J=2,3, 1H), 8,30 (d, J=2,4, 1H), 7,79 (d, J=5,3, 1H), 7,28 (d, J=3,5, 1H), 7,21 (s, 2H), 6,97 (d, J=3,5, 1H), 3,68 (s, 2H), 3,62-3,55 (m, 4H), 2,44 (s, 4H).
2-Amino-5-(1H-pirazol-4-il)-N-piridin-4-il-nicotinamida (“A34”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol, da el compuesto “A34”;
HPLC/EM: 0,89 minutos, [M+H] = 281;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 12,89 (s, 1H, NH), 10,48 (s, 1H, NH), 8,47 (dd, J=10,1, 4,2, 3H), 8,23 (d, J=2,2, 1H), 8,11 (s, 1H), 7,91 (s, 1H), 7,71 (dd, J=10,9, 9,6, 2H), 6,91 (s, 2H);
Metil éster del ácido 4-{[2-amino-5-(5-morfolin-4-ilmetil-tiofen-2-il)-piridin-3-carbonil]-amino}-nicotínico (“A35”)

La reacción de 4-[(2-amino-5-bromo-piridin-3-carbonil)amino]piridin-3-carboxilato de metilo con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico, da el compuesto “A35”; HPLC/EM: 1,36 min, [M+H] = 454;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] (s, 1H, NH), 9,04 (s, 1H), 8,70 (d, J=5,8, 1H), 8,52 (d, J=2,3, 1H), 8,34 (d, J=5,8, 1H), 8,20 (d, J=2,3, 1H), 7,31 (s, 2H), 7,26 (d, J=3,5, 1H), 6,98 (d, J=3,5, 1H), 3,92 (s, 3H), 3,69 (s, 2H), 3,62-3,53 (m, 4H), 2,44 (s, 4H);
N-(2-Acetilamino-piridin-4-il)-2-amino-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida “A36”)

La reacción de N-(2-acetamido-4-piridil)-2-amino-5-bromo-piridin-3-carboxamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico da el compuesto “A36”; HPLC/EM: 1,14 min, [M+H] = 453;
RMN 1H (500 MHz, DMSO-ds) 5 [ppm] 10,63 (s, 1H, NH), 10,37 (s, 1H, NH), 8,43 (d, J=2,3, 1H), 8,38 (d, J=1,2, 1H), 8,23 (d, J=2,3, 1H), 8,20 (d, J=5,6, 1H), 7,60 (dd, J=5,6, 1,9, 1H), 7,26 (d, J=3,5, 1H), 7,13 (s, 2H), 6,96 (d, J=3,5, 1H), 3,67 (s, 2H), 3,61-3,57 (m, 4H), 2,43 (s, 4H), 2,09 (s, 3H);
Ácido 3-[6-Amino-5-piridin-4-ilcarbamoil)-piridin-2-il]-benzoico (“A37")

1. El ácido (3-metoxicarbonilfenil)borónico y 2-amino-5-bromo-N-piridin-4-il-nicotinamida se hacen reaccionar de manera análoga a “A1", etapa 2 para dar 3-[6-amino-5-(4-piridilcarbamoil)-3-piridil]benzoato de metilo.
2. “A37” se obtiene a partir de 3-[6-amino-5-(4-piridilcarbamoil)-3-piridil]benzoato de metilo usando el método descrito para “A31” ;
HPLC/EM: 1,19 min, [M+H] = 335;
RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 13,07 (a, 1H, OH), 11,41 (s, 1H, NH), 8,74 (d, J=7,2, 2H), 8,64 (d, J=2,4, 1H), 8,48 (d, J=2,4, 1H), 8,26 (t, J=1,6, 1H), 8,19 (d, J=7,2, 2H), 8,01-7,88 (m, 2H), 7,62 (t, J=7,8, 1H), 7,38 (s, 2H);
2-amino-N-(3-cloro-piridin-4-il)-5-[1-(2-metoxi-etil-)-1 H-pirazol-4-il]nicotinamida (“A38”)

La reacción de 2-amino-5-bromo-N-(3-cloro-4-piridil)piridin-3-carboxamida con 1-(2-metoxi-etil)-4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1H-pirazol da el compuesto “A38”; HPLC/EM: 1,39 min, [M+H] = 373;
RMN 1H (500 MHz, DMSO-de) 5 [ppm] 10,16 (s, 1H, NH), 8,69 (s, 1H), 8,51 (d, J=5,3, 1H), 8,46 (d, J=2,2, 1H), 8,28 (d, J=2,3, 1H), 8,09 (s, 1H), 7,86 (s, 1H), 7,79 (d, J=5,3, 1H), 7,00 (S, 2H), 4,28 (t, J=5,3, 2H), 3,71 (t, J=5,3, 2H), 3,24 (s, 4H);
2-Amino-N-(4-metoxi-fenil)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A39”)

La reacción de 2-amino-5-bromo-N-(4-metoxifenil)piridin-3-carboxamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico da el compuesto “A39”; HPLC/EM: 1,46 min, [M+H] = 425;
2-Amino-N-(6-metoxi-piridin-3-il)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A40”)

La reacción de 2-amino-5-bromo-N-(6-metoxi-3-piridil)piridin-3-carboxamida con pinacol éster del ácido 5-(4-morfolinilmetil)tiofen-2-borónico da el compuesto “A40”; Hp LC/EM: 1,29 min, [M+H] = 426;
Ejemplo 8
2-Amino-N-(3-metilcarbamoil-piridin-4-il)-5-(5-morfolin-4-ilmetil-tiofen-2-il)-nicotinamida (“A41”)

100 mg de 4-[[2-amino-5-[5-(morfolinometil)-2-tienil]piridin-3-carbonil]amino]piridin-3-carboxilato de metilo (MSC 2392368) se disuelven en 4 ml de THF y se añaden 4 ml de metilamina (40 % en agua). La mezcla se agita durante 2 h a temperatura ambiente. La mezcla se evapora y se trata con éter de petróleo y acetato de etilo. Después de la filtración, se obtienen 79 mg de un sólido de color amarillo amarillo;
HPLC/EM: 1,17 min, [M+H] = 453;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 12,76 (s, 1H), 9,05 (d, J=4,3, 1H), 8,94 (s, 1H), 8,61 (d, J=5,7, 1H), 8,51 (d, J=2,3, 1H), 8,45 (d, J=5,7, 1H), 8,16 (d, J=2,3, 1H), 7,35 (S, 2H), 7,25 (d, J=3,5, 1H), 6,98 (d, J=3,5, 1H), 3,68 (s, 2H), 3,60-3,57 (m, 4H), 2,86 (d, J=4,5, 3H), 2,44 (m, 4H).
Ejemplo 9
2-Amino-5-[1 -(2-hidroxi-etil)-1 H-pirazol-4-il]-N-piridin-4-il-nicotinamida (“A42”)
9.1.2-Amino-5-{1 -[2-(tetrahidro-piran-2-iloxi)-etil]-1 H-pirazol-4-il}-nicotínico:
1,5 g de 1-[2-(tetrahidropiran-2-iloxi)-etil]-4-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-1H-pirazol y 808 mg del ácido 2-amino-5-bromonicotínico se disuelven en 10 ml de DMF. Se añaden 3,3 g de carbonato de potasio a la solución. La mezcla se calienta a 80°C. Se añaden 395 mg de 1,1’-bis(difenilfosfino)ferrocendicloropaladio (II), aducto de diclorometano y la mezcla se calienta durante 2 h. La solución se evapora y el producto en bruto se purifica por cromatografía en gel de sílice usando acetato de etilo/MeOH 9:1; 496 mg del ácido 2-amino-5-{1-[2-(tetrahidro-piran-2-iloxi)-etil]-1 H-pirazol-4-il}-nicotínico como un aceite de color pardo; HPLC/EM: 1,27 min, [M+H] = 333.
9.2. 2-Amino-N-piridin-4-il-5-{1-[2-(tetrahidro-piran-2-iloxi)-etil]-1 H-pirazol-4-il}-nicotinamida:
303 mg del ácido 2-amino-5-{1-[2-(tetrahidro-piran-2-iloxi)-etil]-1H-pirazol-4-il}-nicotínico, 84 mg de 4-aminopiridina y 284 mg de tetrafluoroborato de O-(1H-benzotriazol-1-il)-N,N,N’,N’-tetrametiluronio (TBTU) se disuelven en 10 ml de DMF. Se añaden 0,15 ml de N-etildiisopropilamina y 21 mg de 4-(dimetilamino)-piridina. La mezcla se agita durante la noche a temperatura ambiente. La mezcla de reacción se evapora y se purifica por cromatografía en gel de sílice usando diclorometano/MeOH 9:1.
160 mg de 2-amino-N-piridin-4-il-5-{1 -[2-(tetrahidro-piran-2-iloxi)-etil]-1 H-pirazol-4-il}-nicotinamida se obtienen como un sólido amarillo; HPLC/EM: 1,27 min, [M+H] = 409.
9.3. 2-Amino-5-[1-(2-hidroxi-etil)-1H-pirazol-4-il]-N-piridin-4-il-nicotinamida: 160 mg de 2-amino-N-piridin-4-il-5-{1-[2-(tetrahidro-piran-2-iloxi)-etil]-1 H-pirazol-4-il}-nicotinamida se disuelven en 4 ml de diclorometano. Se añaden 0,4 ml de HCl en dioxano (ca. 4 mol/l). Después de 1 h, el precipitado se filtra y se lava con diclorometano.
99 mg de 2-amino-5-[1-(2-hidroxi-etil)-1H-pirazol-4-il]-N-piridin-4-il-nicotinamida se obtienen como un sólido de color amarillo; HPLC/EM: 0,98 min, [M+H] = 325;
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 10,56 (s, 1H, NH), 8,50 (d, J=6,2, 2H), 8,43 (d, J=2,2, 1H), 8,21 (d, J=2,3, 1H), 8,09 (s, 1H), 7,86 (s, 1H), 7,76 (d, J=6,3, 2H), 6,94 (s, 2H), 4,90 (s, 1H, OH), 4,16 (t, J=5,6, 2H), 3,76 (d, J=4,8, 2H).
Ejemplo 10
Los siguientes compuestos se obtienen de manera análoga al ejemplo 1;
Método HPLC: A- TFA al 0,1 % en H2O, B- TFA al 0,1 % en ACN: Flujo-2,0 ml/min. Columna: X Bridge C8 (50x4,6 mm, 3,5 p).
2-amino-5-(5-morfolin-4-ilmetil-tiofen-3-il)-N-piridin-4-il-nicotinamida (“A43”)

RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 10,53 (s, 1H), 8,53 (d, J = 2,08 Hz, 1H), 8,48 (d, J = 5,00 Hz, 2H), 8,30 (d, J = 2,12 Hz, 1H), 7,69-7,71 (m, 3H), 7,44 (s, 1H), 7,07-7,12 (m, 2H), 3,69 (s, 2H), 3,57-3,59 (m, 4H), 2,49-2,49 (m, 4H); CLEM: Masa encontrada (M+1,396);
HPLC > 97 %, Tr (mín): 1,362.
5-(4-Acetil-1 H-pirrol-2-il)-2-amino-N-piridin-4-il-nicotinamida (“A44”)
RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 11,96 (s, 1H), 10,61 (s, 1H), 8,48-8,53 (m, 1H), 8,47 (d, J = 1,40 Hz, 2H), 8,40 (d, J = 2,04 Hz, 1H), 7,74 (dd, J = 1,48, 4,90 Hz, 2H), 7,68 (dd, J = 1,64, 2,88 Hz, 1H), 7,07 (s, 2H), 6,85 (t, J = 2,24 Hz, 1H), 2,34 (s, 3H);
CLEM: Masa encontrada (M+1,322);
HPLC > 93 %, RTL (min): 1,62.
2-Amino-5-furan-3-il-N-piridin-4-il-nicotinamida (“A45”)

RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 10,54 (s, 1H), 8,44-8,44 (m, 3H), 8,23 (d, J = 2,32 Hz, 1H), 8,12 (d, J = 2,36 Hz, 1H), 7,74 (t, J = 3,36 Hz, 1H), 7,68 (d, J = 4,64 Hz, 2H), 6,97-7,02 (m, 2H);
CLEM: Masa encontrada (M+1,281);
HPLC > 96 %, Tr (mín): 1,64.
2-Amino-5-(1 -bencil-1 H-pirazol-4-il)-N-piridin-4-il-nicotinamida (“A46”)

RMN 1H (400 MHz, DMSO-de) 5 [ppm] 10,49 (s, 1H), 8,43-8,48 (m, 3H), 8,20 (dd, J = 2,28, 6,54 Hz, 2H), 7,90-7,91 (m, 2H), 7,69 (dd, J = 1,52, 4,82 Hz, 2H), 7,25-7,37 (m, 5H), 6,94 (s, 1H), 5,34 (s, 1H);
CLEM: Masa encontrada (M+1,371);
HPLC > 98 %, Tr (mín): 2,34.
2-Amino-5-(1 -metil-1 H-pirazol-3-il)-N-piridin-4-il-nicotinamida (“A47”)
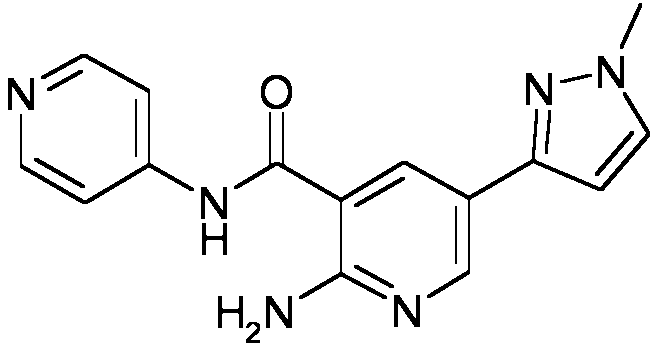
RMN 1H (400 MHz, DMSO-de) 5 [ppm] 10,65 (s, 1H), 8,59 (d, J = 2,20 Hz, 1H), 8,48 (d, J = 6,20 Hz, 2H), 8,36 (d, J = 2,20 Hz, 1H), 7,73 (dd, J = 2,12, 4,42 Hz, 3H), 7,07 (s, 1H), 6,66 (d, J = 2,24 Hz, 1H), 3,87 (s, 3H);
CLEM: Masa encontrada (M+1,295);
HPLC > 99 %, Tr (mín): 1,46.
2-Amino-5-(3,5-dimetil-1 H-pirazol-4-il)-N-piridin-4-il-nicotinamida (“A48”)

RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 12,29 (s, 1H), 10,36 (s, 1H), 8,45 (d, J = 5,24 Hz, 2H), 8,09 (d, J = 2,00 Hz, 1H), 8,00 (d, J = 2,00 Hz, 1H), 7,70 (d, J = 6,24 Hz, 2H), 7,01 (s, 1H), 2,16 (s, 6H);
CLe M: Masa encontrada (M+1,309);
HPLC > 99 %, Tr (mín): 3,71.
2-Amino-N-piridin-4-il-5-tiofen-3-il-nicotinamida (“A49”)

RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 10,53 (s, 1H), 8,57 (d, J = 2,28 Hz, 1H), 8,47 (d, J = 6,24 Hz, 2H), 8,35 (d, J = 2,28 Hz, 1H), 7,80-7,81 (m, 1H), 7,71 (dd, J = 1,48, 4,86 Hz, 2H), 7,66 (dd, J = 2,92, 5,00 Hz, 1H), 7,59 (dd, J = 1,20, 5,00 Hz, 1H), 7,09 (s, 2H);
CLEM: Masa encontrada (m 1,297);
HPLC > 95 %, Tr (mín): 1,88.
2-Amino-5-(1 -metil-1 H-pirrol-2-il)-N-piridin-4-il-nicotinamida (“A50”)

RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 10,45 (s, 1H), 8,46 (d, J = 6,12 Hz, 2H), 8,24 (d, J = 2,16 Hz, 1H), 8,14 (d, J = 2,12 Hz, 1H), 7,71 (d, J = 6,32 Hz, 2H), 7,13 (s, 2H), 6,83 (t, J = 2,00 Hz, 1H), 6,16 (dd, J = 1,84, 3,52 Hz, 1H), 6,05 6,07 (m, 1H), 3,60 (s, 3H);
CLe M: Masa encontrada (m+1,294);
HPLC > 95 %, Tr (mín): 1,84.
2-Amino-5-[1 -(2-morfolin-4-il-etil)-1 H-pirazol-4-il]-N-piridin-4-il-nicotinamida (“A51")
RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 10,51 (s, 1H), 8,47 (dd, J = 1,48, 4,82 Hz, 2H), 8,42 (dd, J = 2,24, Hz, 1H), 8,18 (dd, J = 2,24, Hz, 1H), 8,12 (s, 1H), 7,84-7,85 (m, 1H), 7,70 (dd, J = 1,52, 4,80 Hz, 2H), 6,92 (s, 2H), 4,23 (t, J = 6,64 Hz, 2H), 3,54 (t, J = 4,68 Hz, 4H), 2,73 (t, J = 6,64 Hz, 2H), 2,41 (t, J = 4,40 Hz, 4H);
CLEM: Masa encontrada (M+1,394);
HPLC > 99 %, Tr (mín): 3,84.
2-Amino-5-benzo[b]tiofen-2-il-N-piridin-4-il-nicotinamida (“A52")

RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 11,41 (s, 1H), 8,73 (d, J = 6,52 Hz, 2H), 8,65 (d, J = 2,32 Hz, 1H), 8,15 (s, 1H), 8,15 (d, J = 6,88 Hz, 2H), 7,97 (d, J = 7,52 Hz, 1H), 7,79-7,82 (m, 2H), 7,32-7,43 (m, 4H);
CLe M: Masa encontrada (M+1,347);
HPLC > 94 %, Tr (mín): 2,87.
5-(5-Acetil-tiofen-2-il)-2-amino-N-piridin-4-il-nicotinamida (“A53")

RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 10,62 (s, 1H), 8,60 (d, J = 2,40 Hz, 1H), 8,49 (dd, J = 1,40, 4,86 Hz, 2H), 8,37 (d, J = 2,40 Hz, 1H), 7,95 (d, J = 4,00 Hz, 1H), 7,71 (dd, J = 1,56, 4,82 Hz, 2H), 7,59 (d, J = 3,96 Hz, 1H), 7,39 (s, 2H), 2,48 (s, 3H);
CLe M: Masa encontrada (M+1,339);
HPLC > 96 %, Tr (mín): 2,01.
2-Amino-5-(5-morfolin-4-ilmetil-tiofen-2-il)-N-piridin-4-il-nicotinamida (“A54")
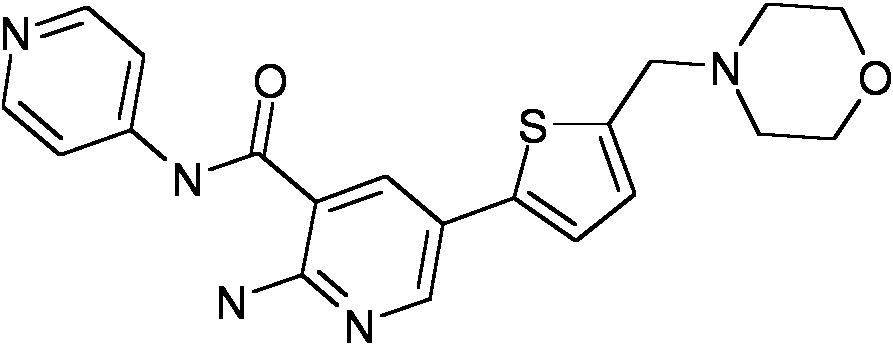
RMN 1H (400 MHz, DMSO-de) 5 [ppm] 8,27-8,34 (m, 3H), 7,60 (s, 2H), 7,06 (d, J = 3,64 Hz, 1H), 6,93 (d, J = 3,36 Hz, 1H), 3,65 (s, 2H), 3,58 (t, J = 4,48 Hz, 4H), 2,41 -2,49 (m, 4H), 1,50 (s, 3H);
CLEM: Masa encontrada (M+1,396);
HPLC > 98 %, Tr (mín): 1,39.
2-Amino-5-(1 H-indol-2-il)-N-piridin-4-il-nicotinamida (“A55”)

RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 11,43 (s, 1H), 10,63 (s, 1H), 8,69 (d, J = 2,04 Hz, 1H), 8,47-8,49 (m, 3H), 7,73 (d, J = 5,12 Hz, 2H), 7,50 (d, J = 7,64 Hz, 1H), 7,37 (d, J = 7,96 Hz, 1H), 7,13 (s, 2H), 6,83-7,08 (m, 3H);
Cl EM: Masa encontrada (M+1,330);
HPLC > 99 %, Tr (mín): 2,60.
2-Amino-5-1 H-indol-3-il)-N-piridin-4-il-nicotinamida (“A56”)

RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 11,33 (s, 1H), 10,55 (s, 1H), 8,46-8,51 (m, 3H), 8,30 (d, J = 2,28 Hz, 2H), 7,83 (d, J = 7,92 Hz, 1H), 7,67-7,82 (m, 2H), 7,44 (d, J = 8,00 Hz, 1H), 7,05-7,16 (m, 2H), 6,93 (s, 2H);
Cl EM: Masa encontrada (M+1,330);
HPLC > 93 %, Tr (mín): 2,23.
2-Amino-N-piridin-4-il-5-(1 H-pirrol-2-il)-nicotinamida (“A57”)

RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 11,15 (s, 1H), 10,54 (s, 1H), 8,46-8,48 (m, 3H), 8,22 (d, J = 2,28 Hz, 1H), 7,71 (dd, J = 1,52, 4,86 Hz, 2H), 6,89 (s, 2H), 6,82 (dd, J = 2,56, 4,02 Hz, 1H), 6,42 (t, J = 3,60 Hz, 1H), 6,10 (dd, J = 2,52, 5,66 Hz, 1H);
CLEM: Masa encontrada (M+1,280);
HPLC > 94 %, Tr (mín): 1,71.
2-Amino-5-(1 H-imidazo[1,2-a]piridin-2-il)-N-piridin-4-il-nicotinamida (“A58”)
RMN 1H (400 MHz, DMSO-de) 5 [ppm] 10,46 (s, 1H), 8,55 (d, J = 6,88 Hz, 1H), 8,47 (d, J = 6,04 Hz, 2H), 8,41 (d, J = 2,12 Hz, 1H), 8,35 (d, J = 2,00 Hz, 1H), 7,79 (s, 1H), 7,74-7,77 (m, 2H), 7,63-7,65 (m, 1H), 7,26-7,30 (m, 3H), 6,92 6,94 (m, 1H).
CLEM: Masa encontrada (M+1, 331);
HPLC > 91 %, Tr (mín): 3,68.
2-Amino-N-piridin-4-il-5-(1 H-pirrol-3-il)-nicotinamida (“A59”)

RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 10,90 (s, 1H), 10,50 (s, 1H), 8,47 (dd, J = 1,52, 4,80 Hz, 2H), 8,40 (dd, J = 2,24, Hz, 1H), 8,15 (dd, J = 2,28, Hz, 1H), 7,72 (dd, J = 1,52, 4,78 Hz, 2H), 7,18 (dd, J = 1,88, 4,20 Hz, 1H), 6,79-6,82 (m, 3H), 6,43-6,45 (m, 1H);
CLEM: Masa encontrada (M+1, 280);
HPLC > 98 %, Tr (mín): 1,43.
2-Amino-5-benzofuran-3-il-N-piridin-4-il-nicotinamida (“A60”)

RMN 1H (400 MHz, DMSO-d6) 5 [ppm] 10,58 (s, 1H), 8,54-8,55 (m, 3H), 8,33-8,36 (m, 2H), 7,95-7,97 (m, 1H), 7,72 (d, J = 3,44 Hz, 2H), 7,65-7,65 (m, 1H), 7,32-7,41 (m, 2H), 7,13 (s, 2H);
CLEM: Masa encontrada (M+1, 331);
HPLC > 93 %, Tr (mín): 2,50.
2-Amino-5-(1 -metil-1 H-pirazol-4-il)-N-piridin-4-il-nicotinamida (“A61”)
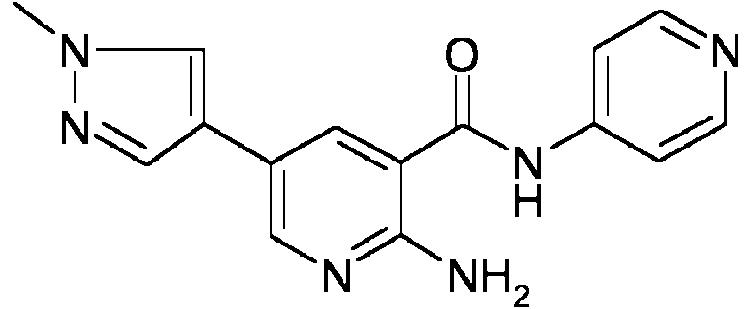
Ejemplo 11
Condiciones de CL-EM
Sistema de la serie de Hewlett Packard HP 1200 con las siguientes características: fuente iónica: electroatomizado (modo positivo); exploración: 100-1000 m/e; voltaje de fragmentación: 100 V; temperatura del gas: 350°C, UV: 220 nm.
Método 1
Condiciones de HPLC/EM:
columna: Chromolith SpeedROD RP-18e, 50-4,6
gradiente: A:B = 96:4 a 0:100
B al 4 % ^ B al 100 %: 0 min bis 2,8 min
B al 100 %: 2,8 min bis 3,3 min
B al 100 % ^ B al 4 %: 3,3 min bis 4 min
caudal: 2,4 ml/min
eluyente A: agua ácido fórmico al 0,05 %
eluyente B: acetonitrilo ácido fórmico al 0,04 %
longitud de onda: 220 nm
Espectroscopia de masa: modo positivo
Método 2
Condiciones de HPLC/EM:
columna: Chromolith SpeedROD RP-18e, 50-4,6
gradiente: A:B = 100:0 a 0:100
B al 0 % ^ B al 0 %: 0 min bis 0,5 min
B al 0 % ^ B al 100 % 0,5 min bis 2,6 min
B al 100 %: 2,6 min bis 3,0 min
B al 100 % ^ B al 0 %: 3,0 min bis 3,1 min
caudal: 2,4 ml/min
eluyente A: agua ácido fórmico al 0,05 %
eluyente B: acetonitrilo ácido fórmico al 0,04 %
longitud de onda: 220 nm
Espectroscopia de masa: modo positivo
2-Amino-5-(3-hidroximetil-fenil)-N-piridin-4-il-nicotinamida (“A62”)

100 mg de 2-amino-5-bromo-N-piridin-4-il-nicotinamida y 62,2 mg del ácido 3-hidroxi-metil)-bencenborónico se disuelven en 4 ml de DMF. Se añaden 90,4 mg de Na2CO3 y 1 ml de agua bajo argón. Se añaden 13,9 mg de [1,1-bis(difenilfosfino)ferroceno] dicloropaladio (II). La mezcla se agita durante 14 h a 100°C.
La mezcla de reacción se enfría a temperatura ambiente y la DMF se evapora y el producto se purifica por cromatografía.
Se obtienen 45 mg de “A62"; método 1: HPLC/EM: 1,11 min, [M+H] = 321;
RMN 1H (400 MHz, DMSO-ds) 5 [ppm] 9,01 (1H, d, J 2,2), 8,88-8,83 (2H, m), 8,67 (1H, d, J 2,3), 8,34-8,29 (2H, m), 7,79 (1H, s), 7,69 (1H, d, J 7,6), 7,52 (1H, t, J 7,6), 7,46 (1H, d, J 7,8), 4,65 (2H, s).
Los siguientes compuestos se obtienen de manera análoga:
2-amino-5-2-hidroximetil-fenil)-N-piridin-4-il-nicotinamida (“A63”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 2-(hidroximetil)-bencenborónico da “A63”; método 1: HPLC/EM: 1,07 min, [M+H] = 321;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 8,88 (1H, d, J 2,1), 8,81 (2H, d, J 7,2), 8,52 (1H, d, J 2,1), 8,31 (2H, d, J 7,3), 7,63 (1H, d, J 7,3), 7,53-7,44 (3H, m), 4,52 (2H, s);
2-Amino-5-(2-ferc-butilsulfamoil-fenil)-N-piridin-4-il-nicotinamida (“A64”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 2-ferc-butil-sulfamoil-bencenborónico da “A64”; método 1: HPLC/EM: 1,41 min, [M+H] = 426;
RMN 1H (400 MHz, DMSO-ds/TFA-d^ 5 [ppm] 8,86-8,80 (3H, m), 8,39 (1H, d, J 2,1), 8,28 (2H, d, J 7,3), 8,15 (1H, dd, J 7,9, 1,2), 7,73 (2H, dtd, J 31,4, 7,6, 1,4), 7,57 (1H, dd, J 7,6, 1,2), 1,09 (9 H, s);
2-Amino-5-(4-amino-fenil)-N-piridin-4-il-nicotinamida (“A65”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con clorhidrato del ácido 4-aminofenil-borónico da “A65” ; método 2: HPLC/EM: 1,53 min, [M+H] = 306;
RMN 1H (400 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,05 (1H, d, J 2,2), 8,87 (2H, d, J 7,5), 8,72 (1H, d, J 2,2), 8,36 (2H, d, J 7,3), 7,99 (2H, d, J 8,5), 7,60-7,55 (2H, m);
2-Amino-5-(2-formil-fenil)-N-piridin-4-il-nicotinamida (“A66”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 2-formilfenil-borónico da “A66” ; método 1:
HPLC/EM: 1,23 min, [M+H] = 319;
RMN 1H (400 MHz, DMSO-ds/TFA-dO 5 [ppm] 10,08 (1H, s), 8,88 (1H, d, J 2,2), 8,81 (2H, d, J 7,3), 8,51 (1H, d, J 2,2), 8,32-8,28 (2H, m), 8,11-8,07 (1H, m), 7,83 (1H, td, J 7,6, 1,5), 7,72 (1H, t, J 7,2), 7,64 (1H, dd, J 7,6, 0,9);
Etil éster del ácido 3-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A67”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 3-etoxicarbonil-fenilborónico da “A67”; método 1: HPLC/EM: 1,48 min, [M+H] = 363;
RMN 1H (400 MHz, DMSO-ds/TFA-dO 5 [ppm] 9,06 (1H, d, J 2,2), 8,85 (2H, d, J 7,3), 8,76 (1H, d, J 2,2), 8,41 (1H, t, J 1,7), 8,33 (2H, d, J 7,3), 8,13-8,03 (2H, m), 7,70 (1H, t, J 7,8), 4,41 (2H, q, J 7,1), 1,39 (3H, t, J 7,1);
2-Amino-5-(4-hidroximetil-fenil)-N-piridin-4-il-nicotinamida (“A68”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 4-(hidroximetil)-fenilborónico; método 1: HPLC/EM: 1,12 min, [M+H] = 321;
RMN 1H (400 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,02 (1H, d, J 2,2), 8,83 (2H, d, J 7,3), 8,65 (1H, d, J 2,2), 8,33 (2H, d, J 7,3), 7,79 (2H, d, J 8,2), 7,54 (2H, d, J 8,4), 4,63 (2H, s);
2-Amino-5-(3-hidroxi-fenil)-N-piridin-4-il-nicotinamida (“A69”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con pinacol éster del ácido 3-hidroxifenil-borónico, da “A70”; método 1: HPLC/EM: 1,12 min, [M+H] = 307.
RMN 1H (400 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,02 (1H, d, J 2,2), 8,82 (2H, d, J 5,1), 8,62 (1H, s), 8,34 (2H, d, J 7,3), 7,36 (1H, t, J 7,8), 7,22 (2H, d, J 8,1), 6,96 (1H, dd, J 7,5, 1,8);
Etil éster del ácido 2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A70”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con etil-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzoato da “A70” ; método 1: HPLC/EM: 1,37 min, [M+H] = 363;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 8,86 (1H, d, J 2,1), 8,79 (2H, d, J 7,1), 8,40 (1H, d, J 2,0), 8,32 (2H, d, J 7,3), 8,12 (1H, dd, J 7,8, 0,9), 7,75 (1H, td, J 7,6, 1,1), 7,64 (1H, td, J 7,7, 0,9), 7,57 (1H, d, J 7,5), 4,23 (2H, q, J 7,1), 1,22 (3H, t);
Etil éster del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A71”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 4-etoxicarbonil-fenilborónico da “A71” ; método 1: HPLC/EM: 1,46 min, [M+H] = 363;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,08 (1H, d, J 2,2), 8,84 (2H, d, J 7,2), 8,77 (1H, d, J 2,1), 8,33 (2H, d, J 7,2), 8,17 (2H, d, J 8,5), 7,97 (2H, d, J 8,4), 4,38 (2H, q, J 7,1), 1,38 (3H, t, J 7,1);
2-Amino-5-(4-hidroxi-fenil)-N-piridin-4-il-nicotinamida (“A72”)
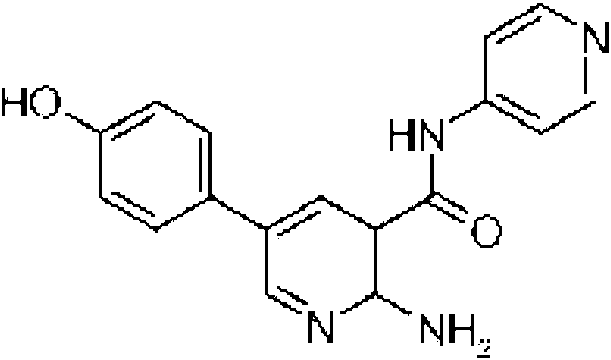
La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 4-hidroxibencen-borónico da “A72” ; método 1: HPLC/EM: 1,07 min, [M+H] = 307;
RMN 1H (500 MHz, DMSO-ds/TFA-dO 5 [ppm] 8,99 (1H, d, J 2,1), 8,78 (2H, dd, J 7,2, 2,4), 8,51 (1H, t, J 2,1), 8,35 (2H, d, J 7,2), 7,61 (2H, dd, J 8,5, 1,8), 7,01 (2H, d, J 8,6);
2-Amino-5-(2-hidroxi-fenil)-N-piridin-4-il-nicotinamida (“A73”)
La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con 2-(4,4,5,5-tetrametil-1 ,3,2-dioxaborolan-2-il)fenol da “A73”; método 1: HPLC/EM: 1,16 min, [M+H] = 307;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 8,95 (1H, d, J 2,1), 8,83 (2H, d, J 7,2), 8,62 (1H, d, J 2,1), 8,30 (2H, d, J 7,2), 7,53 (1H, dd, J 7,7, 1,5), 7,32-7,27 (1H, m), 7,11 -7,07 (1H, m), 7,03-6,98 (1H, m);
2-Amino-5-(3-formil-fenil)-N-piridin-4-il-nicotinamida (“A74”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 3-formilbencen-borónico da “A75”; método 1: HPLC/EM: 1,29 min, [M+H] = 319;
RMN 1H (500 MHz, DMSO-ds/TFA-d^ 5 [ppm] 10,13 (1H, s), 9,13 (1H, d, J 2,2), 8,83-8,75 (3H, m), 8,36 (3H, d, J 7,2), 8,14-8,02 (2H, m), 7,78 (1H, t, J 7,7);
2-Amino-5-(4-formil-fenil)-N-piridin-4-il-nicotinamida (“A75”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 4-formilbencen-borónico da “A75”; método 1: HPLC/EM: 1,29 min, [M+H] = 319;
RMN 1H (500 MHz, DMSO-ds/TFA-d^ 5 [ppm] 10,10 (1H, s), 9,08 (1H, d, J 2,2), 8,86 (2H, d, J 7,2), 8,82 (1H, d, J 2,2), 8,32 (2H, d, J 7,3), 8,12-8,05 (4 H, m);
2-Amino-5-(3-aminometil-fenil)-N-piridin-4-il-nicotinamida (“A76”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con clorhidrato del ácido 3-aminometil-fenilborónico da “A76”; método 1: HPLC/EM: 0,94 min, [M+H] = 320;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 8,99 (1H, d, J 2,3), 8,86 (2H, d, J 7,3), 8,65 (1H, d, J 2,2), 8,31 (2H, d, J 7,3), 7,93 (1H, s), 7,87 (1H, d, J 7,6), 7,61 (2H, dt, J 15,7, 7,7), 4,17 (2H, s);
2-Amino-5-(4-aminometil-fenil)-N-piridin-4-il-nicotinamida (“A77”)
La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con clorhidrato del ácido 4-aminometil-fenilborónico da “A77”; método 1: HPLC/EM: 1,07 min, [M+H] = 320;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,02 (1H, d, J 2,2), 8,86 (2H, d, J 7,3), 8,73 (1H, d, J 2,3), 8,31 (2H, d, J 7,3), 7,91 (2H, d, J 8,4), 7,67 (2H, d, J 8,4), 4,15 (2H, s);
2-Amino-N-piridin-4-il-5-(3-sulfamoil-fenil)-nicotinamida (“A78”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con pinacol éster del ácido bencensulfonamida-3-borónico da “A78” ; método 1: HPLC/EM: 1,13 min, [M+H] = 370;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 9,01 (1H, d, J 2,2), 8,86 (2H, d, J 7,3), 8,74 (1H, d, J 2,3), 8,33-8,28 (3H, m), 8,05 (1H, d, J 7,8), 7,98 (1H, d, J 7,9), 7,77 (1H, t, J 7,9);
2-Amino-5-[3-(4-hidroxi-piperidin-1-sulfonil)-fenil]-N-piridin-4-il-nicotinamida (“A79”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con 1-[3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-bencensulfonil]-piperidin-4-ol da “A79” ; método 1: HPLC/EM: 1,29 min, [M+H] = 454;
RMN 1H (400 MHz, DMSO-d6/TFA-d1) 5 [ppm] 9,01 (1H, d, J 2,3), 8,86 (2H, d, J 7,3), 8,80 (1H, d, J 2,2), 8,30 (2H, d, J 7,3), 8,19-8,13 (2H, m), 7,87-7,80 (2H, m), 3,64-3,57 (1H, m), 3,25-3,17 (2H, m), 2,93-2,89 (2H, m), 1,83-1,75 (2H, m), 1,58-1,44 (2H, m);
Metil éster del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-2-fluoro-benzoico (“A80”)

La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 3-fluoro-4-metoxicarbonil-fenilborónico da “A80” ; método 1: HPLC/EM: 1,48 min, [M+H] = 367;
RMN 1H (400 MHz, DMSO-ds/TFA-d^ 5 [ppm] 9,10 (1H, d, J 2,2), 8,84 (3H, t, J 4,8, 2,5), 8,34 (2H, d, J 7,3), 8,09 (1H, t, J 8,0), 7,87 (1H, dd, J 12,2, 1,6), 7,80 (1H, dd, J 8,2, 1,7), 3,92 (3H, s);
Terc-butil éster del ácido {2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-fenil}-carbámico (“A81”)
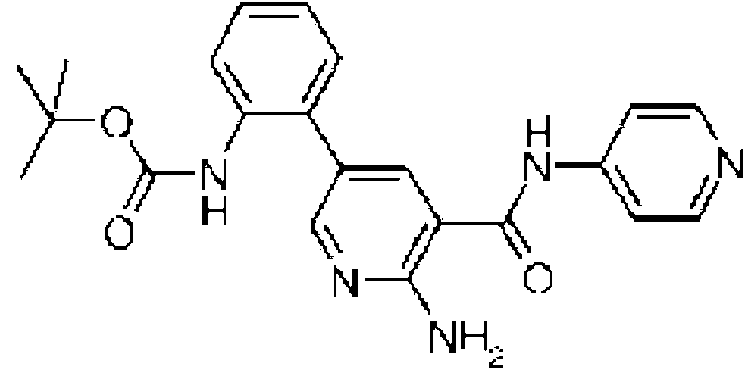
La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con íerc-butil éster del ácido [2-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-fenil]-carbámico da “A81”; método 1: HPLC/EM: 1,38 min, [M+H] = 406;
Terc-butil éster del ácido {3-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-fenil}-carbámico (“A82”)
La reacción de 2-amino-5-bromo-N-piridin-4-il-nicotinamida con el ácido 3-(N-Boc-amino)fenilborónico da “A82” ; método 1: HPLC/EM: 1,56 min, [M+H] = 406.
Ejemplo 12
2-Amino-5-{4-[(2-metoxi-etil)-metil-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A83”)

12.1. Ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A31 ”)
Una solución de etil éster del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (290 mg, 0,08 mmol), THF (5 ml) y NaOH 1 N (4 ml, 25,0) se agita a temperatura ambiente durante 14 h. El THF se retira al vacío y la mezcla se acidifica con HCl 1 N.
El precipitado resultante se filtra, se lava con agua y se seca. Se obtienen 280 mg del producto deseado como un sólido blanco; método 1: HPLC/EM: 1,21 min, [M+H] = 335;
RMN 1H (400 MHz, DMSO-de/TFA-d1) 5 [ppm] 9,13 (1H, s), 8,93-8,70 (4 H, m), 8,38 (2H, d, J 6,6), 7,97 (2H, d, J 8,4), 7,83 (1H, d, J 8,5).
Usando el procedimiento mencionado anteriormente, se obtienen de manera análoga los siguientes compuestos: Ácido 3-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A84”)
La reacción de etil éster del ácido 3-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico da “A84” ; método 1: HPLC/EM: 1,21 min, [M+H] = 335;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,12 (1H, d, J 2,2), 8,85 (2H, d, J 7,2), 8,78 (1H, d, J 2,2), 8,44 (1H, s), 8,37 (2H, d, J 7,2), 8,10 (2H, d, J 7,9), 7,68 (1H, t, J 7,8);
Ácido 2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico (“A85”)
La reacción de etil éster del ácido 2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico da “A85”; método 1: HPLC/EM: 1,15 min, [M+H] = 335;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 8,86 (1H, d, J 2,1), 8,79 (2H, d, J 7,1), 8,40 (1H, d, J 2,0), 8,32 (2H, d, J 7,3), 8,12 (1H, dd, J 7,8, 0,9), 7,75 (1H, td, J 7,6, 1,1), 7,64 (1H, td, J 7,7, 0,9), 7,57 (1H, d, J 7,5);
Ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-2-fluoro-benzoico (“A86”)
La reacción de metil éster del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-2-fluoro-benzoico da “A86”; método 1: HPLC/EM: 1,22 min, [M+H] = 353;
RMN 1H (400 MHz, DMSO-ds/TFA-d^ 5 [ppm] 9,13 (1H, d, J 2,2), 8,83 (3H, t, J 4,7), 8,37 (2H, d, J 7,4), 8,10 (1H, t, J 8,0), 7,85 (1H, dd, J 12,1, 1,7), 7,78 (1H, dd, J 8,2, 1,7).
12.2. 2-Amino-5-{4-[(2-metoxi-etil)-metil-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A83”)
60 mg (0,18 mmol) de 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico y 23,13 gl (0,22 mmol) de N-(metoxietil)metilamina se disuelven en 2 ml de DMF. 102,36 mg de HATU (hexafluorofosfato de (2-(7-aza-1H-benzotriazol-1-il)-1,1,3,3-tetrametil-uronio) y 59,19 gl de N-metilmorfolina se añaden a la solución. La mezcla se agita durante 3 h a temperatura ambiente. La d Mf se evapora y el producto se purifica por cromatografía.
Se obtienen 16 mg de “A83"; método 1: HPLC/EM: 1,22 min, [M+H] = 406;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,05 (1H, d, J 2,2), 8,86 (2H, d, J 7,3), 8,74 (1H, d, J 2,2), 8,31 (2H, d, J 7,3), 7,89 (2H, d, J 8,0), 7,60 (2H, d, J 8,3), 3,75-3,57 (2H, m), 3,53-3,41 (3H, m), 3,38-3,19 (2H, m), 3,04 (3H, s). Usando el procedimiento mencionado anteriormente, se obtienen de manera análoga los siguientes compuestos: 2-Amino-5-(4-dietilcarbamoil-fenil)-N-piridin-4-il-nicotinamida (“A87”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con dietilamina da “A87”; método 1: HPLC/EM: 1,36 min, [M+H] = 390;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,05 (1H, d, J 2,3), 8,86 (2H, d, J 7,3), 8,74 (1H, d, J 2,2), 8,31 (2H, d, J 7,3), 7,90 (2H, d, J 8,4), 7,55 (2H, d, J 8,3), 3,60-3,17 (4 H, m), 1,29-1,00 (6 H, m);
2-Amino-5-(4-carbamoil-fenil)-N-piridin-4-il-nicotinamida (“A88”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con amoníaco da “A88” ; método 1: HPLC/EM: 1,10 min, [M+H] = 334;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,06 (1H, d, J 2,2), 8,86 (2H, d, J 7,2), 8,77 (1H, d, J 2,2), 8,31 (2H, d, J 7,2), 8,11 (2H, d, J 8,4), 7,94 (2H, d, J 8,4);
2-Amino-5-{4-[bis-(2-metoxi-etil)-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A89”)
La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con bis-(2-metoxi-etil)-amina da “A89”; método 1: HPLC/EM: 1,30 min, [M+H] = 450; RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,08 (1H, d, J 2,2), 8,83 (2H, d, J 7,2), 8,72 (1H, d, J 2,1), 8,34 (2H, d, J 7,3), 7,88 (2H, d, J 8,2), 7,59 (2H, d, J 8,3), 3,73-3,26 (14 H, m);
2-Amino-5-{4-[(2-dimetilamino-etil)-metil-carbamoil]-fenil}-N-piridin-4-il-nicotinamida “A90”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con N,N,N’-trimetiletilendiamina da “A90”; método 2: HPLC/e M: 1,55 min, [m H] = 419;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,08 (1H, d, J 2,1), 8,80 (2H, dd, J 7,3, 1,1), 8,71 (1H, d, J 2,0), 8,35 (2H, d, J 7,3), 7,91 (2H, d, J 8,1), 7,69 (2H, d, J 8,1), 3,90 (2H, t), 3,46 (2H, t), 3,05 (3H, s), 2,95 (6 H, s);
2-Amino-5-{4-[(2-dimetilamino-etil)-etil-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A91”)

La reacción de 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con N’-etil-N,N-dimetil-etan-1,2-diamina da “A91 ” ; método 1: HPLC/EM: 1,08 min, [M+H] = 433;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,09 (1H, d, J 2,2), 8,80 (2H, d, J 7,3), 8,71 (1H, d, J 2,2), 8,36 (2H, d, J 7,4), 7,91 (2H, d, J 8,3), 7,64 (2H, d, J 8,3), 3,86 (2H, t), 3,44 (2H, t, J 6,4), 3,41-3,27 (2H, m), 2,98 (6 H, s), 1,14 (3H, t, J 6,6);
2-Amino-5-[4-(2-metoxi-etilcarbamoil)-fenil]-N-piridin-4-il-nicotinamida (“A92”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con 2-metoxi-etilamina da “A92”; método 1: HPLC/EM: 1,25 min, [M+H] = 392;
RMN 1H (500 MHz, DMSO-da/TFA-d1) 5 [ppm] 9,10 (1H, d, J 2,2), 8,81 (2H, d, J 7,3), 8,74 (1H, d, J 2,2), 8,35 (2H, d, J 7,4), 8,11 (2H, d, J 8,5), 7,91 (2H, d, J 8,5), 3,55 (4 H, s), 3,34 (3H, s);
2-Amino-5-{4-[bis-(2-hidroxi-etil)-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A93”)
La reacción de 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con 2-(2-hidroxi-etilamino)-etanol da “A93” ; método 2: HPLC/EM: 1,62 min, [M+H] = 422;
RMN 1H (500 MHz, DMSO-d6) 5 [ppm] 8,57 (1H, d, J 2,3), 8,49 (2H, dd, J 4,8, 1,5), 8,39 (1H, d, J 2,3), 7,77 (2H, d, J 8,3), 7,73 (2H, dd, J 4,8, 1,5), 7,49 (2H, d, J 8,3), 3,64-3,46 (8 H, m);
2-Amino-5-[4-(3-metoxi-propilcarbamoil)-fenil]-N-piridin-4-il-nicotinamida (“A94”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con 3-metoxi-propilamina da “A94”; método 1: HPLC/EM: 1,30 min, [M+H] = 406;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 9,11 (1H, d, J 2,2), 8,76 (2H, d), 8,69 (1H, d, J 2,1), 8,37 (2H, d, J 7,3), 8,07 (2H, d, J 3,3), 7,88 (2H, d, J 8,4), 3,48-3,45 (4 H, m), 3,31 (3H, s), 1,88 (2H, p, J 6,5);
2-Amino-5-{4-[metil-(3-metil-3H-imidazol-4-ilmetil)-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A95”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con metil-(3-metil-3H-imidazol-4-ilmetil)-amina da “A95” ; método 1: HPLC/EM: 1,07 min, [M+H] = 442;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 9,09 (1H, s), 9,08 (1H, d, J 2,1), 8,79 (2H, d, J 7,3), 8,70 (1H, d, J 2,0), 8,35 (2H, d, J 7,3), 7,89 (2H, d, J 8,3), 7,71 (1H, s), 7,68 (2H, d, J 8,3), 3,97 (2H, s), 3,00 (3H, s), 2,79 (3H, s);
Terc-butil éster del ácido 2-{4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoilamino}-etil carbámico (“A96”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con ferc-butil éster del ácido (2-aminoetil)-carbámico da “A96” ; método 1: Hp LC/EM: 1,47 min, [M+H] = 477;
2-Amino-5-{3-[(2-metoxi-etil)-metil-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A97")

La reacción del ácido 3-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con N-(metoxi-etil)-metil-amina “A97” ; método 1: HPLC/EM: 1,23 min, [M+H] = 406;
RMN 1H (500 MHz, DMSO-ds/TFA-d^ 5 [ppm] 9,03 (1H, s), 8,86 (2H, d, J 7,3), 8,74 (1H, s), 8,31 (2H, d, J 7,4), 7,88 (2H, d, J 1,6), 7,62 (1H, t, J 7,9), 7,49 (1H, d, J 7,7), 3,73-2,96 (10 H, m);
2-Amino-5-(3-dietilcarbamoil-fenil)-N-piridin-4-il-nicotinamida (“A98”)

La reacción del ácido 3-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con dietilamina da “A98”; método 1: HPLC/EM: 1,37 min, [M+H] = 390;
RMN 1H (500 MHz, DMSO-ds/TFA-d1) 5 [ppm] 9,04 (1H, d, J 2,1), 8,85 (2H, d, J 7,2), 8,74 (1H, d, J 2,1), 8,31 (2H, d, J 7,2), 7,89 (1H, d, J 8,1), 7,84 (1H, s), 7,62 (1H, t, J 7,7), 7,45 (1H, d, J 7,5), 3,57-3,23 (4 H, m), 1,28-1,01 (6 H, m);
2-Amino-5-(3-carbamoil-fenil)-N-piridin-4-il-nicotinamida (“A99”)

La reacción del ácido 3-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con amoníaco da “A99” ; método 2: HPLC/EM: 1,64 min, [M+H] = 334.
RMN 1H (500 MHz, DMSO-ds/TFA-d^ 5 [ppm] 9,07 (1H, d, J 2,2), 8,85 (2H, d, J 7,3), 8,76 (1H, d, J 2,2), 8,36 (1H, t, J 1,7), 8,32 (2H, d, J 7,3), 8,00 (2H, td, J 8,3, 1,6), 7,65 (1H, t, J 7,8);
2-Amino-5-(2-dietilcarbamoil-fenil)-N-piridin-4-il-nicotinamida (“A100”)
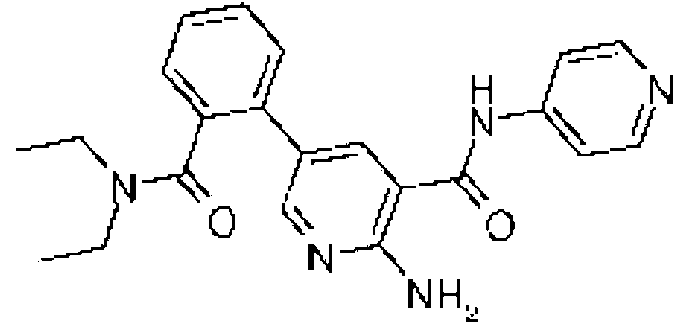
La reacción del ácido 2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con dietilamina da “A100” ; método 2: HPLC/EM: 1,35 min, [M+H] = 390;
RMN 1H (500 MHz, DMSO-ds) 5 [ppm] 8,83 (2H, d, J 7,3), 8,81 (1H, d, J 2,2), 8,35 (1H, d, J 2,1), 8,31 (2H, d, J 7,3),
7,70-7,67 (1H, m), 7,62 (1H, td, J 7,6, 1,3), 7,56 (1H, ddd, J 6,0, 5,5, 1,4), 7,45 (1H, dd, J 7,5, 1,1), 3,47-3,30 (2H, m), 3,03 (2H, q, J 7,0), 1,02-0,91 (3H, m);
2-Amino-5-2-carbamoil-fenil)-N-piridin-4-il-nicotinamida (“A101”)

La reacción del ácido 2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoico con amoníaco da “A101”; método 1: HPLC/EM: 1,05 min, [M+H] = 334;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 8,86 (1H, d, J 2,1), 8,79 (2H, d, J 7,1), 8,40 (1H, d, J 2,0), 8,32 (2H, d, J 7,3), 8,12 (1H, dd, J 7,8, 0,9), 7,75 (1H, td, J 7,6, 1,1), 7,64 (1H, td, J 7,7, 0,9), 7,57 (1H, d, J 7,5);
2-Amino-5-{3-fluoro-4-[(2-metoxi-etil)-metil-carbamoil]-fenil}-N-piridin-4-il-nicotinamida (“A102”)

La reacción del ácido 4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-2-fluoro-benzoico con N-(metoxietil)-metilamina da “A102”; método 1: HPLC/EM: 1,34 min, [M+H] = 424;
RMN 1H (500 MHz, DMSO-do/TFA-dO 5 [ppm] 9,09 (1H, d, J 2,0), 8,81 (2H, d, J 7,2), 8,76 (1H, d, J 2,2), 8,35 (2H, d, J 7,3), 8,08 (1H, s), 7,82-7,69 (1H, m), 7,57 (1H, td, J 7,6, 2,9), 3,72 (1H, t, J 5,5), 3,63 (1H, t, J 5,5), 3,43 (2H, dd, J 10,0, 4,1), 3,36 (2H, s), 3,23 (1H, s), 3,10 (2H, s), 2,97 (1H, s);
Ejemplo 13
2-Amino-5-(2-amino-fenil)-N-piridin-4-il-nicotinamida (“A103”)

13.1. Terc-butil éster del ácido 2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-fenil-carbámico
El compuesto del título se obtiene a partir de 2-amino-5-bromo-N-(2-etoxi-piridin-4-il)-nicotinamida y ferc-butil éster del ácido [2-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-fenil]-carbámico de manera análoga a la preparación de “A62”; método 1: HPLC/EM: 1,34 min, [M+H] = 406.
13.2. 2-Amino-5-(2-amino-fenil)-N-piridin-4-il-nicotinamida
100 mg de ferc-butil éster del ácido {2-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-fenil}-carbámico se disuelven en 2 ml de diclorometano. Se añaden 0,5 ml de HCl en dioxano (4 molar). La mezcla se agita 2 h a temperatura ambiente. La mezcla se filtra y el sólido se lava con diclorometano. Se obtienen 80 mg de “A103", sal de clorhidrato; método 1: HPLC/EM: 1,17 min, [M+H] = 306;
RMN 1H (500 MHz, DMSO-de/TFA-di) 5 [ppm] 8,99 (1H, d, J 2,2), 8,85 (2H, d, J 7,3), 8,49 (1H, d, J 2,1), 8,43 (2H, d, J 7,5), 7,68-7,55 (4 H, m).
Usando el procedimiento mencionado anteriormente, se obtienen de manera análoga los siguientes compuestos:
2-Amino-5-(3-amino-fenil)-N-piridin-4-il-nicotinamida (“A104”)

La solvólisis de íerc-butil éster del ácido {3-[6-amino-5-(piridin-4-McarbamoM)-piridin-3-M]-fenil}-carbámico da “A104”; método 1: HPLC/EM: 0,99 min, [M+H] = 306;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 9,02 (1H, d, J 2,2), 8,86 (2H, d, J 7,3), 8,69 (1H, d, J 2,2), 8,37 (2H, d, J 7,3), 7,94 (1H, d, J 8,1), 7,86 (1H, t, J 1,8), 7,70 (1H, t, J 7,9), 7,54 (1H, dd, J 8,0, 1,2);
2-Amino-5-[4-(2-amino-etilcarbamoil)-fenil]-N-piridin-4-il-nicotinamida (“A105”)

La solvólisis de íerc-butil éster del ácido (2-{4-[6-amino-5-(piridin-4-ilcarbamoil)-piridin-3-il]-benzoilamino}-etil)-carbámico da “A105”; método 2: HPLC/EM: 1,57 min, [M+H] = 377;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 9,18 (1H, d, J 2,2), 8,77 (2H, d, J 7,3), 8,72 (1H, d, J 2,2), 8,42 (2H, d, J 7,3), 8,13 (2H, d, J 8,4), 7,96 (2H, d, J 8,5), 3,67 (2H, t, J 10,6, 4,6), 3,14 (2H, t, J 5,8).
Ejemplo 14
2-Amino-N-piridin-4-il-5-(2-sulfamoil-fenil)-nicotinamida (“A106”)

14.1.2-Amino-5-(2-íerc-butilsulfamoil-fenil)-N-piridin-4-il-nicotinamida.
El compuesto se obtiene por la reacción de 2-amino-5-bromo-N-(2-etoxi-piridin-4-il)-nicotinamida con el ácido 2-íerc-butilsulfamoil-bencenborónico de manera análoga a la etapa 1 para “A62” ; método 1: HPLC/EM: 1,41 min, [M+H] = 426;
RMN 1H (400 MHz, DMSO-d6/TFA-d1) 5 [ppm] 8,86-8,80 (3H, m), 8,39 (1H, d, J 2,1), 8,28 (2H, d, J 7,3), 8,15 (1H, dd, J 7,9, 1,2), 7,73 (2H, dtd, J 31,4, 7,6, 1,4), 7,57 (1H, dd, J 7,6, 1,2), 1,09 (9 H, s).
14.2. 2-Amino-N-piridin-4-il-5-(2-sulfamoil-fenil)-nicotinamida (“A106”)
20 mg de 2-amino-5-(2-ferc-butilsulfamoil-fenil)-N-piridin-4-il-nicotinamida se disuelve en 0,5 ml de ácido trifluoroacético. La mezcla se agita a 80°C durante 14 h. La mezcla de reacción se enfría a temperatura ambiente, se añaden 2 ml de heptano y el disolvente se retira al vacío, el residuo se disuelve en cloruro de metileno y el precipitado resultante se filtra, para dar 17 mg de “A106" como la sal de trifluoroacetato; método 1: HPLC/EM: 1,07min, [M+H] = 370;
RMN 1H (400 MHz, DMSO-d6/TFA-d1) 5 [ppm] 8,86 (1H, d, J 2,2), 8,82 (2H, d, J 7,3), 8,42 (1H, d, J 2,2), 8,33 (2H, d, J 7,5), 8,18 (1H, dd, J 7,8, 1,4), 7,73 (2H, dtd, J 22,4, 7,6, 1,4), 7,58 (1H, dd, J 7,4, 1,4).
Ejemplo 15
-Amino-5-[4-(ferc-butilamino-metil)-fenil]-N-piridin-4-il-nicotinamida (“A107”)

15.1.2-Amino-5-(4-formyl-fenil)-N-piridin-4-il-nicotinamida
El compuesto del título se obtiene mediante la reacción de 2-amino-5-bromo-N-(2-etoxi-piridin-4-il)-nicotinamida con el ácido 4-formilbencenborónico de manera análoga a la etapa 1 para “A62”; método 1: HPLC/EM: 1,29 min, [M+H] = 319;
RMN 1H (500 MHz, DMSO-d6/TFA-d1) 5 [ppm] 10,10 (1H, s), 9,08 (1H, d, J 2,2), 8,86 (2H, d, J 7,2), 8,82 (1H, d, J 2,2), 8,32 (2H, d, J 7,3), 8,12-8,05 (4 H, m).
15.2. 2-Amino-5-[4-(ferc-butilamino-metil)-fenil]-N-piridin-4-il-nicotinamida (“A107”)
70 mg de NaB(OAc)3H se añade a una mezcla de 50 mg de 2-amino-5-(4-formil-fenil)-N-piridin-4-il-nicotinamida, 20,6 pl de ferc-butilamina y 9 pl de ácido acético en 0,5 ml de 1,2-dicloroetano y 0,5 ml de tetrahidrofurano. La suspensión resultante se agita a 50°C durante 14 h. La mezcla de reacción se hace básica con una solución de NaOH 2N y se extrae con diclorometano. La capa orgánica se lava con salmuera, después se separa y se seca sobre Na2SO4. El agente de secado se filtra y el disolvente se retira al vacío. El producto se purifica por cromatografía para producir 10 mg de “A107” como un sólido de color blanco; método 1: HPLC/EM: 1,10 min, [M+H] = 376;
RMN 1H (400 MHz, DMSO-d6/TFA-d1) 5 [ppm] 9,05 (1H, d, J 2,2), 8,84 (2H, d, J 7,3), 8,75 (1H, d, J 2,2), 8,34 (2H, d, J 7,3), 7,94 (2H, d, J 8,4), 7,73 (2H, d, J 8,4), 4,20 (2H, s), 1,44 (9 H, s).
Ejemplo 16
2-Amino-5-(2-etilsulfamoil-fenil)-N-piridin-4-il-nicotinamida (“A108”)

En una atmósfera de argón, se carga un recipiente de reacción con 100 mg de 2-amino-5-bromo-N-piridin-4-ilnicotinamida, 86 mg de bis(pinacolato)diboro, 5 ml de N,N-dimetilformamida desgasificada y 134 mg de acetato de potasio y se agita a temperatura ambiente. El recipiente de reacción se carga con 27 mg de (1,1 ’-bis(difenil-fosfino)-ferrocen)dicloropaladio (ll) y se agita a 80°C durante 14 h. Se añaden a la solución 108 mg de 2-bromo-N-etilbencensulfonamida, 27 mg de (1,1 ’-bis(difenilfosfino)-ferrocen)dicloro-paladio (II) y 200 pl de agua. La mezcla de reacción se agita a 100°C durante 14 h, se enfría a temperatura ambiente y se evapora. El producto se purifica por cromatografía. Se obtienen 8 mg de “A108"; método 1: HPLC/EM: 1,32 min, [M+H] = 398;
RMN 1H (500 MHz, DMSO-ds/TFA-di) 5 [ppm] 8,95 (1H, d, J 2,0), 8,73 (2H, d, J 7,3), 8,38 (1H, d, J 2,1), 8,36 (2H, d, J 7,3), 8,14 (1H, d, J 7,9), 7,76 (1H, t, J 7,5), 7,70 (1H, t, J 7,7), 7,57 (1H, d, J 7,5), 2,85 (2H, q, J 7,2), 1,01 (3H, t, J 7,2).
Valores CI50 de los compuestos de acuerdo con la invención que inhiben TBK1 y IKKs


Los siguientes ejemplos se relacionan con medicamentos:
Ejemplo A: Viales de inyección
Una solución de 100 g de un principio activo de la fórmula I y 5 g de fosfatos ácido de disodio en 3 l de agua bidestilada, se ajusta a pH 6,5 usando ácido clorhídrico 2 N, se filtra estéril, se transfiere en viales de inyección, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada vial de inyección contiene 5 mg de principio activo. Ejemplo B: Supositorios
Una mezcla de 20 g de un principio activo de la fórmula I con 100 g de lecitina de soja y 1400 g de manteca de cacao se funde, se vierte en moldes y se deja enfriar. Cada supositorio contiene 20 mg de principio activo.
Ejemplo C: Solución
Una solución se prepara de 1 g de un principio activo de la fórmula I, 9,38 g de NaH2PÜ4-2 H2O, 28,48 g de Na2HPÜ4 12 H2O y 0,1 g de cloruro de benzalconio en 940 ml de agua bidestilada. El pH se ajusta a 6,8, y la solución se hace hasta 1 l y se esteriliza por irradiación. Esta solución puede usarse en la forma de gotas para los ojos.
Ejemplo D: Ungüento
500 mg de un principio activo de la fórmula I se mezclan con 99,5 g de Vaseline en condiciones asépticas.
Ejemplo E: Comprimidos
Una mezcla de 1 kg de principio activo de la fórmula I, 4 kg de lactosa, 1,2 kg de almidón de papa, 0,2 kg de talco y 0,1 kg de estearato de magnesio se prensa de una manera convencional, para dar los comprimidos, de tal manera que cada comprimido contiene 10 mg de principio activo.
Ejemplo F: Grageas
Los comprimidos se prensan de manera análoga al Ejemplo E y subsecuentemente se recubren de una manera convencional con un recubrimiento de sucrosa, almidón de papa, talco, tragacanto y colorante.
Ejemplo G: Cápsulas
2 kg de principio activo de la fórmula I se introducen en cápsulas de gelatina dura de una manera convencional, de tal manera que cada cápsula contiene 20 mg del principio activo.
Ejemplo H: Ámpollas
Una solución de 1 kg de principio activo de la fórmula I en 60 l de agua bidestilada se filtra estéril, se transfiere en ámpulas, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada ámpolla contiene 10 mg de principio activo.
Claims (11)
1. Compuestos de la fórmula I:

en la que
X representa CH,
R representa Het,
R1 representa piridilo, pirimidilo, piridazinilo, o furo[3,2-b]piridilo, cada uno de los cuales está sin sustituir o monoo disustituido por Hal, A, OR5, CN, COOA, COOH, CON(R5)2 y/o NR5COA';
Het representa furilo, tienilo, pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, piridilo, pirimidinilo, triazolilo, tetrazolilo, tiadiazol, piridazinilo, pirazinilo, indolilo, isoindolilo, bencimidazolilo, indazolilo, quinolilo, 1,3-benzodioxolilo, benzotiofenilo, benzofuranilo o imidazopiridilo, cada uno de los cuales está sin sustituir o mono-, di- o trisustituido por A, COA, (CH2)pHet2, OH, OA, Hal, (CH2)pN(R5)2, NO2, CN, (CH2)pCOOR5, (CH2)pCON(R5)2, NR5COA, (CH2)pCOHet2 y/o (CH2)pfenilo,
Het2 representa dihidropirrolilo, pirrolidinilo, tetrahidroimidazolilo, dihidropirazolilo, tetrahidropirazolilo, dihidropiridilo, tetrahidropiridilo, piperidinilo, morfolinilo, hexahidropiridazinilo, hexahidropirimidinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o mono sustituido por OH y/o A,
A' representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en el que de 1-7 átomos de H pueden reemplazarse por F,
A representa alquilo no ramificado o ramificado que tiene 1-10 átomos de C, en el que uno o dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N, O, S y/o por grupos -CH=CH- y/o además de 1-7 átomos de H pueden reemplazarse por F,
R5 representa H o alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en el que de 1-7 átomos de H pueden reemplazarse por F,
Hal representa F, Cl, Br o I,
n representa 0, 1, 2, 3 o 4,
p representa 0, 1 o 2,
q representa 1, 2, 3 o 4,
y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones.
2. Compuestos de acuerdo con la reivindicación 1, en la que:
Het representa tienilo, pirazolilo, piridilo, cada uno de los cuales está sin sustituir o mono- o disustituido por A, (CH2pHet2, (CH2)pCON(R5)2 y/o (CH2)pfenilo,
y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones.
3. Compuestos de acuerdo a una o más de las reivindicaciones 1-2, en las que:
Het2 representa pirrolidinilo, piperidinilo, morfolinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o monosustituido por OH y/o A,
y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones.
4. Compuestos de cacuerdi a una o más de las reivindicaciones 1 -3, en las que:
A representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que uno o dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N y/u O además de 1-7 átomos de H pueden reemplazarse por F,
y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones.
5. Compuestos de acuerdo a una o más de las reivindicaciones 1-4, en las que:
X representa CH,
R representa Het,
R1 representa piridilo, pirimidilo, piridazinilo o furo[3,2-b]piridilo, cada uno de los cuales está sin sustituir o monosustituido por Hal, A, OR5, COOA, COOH, CON(R5)2 y/o NR5COA’,
Het representa tienilo, pirazolilo, piridilo, cada uno de los cuales está sin sustituir o mono- o disustituido por A, (CH2)pHet2, (CH2)pCON(R5)2 y/o (CH2)pfenilo,
Het2 representa pirrolidinilo, piperidinilo, morfolinilo, [1,3]dioxolanilo, piperazinilo, cada uno de los cuales está sin sustituir o monosustituido por OH y/o A,
A’ representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que de 1-7 átomos de H pueden reemplazarse por F,
A representa alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que uno o dos grupos CH y/o CH2 no adyacentes pueden reemplazarse por átomos de N y/o O y/o además de 1-7 átomos de H pueden reemplazarse por F,
R5 representa H o alquilo no ramificado o ramificado que tiene de 1-6 átomos de C, en la que de 1-7 átomos de H pueden reemplazarse por F,
Hal representa F, Cl, Br o I,
n representa 0, 1, 2, 3 o 4,
p representa 0, 1 o 2,
q representa 1, 2, 3 o 4,
y sales usadas farmacéuticamente, tautómeros y estereoisómeros de las mismas, incluyendo sus mezclas en todas las relaciones.
6. Compuestos de acuerdo con la reivindicación 1, seleccionados entre el grupo



y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo las mezclas de los mismos en todas las relaciones.
7. Proceso para la preparación de compuestos de la fórmula I de acuerdo con las reivindicaciones 1-6 y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, caracterizados porque:
a) un compuesto de la fórmula II:

en la que Y representa Br o I,
X y R1 tienen los significados indicados en la reivindicación 1,
se hace reaccionar con un compuesto de la fórmula III:
R-L III
en la que R tiene el significado indicado en la reivindicación 1 y
L representa un ácido borónico o un grupo éster de ácido borónico,
o
b) un compuesto de la fórmula IV:
en la que R y X tienen los significados indicados en la reivindicación 1 y
L1 representa Cl, Br, I o un grupo OH modificado funcionalmente libre o reactivamente,
se hace reaccionar con un compuesto de la fórmula V
R1-NH2 V
en la que R1 tiene el significado indicado en la reivindicación 1
y/o
una base o ácido de la fórmula I se convierte en una de sus sales.
8. Medicamentos, que comprenden por lo menos un compuesto de la fórmula I de acuerdo con las reivindicaciones 1 -6 y/o sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo mezclas de los mismos en todas las relaciones, y opcionalmente excipientes y/o adyuvantes.
9. Compuestos de acuerdo con las reivindicaciones 1-6 y sales usadas farmacéuticamente, tautómeros y estereoisómeros de los mismos, incluyendo mezclas de los mismos en todas las relaciones, para usarse en el tratamiento de cáncer, choque séptico, glaucoma primario de ángulo abierto (POAG), hiperplasia, artritis reumatoide, psoriasis, arterosclerosis, retinopatía, osteoartritis, endometriosis, inflamación crónica y/o enfermedades neurodegenerativas.
10. Compuestos de la fórmula I de acuerdo con las reivindicaciones 1-6 y/o sales fisiológicamente aceptables, tautómeros y estereoisómeros de los mismos, para usarse en el tratamiento de tumores, en donde una cantidad terapéuticamente eficaz de un compuesto de la fórmula I se administra en combinación con un compuesto del grupo 1) modulador del receptor de estrógeno, 2) modulador del receptor de andrógeno, 3) modulador del receptor retinoide, 4) agente citotóxico, 5) agente antiproliferativo, 6) inhibidor de la proteína prenil-transferasa, 7) inhibidor de HMG-CoA reductasa, 8) inhibidor de VIH proteasa, 9) inhibidor de transcriptasa inversa y 10) inhibidores de angiogénesis adicionales.
11. Compuestos de la fórmula I de acuerdo con las reivindicaciones 1-6 y/o sales fisiológicamente aceptables, tautómeros y estereoisómeros de los mismos, para usarse en el tratamiento de tumores, en donde una cantidad terapéuticamente eficaz de un compuesto de la fórmula I se administra en combinación con radioterapia y un compuesto del grupo 1) modulador del receptor de estrógeno, 2) modulador del receptor de andrógeno, 3) modulador del receptor retinoide, 4) agente citotóxico, 5) agente antiproliferativo, 6) inhibidor de la proteína prenil-transferasa, 7) inhibidor de HMG-CoA reductasa, 8) inhibidor de VIH proteasa, 9) inhibidor de transcriptasa inversa y 10) inhibidores de angiogénesis adicionales.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161488997P | 2011-05-23 | 2011-05-23 | |
| PCT/US2012/032983 WO2012161877A1 (en) | 2011-05-23 | 2012-04-11 | Pyridine-and pyrazine derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2699256T3 true ES2699256T3 (es) | 2019-02-08 |
Family
ID=45976540
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12715279T Active ES2699256T3 (es) | 2011-05-23 | 2012-04-11 | Piridina y derivados de pirazina |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US9273029B2 (es) |
| EP (1) | EP2714677B1 (es) |
| JP (3) | JP6054954B2 (es) |
| KR (1) | KR101581522B1 (es) |
| CN (1) | CN103748086B (es) |
| AU (1) | AU2012259333B2 (es) |
| BR (1) | BR112013029640A2 (es) |
| CA (1) | CA2832605C (es) |
| DK (1) | DK2714677T3 (es) |
| EA (1) | EA023364B1 (es) |
| ES (1) | ES2699256T3 (es) |
| HR (1) | HRP20181884T1 (es) |
| IL (1) | IL229528A (es) |
| LT (1) | LT2714677T (es) |
| MX (1) | MX352975B (es) |
| PL (1) | PL2714677T3 (es) |
| PT (1) | PT2714677T (es) |
| RS (1) | RS58015B1 (es) |
| SG (1) | SG194105A1 (es) |
| SI (1) | SI2714677T1 (es) |
| WO (1) | WO2012161877A1 (es) |
| ZA (1) | ZA201307429B (es) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012161877A1 (en) * | 2011-05-23 | 2012-11-29 | Merck Patent Gmbh | Pyridine-and pyrazine derivatives |
| GB201303109D0 (en) | 2013-02-21 | 2013-04-10 | Domainex Ltd | Novel pyrimidine compounds |
| NZ717552A (en) * | 2013-08-07 | 2021-07-30 | Merck Patent Gmbh | Piperidine urea derivatives |
| US9856223B2 (en) | 2013-12-13 | 2018-01-02 | Dana-Farber Cancer Institute, Inc. | Methods to treat lymphoplasmacytic lymphoma |
| EP3079682A4 (en) | 2013-12-13 | 2017-08-30 | Dana-Farber Cancer Institute, Inc. | Methods to treat lymphoplasmacytic lymphoma |
| CA2939164A1 (en) | 2014-02-12 | 2015-08-20 | Iteos Therapeutics | Novel 3-(indol-3-yl)-pyridine derivatives, pharmaceutical compositions and methods for use |
| PE20170247A1 (es) | 2014-05-15 | 2017-03-29 | Iteos Therapeutics | Derivados de pirrolidina-2,5-diona, composiciones farmaceuticas y metodos para usar como inhibidores ido1 |
| TW201613916A (en) | 2014-06-03 | 2016-04-16 | Gilead Sciences Inc | TANK-binding kinase inhibitor compounds |
| NZ729618A (en) | 2014-09-26 | 2018-07-27 | Gilead Sciences Inc | Aminotriazine derivatives useful as tank-binding kinase inhibitor compounds |
| US9994547B2 (en) | 2014-10-06 | 2018-06-12 | Takeda Pharmaceutical Company Limited | Heteroarylamide inhibitors of TBK1 |
| US10112957B2 (en) | 2014-10-22 | 2018-10-30 | Dana-Farber Cancer Institute, Inc. | Thiazolyl-containing compounds for treating proliferative diseases |
| MA41598A (fr) * | 2015-02-25 | 2018-01-02 | Constellation Pharmaceuticals Inc | Composés thérapeutiques de pyridazine et leurs utilisations |
| WO2016147144A1 (en) | 2015-03-17 | 2016-09-22 | Pfizer Inc. | Novel 3-indol substituted derivatives, pharmaceutical compositions and methods for use |
| EP3334733A1 (en) | 2015-08-10 | 2018-06-20 | Pfizer Inc | 3-indol substituted derivatives, pharmaceutical compositions and methods for use |
| EP3394044A1 (en) | 2015-12-17 | 2018-10-31 | Gilead Sciences, Inc. | Tank-binding kinase inhibitor compounds |
| WO2017102091A1 (en) | 2015-12-18 | 2017-06-22 | Bayer Pharma Aktiengesellschaft | Heteroarylbenzimidazole compounds |
| WO2017207534A1 (en) | 2016-06-03 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Substituted heteroarylbenzimidazole compounds |
| US10710980B2 (en) | 2016-07-20 | 2020-07-14 | Novartis Ag | Aminopyridine derivatives and their use as selective ALK-2 inhibitors |
| DE102016113714A1 (de) | 2016-07-26 | 2018-02-01 | Rosa Karl | Transfektionsverfahren mit nicht-viralen Genliefersystemen |
| GB201702947D0 (en) | 2017-02-23 | 2017-04-12 | Domainex Ltd | Novel compounds |
| TWI771410B (zh) * | 2017-04-27 | 2022-07-21 | 日商石原產業股份有限公司 | N-(4-吡啶基)菸鹼醯胺化合物或其鹽 |
| CN115038443A (zh) | 2019-11-22 | 2022-09-09 | 因西特公司 | 包含alk2抑制剂和jak2抑制剂的组合疗法 |
| JP2023530316A (ja) | 2020-06-16 | 2023-07-14 | インサイト・コーポレイション | 貧血の治療のためのalk2阻害剤 |
| CN116710090A (zh) * | 2020-10-09 | 2023-09-05 | Napa医疗有限公司 | Cd38的杂芳基酰胺抑制剂 |
| CN116099004B (zh) * | 2022-12-30 | 2024-01-30 | 深圳开悦生命科技有限公司 | Rna解旋酶dhx33抑制剂在制备用于治疗膀胱癌的药物中的应用 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5747469A (en) | 1991-03-06 | 1998-05-05 | Board Of Regents, The University Of Texas System | Methods and compositions comprising DNA damaging agents and p53 |
| GB9904387D0 (en) | 1999-02-25 | 1999-04-21 | Pharmacia & Upjohn Spa | Antitumour synergistic composition |
| SE0102438D0 (sv) | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| SE0102439D0 (sv) | 2001-07-05 | 2001-07-05 | Astrazeneca Ab | New compounds |
| SE0203754D0 (sv) | 2002-12-17 | 2002-12-17 | Astrazeneca Ab | New compounds |
| SE0203752D0 (sv) | 2002-12-17 | 2002-12-17 | Astrazeneca Ab | New compounds |
| EP1715867A4 (en) * | 2004-02-12 | 2009-04-15 | Merck & Co Inc | BIPYRIDYLAMIDE AS MODULATORS OF METABOTROPIC GLUTAMATE RECEPTOR-5 |
| TW200800203A (en) * | 2006-03-08 | 2008-01-01 | Astrazeneca Ab | New use |
| EP2016075A1 (en) | 2006-05-03 | 2009-01-21 | AstraZeneca AB | Thiazole derivatives and their use as anti-tumour agents |
| WO2009030890A1 (en) | 2007-09-03 | 2009-03-12 | University Court Of The University Of Dundee | Pyrimidine compounds for the treatment of cancer, septic shock and/or primary open angle glaucoma |
| ATE523508T1 (de) | 2007-10-25 | 2011-09-15 | Astrazeneca Ab | Für die behandlung von zellproliferativen erkrankungen geeignete pyridin- und pyrazinderivate |
| WO2009122180A1 (en) * | 2008-04-02 | 2009-10-08 | Medical Research Council | Pyrimidine derivatives capable of inhibiting one or more kinases |
| ES2663222T3 (es) | 2008-12-19 | 2018-04-11 | Vertex Pharmaceuticals Incorporated | Derivados de pirazina útiles como inhibidores de la quinasa ATR |
| GB0903759D0 (en) * | 2009-03-04 | 2009-04-15 | Medical Res Council | Compound |
| WO2011089416A1 (en) | 2010-01-19 | 2011-07-28 | Astrazeneca Ab | Pyrazine derivatives |
| WO2012161877A1 (en) * | 2011-05-23 | 2012-11-29 | Merck Patent Gmbh | Pyridine-and pyrazine derivatives |
-
2012
- 2012-04-11 WO PCT/US2012/032983 patent/WO2012161877A1/en not_active Ceased
- 2012-04-11 EA EA201301302A patent/EA023364B1/ru not_active IP Right Cessation
- 2012-04-11 PL PL12715279T patent/PL2714677T3/pl unknown
- 2012-04-11 JP JP2014512837A patent/JP6054954B2/ja not_active Expired - Fee Related
- 2012-04-11 ES ES12715279T patent/ES2699256T3/es active Active
- 2012-04-11 DK DK12715279.1T patent/DK2714677T3/en active
- 2012-04-11 SG SG2013074638A patent/SG194105A1/en unknown
- 2012-04-11 CN CN201280025133.3A patent/CN103748086B/zh not_active Expired - Fee Related
- 2012-04-11 KR KR1020137030982A patent/KR101581522B1/ko not_active Expired - Fee Related
- 2012-04-11 LT LTEP12715279.1T patent/LT2714677T/lt unknown
- 2012-04-11 HR HRP20181884TT patent/HRP20181884T1/hr unknown
- 2012-04-11 RS RS20181427A patent/RS58015B1/sr unknown
- 2012-04-11 MX MX2013012979A patent/MX352975B/es active IP Right Grant
- 2012-04-11 SI SI201231465T patent/SI2714677T1/sl unknown
- 2012-04-11 PT PT12715279T patent/PT2714677T/pt unknown
- 2012-04-11 EP EP12715279.1A patent/EP2714677B1/en active Active
- 2012-04-11 US US14/118,845 patent/US9273029B2/en not_active Expired - Fee Related
- 2012-04-11 AU AU2012259333A patent/AU2012259333B2/en not_active Ceased
- 2012-04-11 BR BR112013029640A patent/BR112013029640A2/pt active Search and Examination
- 2012-04-11 CA CA2832605A patent/CA2832605C/en active Active
-
2013
- 2013-10-04 ZA ZA2013/07429A patent/ZA201307429B/en unknown
- 2013-11-21 IL IL229528A patent/IL229528A/en active IP Right Grant
-
2016
- 2016-06-23 JP JP2016124235A patent/JP2016196492A/ja active Pending
-
2018
- 2018-03-28 JP JP2018061990A patent/JP6763904B2/ja not_active Expired - Fee Related
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2699256T3 (es) | Piridina y derivados de pirazina | |
| AU2012306746B2 (en) | Benzonitrile derivatives as kinase inhibitors | |
| ES2622526T3 (es) | Derivados de tiazol | |
| CN104093722B (zh) | 作为TBK1和IKK抑制剂的呋喃并[3,2-b]-和噻吩并[3,2-b]吡啶衍生物 | |
| AU2012342891B2 (en) | 3-Cyanoaryl-1H-pyrrolo[2.3-b]pyridine derivatives | |
| HK1196601B (en) | Pyridine-and pyrazine derivatives | |
| HK1199880B (en) | Benzonitrile derivatives as kinase inhibitors | |
| HK1202550B (en) | Furo [3, 2 - b] - and thieno [3, 2 - b] pyridine derivatives as tbk1 and ikk inhibitors | |
| HK1194067B (en) | Thiazole derivatives |