ES2751325T3 - Hidróxidos dobles con capas de magnesio-hierro, su producción y su utilización para el tratamiento de hiperfosfatemia - Google Patents
Hidróxidos dobles con capas de magnesio-hierro, su producción y su utilización para el tratamiento de hiperfosfatemia Download PDFInfo
- Publication number
- ES2751325T3 ES2751325T3 ES10739987T ES10739987T ES2751325T3 ES 2751325 T3 ES2751325 T3 ES 2751325T3 ES 10739987 T ES10739987 T ES 10739987T ES 10739987 T ES10739987 T ES 10739987T ES 2751325 T3 ES2751325 T3 ES 2751325T3
- Authority
- ES
- Spain
- Prior art keywords
- suspension
- mixed metal
- less
- metal compound
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000011282 treatment Methods 0.000 title claims description 16
- 201000005991 hyperphosphatemia Diseases 0.000 title claims description 14
- 238000004519 manufacturing process Methods 0.000 title abstract description 47
- 150000004679 hydroxides Chemical class 0.000 title description 7
- MHKWSJBPFXBFMX-UHFFFAOYSA-N iron magnesium Chemical compound [Mg].[Fe] MHKWSJBPFXBFMX-UHFFFAOYSA-N 0.000 title description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims abstract description 301
- 150000002736 metal compounds Chemical class 0.000 claims abstract description 252
- 239000000725 suspension Substances 0.000 claims abstract description 229
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 207
- 238000006243 chemical reaction Methods 0.000 claims abstract description 204
- 229910001868 water Inorganic materials 0.000 claims abstract description 198
- 239000000047 product Substances 0.000 claims abstract description 194
- 229910000029 sodium carbonate Inorganic materials 0.000 claims abstract description 149
- 235000017550 sodium carbonate Nutrition 0.000 claims abstract description 149
- 238000001035 drying Methods 0.000 claims abstract description 128
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims abstract description 105
- 239000012043 crude product Substances 0.000 claims abstract description 86
- 239000013078 crystal Substances 0.000 claims abstract description 79
- 150000003839 salts Chemical class 0.000 claims abstract description 58
- 229910052782 aluminium Inorganic materials 0.000 claims abstract description 55
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims abstract description 52
- 239000007787 solid Substances 0.000 claims abstract description 45
- 238000001704 evaporation Methods 0.000 claims abstract description 44
- 230000008020 evaporation Effects 0.000 claims abstract description 44
- 238000003801 milling Methods 0.000 claims abstract description 32
- 238000010438 heat treatment Methods 0.000 claims abstract description 27
- 238000002156 mixing Methods 0.000 claims abstract description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 claims abstract description 10
- 238000000034 method Methods 0.000 claims description 157
- 150000001875 compounds Chemical class 0.000 claims description 142
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 97
- 239000002245 particle Substances 0.000 claims description 90
- 239000011148 porous material Substances 0.000 claims description 57
- 229910052751 metal Inorganic materials 0.000 claims description 41
- 239000002184 metal Substances 0.000 claims description 41
- 150000001450 anions Chemical class 0.000 claims description 24
- 150000002739 metals Chemical class 0.000 claims description 16
- 239000003814 drug Substances 0.000 claims description 13
- 229910000765 intermetallic Inorganic materials 0.000 claims description 11
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- 239000003981 vehicle Substances 0.000 claims description 5
- 239000002671 adjuvant Substances 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- MYLBTCQBKAKUTJ-UHFFFAOYSA-N 7-methyl-6,8-bis(methylsulfanyl)pyrrolo[1,2-a]pyrazine Chemical compound C1=CN=CC2=C(SC)C(C)=C(SC)N21 MYLBTCQBKAKUTJ-UHFFFAOYSA-N 0.000 claims description 2
- VTLYFUHAOXGGBS-UHFFFAOYSA-N Fe3+ Chemical compound [Fe+3] VTLYFUHAOXGGBS-UHFFFAOYSA-N 0.000 abstract description 5
- 238000001914 filtration Methods 0.000 description 89
- 239000000463 material Substances 0.000 description 84
- 239000000243 solution Substances 0.000 description 68
- 239000011777 magnesium Substances 0.000 description 64
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical class [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 63
- 229910052938 sodium sulfate Inorganic materials 0.000 description 61
- 235000011152 sodium sulphate Nutrition 0.000 description 61
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 59
- 239000010410 layer Substances 0.000 description 59
- 239000007832 Na2SO4 Substances 0.000 description 57
- 229910019142 PO4 Inorganic materials 0.000 description 54
- 239000010452 phosphate Substances 0.000 description 54
- 238000010998 test method Methods 0.000 description 54
- 102000006335 Phosphate-Binding Proteins Human genes 0.000 description 45
- 108010058514 Phosphate-Binding Proteins Proteins 0.000 description 45
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 45
- 238000001556 precipitation Methods 0.000 description 45
- 230000008569 process Effects 0.000 description 44
- 238000004458 analytical method Methods 0.000 description 41
- 238000005406 washing Methods 0.000 description 37
- 239000000523 sample Substances 0.000 description 36
- 239000003153 chemical reaction reagent Substances 0.000 description 35
- 239000011734 sodium Substances 0.000 description 31
- 235000011121 sodium hydroxide Nutrition 0.000 description 31
- 239000000203 mixture Substances 0.000 description 30
- 238000007792 addition Methods 0.000 description 28
- 229910052799 carbon Inorganic materials 0.000 description 26
- 239000011229 interlayer Substances 0.000 description 25
- 229910052749 magnesium Inorganic materials 0.000 description 25
- 230000008901 benefit Effects 0.000 description 24
- 230000014759 maintenance of location Effects 0.000 description 24
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 23
- 230000032683 aging Effects 0.000 description 22
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 20
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 20
- 239000000126 substance Substances 0.000 description 20
- 230000012010 growth Effects 0.000 description 19
- KKCBUQHMOMHUOY-UHFFFAOYSA-N Na2O Inorganic materials [O-2].[Na+].[Na+] KKCBUQHMOMHUOY-UHFFFAOYSA-N 0.000 description 18
- 238000002955 isolation Methods 0.000 description 17
- 230000001276 controlling effect Effects 0.000 description 16
- GDVKFRBCXAPAQJ-UHFFFAOYSA-A dialuminum;hexamagnesium;carbonate;hexadecahydroxide Chemical compound [OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Al+3].[Al+3].[O-]C([O-])=O GDVKFRBCXAPAQJ-UHFFFAOYSA-A 0.000 description 16
- 238000009826 distribution Methods 0.000 description 16
- 239000012065 filter cake Substances 0.000 description 16
- 238000003860 storage Methods 0.000 description 16
- 238000002360 preparation method Methods 0.000 description 15
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- 238000013019 agitation Methods 0.000 description 14
- 230000002829 reductive effect Effects 0.000 description 14
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 13
- 230000027455 binding Effects 0.000 description 13
- 229910001701 hydrotalcite Inorganic materials 0.000 description 13
- 229960001545 hydrotalcite Drugs 0.000 description 13
- 229910052742 iron Inorganic materials 0.000 description 13
- -1 phosphate compound Chemical class 0.000 description 13
- 238000001694 spray drying Methods 0.000 description 13
- 239000011230 binding agent Substances 0.000 description 12
- 239000012535 impurity Substances 0.000 description 12
- 239000008186 active pharmaceutical agent Substances 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 229910001385 heavy metal Inorganic materials 0.000 description 11
- RUTXIHLAWFEWGM-UHFFFAOYSA-H iron(3+) sulfate Chemical compound [Fe+3].[Fe+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O RUTXIHLAWFEWGM-UHFFFAOYSA-H 0.000 description 11
- 229910000360 iron(III) sulfate Inorganic materials 0.000 description 11
- 238000002441 X-ray diffraction Methods 0.000 description 10
- 239000012530 fluid Substances 0.000 description 10
- 210000003734 kidney Anatomy 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- 238000012545 processing Methods 0.000 description 10
- 238000000926 separation method Methods 0.000 description 10
- 229910052708 sodium Inorganic materials 0.000 description 10
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 9
- 229940088679 drug related substance Drugs 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 239000007789 gas Substances 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- 239000011324 bead Substances 0.000 description 8
- 238000005119 centrifugation Methods 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 238000000975 co-precipitation Methods 0.000 description 8
- 239000012467 final product Substances 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- 238000007873 sieving Methods 0.000 description 8
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical group [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 239000011575 calcium Substances 0.000 description 7
- 229910052791 calcium Inorganic materials 0.000 description 7
- 150000001768 cations Chemical class 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- 238000005259 measurement Methods 0.000 description 7
- 239000002244 precipitate Substances 0.000 description 7
- 230000005855 radiation Effects 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 239000011651 chromium Substances 0.000 description 6
- 208000020832 chronic kidney disease Diseases 0.000 description 6
- 239000010949 copper Substances 0.000 description 6
- 238000009295 crossflow filtration Methods 0.000 description 6
- 230000007423 decrease Effects 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- 238000005469 granulation Methods 0.000 description 6
- 230000003179 granulation Effects 0.000 description 6
- 238000005342 ion exchange Methods 0.000 description 6
- 230000007774 longterm Effects 0.000 description 6
- 230000000144 pharmacologic effect Effects 0.000 description 6
- 239000002694 phosphate binding agent Substances 0.000 description 6
- 239000006187 pill Substances 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 238000010521 absorption reaction Methods 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 229910052804 chromium Inorganic materials 0.000 description 5
- 229910052802 copper Inorganic materials 0.000 description 5
- 238000000354 decomposition reaction Methods 0.000 description 5
- 238000011026 diafiltration Methods 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 239000000796 flavoring agent Substances 0.000 description 5
- 235000013305 food Nutrition 0.000 description 5
- 230000000704 physical effect Effects 0.000 description 5
- 239000008213 purified water Substances 0.000 description 5
- 239000007921 spray Substances 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 230000003068 static effect Effects 0.000 description 5
- 229910021654 trace metal Inorganic materials 0.000 description 5
- 238000005550 wet granulation Methods 0.000 description 5
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- CPLXHLVBOLITMK-UHFFFAOYSA-N Magnesium oxide Chemical compound [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 238000009825 accumulation Methods 0.000 description 4
- 239000012491 analyte Substances 0.000 description 4
- 125000000129 anionic group Chemical group 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 125000002091 cationic group Chemical group 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 230000018044 dehydration Effects 0.000 description 4
- 238000006297 dehydration reaction Methods 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 238000000502 dialysis Methods 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 229910052500 inorganic mineral Inorganic materials 0.000 description 4
- 229910000358 iron sulfate Inorganic materials 0.000 description 4
- BAUYGSIQEAFULO-UHFFFAOYSA-L iron(2+) sulfate (anhydrous) Chemical compound [Fe+2].[O-]S([O-])(=O)=O BAUYGSIQEAFULO-UHFFFAOYSA-L 0.000 description 4
- 235000010755 mineral Nutrition 0.000 description 4
- 239000011707 mineral Substances 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- 229940127557 pharmaceutical product Drugs 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 238000001179 sorption measurement Methods 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 3
- 208000013038 Hypocalcemia Diseases 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 230000002411 adverse Effects 0.000 description 3
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 235000019634 flavors Nutrition 0.000 description 3
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 3
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 3
- 230000000705 hypocalcaemia Effects 0.000 description 3
- 229910052745 lead Inorganic materials 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 150000002681 magnesium compounds Chemical class 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 230000000877 morphologic effect Effects 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 230000001376 precipitating effect Effects 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 210000002700 urine Anatomy 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- 229910017569 La2(CO3)3 Inorganic materials 0.000 description 2
- 229910025794 LaB6 Inorganic materials 0.000 description 2
- 229910003023 Mg-Al Inorganic materials 0.000 description 2
- 208000001647 Renal Insufficiency Diseases 0.000 description 2
- KHNXRSIBRKBJDI-UHFFFAOYSA-N Sevelamer hydrochloride Chemical compound Cl.NCC=C.ClCC1CO1 KHNXRSIBRKBJDI-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000005475 Vascular calcification Diseases 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- DIZPMCHEQGEION-UHFFFAOYSA-H aluminium sulfate (anhydrous) Chemical compound [Al+3].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O DIZPMCHEQGEION-UHFFFAOYSA-H 0.000 description 2
- 238000005349 anion exchange Methods 0.000 description 2
- 229940069428 antacid Drugs 0.000 description 2
- 239000003159 antacid agent Substances 0.000 description 2
- 229910052785 arsenic Inorganic materials 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 239000010839 body fluid Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229910052793 cadmium Inorganic materials 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 238000005266 casting Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000002485 combustion reaction Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- CVSVTCORWBXHQV-UHFFFAOYSA-N creatine Chemical compound NC(=[NH2+])N(C)CC([O-])=O CVSVTCORWBXHQV-UHFFFAOYSA-N 0.000 description 2
- 239000002178 crystalline material Substances 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000003795 desorption Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 150000004676 glycans Chemical class 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 238000001095 inductively coupled plasma mass spectrometry Methods 0.000 description 2
- 229910010272 inorganic material Inorganic materials 0.000 description 2
- 239000011147 inorganic material Substances 0.000 description 2
- 201000006370 kidney failure Diseases 0.000 description 2
- NZPIUJUFIFZSPW-UHFFFAOYSA-H lanthanum carbonate Chemical compound [La+3].[La+3].[O-]C([O-])=O.[O-]C([O-])=O.[O-]C([O-])=O NZPIUJUFIFZSPW-UHFFFAOYSA-H 0.000 description 2
- 229960001633 lanthanum carbonate Drugs 0.000 description 2
- 239000000395 magnesium oxide Substances 0.000 description 2
- 235000012245 magnesium oxide Nutrition 0.000 description 2
- 230000000873 masking effect Effects 0.000 description 2
- 235000012054 meals Nutrition 0.000 description 2
- 229910021645 metal ion Inorganic materials 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 229910052750 molybdenum Inorganic materials 0.000 description 2
- 238000010606 normalization Methods 0.000 description 2
- 239000003002 pH adjusting agent Substances 0.000 description 2
- 239000006174 pH buffer Substances 0.000 description 2
- 239000012466 permeate Substances 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 238000012805 post-processing Methods 0.000 description 2
- 238000000634 powder X-ray diffraction Methods 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 229910052761 rare earth metal Inorganic materials 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000007916 tablet composition Substances 0.000 description 2
- 238000002604 ultrasonography Methods 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 235000005282 vitamin D3 Nutrition 0.000 description 2
- 239000011647 vitamin D3 Substances 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- GNENVASJJIUNER-UHFFFAOYSA-N 2,4,6-tricyclohexyloxy-1,3,5,2,4,6-trioxatriborinane Chemical compound C1CCCCC1OB1OB(OC2CCCCC2)OB(OC2CCCCC2)O1 GNENVASJJIUNER-UHFFFAOYSA-N 0.000 description 1
- MIDXCONKKJTLDX-UHFFFAOYSA-N 3,5-dimethylcyclopentane-1,2-dione Chemical compound CC1CC(C)C(=O)C1=O MIDXCONKKJTLDX-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- WBZFUFAFFUEMEI-UHFFFAOYSA-M Acesulfame k Chemical compound [K+].CC1=CC(=O)[N-]S(=O)(=O)O1 WBZFUFAFFUEMEI-UHFFFAOYSA-M 0.000 description 1
- 206010000599 Acromegaly Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 208000020084 Bone disease Diseases 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 208000004434 Calcinosis Diseases 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 244000223760 Cinnamomum zeylanicum Species 0.000 description 1
- 235000005979 Citrus limon Nutrition 0.000 description 1
- 244000131522 Citrus pyriformis Species 0.000 description 1
- 240000000560 Citrus x paradisi Species 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 244000303965 Cyamopsis psoralioides Species 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UHFFFAOYSA-N D-OH-Asp Natural products OC(=O)C(N)CC(O)=O CKLJMWTZIZZHCS-UHFFFAOYSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-UHFFFAOYSA-N D-alpha-Ala Natural products CC([NH3+])C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 240000004670 Glycyrrhiza echinata Species 0.000 description 1
- 235000001453 Glycyrrhiza echinata Nutrition 0.000 description 1
- 235000006200 Glycyrrhiza glabra Nutrition 0.000 description 1
- 235000017382 Glycyrrhiza lepidota Nutrition 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000577115 Homo sapiens Monocarboxylate transporter 2 Proteins 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- 208000000038 Hypoparathyroidism Diseases 0.000 description 1
- QNAYBMKLOCPYGJ-UWTATZPHSA-N L-Alanine Natural products C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UWTATZPHSA-N L-Aspartic acid Natural products OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- 229930182844 L-isoleucine Natural products 0.000 description 1
- 239000004395 L-leucine Substances 0.000 description 1
- 235000019454 L-leucine Nutrition 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000030136 Marchiafava-Bignami Disease Diseases 0.000 description 1
- 235000006679 Mentha X verticillata Nutrition 0.000 description 1
- 235000002899 Mentha suaveolens Nutrition 0.000 description 1
- 235000001636 Mentha x rotundifolia Nutrition 0.000 description 1
- 208000029725 Metabolic bone disease Diseases 0.000 description 1
- 229910020068 MgAl Inorganic materials 0.000 description 1
- 102100025272 Monocarboxylate transporter 2 Human genes 0.000 description 1
- 102000007474 Multiprotein Complexes Human genes 0.000 description 1
- 108010085220 Multiprotein Complexes Proteins 0.000 description 1
- 206010033296 Overdoses Diseases 0.000 description 1
- 102000003982 Parathyroid hormone Human genes 0.000 description 1
- 108090000445 Parathyroid hormone Proteins 0.000 description 1
- 108010064851 Plant Proteins Proteins 0.000 description 1
- 244000134552 Plantago ovata Species 0.000 description 1
- 235000003421 Plantago ovata Nutrition 0.000 description 1
- 229920002518 Polyallylamine hydrochloride Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 239000009223 Psyllium Substances 0.000 description 1
- 240000007651 Rubus glaucus Species 0.000 description 1
- 235000011034 Rubus glaucus Nutrition 0.000 description 1
- 235000009122 Rubus idaeus Nutrition 0.000 description 1
- 208000005770 Secondary Hyperparathyroidism Diseases 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 244000098338 Triticum aestivum Species 0.000 description 1
- 244000290333 Vanilla fragrans Species 0.000 description 1
- 235000009499 Vanilla fragrans Nutrition 0.000 description 1
- 235000012036 Vanilla tahitensis Nutrition 0.000 description 1
- 229930003316 Vitamin D Natural products 0.000 description 1
- QYSXJUFSXHHAJI-XFEUOLMDSA-N Vitamin D3 Natural products C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)CCCC(C)C)=C/C=C1\C[C@@H](O)CCC1=C QYSXJUFSXHHAJI-XFEUOLMDSA-N 0.000 description 1
- 235000009754 Vitis X bourquina Nutrition 0.000 description 1
- 235000012333 Vitis X labruscana Nutrition 0.000 description 1
- 240000006365 Vitis vinifera Species 0.000 description 1
- 235000014787 Vitis vinifera Nutrition 0.000 description 1
- 229920002494 Zein Polymers 0.000 description 1
- 239000000619 acesulfame-K Substances 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000002156 adsorbate Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 238000005054 agglomeration Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 229960003767 alanine Drugs 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- VXAUWWUXCIMFIM-UHFFFAOYSA-M aluminum;oxygen(2-);hydroxide Chemical compound [OH-].[O-2].[Al+3] VXAUWWUXCIMFIM-UHFFFAOYSA-M 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000021120 animal protein Nutrition 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000003613 bile acid Substances 0.000 description 1
- 229910001593 boehmite Inorganic materials 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 230000004097 bone metabolism Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 230000002308 calcification Effects 0.000 description 1
- 230000002092 calcimimetic effect Effects 0.000 description 1
- 235000020964 calcitriol Nutrition 0.000 description 1
- 239000011612 calcitriol Substances 0.000 description 1
- GMRQFYUYWCNGIN-NKMMMXOESA-N calcitriol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C GMRQFYUYWCNGIN-NKMMMXOESA-N 0.000 description 1
- 229960005084 calcitriol Drugs 0.000 description 1
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 description 1
- 239000001639 calcium acetate Substances 0.000 description 1
- 235000011092 calcium acetate Nutrition 0.000 description 1
- 229960005147 calcium acetate Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 235000013736 caramel Nutrition 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 229940084030 carboxymethylcellulose calcium Drugs 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 210000000080 chela (arthropods) Anatomy 0.000 description 1
- CEZCCHQBSQPRMU-UHFFFAOYSA-L chembl174821 Chemical compound [Na+].[Na+].COC1=CC(S([O-])(=O)=O)=C(C)C=C1N=NC1=C(O)C=CC2=CC(S([O-])(=O)=O)=CC=C12 CEZCCHQBSQPRMU-UHFFFAOYSA-L 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 235000017803 cinnamon Nutrition 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 239000002734 clay mineral Substances 0.000 description 1
- 230000003749 cleanliness Effects 0.000 description 1
- 230000001149 cognitive effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229960003624 creatine Drugs 0.000 description 1
- 239000006046 creatine Substances 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- PSHMSSXLYVAENJ-UHFFFAOYSA-N dilithium;[oxido(oxoboranyloxy)boranyl]oxy-oxoboranyloxyborinate Chemical compound [Li+].[Li+].O=BOB([O-])OB([O-])OB=O PSHMSSXLYVAENJ-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000002036 drum drying Methods 0.000 description 1
- 238000007580 dry-mixing Methods 0.000 description 1
- 238000010981 drying operation Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 239000012527 feed solution Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 230000004992 fission Effects 0.000 description 1
- 235000013312 flour Nutrition 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000024924 glomerular filtration Effects 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 150000004688 heptahydrates Chemical class 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 235000011167 hydrochloric acid Nutrition 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- FAHBNUUHRFUEAI-UHFFFAOYSA-M hydroxidooxidoaluminium Chemical compound O[Al]=O FAHBNUUHRFUEAI-UHFFFAOYSA-M 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000002354 inductively-coupled plasma atomic emission spectroscopy Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000011872 intimate mixture Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910001608 iron mineral Inorganic materials 0.000 description 1
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N iron oxide Inorganic materials [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- VBMVTYDPPZVILR-UHFFFAOYSA-N iron(2+);oxygen(2-) Chemical class [O-2].[Fe+2] VBMVTYDPPZVILR-UHFFFAOYSA-N 0.000 description 1
- VXWSFRMTBJZULV-UHFFFAOYSA-H iron(3+) sulfate hydrate Chemical compound O.[Fe+3].[Fe+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O VXWSFRMTBJZULV-UHFFFAOYSA-H 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- 210000001503 joint Anatomy 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229910052746 lanthanum Inorganic materials 0.000 description 1
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 229960003136 leucine Drugs 0.000 description 1
- 229940010454 licorice Drugs 0.000 description 1
- 210000003041 ligament Anatomy 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004643 material aging Methods 0.000 description 1
- 239000002905 metal composite material Substances 0.000 description 1
- 229910000000 metal hydroxide Inorganic materials 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000006911 nucleation Effects 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 208000005368 osteomalacia Diseases 0.000 description 1
- 238000000643 oven drying Methods 0.000 description 1
- 238000013021 overheating Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000000199 parathyroid hormone Substances 0.000 description 1
- 229960001319 parathyroid hormone Drugs 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 229960005190 phenylalanine Drugs 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 239000006069 physical mixture Substances 0.000 description 1
- 235000021118 plant-derived protein Nutrition 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 208000006078 pseudohypoparathyroidism Diseases 0.000 description 1
- 229940070687 psyllium Drugs 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000002910 rare earth metals Chemical class 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229960003693 sevelamer Drugs 0.000 description 1
- ZNSIZMQNQCNRBW-UHFFFAOYSA-N sevelamer Chemical compound NCC=C.ClCC1CO1 ZNSIZMQNQCNRBW-UHFFFAOYSA-N 0.000 description 1
- 229960003027 sevelamer hydrochloride Drugs 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 230000002037 soft tissue calcification Effects 0.000 description 1
- 238000003980 solgel method Methods 0.000 description 1
- 239000002195 soluble material Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000012358 sourcing Methods 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical compound [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 239000007885 tablet disintegrant Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- 239000000892 thaumatin Substances 0.000 description 1
- 235000010436 thaumatin Nutrition 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- ZSDSQXJSNMTJDA-UHFFFAOYSA-N trifluralin Chemical compound CCCN(CCC)C1=C([N+]([O-])=O)C=C(C(F)(F)F)C=C1[N+]([O-])=O ZSDSQXJSNMTJDA-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000019166 vitamin D Nutrition 0.000 description 1
- 239000011710 vitamin D Substances 0.000 description 1
- 150000003710 vitamin D derivatives Chemical class 0.000 description 1
- 229940046008 vitamin d Drugs 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 238000001238 wet grinding Methods 0.000 description 1
- 210000000707 wrist Anatomy 0.000 description 1
- 238000009681 x-ray fluorescence measurement Methods 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 239000005019 zein Substances 0.000 description 1
- 229940093612 zein Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/06—Aluminium, calcium or magnesium; Compounds thereof, e.g. clay
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/06—Aluminium, calcium or magnesium; Compounds thereof, e.g. clay
- A61K33/10—Carbonates; Bicarbonates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
- A61K33/26—Iron; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K6/00—Preparations for dentistry
- A61K6/80—Preparations for artificial teeth, for filling teeth or for capping teeth
- A61K6/84—Preparations for artificial teeth, for filling teeth or for capping teeth comprising metals or alloys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/143—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1611—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y30/00—Nanotechnology for materials or surface science, e.g. nanocomposites
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G49/00—Compounds of iron
- C01G49/009—Compounds containing iron, with or without oxygen or hydrogen, and containing two or more other elements
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/20—Two-dimensional structures
- C01P2002/22—Two-dimensional structures layered hydroxide-type, e.g. of the hydrotalcite-type
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/60—Compounds characterised by their crystallite size
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/51—Particles with a specific particle size distribution
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/64—Nanometer sized, i.e. from 1-100 nanometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/11—Powder tap density
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/14—Pore volume
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/80—Compositional purity
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/80—Compositional purity
- C01P2006/82—Compositional purity water content
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Inorganic Chemistry (AREA)
- Organic Chemistry (AREA)
- Nanotechnology (AREA)
- Oral & Maxillofacial Surgery (AREA)
- Crystallography & Structural Chemistry (AREA)
- Physics & Mathematics (AREA)
- Materials Engineering (AREA)
- General Physics & Mathematics (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Composite Materials (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Hematology (AREA)
- Plastic & Reconstructive Surgery (AREA)
- Diabetes (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Obesity (AREA)
- Compounds Of Alkaline-Earth Elements, Aluminum Or Rare-Earth Metals (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Compounds Of Iron (AREA)
- Medicinal Preparation (AREA)
- Manufacture And Refinement Of Metals (AREA)
Abstract
Un método para producir un compuesto metálico mixto de hidróxido doble en capas, que comprende al menos Mg2+ y al menos Fe3+ que tiene una relación molar de Mg2+ a Fe3+ de 2,5:1 a 1,5:1 que tiene un contenido de aluminio de menos de 10.000 ppm, con un tamaño de cristal promedio de menos de 20 nm (200 Å), y con un área superficial de 40 a 80 m2 por gramo de compuesto metálico mixto, que comprende los pasos de: (a) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 11, y en donde el Na2CO3 se proporciona en exceso de 0 a 4,0 moles que se requiere para completar la reacción; (b) someter la suspensión a mezcla en condiciones que proporcionen una potencia por unidad de volumen de 0,03 a 1,6 kW/m3; (c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10% en peso (d) secar el producto crudo calentando el producto crudo a una temperatura no más de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco; y (e) fresar el producto crudo seco para proporcionar un área superficial de 40 a 80 m2 por gramo.
Description
DESCRIPCIÓN
Hidróxidos dobles con capas de magnesio-hierro, su producción y su utilización para el tratamiento de hiperfosfatemia
CAMPO DE LA INVENCIÓN
[0001] La presente invención se refiere a un método para producir meta mixto! Compuestos y Compuestos preparados por estos métodos. Estos Compuestos pueden tener actividad farmacéutica, especialmente como aglutinantes de fosfato. La presente invención se refiere además a nuevos Compuestos metálicos mixtos. Aún más, la invención se refiere a composiciones farmacéuticas que contienen los Compuestos anteriores y al uso farmacéutico de los Compuestos.
ANTECEDENTES DE LA INVENCIÓN
Hiperfosfatemia
[0002] La hiperfosfatemia es un trastorno electrolítico en el que hay un nivel anormalmente elevado de fosfato en la sangre. La hiperfosfatemia se observa con frecuencia en pacientes en diálisis, ya que los regímenes de diálisis estándar no pueden eliminar la carga de fosfato ingerida incluso con una dieta baja en fosfato, y se asocia con un mayor riesgo de muerte y el desarrollo de calcificación vascular. La presencia de hiperfosfatemia conduce a hipocalcemia, hiperparatiroidismo secundario, reducción de 1,25 Vit D3 y enfermedad ósea metabólica progresiva. La hiperfosfatemia es en última instancia responsable del aumento de la calcificación vascular, pero estudios recientes también han sugerido que el proceso puede verse afectado adicionalmente por 1,25 Vit D3 y un producto de fosfato de calcio elevado. Los pacientes con hiperfosfatemia crónica no controlada desarrollan calcificaciones progresivamente extensas de los tejidos blandos debido al depósito de producto de calcio/fosfato en la piel, articulaciones, tendones, ligamentos. También se han descrito depósitos oculares de producto de calcio/fosfato.
[0003] El control de los niveles de fosfato en suero usando aglutinantes de fosfato orales se ha convertido, por lo tanto, en un objetivo terapéutico clave en el tratamiento de pacientes en diálisis. Estos aglutinantes, tomados con alimentos, hacen que el fosfato contenido sea insoluble y, por lo tanto, no absorbible.
Aglutinantes de fosfato
[0004] Históricamente, aglutinantes de fosfato incluyen sales de aluminio. Sin embargo, se descubrió que el uso de sales de aluminio da lugar a complicaciones tóxicas adicionales debido a la acumulación de aluminio, por ejemplo, reducción en la producción de hemoglobina, deterioro en la reparación natural y producción de hueso y posible deterioro de la función neurológica/cognitiva. La enfermedad ósea renal, la osteomalacia y la demencia son las toxicidades más significativas relacionadas con la absorción de aluminio. Se propusieron otros compuestos de aluminio como el hidróxido de óxido de aluminio microcristalino (boehmita) y ciertas hidrotalcitas para este uso, como se describe en Ookubo et al, Journal Pharmaceutical Sciences (noviembre de 1992), 81 (11), 1139-1140. Sin embargo, estos padecen los mismos inconvenientes.
[0005] El carbonato de calcio o acetato de calcio se utilizan como aglutinantes de fosfato. Sin embargo, estos sufren el inconveniente de que tienden a promover la hipocalcemia a través de la absorción de altas cantidades de calcio ingerido y están vinculados a una calcificación cardiovascular acelerada que puede causar efectos secundarios graves. En consecuencia, se requiere un monitoreo frecuente de los niveles de calcio en suero durante la terapia con aglutinantes de fosfato a base de calcio, la Iniciativa de resultados de calidad de la enfermedad renal de la National Kidney Foundation sugiere el uso limitado de sales a base de calcio (Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease, Guía 5, pág. 1 pt 5.5). Los esfuerzos recientes, por lo tanto, se han centrado en el desarrollo de aglutinantes de fosfato libres de calcio. Más recientemente, el carbonato de lantano y el sevelamer HCl se han utilizado como aglutinantes de fosfato sin calcio. El hidrocloruro de sevelamer es un hidrocloruro de polialilamina ligado a craso de hidrogel no absorbido por absorción de agua, pero debido a su estructura también se une a ciertas vitaminas liposolubles y ácidos biliares y, por lo tanto, se informa en V. Autissier et al, Journal of Pharmaceutical Sciences, Vol. 96, No 10, octubre de 2007, para requerir grandes dosis para que sea efectivo porque tiene una mayor propensión a que el fosfato unido sea desplazado por estos aniones competidores. Una gran carga de píldoras o tabletas grandes a menudo se asocian con un cumplimiento deficiente del paciente y este tipo de producto también se considera relativamente costoso para sus contrapartes de calcio. Sevelamer también se ha asociado con efectos adversos de Gl AJ Hutchison et al, Drugs 2003; 63 (6), 577-596.
[0006] El carbonato de lantano es un quelante de fosfato que se ha demostrado ser tan eficaz como carbonato de calcio con una menor incidencia de hipocalcemia. La administración a largo plazo de lantano, un elemento de tierras raras, sigue planteando problemas de seguridad con respecto a la posible acumulación de un metal de tierras raras en el tejido corporal que puede potenciarse en la insuficiencia renal - Tilman B Druke, Seminars in Dialysis, Volumen 20, Número 4, página 329-332, julio/agosto de 2007
[0007] Muchas preparaciones inorgánicas conocidas para el tratamiento de la hiperfosfatemia son aglutinantes de fosfato eficaces solamente en un intervalo de pH limitado. Además, aglutinantes particularmente alcalinos podrían tamponar el pH del estómago hasta un alto nivel en el que no tendrían una capacidad de unión a fosfato.
[0008] Para superar los inconvenientes asociados con aluminio y también problemas de eficacia en un intervalo de pH limitado, el documento WO-A-99/15189 describe el uso de compuestos metálicos mixtos que están libres de aluminio y que tienen una capacidad de enlace a fosfato de al menos 30% en peso del peso total de fosfato presente, en un intervalo de pH de 2-8.
Compuestos metálicos mixtos
[0009] Compuestos metálicos mixtos (compuestos metálicos mixtos) existen como los llamados "hidróxido doble en capas" (LDH) que se utiliza para designar hidróxidos lamelares sintéticos o naturales con dos tipos de cationes metálicos en las capas principales y dominios de capas intermedias que contienen especies aniónicas. Esta amplia familia de compuestos a veces también se conoce como arcillas aniónicas, en comparación con las arcillas catiónicas más habituales cuyos dominios interlamelares contienen especies catiónicas. Las LDH también se han descrito como compuestos similares a la hidrotalcita en referencia a uno de los politipos del mineral a base de [Mg-Al] correspondiente. (Ver "Layered Double Hydroxides: Present and Future", ed, V Rives, 2001 pub. Nova Science).
[0010] Por compuesto metálico mixto, se entiende que la estructura atómica del compuesto incluye los cationes de al menos dos metales diferentes distribuidos uniformemente en toda su estructura. El término compuesto metálico mixto no incluye mezclas de cristales de dos sales, donde cada tipo de cristal solo incluye un catión metálico. Los compuestos metálicos mixtos son típicamente el resultado de la coprecipitación de la solución de diferentes compuestos metálicos individuales en contraste con una mezcla física sólida simple de dos sales metálicas diferentes. Los compuestos metálicos mixtos como se usan en el presente documento incluyen compuestos del mismo tipo de metal pero con el metal en dos estados de valencia diferentes, por ejemplo, Fe (ii) y Fe (III), así como compuestos que contienen más de dos tipos de metales diferentes en un compuesto.
[0011] El compuesto metálico mixto puede comprender también material amorfo (no cristalino). Por el término amorfo se entiende fases cristalinas que tienen tamaños cristalinos por debajo de los límites de detección de las técnicas de difracción de rayos X, o fases cristalinas que tienen cierto grado de orden, pero que no exhiben un patrón de difracción cristalina y/o materiales amorfos verdaderos que exhibe un orden de corto alcance, pero no un orden de largo alcance.
[0012] Compuestos metálicos mixtos presentan desafíos únicos en el uso de material inorgánico para uso farmacéutico y, en particular, para la unión de fosfato y que están libres de Al.
[0013] Por ejemplo, el uso de compuesto metálico mixto para la consecución de los efectos terapéuticos de fosfato (u otro uso funcional farmacéutico) depende de procesos superficiales como la fisisorción (intercambio iónico) y la quimisorción (formación de un enlace químico) que es atípico para un medicamento; la actividad terapéutica de la mayoría de los fármacos se basa en compuestos orgánicos que suelen ser más solubles.
[0014] Aún más, se requieren dosis alta diaria y repetida a largo plazo (crónica) para pacientes renales pero su recuento total de píldoras diarias requiere una carga baja de la tableta debido a la ingesta de fluido restringido. En consecuencia, se requiere una dosis alta de sustancia farmacológica en el producto final (por ejemplo, tableta) y, por lo tanto, el producto final es muy sensible a las propiedades de la sustancia farmacológica de compuestos metálicos mixtos, a diferencia de las formulaciones normales. Esto significa que las propiedades de la tableta, incluidas las propiedades físicas clave, y los procesos de fabricación de la tableta, como la granulación, a menudo están influenciados principalmente por las propiedades de la sustancia activa del compuesto metálico mixto en lugar de únicamente por las de los excipientes. Para poder fabricar un producto farmacéutico que comprenda cantidades tan significativas de compuesto metálico mixto con el control y la consistencia necesarios para el uso farmacéutico, es esencial un medio para controlar una serie de propiedades químicas y físicas opuestas del compuesto metálico mixto.
[0015] Por lo tanto, teniendo en cuenta estos requisitos, la fabricación de tales materiales, en particular a gran escala, presenta problemas significativos se describen un número de estos problemas a continuación.
Envejecimiento
[0016] El proceso de envejecimiento (el crecimiento de cristalitos) aumenta generalmente con aumento del procesamiento y manipulación, (no intencionales) así como por el proceso por el que los cristalitos se cultivan intencionalmente por una combinación de agitación y tratamiento térmico de la suspensión de reacción antes del control de filtración y la prevención del crecimiento de los cristales puede ser difícil.
[0017] Las enseñanzas de compuestos metálicos mixtos MgAI que se fabrican en forma de edad para aplicaciones médicas tales como antiácidos, no abordan los problemas de la fabricación de compuestos metálicos mixtos no envejecidos (en una escala más grande), cuando la forma no envejecida es requerida, por ejemplo, para mantener la
actividad terapéutica de la unión de fosfato. Además, cuando se reemplaza Al por Fe, encontramos que el compuesto metálico mixto cambia propiedades tales como volverse más difícil de lavar y moler a escala comercial.
[0018] Los compuestos metálicos mixtos que contienen Al que se envejecen intencionalmente para aumentar el crecimiento de cristales se han fabricado previamente a gran escala. En contraste, no parece haber ejemplos de fabricación a gran escala de compuestos metálicos mixtos libres de Al no envejecidos.
[0019] El método descrito en el documento WO99/15189 se refiere a compuestos de Al metálicos mixtos libres e incluye ejemplos de materiales no envejecidos y envejecidos. Sin embargo, los productos divulgados en esta publicación se proporcionan a una escala relativamente pequeña, el documento WO99/15189 no aborda los problemas de provisión de producto a escala significativa al tiempo que evita el envejecimiento del producto.
[0020] La fabricación de compuestos no envejecidos metálicos mixtos Mg:Fe (Mg:Fe definidos por la relación molar de aquí en adelante) a gran escala es problemática por varias razones. Por ejemplo, la fabricación de compuestos metales mixtos de Mg.Fe sin envejecer es problemática cuando se utilizan métodos de filtración convencionales. El material no envejecido produce una caída de presión alta a través de la torta del filtro durante el aislamiento, lo que conduce a tasas de filtración bajas o pérdidas de rendimiento durante la filtración convencional. Además, estos tipos de compuestos de Mg:Fe metálicos típicamente tienen un tamaño de partícula de suspensión pequeño y, como tal, es difícil llevar a cabo el aislamiento mientras se minimiza el envejecimiento. Por ejemplo, las partículas pequeñas pueden dar lugar a mayores tiempos de procesamiento y/o problemas de manipulación.
[0021] Por otra parte, el exceso de procesamiento y manipulación (por ejemplo, fresado y secado excesivo) puede presentar cambios que son inaceptables en el Compuesto metal mixto final Mg:Fe. En particular con tales compuestos, es importante secar el material con cuidado ya que es fácil cambiar el área de superficie o el volumen de poro interno y, por lo tanto, cambiar la actividad terapéutica. Estas propiedades morfológicas típicas son características importantes que afectan tanto a la calidad del compuesto metálico final mixto como a los procesos de fabricación posteriores utilizados para producir el producto farmacéutico formulado final que contiene el compuesto metálico mixto.
[0022] Si los compuestos metálicos mixtos incorrectamente procesados pueden ser inaceptablemente difíciles. Esto puede conducir a problemas consecuentes de tasas de fresado disminuidas y mayor entrada de energía para lograr un tamaño de partícula dado. Este efecto de "golpe" en el procesamiento puede afectar el rendimiento del proceso y dar como resultado un trabajo excesivo del material y el consiguiente envejecimiento.
[0023] Los métodos para preparaciones a escala de laboratorio de MgFe LDH se describen en la técnica tales como US 4629626; Duan X, Evans DG, Chem. Commun., 2006,485-496; W. Meng y col., J Chem. Sci., Vol. 117, n.° 6 de noviembre de 2005, págs. 635-639; Carlino, Chemistry between the sheets, Chemistry in Britain, septiembre de 1997, págs. 59-62; Hashi y otros, Clays and Clay Minerals (1983) pp 152-15; Raki y col., Chem Mates., 1995, 7, 221-224; Ookubo y col., Langmuir (1993), 9, págs. 1418-14221; Zhang y col. Inorganic Materials Vol.4 Mar 132-138 (1997), Reichle, Solid States Ionics, 22, pp 135-141 (1986); Ulibarri et al, Kinetics of the Thermal Dehydration of some layered Hydrocycarbonates, Thermochimica Acta, vol. 135, págs. 231-236 (1988); Hansen y col., Applied Clay Science 10 (1995) pp 5-19.
[0024] Estos métodos sólo describen preparaciones a escala de laboratorio. Además, estos materiales se obtienen a través de un proceso que incluye una etapa de envejecimiento (es decir, un proceso deliberado de aumento del crecimiento de cristales que se logra típicamente calentando la suspensión de reacción durante un período prolongado de tiempo, tal como un proceso hidrotérmico). En general, los compuestos de la técnica anterior también contienen sustancialmente más de un tipo de anión en la región de la capa intermedia.
[0025] Los métodos para la fabricación a gran escala de hidrotalcitas de MgAl se describen en la técnica, tales como los documentos US-A-3650704, WO-A-2008/129034 y WO-A-93/22237. Sin embargo, estos describen el proceso para obtener materiales en forma envejecida que dan como resultado un tamaño de cristalita más grande (superior a 200 Angstrom) y no están libres de aluminio. En el proceso descrito en el documento WO2009/050469, los compuestos de hidróxido doble en capas preparados no se someten a una etapa de envejecimiento para evitar que los cristales crezcan en tamaño y mantener un área superficial elevada.
[0026] Los aspectos de la invención se definen en las reivindicaciones adjuntas.
ASPECTOS RESUMIDOS DE LA INVENCIÓN
[0027] En un aspecto, la presente invención proporciona un método de producción de un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+
tiene una relación molar de Mg2+ a Fe3+ de 2,5:1:1 a 1,5:1
con un contenido de aluminio de menos de 10.000 ppm, con un tamaño medio de cristal de menos de 20 nm (200 A) y con un área superficial de 40 a 80 m2 por gramo de compuesto metálico mixto, que comprende los pasos de:
(a) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 11, y en donde el Na2CO3 se proporciona en exceso de 0 a 4,0 moles que se requiere para completar la reacción
(b) someter la suspensión a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,03 a 1,6kW/m3
(c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10% en peso
(d) secar el producto crudo calentando el producto crudo a una temperatura no mayor de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco, y
(e) moler el producto crudo seco para proporcionar un área superficial de 40 a 80 m2 por gramo.
[0028] Las realizaciones preferidas se definen en las reivindicaciones dependientes.
[0029] En un aspecto, la presente divulgación proporciona un método de producción de un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+
que tiene un contenido de aluminio de menos de 10.000 ppm, que tienen un tamaño de cristal promedio de menos de 20 nm (200 A) que comprende los pasos de:
(a) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 11, y en donde el Na2CO3 se proporciona en exceso de 2,0 a 4,0 moles de lo requerido para completar la reacción
(b) someter la suspensión a mezcla en condiciones que proporcionen una potencia por unidad de volumen de 0,03 a 1,6kW/m3
(c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10% en peso
(d) secar el producto crudo ya sea por
(i) calentar el producto crudo a una temperatura no mayor de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco, o
(ii) exponer el producto crudo a un secado rápido en agua velocidad de evaporación de 500 a 50.000 kg de agua por hora por kg de producto seco.
[0030] En un aspecto, la presente divulgación proporciona un método de producción de un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+
que tiene un contenido de aluminio de menos de 10.000 ppm,
con un tamaño medio de cristal de menos de 20 nm (200A);
comprendiendo el método el paso de:
(a) combinar una sal Mg2+ y una sal Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura entre 15 y 30°C, y:
(i) en donde el pH de la suspensión se mantiene de 9,5 a menos de 9,8, y en donde el Na2CO3 se proporciona en un exceso de más de 10 a no más de 5,0 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene a de 9,5 a menos de 10, y en el que el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 4,0 moles que se requiere para completar la reacción; o
en donde el pH de la suspensión se mantiene entre 9,5 y no más de 10,1, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 2,7 moles que el requerido para completar la reacción; o
(iv) en donde el pH de la suspensión se mantiene a de 9,5 a 10,5, y en el que el Na2CO3 se proporciona en un exceso de desde mayor que 1,0 a no más de 2,0 moles que se requiere para completar la reacción; o
(v) en donde el pH de la suspensión se mantiene entre más de 9,5 y no más de 11, y en donde el Na2CO3 se proporciona en un exceso de 00 a no más de 1,0 moles que el requerido para completar el reacción;
o el método que comprende la etapa de:
(b) combinar una sal Mg2+ y una sal Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura de 30 a 60°C, y:
(i) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 11, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles de lo requerido para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción.
[0031] En un aspecto, la presente descripción proporciona un método para producir un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+
con un contenido de aluminio de menos de 10.000 ppm, con un tamaño de cristal promedio de menos de 20 nm (200 A), comprendiendo el método la etapa de:
(a) combinar una sal Mg2+ y una sal Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura entre 15 y 30°C, y:
(i) en donde el pH de la suspensión se mantiene de 9,5 a menos de 9,8, y en donde el Na2CO3 se proporciona en un exceso de más de 20 a no más de 4,0 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene a de 9,5 a menos de 10,3, y en el que el Na2CO3 se proporciona en un exceso de más de 2,0 a menos de 4,0 moles que se requiere para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9.8 y no más de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a menos de 2.7 moles que el requerido para completar la reacción; o
(iv) en donde el pH de la suspensión se mantiene en más de 9.8 a menos de 10,3, y en donde el Na2CO3 se proporciona en un exceso de 1,0 a menos de 4,0 moles que el requerido para completar la reacción,
o el método que comprende la etapa de:
(b) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura de 30 a 65°C, y:
(i) en donde el pH de la suspensión se mantiene a de 9,5 a no más de 10,5, y en el que el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles que se requiere para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene entre 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción.
[0032] En un aspecto, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+ que tiene un contenido de aluminio de menos de 10.000 ppm, con un tamaño medio de cristal de menos de 20 nm (200 A), en donde el compuesto se obtiene u obtiene por un método que comprende las etapas de
(a) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 11, y en donde el Na2CO3 se proporciona en exceso de 0 a 4,0 moles que se requiere para completar la reacción
(b) someter la suspensión a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,03 a 16kW/m3
(c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10% en peso
(d) secar el producto crudo ya sea por
(i) calentar el producto crudo a una temperatura no mayor de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco, o
(ii) exponer el producto crudo a un secado rápido en agua velocidad de evaporación de 500 a 50.000 kg de agua por hora por kg de producto seco.
[0033] En un aspecto, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+ que tiene un contenido de aluminio de menos de 10.000 ppm, con un tamaño medio de cristal de menos de 20 nm (200 A), en donde el compuesto se obtiene por un método que comprende los pasos de:
(a) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 11, y en donde el Na2CO3 se proporciona en exceso de 2,0 a 4,0 moles que se requiere para completar la reacción
(b) someter la suspensión a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 003 a 1,6kW/m3
(c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10% en peso
(d) secar el producto crudo ya sea por
(i) calentar el producto crudo a una temperatura no mayor de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco, o
(ii) exponer el producto crudo a un secado rápido en agua velocidad de evaporación de 500 a 50.000 kg de agua por hora por kg de producto seco.
[0034] En un aspecto, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+ que tiene un contenido de aluminio de menos de 10.000 ppm, con un tamaño medio de cristal de menos de 20 nm (200 A), en donde el compuesto se obtiene u obtiene por un método que comprende los pasos de:
(a) combinar una sal Mg2+ y una sal Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura entre 15 y 30°C, y:
(i) en donde el pH de la suspensión se mantiene entre 9,5 y menos de 9,8, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 50 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene entre 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 4,0 moles de lo que se requiere para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre 9,5 y no más de 10,1, y en donde el Na2CO3 se proporciona en un exceso de más de 10 a no más de 2,7 moles de lo que se requiere para completar el reacción; o
(iv) en donde el pH de la suspensión se mantiene entre 9,5 y 10 5, y en el que el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 2,0 moles que el requerido para completar el reacción; o
(v) en donde el pH de la suspensión se mantiene entre más de 9,5 y no más de 11, y en donde el Na2CO3 se proporciona con un exceso de entre 0,0 y no más de 1,0 moles que el requerido para completar la reacción
o por el método que comprende la etapa de:
(b) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura de 30 a 60°C y
(i) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 11, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles de lo requerido para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción
[0035] En un aspecto, la presente descripción proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+ que tiene un contenido de aluminio de menos de 10.000 ppm, que tiene un tamaño de cristal promedio de menos de 20 nm (200 A), en el que el compuesto es obtenido u obtenible por un método que comprende los pasos de:
(a) combinar una sal Mg2+ y una sal Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura entre 15 y 30°C, y:
(i) en donde el pH de la suspensión se mantiene entre 9,5 y menos de 98, y en el que el Na2CO3 se proporciona en un exceso de más de 2,0 a no más de 4,0 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene a de 9,5 a menos de 10,3, y en el que el Na2CO3 se proporciona en un exceso de más de 2,0 a menos de 4,0 moles que se requiere para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9.8 y no más de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a menos de 2,7 moles que el requerido para completar la reacción; o
(iv) en donde el pH de la suspensión se mantiene a más de 9,8 a menos de 10,3, y en donde el Na2CO3 se proporciona en un exceso de 1,0 a menos de 4,0 moles que el requerido para completar la reacción;
o mediante el método que comprende la etapa de:
(b) combinar una sal Mg2+ y una sal Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura de 30 a 65°C y
(i) en donde el pH de la suspensión se mantiene a de 9,5 a no más de 10,5, y en el que el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles que se requiere para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene entre 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción.
[0036] En un aspecto, la presente descripción proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos
la relación molar de Mg2+ a Fe3+ es 25:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y la partícula promedio d50
el tamaño del compuesto metálico mixto es inferior a 300 |jm.
[0037] En un aspecto la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5: 1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el volumen de poros de agua del compuesto metálico mixto es de 0,25 a 0,7 cm 3/g de compuesto metálico mixto.
[0038] En un aspecto la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 25:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio del cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el contenido de sulfato entre capas del compuesto es de 1,8 a 5% en peso.
[0039] En un aspecto la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5: 1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el contenido de sulfato entre capas del compuesto es de 18 a 3,2% en peso.
[0040] En un aspecto la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5: 1 a 1,5:1,
El compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
El tamaño promedio de los cristales del compuesto metálico mixto es inferior a 20 nm (200 A),
y el contenido de sulfato entre capas del compuesto es de 1,8 a 5% en peso.
[0041] En un aspecto la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5: 1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es inferior a 20 nm (200 A),
y el contenido de sulfato entre capas del compuesto es de 1,8 a 3,2% en peso.
[0042] En un aspecto la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5: 1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño de cristal promedio del compuesto metálico mixto es menos de 20 nm (200 A),
y el área de superficie es de 80 a 145 m2 por gramo de compuesto.
[0043] En un aspecto, la presente invención proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+, en el que
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el mezclado el compuesto metálico tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), el área de superficie es de 40 a 80 m2 por gramo de compuesto,
y opcionalmente: el tamaño de partícula promedio d50 del compuesto metálico mixto es inferior a 100 pm, el volumen de poros de agua del compuesto metálico mixto es de 0,25 a 0,7 cm 3/g de compuesto metálico mixto, y/o el contenido de sulfato entre capas del compuesto es de 2 a 5% en peso.
[0044] En un aspecto, la presente invención proporciona un compuesto metálico mixto como se describe en el presente documento para su uso como un medicamento.
[0045] En un aspecto, la presente divulgación proporciona un compuesto metálico mixto como se describe en este documento para fosfatado de unión.
[0046] En un aspecto, la presente invención proporciona un compuesto metálico mixto como se describe en el presente documento para su uso en el tratamiento de la hiperfosfatemia
[0047] En un aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto metálico mixto como se describe en el presente documento y opcionalmente uno o más adyuvantes, excipientes, diluyentes o vehículos farmacéuticamente aceptables.
Algunas ventajas
[0048] El presente método proporciona un proceso que puede ser operado a gran escala para proporcionar un medicamento de composición constante, que es estable durante el almacenamiento y se puede formular fácilmente y/o envasar enlace a fosfato farmacéutico. Además, el presente método proporciona el control de propiedades clave como el tamaño promedio de cristal, el tamaño de partícula, el área de superficie, otros parámetros de morfología (como el volumen de poros) y el grado de hidratación, todos los cuales son importantes para dicha fabricación.
[0049] Los compuestos metálicos mixtos sin Al que contienen Fe y Mg tienen típicamente una estructura de tipo arcilla. Esto presenta limitaciones en vista de la difícil filtración de tales productos que a su vez afectan la viabilidad de un proceso controlado
[0050] El presente método proporciona un proceso que permite la fabricación de compuestos metálicos mixtos consistentes "libres de Al" adecuados para uso en formulaciones de productos finales (por ejemplo, formulaciones de tabletas, etc.). El efecto terapéutico de los productos finales y la capacidad de procesar los compuestos metálicos mezclados en un producto final de manera consistente, dependen de las propiedades físicas (es decir, de tamaño de partícula y cristalito) y químicas (es decir, composición) del compuesto metálico mixto. El presente proceso proporciona un método para una fabricación consistente de compuestos metálicos mixtos "libres de Al" de grado farmacéutico con un tamaño consistente de partículas y cristalitos.
[0051] En un aspecto, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+, donde la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1, el mezclado El compuesto metálico tiene un contenido de aluminio de menos de 10.000 ppm, el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el contenido de sulfato entre capas del compuesto es de 1,8 a 5% en peso (como de 1,8 a 3,2% en peso).
[0052] Para los compuestos metálicos mixtos, el mantenimiento de la relación objetivo de metal molar (Mg:Fe) durante la reacción, mientras que satisfacen los criterios anteriores es difícil, ya que esta se ve afectada por la forma en que se procesa el material. Descubrimos que la estequiometría correcta no solo está determinada por las relaciones correctas de los materiales de partida, sino también por el pH de la reacción; es decir, cuando un pH está por debajo de pH 9,5, puede producirse una precipitación incompleta de magnesio y un pH demasiado alto (es decir, por encima de pH 11) corre el riesgo de perder hierro.
[0053] Compuestos metálicos mixtos pueden tener más de un tipo de anión dentro de la región entre capas. Esto puede introducir impurezas que no son deseables cuando se considera el uso farmacéutico y descubrimos que también puede afectar la actividad terapéutica (de unión al fosfato). También hemos encontrado, sorprendentemente, que el tipo y la cantidad de aniones presentes en la región de la capa intermedia tiene un efecto marcado en el tiempo necesario para completar la separación y el lavado del producto no envejecido, particularmente para la fabricación a escala comercial. Por ejemplo, hemos encontrado que a niveles bajos (por debajo de 1,8% en peso) de sulfato entre capas, los tiempos de separación y lavado aumentan significativamente.
[0054] Se encontró que cuando la cantidad de sulfato de capa intermedia se mantiene desde 1,8 hasta 5% en peso (tal como 1,8 a 3,2% en peso) de enlace de fosfato de más de 0,60 mmol de compuesto de fosfato/g puede obtenerse mientras se mantiene bajo filtración y tiempos de lavado.
[0055] La combinación de los parámetros del proceso del presente método permite la preparación de compuestos de metales mezclados que hayan controlado los niveles de sulfato (SO4) en la región entre capas.
[0056] SO4 soluble en forma de sal Na2SO4 se puede eliminar fácilmente por lavado, mientras que el sulfato de capa intermedia no se puede eliminar por lavado con agua.
Descubrimos que el sulfato entre capas podría reducirse sin aumentar necesariamente los tiempos de filtración y lavado volviendo a colocar el compuesto seco en una solución que contenga carbonato que permita el intercambio iónico; sin embargo, esto significó una reacción adicional, una etapa de aislamiento y secado, y generalmente condujo a una disminución en la unión de fosfato. Además, esta ruta daría lugar a un tiempo total de fabricación más largo. Por lo tanto, esta ruta para controlar el sulfato entre capas es menos preferida. Alternativamente, el sulfato entre capas puede reducirse lavando o volviendo a colocar la torta de filtración después del aislamiento con una solución que contenga carbonato en lugar de agua. De nuevo, esto conduciría a una etapa de proceso adicional y es menos preferido.
[0057] Se encontró que mediante el control de los parámetros del proceso como se describe en el presente documento, se puede preparar un compuesto metálico mixto que tiene bajos niveles de impurezas y, en particular impurezas de metales pesados, sin la necesidad de realizar la purificación para eliminar tales impurezas la presente divulgación puede proporcionar un proceso para preparar un compuesto metálico mixto que tenga un contenido de plomo de menos de 1 ppm y/o un contenido de cromo de menos de 30 ppm y/o un contenido total de metales pesados de menos de 10 ppm y/o un contenido de sodio expresado como Na2O de menos de 0,5% en peso. En un aspecto, el compuesto metálico mixto tiene un contenido total de metal pesado de menos de 25 ppm, preferiblemente menos de 10 ppm. Por ejemplo, la presente descripción puede proporcionar un proceso para preparar un compuesto metálico mixto que tiene un contenido total de metal pesado de menos de 15 ppm, un contenido de plomo inferior a 10 ppm, un nivel de cromo menos de 35 ppm y un contenido de sodio (expresada como Na2O) de menos de 1% en peso.
[0058] Contenido de metales pesados al que se hace referencia en el presente documento son el grupo que consiste en As, Cd, Pb, Hg, Sb, Mo y Cu Por lo tanto, la referencia a un total de contenido de metales pesados se entiende que significa el contenido combinado de As, Cd, Pb, Hg, Sb, Mo y Cu.
[0059] WO99/15189 enseña la preparación de una suspensión a pH por encima de 10 utilizando una relación de 5 moles de Na2CO3: 12 de NaOH mol, lo que equivale a un exceso de 4 moles de Na2CO3 que el requerido para completar la ecuación de reacción:
4MgSO4 Fe2(SO4)3 12NaOH 5Na2CO3->
Mg4Fe2(OH)12CO3.nH2O 7Na2SO4 4Na2CO3
Hemos encontrado que este exceso de 4 moles de Na2CO3 no se prefiere, especialmente cuando se precipitó a un pH de más de 10 y a temperatura ambiente. Descubrimos que esta combinación da como resultado una solubilidad reducida del carbonato en la solución reactiva a las temperaturas de reacción deseadas, proporciona una filtración pobre y tiempos de lavado. Hemos encontrado que al preparar material no envejecido con una relación molar de Mg:Fe 2:1 según el método del documento WO99/15189, este material era más difícil de separar y lavar cuando se fabricaba a escala mientras se mantenía una buena unión a fosfato. Esto dio como resultado la pérdida de lotes como resultado de que el material estaba fuera de especificación.
[0060] El carbonato de sodio no sólo proporciona el carbonato para los sitios de intercambio aniónico, pero también actúa como un tampón de pH que ayuda a controlar el pH durante la precipitación. La capacidad de mantener un pH de precipitación preciso aumenta considerablemente cuando está presente Na2CO3. Sin embargo, también hemos encontrado que la tasa de filtración aumenta significativamente cuando el Na2CO3 se reduce de un exceso de 2,7 moles a un exceso de cero. Una alta tasa de filtración es ventajosa cuando se busca fabricar una forma no envejecida de los compuestos de metales mixtos de Mg Fe. Dichos materiales pueden ser difíciles de filtrar. La disminución adicional del Na2CO3 adicionalmente, como por debajo del exceso de 2,7 moles, puede dar como resultado un control de pH menos preciso, así como aumentar el nivel de aniones sulfato presentes en la región de la capa intermedia. Como consecuencia de lo anterior, hemos identificado que existe una compleja interrelación entre el pH, el exceso de
moles de Na2CO3 y la temperatura a la que se mantiene la suspensión, todo lo cual es importante para mantener una buena unión y filtración de fosfato y/o lavado veces. En particular, hemos determinado que para obtener una unión a fosfato por encima de 0,60 mmol de fosfato/g de compuesto y mantener una buena filtración y tiempo de lavado, se prefiere producir una suspensión espesa, en la que la temperatura se mantenga entre 15 y 30°C
(i) en donde el pH de la suspensión se mantiene de 9,5 a menos de 9,8, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 5,0 moles que el requerido para completar la reacción; o (ii) en donde el pH de la suspensión se mantiene a de 9,5 a menos de 10, y en el que el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 4,0 moles que se requiere para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre 9,5 y no más de 10,1, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 2,7 moles que el requerido para completar la reacción; o
(iv) en donde el pH de la suspensión se mantiene a de 9,5 a 10,5, y en el que el Na2CO3 se proporciona en un exceso de desde mayor que 1,0 a no más de 2,0 moles que se requiere para completar la reacción; o en donde el pH de la suspensión se mantiene entre más de 9,5 y no más de 11, y en donde el Na2CO3 se proporciona con un exceso de entre 0,0 y no más de 1,0 moles que el requerido para completar la reacción
[0061] Alternativamente, una sal Mg2+ y una sal Fe3+ pueden combinarse con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura de 30 a 60°C
(i) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 11, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2 moles que el requerido para completar la reacción; o (ii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles de lo requerido para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción
[0062] Además, se ha encontrado que con el fin de evitar la presencia de fases cristalinas adicionales en el compuesto (es decir, fases diferentes que las de tipo hidrotalcita como se detecta por polvo de difracción de rayos X) es necesario para lavar el producto a un grado tal que el sulfato no unido (SO4), es decir, en forma de sulfato de sodio (Na2SO4) se mantiene por debajo del 1,5% en peso (cuando se expresa como Na2SO4) y preferiblemente menos del 1% en peso (cuando se expresa como SO4). Por el contrario, esto solo se puede lograr cuando se mantiene una pequeña cantidad de sulfato entre capas para permitir una filtración y lavado efectivos a escala comercial.
[0063] El presente método es un proceso de co-precipitación. Tales procesos fomentan la formación de diferentes fases cristalinas, además de la fase de hidrotalcita. Para su uso como un activo en formulaciones farmacéuticas, existe el requisito de poder identificar y controlar la fase de interés. El presente método proporciona la preparación de compuestos metálicos mixtos que contienen menos de (o están sustancialmente libres de) cualquier otra fase cristalina según lo determinado por la ausencia de líneas de difracción de XRD, excepto las atribuidas a una fase de hidrotalcita. La fase de hidrotalcita tuvo el siguiente análisis de difracción de rayos X por difracción: dA (espacios 'd') 7,74*, 3,86*, 3,20, 2,62*, 2,33*, 1.97*, 1,76, 1,64, 1,55*, 1,52*, 1.48, 1.44*, 1.40, de los cuales los picos marcados * son los ocho picos más intensos típicamente vistos en las muestras no envejecidas. Los cinco picos restantes solo se resuelven en muestras más cristalinas, típicamente como resultado del envejecimiento.
[0064] En resumen, hemos encontrado que un proceso total de producción (de la reacción a secado) se proporciona como para evitar el crecimiento del tamaño de los cristalitos (por encima de tamaño de cristal medio 200 A) a fin de mantener la actividad de unión sin significativamente fosfato obstaculizando el proceso de aislamiento y lavado del compuesto. Esto fue logrado por control cuidadoso de las condiciones del proceso y una selección específica de los mismos, como mediante el control del sulfato entre capas del 1,8 al 5% en peso (como del 1,8 al 3,2% en peso) que a su vez se puede controlar mediante la selección del exceso de Na2CO3, reacción pH y suspensión de la reacción de temperatura.
[0065] En un aspecto, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+, donde la relación molar de Mg2+ a Fe3+ es 1,5:1 a 2,5:1, el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm, el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el tamaño de partícula promedio d50 del compuesto metálico mixto es menos de 300 pm. Sorprendentemente, hemos descubierto que este compuesto no envejecido con un rango de tamaño de cristal promedio y con un tamaño de partícula fresado inferior a 300 micras, tiene las ventajas de una buena unión controlada de fosfato (por encima de 0,60 mmol de fosfato/g de compuesto) mientras mantiene una baja liberación de magnesio (menos de 0,2 mmol de compuesto de magnesio/g). Por encima de 300 micras de tamaño de partícula, hemos encontrado que la unión de fosfato disminuye notablemente y la liberación de magnesio aumenta a más de 0,2 mmol de compuesto de magnesio/g.
[0066] Se ha encontrado que un tamaño de cristal promedio del compuesto metálico mixto de menos de 20 nm (200 A) y área de superficie alta (80-145 m2/g) puede ser fabricado usando un proceso que comprende una etapa de secado
corto de residencia de tal manera que el material resultante tiene un tamaño de cristal medio pequeño y un área de superficie alta, pero también tiene una capacidad de unión a fosfato más alta, así como una menor liberación de magnesio en comparación con un tamaño de partícula promedio d50 similar al del material de área de superficie baja (40-80 m2/g); incluso cuando el material no se muele más. El requisito de no fresar tiene la ventaja de reducir los pasos de procesamiento. Una ventaja adicional es que dicho material puede ser adecuado para procesos de formación de tabletas sin la necesidad de granulación húmeda. Una ventaja adicional es que el material fabricado a través de la ruta de residencia corta puede estar expuesto a temperaturas superiores a 150°C porque el tiempo de residencia (menos de 5 minutos) del producto en el secador es generalmente demasiado corto para permitir cualquier descomposición del compuesto.
[0067] El compuesto metálico mixto que tiene un tamaño medio de cristal de 10 a 20 nm (100 a 200 A) y el área superficial de 40-80 m2/g tiene la ventaja de una buena estabilidad, en particular fosfato de unión, en el almacenamiento. Este producto tiene el beneficio adicional de proporcionar un tamaño de tableta más pequeño (es decir, menos de 500 mm3 para un compuesto de 500 mg) mejorando así la carga de la píldora de la tableta; un problema frecuente en el tratamiento de pacientes renales. El material de superficie de 40-80 m2/g que requirió micronización puede fabricarse a escala comercial, incluida el fresado del material, con un impacto mínimo sobre el envejecimiento del material, como se refleja en el mantenimiento de un tamaño de cristal promedio pequeño por debajo de 200 A Si el tamaño del cristalito es inferior a 100 A presenta dificultades de fresado a partículas de tamaño pequeño, por ejemplo, problemas con impurezas de metales traza, velocidad de fresado y descomposición del producto y secado excesivo del producto. Además, un beneficio sorprendente adicional es que tales materiales tampoco exhiben una reducción significativa en la tasa de absorción de fosfato, a pesar de las áreas superficiales más bajas. Esta faceta puede ser importante cuando se consideran dichos materiales para aplicaciones farmacéuticas en las que la unión del fosfato debe ser rápida, como el cuidado renal. Hemos encontrado que el material descrito anteriormente se une al 80% de fosfato en 10 minutos (Método de prueba 3).
[0068] En cuanto al producto de 10 a 20 nm (100 a 200 A) tamaño de cristal medio y 40-80 m2/g de superficie, el producto de bajo área superficial y volumen de poros bajo por el agua 0,3 a 0,65 cm 3/g tiene el beneficio adicional de proporcionar un tamaño de tableta más pequeño (es decir, menos de 500 mm3 para un compuesto de 500 mg) mejorando así la carga de la píldora de la tableta; problemas frecuentes en el tratamiento de pacientes renales. Además, un material de mayor densidad es más adecuado para la fabricación por granulación húmeda de tabletas compactas.
[0069] Si el producto se seca a menos del 85% en peso de contenido de sólidos secos, se pueden observar problemas de almacenamiento debido a la desorción de agua. Si el producto se seca a menos del 80% en peso de contenido de sólidos secos, el fresado puede ser problemática. Si el producto se seca a más del 99% en peso, la unión de fosfato puede reducirse. Si el producto está demasiado seco, también se pueden observar problemas de almacenamiento debido a la adsorción de agua. Por lo tanto, en una realización, el producto se seca de modo que tenga 80% en peso a 99% en peso de contenido sólido seco, preferiblemente 85% en peso a 99% en peso.
[0070] En un aspecto, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+, donde la relación molar de Mg2+ a Fe3+ es 1,5:1 a 2,5:1, el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm, el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el volumen de poros de agua del compuesto metálico mixto es de 0,3 a 0,65 cm 3/g de compuesto metálico mixto. Sorprendentemente, hemos encontrado que este compuesto de bajo volumen de poros tiene la ventaja de una buena unión de fosfato que esencialmente no cambia tras el almacenamiento durante períodos de hasta años, lo que lo hace viable como material farmacéuticamente activo. Se puede esperar típicamente que se requieran volúmenes de poro significativamente más altos para lograr tal estabilidad.
[0071] Como se usa en el presente documento, 'volumen de poros de agua' el término se refiere al volumen de poros tal como se determina de acuerdo con el Método de Ensayo 15.
[0072] Para facilitar la referencia, estos y otros aspectos de la presente invención se describen ahora en encabezados de sección apropiados. Sin embargo, las enseñanzas de cada sección se pueden combinar y no se limitan necesariamente a cada sección en particular.
ASPECTOS PREFERIDOS
[0073] Como se describe en el presente documento la presente invención proporciona un método para producir un compuesto metálico mixto como se define en la reivindicación 1.
[0074] Se entenderá por un experto en la técnica que por "tamaño de cristal medio" se entiende el tamaño de cristal, medido de acuerdo con el Método de Ensayo 2.
[0075] En un aspecto preferido, en la etapa (a) una sal de Mg2+ y una sal de Fe3+ se combinan con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 10,5. Preferiblemente, el pH de la suspensión se mantiene entre 9,5 y menos de 10,1, preferiblemente el pH de la suspensión se mantiene entre
9,5 y menos de 10. Preferiblemente, el pH de la suspensión se mantiene entre 9,5 y menos de 9,8. Preferiblemente, el pH de la suspensión se mantiene entre 9,6 y 9,9. Más preferiblemente, el pH de la suspensión se mantuvo a aproximadamente 9,8.
[0076] En un aspecto preferido, en la etapa (a) una sal Mg2+ y una sal Fe3+ se combinan con Na2CO3 y NaOH para producir una suspensión, en donde el Na2CO3 se proporciona en un exceso de 2,0 a menos de 4,0 moles, preferiblemente a un exceso de 2,7 a menos de 4,0 moles, preferiblemente a un exceso de 2,7 a menos de 32 moles, preferiblemente a un exceso de 2,7 a menos de 3,0 moles. Más preferiblemente, el Na2CO3 se mantiene en un exceso de aproximadamente 2,7 moles.
[0077] En un aspecto preferido, en la etapa (a), una sal de Mg2+ y una sal de Fe3+ se combinan con Na2CO3 y NaOH para producir una suspensión, en la que la suspensión se mantiene a una temperatura entre 15 y 30°C
(i) en donde el pH de la suspensión se mantiene de 9,5 a menos de 9,8, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 5,0 moles que el requerido para completar la reacción; o (ii) en donde el pH de la suspensión se mantiene entre 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 10 a no más de 4,0 moles que el requerido para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre 9,5 y no más de 10,1, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 2,7 moles que el requerido para completar la reacción; o
(iv) en donde el pH de la suspensión se mantiene de 9,5 a 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 2,0 moles de lo que se requiere para completar la reacción, o
(v) en donde el pH de la suspensión se mantiene entre más de 9,5 y no más de 11, y en donde el Na2CO3 se proporciona en un exceso de entre 0,0 y no más de 10 moles de lo requerido para completar la reacción.
[0078] En un aspecto preferido, en la etapa (a) una sal Mg2+ y una sal Fe3+ se combinan con Na2CO3 y NaOH para producir una suspensión, en el que la suspensión se mantiene a una temperatura entre 15 y 30°C
(i) en donde el pH de la suspensión se mantiene de 9,5 a menos de 9,8, y en el que el Na2CO3 se proporciona en un exceso de más de 2,0 a no más de 4,0 moles que el requerido para completar la reacción; o (ii) en donde el pH de la suspensión se mantiene a de 9,5 a menos de 10,3, y en el que el Na2CO3 se proporciona en un exceso de más de 2,0 a menos de 4,0 moles que se requiere para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9,8 y no más de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a menos de 2,7 moles que el requerido para completar la reacción; o
(iv) en donde el pH de la suspensión se mantiene a más de 9,8 a menos de 10,3, y en donde el Na2CO3 se proporciona en un exceso de 10 a menos de 4,0 moles que el requerido para completar la reacción;
[0079] En un aspecto preferido, en la etapa (a) una sal Mg2+ y una sal Fe3+ se combinan con Na2CO3 y NaOH para producir una suspensión, en el que la suspensión se mantiene a una temperatura de 30 a 60°C
(i) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 11, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2 moles que el requerido para completar la reacción; o (ii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles de lo requerido para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción.
[0080] En un aspecto preferido, en la etapa (a) una sal Mg2+ y una sal Fe3+ se combinan con Na2CO3 y NaOH para producir una suspensión, en el que la suspensión se mantiene a una temperatura de 30 a 65°C
(i) en donde el pH de la suspensión se mantiene a de 9,5 a no más de 10,5, y en el que el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles que se requiere para completar la reacción; o (ii) en donde el pH de la suspensión se mantiene entre 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción.
[0081] En un aspecto preferido, en la etapa (b) la suspensión se somete a la mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,03 a 1,6KW/m3. En un aspecto preferido, en la etapa (b) la suspensión se somete a mezcla en condiciones proporcionando una potencia por unidad de volumen de 0,03 a 0,5kW/m3. En un aspecto preferido, en la etapa (b) la suspensión se somete a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,05 a 0,5kW/m3.
[0082] En un aspecto preferido, en la etapa (b) la suspensión se controla para una distribución de tamaño de partícula
d50 (psd) de al menos 40 |jm. Preferiblemente, la suspensión se controla a una d50 psd de más de 40 |jm. Preferiblemente, la suspensión se controla a una d50 psd de al menos 50 jm . Preferiblemente, la suspensión se controla a una d50 psd de al menos 60 jm . Más preferiblemente, la suspensión se controla a una d50 psd de al menos 70 jm . Preferiblemente, la suspensión se controla a una d50 psd de más de 70 jm . El d50 psd de la suspensión es tal como se mide de acuerdo con el Método de Prueba 9 aquí.
[0083] Como se usa en el presente documento, la 'distribución del tamaño de partícula' término se refiere al d50 o distribución media de tamaño de partícula tal como se determina de acuerdo con el Método de Ensayo 24. D50 se refiere al percentil 50 de ese método de ensayo.
[0084] En un aspecto preferido, en la etapa (b) la suspensión se controla para una psd d50 de al menos 40 jm después de la adición de los reactivos y después de un tiempo de retención inicial de 30 minutos para alcanzar la distribución óptima de tamaño de partícula. Preferiblemente, la suspensión se controla a una d50 psd de al menos 50 jm después de la adición de los reactivos y después de un tiempo de retención inicial de 30 minutos. Preferiblemente, la suspensión se controla a una d50 psd de al menos 60 jm después de la adición de los reactivos y después de un tiempo de retención inicial de 30 minutos. Más preferiblemente, la suspensión se controla a una d50 psd de al menos 70 jm después de la adición de los reactivos y después de un tiempo de retención inicial de 30 minutos.
[0085] En un aspecto preferido, el tiempo de retención de la suspensión antes del aislamiento, tal como antes del paso (c), es menos de 16 horas, preferiblemente menos de 12 horas. Preferiblemente, el tiempo de retención es más de 30 minutos. En un aspecto preferido, el tiempo de retención de la suspensión antes del aislamiento, tal como antes del paso (c), de 30 minutos a 16 horas. En un aspecto preferido, el tiempo de retención de la suspensión antes del aislamiento, tal como antes del paso (c), de 30 minutos a 12 horas. Si el tiempo de retención aumenta a más de 16 horas, el tamaño del cristalito puede aumentar y/o cambiar el tamaño de las partículas.
[0086] En el paso (c) del presente método, el compuesto metálico mixto se separa de la suspensión, para obtener un producto en bruto que tiene un contenido sólido seco de al menos 10% en peso. Preferiblemente, el compuesto metálico mixto se separa de la suspensión, para obtener un producto bruto que tiene un contenido sólido seco de al menos 15% en peso. Más preferiblemente, el compuesto metálico mixto se separa de la suspensión, para obtener un producto bruto que tiene un contenido sólido seco de al menos 20% en peso.
[0087] En el paso (d) del presente método el producto en bruto se seca ya sea por
(i) calentar el producto crudo a una temperatura no mayor de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco. (según la invención), o
(ii) exponer el producto crudo a un secado rápido a una velocidad de evaporación de agua de 500 a 50.000 kg de agua por hora por kg de producto seco (no según la invención).
[0088] Se entenderá por un experto en la técnica que la referencia a "calentar el producto en bruto a una temperatura de no más de X°C" se refiere al calentamiento del producto de tal manera que la temperatura de la masa del producto no es mayor que X°C. Se entenderá que la temperatura a la que está expuesto el producto, por ejemplo, la temperatura del tambor en el caso del secado del tambor, o la temperatura de la cubierta del producto puede ser mayor que X°C cuando la temperatura total del producto es X°C.
[0089] Se entenderá por un experto en la técnica que la referencia a una tasa de evaporación de agua a una velocidad de agua kg por hora por kg de producto seco, debe ser medida de acuerdo con el Método de Ensayo 18.
[0090] En el método inventivo, se sigue el paso d (i), es decir, en el paso (d) el producto crudo se seca calentando el producto crudo a una temperatura no mayor de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco.
[0091] En un procedimiento alternativo (no de acuerdo con la invención), paso d (ii) es seguido, que es en el paso (d) el producto bruto se seca exponiendo el producto en bruto a parpadear de secado o secado por pulverización a una evaporación de agua tasa de 500 a 50.000 kg de agua por hora por kg de producto seco.
[0092] Preferiblemente, cuando se sigue el paso d (i), el producto en bruto se seca por calentamiento del producto en bruto a una temperatura de no más de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1 kg de agua por hora por kg de producto seco, más preferiblemente una velocidad de evaporación de agua de 0,05 a 0,5 kg de agua por hora por kg de producto seco, incluso más preferiblemente una velocidad de evaporación de agua de 0,09 a 0,5 kg de agua por hora por kg de producto seco, lo más preferido un agua velocidad de evaporación de 0,09 a 0,38 kg de agua por hora por kg de producto seco.
[0093] Preferiblemente, cuando se sigue el paso d (i), el producto en bruto se seca por calentamiento del producto en bruto a una temperatura de no más de 150°C, tal como no mayor que 140°C, tal como no más de 130°C, como no más de 120°C, como no más de 110°C, como no más de 100°C, como no más de 90°C, como entre 60 y 150°C, como
desde 70 a 150°C, como 60 a 140°C, como 70 a 140°C, como 60 a 130°C, como 70 a 130°C, como 60 a 120°C, como de 70 a 120°C, como de 60 a 110°C, como de 70 a 110°C, como de 60 a 100°C, como de 70 a 100°C, como de 60 a 90°C, tal como de 70 a 90°C Preferiblemente, cuando se sigue el paso d (i), el producto crudo se seca calentando el producto crudo a una temperatura de 35 a 150°C, tal como de 35 a 140°C, como de 35 a 130°C, como de 35 a 120°C, como de 35 a 110°C, como de 35 a 100°C, como de 35 a 90°C, como de 35 a 80°C, como de 35 a 70°C, tal como de 35 a 60°C, tal como de 35 a 50°C. Preferiblemente, cuando se sigue el paso d (i), el producto crudo se seca calentando el producto crudo a una temperatura de más de 40 a 150°C, tal como de más de 40 a 140°C, tal como de más de 40 a 130°C, tal como de más de 40 a 120°C, tal como de más de 40 a 110°C, tal como de más de 40 a 100°C, tal como de más de 40 a 90°C, tal como desde más de 40 hasta 80°C, tal como desde más de 40 hasta 70°C, como desde más de 40 hasta 60°C, como desde más de 40 hasta 50°C. Hemos encontrado que es particularmente preferido calentar el producto crudo a una temperatura no más de 90°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco. En este aspecto, el tamaño de cristal promedio no aumenta significativamente durante el secado y se pueden proporcionar los tamaños de cristal promedio ventajosos descritos aquí.
[0094] Preferiblemente, cuando se sigue el paso d (i), el producto en bruto se seca por exposición del producto bruto a una temperatura de no más de 150°C, exponiendo preferiblemente el producto en bruto a una temperatura de no más de 140°C, preferiblemente exponiendo el producto crudo a una temperatura no más de 130°C, preferiblemente exponiendo el producto crudo a una temperatura no más de 120°C, preferiblemente exponiendo el producto crudo a una temperatura no más de 110°C, preferiblemente exponiendo el producto crudo a una temperatura no más de 100°C, preferiblemente exponiendo el producto crudo a una temperatura no más de 90°C, preferiblemente exponiendo el producto crudo a una temperatura de 60 a 150°C, preferiblemente exponiendo el producto crudo producto a una temperatura de 70 a 150°C, preferiblemente exponiendo el producto crudo a una temperatura de 60 a 140°C, preferiblemente exponiendo el producto crudo a una temperatura de 70 a 140°C, preferiblemente exponiendo el producto crudo a una temperatura de 60 a 130°C, preferiblemente exponiendo el producto crudo a una temperatura de 70 a 130°C, preferiblemente exponiendo el producto crudo a una temperatura de 60 a 120°C, preferiblemente exponiendo el producto crudo a una temperatura de de 70 a 120°C, preferiblemente exponiendo el producto crudo a una temperatura de 60 a 110°C, preferiblemente exponiendo el producto crudo a una temperatura de 70 a 110°C, preferiblemente exponiendo el producto crudo a una temperatura de 60 a 100°C, preferiblemente exponiendo el producto crudo a una temperatura de 70 a 100°C, preferiblemente exponiendo el producto crudo a una temperatura de 60 a 90°C, preferiblemente de 70 a 90°C.
[0095] Preferiblemente, cuando se sigue el paso d (i), el producto en bruto se seca a entre 5-10% en peso de humedad mediante la exposición del producto en bruto a una temperatura de 35-90°C y suficiente para proporcionar una velocidad de evaporación de agua de 005 a 0,5 kg de agua por hora por kg de producto seco.
[0096] Preferiblemente, cuando se sigue el paso d (ii), el producto crudo se seca exponiendo el producto en bruto a parpadear de secado o secado por pulverización a una velocidad de evaporación de agua 900 a 40000 kg de agua por hora por kg de producto seco.
[0097] Preferiblemente, cuando (ii) es seguido del paso d, el producto crudo se seca exponiendo el producto en bruto a parpadear secado a una velocidad de evaporación de agua desde 1500 hasta 50.000 o exponer el producto a secado por pulverización a una velocidad de evaporación de agua de 500 a 1500 kg de agua por hora por kg de producto seco. Más preferiblemente, ya sea de 20.000 a 50.000 por secado rápido o de 900 a 1100 por secado por pulverización.
[0098] Preferiblemente, cuando (ii) es seguido del paso d, el producto crudo se seca exponiendo el producto en bruto a parpadear de secado o secado por pulverización a una velocidad de evaporación de agua de 500 a 50.000 kg de agua por hora por kg de producto seco, un delta T de 0,30 a 0,80 y tiempo de residencia del producto en la secadora de menos de 5 minutos.
[0099] Antes de la etapa (a) del presente método, después del paso (d) del presente método, entre cualquiera de los pasos (a), (b), (c) y (d) del presente método, se pueden proporcionar uno o más pasos adicionales. Estos pasos adicionales están abarcados por el presente método. Por ejemplo, en un aspecto preferido, el producto bruto se lava antes de el paso (d).
[0100] Un paso de proceso adicional de acuerdo con un aspecto de la presente descripción comprende realizar un intercambio iónico en el compuesto metálico mixto. Esto puede realizarse en cualquier momento durante el proceso, tal como cuando está presente en la suspensión, como un producto crudo o un producto seco. Un intercambio iónico preferido es con respecto al sulfato presente en el compuesto metálico mixto. El intercambio iónico realizado con respecto al sulfato presente en el compuesto metálico mixto se realiza preferiblemente intercambiando sulfato con carbonato, por ejemplo poniendo en contacto el compuesto metálico mixto con una solución que contiene carbonato.
[0101] Por lo tanto, en este aspecto, se proporciona un método para producir un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+ que tiene un contenido de aluminio de menos de 10.000 ppm, que tiene un tamaño de cristal promedio de menos de 20 nm (200Á)
que comprende los pasos de:
(a) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 11, y en donde el Na2CO3 se proporciona en exceso de 0 a 4,0 moles (como un exceso de 2,0 a 4,0 moles) que se requiere para completar la reacción
(b) someter la suspensión a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,03 a 1,6kW/m3
(c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10% en peso
(d) secar el producto crudo ya sea al
(i) calentar el producto crudo a una temperatura no más de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 15 kg de agua por hora por kg de producto seco, o (ii) exponer el producto crudo a un secado rápido a velocidad de evaporación del agua de 500 a 50.000 kg de agua por hora por kg de producto seco.
(e) contactar opcionalmente la suspensión, el producto bruto o el compuesto metálico mixto, con una solución que contiene carbonato para intercambiar sulfato presente en el compuesto metálico mixto con carbonato.
[0102] La separación del producto puede realizarse por cualquier método adecuado. Por ejemplo, el compuesto metálico mixto se puede separar de la suspensión mediante centrifugación. Se pueden utilizar diferentes métodos de filtración, pero se obtiene un aspecto preferido mediante un método de filtración que usa centrifugación que combina filtración seguido de lavado y deshidratación en un solo paso. Otro aspecto preferido se obtiene mediante un método de filtración que usa un filtro de correa que combina filtración seguido de lavado y deshidratación en un solo paso.
[0103] Después del paso (d), el producto puede tratarse adicionalmente. La presente invención abarca productos obtenidos en virtud de un tratamiento adicional. En un aspecto, el producto crudo seco se clasifica tamizando a un tamaño de partícula promedio d50 de menos de 300 pm, más preferiblemente el producto crudo seco se muele a un tamaño medio de partícula d50 de menos de 200 pm, más preferiblemente el crudo seco el producto se muele a un tamaño de partícula promedio d50 de menos de 100 pm, más preferiblemente el producto crudo seco se muele a un tamaño de partícula promedio d50 de 2 a 50 pm, más preferiblemente el producto crudo seco se muele a una partícula promedio d50 tamaño de 2 a 30 pm.
[0104] Preferiblemente, como se ha medido por tamizado, menos de 10% en peso de partículas son mayores que 106 pm de diámetro, más preferiblemente menos de 5%. Lo más preferiblemente, ninguna partícula tiene un diámetro mayor de 106 pm medido por tamizado.
[0105] Después del paso d(i), el producto puede tratarse adicionalmente. La presente invención abarca productos obtenidos en virtud de un tratamiento adicional. En un aspecto, el producto crudo seco se muele. Más preferiblemente, el producto crudo seco se muele a un tamaño de partícula promedio d50 de menos de 10 pm, aún más preferiblemente el producto crudo seco se muele a un tamaño medio de partícula d50 de 2-10 pm, lo más preferido es que el producto crudo seco fresado a un tamaño de partícula promedio d50 de 2 a 7 pm, pero lo más preferido es que el producto crudo seco se muele a un tamaño de partícula promedio d50 de aproximadamente 5 pm.
[0106] Después del paso d (i) se muele el producto crudo secado para proporcionar un área superficial de 40-80 m2/g, más preferiblemente a una superficie de 40-70 m2/g, incluso más preferiblemente a una superficie área de 45-65 m2/g, más preferida a un área de superficie de 50-60 m2/g.
[0107] Preferiblemente, después del paso d (i) el producto bruto seco se muele a un tamaño de partícula medio d50 2-10 pm y un área superficial de 40-80 m2/g de compuesto.
[0108] Preferiblemente, después del paso d (ii) el producto seco no se muele. Preferiblemente, después del paso d (ii), el producto seco tiene un tamaño de partícula promedio d50 de 10-50 pm y un área superficial de 80-145 m2/g de compuesto.
[0109] Preferiblemente, el producto bruto seco tiene un contenido de agua de menos de 15% en peso, preferiblemente el producto bruto seco tiene un contenido de agua de menos de 10% en peso, preferiblemente el producto bruto seco tiene un contenido de agua de 1-15% en peso, preferiblemente el producto crudo seco tiene un contenido de agua de 5-15% en peso, preferiblemente el producto crudo seco tiene un contenido de agua de 5-10% en peso, preferiblemente el producto crudo seco tiene un contenido de agua de 8-15% en peso, preferiblemente el producto crudo seco tiene un contenido de agua de 8-11% en peso, basado en el peso total del producto crudo seco.
[0110] Preferiblemente, el compuesto metálico mixto tiene un contenido sólido seco de al menos 10% en peso. Preferiblemente, el compuesto metálico mixto tiene un contenido sólido seco de al menos 15% en peso. Más preferiblemente, el compuesto metálico mixto tiene un contenido sólido seco de al menos 20% en peso. Cuando se seca, el compuesto metálico mixto tiene un contenido sólido seco de al menos 80% en peso. Preferiblemente, el compuesto metálico mixto seco tiene un contenido sólido seco de más del 85% en peso. Preferiblemente, el compuesto
metálico mixto seco tiene un contenido sólido seco de menos del 99% en peso. Más preferiblemente, el compuesto metálico mixto seco tiene un contenido sólido seco de menos del 95% en peso. El compuesto metálico mixto seco más preferido tiene un contenido sólido seco de 90 a 95% en peso.
[0112] Como se describe en el presente documento, el compuesto tiene un tamaño de cristal promedio de menos de 20 nm (200 A). Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 100 a 200 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 155 a 200 A Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 110 a 195 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 110 a 185 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 115 a 165 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 120 a 185 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 130 a 185 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 140 a 185 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 150 a 185 A. Preferiblemente, el compuesto tiene un tamaño de cristal promedio de 150 a 175 A. Más preferiblemente, el compuesto tiene un tamaño de cristal promedio de 155 a 175 A. Más preferiblemente, el compuesto tiene un tamaño de cristal promedio de 155 a 165 A.
[0113] En una realización preferida adicional, se proporciona la producción de un compuesto metálico mixto que tiene un tamaño de cristal promedio de menos de 13 nm (130A) y una capacidad de unión a fosfato de más de 0,65 mmol de fosfato/g de compuesto metálico mixto.
[0114] En una realización preferida adicional, se proporciona la producción de un compuesto metálico mixto que tiene un tamaño de cristal promedio de menos de 9 nm (90 A) y una capacidad de unión a fosfato de más de 0,70 mmol de fosfato/g de compuesto metálico mixto.
[0115] En un aspecto preferido, la presente descripción proporciona un metal mixto que comprende al menos Mg2+ y al menos Fe3+ en el que la relación molar de Mg2+ a Fe3+ es 2,1:1 a 1,7:1 que tiene un contenido de aluminio de menos de 30 ppm, que tiene un tamaño de cristal promedio de 110-195 A, que tiene un sulfato entre capas de 2,1 a 5% en peso (tal como de 2,1 a 3,2% en peso) que comprende los pasos de
(a) combinando una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene de 9,6 a menos de 10, y en donde el Na2CO3 se proporciona a un exceso de 2,7 moles que se requiere para completar la reacción
(b) someter la suspensión a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,05 a 0,05 kW/m3
(b1) controlar la suspensión a una temperatura de 20 a 25°C
(b2) controlar opcionalmente la suspensión a una d50 psd de al menos 40 pm (preferiblemente controlar la suspensión a una d50 psd de al menos 40 pm)
(c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10 % en peso
(d) (i) secar el producto crudo a 5-10% en peso de humedad exponiendo el producto crudo a una temperatura de 40-90°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 0,5 kg de agua por hora por kg de producto seco
[0116] El compuesto preparado mediante el presente método puede ser cualquier compuesto metálico mixto que comprende Mg2+ y al menos Fe3+. En un aspecto preferido, el compuesto es un compuesto que tiene una estructura de hidrotalcita. Preferiblemente el compuesto es de fórmula
MN1.xMmx(OH)2An-y.mH2O (II)
en donde M" es uno o más metales bivalentes y es al menos Mg2+;
Mm es uno o más metales trivalentes y es al menos Fe3+;
An_ es uno o más aniones n-valente y es al menos CO32';
1,0 < x / Zyn <1,2, 0 < x <0,67, 0 < y <1 y 0 <m <10.
[0117] El método por el que será bien conocido para un experto en la técnica, la fórmula molecular de un compuesto metálico mixto puede ser determinada. Se entenderá que la fórmula molecular puede determinarse a partir del análisis de relación M"/Mm (Método de Ensayo 1), análisis SO4 (Método de Ensayo 5), análisis CO3 (Método de prueba 6) y análisis H2O (Método de Ensayo 12).
[0118] Preferiblemente, 0 < x < 0,4,0 < y < 1 y 0 < m < 10.
[0119] Preferiblemente 1,05 < x /Z yn < 1,2, preferiblemente 1,05 < x /Z yn y n < 1,15. En un aspecto preferido x /Z yn y n = 1.
[0120] En un aspecto preferido 0,1 < x, tal como 02 < x, 0,3 < x, 0,4 < x, o 0,5 < x. En un aspecto preferido 0 < x < 0,5. Se entenderá que x = [Mm]/([MN] [Mm) donde [MN] es el número de moles de M" por mol de compuesto de fórmula I y [Mm] es el número de moles de Mm por mol de compuesto de fórmula I.
[0121] En un aspecto preferido 0 < y < 1 Preferiblemente 0 < y < 0,8. Preferiblemente 0 < y < 0,6. Preferiblemente 0 < y < 0,4. Preferiblemente 0,05 < y < 0,3. Preferiblemente 0,05 < y < 0,2. Preferiblemente 0,1 < y < 0,2. Preferiblemente 0,15 < y < 0,2.
[0122] En un aspecto preferido 0 < m < 10. Preferiblemente 0 < m < 8. Preferiblemente 0 < m < 6. Preferiblemente 0 < m < 4 Preferiblemente 0 < m < 2 Preferiblemente 0 < m < 1. Preferiblemente 0 < m < 0,7. Preferiblemente 0 < m < 0,6. Preferiblemente 01 < m < 0,6. Preferiblemente 0 < m < 0,5. Preferiblemente 0 < m < 0,3. Preferiblemente 0 < m < 0,15 Preferiblemente 0,15 < m < 0,5 El número de moléculas de agua m puede incluir la cantidad de agua que puede ser absorbida en la superficie de los cristalitos, así como el agua entre capas. Se estima que el número de moléculas de agua está relacionado con x de acuerdo con: m = 0,81 - x.
[0123] Se apreciará que cada uno de los valores preferidos de x, y, z y m pueden combinarse
[0124] En un aspecto preferido, el compuesto tiene un contenido de aluminio de menos de 5000 ppm, más preferiblemente menos de 1000 ppm, lo más preferido 100 ppm, lo más preferiblemente 30 ppm.
[0125] En un aspecto preferido, el contenido total de sulfato del compuesto es de 1,8 a 5% en peso. Por contenido total de sulfato se entiende el contenido de sulfato que está presente en el compuesto. Esto puede determinarse mediante métodos bien conocidos y, en particular, según el método de prueba 1. Preferiblemente, el sulfato total es del 2 al 5% en peso, preferiblemente del 2 al 3,7% en peso, preferiblemente del 2 al 5% en peso, preferiblemente del 2 al menos del 5% en peso, preferentemente del 2,1 al 5% en peso preferentemente del 2,1 al menos del 5% en peso, preferentemente del 2,2 al 5% en peso, preferentemente del 2,2 al menos del 5% en peso, preferentemente del 2,3 al 5% en peso, preferentemente de 2,3 a menos de 5% en peso.
[0126] En un aspecto preferido, el contenido de sulfato total del compuesto es de 1,8 a 4,2% en peso. Por contenido de sulfato total significa contenido de sulfato que está presente en el compuesto. Esto puede determinarse mediante métodos bien conocidos y, en particular, según el Método de Prueba 1. Preferiblemente, el sulfato total es del 2 al 4,2% en peso, preferiblemente del 2 al 3,7% en peso, preferiblemente de 2 a 3,2% en peso, preferiblemente de 2 a menos de 3,2% en peso, preferiblemente de 2,1 a 3,2% en peso preferiblemente de 2,1 a menos de 3,2% en peso, preferiblemente de 2,2 a 3,2% en peso, preferiblemente de 2,2 a menos de 3,2% en peso, preferiblemente de 2,3-3,2% en peso, preferiblemente de 2,3 a menos de 3,2% en peso.
[0127] El compuesto también contendrá una cantidad de sulfato que está unida dentro del compuesto. Este contenido de sulfato, el sulfato entre capas, no puede eliminarse mediante un proceso de lavado con agua. Como se usa en el presente documento, las cantidades de sulfato entre capas son la cantidad de sulfato determinada de acuerdo con el Método de Ensayo 5. En un aspecto preferido, el contenido de sulfato entre capas del compuesto es del 1,8 al 5% en peso, preferiblemente del 1,8 al 3,2% en peso, preferiblemente del 2 al 5% en peso, preferiblemente del 2 al menos del 5% en peso, preferiblemente del 2 al 3,2% en peso, preferiblemente del 2 al 3,1% en peso, preferiblemente del 2 al 3,0% en peso Preferiblemente, el contenido de sulfato entre capas del compuesto es de 2,1 a 5% en peso, preferiblemente de 2,1 a 3,2% en peso, preferiblemente de 2,1 a menos de 3,2% en peso. Más preferiblemente el contenido de sulfato entre capas del compuesto es de 2,2 a 5% en peso, preferiblemente de 2,2 a 3,2% en peso, preferiblemente de 2,2 a menos de 3,2% en peso. Aún más preferiblemente, el contenido de sulfato entre capas del compuesto es del 2,3 al 5% en peso, preferiblemente del 2,3 al 3,2% en peso, preferiblemente del 2,3 al menos del 3,2% en peso. Lo más preferiblemente, el contenido de sulfato entre capas del compuesto es del 2,5 al 5% en peso, preferiblemente del 2,5 al 3,2% en peso, preferiblemente del 2,5 al menos del 3,2% en peso. Aún más preferido, el contenido de sulfato entre capas del compuesto es del 2,5 al 3,0% en peso.
[0128] Como se ha discutido en este documento, la presente invención proporciona compuestos nuevos. Como se describe aquí, la presente descripción proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm, el tamaño de cristal promedio del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el tamaño de partícula promedio d50 del compuesto metálico mixto es menos de 300 pmetro. Preferiblemente, el tamaño de partícula promedio d50 del compuesto metálico mixto es inferior a 200 pm.
[0129] Como se ha discutido en este documento, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el volumen de poros de agua del compuesto metálico mixto es de 0,25 a 0,7 cm 3/g de compuesto metálico mixto. Preferiblemente, el volumen de poros en agua del compuesto metálico mixto es de 0,3 a 0,65 cm 3/g de compuesto metálico mixto. Preferiblemente, el volumen de poros en agua del compuesto metálico mixto es,35-0,65 cm 3/g de compuesto metálico mixto. Preferiblemente, el volumen de poros de agua del compuesto metálico mixto es de 0,3 a 0,6 cm 3/g de compuesto metálico mixto.
[0130] En otra forma de realización preferida de este aspecto el volumen de poros de nitrógeno del compuesto metálico mixto es 0,28-0,56 cm3/g, como se usa en el presente documento, el término 'volumen de poros de nitrógeno' se refiere al volumen de poros tal como se determina de acuerdo con Método de prueba 14.
[0131] Cuando el volumen de poros de nitrógeno del compuesto metálico mixto es de 0,28 a 0,56 cm3/g, se ha encontrado que la estrecha correlación con el volumen de poros de agua es tal que no es necesario determinar el volumen de poros de agua Por lo tanto, en un aspecto adicional, la presente descripción proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+, en donde
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), y el volumen de poros de nitrógeno del compuesto metálico mixto es de 0,28 a 0,56 cm3/g.
[0132] Como se ha discutido en este documento, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 25:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño de cristal promedio del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A)
y el contenido de sulfato entre capas del compuesto es de 1,8 a 5% en peso (tal como de 1,8 a 3,2% en peso). Preferiblemente, el tamaño promedio de cristal del compuesto metálico mixto es de 12 a 20 nm (120 a 200 A).
[0133] Como se ha discutido en este documento, la presente divulgación proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño de cristal promedio del compuesto metálico mixto es menor de 20 nm (200 A),
y el contenido de sulfato entre capas del compuesto es de 2,1 a 5% en peso (como 1,8 a 3,2% en peso). Preferiblemente, el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A).
[0134] Como se describe en el presente documento, la presente descripción proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm,
el tamaño de cristal promedio del compuesto metálico mixto es inferior a 20 nm (200 A),
y el área superficial es de 80 a 145 m2 por gramo de compuesto. Preferiblemente, el compuesto tiene un tamaño de partícula promedio d50 de 10 a 350 pm (y preferiblemente en donde el compuesto no ha sido sometido a fresado) Preferiblemente el compuesto tiene un tamaño de partícula promedio d50 de 10 a 300 pm Preferiblemente el compuesto tiene un d50 tamaño de partícula promedio de 10 a 210 pm Preferiblemente, el compuesto tiene un tamaño de partícula promedio d50 de 10 a 100 pm. Preferiblemente, el compuesto tiene un tamaño de partícula promedio d50 de 10 a 50 pm. Preferiblemente, el compuesto tiene un tamaño de partícula promedio d50 de 10 a 35 pm. Preferiblemente, el compuesto libera magnesio en una cantidad inferior a 0,15 mmol de compuesto de magnesio/g. La liberación de magnesio se determina de acuerdo con el Método de Ensayo 3.
[0135] Como se describe aquí, la presente invención proporciona un compuesto metálico mixto que comprende al menos Mg2+ y al menos Fe3+,
en donde
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el compuesto metálico mixto tiene un aluminio contenido de menos de 10.000 ppm,
el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), el área de superficie es de 40 a 80 m2 por gramo de compuesto.
[0136] Preferiblemente, el tamaño medio de partícula d50 del compuesto metálico mixto es menor que 100 pm. Preferiblemente, el tamaño medio de partícula d50 del compuesto metálico mixto es de menos de 50 pm. Preferiblemente, el tamaño de partícula promedio d50 del compuesto metálico mixto es inferior a 20 pm.
Preferiblemente, el tamaño de partícula promedio d50 del compuesto metálico mixto es inferior a 10 |jm. Preferiblemente, el tamaño de partícula promedio d50 del compuesto metálico mixto es de aproximadamente 5 jm . Preferiblemente, el volumen de poros de agua del compuesto metálico mixto es de 0,25 a 0,7 cm3/g de compuesto metálico mixto, preferiblemente el volumen de poros de agua es de 0,3 a 0,65 cm3/g de compuesto metálico mixto, preferiblemente el volumen de poros de agua es de 0,3 a 0,6 cm3/g de compuesto metálico mixto Preferiblemente, el volumen de poro de nitrógeno del compuesto metálico mixto es 0,28-0,56 cm3/g de compuesto metálico mixto.
[0137] En cada uno de los aspectos de la divulgación en el que se proporciona un compuesto metal mixto, preferiblemente
(1) el contenido de sulfato entre capas del compuesto es de 2,2 a 5% en peso (tal como de 1,8 a 3,2% en peso), y/o (2) el compuesto es de la fórmula
M"i.xMmx(OH)2An-y.mH2O
en donde M" es uno o más metales bivalentes y es al menos Mg2+;
Mm es uno o más metales trivalentes y es al menos Fe3+;
An- es uno o más aniones n-valentes y es al menos CO32",
x/Zyn es de 1 a 1,2 (preferiblemente x/Zyn es de 1,05 a 1,15, preferiblemente x/Zyn es 1) 0 < x <0,4,
0 < y < 1 y
0 < m <10, y/o
(3) el compuesto tiene un contenido de aluminio de menos de 100 ppm, preferiblemente un contenido de aluminio de menos de 30 ppm.
(4) el contenido de sulfato entre capas del compuesto es de 1,8 a 5% en peso (tal como de 1,8 a 3,2% en peso), y/o
(5) el compuesto tiene un tamaño de partícula promedio d50 de menos de 100 jm , preferiblemente el compuesto tiene un tamaño de partícula promedio d50 de 5 a 50 jm , preferiblemente el compuesto tiene un tamaño de partícula promedio d50 de aproximadamente 5 my/o (6) el volumen de poros de agua del compuesto metálico mixto es de 0,3 a 0,65 cm3/g de compuesto metálico mixto y/o (7) el compuesto tiene un contenido sólido seco de al menos 20% en peso.
[0138] El compuesto puede tener cualquier grado de porosidad, sujeto a cualquier rango especificado en el presente documento. En un aspecto preferido, el volumen de poros en agua del compuesto metálico mixto es de 0,25 a 0,7 cm3/g de compuesto metálico mixto. En un aspecto preferido, el volumen de poros en agua del compuesto metálico mixto es de 0,3 a 0,65 cm3/g de compuesto metálico mixto.
[0139] Preferiblemente, el compuesto metálico mixto comprende al menos algo de material que es un hidróxido doble en capas (LDH). Más preferiblemente, el compuesto metálico mixto de fórmula (I) es un hidróxido doble en capas. Como se usa en el presente documento, el término "hidróxido doble en capas" se usa para designar hidróxidos laminares sintéticos o naturales con dos tipos diferentes de cationes metálicos en las capas principales y dominios entre capas que contienen especies aniónicas. Esta amplia familia de compuestos a veces también se conoce como arcillas aniónicas, en comparación con las arcillas catiónicas más habituales cuyos dominios interlamelares contienen especies catiónicas. Las LDH también se han descrito como compuestos similares a la hidrotalcita en referencia a uno de los politipos del mineral a base de [Mg-Al] correspondiente.
[0140] Un compuesto metálico mixto particularmente preferido contiene al menos uno de iones carbonato e iones hidroxilo.
[0141] Un compuesto particularmente preferido contiene como M1 y Mm, magnesio y hierro (iii) respectivamente.
PROCESO
[0142] El compuesto metálico mixto o compuestos pueden ser convenientemente hechos por co-precipitación a partir de una solución, seguido por centrifugación o filtración, a continuación, secado, fresado y/o tamizado. El compuesto metálico mixto se puede volver a humedecer como parte del proceso de granulación en húmedo y los gránulos resultantes se secan en un secador de lecho fluido. El grado de secado en el lecho fluido se usa para establecer el contenido de agua deseado de la tableta final.
[0143] Dos métodos de coprecipitación se pueden utilizar, es decir, uno a baja supersaturación, con lo que el pH de la solución de reacción se mantiene constante mediante el control de la adición de una segunda solución de una precipitación alcalina, o alternativamente precipitación a alta sobresaturación, con lo que el pH de la solución de la reacciónse cambia continuamente mediante la adición de la solución de metal mezclado a una solución alcalina ya presente en el recipiente del reactor. Se prefiere el método de precipitación mediante el cual el pH se mantiene
constante, ya que esto evita la formación de compuestos metálicos individuales como las fases M(OH)2 y/o M(OH)3 en lugar de compuestos metálicos mixtos.
[0144] Otros métodos de precipitación del compuesto metálico mixto también pueden ser posibles si el tamaño del cristalito se controla a menos de 200 A. Por ejemplo, un método de precipitación que implica etapas separadas de nucleación y envejecimiento, un método de hidrólisis de urea, un método de hidrólisis inducida, un método de óxido de sal, un método de sol-gel, un método de electrosíntesis, una oxidación in situ del ion metálico divalente a un ión metálico trivalente, un método denominado "Chimie Douce" o un método en el que el compuesto metálico mixto se puede formar calentando una mezcla íntima de sales metálicas individuales finamente divididas a una temperatura en la que puede ocurrir una reacción sólido-sólido, lo que lleva a una mezcla formación de compuestos metálicos.
[0145] Los métodos de síntesis de correos que tienden a promover el envejecimiento son menos preferidos pero se pueden utilizar si el tamaño de los cristalitos se controla a menos de 200 A. Ejemplos de posibles pasos de tratamiento térmico post síntesis incluyen hidrotermal, microondas y ultrasonido.
[0146] Una variedad de métodos se puede utilizar para separar el compuesto metálico mixto de la suspensión de reacción. También son posibles diferentes métodos de lavado, secado y fresado donde el tamaño del cristalito es inferior a 200 A.
[0147] Las sustancias de la invención preparadas por tratamiento de un material de partida adecuado tal como se describe anteriormente en este documento pueden prepararse al proporcionar una primera solución de un compuesto soluble en agua de metal M1 y un compuesto soluble en agua de metal Mm, eligiéndose los aniones para no provocar precipitaciones de la primera solución (A). También se proporciona una segunda solución (B), de un hidróxido soluble en agua (por ejemplo, NaOH) y una sal soluble en agua de anión An' (el catión se elige para que no precipite con el hidróxido o el anión con el metal del hidróxido), las dos soluciones se combinan y el material de partida compuesto metálico mixto se forma por coprecipitación. Por ejemplo, la solución A se compone disolviendo sulfato de magnesio y sulfato férrico en agua purificada. La solución B se forma disolviendo carbonato de sodio e hidróxido de sodio en agua purificada. Se agrega un talón de agua purificada a un reactor, las soluciones A y B se alimentan de forma controlada por la relación. Después de que el producto se forma en el reactor, puede comprender material cristalino sólido, generalmente también con la presencia de algún material amorfo sólido. Preferiblemente, al menos parte del material así formado es de una doble capa de hidróxido y/o de una estructura de hidrotalcita, generalmente también con algo de material amorfo y/o poco cristalino, preferiblemente después de la coprecipitación, el material se filtra o centrifuga, lavado luego secado por calentamiento. El secado se lleva a cabo ya sea (i) exponiendo el producto crudo a una temperatura no superior a 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco, o (ii) exponer el producto crudo a secado instantáneo o secado por pulverización a una velocidad de evaporación de agua de 500 a 50.000 kg de agua por hora por kg de producto seco, por ejemplo mediante secado en horno, secado por pulverización o secado en lecho fluido.
[0148] Opcionalmente, el material seco puede clasificarse primero, para eliminar partículas de gran tamaño mediante fresado y/o tamizado y/o cualquier otra técnica adecuada, por ejemplo, para restringir el material a tratar a partículas que no son sustancialmente mayores de 300 pm de diámetro.
Reacción
[0149] Hay una gran cantidad de opciones disponibles para llevar a cabo la reacción. Estas pueden controlarse para llevar a cabo la reacción de la manera deseada. Por ejemplo, el tipo de equipo reactivo, la composición de la corriente del reactivo, la temperatura y el pH, el modo de adición de reactivo, el sistema de agitación y el tiempo de retención pueden especificarse para producir la reacción deseada.
[0150] Varios tipos de reactores son comunes en la industria farmacéutica, estos incluyen reactores discontinuos y continuos.
[0151] La estructura del material y el tamaño del cristalito se pueden determinar significativamente en la etapa de reacción mediante un control estricto de la concentración de la solución de reactivo, la temperatura de reacción, el tiempo que se mantiene la masa de reacción después de la precipitación y el modo de adición del reactivo. Además, para lograr la estructura de material preferida con un tamaño de cristalito pequeño, una alta concentración de solución (al final del período de reacción de 4,8 - 5,4% en peso), baja temperatura de reacción (15 - 25°C) y un tiempo de retención corto (típicamente < 12 horas) son preferidos.
[0152] Para preparar los compuestos metálicos mixtos a una relación molar preferida Mg:Fe, por ejemplo de 1,5:1 a 2,5:1, se prefiere un control preciso de pH durante el proceso de precipitación. El control preciso del pH generalmente se logra mediante la calibración frecuente de electrodos de pH y el monitoreo y ajuste del pH a lo largo del proceso de precipitación. El pH de la reacción puede controlarse variando la velocidad relativa de adición de la Solución A a la Solución B añadida al reactor. Hemos encontrado que solo se prefiere la variación de la Solución B ya que esto mantiene la concentración de precipitado en la mezcla de reacción a un nivel constante. La variación en el caudal de la Solución B se puede llevar a cabo manualmente o utilizando un algoritmo de control adecuado.
[0153] Hemos encontrado una composición reactante óptima, por lo que se cumplen los requisitos opuestos de mantener una temperatura de reacción relativamente baja, pero lograr una tasa de filtración razonable y un buen control del pH.
[0154] Por ejemplo, la relación molar M2+:M3+ preferida entre 1,5:1 a 2,5:1 del compuesto metálico mixto se puede lograr manteniendo las corrientes de reactivo en solución incluso a temperaturas relativamente bajas, limitando así la reacción temperatura. La temperatura de reacción puede ser importante para determinar la extensión del crecimiento de cristalitos y, por lo tanto, la actividad de unión al fosfato.
[0155] El método preferido permite lograr buenas tasas de filtración, esto nuevamente puede ser importante para limitar el tiempo de almacenamiento en masa de la reacción, que en términos conocidos ayuda a determinar la extensión del crecimiento de cristalitos y, por lo tanto, la actividad de unión al fosfato.
[0156] Además, las condiciones de reacción y la receta preferidas ayudan a mantener un buen control del pH. Se requiere un buen control del pH para alcanzar el pH diana.
[0157] El modo de adición de la corriente de reactivo puede ser importante para definir la calidad del producto de reacción. Son posibles diferentes combinaciones de modo de adición y pueden incluir la adición de una corriente de reactivo en un exceso del otro reactivo (cualquiera de los reactivos podría seleccionarse como la corriente añadida).
[0158] Hemos encontrado que la adición simultánea de una corriente de reactivo de pH alto que contiene iones de carbonato e hidróxido, y una corriente de reactivo de pH bajo que contiene iones de metal y sulfato en un talón proporciona un control de pH más preciso de la suspensión de reacción. Por lo tanto, en una realización preferida, la reacción se lleva a cabo mediante la adición simultánea (coprecipitación) de una corriente de reactivo que contiene iones de carbonato e hidróxido, y una corriente de reactivo que contiene iones de metal y sulfato.
[0159] De manera similar, el producto se puede eliminar de forma continua a medida que se añaden las corrientes de reactivo, o al final de un período definido.
[0160] Se logran buenas características de filtración apuntando a un tamaño de partícula relativamente grande, tal como de al menos 40 pm, controlando la potencia por unidad de volumen de 0,03 a 1,6 kW/m3. Hemos identificado que la potencia por unidad de volumen de 0,05 a 05 kW/m3 usando impulsores configurados para agitación de flujo axial, ayuda a producir una suspensión con características de filtración mejoradas (como menor tiempo de filtración y lavado, y alto contenido final de sólidos en la torta)
[0161] Por lo tanto, en una realización preferida, la agitación se usa para someter la suspensión a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,03 a 1,6 kW/m3 proporcionada por mezcladores estáticos, agitadores de impulsor, bomba, mezclador de chorro o dinámica en mezclador de línea. Por lo tanto, en una realización preferida adicional, la agitación de flujo axial se usa para someter la suspensión a mezcla en condiciones que proporcionan una potencia por unidad de volumen de 0,03 a 1,6 kW/m3 proporcionada por un agitador de impulsor. Más preferiblemente una potencia por unidad de volumen de 0,05 a 0,5 kW/m3 suministrada por agitación del impulsor. Esto proporciona una suspensión de reacción con las características de filtración preferidas.
[0162] Por lo tanto, en una realización separada, la reacción se agitó usando medios distintos de un agitador impulsor convencional.
[0163] Un tiempo óptimo de retención ha sido identificado como de 30 minutos a 12 horas. Para tiempos de retención de más de 16 horas, la filtración se vuelve difícil debido a una reducción en el tamaño de partícula durante el uso de la agitación en el tiempo de retención y se produce el envejecimiento.
Filtración
[0164] Hay una gran cantidad de opciones disponibles para llevar a cabo los pasos de aislamiento y lavado del producto, sin embargo, el tipo de equipo de filtración y los parámetros del proceso operativo deben definirse y controlarse cuidadosamente.
[0165] Por ejemplo, para limitar el tiempo total de retención de la suspensión de reacción (y, por lo tanto, el crecimiento de cristalitos), es beneficioso minimizar el tiempo para el aislamiento de la torta y el tiempo de lavado. También es preferible un alto contenido de sólidos de la torta ya que esto reduce el tiempo de secado y de ahí la propensión al crecimiento de cristalitos durante la etapa de secado.
[0166] Se usan varios tipos de filtros en la industria farmacéutica, estos incluyen: filtros Neutsche, secadores de filtro, centrifugadoras filtrantes, filtros de correa, filtros de placa y marco.
[0167] Al aislar el compuesto metálico mixto no envejecido, la tasa de filtración global puede ser extremadamente baja
debido a la dificultad relativa de aislar y lavar estos compuestos metálicos mixtos de tipo arcilla no envejecidos, lo que hace que esto sea económicamente poco atractivo si no se controla a las condiciones preferidas. El material no envejecido de tamaño de cristalito de menos de 200 A, tiene la tendencia a dar como resultado el "cegamiento" de los medios de filtración y/o las propiedades de tipo arcilla tienen una tendencia a formar una torta más impermeable.
[0168] En una realización preferida, hemos producido una alta tasa de filtración usando un filtro de correa.
Secado
[0169] Hay una gran cantidad de opciones disponibles para llevar a cabo la operación de secado, estas deben definirse y controlarse para llevar a cabo el paso de secado de la mejor manera.
[0170] Por ejemplo, el tipo de secado, el modo de secado y la velocidad de secado debe especificarse y controlarse de modo que el producto crudo se seque ya sea (i) exponiendo el producto crudo a una temperatura no más de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco, o (ii) exponiendo el producto crudo a secado rápido o secado por pulverización a una velocidad de evaporación de agua de 500 a 50.000 kg de agua por hora por kg de producto seco. La velocidad de secado se ve afectada por factores que incluyen el modo de secado, la temperatura de la superficie calentada/temperatura del medio de calentamiento, el grado de agitación, el nivel de vacío (si lo hay), etc. La temperatura del producto debe limitarse a no más de 150°C para evitar daños a la sustancia farmacológica.
[0171] Varios tipos de secadores son comunes en la industria farmacéutica, estos incluyen secadores de tiempo de residencia largos (caracterizados típicamente por un tiempo de residencia de hasta 20 h) tales como secadores esféricos, cónicos, de doble cono, de bandeja (vacío, presión ambiental) y secadores de tiempo de residencia cortos (caracterizados típicamente por un tiempo de residencia de hasta varios minutos) incluyen; Spray, Spin Flash, etc.
[0172] Hemos encontrado que de los diversos diseños de secado por lotes, un secador esférico agitado ofrece una gran área de superficie calentada al producto. Por lo tanto, una tasa de producto más alta por unidad de área de 1 a 2,1 kg de producto/(m2 h) y, por lo tanto, son posibles tasas de transferencia de calor y secado altas. Dado que el envejecimiento (crecimiento de cristalitos) puede ocurrir durante el secado, es importante minimizar el tiempo de secado/maximizar la velocidad de secado. Para evitar la descomposición de la sustancia farmacológica, cuando se usa el calentamiento de la superficie, la temperatura de la superficie de secado se limita típicamente a 150°C para el secado por lotes y preferiblemente a 90°C o menos para evitar el crecimiento de tamaño de cristal promedio por encima de 200 A. La evacuación parcial del secador deprime el punto de ebullición del agua en la masa de secado, lo que limita el crecimiento de cristales, esta depresión también sirve para maximizar la velocidad de secado. La velocidad de secado se manipula maximizando el vacío del secador y aumentando la temperatura de la carcasa hasta 120°C durante una fase de secado inicial, para eliminar el agua a la velocidad más alta posible, y luego reduciendo la velocidad al reducir la temperatura de la carcasa a menos de 90°C, a fin de alcanzar con precisión un punto final de humedad definido mientras se mantiene un tamaño de cristalito inferior a 200 A. El punto final de la humedad se puede inferir mediante el control de la masa de evaporación, o medir directamente, mediante el análisis de los contenidos secos u otros métodos adecuados.
Secadores cónicos (por ejemplo, tipo Nautamixer) ofrecen beneficios similares al secador esférico.
[0173] Se encontraron secador de bandejas a vacío para producir un producto de calidad aceptable, sin embargo este tipo de secador puede requerir intervención manual (por ejemplo, redistribución de sólidos) para el secado uniforme, y tiene rendimiento limitado.
[0174] Los secadores discontinuos de tiempo de residencia largos descritos (esféricos, cónicos, al vacío) tienen tasas de secado relativamente bajas (expresadas en términos normalizados como kg de evaporación por kg de producto por hora) en comparación con los métodos de secado de tiempo de residencia corto. Hemos encontrado que la temperatura de la sustancia farmacológica durante el secado y la velocidad de secado pueden influir significativamente en el tamaño del cristalito y la morfología de la sustancia farmacológica.
[0175] Por ejemplo, el secado por lotes de tiempo de residencia prolongado tiende a producir un tamaño de cristal promedio relativamente grande y un volumen de poro y un área superficial relativamente bajos, mientras que se ha descubierto que los métodos de residencia cortos tales como el secado por pulverización y el secado rápido por rotación producen cristales relativamente más pequeños volumen de poro relativamente grande y área de superficie. El material producido usando un tiempo de secado corto puede mostrar un rendimiento mejorado de unión a fosfato; Esto puede deberse a las diferentes propiedades cristalinas y morfológicas. El material secado por pulverización tiene la ventaja adicional de la granulación solo para la mezcla en seco (por ejemplo, para la fabricación de tabletas) y puede llevarse a cabo sin fresado previa de la sustancia farmacológica.
[0176] Los métodos de tiempo de secado de larga residencia se definen típicamente por tener un tiempo de residencia igual o superior a 3 horas. Ejemplos de estos incluyen: secado de bandejas, secado de calderas, secado de bandejas, secado rotativo (envoltura), secado rotativo (interno), secado de doble cono.
[0177] Hemos encontrado velocidades de evaporación promedio de entre 9 y 29 kg de agua/(h.m2) que se pueden lograr usando un secador esférico de vacío agitado. Dicho de manera alternativa, esto es equivalente a una velocidad de evaporación de aproximadamente 0,05 a 0,5 kg de agua por hora por kg de producto seco. El tamaño del cristalito del producto producido en este rango de velocidades de evaporación es típicamente entre 100 y 200 Angstroms. Para un producto seco de calidad consistente, el secador debe ser alimentado con una torta húmeda (típicamente >20% en peso de sólidos).
[0178] Cada uno de los secadores anteriores funciona por lotes.
[0179] Los ejemplos de métodos de tiempo de secado de residencia corto típicamente tienen tiempos de residencia mucho más bajos. Estos difieren dependiendo de los tipos de tecnología y se definen como: secado por centrifugado rápido, tiempo de residencia típico de 5 a 500 segundos/secado por pulverización, tiempo de residencia típico de hasta 60 segundos.
[0180] Las velocidades de evaporación típicas se definen como: centrifugado rápido 70 - 300 kg de agua/(h.m3) volumen del recipiente, pulverización 5 - 25 kg de agua/(h.m3) volumen del recipiente. Las tasas de evaporación para secado rápido y secado por atomización también se calculan como 500 a 50.000 kg de agua/(h.kg de fármaco). El tamaño del cristalito del producto producido en este rango de tasas de evaporación es típicamente menor de 140 Angstroms. El secador por rotación instantánea puede alimentarse con torta húmeda (típicamente >20% en peso de sólidos), mientras que el secador por pulverización debe alimentarse con una suspensión fluida a una concentración más baja (típicamente hasta 10% en peso de sólidos)
[0181] Todos los secadores de tiempo de residencia cortos anteriores pueden funcionar de manera continua.
[0182] Se pueden usar varias tecnologías de tiempo de residencia 'medio' que dependen principalmente de los transportadores de uso y son operados continuamente. Estos pueden ser menos preferidos si se producen problemas para garantizar una calidad constante del producto (contenido de humedad variable) y limpieza para la producción farmacéutica. Ejemplos de tecnologías de tiempo de residencia medio se enumeran como: estante giratorio, vibrador de canal, tipo turbo.
USOS
[0183] Preferiblemente, el compuesto se utiliza en la fabricación de un medicamento para la profilaxis o el tratamiento de la hiperfosfatemia.
[0184] En un aspecto adicional, la presente descripción proporciona el uso de un compuesto de la presente invención u obtenido/obtenible de acuerdo con la presente invención en la fabricación de un medicamento para la profilaxis o el tratamiento de cualquiera de hiperfosfatemia, insuficiencia renal, hipoparatiroidismo, seudohipoparatiroidismo, acromegalia aguda no tratada, enfermedad renal crónica y sobremedicación de sales de fosfato.
[0185] Ejemplos de uno o más de los síntomas que pueden indicar el riesgo de presencia de ERC una concentración de creatina superior a 1,6 mg/dL, un nivel de fosfato en sangre superior a 4,5 mg/dL, cualquier sangre detectable en orina, orina concentración de proteína superior a 100 mg/dL, una concentración de albúmina en orina superior a aproximadamente 100 mg/dL, una tasa de filtración glomerular (TFG) inferior a 90 ml/min/1,73 m2 o una concentración de hormona paratiroidea en la sangre superior a 150 pg/ml. Los síntomas también están definidos por la Iniciativa de Calidad de Resultados de la Enfermedad de Fidney de la Fundación Nacional del Riñón "NKF-K/DOQI" o "K/DOQI".
[0186] En un aspecto preferido, la enfermedad renal crónica (ERC) tratada de acuerdo con la invención de presencia es ERC que tiene paso uno a cinco.
[0187] El medicamento puede usarse en animales, preferiblemente seres humanos.
COMPOSICIONES FARMACÉUTICAS
[0188] Un vehículo farmacéuticamente aceptable puede ser cualquier material con el que se formule la sustancia de la invención para facilitar su administración. Un vehículo puede ser un sólido o un líquido, incluido un material que normalmente es gaseoso pero que se ha comprimido para formar un líquido, y se puede usar cualquiera de los vehículos que se usan normalmente en la formulación de composiciones farmacéuticas. Preferiblemente, las composiciones de acuerdo con la invención contienen 0,5% a 95% en peso de ingrediente activo. El término vehículo farmacéuticamente aceptable abarca diluyentes, excipientes o adyuvantes.
[0189] Cuando las sustancias de la invención son parte de una composición farmacéutica, pueden formularse en cualquier forma de composición farmacéutica adecuada, por ejemplo, polvos, gránulos, granulados, bolsitas, cápsulas, paquetes de palo, batallas, tabletas, pero especialmente en una forma adecuada para administración oral, por ejemplo, en forma de dosis unitaria sólida, como tabletas, cápsulas, o en forma líquida, como suspensiones líquidas,
especialmente suspensiones acuosas o formulaciones semisólidas, por ejemplo, geles, barra masticable, dosificación dispersante, forma de dosificación masticable o bolsita comestible. Adición directa a la comida también puede ser posible.
[0190] También son posibles formas de dosificación adaptadas para administración extracorpórea o incluso intravenosa. Se pueden producir formulaciones adecuadas mediante métodos conocidos usando vehículos sólidos convencionales tales como, por ejemplo, lactosa, almidón o talco o vehículos líquidos como, por ejemplo, agua, aceites grasos o parafinas líquidas. Otros vehículos que pueden usarse incluyen materiales derivados de proteínas animales o vegetales, como las gelatinas, dextrinas y proteínas de semillas de soja, trigo y psilio; gomas tales como acacia, guar, agar y xantano; polisacáridos; alginatos; carboximetilcelulosas; carragenanos; dextranos; pectinas polímeros sintéticos tales como polivinilpirrolidona, complejos de polipéptidos/proteínas o polisacáridos tales como complejos de gelatina-acacia, azúcares tales como manitol, dextrosa, galactosa y trehalosa, azúcares cíclicos tales como ciclodextrina; sales inorgánicas tales como fosfato de sodio, cloruro de sodio y silicatos de aluminio; y aminoácidos que tienen de 2 a 12 átomos de carbono tales como glicina, L-alanina, ácido L-aspártico, ácido L-glutámico, L-hidroxiprolina, L-isoleucina, L-leucina y L-fenilalanina
[0191] Componentes auxiliares tales como desintegrantes de tabletas, solubilizantes, conservantes, antioxidantes, tensioactivos, potenciadores de la viscosidad, agentes colorantes, agentes aromatizantes, modificadores del pH, edulcorantes o agentes para enmascarar el sabor también pueden incorporarse en la composición. Los agentes colorantes adecuados incluyen óxidos de hierro rojo, negro y amarillo y colorantes FD & C, tales como azul FD & C N° 2 y rojo FD & C N° 40 disponibles de Ellis & Everard. Los agentes aromatizantes adecuados incluyen sabores de menta, frambuesa, regaliz, naranja, limón, pomelo, caramelo, vainilla, cereza y uva y combinaciones de estos. Los modificadores de pH adecuados incluyen hidrogenocarbonato de sodio, ácido cítrico, ácido tartárico, ácido clorhídrico y ácido maleico. Los edulcorantes adecuados incluyen aspartamo, acesulfamo K y taumatina. Los agentes de enmascaramiento del sabor adecuados incluyen hidrogenocarbonato de sodio, resinas de intercambio iónico, compuestos de inclusión de ciclodextrina, adsorbatos o principios activos microencapsulados.
[0192] Para el tratamiento y la profilaxis de la hiperfosfataernia, se administran diariamente cantidades de 0,1 a 500, más preferiblemente de 1 a 200, mg/kg de peso corporal de la sustancia de la invención como compuesto activo para obtener los resultados deseados. puede ser necesario de vez en cuando para apartarse de las cantidades mencionadas anteriormente, dependiendo del peso corporal del paciente, el método de aplicación, la especie animal del paciente y su reacción individual al medicamento o el tipo de formulación o el tiempo o intervalo en el que se aplica el medicamento En casos especiales, puede ser suficiente usar menos de la cantidad mínima indicada anteriormente, mientras que en otros casos puede ser necesario exceder la dosis máxima. Para una dosis mayor, puede ser aconsejable dividir la dosis en varias dosis individuales más pequeñas. Finalmente, la dosis dependerá de la discreción del médico encargado. Por lo general, se preferirá la administración antes de las comidas, por ejemplo, dentro de una hora antes de una comida o tomada con alimentos
[0193] Una dosis unitaria sólida única típica para la administración adulta humana puede comprender de 1 mg a 1 g, preferiblemente de 10 mg a 800 mg de la sustancia de la invención.
[0194] Una forma de dosis unitaria sólida también puede comprender un aditivo que controla la velocidad de liberación. Por ejemplo, la sustancia de la invención puede mantenerse dentro de una matriz polimérica hidrófoba de modo que se lixivie gradualmente de la matriz al entrar en contacto con fluidos corporales. Alternativamente, la sustancia de la invención puede mantenerse dentro de una matriz hidrófila que se disuelve gradual o rápidamente en presencia de fluido corporal. La tableta puede comprender dos o más capas que tienen diferentes propiedades de liberación. Las capas pueden ser hidrófilas, hidrófobas o una mezcla de capas hidrófilas e hidrófobas. Las capas adyacentes en una tableta multicapa pueden estar separadas por una capa de barrera insoluble o una capa de separación hidrófila. Una capa de barrera insoluble puede estar formada por materiales utilizados para formar la carcasa insoluble. Se puede formar una capa de separación hidrófila a partir de un material más soluble que las otras capas del núcleo de la tableta, de modo que a medida que la capa de separación se disuelve, las capas de liberación del núcleo de la tableta quedan expuestas.
[0195] Los polímeros que controlan la velocidad de liberación adecuados incluyen polimetacrilatos, etilcelulosa, hidroxipropilmetilcelulosa, metilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa de sodio, carboximetilcelulosa de calcio, polímero de ácido acrílico, polietilenglicol, óxido de polietileno, carragenano, acetato de celulosa, zein, etc.
[0196] Los materiales adecuados que se hinchan en contacto con líquidos acuosos incluyen los materiales poliméricos incluyen carboximetilcelulosa de sodio reticulada, hidroxipropilcelulosa reticulada, hidroxipropilcelulosa de alto peso molecular, carboximetilamida, copolímero de divinilbenceno y metacrilato de potasio, polimetilmetacrilato, polivinilpirrolidona reticulada y polivinilalcoholes de alto peso molecular.
[0197] Las formas de dosis unitarias sólidas que comprenden una sustancia de la invención pueden empaquetarse juntas en un recipiente o presentarse en tiras de aluminio, envases de blíster o similares, por ejemplo, marcados con días de la semana contra las dosis respectivas, para orientación del paciente.
[0198] También existe la necesidad de formulaciones que puedan mejorar el cumplimiento del paciente, por ejemplo en el caso de pacientes de edad avanzada o pediátricos. Una formulación en forma de dosis en polvo podría diluirse en agua, reconstituirse o dispersarse.
COMBINACIONES
[0199] El compuesto de la presente invención pueden usarse como el único ingrediente activo o en combinación con otro agente de unión a fosfato. También puede usarse en combinación con un calcimimético tal como cinacalet, vitamina D o calcitriol.
[0200] En otro aspecto, la presente descripción proporciona el uso de un compuesto de la presente invención u obtenido/obtenible de acuerdo con la presente invención en la fabricación de un medicamento para la profilaxis o el tratamiento de la hiperfosfatemia.
EJEMPLOS
Descripción general de la reacción.
[0201] El compuesto metálico mixto se forma por la reacción de una mezcla acuosa de sulfato de magnesio y sulfato férrico con una mezcla acuosa de hidróxido de sodio y carbonato de sodio. La precipitación se lleva a cabo a un pH de alrededor de 9,8 y una temperatura de reacción a partir de alrededor 22°C y subiendo hasta 30°C tras la adición de reactivos. El precipitado resultante se filtra, se lava, se seca y se muele.
[0202] La reacción de síntesis se representa así.
4MgSO4 Fe2(SO4)3 12NaOH (XS+1)Na2COa ^ Mg4Fe2(OH)i2.COa.nH2O 7Na2SO4 XSNa2CO3.
[0203] Esto genera un compuesto metálico mixto con una relación molar de Mg:Fe de típicamente 2:1 y el subproducto de reacción sulfato de sodio. El exceso de carbonato de sodio (XS) añadido a la mezcla de reacción junto con el sulfato de sodio se elimina del precipitado.
[0204] Al cambiar la relación molar de los cationes M":Mm a 1:1, 2:1, 3:1, 4:1 se lograron diferentes materiales de composición. El exceso de carbonato de sodio y el pH de reacción también se cambiaron en experimentos separados.
[0205] La fórmula molecular de los hidróxidos dobles laminares se pueden medir mediante diferentes métodos El método actual utilizado para determinar la fórmula molecular de los ejemplos en el presente documento se determinó a partir del análisis de M"/Mm ratio (Método 1), análisis SO4 (Método 5), análisis CO3 (Método 6) y análisis H2O (Método 12). La fórmula [MN1-xMm(OH)2][(CO3)y-i(SO4)y2.mH2O][Na2SO4]z se usó para describir la composición de los ejemplos (1-66) que se muestra en el presente documento a continuación con más detalle para un compuesto metálico mixto en el que:
x = [MIN]/([MN] [Mm]) donde [M1] es el número de moles de metal bivalente M1 por mol de compuesto de fórmula I y [Mm] es el número de moles de metal trivalente Mm por mol de compuesto de fórmula I.
Zy'= suma de los moles aniones entre capas y1' (CO32-) y2'(SO42-) o cualquier otro anión en el que
y1' = % en peso CO32- / Mw CO32-y2' = (% en peso SO42- total/Mw SO42-) -(% en peso Na2O/Mw Na2O)
[0206] Los aniones entre capas también se definen como aniones unidos o aniones que no se pueden eliminar lavando con agua.

Zy = es la suma de moles de aniones entre capas corregida con el factor de normalización de fórmula (f).
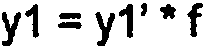
f = x / (2 * % en peso Mm2O3 / Mw Mm2O3) = factor de normalización de fórmula
m' = % en peso H2O / (Mw H2O)
m = m' * f
% en peso de H2O = LOD (pérdida en el secado medida a 105°C)
z = z' * f z = cantidad de sulfato restante que puede eliminarse mediante lavado y se calcula a partir de la cantidad de Na2O, cuyo total se supone que está asociado con SO42- como Na2SO4 soluble
z' = % en peso Na2O/Mw Na2O
[0207] La relación x / Zyn se puede calcular a partir de los valores de x y la suma de aniones entre capas (Zyn) cuyos datos se insertan a continuación en la fórmula molecular [Mg1-xFex(OH)2][(CO3)y-i(SO4)y2mH2O][Na2SO4]z.
Ejemplo 1 (no según la invención)
[0208] Preparado por el método descrito a continuación para la preparación de aproximadamente 250 gramos de producto seco dirigido a tener una relación molar Mg:Fe de 1:1.
[0209] La fórmula molecular real encontrada por análisis fue:

En donde x = 0,5, y1 = 0,14, y2 = 0,02, m = 0,4, z = 0.
[0210] Se prepararon dos materiales de partida, designados solución 1 y solución 2 por el método que se describe a continuación, para proporcionar un exceso de Na2CO3 de 2,7 moles (en la ecuación de reacción 1).
[0211] Se disolvieron sulfato de magnesio y sulfato de hierro en agua AnalaR™ para preparar la solución 1. En un recipiente separado se disolvieron carbonato de sodio e hidróxido de sodio en agua AnalaR™ para preparar la solución 2. Los pesos utilizados se calcularon para dar la proporción deseada de cationes metálicos.
[0212] Para la preparación de la solución 1, se pesó agua AnalaR™ en un recipiente y se agitó usando un mezclador superior, en el que se disolvió una cantidad apropiada de hidrato de sulfato férrico (grado GPR). Una vez disuelto, sulfato de magnesio (sal de Epsom)) se transfirió cuantitativamente a la solución agitada de sulfato de hierro y se dejó disolver.
[0213] Para la preparación de la solución 2, se pesó agua AnalaR™ en un recipiente y se agitó usando un mezclador superior, en el que se disolvió una cantidad apropiada de carbonato de sodio (Pharmakarb). Una vez disuelto, el hidróxido de sodio (Soda cáustica perlada) se transfirió cuantitativamente a la solución agitada de carbonato de sodio y se dejó disolver.
[0214] Las soluciones se agregaron simultáneamente a agua de talón agitada de 1100 cm3 a velocidades de flujo controladas suficientes para mantener un pH de 10,3 en la mezcla de reacción (+/- 0,2 unidades de pH) a una temperatura de reacción que no exceda los 30°C. La concentración final de la suspensión fue de alrededor del 5,1% en peso del compuesto.
[0215] Cuando se completaron las adiciones, la mezcla de reacción se mezcló durante otros 30 minutos y luego se filtró usando un sistema de filtración de Buchner. La suspensión del producto se filtró usando una bomba de vacío y un embudo Buchner con un papel de filtro sin cenizas endurecido Whatman® (No 541). Después de filtrarse, la torta del filtro se lavó con porciones de agua AnalaR™.
[0216] El producto filtrado se lavó a continuación con 220 cm3 porciones de agua AnalaR™ fría Después del aislamiento del producto se secó usando un horno precalentado.
[0217] Se colocó un peso de agua AnalaR™ en un recipiente. Se usaron unidades de control de flujo para suministrar los caudales apropiados de las soluciones de sulfato de metal y carbonato alcalino.
[0218] Después del aislamiento, el producto lavado se transfirió a un recipiente y se secó en un horno precalentado a 120°C durante tres horas.
[0219] La muestra del producto para análisis se molió usando un molino de bolas (Retsch PM 100). Los parámetros de fresado se establecieron en función de las propiedades del producto.
[0220] La muestra de producto para el análisis se molió a través de un tamiz de acero inoxidable de 200 mm de
diámetro y 106 |jm, utilizando un agitador de tamiz (Retsch AS-200). El material de gran tamaño se devolvió a la muestra seca de existencias para volver a moler, hasta que todo el material esté < 106 jm .
Ejemplo 2
[0221] Método de preparación como para el Ejemplo 1 pero dirigido a tener una relación molar Mg:Fe de 2:1 La fórmula molecular real encontrada por análisis fue
[Mgo,64Feo,36(OH)2][(CO3)y1(SO4)o,o3,0,20H2O][Na2SO4]z,
Ejemplo 3 (no según la invención)
[0222] Método de preparación como para el Ejemplo 1 pero dirigido a tener una relación molar Mg:Fe de 3:1.
Ejemplo 4 (no de acuerdo con la invención)
[0223] Método de preparación como para el Ejemplo 1 pero dirigido a tener una relación molar Mg:Fe de 4:1 Ejemplo 5 y 6
[intencionalmente en blanco]
Ejemplo 7
[0224] Método de preparación como para el Ejemplo 1 pero dirigido a tener una relación molar Mg:Fe de 2:1 y envejecimiento previsto (aumento en el tamaño del cristalito) mediante la introducción de un paso de método adicional inmediatamente después de la precipitación en el que la suspensión de reacción se envejece por tratamiento térmico. La suspensión se calienta a reflujo durante 4 horas usando una placa caliente y un condensador Liebig para refluir la muestra en un matraz sellado. La muestra se filtró inmediatamente usando un embudo Buchner al vacío. El compuesto envejecido se aisló luego usando el mismo método descrito para el Ejemplo 1
[0225] La fórmula molecular real encontrada mediante análisis fue:
[Mgo,66Feo,34(OH)2][(CO3)y1(SO4)o,o1,0,09H2O][Na2SO4]z
Ejemplo 8-24
[0226] Método de preparación como para el Ejemplo 2 pero con una fuente férrica líquida de 4o,4 a 42,9% en peso de sulfato férrico de agua, el estándar de la industria adecuado para el consumo humano cumple con la norma b S EN 89o: 2oo4). El método se varió porque se realizaron a pH de precipitación diferente, exceso de Na2CO3 diferente y temperatura de reacción diferente, es decir, no envejecida (es decir, a una temperatura de reacción relativamente baja de 15, 3o o 65 grados Celsius) o envejecida (a 9o grados Celsius) de acuerdo con los ejemplos descritos a continuación. Cuando los ejemplos no estaban envejecidos, las soluciones se enfriaron (a 15 Celsius) o no se produjo tratamiento térmico de la suspensión de reacción (a 3o Celsius) o algo de calentamiento suave (a 65 Celsius); mientras que al envejecer, el tratamiento térmico de la suspensión de reacción se produjo mediante el uso de un vaso de precipitados de vidrio sellado con un condensador colocado en una placa calefactora y una suspensión de reacción calentada a 9o°C durante 4 horas). Cuando no se menciona la temperatura de reacción, la reacción se realizó al estándar temperatura ambiente de aproximadamente 25-3o Celsius. La suspensión de reacción se enfrió a 15 grados Celsius colocando el vaso de precipitados metálico en un baño de agua con hielo; La temperatura se controló mediante un termómetro y se controló mediante la adición y eliminación de hielo. La suspensión de reacción se calentó a 65 grados Celsius colocando el vaso de precipitados metálico en un baño de agua termostáticamente controlado Grant W38. La temperatura se controló mediante un termómetro. La suspensión de reacción conducida a 3o grados Celsius comenzó a temperatura ambiente pero gradualmente se elevó a una temperatura final de 3o grados Celsius después de la adición de los reactivos. Después de la adición de los reactivos, la suspensión se mezcló durante 3o minutos antes de la filtración con la excepción del ejemplo 21 que se mezcló durante 96o minutos.
[0227] La fórmula molecular real determinada por análisis, tamaño de los cristalitos, precipitación de pH, tratamiento en suspensión, el exceso de moles de Na2CO3 en la receta se enumeran a continuación para cada ejemplo. Los resultados de los ejemplos 8-24 se muestran en la Tabla 3 y la Figura 1.
Ejemplo
[0228]
8 (no según la invención)
[Mgo,2Feo,8(OH)2][(COa)o,16(SO4)o,oo,42H2O].[Na2SO4]o,co
Tamaño del cristalito: no determinado (nd)
pH de precipitación = 8,0; la temperatura de reacción es de 30 Celsius: 2,7 moles de exceso de Na2CO3
(no según la invención)
[Mgo,5Feo,5(OH)2][(CO3)o,i4(SO4)o,o20,39H2O].[Na2SO4]o,oo
Tamaño del cristalito: >2oo A pH de precipitación = 9,8; la temperatura de reacción es 9o Celsius, 2,7 moles de exceso de Na2CO3
b (no según la invención)
[Mgo,5Feo,5(OH)2][(CO3)o,i4(SO4) o,o2 o,39H2O][Na2SO4]o,oo
Tamaño del cristalito: >2oo A
pH de precipitación = 9,8; la temperatura de reacción es de 9o grados Celsius; 4 moles de exceso de Na2CO3 0 [Mgo,67Feo,38(OH)2][(CO3)o,i4(SO4)o,oi,o,23H2O].[Na2SO4]o,oo
Tamaño del cristalito. no determinado (nd)
pH de precipitación = io,1, la temperatura de reacción es 65°C; 2,7 moles de exceso de Na2CO3
1 [Mgo,67Feo,33(OH)2][(CO3)o,i4(SO4)o,oi,o,25H2O].[Na2SO4]o,oo
Tamaño del cristalito: no determinado (nd)
PH de precipitación = 9,8; la temperatura de reacción es 65°C; 2,7 moles de exceso de Na2CO3
2 [Mgo,67Feo,33(OH)2][(CO3)o,i4(SO4)o,oi. o,39H2O][Na2SO4]o,oo
Tamaño del cristalito: no determinado (nd)
pH de precipitación = i i ; la temperatura de reacción es 3o Celsius, 4 moles de exceso de Na2CO3 3 [Mgo,66Feo,34(OH)2][(CO3)o,i5(SO4)o,o2.o,39H2O].
[Na2SO4]o,oo Tamaño del cristalito: no determinado (nd)
pH de precipitación = io,5; la temperatura de reacción es de 3o Celsius; 2,7 moles de exceso de Na2CO3 4 [Mgo,66Feo,34(OH)2][(CO3)o,i4(SO4)o,o2.o,23H2O].
[Na2SO4]o,oo Tamaño del cristalito. no determinado (nd)
pH de precipitación = io,3; la temperatura de reacción es de 3o Celsius, 2,7 moles de exceso de Na2CO3 5 [Mgo,67Feo,33(OH)2][(CO3)o,i3(SO4)o,o2.o,73H2O].
[Na2SO4]o,oo Tamaño del cristalito: no determinado (nd)
pH de precipitación = io,5; la temperatura de reacción es de 3o Celsius; i moles de exceso de Na2CO3 6 [Mgo,67Feo,33(OH)2][(CO3)o,i3(SO4)o,o2.o,38H2O].[Na2SO4]o,oo
Tamaño del cristalito: no determinado (nd)
pH de precipitación = io , i; la temperatura de reacción es de 3o Celsius; 2,7 moles de exceso de Na2CO3
7 [Mgo,66Feo,34(OH)2][(CO3)o,i6(SO4)o,o2.o,37H2O].[Na2SO4]o,oo
Tamaño del cristalito: < ioo A
pH de precipitación = 9,8; la temperatura de reacción es 3o Celsius 2,7 moles de exceso de Na2CO3
8 [Mgo,66Feo,34(OH)2][(CO3)o,i2(SO4)o,o3,o,65H2O][Na2SO4]o
Tamaño de cristalito: no determinado (nd)
pH de precipitación = 9,8; la temperatura de reacción es de 3o Celsius; 4 moles de exceso de Na2CO3
9 [Mgo,67Feo,33(OH)2][CO3)o,i3(SO4)o,o3,o,38H2O][Na2SO4]o,oo
Tamaño del cristalito: no determinado (nd)
pH de precipitación = i i ; la temperatura de reacción es de i5°C; i moles de exceso de Na2CO3
0 [Mgo,64Feo,36(OH)2][(CO3)o,i2(SO4)o,o3.ndH2O].
[Na2SO4]o,oo Tamaño del cristalito: no determinado (nd)
pH de precipitación = 9,6; la temperatura de reacción es de 3o Celsius; 2,7 moles de exceso de Na2CO3
1 [Mgo,64Feo,36(OH)2][(CO3)o,i3(SO4)o,o4,o,56H2O].
[Na2SO4]o,oo Tamaño del cristalito: no determinado (nd)
pH de precipitación = 9,5; la temperatura de reacción es de 3o Celsius; 2,7 moles de exceso de Na2CO3
2 [Mgo,62Feo,38(OH)2][(CO3)o,i2(SO4)o,o4o,49H2O][Na2SO4]o,oo
Tamaño del cristalito: no determinado (nd)
pH de precipitación = 9,5; la temperatura de reacción es de 3o Celsius; 4 moles de exceso de Na2CO3
3 [Mgo,64Feo,36(OH)2][(CO3)o,i2(SO4)o,o4,o,52H2O][Na2SO4]o,oo
Tamaño del cristalito: no determinado (nd)
pH de precipitación = 95, temperatura de reacción es 3o Celsius, i moles de exceso de Na2CO3
4 (no según la invención)
Mgo,58Feo,42(OH)2][(CO3)o,i(SO4)o,o5,o,43H2O][Na2SO4]o,oo
Tamaño del cristalito: no determinado (nd)
pH de precipitación = 95; la temperatura de reacción es de i5°C; i moles de exceso de Na2CO3
Ejemplo 25-27
[0229] El método de preparación y la relación Mg:Fe como para el Ejemplo 2 con una fuente férrica de 40,4 a 42,9% en peso de sulfato férrico de agua, el estándar de la industria adecuado para consumo humano se ajusta a BS EN 890: 2004 y el pH de precipitación varía de acuerdo con la Tabla 4 y filtrado.
Ejemplo La fórmula molecular real encontrada por análisis fue:
25 [Mg0,65Fe0,35(OH)2][(CO3)yi(SO4)y2 mH2O].[Na2SO4]0,00
26 [Mg0,65Fe0,35(OH)2][(CO3)y1(SO4)y2. mH2O][Na2SO4]z
27 [Mg0,65Fe0,35(OH)2][(CO3)0,13(SO4) 0,02 m H ^ . ^ S O ^
Ejemplo 28
[0230] Para preparar 163 kg del compuesto metálico mixto (base seca) se prepararon dos soluciones de partida designadas solución A y solución B. Para preparar la solución A, 138 kg (base seca), sulfato de hierro de 40,4 a 42,9% en peso de sulfato férrico del estándar de la industria del agua adecuado para el consumo humano que cumple con la norma BS EN 890: 2004, 166 kg (base seca) de sulfato de magnesio (agregado como hidrato de hepta) se disolvieron en un total de 1034 kg de agua donde esta cantidad total de agua incluye el agua asociada con la solución de sulfato férrico. Para preparar la solución B, se disolvieron 173 kg de hidróxido de sodio y 129 kg de carbonato de sodio en 948 kg de agua para proporcionar soluciones homogéneas. El recipiente de reacción del talón de agua fue de 840 kg. El agua suministrada al talón era el 30% del agua total suministrada.
[0231] Las temperaturas de la solución de reactivo se ajustan a alrededor de 22°C antes de la adición. Las corrientes reactivas (soluciones A y B) se alimentan simultáneamente al recipiente de reacción a una velocidad tal que se mantenga un pH de reacción de 9,8. Se aplica enfriamiento del contenido del recipiente para mantener una temperatura de 20-25°C. Se introduce un tacón de agua purificada antes de la introducción de las corrientes de reactivo para permitir la agitación del recipiente en la fase de reacción inicial y proporcionar una concentración final de la suspensión de alrededor del 5,1% en peso de compuesto.
[0232] El recipiente se agita utilizando un agitador de flujo axial de alto cizallamiento y bajo cizallamiento que funciona a una potencia por unidad de volumen de 0,1 kW/m3 y donde las soluciones reactivas se entregan a un área de alto volumen de rotación El reactor se deflecta para promover una buena mezcla.
[0233] La suspensión de precipitado se mantiene en el recipiente de reacción (también denominado tiempo de retención) durante hasta 12 horas y se transfiere en alícuotas a una centrífuga de filtración vertical para aislamiento y lavado, usando agua purificada para proporcionar la velocidad máxima del producto. El lavado se termina para conseguir un contenido de sodio residual (expresada como Na2O en el producto seco) de menos de 0,40% en peso.
[0234] La torta húmeda se descarga de la centrífuga y se seca en un secador de vacío esférico, agitado. La temperatura del vacío y la carcasa se ajustan para proporcionar una temperatura del producto en la secadora de aproximadamente 72°C. La velocidad de secado fue de 0,26 kg de agua/(kg de producto seco h), un tiempo de residencia de 12 horas y una velocidad de producto por unidad de área de 4,6 kg de producto/(m2 h).
[0235] El producto seco se muele primero en bruto usando un molino de desmoldeo a una distribución de tamaño de partícula (Método de Ensayo 24) de típicamente 200 micras (D50) seguido de micronización final a un tamaño de partícula de típicamente 5 micras (D50).
Ejemplo La fórmula molecular real encontrada por análisis fue:
28 [MgQ,66FeQ,34(OH)2][(CO3)0,12(SO4)0,03,0,20 H2O].
Ejemplo 28b
[0236] distribución del tamaño de partícula se midió en la suspensión de reacción después de la adición de los reactivos y después de un tiempo de espera de 4 horas y la tasa de filtración medido durante la etapa de aislamiento
Ejemplo 28c - Sistemas de reacción alternativos
[0237] Se aplicó CFD (Método de Ensayo 27) al ejemplo 28 para obtener una potencia de mezcla por unidad de volumen en el punto de adición de las corrientes de reactivo.
[0238] Los cálculos se basaron en el sistema que tiene
Densidad de la suspensión. 1200 kg.m-3
Viscosidad de la suspensión. 20 cP (002 Pa.s)
Velocidad del eje agitador: 100 rpm
Deflector: placas planas
El patrón de mezcla se demuestra a través de pistas de partículas de cada una de las dos entradas de corriente reactante y muestra que las corrientes permanecen segregadas entre sí de manera efectiva antes de que el fluido se haya dispersado ampliamente en el volumen. También se generaron contornos de concentración y confirman que el mezclado tiene lugar muy rápidamente.
[0239] Para derivar los requisitos para la fabricación de compuestos metálicos mixtos para sistemas de mezcla alternativos (por ejemplo, mezcladores estáticos), se han aplicado los resultados para el sistema convencional de agitación de bajo cizallamiento (descrito anteriormente).
[0240] Los sistemas de mezcla alternativos tales como mezcladores estáticos, mezcladores de chorro o mezcladores dinámicos en línea y, en particular, un mezclador estático Kenics KM pueden ser adecuados para proporcionar un volumen en el que pueda tener lugar la reacción y adecuados para proporcionar el régimen de mezcla necesario. Por ejemplo, el mezclador estático Kenics KM que utiliza un volumen de zona de alimentación nocional de 5x10-4 m3, para proporcionar una relación de potencia a masa (1 28 W/kg - equivalente a 1,54 kW/m3) y tiempo de residencia (1,25 segundos). la longitud fue fijada por la longitud mínima recomendada de 4 elementos (por lo tanto, L/D = 6). El diámetro resultante fue de 100 mm y el caudal de 280 litros/min.
[0241] En resumen, para unos sistemas de reacción convencionalmente agitados una potencia por unidad de volumen gama (intensidad de mezclado) de 003 a 0,5 kW/m3 se ha establecido como óptimo. Utilizando equipos de mezcla alternativos, se ha establecido un rango de potencia por unidad de volumen (intensidad de mezcla) de 0,03 a 1,6 kW/m3 como óptimo.
Ejemplo 29
Como por ejemplo 28 pero con un pH de reacción de 10,3.
Ejemplo 30
[0242] Como por ejemplo 28 pero con un tamaño de lote de 7 kg usando una alimentación de una sola alícuota al filtro Neutsch en lugar de centrifugar de modo que la masa de reacción se aísle y lave dentro de un período de tiempo de no más de 16 horas, una bandeja horno en lugar de un secador esférico y fresado.
[0243] La torta húmeda se descarga del filtro Neutsch y se seca en un horno de bandeja de vacío donde las paredes del horno se calientan a 120 a 130°C y se lleva a cabo una redistribución manual regular de la masa de secado. La velocidad de secado fue de 0,38 kg de agua/(kg de producto seco h) con un tiempo de secado total de 12 a 16 horas y una velocidad de producto por área de superficie de secado calentada de 0,2 kg de producto/(m2.h).
[0244] El producto seco se microniza usando un molino de impacto hasta un tamaño de partícula de típicamente 5 micras (D50, Método de Ensayo 24).
Ejemplo La fórmula molecular real encontrada por análisis fue:
30 [Mg0,65Fe0,36(OH)2][(CO3)0,11(SO4)y2,0,49H2O].[Na2SO4]z
Ejemplo 31
[0245] Como por ejemplo 30 pero con un pH de reacción de 10,3.
Ejemplo La fórmula molecular real encontrada por análisis fue:
31 [Mg0,66Fe0,34(OH)2][(CO3)0,12(SO4)0,020,28 H2O].[Na2SO4]0,0
Ejemplo 32
[0246] Como por ejemplo 28 pero con un filtro de cinta de vacío en lugar de centrífuga. Las muestras de torta húmeda descargadas del filtro de la correa de vacío se secaron en el horno de laboratorio a 120°C durante tres horas.
[0247] de la muestra del producto para el análisis se molieron utilizando un molino de bolas para permitir que pase a través de un 106 pm tamiz.
Ejemplo 33
[0248] Una suspensión de reacción se preparó de acuerdo con el método del ejemplo 2 pero con un pH de reacción de 9,6, una fuente férrico líquido (una solución de 40,4 a sulfato férrico 42,9% en peso de estándar de la industria de
agua apta para el consumo humano se ajusta a BS EN 890:2004) y un tamaño de lote nominal de 400 cm3 La suspensión de reacción se lavó usando filtración de flujo tangencial (Sartorius Slice sistema superior 200 banco con 200 cm2 área de filtración, PESU 0,1 micras membrana) operada en modo de velocidad constante. El sistema se lavó abundantemente y se llena con agua DI antes de la filtración, la tasa de permeado fue regulada para evitar filtro filtración de bloqueo con adición de agua de lavado (diafiltración) se llevó a cabo para conseguir un contenido de sodio residual (expresado como Na2O en el producto seco) de menos de 0,40% en peso. La suspensión lavada se concentró luego usando filtración al vacío convencional y se secó en un horno de laboratorio.
Ejemplo La fórmula molecular real encontrada por análisis fue:
33 [Mg0,65Fe0,35(OH)2][(CO3)y1(SO4)y2mH2O].[Na2SO4]z
Ejemplo 34
[0249] Como por ejemplo 33 pero con un pH de reacción de 10,1
Ejemplo 35
[0250] Como por ejemplo 33 pero con un pH de reacción de 10,3.
Ejemplo La fórmula molecular real encontrada por análisis fue:
35 [MgQ,65FeQ,35(OH)2][(CO3)y1(SO4)y2mH2O].[Na2SO4]z
Ejemplo 36
[0251] Se preparó una suspensión de reacción para el procesamiento de acuerdo con el método del Ejemplo 28, sin embargo, posteriormente se procesaron 620 kg de suspensión de reacción usando filtración de flujo tangencial en lugar de centrifugación. Se usó un pH de reacción de 9,8.
[0252] Antes de la filtración, para reducir el riesgo de bloqueo de la membrana, la suspensión de reacción se hizo circular a través de un molino coloidal húmedo para reducir el tamaño de partícula D50 (Método de Ensayo 9) de 60 a 51 micras.
[0253] Se usó una unidad de filtración Sartorius Sartoflow Beta con once membranas Sartocon II dando un área de filtración total de 7,7 m2. El sistema se enjuagó y se llenó con agua DI antes de la filtración, la velocidad de permeado se reguló para evitar el bloqueo del filtro. Se usó una bomba de lóbulo giratorio para hacer circular la suspensión a través del sistema a una presión de entrada de entre 2 y 3,5 bares y un flujo de retención típico de 3400 l/h. La filtración con la adición de agua de lavado (diafiltración) se llevó a cabo hasta alcanzar un contenido de sodio residual (expresado como Na2O en el producto seco) de menos de 0,40% en peso.
[0254] Se tomó una muestra de una cantidad representativa de suspensión, se aisló y se secó de acuerdo con el método del Ejemplo 1 pero sin lavado adicional de la torta
Ejemplo 37
[0255] Como por ejemplo 36 pero con un pH de reacción de 10,3. El tamaño de partícula (D50, Método de Ensayo 9) se redujo mediante fresado en húmedo de 47 a 44 micras.
Ejemplo 38
[0256] Como por ejemplo 28, pero el método se varió porque se llevaron a cabo con condiciones de secado ligeramente diferentes. La tasa de secado fue de aproximadamente 0,27 kg de agua/(kg de producto seco.h), un tiempo de residencia de 13 horas, una tasa de producto por unidad de área de 1,4 kg de producto/(m2 h), y la temperatura máxima de secado alcanzada es aproximadamente 85°C
Ejemplo La fórmula molecular real encontrada por análisis fue:
38 [Mg0,66Fe0,34(OH)2][(CO3)0,13(SO4)0,03.0,36 H2O].[Na2SO4]0,00
Ejemplo 39
[0257] Como por ejemplo 28, pero el método se varió porque se llevaron a cabo con diferentes condiciones de secado. La velocidad de secado fue de aproximadamente 0,38 kg de agua/(kg de producto seco h), un tiempo de residencia de 9 horas y una velocidad de producto por unidad de área de 1,2 kg de producto/(m2 h).
Ejemplo La fórmula molecular real encontrada por análisis fue:
39 [Mg0,67Fe0,33(OH)2][(CO3)0,13(SO4)0,02.0,26 H2O].[Na2SO4]0,00
Ejemplo 40
[0258] Como por ejemplo 30, pero el método se varió porque se llevaron a cabo con diferentes condiciones de secado. La tasa de secado fue de aproximadamente 0,21 kg de agua/(kg de producto seco.h) un tiempo de residencia de 18 horas y una tasa de producto por unidad de área de 0,1 kg de producto/(m2 h)
Ejemplo 41
[0259] Como por ejemplo 30 pero el método se varió porque se llevaron a cabo con diferentes condiciones de secado. La tasa de secado fue de aproximadamente 0,27 kg de agua/(kg de producto seco.h) un tiempo de residencia de 16 horas y una tasa de producto por unidad de área de 0,2 kg de producto/(m2 h)
Ejemplo La fórmula molecular real encontrada por análisis fue:
41 [Mg0,66Fe0,34(OH)2][(CO3)0,13(SO4)0,02.mH2O].[Na2SO4]0,00
Ejemplo 42-47
[0260] Como por ejemplo 28, pero el método se varió porque se llevaron a cabo con diferentes condiciones de secado como se describe en la Tabla 7 cuando se secaron con un secador de vacío esférico, agitado (secador de larga residencia)
Ejemplo Ejemplo La fórmula molecular real encontrada por análisis fue:
43 [Mg0,66Fe0,34(OH)2][(CO3)0,13(SO4)0,02.0,53 H2O].[Na2SO4]0,01
44 [Mg0,65Fe0,35(OH)2][(CO3)0,12(SO4)0,03.mH2O].[Na2SO4]0,00
45 [Mg0,67Fe0,33(OH)2][(CO3)0,13(SO4)0,03.mH2O].[Na2SO4]0,00
46 [Mg0,66Fe0,34(OH)2][(CO3)0,12(SO4)0,03,0,29 H2O].[Na2SO4]0,00
47 [Mg0,65Fe0,35(OH)2][(CO3)0,13(SO4)0,03.mH2O].[Na2SO4]0,00
Ejemplo 48-49 (no según la invención)
[0261] Como por ejemplo 28 (centrifugación) pero luego el método se varió porque la torta del filtro se secó usando un secador de tipo de residencia corta (Spin-Flash Drier, fabricante/modelo; Anhidro/SFD51) en donde el delta T era 0,40 (Ejemplo 48 ) o un delta T de 0,66 (Ejemplo 49).
Condiciones secador por pulverización
Ejemplo Tin (°C) delta T Velocidad Tasa de rotatoria (%) producto
(kg/h)
48 250 150 0,40 90 8
49 350 120 0,66 45 20
Delta T — (Tin — T out) / Tin
Ejemplo La fórmula molecular real encontrada por análisis fue:
48 [Mg0,65Fe0,35(OH)2][(CO3)0,15(SO4)0,02.0,19H2O].[Na2SO4]0,00
49 [Mg0,66Fe0,34(OH)2][(CO3)0,14(SO4)0,02.0,16H2O].[Na2SO4]0,00
Ejemplo 50 (no según la invención)
[0262] Como por ejemplo 36 (filtración de flujo tangencial) pero luego se varió el método en que la suspensión se secó usando un secador de tipo de residencia corta (Secador por pulverización, fabricante/modelo; Anhidro/CSD71) con un delta T de 0,69.
Ejemplo Tin (°C)  delta T Velocidad de
delta T Velocidad de
inclinación (%)
50 350 110 0,40 208,3
Delta T = (Tin — T out) / Tin
Ejemplo La fórmula molecular real encontrada por análisis fue:
50 [Mg0,66Fe0,34(OH)2][(CO3)0,14(SO4)0,02.0,37 H2O].[Na2SO4]0,01
Ejemplo 51-52 (no según la invención)
[0263] Como por ejemplo 28 (centrifugación) pero luego el método se varió porque la torta del filtro se diluyó primero para proporcionar una suspensión del 10,1% en peso y luego se secó usando un secador de tipo de residencia corta (secador por pulverización, fabricante/modelo, Anhidro/CSD71) en donde el delta T era 0,74 (Ejemplo 51) o un delta T de 0,76 (Ejemplo 52)
Condiciones de secado por pulverización
Ejemplo Tin (°C)  delta T Velocidad de inclinación (Hz)
delta T Velocidad de inclinación (Hz)
51 350 110 0,74 208,3 Hz
52 325 120 0,76 208,3 Hz
Ejemplo La fórmula molecular real encontrada por análisis fue:
51 [Mg0,66Fe0,34(OH)2][(CO3)0,14(SO4)0,01.0,34H2O].[Na2SO4]0,01
52 [Mg0,65Fe0,35(OH)2][(CO3)0,14(SO4)0,03.ü,38H2O].[Na2SO4]0,00
Ejemplo 53-59
[0264] Como por ejemplo 28 (centrifugación) pero luego se varió el método en que en lugar de micronización, el producto seco solo se molió grueso a 343 micras (pm) (D50) y se separó a continuación en 6 fracciones de tamaño de partícula diferentes por tamizado. Se utilizaron seis tamices diferentes con un tamaño de parámetro de tamiz de respectivamente; base, 20 micras, 75 micras, 106 micras, 180 micras, 355 micras. Las fracciones de tamiz se obtuvieron mediante tamizado manual. Las 6 fracciones de tamiz diferentes (Ejemplo 53-58) obtenidas por este método se describen en la Tabla 9.
Ejemplo La fórmula molecular real encontrada por análisis fue:
53 [Mg0,65Fe0,35(OH)2][(CO3)y1(SO4)0,03.mH2O].[Na2SO4]0,00 Fracción de tamiz: >355 pm
54 [Mg0,65Fe0,as(OHM(cO3)y1(sO4)0,c0.mH2O].[Na2SO4]0,66 Fracción del tamiz: 180 - 355 55 pm
56 [Mg0,65Fe0,35(OH)2][(CO3)y1(SO4)0,03.mH2O].[Na2SO4]0,00 Fracción del tamiz: 106 - 180 57 pm
58 [Mg0,65Fe0,35(OH)2][(CO3)y1(SO4)0,03.mH2O].[Na2SO4]0,00 Fracción de tamiz: 75 -106 pm
58 [Mg0,65Fe0,35(oH)2][(cO3)y1(sO4)0,03.mH2O][Na2SO4]0,00 Fracción de tamiz: < 106 pm
Ejemplo 60
[0265] Como por ejemplo 28, pero el método se varió porque se llevaron a cabo con diferentes condiciones de secado. La tasa de secado fue de aproximadamente 0,33 kg de agua/(kg de producto seco.h), un tiempo de residencia de 9,8 horas y una tasa de producto por unidad de área de 1,6 kg de producto/(m2.h) y la temperatura máxima de secado alcanzada es aproximadamente 76°C.
Ejemplo La fórmula molecular real encontrada por análisis fue:
60 [Mg0,65Fe0,35(OH)2][(CO3)0,12(SO4)0,03.mH2O].[Na2SO4]0,00
Ejemplo 61
[0266] Como por ejemplo 28, pero el método se varió porque se llevaron a cabo con diferentes condiciones de secado. La tasa de secado fue de aproximadamente 0,28 kg de agua/(kg de producto seco.h), un tiempo de residencia de 10,3 horas y una tasa de producto por unidad de área de 1,5 kg de producto/(m2.h) y la temperatura del producto alcanzada es de aproximadamente 64°C.
Ejemplo La fórmula molecular real encontrada por análisis fue:
61 [Mg0,66Fe0,34(OH)2][(CO3)0,13(SO4)0,03.mH2O].[Na2SO4]0,00
Ejemplo comparativo 62 y ejemplos 63-65
[0267] Como para el Ejemplo 2, pero el método se varió porque se llevaron a cabo a diferentes pH, diferente exceso de Na2CO3 y diferente fuente férrica de acuerdo con la Tabla 12. Además, el Ejemplo Comparativo 62 se preparó con sulfato de aluminio en lugar de sulfato de hierro.
[0268] Se usaron dos fuentes férricas diferentes designadas A y B:
A: de grado GPR Rectapur
B: una fuente férrica más pura, como una solución de 40,4 a 42,9% en peso de sulfato férrico de agua estándar de la industria adecuada para consumo humano conforme a BS e N 890:2004
Ejemplo La fórmula molecular real encontrada por análisis fue:
62 [Mg0,79Al0,21(OH)2][(CO3)0,16(SO4)0,02.mH2O].[Na2SO4]0,00
(Comparativo) [Mg0,66Fe0,34(OH)2|(CO3)0,14(SO4)0,02.ü,22H2O].[Na2SO4]0,00 63 [Mg0,66Fe0,34(OH)2][(CO3)y1(SO4)0,02.mH2O].[Na2SO4]0,00
64 [Mg0,66Fe0,34(OH)2][(CO3)y1 (SO4)0,01.mH2O].[Na2SO4]0,00
65
Ejemplo 66 (no según la invención)
[0269] Como en el ejemplo 28, pero el método se varió entonces porque la torta de filtro se secó usando un tipo de secador de residencia corta (Spin-Flash Drier, fabricante/modelo; Anhidro/SFD51) en donde el delta T era 0,48.
Ejemplo Tin(°C) Tout (°C) delta T Velocidad de atomizador, Hz
66 250 130 0,48 90
Delta T — (Tin — T out) / Tin
Ejemplo La fórmula molecular real encontrada por análisis fue:
66 [Mg0,65Fe0,3s(OHM(CO3fo14(SO4)0,c0.0,31H2OMNa2SO4]0
METODOS
Método de Ensayo 1 Análisis XRF
[0270] El análisis XRF del producto se realizó usando un espectrómetro XRF dispersivo de longitud de onda Philips PW2400. La muestra se fusionó con 50:50 de litio tetra/metaborato (alta pureza) y se presentó al instrumento como una perla de vidrio. Todos los reactivos utilizados fueron de grado analítico o equivalente a menos que se especifique. Agua AnalaR™, tetraborato de litio, 50% de metaborato, 50% de flujo (ICPH Fluore-X 50 de alta pureza). Se utilizó un horno de mufla capaz de 1025°C, pinzas extendidas, pinzas de mano, bandeja de fundición Pt/5% Au y plato Pt/5%/Au. Se pesaron con precisión 1,5 g (+/- 0,0002 g) de muestra y 7,5000 g (+/- 0,0002 g) de tetra/metaborato en un plato de Pt/5%/Au. Los dos componentes se mezclaron ligeramente en el plato usando una espátula, antes de colocarlos en el horno preestablecidos a 1025°C durante 12 minutos. El plato se agitó a los 6 minutos y 9 minutos para asegurar la homogeneidad de los minutos. El plato se agitó a los 6 minutos y 9 minutos para garantizar la homogeneidad de la muestra. También a los 9 minutos, la bandeja de colada se colocó en el horno para permitir el equilibrio de la temperatura. Después de 12 minutos, la muestra fundida se vertió en la bandeja de colada, que se retiró del horno y se dejó enfriar. La composición del cordón se determinó usando el espectrofotómetro.
[0271] Se utilizó el método XRF para determinar el Al, Fe, Mg, Na y el contenido total de sulfato del compuesto, así como la relación de MII a MIII.
Método de Ensayo 2 Mediciones de difracción de rayos X (DRX)
[0272] Los datos se recogieron para muestras de partículas finas 2-70° 20 en un difractómetro de rayos X de polvo automático de Philips usando radiación alfa de cobre K generada a 40 kV y 55 mA.
[0273] Los datos de difracción de rayos X en polvo (XRD) se recogieron desde 2-70 grados 2theta en un difractómetro de rayos X en polvo automático Philips PW 1800 usando radiación alfa de cobre K generada a 40kV y 55mA, un
tamaño de paso de 0,02 grados 2theta con un tiempo de conteo de 4 segundos por paso, una ranura de divergencia automática que proporciona una radiación irradiada. Se utilizó un área de muestra de 15 x 20 mm, junto con una ranura de recepción de 0,3 mm y un monocromador de haz difractado.
[0274] El volumen medio de cristalito de volumen aproximado puede determinarse a partir del ancho, a la mitad de la altura del pico, del pico de difracción de rayos X en polvo a aproximadamente 11,5 grados 2 theta (el pico está típicamente en el intervalo de 8 a 15 grados 2 theta para materiales de tipo hidrotalcita) usando la relación dada en la Tabla 1 que se deriva usando la ecuación de Scherrer. La contribución al ancho del pico de la ampliación de la línea de instrumentos fue de 0,15 grados, determinada midiendo el ancho del pico a aproximadamente 21,4 grados 2 theta de una muestra de LaB6 (NIST SRM 660) en las mismas condiciones,
Tabla 1 Conversión de ancho de pico XRD a tamaño de cristalito usando la ecuación de Scherrer

(continúa)

[0275] Los valores en la Tabla 1 se calcularon usando la ecuación de Scherrer:
D = K * X / p * cos© Ecuación 1
Dónde:
D = tamaño de cristalito (A)
K = factor de forma
X = longitud de onda de radiación utilizada (en A)
p = ancho de pico medido como FWHM (ancho completo a la mitad de la altura máxima) y corregido para el ensanchamiento de la línea de instrumentos (expresado en radianes)
© = el ángulo de difracción (mitad de la posición máxima 2©, medida en radianes)
Factor de forma
[0276] Este es un factor para la forma de la partícula, típicamente entre 0,8 y 1,0, se usa un valor de 0,9.
Longitud de onda de la radiación
[0277] Esta es la longitud de onda de la radiación utilizada. Para la radiación de cobre K alfa, el valor utilizado es 1,54056 A.
Ancho de pico
[0278] El ancho de un pico es la suma de dos conjuntos de factores: instrumental y muestra.
[0279] Los factores instrumentales se miden típicamente midiendo el ancho del pico de una muestra altamente cristalina (picos muy estrechos). Como no se dispone de una muestra altamente cristalina del mismo material, se ha utilizado LaB6. Para las mediciones actuales se ha utilizado un valor de instrumento de 015 grados.
[0280] Por lo tanto, para la medida más precisa del tamaño de cristalito usando la ecuación de Scherrer, el ancho del pico debido a factores instrumentales se debe restar del ancho del pico medido, es decir:
p = B(medida) b(instrumental)
[0281] El ancho del pico se expresa en radianes en la ecuación de Scherrer.
[0282] El ancho del pico (como FWHM) se ha medido ajustando una parábola u otro método adecuado al pico después de restar un fondo adecuado.
Posición de pico
[0283] Se ha utilizado un valor de 11,5°2© que proporciona un ángulo de difracción de 5,75°. Corresponde a 0,100 radianes.
Método de Ensayo 3 Capacidad de unión a fosfato y liberación de Mg
[0284] Tampón de fosfato (pH = 4) se preparó pesando 5,520 g (+/- 0001 g) de dihidrogenofosfato de sodio, seguido por adición de agua AnalaR™ y transferir a un matraz volumétrico de 1ltr
Al matraz volumétrico de 1 litro se añadió entonces 1 M HCl gota a gota para ajustar el pH a pH 4 (+/- 0,1) mezclando entre adiciones. El volumen se completó con precisión hasta 1 litro usando agua AnalaR™ y se mezcló completamente. Se añadieron 0,5 g (+/- 0,005 g) de cada muestra a un matraz volumétrico (50 ml) que contenía una solución tampón de fosfato 40 mM (12,5 ml) a 37,5°C en un agitador orbital Grant OLS 200. Todas las muestras se prepararon por duplicado. Los vasos se agitaron en el agitador orbital durante 30 minutos. La solución se filtró luego usando un filtro de jeringa de 0,45 pm. 2,5 cm3 partes alícuotas de sobrenadante se pipetearon del sobrenadante y se transfieren a un fresco tubos de recogida de sangre. 7,5 cm3 de agua AnalaR™ se pipetearon a cada 2,5 cm3 alícuota y el tapón de rosca equipado y se mezcla a fondo. Las soluciones se analizaron luego en un ICP-OES calibrado.
[0285] El fosfato capacidad de unión se determinó por:
Enlace de fosfato (mmol/g) = Sp (mmol/l) - Tp (mmol/l) / W (g/l) dónde:
Tp = valor de analito para fosfato en la solución de fosfato después de la reacción con aglutinante de fosfato = solución P (mg/l) * 4 / 30,97.
Sp = Valor de analito para fosfato en la solución de fosfato antes de la reacción con aglutinante de fosfato. W = aglutinante de concentración (g/l) utilizado en el Método de Ensayo (es decir, 0,4 g/10 cm3 = 40 g/l) [0286] La liberación de magnesio fue determinada por:
Liberación de magnesio (mmol/g) = TMg (mmol/l) - SMg (mmol/l) / W (g/l)
dónde:
TMg = Valor de analito para magnesio en la solución de fosfato después de la reacción con aglutinante de fosfato = solución Mg (mg/l) * 4/24,31.
SMg = Valor de analito para magnesio en la solución de fosfato antes de la reacción con aglutinante de fosfato.
[0287] No se informó la liberación de Fe ya que la cantidad de hierro liberada del compuesto era demasiado pequeña y estaba por debajo del límite de detección.
Método de Ensayo 4 Enlace de fosfato y liberación de magnesio en suspensión de alimentos
[0288] El péptido MCT2 , suplemento alimenticio (SHS International) se mezcló para formar una suspensión del 20% (p/v) en HCl 0 01 M. Se mezclaron partes alícuotas separadas de 0,05 g de compuesto seco con 5 cm3 de la suspensión de alimentos y se agitaron constantemente durante 30 minutos a temperatura ambiente. Se extrajo una parte alícuota de 3 cm3y se centrifugó a 4000 rpm durante 10 minutos, y se disolvieron el fosfato y se midió el magnesio en solución.
Método de Ensayo 5 Determinación de sulfato
Sulfato total en el compuesto
[0289] El sulfito (SO3) se mide en el compuesto por medición de XRF (Método de Ensayo 1) y se expresa como sulfato total (SO4) de acuerdo con:
Total SO4(% en peso) = (SO3) x 96/80
Total SO4 (mol) = SO4 total (% en peso) / peso molecular SO4
Sulfato de sodio (forma soluble de sulfato presente en el compuesto)
[0290] Na2O se mide en el compuesto por medición XRF (Método de Ensayo 1).
Se supone que el Na2O está asociado con la forma más soluble de SO4 en forma de Na2SO4 presente en el compuesto.
En consecuencia, el número de moles de Na2O se supone igual a la de la forma soluble de sulfato y por lo tanto se calcula como:
SO4 soluble (mol) = Na2O (mol) = % en peso Na2O / peso molecular Na2O Sulfato entre capas (forma insoluble de sulfato presente en el compuesto también denominado sulfato unido)
[0291] El sulfato entre capas se calcula de acuerdo con
SO4 entre capas (mol) = SO4 total (mol) - SO4 soluble (mol)
SO4 entre capas (% en peso) = SO4 entre capas (mol) x peso molecular SO4 Método de Ensayo 6 Análisis de contenido de carbono por el método Leco
[0292] Este método se usó para determinar los niveles de contenido de carbono (indicativo de la presencia del anión carbonato presente en el compuesto metálico mixto).
Una muestra de masa conocida se quema a aproximadamente 1350°C en un horno en una atmósfera de oxígeno puro. Cualquier carbono en la muestra se convierte en CO2 que se pasa a través de una trampa de humedad antes de ser medida por un detector infrarrojo. Al comparar con un estándar de concentración conocida, se puede encontrar el contenido de carbono de la muestra. Se usó un analizador de carbono y azufre Leco SC-144DR, con suministro de oxígeno, botes de combustión de cerámica, lanza para botes y tenazas. Se pesaron 0,2 g (+/- 0,01 g) de muestra en un bote de combustión. Luego se colocó el bote en el horno Leco y se analizó el contenido de carbono. El análisis se realizó por duplicado.
[0293] El % C fue determinado por:
%C (muestra) = (%C1 %C2)/2
Donde C1 y C2 son resultados individuales de carbono
[0294] Los resultados de las mediciones de contenido de carbono se ven en la Tabla 3 y la Figura 1 y se expresaron como % CO2 =% C x 44/12
Método de Ensayo 7 Tiempo de lavado
[0295] El tiempo de lavado se midió en minutos redondeados al minuto más cercano, fue el tiempo que tardó en pasar un lavado (es decir, un volumen de agua de lavado) a través del filtro. La torta húmeda no se dejó secar ni agrietar durante este período. El tiempo se midió usando un cronómetro.
Método de Ensayo 8 Tiempo de filtración (escala de laboratorio)
[0296] El tiempo de filtración se midió en minutos redondeados al minuto más cercano, fue el tiempo que llevó la lechada a pasar por el filtro, pero la torta húmeda resultante no se dejó secar. El tiempo se midió usando un reloj de parada.
Método de Ensayo 9 Distribución del tamaño de partícula (PSD) por Lasentech
[0297] En el proceso de distribución de tamaño de partícula en la suspensión se midió usando una sonda Lasentech El tamaño medio de partícula d50, se obtiene como parte de esta técnica analítica.
Método de Ensayo 10 Velocidad de filtración (ml/min)
[0298] Definido como la cantidad de filtrado obtenido en un tiempo dado
Método de Ensayo 11 Velocidad de filtración (kg de producto seco/m2.h)
[0299] La velocidad de filtración (kg de producto seco/m2.h) se define como la masa de la torta húmeda, expresada como compuesto seco, aislado, lavado, deshidratado y descargado por hora, dividido por el área del filtro utilizado. Método de Ensayo 12 Contenido de humedad
[0300] El contenido de humedad del compuesto metálico mixto se determina a partir de la pérdida de peso (LOD) después del secado a 105°C durante cuatro horas a presión ambiente en un horno de laboratorio
Método de Ensayo 13 ídeiado en blanco intencionalmentel
Método de Ensayo 14 Área de superficie y volumen de poro (Método de nitrógeno -N?)
[0301] Mediciones de área superficial y el volumen de poro se obtuvieron usando adsorción de gas nitrógeno en un intervalo de presiones relativas usando un Micromeritics Tristar ASAP 3000. Las muestras se desgasificaron bajo vacío durante 4 horas a 105°C antes del comienzo de las mediciones. Típicamente, se obtuvo un vacío de <70 mTorr después de la desgasificación.
[0302] Las áreas superficiales fueron calculadas mediante la aplicación de Teller (BET) de teoría usando datos de adsorción de nitrógeno de Brunauer, Emmett y obtenido en el intervalo de presión relativa de 0,08 hasta 0,20 P/Po.
[0303] El volumen de poro se obtuvo del circuito de desorción de la isoterma de adsorción de nitrógeno, usando el volumen de gas adsorbido a una presión relativa (P/Po) de 098. La cantidad de gas adsorbido a 0,98 presión relativa (en cc/g en STP) se convierte en un volumen equivalente líquido multiplicando por el factor de conversión de densidad de 0,0015468. Esto da la cifra del volumen de poro reportado en cm3/g.
P = presión de vapor parcial de nitrógeno en equilibrio con la muestra a 77K
Po = presión saturada de gas nitrógeno.
Método de Ensayo 15 Volumen de poros (método del agua)
Volumen de poros de agua
Objetivo
[0304] Para llenar los poros internos de una muestra (en forma de polvo) con agua de modo que, cuando se llenan todos los poros, la tensión superficial del líquido hace que la mayoría de la muestra forme un agregado que se adhiere a un frasco de vidrio en inversión de la jarra.
Equipo
[0305]
(1) Tarro de polvo de cuello ancho (30 mm) de vidrio transparente 120 cm3 con tapón de rosca. Dimensiones: Altura 97 mm. Diámetro exterior 50 mm (número de pieza Fisher BTF-600-080)
(2) 10 cm3 Bureta de grado A
(3) Agua desionizada
(4) Tapón de goma de 74 mm de diámetro superior cónico a 67 mm. Altura total 49 mm
(5) Balance de 4 decimales calibrado
Procedimiento
[0306]
(1) una muestra de 5,00 g (±0,01) en el frasco de vidrio, agregue una alícuota de agua de 1 cm3.
(2) Después de esta adición, golpee vigorosamente el extremo inferior del frasco sellado contra el tapón de goma 4 veces.
(3) Con un movimiento brusco del brazo, mueva el frasco con la muñeca para invertir el frasco y verifique la muestra:
a Si la muestra se aglomera y la mayoría (> 50%) de la muestra se adhiere al frasco, este es el punto final (vaya a la sección de resultados a continuación). Si se observa agua libre con la muestra, se ha excedido el punto final y la prueba debe descartarse y comenzar de nuevo con una nueva muestra.
b Si la muestra se desprende del frasco (incluso si la aglomeración es evidente), agregue otros 0,1 cm3 de agua y repita los pasos (2) a (3) anteriores hasta llegar al punto final (3a)).
[0307] Resultados
[0308] El volumen de poros de agua se calcula de la siguiente manera:
Volumen de poros de agua (cm3/g) = Volumen de agua añadida (cm3) / peso de muestra (g) Método de Ensayo 16 Agua total agregada/kg APl - punto de granulación
[0309] Esta es la cantidad de agua añadida a una mezcla seca de 80% en peso de compuesto metálico mixto y 20% en peso de excipientes para formar granulados (es decir, hasta que se alcanza un punto de granulación).
Método de Ensayo 17 Volumen de la tableta
[0310] El volumen de la tableta se calcula a partir de las dimensiones de la tableta usando un paquete de diseño de computadora (iHolland Ltd).
Método de Ensayo 18 Tasa de secado
[0311] Para el cálculo de la velocidad de masa secada (kg de agua/(kg de producto seco.h)) de agua eliminada durante el secado por unidad de tiempo se dividió por la masa del producto seco producido. El tiempo utilizado para calcular la velocidad de secado es el tiempo de residencia del secador definido en el Método de Ensayo 19.
Método de Ensayo 19 Tiempo de residencia en la secadora
[0312] Para secadores de residencia larga, el tiempo de residencia es el tiempo durante el cual se elimina el agua del material que se está secando.
[0313] Para secadores de residencia corta como el secado por pulverización, el tiempo de residencia se calcula de la siguiente manera.
[0314] Primero se determina el volumen interno del secador. El tiempo de residencia del aire o gas alimentado a la secadora es entonces calculado dividiendo el volumen del intervalo por el flujo de aire o gas. Se supone, dado que no se produce una acumulación significativa de sólidos dentro del secador, que el tiempo promedio de residencia de partículas es aproximadamente igual al tiempo de residencia de aire o gas.
Método de Ensayo 20 Tasa de producto por unidad de área
[0315] La tasa de producto por unidad de área kg producto/(m2 h) se puede calcular dividiendo la masa del producto seco producido por unidad de tiempo con el área de superficie utilizada para calentar.
Método de Ensayo 21 Delta T
[0316] La temperatura delta se define para procesos de secado de residencia corta como (Tin - Tout)/Tin donde Tin es la temperatura del gas de entrada, °C
Tout es la temperatura del gas de salida o la temperatura del producto,°C (suponiendo que el gas y el producto está a la misma temperatura)
Método de Ensayo 22 Densidad a granel aprovechada
[0317] La densidad a granel aprovechada se determinó usando un recolector automático Copley JV1000. La medición se realizó mediante la adición del producto (50,0g, /- 5,0g) en un cilindro de medición limpio (dedicado para el aparato). Se anotó el peso exacto. Se anotó el volumen inicial. Luego se colocó el cilindro en el recolector automático y la máquina se configuró para 3750 toques ingresando el número de toques requeridos y luego presionando inicio. El volumen del cilindro se volvió a tomar cuando se completó el número total de tomas (volumen final). La densidad a granel aprovechada obtenida se calculó de la siguiente manera:
Densidad a granel aprovechada (g/ml) = peso (g) / volumen final (ml) Método de Ensayo 23 Índice de fluidez de Carr
[0318] El índice Carr se calculó usando la siguiente fórmula y los datos disponibles de la prueba de densidad a granel aprovechada,
Indice de Carr (%) = 100 * ((volumen inicial (ml) - volumen final (ml)) / volumen inicial (ml)) Un resultado superior al 25% indica una capacidad de flujo deficiente y menos del 15% indica una buena capacidad de flujo.
Método de Ensayo 24 Distribución promedio del tamaño de partícula (d50 PSD) de polvos
[0319] El tamaño de partícula se determinó usando un Mastersizer 'S' equipado con una lente 300Rf y una unidad de dispersión DIF 2012. Los datos fueron interpretados y analizados utilizando el software Malvern Mastersizer. El Malvern estaba conectado para procesar el suministro de agua. Se utilizaron los siguientes parámetros del programa, 80% de velocidad de la bomba, 80% de velocidad del agitador, 50% de ultrasonidos y 3 minutos de tiempo de residencia. Se comprobó que el nivel de fondo era inferior a 100 unidades. Cuando el programa lo solicitó, la muestra se agregó en porciones para alcanzar entre 15% y 25% de oscurecimiento. El análisis comenzó automáticamente. Se comprobó que el residuo era inferior al 1%. La muestra se analizó por duplicado. Los resultados se calcularon usando el software tomando el % de volumen bajo los tamaños de partícula entre 1,85 y 184 micras. Esto se expresó como resultados percentiles con el tamaño medio de partícula (D50, 50° percentil), 90° percentil (D90) y 10° percentil (D10). Método de Ensayo 25 Análisis de metales de Al, Cr, Pb
[0320] Las muestras se acidificaron, diluyeron y los metales especificados se analizaron usando ICP-MS. Las muestras fueron analizadas por duplicado.
Método de Ensayo 26 Contenido total de metales pesados
[0321] Los metales se determinaron acidificando las muestras primero seguido de análisis usando ICP-MS. El contenido total de metales pesados (ppm) se calculó sumando los siguientes metales: como (ppm) Cd (ppm) Pb (ppm) Hg (ppm) Sb (ppm) Mo (ppm) Cu (ppm)
Método de Ensayo 27 - Potencia por unidad de volumen
[0322] La aplicación de software dinámica de fluidos computacionales (CFD) se utilizó para simular el flujo de fluido dentro del recipiente de reacción para establecer los requisitos en el equipo de mezcla.
RESULTADOS Y DISCUSIÓN
[0323] Hemos encontrado problemas críticos con el proceso de fabricación a mayor escala (definido como desde la reacción hasta las etapas de secado) cuando intentamos evitar un aumento en el tamaño del cristalito. Esto se describe en más detalle a continuación
ENLACE DE FOSFATO
[0324] Para dosis altas diarias y repetidas a largo plazo (crónicas) requeridas para pacientes con riñón, el recuento diario total de píldoras requiere una carga baja de tabletas debido a la ingesta restringida de líquidos, en consecuencia,
una dosis alta de sustancia farmacológica (compuesto metálico mixto) de hasta 80% en peso se requiere en el producto final (es decir, tableta) mientras se mantiene una buena actividad terapéutica (tal como unión a fosfato) y estabilidad de almacenamiento. Hemos encontrado que el producto final es, por lo tanto, muy sensible a una serie de propiedades químicas y físicas opuestas del compuesto metálico mixto, como la composición (relación Mg:Fe, sulfato), tamaño de cristalito, propiedades morfológicas (área de superficie, tamaño de partícula, volumen de poro) de los compuestos metálicos mixtos. Esto es diferente a los requisitos normales impuestos a los productos farmacéuticos que típicamente contienen sustancias farmacéuticas de tipo orgánico más solubles en concentraciones más bajas que dependen menos de una morfología particular.
[0325] Las variantes de la estructura de hidrotalcita Mg:Fe que tenían diferentes relaciones molares Mg:Fe de 2:1, 3:1 y 4:1 se compararon para el rendimiento de unión a fosfato y la liberación de magnesio (Tabla 2). La liberación del magnesio, asociada con el uso farmacéutico de compuestos metálicos mixtos, puede reducirse seleccionando una relación molar adecuada de Mg:Fe. Los datos mostraron que el material con una proporción de 2:1 tenía la mayor unión de fosfato por mol de magnesio liberado en una prueba de unión de fosfato en presencia de una suspensión de harina. Los datos también muestran que una relación molar Mg:Fe de 2:1 no tiene la presencia de ninguna otra fase no hidrotalcita. Además, hemos encontrado que los compuestos metálicos mixtos no envejecidos con un tamaño de cristalito inferior a 200 angstrom (A) proporcionan una mayor unión al fosfato que los compuestos de edad que normalmente tienen un tamaño de cristalito muy superior a 200 A.
Tabla 2 Selección de la relación molar de Mg:Fe preferida y tamaño de cristalito

[0326] Sin embargo, hemos encontrado que si se procesa incorrectamente, el tamaño del cristalito de los compuestos metálicos mezclados continuará creciendo en tamaño y es difícil de filtrar, particularmente a gran escala, esto presenta problemas significativos. Hemos descubierto un nuevo proceso para el control de diferentes etapas de producción (desde la reacción hasta el secado), como evitar el crecimiento del tamaño del cristalito por encima de 200 A para mantener la actividad de unión al fosfato sin dificultar significativamente el proceso de aislamiento, lavado y secado del compuesto. Esto se logró mediante una cuidadosa selección y control de las condiciones específicas del proceso. Nuestro enfoque se describe con más detalle en los siguientes ejemplos.
PRECIPITACIÓN
[0327] Hemos encontrado que las ventajas del compuesto metálico mixto de la relación molar de Mg:Fe de aproximadamente 2:1, tales como una buena unión a fosfato, no solo están determinadas por el tamaño del cristalito sino también preferiblemente por los bajos niveles de sulfato entre capas y el método de fabricación (Tabla 3 y Figura 1). Además, en todo el intervalo de pH considerado, la filtración es difícil debido a la estructura típica del material similar a la arcilla.
[0328] Cuando se prepara el compuesto metálico mixto con el anión carbonato, puede ser posible la presencia de un segundo tipo aniónico. La presencia de un solo tipo aniónico puede considerarse más deseable que una mezcla de aniones. Sorprendentemente, descubrimos que no es necesariamente óptimo no tener sulfato, sino que debe existir una pequeña cantidad de sulfato como sulfato entre capas (unido) para aumentar las tasas de filtración de la estructura tipo arcilla, mientras que el sulfato en forma de sales solubles como Na3SO4 deben eliminarse. Descubrimos que la mayoría de la sal de Na2SO4 soluble se puede eliminar fácilmente mediante lavado, mientras que el sulfato entre capas es menos soluble y sus niveles se controlan principalmente por la cantidad de exceso de Na2CO3 en la receta, el pH de la reacción y el grado de envejecimiento en la suspensión de reacción (es decir, temperatura de la suspensión de reacción). Por ejemplo, hemos encontrado que el sulfato entre capas disminuye cuando: aumenta la temperatura de reacción de la suspensión, aumenta el exceso de Na2CO3, aumenta el pH.
[0329] Opcionalmente, si el sulfato entre capas necesita reducirse aún más para lograr una pureza aún mayor (es decir, menos del 10% en peso de sulfato entre capas) y no se deben reducir las tasas iniciales de aislamiento y lavado, puede ser posible lavar la torta del filtro de nuevo pero esta vez con una solución de Na2CO3 (preferiblemente hasta
una concentración de 1 M) seguido de lavado con agua. Este proceso puede reducir o eliminar el sulfato entre capas restante sin necesariamente reducir las tasas de filtración o la unión de fosfato. Sin embargo, se prefiere si la mayoría del sulfato entre capas se elimina durante la etapa de reacción en lugar de requerir la necesidad de lavar con carbonato. Los pasos adicionales del proceso pueden disminuir rendimiento, así como alentar el crecimiento del tamaño de cristalita.
[0330] Por lo tanto, en otro aspecto de la descripción, el compuesto se lava primero con agua para eliminar el SO4 soluble y el sodio, seguido de un lavado con una solución de Na2CO3 para eliminar el sulfato entre capas, seguido de un lavado final con agua para eliminar los iones solubles restantes. Preferiblemente, el compuesto se pone en suspensión con un poco de solución Na2CO3 para un máximo de 1 hora para permitir el intercambio del sulfato entre capas intermedio para el carbonato Se cree que el lavado con un exceso de Na2CO3 alentaría eliminación de sulfato restante de la región de capa intermedia. En este aspecto, después del intercambio de la capa intermedia de sulfato por el carbonato, se puede proporcionar un compuesto metálico mixto libre de Al con menos del 1% en peso de sulfato entre capas (preferiblemente menos del 05% en peso) y menos del 0,5% en peso de sulfato soluble.
[0331] Cuando el producto no se lava con solución de Na2CO3, también hemos encontrado que la unión a fosfato varía en función del nivel de sulfato, por ejemplo, existe un nivel óptimo de sulfato entre capas de entre 1,8 y 5% en peso, en donde una buena unión a fosfato y se mantiene la filtración. La unión de fosfato disminuye por debajo del 1,8% en peso. Por encima del 5% en peso, la capa intermedia de SO4 se vuelve más variable y el nivel de SO4 es demasiado alto para ser aceptable y aumenta el tiempo de lavado y filtración. Los mejores resultados se obtuvieron entre 2,5 y 5% en peso de sulfato entre capas.

SEPARACIÓN
[0332] Las características de los compuestos metálicos mixtos libres de Al que resultan de su estructura similar a la arcilla, que reemplaza Al con Fe y su forma no envejecida presentan limitaciones cuando se fabrican a escala comercial. Deben resolverse las limitaciones, como la filtración difícil y la dureza del material, manteniendo al mismo tiempo un proceso a escala y compuestos metálicos mixtos con buena unión al fosfato, estabilidad de almacenamiento y que no afecten negativamente los procesos de fabricación posteriores utilizados para producir el producto farmacéutico formulado final que contiene el compuesto metálico mixto.
[0333] Durante el escalado descubrimos que era difícil preparar este material cuando se usaban técnicas de filtración tradicionales tales como un filtro de correa, filtro de presión Neutsche. Incluso un método de centrifugación no funcionó eficazmente a esta gran escala.
[0334] Resolvimos este problema seleccionando rangos específicos de uno o más de los siguientes: (i) selección del rango de sulfato entre capas (de 1,8 a 5% en peso) mediante el control de Na2CO3 y la selección de pH (ii) una psd preferida de la suspensión de reacción (D50> 40 micras, preferiblemente superior a 70 micras) y el contenido de humedad de la suspensión de reacción (más del 90% en peso) y la torta de filtración (menos del 80% en peso) (iii) selección de un régimen de agitación específico (una potencia por unidad de volumen de 0,03 a 1,6 kW/m3), (iv) selección de un método de filtración preferido y su funcionamiento (centrífuga). En un aspecto altamente preferido, seleccionamos cada uno de los siguientes: (i) selección del rango de sulfato entre capas (de 2 a 5% en peso) mediante el control de Na2CO3 y pH (ii) selección de una psd preferida de la suspensión de reacción (D50> 40 micras, preferiblemente más de 70 micras) y contenido de humedad de la suspensión de reacción (más del 90% en peso) y torta de filtro (menos del 80% en peso) (iii) selección de un régimen de agitación específico (una potencia por unidad de volumen de 0,03 a 1,6 kW/m3), (iv) selección de un método de filtración preferido (centrífuga).
(i) Sulfato entre capas
[0335] Una alta tasa de filtración y un bajo tiempo de lavado son ventajosos cuando se trata de fabricar los compuestos metálicos mixtos de MgFe a escala comercial y para evitar el crecimiento de cristalitos. Sin embargo, los compuestos metálicos mixtos que consisten en niveles bajos de sulfato entre capas (menos del 1,8% en peso) son más difíciles de filtrar y lavar, mientras que si el contenido de sulfato es demasiado alto (superior al 5% en peso), el tiempo de lavado aumenta nuevamente (Tabla 3 y Figura 1). Encontramos que los niveles de sulfato entre capas se pueden mantener entre 2-5% en peso controlando la temperatura de la suspensión de reacción, el pH durante la reacción y un rango de exceso de Na2CO3 (XS) de cualquiera de las siguientes combinaciones que se muestran a continuación.
[0336] Cuando la suspensión se mantiene a una temperatura entre 15 y 30°C en donde el Na2CO3 se proporciona en un exceso del requerido para completar la reacción y un pH a:
(i) 9,5 < pH <11 y 0 < Na2CO3 < 1 moles
(ii) 9,5 < pH < 10,5 y 1 < Na2CO3 < 2 moles
(iii) 95 < pH < 10,1 y 1 < Na2CO3 < 2,7 moles
(iv) 9,5 < pH < 10 y 1 < Na2CO3 < 4 moles
(v) 9,5 < pH < 9,8 y 1 < Na2CO3 < 5 moles
[0337] Cuando la suspensión se mantiene a una temperatura de 30 a 60°C, en donde el Na2CO3 se proporciona en un exceso del requerido para completar la reacción y un pH a:
(i) 9,5 < pH < 11 y 0 < Na2CO3 < 2
(ii) 9,5 < pH < 10,5 y 0 < Na3CO3 < 2,7 moles
(iii) 9,5 < pH < 10 y 0 < Na3 CO3 < 4 moles
[0338] El exceso de Na3CO3 (XS) se define como el exceso del que se requiere para completar la reacción de 4 MgSO4 Fe2(SO4)3 12 NaOH (XS+1) Na2CO3 -> Mg4Fe2(OH)-,2.CO3.nH2O 7 Na2SO4 (XS) Na2CO3.
[0339] Para compuestos metálicos mixtos, mantener la relación molar del metal objetivo (Mg:Fe) en aproximadamente 2 (1,8 a 2 ,2 , preferiblemente 1,7 a 2 ,1 ) durante la reacción mientras se controla el sulfato entre capas es difícil ya que ambos se ven afectados de manera opuesta por la manera en que se procesa el material. Además, descubrimos que la estequiometría correcta no solo está determinada por las relaciones correctas de los materiales de partida, sino también por el pH de la reacción. Por ejemplo, cuando el pH es demasiado bajo (pH inferior a 9,5) puede producirse una precipitación incompleta de magnesio, por lo que la relación molar Mg:Fe cae muy por debajo del valor objetivo de 2 y tampoco está libre de fases cristalinas no hidrotalcita. Por lo tanto, se prefiere mantener el pH entre 9,5 y 11 y preferiblemente entre un rango de pH aún más estrecho de 9,5-10 y más preferiblemente a 9,8 para proporcionar la relación óptima de magnesio: hierro (1,8 a 2,2, preferiblemente 1,7 a 2,1) mientras mantener buenas tasas de filtración durante la fabricación a escala, mantener una buena unión de fosfato y prevenir el crecimiento de cristales mediante el control de la distribución del tamaño de partícula y el sulfato entre capas.
[0340] La cantidad total de anión (Ccaic) predicha para un compuesto metálico mixto si fuera de una fase de tipo hidrotalcita ideal de una relación molar M2+M3+ de 2:1 puede calcularse mediante la siguiente fórmula: Ccaic = (M3+/(M2+ M3+))/n en donde n es la carga del anión. Por ejemplo, una relación molar M2+:M3+ de 2:1 y una carga de aniones supuesta n = 2 (es decir, como para CO32- o SO42- daría como resultado un valor predicho para (Ccalc) de 0,17. El valor experimental para Cexp se puede determinar a partir de la suma de la cantidad (equivalente molar) de anión sulfato y carbonato. El A se define como la diferencia entre el C calculado y el experimental en el que un valor A más bajo indica una fase de hidrotalcita más pura. El valor A más pequeño se observa cuando se precipita por encima del pH 9,5.
[0341] Los datos de la Tabla 3 (mostrados en la Figura 1) y la descripción de los ejemplos 8-24 en la sección de ejemplos muestran que la mejor calidad general (es decir, buena unión a fosfato, altas tasas de filtración, bajos tiempos de lavado, una relación molar de aproximadamente 20, no se obtienen fases cristalinas que no sean hidrotalcita y un A pequeño para aquellas muestras en las que los niveles de sulfato entre capas están entre 2 y 5% en peso, y preferiblemente una relación molar de sulfato a carbonato de entre 0,14 a 0,26. La cantidad total de anión (sulfato y carbonato) es preferiblemente de 0,15 a 0,20 más preferiblemente es de 0,19 equivalentes molares.
[0342] Para controlar el sulfato entre capas por debajo del 3%, el Na2CO3 tenía que tener un exceso de más de 2 moles. Para mantener el sulfato entre capas por encima del 2% en peso, el pH de la precipitación tenía que ser inferior a 10 y un exceso de 2,7 moles de Na2CO3 o menos.
[0343] El carbonato de sodio no solo proporciona el carbonato para los sitios de intercambio aniónico, sino que también actúa como un tampón de pH que ayuda al control del pH durante la precipitación. La capacidad de mantener un pH de precipitación preciso aumenta considerablemente cuando está presente Na2CO3 y, por esa razón, se prefiere un exceso de Na2CO3 de más de 2. Sin embargo, encontramos que un exceso de Na2CO3 de 4 o superior es menos preferido porque esto podría resultar en un mayor riesgo de disolución incompleta de Na2CO3 en la solución reactiva a las temperaturas de reacción preferidas (de menos de 25°C) al preparar compuestos metálicos mixtos no envejecidos.
[0344] Por ejemplo, durante la disolución del hidróxido de sodio y el carbonato de sodio en las soluciones de alimentación, la temperatura de la solución puede aumentar a 65°C y encontramos que un exceso en Na2CO3 de 4 o más se disuelve; sin embargo, se requirió enfriamiento y/o presurización durante la disolución para limitar la evaporación y reducir la temperatura al mismo valor que el requerido para que la reacción evite el envejecimiento. Cuando la solución de Na2CO3 (de más de 4 moles en exceso) se enfría, se produce una precipitación parcial de 65 a 25°C del Na2CO3.
[0345] Por consiguiente, era preferible mantener el Na2CO3 a exceso de 4 moles o menos. Descubrimos que era posible reducir el exceso de Na2CO3 de 4 a 2,7 moles sin afectar el control del pH.
[0346] En resumen, los datos de la Tabla 3 sugieren que cuando fuera de un intervalo de sulfato de capa intermedia 1,8 a 5%, unión a fosfato o bien disminuye y/o la relación molar Mg:Fe de 2,0 no se mantiene y/o separación de la suspensión es más difícil de lograr. Se apuntó una relación molar de Mg:Fe de 2,0 para obtener la mayor unión de fosfato por mol de magnesio liberado. Un intervalo preferido de sulfato de capa intermedia entre 1,8 a 5% se logró mediante la selección de pH y exceso de Na2CO3 dentro de un rango estrecho.
(ii) Suspensión de reacción psd y contenido de humedad de la torta del filtro
[0347] Distribución del tamaño de partícula (PSD): la distribución del tamaño de partícula es un parámetro importante del material que influye en el tiempo de filtración de la suspensión de precipitado de reacción. En el laboratorio con una concentración de reactivo similar, velocidad de adición de reactivo, temperatura de reacción y pH y volumen del talón de agua, las diferentes configuraciones de agitación produjeron diferentes PSD. Por lo tanto, la PSD del precipitado de reacción está fuertemente influenciada por el régimen de agitación, la configuración del recipiente y el modo de adición del reactivo. Hemos identificado las condiciones de agitación a escala comercial para permitir condiciones óptimas de filtración y lavado, asegurando al mismo tiempo que el producto final permanezca esencialmente igual al de las toneladas bajas por año.
[0348] Sin limitarse a la teoría, se postula que un pH elevado/mezcla subóptima puede dar como resultado un tamaño de partícula pequeño que puede bloquear la tela del filtro, reducir la velocidad de filtración a través de la torta y limitar el contenido de sólidos finales del filtro torta. Encontramos que hay un aumento significativo en el tiempo de filtración cuando la suspensión de reacción psd (d50) se reduce a menos de 70 micras. Las investigaciones (datos mostrados en la Tabla 4) demostraron que el control del tamaño de partícula por encima de aproximadamente 70 micras es preferible para mantener una alta tasa de filtración adecuada para el uso de métodos de separación a escala comercial, tales como centrífuga, Neutsch y filtros de correa. Además, descubrimos que el crecimiento no deseado de cristales (envejecimiento) puede minimizarse si el tiempo de filtración se mantiene al mínimo. También encontramos que una reducción en el tamaño de partícula a menos de 70 micras conduce a un aumento en el contenido de humedad de la torta de filtro a más del 80% en peso. Esta torta de filtro es más pegajosa y, por lo tanto, es más difícil de quitar del equipo de filtración y tenderá para sostenerse en dispositivos mecánicos o recipientes durante la manipulación. Un
contenido de humedad preferido de la torta de filtro es, por lo tanto, inferior al 80% en peso. Por consiguiente, también se prefiere separar el compuesto metálico mixto de la suspensión de reacción de más del 90% en peso de contenido de humedad.
[0349] Por lo tanto, existe una combinación preferida de una torta de filtro de contenido de humedad (menos del 80% en peso) y una PSD (de más de 70 micras) para permitir la fabricación en una escala más grande de composiciones libres de aluminio.
(iii) Régimen de agitación
[0350] Los resultados descritos en el presente documento demuestran que la PSD preferida de la suspensión de reacción se puede obtener al mantener el pH de la reacción entre pH 9,5-11 (preferiblemente a pH 9,5 a 10, más preferiblemente 9,8). En general, las enseñanzas del documento WO99/15189 no permitirían la separación del compuesto a escala comercial. Además, encontramos que se prefiere el método de agitación (potencia por unidad de volumen de 0,03 a 1,6 kW/m3) durante la precipitación. Agitación lenta (es decir, suficiente para mantener la solución homogénea) luego se mantuvo durante el tiempo de retención. Encontramos que el tiempo de filtración aumentó significativamente cuando la suspensión se agitó durante un período prolongado durante el tiempo de retención. Por ejemplo, descubrimos que se prefiere un tiempo de retención de más de 30 minutos pero menos de 12 horas y que la suspensión durante el tiempo de retención debe agitarse suavemente. El tiempo de retención de la suspensión se define como el período de tiempo entre el momento en que cesa la adición de las soluciones A y B (finaliza la fase de reacción) y la última parte alícuota de la suspensión se agrega al equipo de filtración. En la planta piloto y a gran escala comercial donde se utilizan centrifugadoras, el tiempo de retención de la suspensión es típicamente de hasta 12 horas, ya que se aíslan, lavan, desecan y descargan múltiples alícuotas de masa de reacción como torta húmeda.
[0351] La configuración de agitación de reacción específica para mantener condiciones de cizallamiento bajo mientras que al mismo tiempo permite una mezcla suficiente también resultó útil para obtener una psd preferida de más de 70 micras (cuando se mide al final del tiempo de retención). La entrada de potencia específica debe controlarse para evitar una velocidad que sea demasiado baja pero no a una velocidad que descomponga las partículas en partículas muy finas de psd de menos de 70 micras. La evaporación del agua de la suspensión de reacción y el envejecimiento de los cristalitos fue prevenido manteniendo la temperatura de reacción por debajo de 30 Celsius y típicamente no fue inferior a 15 Celsius para evitar una reducción inaceptable en la solubilidad de la corriente de alimentación del reactivo.

(iv) Selección de un método de filtración preferido y su funcionamiento.
[0352] En general, se usa un método de filtración para aislar el producto de la forma de suspensión, lavar a un punto final de impureza predeterminado y deshidratar la torta para obtener un material con un contenido de sólidos suficientemente alto para facilitar el manejo y para un secado eficiente. En el caso de los equipos de filtración de laboratorio, el deshidratado generalmente no se lleva a cabo debido a las limitaciones del equipo utilizado y a las pequeñas cantidades manipuladas.
Filtración de flujo tangencial (TFF)
[0353] Un método por el cual la velocidad de filtración aumenta cuando la psd es inferior a 70 micras es el método de filtración de flujo tangencial (diafiltración) (tablas 4 y 5); sin embargo, la diafiltración en general tiene tasas de filtración significativamente más bajas (en todos los rangos de psd) y solo es adecuada para la filtración de suspensiones diluidas de más del 94% en peso de contenido de humedad. La suspensión de reacción se puede lavar, pero no concentrar, ya que los contenidos de humedad por debajo de aproximadamente 94% en peso, conduciría al bloqueo del TFF. La suspensión lavada requeriría un aporte de energía mucho mayor durante el secado. En consecuencia, la separación por diafiltración es menos preferida.
Filtro Neutsche
[0354] El aislamiento y lavado de la sustancia farmacológica también se llevó a cabo usando un filtro Neutsche a una escala de producción de 7 kg. Este equipo proporcionó una buena separación y lavado del producto, pero la tasa de filtración específica (kg de producto/m2 h) fue extremadamente lenta y la torta del filtro contenía hasta 85% en peso de contenido de humedad que requería un uso de energía mucho mayor durante el secado. Se logró una profundidad de la torta de < 7 cm en el filtro Neutsche y 10 cm en un filtro/secador, mientras que las profundidades de la torta de 30 cm o más no son infrecuentes para filtros/secadores en otras aplicaciones, Separación, por filtro Neutsch que encontramos menos preferidos para fabricación a escala comercial debido a la menor tasa de filtración y las limitaciones en el manejo de grandes cantidades del producto similar a la arcilla.
Filtro de correa
[0355] Se prefiere un filtro de correa ya que descubrimos que estos podrían funcionar con un rango de profundidad de la torta de 15-25 mm a velocidades de filtración relativamente altas. Este método de filtración proporciona velocidades de filtración altas cuando el psd se mantiene por encima de 70 micras.
Centrífugo
[0356] Se ensayaron diferentes métodos de filtración, pero se obtuvieron los mejores resultados con un método de filtración usando una centrífuga que combina filtración seguido de lavado y deshidratación en un solo paso. Se prefiere la filtración centrífuga y proporciona ventajas de alta tasa de filtración y contenido de humedad preferido de la torta del filtro mientras se mantiene la calidad del producto (Tablas 4 y 5).
Tabla 5 Selección de métodos de filtración.

EL SECADO
[0357] Encontramos que demasiado procesamiento y manipulación; por ejemplo, como el secado excesivo puede presentar cambios (como el crecimiento del tamaño de cristalito) que son inaceptables en el compuesto final de Mg:Fe de metal mezclado. En la Tabla 6 se muestra cómo la morfología API (vi) afecta la estabilidad de almacenamiento y
el procesamiento posterior. El control de secado para lograr la porosidad requerida (vii) y el tamaño de cristalito (viii) se describe con más detalle en las Tablas 7 y 8
(vi) Morfología
[0358] Se requieren altas dosis diarias repetidas a largo plazo (crónicas) y una ingesta restringida de líquidos para pacientes renales. En consecuencia, se requiere una alta dosis de sustancia farmacológica en el producto final (es decir, tableta) y la fabricación y cualidades del producto final. Es, por lo tanto, sensible a las propiedades de forma y morfología de la sustancia farmacológica de compuestos metálicos mixtos, a diferencia de las formulaciones más típicas. Esto significa que las propiedades de la tableta, incluidas las propiedades físicas clave, y los procesos de fabricación de la tableta, como la granulación, a menudo están influenciados principalmente por las propiedades de la sustancia activa del compuesto metálico mixto en lugar de las de los excipientes, como es más típico caso. Para poder fabricar un producto farmacéutico que comprenda cantidades tan significativas de compuestos metálicos mixtos con el control y la consistencia necesarios para el uso farmacéutico, es esencial un medio para controlar una serie de estas propiedades físicas de los compuestos metálicos mixtos.
[0359] Es importante secar el material con cuidado ya que es fácil cambiar el área de superficie o el volumen de poro interno y, por lo tanto, cambiar la actividad terapéutica (Tabla 6).

[0360] La Tabla 6 muestra cómo el volumen de poros y el área de superficie afectan el control de la capacidad de unión a fosfato, la estabilidad de almacenamiento, el proceso de granulación y la producción de tabletas. Como regla general, se puede esperar que los materiales de tipo hidrotalcita de un área superficial más alta tengan una mayor capacidad de intercambio iónico y, por lo tanto, una mayor unión a fosfato; esto se puede ver en el Ejemplo 66, que tiene un área superficial alta de 119 m2/g y también un alto valor de unión a fosfato. Sin embargo, el material con el área superficial más alta de 119 m2/g resultó ser menos estable tras el almacenamiento porque la actividad de unión a fosfato disminuyó en un 12%. Hemos encontrado que se prefiere un rango de área superficial más bajo de entre 40 y 80 m2/g, ya que tiene la ventaja de mantener una buena unión a fosfato (más de 0,6 mmol/g API) que es importante también esencialmente sin cambios (solo 5% o menos cambio) tras el almacenamiento durante períodos de hasta años, lo que lo hace más viable como material farmacéutico activo. Por lo general, se puede esperar que se requieran áreas de superficie significativamente más altas para lograr una unión estable de fosfato, tales materiales de superficie inferior áreas (por N2) de entre 40-80 m2/g y tienen un volumen de poros (por N2) de 0,10-0,28 cm3/g y un volumen de poros (por agua) de 0,3-0,6 cm3/g Se puede esperar que g tenga una mayor sensibilidad a cualquier cambio en la estructura interna que resulte en la inhibición del acceso de los iones de fosfato al material y la consiguiente reducción en la capacidad de unión al fosfato. Sorprendentemente, los datos presentados en la Tabla 6 muestran que todos estos ejemplos de compuesto metálico mezclado de áreas de superficie inferior son estables al almacenamiento y mantienen una buena unión al fosfato. Además, los materiales de áreas de superficie más bajas, en el intervalo de entre 40-70 m2/g y un bajo volumen de poros (agua) de 0,3-0,6 cm3/g ofrecen la ventaja de un material más denso que luego puede procesarse en una forma de dosificación que es más pequeña (es decir, como se puede ver en la Tabla 6, volumen de tableta de menos de 500 mm3), mejorando así la carga de la tableta; Un problema frecuente en el tratamiento de pacientes renales. Además, un beneficio sorprendente adicional es que dichos materiales tampoco exhiben una reducción significativa en la tasa de absorción de fosfato, a pesar de las áreas de superficie más bajas. Esta faceta puede ser importante cuando se consideran dichos materiales para aplicaciones farmacéuticas en las que la unión del fosfato debe ser rápida, como el cuidado renal. También hemos encontrado que el material de cristalito de tamaño inferior a 200 A se une a más del 80% de fosfato en 10 minutos (de acuerdo con el Método de Ensayo 3 pero medido a diferentes intervalos de tiempo) cuando se mantiene a un tamaño de partícula promedio inferior a 100 |jm, preferiblemente menos de 50 jm , lo más preferido es menos de 10 jm y un área de superficie de más de 40 m2/g.
(vii) Fabricación de compuestos metálicos mixtos porosos no envejecidos (secado)
[0361] Hemos encontrado que el área de superficie de la sustancia farmacológica se determina mediante una combinación de velocidad de secado, tiempo de residencia, velocidad de producto por unidad de área y delta T (Tabla 7). La velocidad de secado se ve afectada tanto por el modo de secado como por otros parámetros del proceso, como la temperatura del producto, la superficie de calentamiento/temperatura del gas.
[0362] Un producto de tamaño de cristalito entre 90 y 200 A y un área de superficie (por N2) de entre 40-80 m2/g, y/o volumen de poro (por N2) de entre 0,10-0,28 cm3/g y/o volumen de poro (por agua) de entre 0,3-0,6 cm3/g puede ser logrado al exponer el producto crudo a una temperatura del producto de más de 80 pero no más de 150°C y proporcionar una velocidad de secado (velocidad de evaporación del agua) de entre 0,05 a 0,5 kg de agua por hora por kg de producto seco y/o proporcionar un tiempo de residencia de la secadora de entre 10 minutos a 30 horas y/o una tasa de producto por unidad de área de entre 0-7 kg de producto/(m2.h) típicamente lograda mediante el uso de una secadora de alto tiempo de residencia bajo un vacío de presión de 400 mbar (absoluto) o menos. Se puede obtener un producto con un rango de volumen de poro bajo (por agua) de 0,3-0,6 cm3/g mediante una combinación de la centrífuga y el uso del método de secado esférico agitado.
[0363] Alternativamente, un producto de cristalito de tamaño inferior a 140 A y un área de superficie (por N2) de entre 80-150 m2/g y volumen de poro (por N2) de entre 0,28-1,0 cm3/g, y/o un volumen de poro (por agua) de entre 0,6-1,2 cm3/g puede lograrse exponiendo el producto crudo a una temperatura del producto de más de 35°C pero no más de 150°C y proporcionar una tasa de secado (velocidad de evaporación del agua) de entre 500 a 50.000 kg de agua por hora por kg de producto seco y/o proporcionar un tiempo de residencia de la secadora de menos de 10 minutos y/o un delta T de entre 0,2 a 1,0, típicamente logrado mediante el uso de un secador de tiempo de residencia corto

(viii) Efecto de la velocidad de secado sobre el tamaño del cristalito
[0364] La Tabla 8 muestra que el secado debe ser lo suficientemente rápido como para minimizar el crecimiento de cristales, sin embargo, se deben evitar temperaturas de producto a granel superiores a 150°C para evitar daños a la estructura característica del material. Factores como la agitación durante el secado en secadores de tiempo de residencia prolongado también afectaron el tamaño del cristalito. Por ejemplo, las muestras secas (como las obtenidas por Neutsche/Tray Oven) por las cuales no se aplica agitación continua tienden a mostrar un tamaño de cristalito más pequeño que las obtenidas por un secador esférico. Por lo tanto, existe un régimen de secado óptimo.


[0365] Para aquellos compuestos en los que el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A) preferiblemente de 12 a 20 nm, el área superficial (por N2) está entre 40-80 m2/g, el volumen de poros (por N2) está entre 0,10-0,28 cm3/g y el volumen de poros (agua) es de 0,3-0,6 cm3/g Los compuestos de tamaño promedio de cristal entre 100 y 200 A se obtienen preferiblemente por uso de un proceso de secado agitado de larga residencia tal como un secador esférico agitado en el que la velocidad de secado está entre 0,09 a 0,31 kg de agua/(kg de producto seco.h), más preferiblemente una velocidad de secado entre 0,24 a 0,31 kg de agua/(kg de secado producto.h) y/o una tasa de producto por unidad de área (kg producto/m2 h) entre 1-10, más preferiblemente entre 2 7.
[0366] Hemos encontrado, sorprendentemente, que las ventajas del producto de área superficial inferior de tamaño cristalino entre 100 y 200 A y/o área superficial baja 40-80 m2/g y/o bajo volumen de poro (agua) 0,3-0,6 cm3/g preparado mediante un proceso de secado de larga residencia son una buena unión a fosfato, estabilidad de almacenamiento y un material más denso que puede procesarse en una forma de dosificación que es más pequeña, mejorando así la carga de la píldora en tabletas. El tamaño promedio de cristal de menos de 200 A tiene la ventaja de una buena unión controlada de fosfato. Se prefiere un tamaño de cristal promedio de entre 120-200 A si el producto se seca a un contenido de humedad inferior al 15% en peso (preferiblemente entre 5-10% en peso) de un tamaño de lote de entre 50 y 1000 kg cuando se seca mediante un proceso de secado de larga residencia con un secador esférico agitado y cuando se preparan materiales de baja superficie de entre 40 y 80 m2 por gramo y/o volumen de poro (agua) de 0,3-0 6 cm3/g. El producto de baja porosidad se prepara preferiblemente a partir de tamaños de lote entre 50 y 1000 kg con un tiempo de residencia del secador de entre 3 y 30 horas, más preferiblemente entre 5 y 30 horas y lo más preferido entre 9 y 30 horas.
FRESADO
[0367] Opcionalmente, después del paso de secado, el material seco puede clasificarse primero para eliminar partículas de gran tamaño mediante fresado y/o tamizado
(ix) Efectos de la PSD sobre la unión de fosfato y la liberación de magnesio.
[0368] Hemos encontrado que la liberación de Mg permanece constante mientras que la unión de fosfato cambia en función del tamaño de partícula (Tabla 9). Este es un resultado sorprendente, ya que se podría esperar que una distribución de tamaño de partícula más pequeña tenga un área superficial más grande y, por lo tanto, sea más susceptible a la liberación de Mg. Sin embargo, la liberación de magnesio no parece verse afectada significativamente por los cambios en el área de la superficie cuando se mantiene a menos de 80 m2/g a pesar de ser fresada a una distribución de tamaño de partícula (psd) con un (d50) de menos de 60 micras cuando se usa esta ruta. La liberación constante de Mg permite una selección más amplia de un rango de psd preferido para mejorar la unión de fosfato sin comprometer la liberación de Mg. Los datos de la Tabla 9 muestran que un tamaño de partícula preferido d50 es inferior a 177 micrómetros, más preferiblemente inferior a 114, lo más preferido inferior a 60 micrómetros.
Tabla 9 Efecto de la distribución del tamaño de partícula del producto seco sobre la unión de fosfato y la liberación de magnesio

[0369] Si se procesan compuestos metálicos mezclados incorrectamente, pueden volverse inaceptablemente duros. Esto puede conducir a problemas consecuentes de velocidades de fresado disminuidas y mayor aporte de energía para lograr un tamaño de partícula preferido. La consecuencia de esto es que para lograr un tamaño de partícula dado es esencial que el tamaño del cristalito no sea demasiado bajo.
[0370] La Tabla 10 muestra que si el tamaño del cristalito es demasiado bajo (es decir, de 120 A o menos), la velocidad de fresado se reducirá en más del 50% en comparación con el tamaño del cristalito de 195 A, lo que presentará
dificultades a escala cuando fresado a una distribución de tamaño de partícula con un d50 de menos de 114 mieras. Por ejemplo, problemas con la aparición de no hidrotalcita pueden ocurrir fases tales como periclasa de MgO, velocidad de fresado reducida o descomposición del producto debido al sobrecalentamiento del producto. Para aquellos compuestos metálicos mixtos en los que el tamaño del cristalito es inferior a 120 A, se prefiere usar la ruta de secado de residencia corta que no requiere fresado,
Tabla 10 Efecto del tamaño del cristalito sobre la velocidad de fresado y la unión a fosfato

[0371] Si el pH de la reacción sube por encima de pH 11 (y hasta cierto punto por encima de pH 10), hemos encontrado que los compuestos metálicos mixtos resultantes son un material mucho más duro. Por lo tanto, es posible preparar un material más blando por precipitación a pH 9,8 que a valores de pH más altos. En consecuencia, no solo la precipitación a un pH de 9,8 proporciona la ventaja de una mayor capacidad de filtración, también hemos demostrado que esto es beneficioso para lograr mayores tasas de fresado.
[0372] El control de la dureza del material también es importante porque esto también puede aumentar el potencial de recogida de niveles bajos de metales traza del equipo de fresado. Cuando el material es más duro, también debe ser fresado más duro, lo que a su vez puede generar temperaturas más altas durante el fresado, lo que proporciona un material fresado que puede contener productos de descomposición o puede estar demasiado seco (menos del 5% en peso de contenido de humedad según lo determinado por LOD) que a su vez puede generar problemas con el manejo y el procesamiento posterior.
(xi) Métodos de micronización
[0373] Si el contenido de humedad del producto no fresado es superior al 10% en peso, entonces el producto puede volverse demasiado pegajoso para el fresado, mientras que si es inferior al 5% en peso, el producto después del fresado estará demasiado seco y será menos estable tras el almacenamiento y/o proporcionar problemas en el procesamiento en formulaciones de tabletas. Descubrimos que el proceso de fresado da como resultado una pérdida adicional de humedad del 2% en peso que da como resultado un producto fresado. Por lo tanto, generalmente buscamos un contenido de humedad de entre 7 y 10% en peso para el material no fresado
[0374] Las propiedades químicas (es decir, la relación molar de Mg:Fe de 2,1) y físicas (es decir, área de superficie y tamaño de partícula) de la composición de compuestos metálicos mixtos favorecen el equilibrio a un contenido de humedad de 5-8% en peso y, como tal, pueden ser menores estable tras el almacenamiento (es decir, tiende a rehidratarse) si se fabrica con un contenido de humedad inferior al 5% en peso.
[0375] Hemos encontrado que este compuesto puede fabricarse usando un proceso que comprende una etapa de secado de residencia corta de manera que el material representativo resultante tiene un tamaño de cristal medio pequeño y un área de superficie alta, pero también presenta una unión de fosfato importante y sorprendente, incluso cuando el material no se muele más. El requisito de no fresar tiene la ventaja de reducir los pasos de procesamiento. Una ventaja adicional es que dicho material puede ser adecuado para procesos de formación de tabletas sin la necesidad de granulación húmeda debido a las propiedades ventajosas de flujo. Por lo tanto, en un aspecto, la presente invención proporciona un compuesto metálico mixto en el que el tamaño medio de cristal del compuesto metálico mixto es inferior a 20 nm (200 A); en este aspecto, preferiblemente el área superficial es de 80 a 145 m2 por gramo. Los datos de la Tabla 11 muestran que para un compuesto metálico mixto con un área superficial de 80 a 145 m2 por gramo, el tamaño de partícula preferido d50 es inferior a 343 micras, más preferiblemente inferior a 296, incluso más preferiblemente inferior a 203, lo más preferido menos de 31 micras.
Tabla 11

[0376] El compuesto de la mayor área superficial de 80-145 m2 puede ser fabricado usando un proceso que comprende una etapa de secado corto de residencia de tal manera que el material representativo resultante tiene tanto pequeño tamaño de cristal medio como alta área superficial sino que también exhibe importante y sorprendentemente alta fijación de fosfato incluso cuando el material no se muele más. El requisito de no fresar tiene la ventaja de reducir los pasos de procesamiento y evita cualquier problema de dureza. Una ventaja adicional es que dicho material puede ser adecuado para procesos de formación de tabletas sin la necesidad de granulación húmeda.
IMPUREZA
[0377] Los compuestos metálicos mixtos pueden sintetizarse mediante diversas técnicas; sin embargo, es difícil controlar los niveles de impurezas de los compuestos cuando se aíslan en forma no envejecida, a un grado farmacéutico y cuando se preparan sin AI, especialmente cuando se considera que los compuestos metálicos mixtos se preparan a partir de minerales que contienen niveles significativos de impurezas de metales traza, algunos de los cuales pueden estar en forma de metales pesados. En particular, los compuestos preparados a partir de minerales de hierro se mezclan considerablemente con otros tipos de metales, ya que estos se derivan de minerales que existen en la naturaleza. Algunos de estos metales pueden competir con el magnesio y el hierro para la formación del compuesto metálico mixto y quedar atrapados en la fase de hidrotalcita en lugar de formar sales más solubles que se eliminan fácilmente durante el proceso de lavado. Por lo tanto, es necesario controlar los niveles de impurezas de metales traza seleccionando las condiciones preferidas y la receta durante la etapa de precipitación; esto para cumplir con las pautas regulatorias mientras se obtiene a través de un proceso de fabricación que puede entregar esto a escala.
[0378] Otras impurezas, como el sodio y el sulfato, deben controlarse para que la sustancia farmacológica sea de calidad aceptable para el consumo humano. La concentración de sodio se controla mediante el lavado de la torta de la sustancia farmacológica aislada.
[0379] Durante la etapa de filtración y lavado del proceso de fabricación, la suspensión se forma en una torta (con la eliminación del exceso de licor madre). La torta resultante se lava luego con agua para eliminar el exceso de sodio, sulfato y carbonato hasta niveles aceptables para el uso final del material.
[0380] Para uso farmacéutico, es importante poder identificar y controlar la fase cristalina de interés. La forma en que se procesa el material influye en esto, cuando se prepara un compuesto a partir de 2 tipos de metales diferentes, es posible que precipite como una mezcla de compuestos metálicos individuales en lugar de un compuesto metálico mixto. Los compuestos metálicos mixtos se fabrican mediante coprecipitación, lo que puede fomentar la formación de diferentes fases cristalinas además de la fase de hidrotalcita. Por lo tanto, existe la necesidad de compuestos metálicos mixtos libres de Al que también estén libres de cualquier otra fase cristalina según lo determinado por la ausencia de líneas de difracción de XRD, excepto las atribuidas a la fase de hidrotalcita Cuando se prepara de acuerdo con el proceso definido para las muestras no envejecidas de tamaño de cristalito inferior a 200 A hemos encontrado que la fase de hidrotalcita tiene el siguiente análisis de difracción de rayos X de difracción sin la presencia de ninguna otra fase cristalina: dA ('d' espaciamientos) 7,74, 3,86, 2,62, 2,33, 1,97, 1,55, 1,52, 1,44. Cinco picos adicionales en dA 3,20, 1,76, 1,64, 1,48, 1,40 solo se resuelven en muestras más cristalinas, es decir, de un tamaño de cristalito superior a 200 A, típicamente como resultado del envejecimiento.
[0381] Impurezas de metales traza deben ser controladas con el fin de que la sustancia del fármaco es de calidad aceptable para el consumo humano. Encontramos, sorprendentemente, que las concentraciones de trazas de metales pueden controlarse mediante el pH de la reacción, tiempo de retención de la reacción (envejecimiento) y no solo como
se esperaría mediante la selección de materias primas de calidad adecuada o lavado. Por ejemplo, la Tabla 12 muestra cómo hemos podido reducir aún más los niveles de aluminio (Al) y plomo (Pb) mediante el control del pH, el control del exceso de carbonato de sodio y el control del envejecimiento.
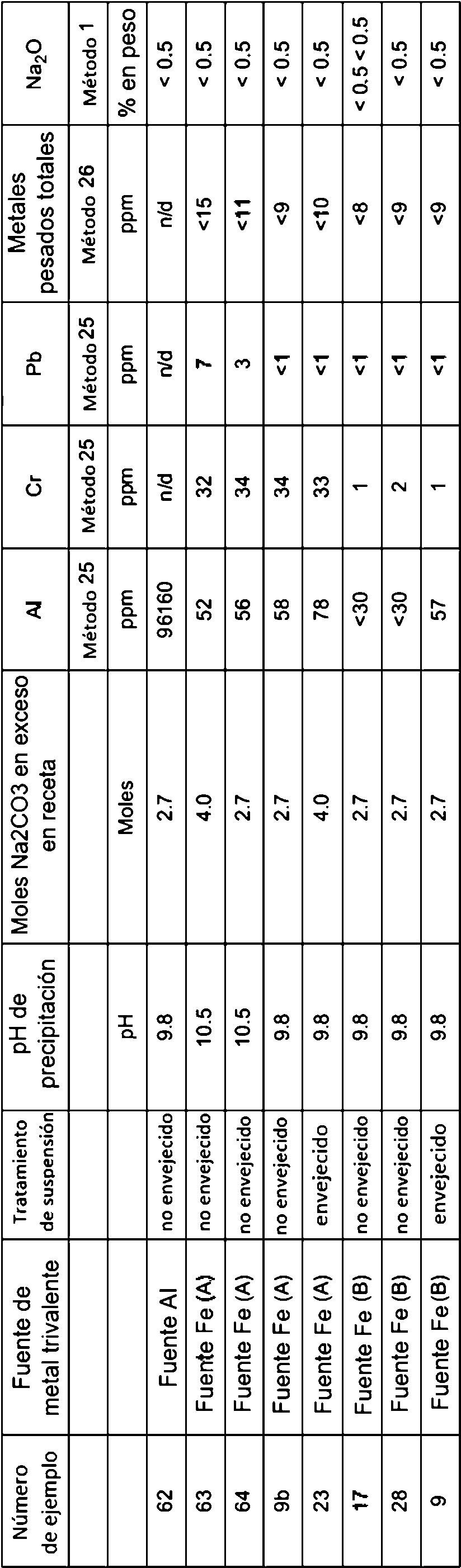
[0382] De la Tabla 12 es posible concluir que incluso cuando se cambia de una solución (A) de Rectapur de grado GPR a una fuente férrica más pura (B) tal como una solución (40,4 a 42,9% en peso de sulfato férrico del estándar de la industria del agua adecuado para consumo humano conforme a BS EN 890: 2004), los niveles de aluminio pueden reducirse aún más (es decir, a menos de 30 ppm) evitando el envejecimiento excesivo (es decir, en donde el tamaño del cristalito es > 200 A).
[0383] El ejemplo 62 se preparó con sulfato de aluminio sólido de Alfa Aesar 98% CAS 17927-65-0 en lugar de sulfato férrico. Todas las demás materias primas eran de la misma fuente.
[0384] Todas las muestras mostradas en la Tabla 12 se lavaron igualmente como se indica por niveles bajos (<0,5% en peso) y similares Na2O. El proceso de lavado se desarrolló para proporcionar los niveles de Na2O requeridos.
[0385] Como se describe en el presente documento, los niveles de aluminio del compuesto metálico mixto son inferiores a 10.000 ppm. Este nivel se considera exposición adecuada a Al para un individuo sano y es típico de los compuestos de grado farmacéutico (es decir, con una pureza del 99%). En contraste, los compuestos metálicos mixtos disponibles comercialmente como antiácidos en forma de una relación molar Mg:Al de 3:1, típicamente contienen diez veces más Al (es decir, hasta 100.000 ppm de aluminio) y, por lo tanto, no son adecuados para uso a largo plazo. Los pacientes renales son propensos a la acumulación de aluminio, por lo tanto, es más preferible si el contenido de aluminio es inferior a 2000 ppm (> 99,8% de pureza) en base a una ingesta diaria total de 6 g/día y orientación normativa general.
[0386] Hemos encontrado que se puede alcanzar un nivel de Al de 1000 ppm (99,9% de pureza) cuando se utiliza un proceso a gran escala para la fabricación de materiales no envejecidos. Para pacientes renales, se prefiere un contenido de aluminio lo más bajo posible y, por lo tanto, es más preferible un contenido de aluminio de menos de 1000 ppm. Con nuestro proceso, generalmente podemos alcanzar niveles de aluminio inferiores a 100 ppm; por lo tanto, los niveles de aluminio inferiores a 100 ppm son aún más preferidos. Mediante un control cuidadoso de las condiciones de reacción, podemos alcanzar niveles de aluminio inferiores a 30 ppm, que es el más preferido.
[0387] Los datos de la Tabla 12 también demuestran que es posible mantener los niveles de plomo (Pb) por debajo del límite de detección de < 1 ppm cuando se precipita a pH 9,8 y usando un exceso de 2,7 moles de Na2CO3 en la receta en lugar de precipitar el compuesto metálico mixto a pH 10,5 y usar un exceso de 4,0 moles de Na2CO3 incluso cuando se usa una fuente más impura de sulfato férrico. También descubrimos que el contenido total de metales pesados podría mantenerse a menos de 10 ppm de metales pesados totales (Método de Ensayo 26) cuando se usa la receta preferida de pH 9,8 y un exceso de 2,7 moles de Na2CO3.
[0388] Se requieren niveles de cromo (Cr) que se han de limita a < 25 ppm de acuerdo con la directriz de reactivos de metal para compuestos medicinales CHMP/CWP/QWP/4446/00. La Tabla 12 demuestra que hemos podido reducir los niveles de cromo de aproximadamente 35 ppm a menos del límite de detección (menos de 1 ppm).
[0389] Varias modificaciones y variaciones de la presente invención serán evidentes para los expertos en la técnica sin apartarse del alcance de la invención. Aunque la invención se ha descrito en relación con realizaciones preferidas específicas, debe entenderse que la invención como se reivindica no debe limitarse indebidamente a tales realizaciones específicas. De hecho, diversas modificaciones de los modos descritos para llevar a cabo la invención que son obvios para los expertos en química, biología o campos relacionados están destinados a estar dentro del alcance de las siguientes reivindicaciones.
Claims (10)
1. Un método para producir un compuesto metálico mixto de hidróxido doble en capas, que comprende al menos Mg2+ y al menos Fe3+ que tiene una relación molar de Mg2+ a Fe3+ de 2,5:1 a 1,5:1 que tiene un contenido de aluminio de menos de 10.000 ppm, con un tamaño de cristal promedio de menos de 20 nm (200 A), y con un área superficial de 40 a 80 m2 por gramo de compuesto metálico mixto, que comprende los pasos de:
(a) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde el pH de la suspensión se mantiene entre 9,5 y 11, y en donde el Na2CO3 se proporciona en exceso de 0 a 4,0 moles que se requiere para completar la reacción;
(b) someter la suspensión a mezcla en condiciones que proporcionen una potencia por unidad de volumen de 0,03 a 1,6 kW/m3;
(c) separar el compuesto metálico mixto de la suspensión, para obtener un producto crudo que tiene un contenido sólido seco de al menos 10% en peso
(d) secar el producto crudo calentando el producto crudo a una temperatura no más de 150°C y suficiente para proporcionar una velocidad de evaporación de agua de 0,05 a 1,5 kg de agua por hora por kg de producto seco; y
(e) fresar el producto crudo seco para proporcionar un área superficial de 40 a 80 m2 por gramo.
2. Un método de acuerdo con la reivindicación 1, en donde el compuesto es de la fórmula
MN1-xMmx(OH)2A%.mH2O (II)
en donde MII es uno o más metales bivalentes y es al menos Mg2+;
MIII es uno o más metales trivalentes y es al menos Fe3+;
An- es uno o más aniones n-valentes y es al menos CO3(i) 2-;
x/Zyn es de 1 a 1,2
0 < x <0,4,
0 < y < 1 y
0 < m <10.
3. Un método de acuerdo con una cualquiera de las reivindicaciones anteriores, en donde el pH de la suspensión se mantiene entre 9,5 y 10,5.
4. Un método de acuerdo con una cualquiera de las reivindicaciones anteriores, en el que el paso (a) comprende:
(A) combinar una sal Mg2+ y una sal Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura entre 15 y 30°C, y:
(i) en donde el pH de la suspensión se mantiene de 9,5 a menos de 9,8, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 5,0 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene a de 9,5 a menos de 10, y en el que el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 4,0 moles que se requiere para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre 9,5 y no más de 10,1, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 2,7 moles de lo que se requiere para completar la reacción; o
(iv) en donde el pH de la suspensión se mantiene de 9,5 a 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 1,0 a no más de 2,0 moles que el requerido para completar la reacción; o (v) en donde el pH de la suspensión se mantiene entre más de 9,5 y no más de 11, y en donde el Na2CO3 se proporciona en un exceso de 0,0 a no más de 1,0 moles que el requerido para completar el reacción;
o el paso (a) comprende:
(B) combinar una sal de Mg2+ y una sal de Fe3+ con Na2CO3 y NaOH para producir una suspensión, en donde la suspensión se mantiene a una temperatura de 30 a 60°C, y :
(i) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 11, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2 moles que el requerido para completar la reacción; o
(ii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10,5, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 2,7 moles de lo requerido para completar la reacción; o
(iii) en donde el pH de la suspensión se mantiene entre más de 9,5 y menos de 10, y en donde el Na2CO3 se proporciona en un exceso de más de 0 a menos de 4 moles que el requerido para completar la reacción.
5. Método según la reivindicación 1, en el que la velocidad de evaporación del agua es de 0,05 a 0,5 kg de agua por hora por kg de producto seco.
6. Un compuesto metálico mixto de hidróxido doble en capas que comprende al menos Mg2+ y al menos Fe3+, en donde
la relación molar de Mg2+ a Fe3+ es 2,5:1 a 1,5:1,
el compuesto metálico mixto tiene un contenido de aluminio de menos de 10.000 ppm, el tamaño promedio de cristal del compuesto metálico mixto es de 10 a 20 nm (100 a 200 A), el área de superficie es de 40 a 80 m2 por gramo de compuesto metálico mixto,
y opcionalmente:
el tamaño de partícula promedio d50 del compuesto metálico mixto es inferior a 100 pm, el volumen de poros de agua del compuesto metálico mixto es de 0,25 a 0,7 cm3/g de compuesto metálico mixto, y/o el contenido de sulfato entre capas del compuesto es de 2 a 5% en peso.
7. Un compuesto metálico mixto de acuerdo con la reivindicación 6, en donde el volumen de poros de agua del compuesto metálico mixto es de 0,25 a 0,7 cm3/g de compuesto metálico mixto.
8. Un compuesto metálico mixto de acuerdo con la reivindicación 6 o la reivindicación 7 para uso como medicamento.
9. Un compuesto metálico mixto de acuerdo con una cualquiera de las reivindicaciones 6 o 7 para uso en el tratamiento de la hiperfosfatemia.
10. Una composición farmacéutica que comprende un compuesto metálico mixto como se define en las reivindicaciones 6 o 7 y opcionalmente uno o más adyuvantes, excipientes, diluyentes o vehículos farmacéuticamente aceptables.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0913525.2A GB0913525D0 (en) | 2009-08-03 | 2009-08-03 | Method |
| PCT/GB2010/051271 WO2011015859A1 (en) | 2009-08-03 | 2010-08-02 | Method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2751325T3 true ES2751325T3 (es) | 2020-03-31 |
Family
ID=41129559
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES10739987T Active ES2751325T3 (es) | 2009-08-03 | 2010-08-02 | Hidróxidos dobles con capas de magnesio-hierro, su producción y su utilización para el tratamiento de hiperfosfatemia |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US9314481B2 (es) |
| EP (2) | EP3611131A1 (es) |
| JP (3) | JP5894073B2 (es) |
| CN (3) | CN102498068B (es) |
| AR (2) | AR077500A1 (es) |
| DK (1) | DK2462062T3 (es) |
| ES (1) | ES2751325T3 (es) |
| GB (1) | GB0913525D0 (es) |
| PT (1) | PT2462062T (es) |
| TW (3) | TWI649267B (es) |
| WO (1) | WO2011015859A1 (es) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9720061D0 (en) | 1997-09-19 | 1997-11-19 | Crosfield Joseph & Sons | Metal compounds as phosphate binders |
| MY157620A (en) | 2006-01-31 | 2016-06-30 | Cytochroma Dev Inc | A granular material of a solid water-soluble mixed metal compound capable of binding phosphate |
| GB0714670D0 (en) * | 2007-07-27 | 2007-09-05 | Ineos Healthcare Ltd | Use |
| GB0720220D0 (en) * | 2007-10-16 | 2007-11-28 | Ineos Healthcare Ltd | Compound |
| GB0913525D0 (en) | 2009-08-03 | 2009-09-16 | Ineos Healthcare Ltd | Method |
| GB201001779D0 (en) | 2010-02-04 | 2010-03-24 | Ineos Healthcare Ltd | Composition |
| CN104168955A (zh) * | 2011-11-10 | 2014-11-26 | 凯伊药品公司 | 拟钙剂及其使用方法 |
| WO2013128474A1 (en) * | 2012-03-02 | 2013-09-06 | Council Of Scientific & Industrial Research | Double fortified salt composition containing iron and iodine and process for the preparation thereof |
| CN103214075B (zh) * | 2013-05-20 | 2014-08-13 | 章方军 | 一种复合絮凝剂及其制备方法和用途 |
| PT3073997T (pt) * | 2013-11-27 | 2020-09-08 | Vifor Int Ag | Composição farmacêutica, compreendendo partículas de ligante de fosfato |
| US9592253B1 (en) | 2015-10-14 | 2017-03-14 | ZS Pharma, Inc. | Extended use zirconium silicate compositions and methods of use thereof |
| JP7547048B2 (ja) | 2016-10-12 | 2024-09-09 | ボナファイド・ヘルス,エルエルシー | ピコリン酸マグネシウム組成物および使用方法 |
| US10392267B2 (en) | 2017-04-18 | 2019-08-27 | King Abdulaziz University | Azo dye intercalated Fe(II)/Fe(III) layered double hydroxide for water purification |
| CN113164515A (zh) * | 2018-10-25 | 2021-07-23 | 艾尔金制药有限公司 | 用混合金属化合物治疗的方法 |
| JP7264397B2 (ja) * | 2019-11-13 | 2023-04-25 | 日機装株式会社 | 乳酸吸着剤および乳酸の除去方法 |
| EP4049040A4 (en) * | 2019-12-12 | 2022-10-26 | Hewlett-Packard Development Company, L.P. | OBJECT TRACKING BASED ON THE FLOW DYNAMICS OF A FLOW FIELD |
| US12329778B2 (en) | 2020-01-16 | 2025-06-17 | Vifor Fresenius Medical Care Renal Pharma Ltd | Particles of a mixture of iron(III)-oxyhydroxide, sucrose and one or more starches, preferably of sucroferric oxyhydroxide |
| FR3112968A1 (fr) * | 2020-07-28 | 2022-02-04 | IFP Energies Nouvelles | Procédé de préparation d'un support de catalyseur Fischer-Tropsch comprenant une étape de séchage spin flash |
| JP7722697B2 (ja) | 2021-11-02 | 2025-08-13 | 株式会社Kyulux | 化合物、組成物、ホスト材料および有機発光素子 |
| WO2024177075A1 (ja) * | 2023-02-21 | 2024-08-29 | 神島化学工業株式会社 | 層状複水酸化物及び吸着材 |
Family Cites Families (146)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1001430A (en) | 1910-04-16 | 1911-08-22 | Samuel W Nesbitt | Device for dressing grinding-wheels. |
| US2222924A (en) | 1936-12-10 | 1940-11-26 | Degussa | Preparation of cerium sulphate |
| US2812344A (en) | 1956-02-13 | 1957-11-05 | Ortho Pharma Corp | Ferrous calcium citrate complex |
| FR1214473A (fr) | 1959-01-07 | 1960-04-08 | Procédé de préparation d'un catalyseur à base d'hydroxyde de cérium | |
| BE590192A (es) | 1959-04-27 | |||
| US3395211A (en) | 1965-12-10 | 1968-07-30 | Carter Wallace | Tableting process |
| US3539306A (en) | 1966-07-25 | 1970-11-10 | Kyowa Chem Ind Co Ltd | Process for the preparation of hydrotalcite |
| US3796792A (en) | 1969-12-12 | 1974-03-12 | Kyowa Chem Ind Co Ltd | Composite metal hydroxides |
| SU414849A1 (ru) | 1971-05-21 | 1977-09-05 | Харьковский домостроительный комбинат | Бетонна смесь |
| BE792032A (fr) | 1971-12-23 | 1973-03-16 | Baxter Laboratories Inc | Dispositif de transfert de masse ayant une membrame semi-permeable disposee en spirale et presentant un faible volume de fluide |
| NL174720C (nl) | 1973-12-13 | 1984-08-01 | Philips Nv | Beeldweergeefcel en methode voor de vervaardiging van een vloeibaar kristallijn p-n.butyl-p'-alkoxyazobenzeen. |
| FR2435442A1 (fr) | 1978-09-07 | 1980-04-04 | Om Lab Sa | Silicate de magnesium et d'aluminium synthetique, procede de fabrication de celui-ci et compositions pharmaceutiques comprenant celui-ci |
| US4192900A (en) | 1978-10-12 | 1980-03-11 | Merck & Co., Inc. | Texturized starch products |
| DE2845326C2 (de) | 1978-10-18 | 1985-05-23 | Beiersdorf Ag, 2000 Hamburg | Verwendung einer spezifischen mikrodispersen, amorphen, porösen Kieselsäure zur Herstellung von Digoxin enthaltenden Tabletten mit stark beschleunigter Wirkstoff-Freisetzung |
| DE3039997A1 (de) | 1980-10-23 | 1982-06-03 | Bayer Ag, 5090 Leverkusen | Phosphonohydroxyacetonitril, ein verfahren zu seiner herstellung und seine verwendung als zwischenprodukt fuer die herstellung von arzneimitteln |
| US4351814A (en) | 1980-12-18 | 1982-09-28 | Kyowa Chemical Industry Co., Ltd. | Hydrotalcites having a hexagonal needle-like crystal structure and process for production thereof |
| JPS57156419A (en) | 1981-03-24 | 1982-09-27 | Kyowa Chem Ind Co Ltd | Remedy for sideropenia |
| US4514389A (en) | 1981-05-06 | 1985-04-30 | Kyowa Chemical Industry Co. Ltd. | Gastric antacid and method for controlling pH of gastric juice |
| CA1198674A (en) | 1981-11-23 | 1985-12-31 | Rorer International (Holdings) Inc. | Antacid compositions |
| US4458026A (en) | 1982-06-02 | 1984-07-03 | Union Carbide Corporation | Catalysts for aldol condensations |
| US4582705A (en) | 1982-07-12 | 1986-04-15 | Leonard Primes | Composition for detoxification |
| JPS606619A (ja) | 1983-06-27 | 1985-01-14 | Kyowa Chem Ind Co Ltd | 鉄分欠乏症処置剤及びその製法 |
| GB8317595D0 (en) | 1983-06-29 | 1983-08-03 | Reckitt & Colmann Prod Ltd | Pharmaceutical compositions |
| US4609543A (en) | 1983-11-14 | 1986-09-02 | Nabisco Brands, Inc. | Soft homogeneous antacid tablet |
| JPS60131604A (ja) | 1983-12-20 | 1985-07-13 | Victor Co Of Japan Ltd | 磁気記録再生素子 |
| DE3346943A1 (de) | 1983-12-24 | 1985-07-04 | Bayer Ag, 5090 Leverkusen | Herstellung von hydrotalcit mit verbesserten eigenschaften |
| DE3402878A1 (de) | 1984-01-27 | 1985-08-01 | Algina AG, Zug | Arzneimittel und die verwendung schwerloeslicher calcium- und/oder magnesiumverbindungen als arzneimittel |
| FR2559755A1 (fr) | 1984-02-20 | 1985-08-23 | Rhone Poulenc Spec Chim | Oxyde cerique a nouvelles caracteristiques morphologiques et son procede d'obtention |
| JPS6136222A (ja) | 1984-07-26 | 1986-02-20 | Chugai Pharmaceut Co Ltd | 高リン酸血症治療剤 |
| US4566986A (en) | 1984-08-31 | 1986-01-28 | Waldmann John J | Flocculating agents and processes for making them |
| DE3520108A1 (de) | 1985-06-05 | 1986-12-11 | Varta Batterie Ag, 3000 Hannover | Positive sammlerelektrode fuer akkumulatoren mit alkalischem elektrolyten |
| JPH06705B2 (ja) | 1985-12-19 | 1994-01-05 | 旭化成工業株式会社 | リン酸イオンの固定化剤 |
| US6749864B2 (en) | 1986-02-13 | 2004-06-15 | Takeda Chemical Industries, Ltd. | Stabilized pharmaceutical composition |
| US4871392A (en) | 1986-05-23 | 1989-10-03 | American Cyanamid Company | Aqueous suspension concentrate compositions of pendimethalin |
| US4801454A (en) | 1986-07-17 | 1989-01-31 | The Proctor & Gamble Company | Processes for making colored pharmaceutical compositions |
| EP0300853B1 (fr) | 1987-06-29 | 1991-09-18 | Rhone-Poulenc Chimie | Procédé d'obtention d'un oxyde cérique |
| US4994283A (en) | 1987-07-02 | 1991-02-19 | The Procter & Gamble Company | Iron-calcium mineral supplements with enhanced bioavailability |
| US4786510A (en) | 1987-07-02 | 1988-11-22 | The Procter & Gamble Company | Calcium-iron mineral supplements |
| DE3801382A1 (de) | 1988-01-19 | 1989-08-03 | Ebner Anlagen & Apparate | Verfahren und anlage zur umwandlung von natriumsulfat in natriumhydroxid |
| DE68916983T2 (de) | 1988-02-25 | 1995-01-19 | Yamanouchi Europ Bv | Verfahren zur Herstellung eines pharmazeutischen Granulats. |
| HU201880B (en) | 1988-06-21 | 1991-01-28 | Semmelweis Orvostudomanyi Egye | Process for producing pharmaceutical compositions for profilacting and treating renal diseases and for diminishing frequency of extracorporalis dialysis |
| GB8826333D0 (en) | 1988-11-10 | 1988-12-14 | Unilever Plc | Oral compositions |
| US5153156A (en) | 1989-04-18 | 1992-10-06 | Aristech Chemical Corporation | Process for making efficient anionic clay catalyst, catalysts made thereby, and method of making isophorone |
| FR2647433B1 (fr) | 1989-05-23 | 1991-10-31 | Lafarge Coppee Da | Procede de preparation par voie aqueuse de sulfate de calcium purifie |
| US4970079A (en) | 1989-06-05 | 1990-11-13 | Purdue Research Foundation | Method and composition of oxy-iron compounds for treatment of hyperphosphatemia |
| US5112604A (en) | 1989-09-01 | 1992-05-12 | Riker Laboratories, Inc. | Oral suspension formulation |
| ES2018952A6 (es) | 1989-10-17 | 1991-05-16 | Walton Sa | Procedimiento de preparacion de composiciones antiacidas con tiempo prolongado de permanencia gastrica. |
| JP2918619B2 (ja) * | 1990-04-06 | 1999-07-12 | 花王株式会社 | 金属磁性粉末の製造方法及び磁気記録媒体用塗膜 |
| ATE136869T1 (de) | 1990-06-08 | 1996-05-15 | Commw Scient Ind Res Org | Sorptionsmittel für ethylen |
| US5300302A (en) | 1990-10-04 | 1994-04-05 | Nestec S.A. | Pharmaceutical composition in gel form in a dispensing package |
| DE4036757A1 (de) | 1990-11-17 | 1992-05-21 | Bayer Ag | Antazida-zubereitung mit verlaengerter magenverweilzeit |
| GB2253164B (en) | 1991-02-22 | 1994-10-05 | Hoechst Uk Ltd | Improvements in or relating to electrostatic coating of substrates of medicinal products |
| GB2254556B (en) | 1991-04-11 | 1995-04-12 | Fisons Plc | Formulations containing linolenic acid |
| US5246899A (en) | 1991-08-16 | 1993-09-21 | Amoco Corporation | Simplified preparation of hydrotalcite-type clays |
| JPH05155776A (ja) | 1991-12-02 | 1993-06-22 | Otsuka Pharmaceut Factory Inc | 高リン血症治療剤 |
| JPH05208816A (ja) | 1992-01-28 | 1993-08-20 | Sumitomo Metal Mining Co Ltd | 水酸化セリウム及び酸化セリウムの製造方法 |
| WO1993022237A1 (en) | 1992-04-30 | 1993-11-11 | J.M. Huber Corporation | Method for production of synthetic hydrotalcite |
| US5380542A (en) | 1992-06-19 | 1995-01-10 | Rhone-Poulenc Specialties Chemical Co. | Dietary fiber compositions for use in foods |
| US5273767A (en) | 1992-09-09 | 1993-12-28 | Merck & Co., Inc. | Rapidly hydrating gums |
| US5364828A (en) * | 1992-10-21 | 1994-11-15 | Minerals Technologies | Spheroidal aggregate of platy synthetic hydrotalcite |
| JPH08502976A (ja) | 1992-10-29 | 1996-04-02 | シー−ピー テクノロジー リミテッド パートナーシップ | カルシウムおよび硫酸塩を含有した組成物および複合体、その合成法、これを用いた治療法、これを含んでなる化粧用調製物 |
| NL9201907A (nl) | 1992-11-02 | 1994-06-01 | Tno | Peristaltisch mengende reactor en peristaltische kleppenpomp. |
| DE4239442C2 (de) | 1992-11-24 | 2001-09-13 | Sebo Gmbh | Verwendung eines mit polynuklearen Metalloxidhydroxiden modifizierten Adsorptionsmaterials zur selektiven Elimination von anorganischem Phosphat aus proteinhaltigen Flüssigkeiten |
| CA2110313C (en) | 1992-12-01 | 2004-10-26 | Edward John Roche | Pharmaceutical compositions containing a guanidinothiazole compound and antacids |
| JP3395254B2 (ja) * | 1993-05-28 | 2003-04-07 | 住友金属鉱山株式会社 | 非焼結式アルカリ蓄電池用水酸化ニッケルの製造方法 |
| US5506248A (en) | 1993-08-02 | 1996-04-09 | Bristol-Myers Squibb Company | Pharmaceutical compositions having good dissolution properties |
| TW397686B (en) | 1993-08-11 | 2000-07-11 | Takeda Chemical Industries Ltd | Antacid compositions and pharmaceutical compositions |
| WO1995011033A1 (en) | 1993-10-22 | 1995-04-27 | Commonwealth Scientific And Industrial Research Organisation | Polyoxometallates in the treatment of flavivirus infections |
| TW390813B (en) | 1994-04-29 | 2000-05-21 | Merck & Co Inc | Wet granulation formulation for bisphosphonic acids |
| CZ289893B6 (cs) | 1994-09-21 | 2002-04-17 | Unilever N. V. | Orální prostředek |
| DE19503522A1 (de) * | 1995-02-03 | 1996-08-08 | Rwe Dea Ag | Herstellung gemischter schichtförmig aufgebauter Metallhydroxide sowie deren Metalloxide |
| GB9506126D0 (en) | 1995-03-25 | 1995-05-10 | Johnson Matthey Plc | Pharmaceutical composition and method |
| EP1672061B1 (en) | 1995-09-20 | 2008-09-10 | University of Queensland | Nucleotide sequences encoding ACC synthase enzymes of papaya |
| US5571336A (en) | 1995-09-29 | 1996-11-05 | Wurzburger; Stephen R. | Base solution for cleaning aluminum |
| DE19547356A1 (de) | 1995-12-19 | 1997-06-26 | Vifor Int Ag | Adsorbens für Phosphat aus wäßrigem Medium, dessen Herstellung und Verwendung |
| DE921726T1 (de) | 1996-01-25 | 1999-12-09 | Colin Leslie Young | Molluskizid mit aktion im magen |
| SE9602442D0 (sv) | 1996-06-20 | 1996-06-20 | Astra Ab | Administration of pharmaceuticals |
| PT922464E (pt) | 1996-07-12 | 2005-07-29 | Daiichi Seiyaku Co | Materiais moldados por compressao rapidamente desintegraveis e processo para a sua producao |
| US5654011A (en) | 1996-07-30 | 1997-08-05 | Energetics, Inc. | Dietary supplements |
| JPH1059842A (ja) | 1996-08-13 | 1998-03-03 | Lion Corp | 錠剤用組成物及び打錠方法 |
| JPH10101569A (ja) | 1996-10-01 | 1998-04-21 | Eisai Co Ltd | 制酸剤 |
| JPH10236960A (ja) | 1997-02-24 | 1998-09-08 | Lion Corp | 制酸剤含有組成物 |
| TW592727B (en) | 1997-04-04 | 2004-06-21 | Chugai Pharmaceutical Co Ltd | Phosphate-binding polymer preparations |
| GB9720061D0 (en) | 1997-09-19 | 1997-11-19 | Crosfield Joseph & Sons | Metal compounds as phosphate binders |
| US6028023A (en) | 1997-10-20 | 2000-02-22 | Bulldog Technologies U.S.A., Inc. | Process for making, and use of, anionic clay materials |
| CA2322315C (en) | 1998-03-06 | 2008-09-16 | Eurand International S.P.A. | Fast disintegrating tablets |
| JPH11335269A (ja) | 1998-05-19 | 1999-12-07 | Hisamitsu Pharmaceut Co Inc | 遺伝子関連医薬の経口投与固形製剤 |
| JP2000086537A (ja) | 1998-09-11 | 2000-03-28 | Fuji Chem Ind Co Ltd | 無機化合物糖類組成物、賦形剤、速崩壊性圧縮成型物及びその製造方法 |
| SA99191255B1 (ar) | 1998-11-30 | 2006-11-25 | جي دي سيرل اند كو | مركبات سيليكوكسيب celecoxib |
| KR100331529B1 (ko) | 1999-06-16 | 2002-04-06 | 민경윤 | 난용성 항진균제의 경구투여용 조성물 및 그의 제조 방법 |
| US6448323B1 (en) | 1999-07-09 | 2002-09-10 | Bpsi Holdings, Inc. | Film coatings and film coating compositions based on polyvinyl alcohol |
| US6177581B1 (en) | 1999-10-12 | 2001-01-23 | The Dow Chemical Company | Mixed-metal chelates and process for the preparation thereof |
| US6733780B1 (en) | 1999-10-19 | 2004-05-11 | Genzyme Corporation | Direct compression polymer tablet core |
| DE19958007A1 (de) | 1999-12-02 | 2001-06-07 | Roehm Gmbh | Spritzgußverfahren für (Meth)acrylat-Copolymere mit teritiären Ammoniumgruppen |
| WO2001049302A1 (en) | 2000-01-06 | 2001-07-12 | Marantech Holding, Llc | Compositions and methods for facilitating skin growth and managing skin conditions |
| AUPQ533700A0 (en) | 2000-01-28 | 2000-02-17 | Commonwealth Scientific And Industrial Research Organisation | Soil treatment method |
| JP4439067B2 (ja) * | 2000-02-23 | 2010-03-24 | 三菱原子燃料株式会社 | 鉄系複水酸化物の製造方法及び該複水酸化物を用いた水処理材 |
| JP4762399B2 (ja) * | 2000-05-09 | 2011-08-31 | ライオン株式会社 | アルコキシル化用触媒およびその製造方法並びにこの触媒を用いるアルキレンオキサイド付加物の製造方法 |
| FR2809407B1 (fr) | 2000-05-26 | 2002-08-30 | Rhodia Chimie Sa | Utilisation d'hydrotalcite comme charge dans des compositions de polymeres |
| US20030185886A1 (en) | 2000-05-26 | 2003-10-02 | Hanmi Pharm. Co., Ltd. | Process for the preparation of rapidly disintegrating tablet |
| US6720005B1 (en) | 2000-07-07 | 2004-04-13 | The State Of Oregon Acting By And Through The State Board Of Higher Education On Behalf Of Oregon State University | Coated, platform-generating tablet |
| US6576665B2 (en) | 2001-04-03 | 2003-06-10 | Braintree Laboratories, Inc. | Encapsulated calcium acetate caplet and a method for inhibiting gastrointestinal phosphorous absorption |
| US20040247696A1 (en) | 2001-07-05 | 2004-12-09 | Antelman Marvin S. | Methods of using electron active compounds for managing conditions afflicting mammals |
| UA80682C2 (en) | 2001-08-06 | 2007-10-25 | Pharmacia Corp | Orally deliverable stabilized oral suspension formulation and process for the incresaing physical stability of thixotropic pharmaceutical composition |
| KR20090065552A (ko) | 2001-08-27 | 2009-06-22 | 교와 가가꾸고교 가부시키가이샤 | 제산 ·완하용 정제 |
| AR036354A1 (es) | 2001-08-31 | 2004-09-01 | Takeda Chemical Industries Ltd | Preparacion solida |
| US20050202082A1 (en) | 2001-09-28 | 2005-09-15 | Tsuneyuki Hibino | Press-coated molded article undergoing quick disintegration |
| GB0204771D0 (en) | 2002-02-28 | 2002-04-17 | Phoqus Ltd | Fast disintegrating tablets |
| US7300670B2 (en) | 2002-04-03 | 2007-11-27 | Unilab Pharmatech, Ltd. | Oral suspension formulation |
| PT1501484E (pt) | 2002-04-29 | 2006-10-31 | Egyt Gyogyszervegyeszeti Gyar | Processo para a preparacao de comprimidos de substancias farmaceuticamente activas possuindo propriedades de compressao desfavoraveis com um liquido de granulacao compreendendo celulose microcristalina. |
| US20040161474A1 (en) | 2002-05-24 | 2004-08-19 | Moerck Rudi E. | Rare earth metal compounds methods of making, and methods of using the same |
| WO2004000885A2 (en) | 2002-06-25 | 2003-12-31 | Rhodia, Inc. | Molecular weight reduction of polysaccharides by electron beams |
| DE60310950T2 (de) | 2002-08-09 | 2007-11-15 | Albemarle Netherlands B.V. | Fischer-tropsch-verfahren unter verwendung eines eisenhaltigen schichtförmigen materiales |
| JP4348426B2 (ja) | 2002-08-29 | 2009-10-21 | 独立行政法人産業技術総合研究所 | 高選択性脱リン剤及びその製造方法 |
| WO2005014160A1 (en) | 2003-01-28 | 2005-02-17 | The University Of Wyoming Research Corporation D/B/A Western Research Institute | Bioagent air filtration systems |
| CN1798699B (zh) * | 2003-05-30 | 2010-11-10 | 杰富意矿物股份有限公司 | 金属复合氧化物的制造方法 |
| AU2003243054A1 (en) * | 2003-07-07 | 2005-01-21 | Instituto Mexicano Del Petroleo | Method of obtaining multimetallic oxides derived from hydrotalcite-type compounds |
| AR045068A1 (es) | 2003-07-23 | 2005-10-12 | Univ Missouri | Formulacion de liberacion inmediata de composiciones farmaceuticas |
| MXPA06002257A (es) | 2003-08-26 | 2006-05-17 | Shire Holdings Ag | Farmaceutica que comprende compuestos de lantano. |
| US7381428B2 (en) | 2003-08-26 | 2008-06-03 | Shire International Licensing B.V. | Stabilized lanthanum carbonate compositions |
| WO2005027876A1 (en) | 2003-08-28 | 2005-03-31 | Ranbaxy Laboratories Limited | Pharmaceutical compositions of benzimidazole and processes for their preparation |
| JP4702512B2 (ja) * | 2003-09-19 | 2011-06-15 | 戸田工業株式会社 | ハイドロタルサイト類化合物粒子粉末及び該ハイドロタルサイト類化合物粒子粉末を含有する水系分散体 |
| WO2005087664A1 (ja) * | 2004-03-16 | 2005-09-22 | Waseda University | ハイドロタルサイト様物質およびその製造方法、ならびに有害物質の固定化方法 |
| US20050260271A1 (en) | 2004-05-20 | 2005-11-24 | Eastman Kodak Company | Composition comprising layered host material with intercalated functional-active organic compound |
| US8906940B2 (en) | 2004-05-25 | 2014-12-09 | Santarus, Inc. | Pharmaceutical formulations useful for inhibiting acid secretion and methods for making and using them |
| US20060177415A1 (en) | 2004-11-01 | 2006-08-10 | Burke Steven K | Once a day formulation for phosphate binders |
| WO2006066314A1 (en) * | 2004-12-23 | 2006-06-29 | Commonwealth Scientific And Industrial Research Organisation | Heat treatment of aluminium alloy high pressure die castings |
| US20090108233A1 (en) * | 2004-12-24 | 2009-04-30 | The Univeristy Of Queensland | Preparation of suspensions |
| GB0502787D0 (en) * | 2005-02-10 | 2005-03-16 | Ineos Silicas Ltd | Pharmaceuticlly active compounds, their manufacture, compositions containing them and their use |
| JP4823547B2 (ja) | 2005-03-28 | 2011-11-24 | 株式会社ジーシー | ペースト状歯科用アルギン酸塩印象材組成物 |
| US20090175959A1 (en) | 2005-12-28 | 2009-07-09 | Takeda Pharmaceutical Company Limited | Controlled Release Solid Preparation |
| MY157620A (en) | 2006-01-31 | 2016-06-30 | Cytochroma Dev Inc | A granular material of a solid water-soluble mixed metal compound capable of binding phosphate |
| JP4899182B2 (ja) * | 2006-03-22 | 2012-03-21 | 独立行政法人産業技術総合研究所 | アニオン交換体の製造方法 |
| CA2645205C (en) | 2006-05-19 | 2012-12-04 | Norbrook Laboratories Limited | Stable aqueous suspension having palatable taste |
| PT2319804E (pt) | 2006-12-14 | 2014-11-24 | Novartis Ag | Adsorvente de fosfato à base de ferro (iii)-carboidrato |
| DK2319804T3 (en) | 2006-12-14 | 2015-01-19 | Novartis Ag | Iron (III) -carbohydrat-based phosphatadsorbens |
| CN101049953A (zh) * | 2007-01-12 | 2007-10-10 | 北京化工大学 | 一种层状双羟基复合金属氧化物微球及其制备方法 |
| EP1946750A1 (en) | 2007-01-18 | 2008-07-23 | The Jordanian Pharmaceutical Manufacturing Co. | Co-precipitate, method for preparing the same and uses thereof |
| WO2008129034A1 (en) | 2007-04-20 | 2008-10-30 | Euro Support Catalyst Group Bv | Hydrotalcite-like layered double hydroxide (ldh) composition and process of making same |
| GB0714670D0 (en) * | 2007-07-27 | 2007-09-05 | Ineos Healthcare Ltd | Use |
| JP2009034564A (ja) * | 2007-07-31 | 2009-02-19 | Jdc Corp | アルカリ溶液中でイオン化する物質xの吸着剤および分離方法 |
| GB0720220D0 (en) | 2007-10-16 | 2007-11-28 | Ineos Healthcare Ltd | Compound |
| DE102007059990A1 (de) * | 2007-12-13 | 2009-06-18 | Süd-Chemie AG | Verfahren zur Herstellung nanokristalliner Hydrotalcitverbindungen |
| AR076070A1 (es) * | 2009-03-02 | 2011-05-18 | Vifor Int Ag | Absorbente de fosfato para el tratamiento de la hiperfosfatemia |
| GB0913525D0 (en) | 2009-08-03 | 2009-09-16 | Ineos Healthcare Ltd | Method |
| GB201001779D0 (en) | 2010-02-04 | 2010-03-24 | Ineos Healthcare Ltd | Composition |
-
2009
- 2009-08-03 GB GBGB0913525.2A patent/GB0913525D0/en not_active Ceased
-
2010
- 2010-07-09 TW TW106136737A patent/TWI649267B/zh not_active IP Right Cessation
- 2010-07-09 TW TW099122681A patent/TWI617513B/zh not_active IP Right Cessation
- 2010-07-09 TW TW105116495A patent/TWI619677B/zh not_active IP Right Cessation
- 2010-07-26 AR ARP100102697A patent/AR077500A1/es active IP Right Grant
- 2010-08-02 PT PT107399875T patent/PT2462062T/pt unknown
- 2010-08-02 WO PCT/GB2010/051271 patent/WO2011015859A1/en not_active Ceased
- 2010-08-02 EP EP19186506.2A patent/EP3611131A1/en active Pending
- 2010-08-02 CN CN201080040879.2A patent/CN102498068B/zh active Active
- 2010-08-02 ES ES10739987T patent/ES2751325T3/es active Active
- 2010-08-02 EP EP10739987.5A patent/EP2462062B1/en active Active
- 2010-08-02 JP JP2012523389A patent/JP5894073B2/ja not_active Expired - Fee Related
- 2010-08-02 CN CN201910277263.XA patent/CN110357165B/zh active Active
- 2010-08-02 US US13/388,476 patent/US9314481B2/en active Active
- 2010-08-02 CN CN201610371464.2A patent/CN106115789B/zh active Active
- 2010-08-02 DK DK10739987.5T patent/DK2462062T3/da active
-
2014
- 2014-03-24 US US14/224,040 patent/US9066917B2/en active Active
-
2015
- 2015-05-07 JP JP2015095095A patent/JP6049801B2/ja active Active
-
2016
- 2016-04-19 US US15/133,206 patent/US20160296557A1/en not_active Abandoned
- 2016-11-22 JP JP2016226924A patent/JP6333926B2/ja not_active Expired - Fee Related
-
2018
- 2018-03-28 AR ARP180100782A patent/AR111146A2/es not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| US20120201864A1 (en) | 2012-08-09 |
| GB0913525D0 (en) | 2009-09-16 |
| CN110357165A (zh) | 2019-10-22 |
| EP2462062B1 (en) | 2019-07-17 |
| US20140205667A1 (en) | 2014-07-24 |
| US9066917B2 (en) | 2015-06-30 |
| CN106115789A (zh) | 2016-11-16 |
| AR111146A2 (es) | 2019-06-05 |
| TWI617513B (zh) | 2018-03-11 |
| TW201114691A (en) | 2011-05-01 |
| TWI649267B (zh) | 2019-02-01 |
| EP3611131A1 (en) | 2020-02-19 |
| JP2015171998A (ja) | 2015-10-01 |
| DK2462062T3 (da) | 2019-10-21 |
| JP2017061410A (ja) | 2017-03-30 |
| TW201630818A (zh) | 2016-09-01 |
| JP6333926B2 (ja) | 2018-05-30 |
| TWI619677B (zh) | 2018-04-01 |
| TW201829317A (zh) | 2018-08-16 |
| CN102498068B (zh) | 2016-06-29 |
| US9314481B2 (en) | 2016-04-19 |
| WO2011015859A1 (en) | 2011-02-10 |
| CN102498068A (zh) | 2012-06-13 |
| US20160296557A1 (en) | 2016-10-13 |
| CN110357165B (zh) | 2022-05-13 |
| JP6049801B2 (ja) | 2016-12-21 |
| CN106115789B (zh) | 2019-05-03 |
| PT2462062T (pt) | 2019-11-04 |
| AR077500A1 (es) | 2011-08-31 |
| EP2462062A1 (en) | 2012-06-13 |
| JP2013500933A (ja) | 2013-01-10 |
| JP5894073B2 (ja) | 2016-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2751325T3 (es) | Hidróxidos dobles con capas de magnesio-hierro, su producción y su utilización para el tratamiento de hiperfosfatemia | |
| ES2545388T3 (es) | Compuestos metálicos mixtos para el tratamiento de hiperfosfatemia | |
| ES2599152T3 (es) | Aglutinantes de fosfato farmacéuticamente activos, su fabricación, composiciones que los contienen y su uso | |
| CN113164515A (zh) | 用混合金属化合物治疗的方法 | |
| HK1142525B (en) | Mixed metal compounds for treatment of hyperphosphataemia |