ES2797754T3 - Enlazadores moduladores de la estabilidad para su uso con conjugados de fármaco y anticuerpo - Google Patents
Enlazadores moduladores de la estabilidad para su uso con conjugados de fármaco y anticuerpo Download PDFInfo
- Publication number
- ES2797754T3 ES2797754T3 ES15777735T ES15777735T ES2797754T3 ES 2797754 T3 ES2797754 T3 ES 2797754T3 ES 15777735 T ES15777735 T ES 15777735T ES 15777735 T ES15777735 T ES 15777735T ES 2797754 T3 ES2797754 T3 ES 2797754T3
- Authority
- ES
- Spain
- Prior art keywords
- seq
- phenyl
- ala
- mmol
- phe
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC*(C)[C@@]([C@](C)CC1OC1N(CCC1)[C@@]1[C@](C)C(C)C(N)=O)N(C)C([C@](C(C)=C)NC(C(C)(C)NC(O)=O)=O)=O Chemical compound CC*(C)[C@@]([C@](C)CC1OC1N(CCC1)[C@@]1[C@](C)C(C)C(N)=O)N(C)C([C@](C(C)=C)NC(C(C)(C)NC(O)=O)=O)=O 0.000 description 4
- MFYCYVRJMDQBGJ-UHFFFAOYSA-N C(c1ccccc1)C#Cc1ncc[s]1 Chemical compound C(c1ccccc1)C#Cc1ncc[s]1 MFYCYVRJMDQBGJ-UHFFFAOYSA-N 0.000 description 1
- GCDOESNSPDNZJH-VKCLLPTISA-N CC[C@H](C)[C@@H]([C@@H](CC(N(CCC1)[C@@H]1[C@@H]([C@@H](C)C(N[C@@H](Cc1ccccc1)c1ncc[s]1)=O)OC)=O)OC)N(C)C([C@H](C(C)C)NC(C(C)(C)NC(OCc(cc1)ccc1NC([C@H](CCCNC(N)=O)NC([C@H](C(C)C)NC([C@H](CCCCN)NC(C(C)(C)N)=O)=O)=O)=O)=O)=O)=O Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(N(CCC1)[C@@H]1[C@@H]([C@@H](C)C(N[C@@H](Cc1ccccc1)c1ncc[s]1)=O)OC)=O)OC)N(C)C([C@H](C(C)C)NC(C(C)(C)NC(OCc(cc1)ccc1NC([C@H](CCCNC(N)=O)NC([C@H](C(C)C)NC([C@H](CCCCN)NC(C(C)(C)N)=O)=O)=O)=O)=O)=O)=O GCDOESNSPDNZJH-VKCLLPTISA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/35—Fusion polypeptide containing a fusion for enhanced stability/folding during expression, e.g. fusions with chaperones or thioredoxin
Landscapes
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cell Biology (AREA)
- Transplantation (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Un compuesto de Fórmula (I): **(Ver fórmula)** o una sal o solvato farmacéuticamente aceptable del mismo; en la que: M es un modulador de la estabilidad en el que M es -M1-M2; en el que M1 es -NR1-C(O)-, -NR1-S(O)2-, o está ausente, y M2 se selecciona entre el grupo que consiste en: **(Ver fórmula)** P es una secuencia de péptidos que incluye uno o más residuos de glutamina; Q es un residuo de glutamina presente en P; cada E se selecciona independientemente entre el grupo que consiste en:-C(R1)2-, -O-C(R1)2-C(R1)2- en el que cuando E es -O-C(R1)2-C(R1)2 r es al menos 2, y -C(R1)2-C(R1)2-O- en el que cuando E es -C(R1)2-C(R1)2-O- s es al menos 1; cada R1 se selecciona independientemente entre el grupo que consiste en: H y alquilo C1-C6 lineal o ramificado; en la que -Xm-Yn- es -X-Y- y en la que X-Y se selecciona entre el grupo que consiste en Gly, β-Ala, Val-Cit, Phe- Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg, Phe-N9-Nitro-Arg, Val-Ala y Ala-Ala-Asn; cada Z es PABC (p-aminobencil-carbamoílo); p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y D es un agente citotóxico.
Description
DESCRIPCIÓN
Enlazadores moduladores de la estabilidad para su uso con conjugados de fármaco y anticuerpo
Campo
La presente invención se refiere a enlazadores usados en relación con conjugados anticuerpo-fármaco (CAF) que son capaces de modular por aumento y por disminución la estabilidad extracelular y/o intracelular de conjugados anticuerpo-fármaco. La invención también se refiere a usos terapéuticos y a regímenes de tratamiento que emplean tales conjugados de fármaco-anticuerpo de estabilidad modulada clínicamente ventajosos. Finalmente, se desvelan procedimientos para preparar enlazadores moduladores de la estabilidad y conjugados de anticuerpo-fármaco de estabilidad modulada.
Referencia al listado de secuencias
La presente solicitud se presenta electrónicamente a través de EFS-Web e incluye un listado de secuencias presentada electrónicamente en formato .txt. El archivo .txt contiene un listado de secuencias titulada "PC72108A_SEQ_LISTING_ST25.txt" creada el 10 de agosto de 2015 y que tiene un tamaño de 10 KB. El listado de secuencias contenida en este archivo .txt es parte de la especificación.
Antecedentes
La terapia con anticuerpos proporciona un tratamiento terapéutico dirigido en pacientes con diversos trastornos, tales como cánceres y enfermedades inmunológicas, y, por lo tanto, ha desempeñado un papel importante en la investigación biológica. Se han explorado diferentes enfoques de la terapia con anticuerpos dirigida, incluidos los conjugados anticuerpo-fármaco (CAF). Chari, R. V., Miller, M. L. y Widdison, W. C. (2014) Antibody-drug conjugates: an emerging concept in cancer therapy. Angewandte Chemie 53, 3796-827; Senter, P. D. y Sievers, E. L. (2012) The discovery and development of CAFETRIS® (brentuximab vedotin) for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nature biotechnology 30, 631-7; Lambert, J. M. (2013) Drug-conjugated antibodies for the treatment of cancer. British journal of clinical pharmacology 76, 248-62.
En el caso de los CAF (también llamados inmunoconjugados en ciertos contextos) "cargas útiles" de moléculas pequeñas, que a menudo son pequeñas moléculas citotóxicas (restos de fármaco), están unidos covalentemente (conjugados) a anticuerpos para la liberación local dirigida de los restos de fármaco a los tumores. Los procedimientos de conjugación convencionales para CAF incluyen modificación química a través de las aminas de cadena lateral de lisina, o a través de los grupos cisteína sulfhidrilo activados mediante la reducción de los enlaces disulfuro intracatenarios. Brentuximab vedotin y KCAFYLA® (ado-trastuzumab emtansina) son dos ejemplos de CAF que utilizan estos procedimientos convencionales.
También se han explorado enfoques enzimáticos que usan una transglutaminasa para preparar CAF. Las transglutaminasas pertenecen a una familia de enzimas que catalizan la adición de acilo a una amina primaria. La conjugación con una transglutaminasa proporciona las ventajas de procedimientos de reacción simplificados de ata selectividad y condiciones de reacción suaves. Véase, por ejemplo, Strop y col., Chemistry & Biology, 20:161-167 (2013); y Farias y col., Bioconj. Chem. 25(2):240-250 (2014). Los documentos US2013-0230543 y US2013-0122020 describen la conjugación específica de sitio mediada por transglutaminasa de anticuerpos y moléculas pequeñas.
Los procedimientos de conjugación de CAF convencionales dan como resultado que los CAF tengan una estabilidad inherente en un sistema biológico dado. La liberación de la carga útil en el sitio deseado, típicamente un tumor, depende de las propiedades de la carga útil, el enlazador covalente, el anticuerpo y el sistema biológico en el que se introduce el CAF. A menudo, se libera una cantidad significativa de la carga útil prematuramente desde un CAF menos que óptimamente estable, por ejemplo, en el plasma y, por lo tanto, necesita niveles de dosificación de CAF más altos para lograr la exposición deseada a la carga útil en el sitio del tumor. En otras circunstancias, el uso de un CAF demasiado estable da como resultado una actividad de liberación de carga útil inferior a la óptima dentro de las células tumorales objetivo.
Los enlaces basados en péptidos proteolíticamente escindibles que emplean el elemento de inmolación de paminobenciloxicarbonilo (PABC) se han usado ampliamente en la investigación de conjugados de anticuerpo-fármaco (CAF) desde su introducción en 2002 (Dubowchik, G.M. y col., Bioconjugate Chem. 2002, 13, 855-869). Dichos enlaces supuestamente sufren escisión tras la internalización en células dirigidas a antígenos y la exposición del CAF al entorno proteolítico degradante que se encuentra en los orgánulos endosómicos y lisosómicos. Las variantes unidas a cisteína de estos enlazadores dipeptídicos-PABC han sido patentadas (documento US 6.214.345 B2) y se ha demostrado que las versiones que contienen amina de estos y otros enlaces son apropiadas para conjugaciones específicas de sitio a restos de glutamina usando conjugaciones enzimáticas estimuladas por transglutaminasa microbiana, así como el uso de anticuerpos aglicosilados y variantes de anticuerpos modificados genéticamente que experimentan conjugaciones eficientes utilizando este enfoque de conjugación enzimática (documento WO2012/059882 A2).
El documento WO2013/072813 (número de solicitud PCT PCT/IB2012/056224, publicado el 23 de mayo de 2013)
desvela pentapéptidos citotóxicos, conjugados de anticuerpo-fármaco de los mismos, y procedimientos para usar los mismos para tratar el cáncer.
El documento WO2012/059882 (número de solicitud PCT PCT/IB2011/054899, publicado el 10 de mayo de 2012) desvela conjugados polipeptídicos diseñados y procedimientos para prepararlos usando transglutaminasa.
Sumario
La presente invención se refiere en general a CAF, típicamente preparados empleando una conjugación mediada por transglutaminasa, que tienen propiedades de estabilidad que están alteradas por la presencia de un resto modulador en los enlazadores de CAF. Los inventores han descubierto, sorprendentemente, que dicho resto modulador colocado en una posición específica en un enlazador de CAF y, más específicamente, ciertas características químicas incorporadas en estos restos moduladores, tiene la capacidad de aumentar o disminuir la estabilidad del CAF, según se desee, de modo que se libere una proporción mayor o menor de la carga útil del CAF en el sitio de acción deseado.
Por tanto, en el presente documento se proporcionan sustituciones específicas en el asa de conjugación que contiene amina, que a menudo son derivados acilados de lisina, que pueden tener un profundo efecto sobre la estabilidad en plasma in vitro y la exposición in vivo de los CAF resultantes. Se ha demostrado que la estabilidad del conjugado puede modularse variando la naturaleza de los sustituyentes presentes en las asas de conjugación que contienen amina y, adicionalmente, que el sustituyente se puede elegir para optimizar la estabilidad de cada sitio de conjugación único. La presente invención, por tanto, permite el ajuste de la estabilidad del CAF in vivo y, por tanto, permite la modulación de parámetros que afectan in vivo tanto a la eficacia como a la seguridad y, por tanto, permite la optimización del índice terapéutico de los CAF.
Breve descripción de los dibujos
Las Figuras 1A a 1I muestran estudios de citotoxicidad in vitro de anticuerpos quiméricos anti-Trop2 conjugados en un rango de sitios con la carga útil citotóxica de amino-caproil (C6) vcAur0101.
Las Figuras 1J a 1O muestran estudios de citotoxicidad in vitro de anticuerpos quiméricos anti Trop2 conjugados en los sitios LCQ04 y L11B con las cargas útiles del enlazador de los Ejemplos 2, 3 y 5.
La figura 2 muestra la estabilidad in vivo de los compuestos de CAF del ejemplo 12 y 14 (figura 2A); los compuestos de CAF del ejemplo 16 y 13 (Fig. 2B); y los compuestos de CAF del Ejemplo 11, 18 y 17 (Fig. 2C), unidos a través del sitio LCQ04 en el anticuerpo Trop2. Las líneas discontinuas representan la concentración de la carga útil, las líneas continuas representan la concentración de anticuerpos.
La figura 3 proporciona la confirmación de la carboxilesterasa 1c de ratón como la enzima responsable de la escisión del enlazador vc-pabc en el plasma en comparación con la cepa defectiva de ratón c56/bI6 ces1c -/-, la cepa heterocigota c56/bI6 ces1c /-, y la cepa de tipo salvaje c56/bI6 ces1c /+.
Descripción detallada
La presente invención se refiere en general a conjugados anticuerpo-fármaco (ADC) que tienen un grado deseado de estabilidad. Específicamente, la invención proporciona compuestos de Fórmula (I):

en la que:
M es un modulador de la estabilidad en el que M es -M1-M2 ;
en el que M1 es -NR1-C(O)-, -NR1-S(O)2-, o está ausente, y
M2 se selecciona entre el grupo que consiste en:

cada E se selecciona independientemente entre el grupo que consiste en:-C(R1)2-, -O-C(R1)2-C(R1)2- en el que cuando E es -O-C(R1)2-C(R1)2 r es al menos 2, y -C(R1)2-C(R1)2-O- en el que cuando E es -C(R1)2-C(R1)2-O- s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H y alquilo C1-C6 lineal o ramificado; en la que -Xm-Yn- es -X-Y- y en la que X-Y se selecciona entre el grupo que consiste en Gly, p-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg, Phe-N9-Nitro-Arg, Val-Ala y Ala-Ala-Asn;
cada Z es PABC (p-aminobencil-carbamoílo);
p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y
D es un agente citotóxico.
Las realizaciones de la invención también incluyen compuestos de Fórmula (II):

en la que:
M es un modulador de la estabilidad en el que M es -M1-M2;
en el que M1 es -NR1-C(O)-, -NR1-S(O)2-, o está ausente, y
M2 se selecciona entre el grupo que consiste en:

P es una secuencia de péptidos que incluye uno o más residuos de glutamina;
Q es uno de dichos residuos de glutamina presentes en P;
cada E se selecciona independientemente entre el grupo que consiste en: -C(R1)2-, -O-C(R1)2-C(R1)2- en el que cuando E es -O-C(R1)2-C(R1)2 r es al menos 2, y -C(R1)2-C(R1)2-O- en el que cuando E es -C(R1)2-C(R1)2-O- s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H y alquilo C1-C6 lineal o ramificado; en la que -Xm-Yn- es -X-Y- y en la que X-Y se selecciona entre el grupo que consiste en Gly, p-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg, Phe-N9-Nitro-Arg, Val-Ala y Ala-Ala-Asn;
cada Z es PABC (p-aminobencil-carbamoílo);
p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y
D es un agente citotóxico.
Realizaciones adicionales incluyen compuestos de Fórmula (III):
útiles para la fabricación de conjugados de anticuerpo-fármaco, en la que:
M es un modulador de la estabilidad en el que M es -M1-M2 ;
en el que M1 es -NR1-C(O)-, -NR1-S(O)2-, o está ausente, y
M2 se selecciona entre el grupo que consiste en:

cada E se selecciona independientemente entre el grupo que consiste en: -C(R1)2,-O-C(R1 )2-C(R1)2- en el que cuando E es -O-C(R1)2-C(R1 )2 r es al menos 2, y -C(R1)2-C(R1 )2-O- en el que cuando E es -C(R1)2-C(R1 )2-O- s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H y alquilo C1-C6 lineal o ramificado; en la que -Xm-Yn- es -X-Y- y en la que X-Y se selecciona entre el grupo que consiste en Gly, p-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg, Phe-N9-Nitro-Arg, Val-Ala y Ala-Ala-Asn;
cada Z es PABC (p-aminobencil-carbamoílo);
p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y
D es un agente citotóxico.
En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:
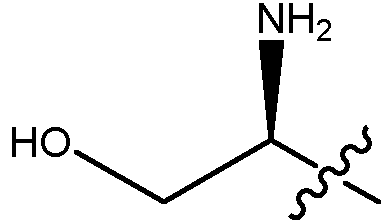
En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:
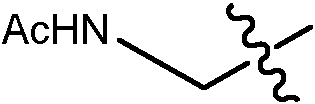
En algunas realizaciones, el ADC de la presente invención comprende un sistema enlazador basado en lisina con un resto modulador de acilo modificado en el que el resto modulador de acilo modificado es:

También se desvela un procedimiento para modular la estabilidad in vivo de un CAF, comprendiendo dicho procedimiento las etapas de: seleccionando el modulador M capaz de modular la estabilidad extracelular de dicho compuesto; incorporar dicho modulador M en dicho conjugado; y administrar dicho conjugado a un paciente.
La estabilidad puede ser modulada de manera tal que se incremente la proporción de agente citotóxico liberado fuera de las células objetivo en comparación con el agente citotóxico liberado dentro de las células objetivo.
La estabilidad puede ser modulada de manera que la proporción de agente citotóxico liberado fuera de las células objetivo en comparación con el agente citotóxico liberado dentro de las células objetivo disminuya.
La estabilidad puede ser modulada de tal manera que aumente la proporción de agente citotóxico liberado dentro de las células objetivo en comparación con el agente citotóxico liberado fuera de las células objetivo.
La estabilidad puede ser modulada de manera que la proporción de agente citotóxico liberado dentro de las células objetivo en comparación con el agente citotóxico liberado fuera de las células objetivo disminuya.
Los procedimientos desvelados en el presente documento comprenden además una etapa de purificación, en los que el CAF se purifica por una etapa de cromatografía.
En algunas realizaciones, la transglutaminasa es una transglutaminasa microbiana purificada o diseñada.
En otro aspecto, la invención proporciona compuestos de la invención para su uso en el tratamiento del cáncer.
También se desvela un procedimiento para inhibir el crecimiento o la progresión tumoral en un sujeto que lo necesita, que comprende administrar al sujeto una cantidad eficaz de una composición farmacéutica que comprende el CAF como se describe en el presente documento.
También se desvela un procedimiento para diagnosticar cáncer en un sujeto sospechoso de padecer cáncer, que comprende a) contactar una muestra del sujeto con el CAF como se describe en el presente documento en condiciones que dan como resultado la unión del CAF con una proteína relacionada con el cáncer, y b ) determinar la unión del CAF a la proteína relacionada con el cáncer.
En algunas realizaciones, el anticuerpo en el CAF como se describe en el presente documento es un anticuerpo monoclonal, un anticuerpo policlonal, un anticuerpo humano, un anticuerpo humanizado, un anticuerpo quimérico, un anticuerpo biespecífico, un minicuerpo, un diacuerpo o un fragmento de anticuerpo.
Las realizaciones de la invención incluyen compuestos como se describen en el presente documento en los que el agente citotóxico D se selecciona del grupo que consiste en: una antraciclina, una auristatina, una espliceostatina, un dímero CBI/CPI (que incluye dímeros "mixtos" que comprenden componentes CBI y CPI, como se describe en la solicitud de patente provisional de Estados Unidos 61/932,118), una caliqueamicina, una duocarmicina, una enediina, una geldanamicina, una maytansina, una puromicina, un taxano, un alcaloide de la vinca, SN-38, una tubulisina, una hemiasterlina, una camptotecina, una combretastatina, una dolastatina, un dímero de indolino-benzodiazepina, un dímero de pirrolobenzodiazepina y una pladienolida, y estereoisómeros, isoésteres, análogos o derivados de los mismos. Por ejemplo, realizaciones en las que el agente citotóxico D es una auristatina seleccionada del grupo que consiste en dolestatina, MMAD, MMAE, MMAF, PF-06380101, PF-06463377 y PF-06456780.
Las realizaciones adicionales de la invención incluyen compuestos en los que P es un péptido que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en: Q, LQG, LLQGG (s Eq ID NO:1), LLQG (SEQ ID NO:2), LSLSQG (SEQ ID NO:3), GGGLLQGG (SEQ ID NO:4), GLLQG (SEQ ID NO:5), LLQ, GSPLAQSHGG (SEQ ID NO:6), GLLQGGG (SEQ ID NO:7), GLLQGG (SEQ ID NO:8), GLLQ (SEQ ID NO:9), LLQLLQGA (SEQ ID NO:10), LLQGA (SEQ ID NO:11), LLQYQGA (SEQ ID NO:12), LLQGSG (SEQ ID NO:13), LLQYQG (SEQ ID NO:14), LLQLLQG (SEQ ID NO:15), SLLQG (SEQ ID NO:16), LLQLQ (SEQ ID NO:17), LLQLLQ (SEQ ID NO:18), LLQGR (SEQ ID NO:19), LLQGPP (SEQ ID NO:20), LLQGPA (SEQ ID NO:21), GGLLQGPP (SEQ ID NO:22), GGLLQGA (SEQ ID NO:23), LLQGA (SEQ ID NO:24), LLQGPGK (SEQ ID NO:25), LLQGPG (SEQ ID NO:26), LLQGP (SEQ ID NO:27), LLQP (SEQ ID NO:28), LLQPGK (SEQ ID NO:29), LLQAPGK (SEQ ID NO:30), LLQGAPG (SEQ ID NO:31), LLQGAP (SEQ ID NO:32), LLQGPA (SEQ ID NO:33), LLQGPP (SEQ ID NO:34), GGLLQGPP (SEQ ID NO:35) y LLQLQG (SEQ ID NO:36).
Las realizaciones de la invención también incluyen compuestos como se describe en el presente documento en los que P es un péptido que comprende la secuencia de aminoácidos XXQX (SEQ ID NO: 37).
Las realizaciones de la invención incluyen compuestos como se describe en el presente documento en los que la estabilidad de dicho compuesto disminuye al menos 1, 5, 10, 25, 50, 75 o 100 veces, con respecto al compuesto correspondiente que carece de dicho modulador M.
Las realizaciones de la invención incluyen compuestos como se describe en el presente documento en los que la estabilidad de dicho compuesto aumenta al menos 1, 5, 10, 25, 50, 75 o 100 veces, con respecto al compuesto correspondiente que carece de dicho modulador M.
Las realizaciones de la invención incluyen compuestos como se describe en el presente documento en los que P se selecciona del grupo que consiste en: un anticuerpo monoclonal, un anticuerpo policlonal, un anticuerpo humano, un anticuerpo humanizado, un anticuerpo quimérico, un anticuerpo biespecífico, un minicuerpo y un fragmento de anticuerpo.
Las realizaciones de la invención incluyen compuestos como se describe en el presente documento en los que el anticuerpo se selecciona de: trastuzumab, mutantes de trastuzumab (por ejemplo, los mutantes de trastuzumab
desvelados en el presente documento o en la solicitud de patente internacional PCT/IB2012/056234), oregovomab, edrecolomab, cetuximab, un anticuerpo monoclonal humanizado contra el receptor de vitronectina (avp3), alemtuzumab, anticuerpos anti-HLA-DR que incluyen un anticuerpo anti-HLA-DR humanizado para el tratamiento del linfoma no Hodgkin, 1311 Lym-1, anticuerpos anti-HLA-Dr10 que incluyen un anticuerpo murino anti-HLA-Dr10 para el tratamiento del linfoma no Hodgkin, anticuerpos anti-cd33, anticuerpos anti-cd22 que incluyen un mAb anti-CD22 humanizado para el tratamiento de la enfermedad de Hodgkin o el linfoma no Hodgkin, labetuzumab, bevacizumab, ibritumomab tiuxetan, ofatumumab, panitumumab, rituximab, tositumomab, ipilimumab y gemtuzumab.
Las realizaciones adicionales de la invención incluyen compuestos como se describe en el presente documento en los que cada Q es, independientemente, un resto de una glutamina endógena a una secuencia peptídica P, o un resto de una glutamina proporcionada en una secuencia marcadora diseñada en dicha secuencia de péptido P (en la que el marcador puede ser una secuencia de aminoácidos múltiples que contiene glutamina, o simplemente glutamina).
En otro aspecto más de la invención, el anticuerpo sirve como vehículo macromolecular, con o sin una función de liberación específica, que facilita la liberación controlada de la carga D a través de un enlazador escindible con modulación de estabilidad.
Las realizaciones de la invención incluyen compuestos como se describe en el presente documento en el que cada Q es un resto de una glutamina endógena a dicha secuencia de péptidos P.
Las realizaciones de la invención incluyen compuestos como se describe en el presente documento, cada Q es un resto de una glutamina proporcionada en una secuencia marcadora diseñada en dicha secuencia de péptidos P (nuevamente, en la que el marcador puede ser una secuencia de aminoácidos múltiples que contiene glutamina, o simplemente glutamina).
También se desvelan procedimientos de modulación de la estabilidad in vivo de un conjugado anticuerpo-fármaco de Fórmula (II):

M es un modulador de la estabilidad;
P es una secuencia de péptidos que incluye uno o más residuos de glutamina;
Q es uno de dichos residuos de glutamina presentes en P;
cada E se selecciona independientemente entre el grupo que consiste en: - C(R1)2-, -O-C(R1)2-C(R1)2- en el que r es al menos 2, y -C(R1)2-C(R1)2-O- en el que s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H, alquilo C1-C6 lineal o ramificado, alquenilo C2-C6 lineal o ramificado y alquinilo C2-C6 lineal o ramificado;
cada X es independientemente un aminoácido, en el que cada aminoácido X es igual o diferente;
cada Y es independientemente un aminoácido, en el que cada aminoácido Y es igual o diferente;
cada Z es independientemente un elemento espaciador, en el que cada elemento espaciador es igual o diferente; m es 0-5, n es 1-5, p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y
D es un agente citotóxico;
comprendiendo dicho procedimiento las etapas de:
seleccionar el modulador M capaz de modular la estabilidad extracelular de dicho compuesto; incorporar dicho modulador M en dicho conjugado; y
administrar dicho conjugado a un paciente.
También se desvelan procedimientos de tratamiento como se describe en el presente documento en los que se incrementa la proporción de agente citotóxico liberado fuera de las células objetivo en comparación con el agente citotóxico liberado dentro de las células objetivo.
También se desvelan procedimientos de tratamiento tal como se ha descrito en los que la proporción de agente citotóxico liberado fuera de las células objetivo en comparación con el agente citotóxico liberado dentro de las células objetivo disminuye.
También se desvelan procedimientos de tratamiento como se describe en el presente documento en los que se incrementa la proporción de agente citotóxico liberado dentro de las células objetivo en comparación con el agente citotóxico liberado fuera de las células objetivo.
También se desvelan procedimientos como los descritos en el presente documento en los que disminuye la proporción de agente citotóxico liberado dentro de las células objetivo en comparación con el agente citotóxico liberado fuera de las células objetivo.
También se desvelan procedimientos de síntesis de un compuesto de Fórmula (II):

M es un modulador de la estabilidad;
P es una secuencia de péptidos que incluye uno o más residuos de glutamina;
Q es uno de dichos residuos de glutamina presentes en P;
cada E se selecciona independientemente entre el grupo que consiste en:-C(R1)2-, -O-C(R1)2-C(R1)2- en el que r es al menos 2, y -C(R1)2-C(R1)2-O- en el que s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H, alquilo C1-C6 lineal o ramificado, alquenilo C2-C6 lineal o ramificado y alquinilo C2-C6 lineal o ramificado;
cada X es independientemente un aminoácido, en el que cada aminoácido X es igual o diferente;
cada Y es independientemente un aminoácido, en el que cada aminoácido Y es igual o diferente;
cada Z es independientemente un elemento espaciador, en el que cada elemento espaciador es igual o diferente; m es 0-5, n es 1-5, p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y D es un agente citotóxico; que comprende las etapas de:
proporcionar una cantidad de un primer compuesto de la estructura

proporcionar una cantidad de un segundo compuesto que comprende una secuencia de péptidos que incorpora una glutamina; y,
hacer reaccionar dichas cantidades del primer y segundo compuestos en presencia de transglutaminasa.
El término "alquilo" por sí mismo o como parte de otro término se refiere a un hidrocarburo de cadena lineal o ramificada, saturado, que tiene el número indicado de átomos de carbono (por ejemplo, alquilo "C1-C8" se refiere a un grupo alquilo que tiene de 1 a 8 átomos de carbono). Cuando la cantidad de átomos de carbono no está indicada, el grupo alquilo tiene de 1 a 8 átomos de carbono. Los alquilos C1-C8 de cadena lineal representativos incluyen, pero sin limitación, metilo, etilo, n-propilo, n-butilo, n-pentilo, n-hexilo, n-heptilo y n-octilo; mientras que los alquilos C1-C8 ramificados incluyen,, pero sin limitación, -isopropilo, -sec-butilo, -isobutilo, -terc-butilo, -isopentilo y -2-metilbutilo; entre los alquilos C2-C8 insaturados se incluyen, pero sin limitación, vinilo, alilo, 1-butenilo, 2-butenilo, isobutilenilo, 1-pentenilo, 2-pentenilo, 3-metil-1-butenilo, 2-metil-2-butenilo, 2,3-dimetil-2-butenilo, 1-hexilo, 2-hexilo, 3-hexilo, acetilenilo, propinilo, 1-butinilo, 2-butinilo, 1-pentinilo, 2-pentinilo y 3-metil-1-butinilo.
El término "heteroalquilo" por sí mismo o junto con otro término significa, a menos que se señale otra cosa, un
hidrocarbono estable de cadena lineal o ramificada, o sus combinaciones, totalmente saturado o que contiene de 1 a 3 grados de insaturación, que consiste en el número indicado de átomos de carbono y de uno a tres heteroátomos seleccionados entre el grupo que consiste en O, N, Si y S, y en el que los átomos de nitrógeno y azufre pueden estar opcionalmente oxidados y el heteroátomo de nitrógeno puede estar opcionalmente cuaternizado. El heteroátomo o heteroátomos O, N y S pueden estar situados en cualquier posición interior del grupo heteroalquilo. El heteroátomo Si puede estar situado en cualquier posición del grupo heteroalquilo, incluyendo la posición en la que el grupo alquilo está unido al resto de la molécula. Hasta dos heteroátomos pueden ser consecutivos.
El término "arilo", por sí mismo o una parte de otro término, significa, a menos que se indique otra cosa, un radical hidrocarburo aromático, carbocíclico, monovalente, sustituido o sin sustituir, de 6-20, preferentemente 6-14, átomos de carbono obtenido a partir de la eliminación de un átomo de hidrógeno de un átomo de carbono individual de un sistema de anillo aromático parental. Los grupos arilo típicos incluyen, pero sin limitación, radicales derivados de benceno, benceno sustituido, naftaleno, antraceno, bifenilo y similares. Un grupo arilo puede estar sustituido con uno o más, preferentemente de 1 a 5, de los siguientes grupos: alquilo C1-C8, -O-(alquilo C1-C8), -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(O)2R', -S(O)R', -OH, halógeno, -N3, -NH2, -NH(R'), -N(R')2 y -CN; en los que cada R' se selecciona independientemente entre -H, alquilo C1-C8 y arilo no sustituido. El término "heterociclilo" por sí mismo o como parte de otro término, a menos que se indique otra cosa, se refiere a un sistema de anillo monocíclico, bicíclico o tricíclico, aromático o no aromático, sustituido o sin sustituir, monovalente, que tiene de 1 a 10, preferentemente de 3 a 8, átomos de carbono (también denominados miembros de anillo) y de uno a cuatro miembros heteroátomos en el anillo seleccionados independientemente entre N, O, P o S, y obtenidos a partir de la eliminación de un átomo de hidrógeno de un átomo del anillo de un sistema de anillo parental. Uno o más de los átomos de N, C o S en el heterociclilo puede estar oxidado. El anillo que incluye el heteroátomo puede ser aromático o no aromático. A menos que se mencione otra cosa, el heterociclilo está unido a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. Los ejemplos representativos de un heterociclilo C1-C10 incluyen, pero sin limitación, tetrahidrofuranilo, oxetanilo, piranilo, pirrolidinilo, piperidinilo, piperazinilo, benzofuranilo, benzotiofeno, benzotiazolilo, indolilo, benzopirazolilo, pirrolilo, tiofenilo (tiofeno), furanilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, quinolinilo, incluyendo restos tales como 1,2,3,4-tetrahidro-quinolinilo, pirimidinilo, piridinilo, piridonilo, pirazinilo, piridazinilo, isotiazolilo, isoxazolilo, tetrazolilo, epóxido, oxetano y BODIPY (sustituido o sin sustituir). Un heterociclilo C1-C10 puede estar sustituido hasta con siete grupos incluyendo, pero sin limitación, alquilo C1-C8, heteroalquilo C1-C8, -OR', arilo, -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S( =O)2R', -S(O)R', halógeno, -N3, -NH2, -NH(R'), -N(R')2 y -CN; en los que cada R' se selecciona independientemente entre -H, alquilo C1-C8, heteroalquilo C1-C8 y arilo. En algunas realizaciones, un heterociclilo sustituido también puede incluir uno o más de: -NHC(=NH)NH2, -NHCONH2, -S(=O)2R' y -SR'.
El término "carbociclilo", por sí mismo o como parte de otro término, a menos que se indique otra cosa, se refiere a un anillo carbocíclico, monocíclico o bicíclico, no aromático, saturado o insaturado, sustituido o sin sustituir, monovalente, de 3, 4, 5, 6, 7 u 8 miembros, obtenido a partir de la eliminación de un átomo de hidrógeno de un átomo del anillo de un sistema de anillo parental. Los carbociclilos representativos incluyen, pero sin limitación, ciclopropilo, ciclobutilo, ciclopentilo, ciclopentadienilo, ciclohexilo, ciclohexenilo, 1,3-ciclohexadienilo, 1,4-ciclohexadienilo, cicloheptilo, 1,3-cicloheptadienilo, 1,3,5-cicloheptatrienilo, ciclooctilo, ciclooctadienilo, biciclo(1.1.1)pentano y biciclo(2.2.2)octano. Un grupo carbociclilo puede estar sin sustituir o sustituido hasta con siete grupos incluyendo, pero sin limitación, alquilo C1-C8, heteroalquilo C1-C8, -OR', arilo, -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S( =O)2R', -S(=O)R', -OH, -halógeno, -N3, -NH2, -NH(R'), -N(R')2 y - c N; en el que cada R' es como se ha definido anteriormente.
El término "alquenil-", a menos que se indique otra cosa, se refiere a un hidrocarburo insaturado, de cadena lineal o ramificada, que contiene al menos un doble enlace y preferentemente de 2 a 10 átomos de carbono. Los ejemplos de un grupo alquenilo incluyen, pero sin limitación, etileno, propileno, 1 -butileno, 2-butileno, isobutileno, sec-butileno, 1-penteno, 2-penteno, isopenteno, 1-hexeno, 2-hexeno, 3-hexeno, isohexeno, 1-hepteno, 2-hepteno, 3-hepteno, 1-octeno, 2-octeno, 3-octeno, 4-octeno, 1-noneno, 2-noneno, 3-noneno, 4-noneno, 1-deceno, 2-deceno, 3-deceno, 4-deceno y 5-deceno. Un grupo alquenilo puede estar sin sustituir o sustituido.
El término "alcoxi-" se refiere al grupo alquil-O- en el que el alquilo es como se define en el presente documento. Los grupos alcoxi ejemplares incluyen, pero sin limitación, metoxi, etoxi, n-propoxi, 1-propoxi, n-butoxi y t-butoxi. Un grupo alcoxi puede estar sin sustituir o sustituido.
El término "alquinil-" se refiere a un hidrocarburo insaturado de cadena lineal o ramificada que contiene al menos un triple enlace y preferentemente de 2 a 10 átomos de carbono. Los ejemplos de un grupo alquinil- incluyen, pero sin limitación, acetileno, propino, 1-butino, 2-butino, isobutino, sec-butino, 1-pentino, 2-pentino, isopentino, 1-hexino, 2-hexino, 3-hexino, isohexino, 1-heptino, 2-heptino, 3-heptino, 1-octino, 2-octino, 3-octino, 4-octino, 1-nonino, 2-nonino, 3-nonino, 4-nonino, 1-decino, 2-decino, 3-decino, 4-decino y 5-decino. Un grupo alquinilo puede estar sin sustituir o sustituido.
El término "heteroaril-" se refiere a grupos aromáticos, mono o bicíclicos, de 5-10 miembros, que contienen al menos un heteroátomo seleccionado entre oxígeno, azufre y nitrógeno, y normalmente de 1 a 9 átomos de carbono. Los ejemplos de radicales heteroarilo monocíclicos incluyen, pero sin limitación, oxazinilo, tiazinilo, diazinilo, triazinilo, tiadiazoílo, tetrazinilo, imidazolilo, tetrazolilo, isoxazolilo, furanilo, furazanilo, oxazolilo, tiazolilo, tiofenilo, pirazolilo,
triazolilo, pirimidinilo, N-piridilo, 2-piridilo, 3-piridilo y 4-piridilo. Los ejemplos de radicales heteroarilo bicíclicos incluyen, pero sin limitación, benzoimidazolilo, indolilo, isoquinolinilo, benzofuranilo, benzotiofenilo, indazolilo, quinolinilo, quinazolinilo, purinilo, benzoisoxazolilo, benzoxazolilo, benzotiazolilo, benzodiazolilo, benzotriazolilo, isoindolilo e indazolilo. Un grupo heteroarilo puede estar sin sustituir o sustituido.
El término "carboxil-" se refiere a un grupo, normalmente un grupo alquilo como se ha definido en el presente documento, que está unido a la estructura parental a través del átomo de oxígeno de una funcionalidad carboxilo (C(O)-O-). Los ejemplos de grupos carboxilo incluyen acetoxi, propionoxi, propilcarboxilo e isopentilcarboxilo.
"Sustituido", por ejemplo, con respecto a un resto químico tal como "alquilo sustituido", a menos que se especifique otra cosa, significa un resto en el que uno o más átomos de hidrógeno están cada independientemente reemplazado independientemente por un sustituyente. Entre los sustituyentes típicos se incluyen, pero sin limitación, -X, -R, -O-, -OR, -SR, -S-, -NR2, -NR3, =NR, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NRC(=O)R, -C(=O)NR2, -SO3-, -SO3H, -S(=O)2R, -OS(=O)2OR, -S(=O)2NR, -S(=O)R, -OP(=O)(OR)2, -P(=O)(OR)2, -PO32", PO3H2, -AsO2H2, -C(=O)R, -C(=O)X, -C(=S)R, -CO2R, -CO2-, -C(=S)OR, -C(O)SR, -C(=S)SR, -C(=O)NR2, -C(=S)NR2 o -C(=NR)NR2, en los que cada X es independientemente un halógeno: -F, -CI, -Br o -I; y cada R es independientemente -H, alquilo C1-C20, heteroalquilo C1-C20, arilo C6-C20, heterociclilo C1-C10, un grupo protector o un resto de profármaco.
También se desvelan conjugados para su uso para tratar un cáncer, inhibir el crecimiento o progresión de un tumor, inhibir de la metástasis de células cancerosas de tumores o inducir la regresión del tumor en un sujeto que lo necesita, que comprende administrar al sujeto una cantidad eficaz de la composición farmacéutica que comprende los ADC como se describen en el presente documento.
Definiciones y técnicas generales
A menos que se defina lo contrario en el presente documento, los términos científicos y técnicos usados en relación con la presente invención tendrán los significados que entienden los expertos en la técnica de forma habitual. Además, a menos que el contexto requiera lo contrario, los términos en singular incluyen pluralidades y los términos en plural incluyen el singular. Generalmente, la nomenclatura utilizada en relación con, y técnicas de, cultivo celular y tisular, biología molecular, inmunología, microbiología, genética y química proteica y de ácidos nucleicos e hibridación descritas en el presente documento son las bien conocidas y comúnmente utilizadas en la técnica.
Los procedimientos y técnicas de la presente invención se realizan generalmente de acuerdo con procedimientos convencionales bien conocidos en la técnica y como se describe en diversas referencias generales y más específicas que se citan y se tratan a lo largo de la presente memoria descriptiva a menos que se indique lo contrario. Véase, por ejemplo, Sambrook J. y Russell D. Molecular Cloning: A Laboratory Manual, 3a ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (2000); Ausubel y col., Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology, Wiley, John & Sons, Inc. (2002); Harlow y Lane Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1998); y Coligan y col., Short Protocols in Protein Science, Wiley, John & Sons, Inc. (2003). Las reacciones enzimáticas y las técnicas de purificación se realizan de acuerdo con las especificaciones del fabricante, como se logra habitualmente en la técnica o como se describe en el presente documento. La nomenclatura empleada junto con, y los procedimientos de laboratorio y técnicas de, biología molecular, bioquímica, inmunología, química analítica, química orgánica sintética y química médica y farmacéutica descritas en el presente documento son aquellas bien conocidas y utilizadas habitualmente en la materia. A lo largo de esta memoria y reivindicaciones, la palabra "comprende" o variaciones tales como "comprende" o "que comprende" se entenderá que implican la inclusión de un número entero o grupo de números enteros indicados, pero no la exclusión de ningún otro número entero o grupo de números enteros.
La expresión "marcador que contiene glutamina", "marcador de glutamina" "marcador que contiene Q", "marcador Q" o "marcador de transglutaminasa" como se usa en el presente documento se refiere a un polipéptido o una proteína que contiene uno o más restos de Gln, o puede referirse a un solo resto de glutamina o glutamina.
Tal como se usa en el presente documento, la expresión "especificidad del sitio" "conjugado específicamente en el sitio," o "reticulado específicamente en el sitio" se refiere a la conjugación o reticulación específica del resto que contiene amina con el anticuerpo en un sitio específico (por ejemplo, en varias posiciones enumeradas en la Tabla 1) a través de un marcador que contiene glutamina, glutamina endógena, y/o una glutamina endógena reactiva mediante ingeniería de anticuerpos o una transglutaminasa modificada por ingeniería genética. La especificidad del sitio se puede medir mediante diversas técnicas, incluyendo, pero sin limitación, espectrometría de masas (por ejemplo, espectrometría de masas por ionización por desorción láser asistida por matriz (MALDI-MS), espectrometría de masas por ionización por electropulverización (ESI-MS), espectrometría de masas en tándem (MS-MS) y espectrometría de masas de tiempo de vuelo (TOF-MS), cromatografía de interacción hidrófoba, cromatografía de intercambio iónico, por mutagénesis dirigida, marcado de fluorescencia, cromatografía de exclusión por tamaño y cristalografía de rayos X.
Los términos "carga" o "carga de fármaco" o "carga útil" representan o se refieren al número promedio de cargas útiles ("carga útil" y "cargas útiles" se usan de forma intercambiable en el presente documento con "fármaco" y "fármacos") por anticuerpo en una molécula de CAF. La carga de fármaco puede variar de 1 a 20 fármacos por anticuerpo. Esto a veces se conoce como DAR, o relación de fármaco a anticuerpo. La relación fármaco-anticuerpo (DAR) del CAF de la
presente invención es de aproximadamente 1 a aproximadamente 60. En algunas realizaciones, la DAR es al menos aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45,46, 47, 48,49, 50, 51, 52, 53, 54, 55,56,57, 58, 59 y 60. El número promedio de medicamentos por anticuerpo, o valor DAR, puede caracterizarse por medios convencionales, tal como espectroscopía UV/visible, espectrometría de masas, ensayo ELISA y HPLC. El valor de DAR cuantitativo también se puede determinar. En algunos casos, la separación, purificación y caracterización de CAF homogéneos que tienen un valor DAR particular se puede lograr por medios tales como HPLC de fase inversa o electroforesis. DAR puede estar limitado por el número de sitios de unión en el anticuerpo. Por ejemplo, en el que la unión es una glutamina como en la presente invención, un anticuerpo puede tener solo uno o varios restos de glutamina adecuados a través de los cuales puede unirse una unidad enlazadora. Habitualmente, menos del máximo teórico de los restos farmacológicos está conjugado con un anticuerpo durante una reacción de conjugación.
Tal como se usa en el presente documento, la expresión "una glutamina endógena (Q) hecha reactiva" se refiere a una glutamina endógena que se ha hecho accesible, se ha expuesto o es reactiva al resto que contiene amina en presencia de una transglutaminasa por ingeniería de anticuerpos (por ejemplo, desglicosilación enzimática y/o modificación de aminoácidos) o por una transglutaminasa modificada por ingeniería genética.
Tal como se usa en el presente documento, la expresión "polímero biocompatible" se refiere a un polímero (por ejemplo, unidades monoméricas o estructurales repetitivas) que es adecuado para el tratamiento terapéutico o médico en un receptor (por ejemplo, humano) sin provocar ningún efecto local o sistémico indeseable en el receptor. Un polímero biocompatible (sintético, recombinante o nativo) puede ser un polímero soluble en agua o insoluble en agua. Un polímero biocompatible también puede ser un polímero lineal o ramificado.
Tal como se usa en el presente documento, el término "anticuerpo" es una molécula de inmunoglobulina capaz de unirse de manera específica a una diana, tal como un hidrato de carbono, polinucleótido, lípido, polipéptido, etc., a través de al menos un sitio de reconocimiento de antígeno, localizado en la región variable de la molécula de inmunoglobulina. Tal como se usa en el presente documento, salvo que se indique otra cosa por el contexto, el término pretende abarcar no solo anticuerpos policlonales o monoclonales intactos, sino también fragmentos de los mismos (tales como Fab, Fab', F(ab')2, Fv), anticuerpos de cadena sencilla (ScFv) y anticuerpos de dominio, incluyendo anticuerpos de tiburón y de camélido), y proteínas de fusión que comprenden una parte de anticuerpo, anticuerpos multivalentes (por ejemplo, COVX-BODY ™), anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos siempre y cuando presenten la actividad biológica deseada) y fragmentos de anticuerpos tal como se describe en el presente documento, y cualquier otra configuración modificada de la molécula de inmunoglobulina que comprende un sitio de reconocimiento de antígeno. Un anticuerpo incluye un anticuerpo de cualquier clase, tal como IgG, IgA o IgM (o subclases de los mismos), y el anticuerpo no necesita ser de ninguna clase en particular. En función de la secuencia de aminoácidos del dominio constante de sus cadenas pesadas, se pueden asignar inmunoglobulinas a las diferentes clases. Hay cinco clases principales de inmunoglobulinas: IgA, IgD, IgE, IgG e IgM, y varias de estas se pueden dividir adicionalmente en subclases (isotipos), por ejemplo, IgG1, IgG2, IgG3, IgG4, IgA1 y IgA2. Los dominios constantes de la cadena pesada que se corresponde con las diferentes clases de inmunoglobulinas se denominan alfa, delta, épsilon, gamma y mu, respectivamente. Las estructuras de la subunidad y las configuraciones tridimensionales de las diferentes clases de inmunoglobulinas son bien conocidas. En un aspecto, la inmunoglobulina es una inmunoglobulina humana, murina, de mono o de conejo.
La expresión "polipéptido que contiene Fab" como se usa en el presente documento se refiere a un polipéptido que comprende un fragmento Fab, fragmento Fab' o "fragmento (Fab')2". Un polipéptido que contiene Fab puede comprender parte o la totalidad de una secuencia de bisagra de tipo salvaje (generalmente en el extremo carboxilo de la porción Fab del polipéptido). Un polipéptido que contiene Fab puede obtenerse o derivarse de cualquier inmunoglobulina adecuada, tal como de al menos uno de los diversos subtipos IgG1, IgG2, IgG3 o IgG4, o de IgA, IgE, IgD o IgM. Un polipéptido que contiene Fab puede ser un polipéptido de fusión que contiene Fab, en l que uno o más polipéptidos están unidos a un polipéptido que contiene Fab. Una fusión Fab combina el polipéptido Fab de una inmunoglobulina con un compañero de fusión, que en general puede ser cualquier proteína, polipéptido o molécula pequeña. Prácticamente cualquier proteína o molécula pequeña se puede unir al polipéptido Fab para generar un polipéptido de fusión que contiene Fab. Los compañeros de fusión que contienen Fab pueden incluir, pero sin limitación, la región de unión al objetivo de un receptor, una molécula de adhesión, un ligando, una enzima, una citocina, una quimiocina, o alguna otra proteína o dominio de proteínas.
Un "fragmento Fab" está compuesto por una cadena ligera y las regiones CH1 y variable de una cadena pesada. La cadena pesada de una molécula Fab no puede formar un puente disulfuro con otra molécula de la cadena pesada.
Un "fragmento Fab'" contiene una cadena ligera y una porción de una cadena pesada que contiene el dominio VH y el dominio CH1 y también la región entre los dominios c H1 y CH2, de forma tal que se puede formar un enlace disulfuro intercatenario entre las dos cadenas pesadas de dos fragmentos Fab' para formar una molécula F(ab')2.
Un "fragmento F (ab')2" contiene dos cadenas ligeras y dos cadenas pesadas que contienen una porción de la región constante entre los dominios CH1 y CH2, de forma tal que se forma un enlace disulfuro intercatenario entre las dos cadenas pesadas. Por tanto, un fragmento F(ab')2 está compuesto por dos fragmentos Fab' que se mantienen juntos a través de un puente disulfuro entre las dos cadenas pesadas.
Los "fragmentos de anticuerpos" como se usa en el presente documento comprenden solo una porción de un anticuerpo intacto, en la que la porción preferentemente conserva al menos una, preferentemente la mayoría o todas, las funciones que normalmente se asocian con esa porción cuando está presente en un anticuerpo intacto.
Un "anticuerpo multiespecífico" es uno que se dirige a más de un antígeno o epítopo. Un anticuerpo "biespecífico", "específico dual" o "bifuncional" es un anticuerpo híbrido que tiene dos sitios de unión a antígeno diferentes. Los anticuerpos biespecíficos son una especie de anticuerpo multiespecífico y pueden producirse mediante diversos procedimientos, que incluyen, pero sin limitación, fusión de hibridomas, enlace de fragmentos Fab' o mutaciones en la bisagra del anticuerpo y los dominios CH3. Véase, por ejemplo, Songsivilai y Lachmann, Clin. Exp. Immunol. 79:315-321 (1990); Kostelny y col., J. Immunol. 148:1547-1553 (1992); y Strop y col., J. Mol. Biol. 420(3):204-219 (2012). Los dos sitios de unión de un anticuerpo biespecífico se unirán a dos epítopos diferentes, que pueden residir en los mismos objetivos proteicos o en otros diferentes.
La expresión "anticuerpo monoclonal" tal como se utiliza en el presente documento, se refiere a un anticuerpo obtenido a partir de una población de anticuerpos sustancialmente homogéneos, es decir, los anticuerpos individuales que comprenden la población son idénticos excepto para posibles mutaciones que se producen de forma natural que pueden estar presentes en cantidades menores. Los anticuerpos monoclonales son muy específicos, estando dirigidos contra un solo antígeno. Además, a diferencia de las preparaciones de anticuerpos policlonales que normalmente incluyen anticuerpos diferentes dirigidos contra determinantes diferentes (epítopos), cada anticuerpo monoclonal se dirige contra un único determinante del antígeno.
Los anticuerpos monoclonales en el presente documento pueden, en determinadas realizaciones, los anticuerpos "quiméricos" incluyen específicamente anticuerpos en los que una porción de la cadena pesada y/o ligera es idéntica u homóloga a las correspondientes secuencias en los anticuerpos derivados de una especie concreta o pertenecientes a una clase o subclase de anticuerpo concreto, mientras que el grupo de la(s) cadena(s) es idéntico u homólogo a las secuencias correspondientes en los anticuerpos derivados de otra especie o pertenecientes a otra clase o subclase de anticuerpos, así como fragmentos de dichos anticuerpos, en tanto que muestren la actividad biológica deseada (patente de Estados Unidos n.° 4,816,567; y Morrison y col., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)).
Las formas "humanizadas" de anticuerpos no humanos (por ejemplo, murinos) son anticuerpos quiméricos que contienen una secuencia mínima derivada de inmunoglobulina no humana. En su mayoría, los anticuerpos humanizados son inmunoglobulinas humanas (anticuerpo receptor) en la que los restos de una región hipervariable del receptor son sustituidos por restos de una región hipervariable de una especie no humana (anticuerpo donante), tal como ratón, una rata, conejo o primate no humano que tiene la especificidad, afinidad y capacidad deseadas. En algunos casos, los restos de la región estructural (FR) de la inmunoglobulina humana están sustituidos por los correspondientes restos no humanos. Los anticuerpos humanizados pueden, también, comprender restos que no se encuentran en el anticuerpo receptor o en el anticuerpo donante. Estas modificaciones se efectúan para refinar adicionalmente el funcionamiento del anticuerpo. En general, el anticuerpo humanizado comprenderá sustancialmente la totalidad de al menos uno, y normalmente dos, dominios variables, en los que todos o sustancialmente todos los bucles hipervariables corresponden a los de una inmunoglobulina no humana o todas o sustancialmente todas las regiones FR son las de una secuencia de inmunoglobulina humana. El anticuerpo humanizado también comprenderá al menos una porción de una región constante de inmunoglobulina (Fc), normalmente de una inmunoglobulina humana. Para más detalles, véase Jones y col., Nature 321:522-525 (1986); Riechmann y col., Nature 332:323-329 (1988); y Presta, Curr. Op. Struct. Biol. 2: 593-596(1992). Véanse también los siguientes artículos de revisión y referencias citadas en los mismos: Vaswani y Hamilton, Ann. Allergy, Asthma & Immunol. 1:105-115 (1998); Harris, Biochem. Soc. Transactions 23:1035-1038 (1995); Hurle y Gross, Curr. Op. Biotech. 5: 428-433(1994).
Un "anticuerpo humano" es uno que posee una secuencia de aminoácidos que corresponde a la de un anticuerpo producido por un ser humano y/o que ha sido fabricado usando cualquiera de las técnicas para producir anticuerpos humanos como se desvela en el presente documento. Esta definición de un anticuerpo humano excluye específicamente un anticuerpo humanizado que comprenda restos de unión a antígeno no humanos.
La "región bisagra" "secuencia bisagra" y variaciones de las mismas, tal como se usa en el presente documento, incluye el significado conocido en la técnica, que ilustra, por ejemplo, Janeway y col., ImmunoBiology: the immune system in health and disease, (Elsevier Science Ltd., NY) (4a ed., 1999); Bloom y col., Protein Science (1997), 6:407-415; Humphreys y col., J. Immunol. Methods (1997), 209:193-202.
La expresión "polipéptido que contiene Fc" como se usa en el presente documento se refiere a un polipéptido (por ejemplo, un anticuerpo o una inmunoadhesina) que comprende las secuencias polipeptídicas del extremo carboxilo de una cadena pesada de inmunoglobulina. El polipéptido que contiene Fc puede comprender regiones Fc nativas o variantes (es decir, secuencias). La región Fc de una inmunoglobulina generalmente comprende dos dominios constantes, un dominio CH2 y un dominio CH3, y, opcionalmente, comprende un dominio CH4. Un polipéptido que contiene Fc puede comprender parte o la totalidad de una secuencia bisagra de tipo salvaje (generalmente en el extremo amino del polipéptido que contiene Fc). Un polipéptido que contiene Fc también puede ser un dímero. Un polipéptido que contiene Fc puede obtenerse o derivarse de cualquier inmunoglobulina adecuada, tal como de al menos uno de los diversos subtipos IgG1, IgG2, IgG3 o IgG4, o de IgA, IgE, IgD o IgM. Aunque los límites de la región Fc de una cadena pesada de inmunoglobulina pueden variar, por ejemplo, la región Fc de la cadena pesada de IgG
humana se define generalmente de modo que abarca desde un resto de aminoácido en la posición Glu216, o desde Ala231, al carboxilo terminal de la misma. La numeración de los restos en la región Fc es la del índice de UE de Kabat. Kabat y col., Sequences of Proteins of Immunological Interest, 5a Ed. Public Health Service, National Institutes of Health, Bethesda, Md., 1991.
Un polipéptido que contiene Fc puede ser un polipéptido de fusión que contiene Fc, en el que uno o más polipéptidos están unidos a un polipéptido que contiene Fc. Una fusión Fc combina el polipéptido Fc de una inmunoglobulina con un compañero de fusión, que en general puede ser cualquier proteína, polipéptido o molécula pequeña. Prácticamente cualquier proteína o molécula pequeña se puede unir a la región Fc para generar un polipéptido de fusión que contiene Fc. Los compañeros de fusión que contienen Fc pueden incluir, pero sin limitación, la región de unión al objetivo de un receptor, una molécula de adhesión, un ligando, una enzima, una citocina, una quimiocina, o alguna otra proteína o dominio de proteínas.
Tal como se usa en el presente documento, el término "inmunoadhesina" designa moléculas similares a anticuerpos o similares a inmunoglobulinas que combinan el "dominio de unión" de una proteína heteróloga (una "adhesina", por ejemplo, un receptor, ligando o enzima) con el componente efector de dominios constantes de inmunoglobulina (es decir, dominio Fc). Estructuralmente, las inmunoadhesinas comprenden una fusión de la secuencia de aminoácidos de la adhesina con la especificidad de unión deseada que no es el sitio de reconocimiento y unión al antígeno (sitio de combinación del antígeno) de un anticuerpo (es decir, es "heterólogo") y una secuencia de dominio constante de inmunoglobulina. La secuencia de dominio constante de inmunoglobulina en la inmunoadhesina se puede obtener de cualquier inmunoglobulina, tales como los subtipos IgG1, IgG2, IgG3 o IgG4, IgA, IgE, IgD o IgM.
Los términos "polipéptido", "oligopéptido", "péptido" y "proteína" se utilizan indistintamente en el presente documento para hacer referencia a cadenas de aminoácidos de cualquier longitud, preferentemente, relativamente cortas (por ejemplo, 10-100 aminoácidos). La cadena puede ser lineal o ramificada, puede comprender aminoácidos modificados y/o puede estar interrumpida por no aminoácidos. Los términos también abarcan una cadena de aminoácidos que se ha modificado de manera natural o por intervención; por ejemplo, formación de enlaces disulfuro, glucosilación, lipidación, acetilación, fosforilación o cualquier otra manipulación o modificación, como la conjugación con un componente de etiquetado. Dentro de la definición también se incluyen, por ejemplo, polipéptidos que contienen uno o más análogos de un aminoácido (incluidos, por ejemplo, aminoácidos no naturales, etc.), así como otras modificaciones conocidas en la técnica. Se entiende que los polipéptidos pueden presentarse como cadenas individuales o cadenas asociadas.
Tal como se usa en el presente documento, la expresión "aminoácido de tipo salvaje" "IgG de tipo salvaje" o "mAb de tipo salvaje" se refiere a una secuencia de aminoácidos o ácidos nucleicos que se encuentra naturalmente dentro de una determinada población (por ejemplo, ser humano, ratones, ratas, células, etc.).
Tal como se usa en el presente documento, la expresión "eficiencia de conjugación" o "eficiencia de reticulación" es la relación entre las cantidades de CAF medidas experimentalmente como se describe en el presente documento dividida por la cantidad máxima de CAF esperada. La eficiencia de conjugación o la eficiencia de reticulación se pueden medir mediante diversas técnicas bien conocidas por los expertos en la materia, tal como cromatografía de interacción hidrofóbica. La eficiencia de conjugación también se puede medir a diferentes temperaturas, tales como temperatura ambiente o 37 °C.
La expresión "función efectora" se refiere a las actividades biológicas atribuibles a la región Fc de un anticuerpo. Los ejemplos de funciones efectoras de anticuerpos incluyen, pero sin limitación, citotoxicidad mediada por células dependiente de anticuerpos (CCDA), unión a receptor Fc, citotoxicidad dependiente del complemento (CDC), fagocitosis, Unión de C1q y regulación por disminución de los receptores de la superficie celular (por ejemplo, receptor de linfocitos B; BCR). Véase, por ejemplo, la patente de Estados Unidos n.° 6,737,056. Dichas funciones efectoras generalmente requieren que la región Fc se combine con un dominio de unión (por ejemplo, un dominio variable de anticuerpo) y puede evaluarse usando varios ensayos conocidos en la técnica para evaluar tales funciones efectoras de anticuerpos. Una medición de ejemplo de la función efectora es a través de la unión de F c y 3 y/o C1q.
Tal como se usa en el presente documento, "citotoxicidad mediada por células dependiente de anticuerpos" y "CCDA" se refieren a una reacción mediada por células en la que las células citotóxicas no específicas que expresan receptores Fc (FcR) (por ejemplo, células citolíticas naturales (N k ), neutrófilos y macrófagos) reconocen el anticuerpo unido en una célula objetivo y posteriormente causan lisis de la célula objetivo. La actividad CCDA de una molécula de interés puede evaluarse utilizando un ensayo de CCDA in vitro, tal como se describe en la patente de Estados Unidos n° 5.500.362 o 5.821.337. Las células efectoras útiles para tales ensayos incluyen células mononucleares de sangre periférica (PBMC) y células NK. Como alternativa o además, la actividad de CCDA de la molécula de interés puede evaluarse in vivo, por ejemplo, en un modelo animal como se desvela en Clynes y col., 1998, PNAS (USA), 95:652-656.
"Citotoxicidad dependiente del complemento" y "CDC" se refieren a la lisis de una diana en presencia del complemento. La ruta de activación del complemento se inicia por la unión del primer componente del sistema del complemento (C1q) con una molécula (por ejemplo, un anticuerpo) que forma un complejo con un antígeno análogo. Para evaluar la activación del complemento, se puede realizar un ensayo de CDC, por ejemplo, como se describe en Gazzano-Santoro y col., J. Immunol. Methods, 202:163 (1996).
Tal como se usa en el presente documento, "receptor de Fc" y "FcR" describen un receptor que se une a la región Fc de un anticuerpo. El FcR preferido es una secuencia nativa de FcR humano. También, un FcR preferido es aquel que se une a un anticuerpo IgG (un receptor gamma) e incluye receptores de las subclases FcyRI, FcyRII, FcyRIII y FcyRIV, incluyendo las variantes alélicas y formas empalmadas alternativamente de estos receptores. Los receptores FcyRII incluyen FcyRIIA (un "receptor activador") y FcyRIIB (un "receptor inhibidor"), que tienen secuencias de aminoácidos similares que difieren principalmente en los dominios citoplasmáticos de las mismas. Los FcR se revisan en Ravetch y Kinet, 1991, Ann. Rev. Immunol., 9:457-92; Capel y col., 1994, Immunomethods, 4:25-34; de Haas y col., 1995, J. Lab. Clin. Med., 126:330-41; Nimmerjahn y col., 2005, Immunity 23:2-4. "FcR" también incluye el receptor neonatal, FcRn, que es responsable de la transferencia de IgG maternas al feto (Guyer y col., 1976, J. Immunol., 117:587; y Kim y col., 1994, J. Immunol., 24:249).
El término "purificar" y variaciones gramaticales del mismo, se utiliza para la eliminación, ya sea total o parcialmente, de al menos una impureza de una mezcla que contiene el CAF y una o más impurezas, que, de ese modo, mejora el nivel de pureza del CAF en la composición (es decir, al disminuir la cantidad (ppm) de impurezas en la composición).
La referencia a "aproximadamente" un valor o parámetro en el presente documento incluye (y describe) realizaciones que se refieren a ese valor o parámetro per se. Por ejemplo, la descripción que hace referencia a "aproximadamente X" incluye la descripción de "X". Los intervalos numéricos incluyen los números que definen el intervalo.
Un "individuo" o un "sujeto" es un mamífero, más preferentemente, un ser humano. Los mamíferos también incluyen, pero sin limitación, animales de granja, animales de deporte, mascotas, primates, caballos, perros, gatos, ratones y ratas.
Se entiende que dondequiera que se describan realizaciones en el presente documento con el lenguaje "que comprende" por lo demás, también se proporcionan realizaciones análogas descritas en términos de "que consiste en" y/o "que consiste esencialmente en".
Cuando se describen aspectos o realizaciones de la invención en términos de un grupo de Markush u otro grupo de alternativas, la presente invención abarca no solo el grupo completo enumerado como una totalidad, sino cada miembro del grupo individualmente y todos los subgrupos posibles del grupo principal, sino también el uno o más miembros ausentes del grupo del grupo principal. La presente invención también prevé la exclusión explícita de uno o más de cualquiera de los miembros del grupo en la invención reivindicada.
Las designaciones de restos en la presente solicitud se basan en el esquema de numeración de la UE del dominio constante (Edelman y col., Proc. Natl. Acad. Sci. USA, 63(1):78-85 (1969).
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que el comprendido habitualmente por un experto en la materia a la que pertenece la presente invención. Los procedimientos y materiales de ejemplo se describen en el presente documento, aunque los procedimientos y materiales similares o equivalentes a los descritos en el presente documento también se pueden utilizar en la práctica o el ensayo de la presente invención. Las publicaciones y otras referencias se mencionan en el presente documento. En caso de conflicto, la presente memoria descriptiva, incluyendo las definiciones, regirá. Aunque en el presente documento se citan varios documentos, esta cita no constituye una admisión de que alguno de estos documentos forme parte del conocimiento general común en la técnica. A lo largo de esta memoria y reivindicaciones, la palabra "comprende" o se entenderá que variaciones como "comprende" o "que comprende" implican la inclusión de un número entero o grupo de números enteros, pero no la exclusión de ningún otro número entero o grupo de números enteros. A menos que el contexto requiera lo contrario, los términos en singular incluyen pluralidades y los términos en plural incluyen el singular. Los materiales, procedimientos y ejemplos son solamente ilustrativos y no pretenden ser limitantes.
Las transglutaminasas son proteínas-glutamina Y-glutamiltransferasas, que típicamente catalizan la transamidación dependiente del pH de los restos de glutamina con restos de lisina. La transglutaminasa utilizada en la invención descrita en el presente documento puede obtenerse o elaborarse a partir de diversas fuentes, o diseñarse para catalizar la transamidación de uno o más restos de glutamina endógenos con uno o más restos de lisina u otro resto que contiene amina. En algunas realizaciones, la transglutaminasa es una transglutaminasa dependiente de calcio que requiere calcio para inducir cambios de conformación enzimática y permitir la actividad enzimática. Por ejemplo, la transglutaminasa puede proceder de hígado de cobaya y obtenerse a través de fuentes comerciales (por ejemplo, Sigma-Aldrich (St Louis, MO) y MP Biomedicals (Irvine, CA)). En algunas realizaciones, la transglutaminasa es una transglutaminasa independiente del calcio que no requiere calcio para inducir cambios conformacionales de la enzima y permitir la actividad enzimática. En algunas realizaciones, la transglutaminasa es una transglutaminasa microbiana derivada de un genoma microbiano, tales como transglutaminasa de Streptoverticillium o Streptomyces (por ejemplo, Streptomyces mobarensis o Streptoverticillium mobarensis). La transglutaminasa independiente del calcio disponible comercialmente, tal como ACTIVA™ (Ajinomoto, Japón) es adecuada para la presente invención. En algunas realizaciones, la transglutaminasa es una proteína de mamífero (por ejemplo, transglutaminasa humana), una proteína bacteriana, una proteína vegetal, una proteína de hongos (por ejemplo, transglutaminasas de Oomycetes y Actinomycetes), o una proteína procariota. En algunas realizaciones, la transglutaminasa es de Micrococcus, Clostridium, Turolpsis, Rhizopus, Monascus o Bacillus.
En algunas realizaciones, la transglutaminasa utilizada en la invención descrita en el presente documento es una transglutaminasa modificada genéticamente que cataliza la transamidación de uno o más restos de glutamina endógenos en el anticuerpo con uno o más restos de lisina u otro resto que contiene aminas. Por ejemplo, uno o más restos de aminoácidos de tipo salvaje en la transglutaminasa natural pueden delecionarse o reemplazarse o sustituirse con uno o más restos de aminoácidos para hacer la transglutaminasa modificada.
En algunas realizaciones, la transglutaminasa usada en la invención descrita en el presente documento también puede ser una proteína recombinante producida usando técnicas recombinantes conocidas por los expertos en la materia. En algunas realizaciones, la transglutaminasa usada en la invención descrita en el presente documento puede ser una proteína purificada. Por ejemplo, la transglutaminasa purificada tiene al menos aproximadamente un 50 % de pureza. Tal como se usa en el presente documento, la proteína "pura" o "purificada" se refiere a una proteína (por ejemplo, transglutaminasa) libre de otros contaminantes proteicos. En algunas realizaciones, la transglutaminasa purificada tiene al menos aproximadamente cualquiera de 55 %-60 %, 60 %/-65 %, 65 %/-70 %, 70 %/-75 %, 75 %/-80 %, 80 %/-85 %, 85 %/-90 %, 90 %/-95 %, 95%-98 % o 99 % de pureza. En algunas realizaciones, la transglutaminasa purificada tiene aproximadamente un 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 96 %, 97 %, 98 %, 99 % o 100 % de pureza.
En algunas realizaciones, el CAF de la presente invención comprende al menos 1 glutamina endógena que se hace reactiva en una reacción de transamidación por ingeniería de anticuerpos o por una transglutaminasa modificada por ingeniería genética. En algunas realizaciones, la ingeniería de anticuerpos es desglicosilación de anticuerpos (por ejemplo, desglicosilación enzimática); o modificación de aminoácidos, incluyendo deleción de aminoácidos, inserción, sustitución, mutación, o cualquier combinación de las mismas en el anticuerpo. Por ejemplo, el aminoácido de tipo salvaje Asn (N) en la posición 297 en un anticuerpo se sustituye o reemplaza con el aminoácido Ala (A), que da como resultado aglicosilación en la posición 297 y glutamina endógena reactiva (Q) en la posición 295. En otro ejemplo, la modificación de aminoácidos en el anticuerpo es una sustitución de aminoácidos de N a Q en la posición 297, que da como resultado aglicosilación en la posición 297, Q endógena reactivo en la posición 295, y conjugación específica de sitio entre N297Q y Q295 y uno o más restos que contienen amina en estos dos sitios en presencia de una transglutaminasa.
En algunas realizaciones, el CAF de la presente invención comprende un marcador que contiene glutamina diseñada en al menos una o más posiciones, que incluyen, pero sin limitaciones, 1) carboxilo terminal de una cadena ligera, una cadena pesada, o tanto la cadena ligera como la cadena pesada; 2) amino terminal de una cadena ligera, una cadena pesada, o tanto la cadena ligera como la cadena pesada; y 3) S60-R61, R108, T135, S160, S168, S190-S192, P189-S192, G200-S202, K222-T225, K222-T223, T223, L251-S254, M252-I253, E294-N297, E293-N297, N297 y/o G385, en la que el marcador que contiene glutamina se inserta en el anticuerpo o reemplaza uno o más aminoácidos endógenos en el anticuerpo.
Los ejemplos del marcador que contiene glutamina específico y su posición de ingeniería correspondiente se proporcionan en la Tabla 1, y se describen en los documentos WO2012/059882 y WO2015/015448.
Tabla 1

continuación

En algunas realizaciones, el anticuerpo del CAF comprende una modificación de aminoácidos en la posición 222, 340 o 370 (numeración de la UE) con respecto al anticuerpo de tipo salvaje en la misma posición. En algunas realizaciones, la modificación es una deleción de aminoácidos, inserción, sustitución, mutación, o cualquier combinación de las mismas. En algunas realizaciones, la sustitución comprende reemplazar un aminoácido de tipo salvaje con otro (por ejemplo, un aminoácido de tipo no salvaje). En algunas realizaciones, el otro (por ejemplo, de tipo no salvaje) o el aminoácido insertado es Arg (por ejemplo, K222R, K340R o K370R). En algunas realizaciones, la inserción comprende insertar uno o más aminoácidos (por ejemplo, inserción de uno, dos, tres o más aminoácidos). En algunas realizaciones, el otro aminoácido (por ejemplo, de tipo no salvaje) es Ala, Asn, Asp, Cys, Glu, Gln, Gly, His, Ile, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr o Val.
Por consiguiente, en algunas realizaciones, el CAF de la presente invención comprende a) sustituciones de aminoácidos en las posiciones N297Q y K222R, en el que el resto que contiene amina se conjuga específicamente en el sitio con la glutamina endógena en la posición 295 y la glutamina sustituida en la posición 297; y/o b) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga específicamente en el sitio con el o los marcadores que contienen glutamina en un extremo carboxilo de una cadena ligera del anticuerpo. En algunas realizaciones, la relación fármaco-anticuerpo (DAR) es de aproximadamente 3-9. En algunas realizaciones,
el marcador que contiene glutamina en el extremo carboxilo de la cadena ligera del anticuerpo es GGLLQGA (SEQ ID NO:23).
En algunas realizaciones, el CAF de la presente invención comprende a) sustituciones de aminoácidos en las posiciones N297Q y K222R, en el que el resto que contiene amina se conjuga específicamente en el sitio con la glutamina endógena en la posición 295 y la glutamina sustituida en la posición 297; b) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga específicamente en el sitio con el o los marcadores que contienen glutamina en un terminal carboxilo de una cadena ligera del anticuerpo; y/o c) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga además específicamente en el sitio con el marcador que contiene glutamina en una o más posiciones seleccionadas del grupo que consiste en S60-R61, R108, T135, S160, S168, S190-S192, P189-S192, G200-S202, K222-T225, K222-T223, T223, L251-S254, M252-I253, E294-N297, E293-N297, N297 y G385 en el anticuerpo, en la que el marcador que contiene glutamina se inserta en el anticuerpo o reemplaza uno o más aminoácidos endógenos en el anticuerpo. En algunas realizaciones, la relación fármaco-anticuerpo es al menos aproximadamente 6. Por ejemplo, en algunas realizaciones, el CAF de la presente invención también comprende a) sustituciones de aminoácidos en las posiciones N297Q y K222R, en el que el resto que contiene amina se conjuga específicamente en el sitio con la glutamina endógena en la posición 295 y la glutamina sustituida en la posición 297; b) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga específicamente en el sitio con el o los marcadores que contienen glutamina en un terminal carboxilo de una cadena ligera del anticuerpo; c) uno o más marcadores de glutamina, en el que el resto que contiene amina se conjuga específicamente en el sitio además con el marcador que contiene glutamina en un extremo carboxilo de una cadena ligera del anticuerpo; y en el que la relación fármaco-anticuerpo (DAR) es de aproximadamente 6-9. En algunas realizaciones, el marcador que contiene glutamina en el extremo carboxilo de la cadena ligera del anticuerpo es GGLLQGA (SEQ ID NO: 23); y el marcador que contiene glutamina en el extremo carboxilo de la cadena pesada del anticuerpo es LLQGA (SEQ iD No : 11).
En una variación, el CAF de la presente invención comprende a) sustituciones de aminoácidos en las posiciones N297Q y K222R, en el que el resto que contiene amina se conjuga específicamente en el sitio con la glutamina endógena en la posición 295 y la glutamina sustituida en la posición 297; b) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga específicamente en el sitio con el o los marcadores que contienen glutamina en un terminal carboxilo de una cadena ligera del anticuerpo; c) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga además específicamente en el sitio con el marcador que contiene glutamina insertada después de la posición del aminoácido T135 en una cadena pesada del anticuerpo; y en el que la relación fármaco-anticuerpo (DAR) es de aproximadamente 6-9. En algunas realizaciones, el marcador que contiene glutamina en el extremo carboxilo de la cadena ligera del anticuerpo es GGLLQGA (SEQ ID NO: 23); y el marcador que contiene glutamina insertada después de T135 en la cadena pesada del anticuerpo es LLQG (SEQ ID NO: 2).
En otra variante, el CAF de la presente invención comprende a) sustituciones de aminoácidos en las posiciones N297Q y K222R, en el que el resto que contiene amina se conjuga específicamente en el sitio con la glutamina endógena en la posición 295 y la glutamina sustituida en la posición 297; b) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga específicamente en el sitio con el o los marcadores que contienen glutamina en un terminal carboxilo de una cadena ligera del anticuerpo; c) uno o más marcadores que contienen glutamina, en el que el resto que contiene amina se conjuga además específicamente en el sitio con el marcador que contiene glutamina en las posiciones de aminoácidos G200-S202 en una cadena pesada del anticuerpo; y en el que la relación fármaco-anticuerpo (DAR) es de aproximadamente 6-9. En algunas realizaciones, el marcador que contiene glutamina en el extremo carboxilo de la cadena ligera del anticuerpo es GGLLQGA (SEQ ID NO: 23); y el marcador que contiene glutamina insertada después de T135 en la cadena pesada del anticuerpo es LLQG (SEQ ID NO: 2).
En algunas realizaciones, el marcador que contiene glutamina para el CAF descrito en el presente documento comprende una secuencia de aminoácidos XXQX (SEQ ID NO: 37), en la que X puede ser un aminoácido convencional 0 no convencional, tal como se describe en el presente documento. Por ejemplo, en algunas realizaciones, X es L (Leu), A (Ala), G (Gly), S (Ser), V (Val), F (Phe), Y (Tyr), H (His), R (Arg), N (Asn), E (Glu), D (Asp), C (Cys), Q (Gln), 1 (Ile), M (Met), P (Pro), T (Thr), K (Lys) o W (Trp).
En algunas realizaciones, el marcador que contiene glutamina comprende una secuencia de aminoácidos seleccionada del grupo que consiste en Q, Lq G, LLQGG (SEQ ID NO:1), LLQG (SEQ ID NO:2), LSLSQG (SEQ ID NO:3), GGGLLQGG (SEQ ID NO:4), GLLQG (SEQ ID NO:5), LLQ, GSPLAQSHGG (SEQ ID NO:6), GLLQGGG (SEQ ID NO:7), GLLQGG (SEQ ID NO:8), GLLQ (SEQ ID NO:9), LLQLLQGA (SEQ ID NO:10), LLQGA (SEQ ID NO:11), LLQYQGA (SEQ ID NO:12), LLQGSG (SEQ ID NO:13), LLQYQG (SEQ ID NO:14), LLQLLQG (SEQ ID NO:15), SLLQG (SEQ ID NO:16), LLQLQ (SEQ ID NO:17), LLQLLQ (SEQ ID NO:18), LLQGR (SEQ ID NO:19), LLQGPP (SEQ ID NO:20), LLQGPA (SEQ ID NO:21), GGLLQGPP (SEQ ID NO:22), GGLLQGA (SEQ ID NO:23), LLQGA (SEQ ID NO:24), LLQGPGK (SEQ ID NO:25), LLQGPG (SEQ ID NO:26), LLQGP (SEQ ID NO:27), LLQP (SEQ ID NO:28), LLQPGK (SEQ ID NO:29), LLQAPGK (SEQ ID NO:30), LLQGAPG (SEQ ID NO:31), LLQGAP (SEQ ID NO:32), LLQGPA (SEQ ID NO:33), LLQGPP (SEQ ID NO:34), GGLLQGPP (SEQ ID NO:35) y LLQLQG (SEQ ID NO:36).
En algunas realizaciones, el marcador que contiene glutamina comprende una secuencia de aminoácidos LLQGA (SEQ ID NO: 24), LQG, GGLLQGA (SEQ ID NO:23), LLQGPA (SEQ ID NO:33), LLQGPP (SEQ ID NO:34),
GGLLQGPP (SEQ ID NO:35), LLQGSG (SEQ ID NO:13), LLQG (SEQ ID NO:2), LLQYQG (SEQ ID NO:14), LLQLLQG (SEQ ID NO:15), LLQLQG (SEQ ID NO:36), LLQLLQ (SEQ ID NO:18), LLQLQ (SEQ ID NO:17), LLQGR (SEQ ID NO:19), LLQYQGA (SEQ ID NO:12), SLLQG (SEQ ID NO:16) o LLQLLQGA (SEQ ID NO:10).
En algunas realizaciones, el marcador que contiene glutamina no comprende una secuencia de aminoácidos seleccionada del grupo que consiste en LGGQGGG (SEQ ID NO:41), GGGQGGL (SEQ ID NO: 42), GXGQGGG (SEQ ID NO: 43), GGXQGGG (SEQ ID NO: 44), GGGQXGG (SEQ ID NO: 45) y GGGQGXG (SEQ ID NO: 46), en la que X es G, A, S, L, V, F, Y, R, N o E). También se describen otros marcadores de ejemplo, por ejemplo, en los documentos US2013/0230543 y US2013/0122020.
En algunas realizaciones, el anticuerpo de los CAF como se describe en el presente documento comprende una modificación de aminoácidos en la última posición de aminoácidos en el extremo carboxilo en relación con un anticuerpo de tipo salvaje en la misma posición. En algunas realizaciones, la modificación es una deleción de aminoácidos, inserción, sustitución, mutación, o cualquier combinación de las mismas. En algunas realizaciones, la sustitución comprende reemplazar un aminoácido de tipo salvaje con otro (por ejemplo, un aminoácido de tipo no salvaje). En algunas realizaciones, la inserción comprende insertar uno o más aminoácidos (por ejemplo, inserción de uno, dos, tres o más aminoácidos). En algunas realizaciones, el otro (por ejemplo, de tipo no salvaje) o el aminoácido insertado es Arg. En algunas realizaciones, el otro aminoácido (por ejemplo, de tipo no salvaje) es Ala, Asn, Asp, Cys, Glu, Gln, Gly, His, Ile, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr o Val. Por ejemplo, en algunas realizaciones, el último aminoácido en el extremo carboxilo del anticuerpo (por ejemplo, la cadena pesada de un anticuerpo) puede delecionarse y el marcador que contiene glutamina diseñada para el extremo C del polipéptido comprende la secuencia de aminoácidos LLQGA (SEQ ID NO: 11) o GGLLQGA (SEQ ID NO: 23).
En algunas realizaciones, el anticuerpo comprende una modificación de aminoácidos en la primera posición de aminoácidos en el término amino en relación con un anticuerpo de tipo salvaje en la misma posición. En algunas realizaciones, la modificación es una deleción de aminoácidos, inserción, sustitución, mutación, o cualquier combinación de las mismas. En algunas realizaciones, la sustitución comprende reemplazar un aminoácido de tipo salvaje con otro aminoácido (por ejemplo, de tipo no salvaje). En algunas realizaciones, la inserción comprende insertar un aminoácido. En algunas realizaciones, el aminoácido de tipo no salvaje o insertado es Arg. En algunas realizaciones, el otro aminoácido (de tipo no salvaje o insertado) es Ala, Asn, Asp, Cys, Glu, Gln, Gly, His, Ile, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr o Val.
En algunas realizaciones, el CAF descrito en el presente documento comprende una cadena pesada de anticuerpo de longitud completa y una cadena ligera de anticuerpo. En algunas realizaciones, el anticuerpo descrito en el presente documento es un anticuerpo monoclonal, un anticuerpo policlonal, un anticuerpo humano, un anticuerpo humanizado, un anticuerpo quimérico, un anticuerpo biespecífico, un minicuerpo, un diacuerpo o un fragmento de anticuerpo. En algunas realizaciones, el anticuerpo es una IgG. En algunas realizaciones, la IgG se selecciona del grupo que consiste en IgG1, IgG2, IgG3 y IgG4. En algunas realizaciones, el anticuerpo es una IgA, IgE, IgD o IgM. En algunas realizaciones, la función efectora (por ejemplo, medida por la unión de Fcy3 y/o C1q) de los CAF descritos en el presente documento no disminuye más de aproximadamente 1 vez, 2 veces, 3 veces, 4 veces o 5 veces en relación con un anticuerpo de tipo salvaje. En algunas realizaciones, el anticuerpo del CAF es una IgG, en el que la función efectora de la IgG disminuye no más de aproximadamente 2 veces con respecto a una IgG de tipo salvaje. En otras realizaciones, la función efectora de la IgG disminuye aproximadamente 2 veces con respecto a una IgG de tipo salvaje. En otras realizaciones, la función efectora de la IgG disminuye más de aproximadamente 2 veces con respecto a una IgG de tipo salvaje. En algunas realizaciones, el anticuerpo del CAF es una IgG, en el que la función efectora de la IgG disminuye no más de aproximadamente 1 veces con respecto a una IgG de tipo salvaje. En otras realizaciones, la función efectora de la IgG disminuye aproximadamente 1 veces con respecto a una IgG de tipo salvaje. En algunas realizaciones, la función efectora de la IgG disminuye más de aproximadamente cualquiera de 1 vez, 3 veces, 4 veces o 5 veces en relación con una IgG de tipo salvaje.
El número de agentes de restos que contienen amina que pueden conjugarse con el anticuerpo depende de 1) el número de marcadores que contienen glutamina que están unidos/insertados al/en el anticuerpo, así como el número de glutaminas en el marcador que contiene glutamina; y/o 2) el número de glutaminas endógenas en el anticuerpo (es decir, glutaminas nativas sin modificar, tales como las glutaminas en los dominios variables, CDR, etc.) y/o 3) el número de glutaminas endógenas que se hacen reactivas mediante ingeniería de anticuerpos como se describe en el presente documento o una transglutaminasa modificada por ingeniería genética. Por ejemplo, dos restos que contienen amina pueden estar conjugados específicamente en el sitio con un anticuerpo en el extremo carboxilo de las dos cadenas ligeras, y cuatro restos que contienen amina pueden estar conjugados específicamente en el sitio con el anticuerpo en las posiciones Q295 y N297Q. En algunas realizaciones, el resto que contiene amina puede ser igual o diferente en cada posición de conjugación.
Los ejemplos de un agente citotóxico incluyen, pero sin limitación, una antraciclina, una auristatina (por ejemplo, dolestatina, MMAD, MMAE, MMAF, PF-06380101, PF-06463377 y PF-06456780), un espliceostatina, un dímero CBI/CPI (que incluye dímeros "mixtos" que comprenden componentes CBI y CPI, como se describe en la solicitud de patente provisional de Estados Unidos 61/932,118), una caliqueamicina, una duocarmicina, una enediina, una geldanamicina, una maytansina, una puromicina, un taxano, un alcaloide de la vinca, SN-38, una tubulisina, una hemiasterlina, una camptotecina, una combretastatina, una dolastatina, un dímero de indolino-benzodiazepina, un
dímero de pirrolobenzodiazepina y una pladienolida, y estereoisómeros, isoésteres, análogos o derivados de los mismos.
Las antraciclinas derivan de la bacteria Streptomyces y se han utilizado para tratar una amplia gama de cánceres, tales como leucemias, linternas, mama, uterino, cánceres de ovario y de pulmón. Las antraciclinas de ejemplo incluyen, pero sin limitación, daunorubicina, doxorrubicina (es decir, adriamicina), epirrubicina, idarrubicina, valrubicina y mitoxantrona.
Las dolastatinas y sus análogos peptídicos y derivados, auristatinas, son agentes antimitóticos altamente potentes que han demostrado tener actividad anticancerosa y antifúngica. Véase, por ejemplo, la patente de Estados Unidos n.°5,663,149 y Pettit y col., Antimicrob. Agents Chemother. 42:2961-2965, 1998. Los ejemplos de dolastatinas y auristatinas incluyen, pero sin limitación, dolastatina 10, auristatina E, auristatina EB (AEB), auristatina EFP (AEFP), MMAD (monometil auristatina D o monometil dolastatina 10), MMAF (monometil auristatina F o N-metilvalina-valinadolaisoleuina-dolaproina-fenilalanina), MMAE (monometil auristatina E o N-metilvalina-valina-dolaisoleuinadolaproina-norefedrina), éster de ácido 5-benzoilvalérico-AE (AEVB) y otras auristatinas novedosas (como las descritas en la publicación de EE. UU. n.° 2013/0129753). En algunas realizaciones, la auristatina es 0101 (2-metilalanil-W-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo -3-{[(1 S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-5-metil-1 -oxoheptan-4-il]-W-metil-L-valinamida) que tiene la estructura siguientes:

En algunas realizaciones, la auristatina es 3377 (N, 2-dimetilalanil-N-{(1S, 2R)-4-{(2S)-2-[(1R, 2R)-3-{[(1S)-1-carboxil-2- feniletil]amino}-1-metoxi-2-metil-3-oxopropil]pirrolidin-1-il}-2-metoxi-1-[(1S)-1-metilpropil]-4-oxobutil}-N-metil-L-valinamida) que tiene la siguiente estructura:

En otras realizaciones, la auristatina es 0131 (2-metil-L-prolil-N-[(3R, 4S, 5S) -1-{(2S)-2-[(1R, 2R)-3-{[(1S)-1-carboxi-2-feniletil]amino}-1-metoxi-2-metil-3-oxopropil]pirrolidin-1-il}-3-metoxi-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida) que tiene la siguiente estructura:

En otras realizaciones, la auristatina es 0121 (2-metil-L-prolil-N-[(3R, 4S, 5S) -1-{(2S)-2-[(1R, 2R)-3-{[(2S)-1-metoxi-1-oxo-3-fenilpropan-2-il]amino}-1-metoxi-2-metil-3-oxopropil]pirrolidin-1-il}-3-metoxi-5-metil-1-oxoheptán-4-il]-N-metil-L-valinamida) que tiene la siguiente estructura:
La camptotecina es un alcaloide de quinolina citotóxico que inhibe la enzima topoisomerasa I. Los ejemplos de camptotecina y sus derivados incluyen, pero sin limitación, topotecán e irinotecán, y sus metabolitos, tal como SN-38.
Las combretastatinas son fenoles naturales con propiedades de alteración vascular en los tumores. Los ejemplos de combretastatinas y sus derivados incluyen, pero sin limitación, combretastatina A-4 (CA-4) y ombrabulina.
Duocarmicina y CC-1065 son agentes alquilantes de ADN con potencia citotóxica. Véase Boger y Johnson, PNAS 92:3642-3649 (1995). Los ejemplos de dolastatinas y auristatinas incluyen, pero sin limitación, (+) - docarmicina A y (+) - duocarmicina SA, y (+) - c C - 1 0 6 5 .
Las enediinas son una clase de productos bacterianos antitumorales caracterizados por anillos de nueve y diez miembros o por la presencia de un sistema cíclico de enlaces triples, dobles, triples conjugados. Los enediinas de ejemplo incluyen, pero sin limitación, calicamicina, esperamicina, uncialamicina, dinemicina y sus derivados.
La hemiasterlina y sus análogos (por ejemplo, HTI-286) se unen a la tubulina, interrumpen la dinámica normal de los microtúbulos y, en cantidades estequiométricas, despolimerizan los microtúbulos.
Las geldanamicinas son antibióticos de benzoquinona ansamicina que se unen a Hsp90 (proteína del shock térmico 90) y se han utilizado medicamentos antitumorales. Los ejemplos de geldanamicinas incluyen, pero sin limitación, 17-AAG (17-N-alilamino-17-demetoxigeldanamicina) y 17-DMAG (17-Dimetilaminoetilamino-17-demetoxigeldanamicina).
Las maytansinas o sus derivados maytansinoides inhiben la proliferación celular al inhibir la formación de microtúbulos durante la mitosis a través de la inhibición de la polimerización de la tubulina. Véase Remillard y col., Science 189:1002-1005 (1975). Los ejemplos de maytansinas y maytansinoides incluyen, pero sin limitación, mertansina (DM1) y sus derivados, así como ansamitocina.
Los dímeros de pirrolobenzodiazepina (PBD) y los dímeros de indolinobenzodiazepina (IGN) son agentes antitumorales que contienen uno o más grupos funcionales inminentes, o sus equivalentes, que se unen al ADN dúplex. Las moléculas PBD e IGN se basan en el producto natural de la atramicina e interactúan con el ADN de una manera selectiva de secuencia, con preferencia por las secuencias purina-guanina-purina. Los PBD de ejemplo y sus análogos incluyen, pero sin limitación, SJG-136.
Las espliceostatinas y pladienolidas son compuestos antitumorales que inhiben el empalme e interaccionan con el espliceosoma, SF3b. Los ejemplos de espliceostatinas incluyen, pero sin limitación, espliceostatina A, FR901464. Los ejemplos de pladienolidas incluyen, pero sin limitación, pladienolida B, pladienolida D y E7107.
Los taxanos son diterpenos que actúan como agentes anti-tubulina o inhibidores mitóticos. Los taxanos de ejemplo incluyen, pero sin limitación, paclitaxel (por ejemplo, TAXOL®) y docetaxel (TAXOTERE®).
Los alcaloides de vinca también son agentes anti-tubulina. Los ejemplos de alcaloides de la vinca incluyen, pero sin limitación, vincristina, vinblastina, vindesina y vinorelbina.
En algunas realizaciones, el resto agente es un polipéptido de toxina (o una proteína de toxina). Los ejemplos de un polipéptido de toxina incluyen, pero sin limitación, cadena A de la difteria, los fragmentos activos no ligantes de la toxina de difteria, cadena de exotoxina A, la cadena A de ricina, la cadena A de abrina, la cadena A de modeccina, sarcina alfa, las proteínas de Aleurites fordii, las proteínas de diantina, las proteínas de Phytolaca americana (PAPI, PAPII y PAP-S), el inhibidor de Momordica charantia, curcina, crotina, el inhibidor de Sapaonaria officinalis, gelonina, mitogelina, restrictocina, fenomicina, enomicina, tricotecenos, péptidos inhibidores del nudo de cistina (ICK) (por ejemplo, ceratotoxinas) y conotoxina (por ejemplo, KIIIA o SmIIIa).
Procedimientos para conjugar CAF
La conjugación de anticuerpos anti-Trop2 con las cargas útiles de enlazador de la presente invención se logra cuando una cantidad de anticuerpo se ajusta a 5 mg/ml en tampón que contiene Tris-HCI 25 mM, pH 8,0-8,5. Se añade una cantidad de carga útil del enlazador en un exceso molar de 5-50 veces o 5-500 veces, sobre el anticuerpo y la reacción enzimática iniciada mediante la adición de transglutaminasa bacteriana al 2 % (p/v) (Ajinomoto Activa TI, Japón), y se incubó con agitación suave a 37 °C durante 16-24 horas. El CAF se purifica utilizando Butyl Sepharose™ High Performance (Butyl HP, GE Healthcare Biosciences) ajustando la mezcla de reacción para obtener una composición tampón de sulfato de amonio 0,75 M, fosfato potásico 25 mM, pH 7 (tampón A). El material se aplica a un buti1HP, se lava con tampón A de 5 CV y se eluye con un gradiente lineal de 20 CV en fosfato de potasio 25 mM, pH 7. Las fracciones que contienen el CAF se combinan, se dializan contra PBS, se concentran usando una unidad de filtro centrífugo Amicon Ultra de 10 kDa (Millipore Corporation), y se esterilizan por filtración a través de un filtro de 0,2 pm. Como alternativa, el CAF se purifica utilizando resina de proteína A MabSelect (GE Healthcare Biosciences) siguiendo protocolos estándar, y el tampón se intercambia en PBS. Se conocen otros procedimientos para conjugar CAF, incluidos los procedimientos descritos en el documento WO2012/059882.
Procedimiento de uso de conjugados anticuerpo-fármaco de la invención
Los CAF de la presente invención son útiles en diversas aplicaciones, que incluyen, pero sin limitación, procedimientos
de tratamiento terapéutico y procedimientos de tratamiento de diagnóstico.
En un aspecto, la invención proporciona compuestos de la invención para su uso en el tratamiento de un cáncer en un sujeto. Por consiguiente, algunas realizaciones, comprende administrar al sujeto una cantidad efectiva de una composición (por ejemplo, composición farmacéutica) que comprende el CAF como se describe en el presente documento. Tal como se usa en el presente documento, los cánceres incluyen, pero sin limitación, un cáncer sólido (tal como, cáncer de vejiga, mama, cervical, coriocarcinoma, colon, esofágico, gástrico, glioblastoma, cabeza y cuello, riñón, hígado, de pulmón (por ejemplo, cáncer de pulmón no microcítico (CPNM)), oral, de ovarios, pancreático, de próstata y de piel); y un cáncer líquido (tal como leucemia linfoblástica aguda (LLA), leucemia linfocítica crónica (LLC), leucemia mielógena aguda (LMA), leucemia mielógena crónica (LMC), leucemia de células pilosas (LCP), leucemia prolinfocítica de linfocitos T (T-PLL), leucemia linfocítica granular grande, leucemia de linfocitos T en adultos y mieloma múltiple).
Se desvela un procedimiento para inhibir el crecimiento o la progresión tumoral en un sujeto que lo necesita, que comprende administrar al sujeto una cantidad eficaz de una composición que comprende los CAF como se describe en el presente documento. Se desvela un procedimiento para inhibir la metástasis de células cancerosas o tumores (por ejemplo, tumores sólidos o líquidos) en un sujeto que lo necesita, que comprende administrar al sujeto una cantidad eficaz de una composición que comprende los c A f como se describe en el presente documento. Se desvela un procedimiento para inducir la regresión tumoral en un sujeto que lo necesita, que comprende administrar al sujeto una cantidad eficaz de una composición que comprende los CAF como se describe en el presente documento.
También se desvela que el resto de agente en los CAF como se describe en el presente documento puede ser un resto detectable, tal como un agente de obtención de imágenes y un marcador de sustrato enzimático. Los CAF como se describe en el presente documento también se pueden usar para ensayos de diagnóstico in vivo, tales como obtención de imágenes in vivo (por ejemplo, PET o SPECT), o un reactivo de tinción.
Algunas realizaciones descritas en el presente documento comprenden además una etapa de tratar a un sujeto con una forma adicional de terapia. En algunas realizaciones, la forma adicional de terapia es una terapia anticancerosa adicional que incluye, pero sin limitación, quimioterapia, radiación, cirugía, terapia hormonal y/o inmunoterapia adicional.
En algunas realizaciones, la forma adicional de terapia comprende administrar uno o más agentes terapéuticos además de los CAF como se describe en el presente documento. Los agentes terapéuticos incluyen, pero sin limitación, un segundo CAF (por ejemplo, CAF convencional, tal como brentuximab vedotin (CAFETRIS®) y adotrastuzumab emtansina (KCAFYLA®)), un anticuerpo (por ejemplo, un anticuerpo anti-VEGF, un anticuerpo anti-HER2, anticuerpo anti-CD25 y/o un anticuerpo anti-CD20), un inhibidor de la angiogénesis, un agente citotóxico (por ejemplo, docetaxel, cisplatino, doxorrubicina, mitomicina, tamoxifeno o fluorouracilo) y un agente antiinflamatorio (por ejemplo, prednisona y progesterona).
Composiciones farmacéuticas
La presente invención también proporciona una composición farmacéutica que comprende los CAF como se describe en el presente documento en un excipiente o vehículo farmacéuticamente aceptable. Los CFA se pueden administrar solos o en combinación con uno o más CFA distintos de la invención o en combinación con uno o más fármacos distintos (o como cualquier combinación de los mismos). Por ejemplo, los CAF de la presente invención pueden administrarse en combinación con los CAF convencionales (es decir, no diseñados con características de modulación de estabilidad). Los procedimientos y usos de la invención, por lo tanto, también abarcan realizaciones de combinaciones (coadministración) con otros agentes activos, como se detalla a continuación.
Tal como se usa en el presente documento, se pretende que el término "coadministración" "administrado conjuntamente" o "en combinación con" signifique y haga referencia a lo siguiente: (i) administración simultánea de una combinación de un CAF desvelado en el presente documento y uno o más agentes terapéuticos a un paciente que necesita tratamiento, cuando dichos componentes se formulan juntos en una única forma de dosificación que libera dichos componentes sustancialmente al mismo tiempo a dicho paciente; (ii) administración sustancialmente simultánea de tal combinación de un CAF desvelado en el presente documento y uno o más agentes terapéuticos a un paciente que necesita tratamiento, cuando dichos componentes se formulan separados entre sí en formas de dosificación separadas que dicho paciente toma sustancialmente al mismo tiempo, con lo cual dichos componentes se liberan sustancialmente al mismo tiempo a dicho paciente; (iii) administración secuencial de tal combinación de un CAF desvelado en el presente documento y uno o más agentes terapéuticos a un paciente que necesita tratamiento, cuando dichos componentes se formulan separados entre sí en formas de dosificación separadas que dicho paciente toma en momentos consecutivos con un intervalo de tiempo significativo entre cada administración, con lo cual dichos componentes se liberan en tiempos sustancialmente diferentes a dicho paciente; y (iv) administración secuencial e tal combinación de un CAF desvelado en el presente documento y uno o más agentes terapéuticos a un paciente que necesita tratamiento, cuando dichos componentes se formulan juntos en una sola forma de dosificación que libera dichos componentes de manera controlada, con lo cual se liberan en dicho paciente de forma concurrente, consecutiva y/o solapada al mismo tiempo y/o a tiempos diferentes, en el que cada parte puede administrarse por la misma ruta o por una diferente.
Generalmente, los CAF desvelados en el presente documento son adecuados para administrarse como una formulación en asociación con uno o más excipientes farmacéuticamente aceptables. El término "excipiente" se usa en el presente documento para describir un ingrediente distinto del compuesto o compuestos de la invención. La elección del excipiente o excipientes dependerá en gran medida de factores tales como el modo particular de administración, el efecto del excipiente en la solubilidad y la estabilidad y la naturaleza de la forma de dosificación. Tal como se usa en el presente documento, "excipiente farmacéuticamente aceptable" incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y agentes retardantes de la absorción, y similares, que son fisiológicamente compatibles. Algunos ejemplos de excipientes farmacéuticamente aceptables son agua, solución salina, solución salina tamponada con fosfato, dextrosa, glicerol, etanol y similares, así como combinaciones de las mismas. En algunas realizaciones, agentes isotónicos, incluyendo, pero sin limitación, azúcares, polialcoholes (por ejemplo, manitol, sorbitol) o cloruro de sodio están incluidos en la composición farmacéutica. Los ejemplos adicionales de vehículos farmacéuticamente aceptables incluyen, pero sin limitación, agentes humectantes o cantidades menores de sustancias auxiliares, tales como agentes humectantes o emulsionantes, conservantes o tampones, que mejoran la vida útil o la efectividad del anticuerpo.
En algunas realizaciones, los CAF descritos en el presente documento pueden desinmunizarse para reducir la inmunogenicidad tras la administración a un sujeto que demanda técnicas conocidas como las descritas, por ejemplo, en la publicación PCT WO98/52976 y el documento WO00/34317.
Las composiciones farmacéuticas de la presente invención y los procedimientos para su preparación son fácilmente evidentes para los expertos en la materia. Dichas composiciones y procedimientos para su preparación pueden encontrarse, por ejemplo, en Remington's Pharmaceutical Sciences, 22a Edición (Mack Publishing Company, 2012). Las composiciones farmacéuticas se fabrican, preferentemente en condiciones de GMP.
Una composición farmacéutica de la invención se puede preparar, envasar o vender a granel, como dosis unitaria sencilla o como una pluralidad de dosis unitarias sencillas. Tal como se usa en el presente documento, una "dosis unitaria" es una cantidad pequeña de la composición farmacéutica que comprende una cantidad predeterminada del principio activo. La cantidad del principio activo es generalmente igual a la dosis del principio activo que se administraría a un sujeto o una fracción conveniente de una dosis tal como, por ejemplo, la mitad o un tercio de dicha dosis. Cualquier procedimiento para administrar péptidos, proteínas o anticuerpos aceptados en la técnica pueden emplearse adecuadamente para los conjugados de polipéptidos diseñados desvelados en el presente documento.
Las composiciones farmacéuticas de la invención son típicamente adecuadas para administración parenteral. La administración parenteral de una composición farmacéutica incluye cualquier vía de administración caracterizada por la rotura física de un tejido de un sujeto y la administración de la composición farmacéutica a través de la rotura en el tejido, lo que generalmente da como resultado la administración directa en el torrente sanguíneo, en el músculo o en un órgano interno. Por ejemplo, la administración parenteral incluye, pero sin limitación, la administración de una composición farmacéutica por inyección de la composición, por aplicación de la composición a través de una incisión quirúrgica, por aplicación de la composición a través de una herida no quirúrgica que penetra en el tejido, y similares. En particular, se contempla que la administración parenteral incluya, pero sin limitación, subcutánea, intraperitoneal, intramuscular, intraesternal, intravenosa, intraarterial, intratecal, intraventricular, intrauretral, intracraneal, inyección intramuscular o infusiones; y técnicas de infusión dialítica renal. En algunas realizaciones, la administración parenteral es la vía intravenosa o subcutánea.
Las formulaciones de una composición farmacéutica adecuada para administración parenteral generalmente comprenden el principio activo combinado con un vehículo farmacéuticamente aceptable, tal como agua estéril o solución salina isotónica estéril. Dichas formulaciones pueden prepararse, envasarse o venderse en una forma adecuada para administración en bolo o para administración continua. Se pueden preparar formulaciones inyectables, envasar o vender en forma de dosificación unitaria, tal como en ampollas o en envases multidosis que contienen un conservante. Las formulaciones para administración parenteral incluyen, pero sin limitación, suspensiones, soluciones, emulsiones en vehículos aceitosos o acuosos, pastas y similares. Dichas formulaciones pueden comprender además uno o más ingredientes adicionales que incluyen, pero sin limitación, agentes de suspensión, estabilizantes o dispersantes. En una realización de una formulación para administración parenteral, el principio activo se proporciona en forma seca (es decir, en polvo o granular) para su reconstitución con un vehículo adecuado (por ejemplo, agua libre de pirógenos estéril) antes de la administración parenteral de la composición reconstituida. Las formulaciones parenterales también incluyen soluciones acuosas que pueden contener excipientes, tales como sales, carbohidratos y agentes tamponantes (preferentemente a un pH de 3 a 9) pero, para algunas aplicaciones, se pueden formular de una manera más adecuada en forma de una solución no acuosa estéril o como una forma desecada que se va a usar junto con un vehículo adecuado tal como agua apirógena estéril. Las formas de administración parenteral de ejemplo incluyen soluciones o suspensiones en soluciones acuosas estériles, por ejemplo, soluciones acuosas de propilenglicol o dextrosa. Dichas formas de dosificación se pueden tamponar adecuadamente, si se desea. Otras formulaciones administrables por vía parenteral que son útiles incluyen las que comprenden el principio activo en forma microcristalina, o en una preparación liposomal. Las formulaciones para administración parenteral pueden formularse para que sean de liberación inmediata y/o modificada. Las formulaciones de liberación modificada incluyen formulaciones de liberación controlada, retardada, sostenida, pulsada, dirigida y programada. Por ejemplo, en un aspecto, las soluciones inyectables estériles se pueden preparar incorporando el polipéptido diseñado que contiene Fc, por ejemplo, conjugado anticuerpo-fármaco o anticuerpo biespecífico, en la cantidad requerida en un disolvente
apropiado con uno o una combinación de ingredientes enumerados anteriormente, según se requiera, seguido de esterilización por filtración. Generalmente, las dispersiones se preparan mediante la incorporación del compuesto activo en un vehículo estéril que contiene un medio de dispersión básico y los otros ingredientes requeridos de los enumerados anteriormente. En el caso de polvos estériles para la preparación de soluciones inyectables estériles, los procedimientos de preparación preferidos son secado al vacío y liofilización, que proporcionan un polvo del principio activo más cualquier ingrediente adicional deseado a partir de una solución de los mismos previamente esterilizada por filtración. Se puede mantener la fluidez adecuada de una solución, por ejemplo, mediante la utilización de un recubrimiento como la lecitina, manteniendo el tamaño de partícula necesario en el caso de la dispersión y/o mediante el uso de tensioactivos. Puede lograrse la absorción prolongada de las composiciones inyectables, incluyendo en la composición un agente que retrase la absorción, por ejemplo, sales de monoestearato y gelatina.
Los regímenes de dosificación se pueden ajustar para proporcionar la respuesta deseada óptima. Por ejemplo, se puede administrar un único bolo, se pueden administrar varias dosis divididas a lo largo del tiempo o se puede reducir o aumentar proporcionalmente la dosis según se indique por las exigencias de la situación terapéutica. Es especialmente ventajoso formular las composiciones parenterales en forma de dosis unitaria para facilitar la administración y la uniformidad de la dosis. La forma de unidad de dosificación, tal como se usa en el presente documento, se refiere a unidades físicamente discretas adecuadas como dosis unitarias para los pacientes/sujetos a tratar; conteniendo cada unidad una cantidad predeterminada de compuesto activo calculada para producir el efecto terapéutico deseado en asociación con el vehículo farmacéutico necesario. La especificación para las formas de unidad de dosificación de la invención está dictada generalmente y depende directamente de (a) las características únicas del resto de agente (por ejemplo, moléculas pequeñas tal como un agente citotóxico) y el efecto terapéutico o profiláctico concreto que se debe conseguir y (b) de las limitaciones inherentes en la técnica de formar tal compuesto activo para el tratamiento de la sensibilidad en individuos.
Por tanto, el técnico experto apreciará, conforme la divulgación proporcionada en el presente documento, que la dosis y el régimen de dosificación se ajustan de acuerdo con procedimientos bien conocidos en las técnicas terapéuticas. Esto es, la dosis máxima tolerable se puede establecer fácilmente y también se puede determinar la cantidad efectiva que proporciona un beneficio terapéutico detectable para un paciente, al igual que los requisitos temporales para administrar cada agente para proporcionar un beneficio terapéutico detectable para el paciente. Por consiguiente, aunque en el presente documento se ilustran ciertos regímenes de dosis y administración, estos ejemplos no limitan de manera alguna la dosis y el régimen de administración que se puede proporcionar a un paciente en la práctica de la presente invención.
Cabe señalar que los valores de dosificación pueden variar con el tipo y la gravedad de la afección a aliviar y pueden incluir dosis únicas o múltiples. También debe entenderse que para cualquier sujeto concreto, los regímenes de dosificación específicos deben ajustarse en el tiempo de acuerdo con la necesidad individual y el juicio profesional de la persona que administra o supervisa la administración de las composiciones, y los intervalos de dosificación indicados en el presente documento son únicamente ejemplos y no están destinados a limitar el ámbito o la práctica de la composición reivindicada. Además, el régimen de dosificación con las composiciones de la presente invención puede basarse en diversos factores, incluyendo el tipo de enfermedad, la edad, el peso, el sexo, la afección médica del paciente, la gravedad de la afección, la vía de administración y el anticuerpo en particular empleado. Por tanto, el régimen de dosificación puede variar ampliamente, pero puede determinarse rutinariamente usando procedimientos estándar. Por ejemplo, las dosis pueden ajustarse según los parámetros farmacocinéticos o farmacodinámicos, que pueden incluir efectos clínicos, tales como efectos tóxicos y/o valores de laboratorio. Por tanto, la presente invención abarca el aumento de la dosis dentro del mismo paciente según lo determine el experto en la materia. La determinación de las dosis y los regímenes apropiados se conoce bien en la técnica relevante y se entenderá que están abarcados por el técnico experto una vez que se hayan proporcionado las enseñanzas desveladas en el presente documento.
Para la administración a sujetos humanos, la dosis mensual total de un CAF desvelado en el presente documento está típicamente en el intervalo de aproximadamente 0,01 mg a aproximadamente 1.200 mg por paciente, dependiendo, por supuesto, del modo de administración. Por ejemplo, una dosis mensual intravenosa puede requerir de aproximadamente 1 a aproximadamente 1.000 mg/paciente. La dosis mensual total puede administrarse en dosis únicas o divididas y puede, según el criterio del médico, desviarse respecto del intervalo típico proporcionado en el presente documento.
Un intervalo no limitante de ejemplo para una cantidad terapéutica o profilácticamente efectiva de un CAF como se desvela en el presente documento es de aproximadamente 0,01 a aproximadamente 1.000 mg/paciente/mes. En determinadas realizaciones, el CAF puede administrarse a aproximadamente 1 a aproximadamente 200 o aproximadamente 1 a aproximadamente 150 mg/paciente/mes. En algunas realizaciones, el paciente es un ser humano.
Kits
También se desvelan kits (o artículos de fabricación) para su uso en el tratamiento de los trastornos descritos anteriormente. Los kits incluyen uno o más recipientes que comprenden un CAF purificado e instrucciones para usar el conjugado para tratar una enfermedad. Por ejemplo, las instrucciones comprenden una descripción de la administración de CAF para tratar una enfermedad, tal como cáncer (por ejemplo, un cáncer sólido o líquido). El kit
puede comprender además una descripción de seleccionar un individuo adecuado para el tratamiento basado en identificar si ese individuo tiene la enfermedad y el estadio de la enfermedad.
Las instrucciones relacionadas con el uso del CFA generalmente incluyen información sobre la dosificación, la pauta posológica y la vía de administración para el tratamiento previsto. Los envases pueden ser dosis unitarias, envases grandes (por ejemplo, envases de dosis múltiples) o dosis de subunidades. Las instrucciones suministradas en los kits de la invención suelen ser instrucciones escritas en una etiqueta o en un prospecto (por ejemplo, una hoja de papel incluida en el kit), pero también son aceptables las instrucciones legibles por máquina (por ejemplo, las instrucciones que se llevan en un disco de almacenamiento magnético u óptico).
Los kits están en un envase adecuado. Los envases adecuados incluyen, pero sin limitación, viales, frascos, jarros, envases flexibles (por ejemplo, Mylar sellado o bolsas de plástico), y similares. También se contemplan los envases para su uso en combinación con un dispositivo específico, tal como un inhalador, dispositivo de administración nasal (por ejemplo, un atomizador) o un dispositivo de infusión como una minibomba. Un kit puede tener un puerto de acceso estéril (por ejemplo, el recipiente puede ser una bolsa de solución intravenosa o un frasco que tiene un tapón perforable por una aguja de inyección hipodérmica). El recipiente también puede tener un puerto de acceso estéril (por ejemplo, el recipiente puede ser una bolsa de solución intravenosa o un frasco que tiene un tapón perforable por una aguja de inyección hipodérmica). Al menos un agente activo en la composición es el CAF cargado más alto como se describe en el presente documento. El recipiente puede comprender además un segundo agente farmacéuticamente activo.
Los kits pueden proporcionar opcionalmente componentes adicionales tales como tampones e información interpretativa. Normalmente, el kit comprende un recipiente y una etiqueta o prospecto sobre el recipiente o asociado con el mismo.
Ejemplos
Se entiende que los ejemplos y realizaciones descritas en el presente documento son solo para fines ilustrativos.
Condiciones de HPLC y CL-EM usadas para los análisis:
Condiciones analíticas de HPLC: Phenomenex Luna C18 (2), 150 x 3,0 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 0% al 100% de B durante 8,5 minutos, después 100% de B durante 1,5 minutos; Caudal: 1,5 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: 10 pl; Instrumento: Agilent 1100 HPLC. Condiciones analíticas de CL-EM: Waters Acquity UPLC HSS T3, C18, 2,1 x 50 mm, 1,7 pm; Fase móvil A: ácido fórmico al 0,1% en agua (v/v); Fase móvil B: ácido fórmico al 0,1% en acetonitrilo (v/v); Gradiente: 5% de B durante 0,1 minutos, del 5 % al 95 % de B durante 2,5 minutos, 95% de B durante 0,35 minuto; Caudal: 1,25 ml/minuto. Temperatura: 60 °C; Detección: 200-450 nm; intervalo de EM (+) de 100-2000 daltons; Volumen de inyección: 5 pl; Instrumento: Waters Acquity.
Condiciones de HPLC preparativa usadas para las purificaciones:
Procedimiento A: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 5% al 50% de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento B: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 20 % al 80 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento C: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 5% al 70 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento D: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 10 % al 70 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento E: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 30 % al 85 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento F: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al
0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 10 % al 90 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento G: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 10 % al 95 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento H: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 25 % al 65 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento I: Phenomenex Synergi Max C18, 250 x 50 mm, columna de 10 um; Fase móvil A: ácido fórmico al 0,2 % en agua (v/v); Fase móvil B: acetonitrilo; Gradiente: del 33,0% al 58% de B en 25 minutos, aumentando después hasta 100% de B en 2 minutos, mantenido durante 5 minutos; Caudal: 80 ml/minuto. Temperatura: no controlada; Detección: DAD 220 nm.
Procedimiento J: DIKMA Diamonsil(2) C18, 200 x 20 mm, columna de 5 um; Fase móvil A: ácido fórmico al 0,225 % en agua (v/v); Fase móvil B: acetonitrilo; Gradiente: del 53 % al 73 % de B durante 11 minutos, aumentando después hasta 100% de B en 0,5 minutos, mantenido durante 2 minutos; Caudal: 35 ml/minuto. Temperatura: no controlada; Detección: DAD 220 nm.
Procedimiento K: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 20 % al 90 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento L: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 10 % al 80 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento M: Phenomenex Luna C18, 150 x 21,2 mm, columna de 5 pm; Fase móvil A: ácido acético al 0,02% en agua (v/v); Fase móvil B: ácido acético al 0,02% en acetonitrilo (v/v); Gradiente: del 1% al 100% de B durante 8,5 minutos y después mantenido en 100% de B durante 2 minutos; Caudal: 27 ml/minuto. Temperatura: 25 °C; Detección: DAD 215 nm, 254 nm; intervalo de EM (+) de 150-2000 daltons; Volumen de inyección: hasta 5 ml. Instrumento: 305 RP Waters FractionLynx LCMS.
Procedimiento N: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 50 % al 100 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento O: Phenomenex Luna C18, 150 x 21,2 mm, columna de 5 pm; Fase móvil A: ácido acético al 0,02% en agua (v/v); Fase móvil B: ácido acético al 0,02% en acetonitrilo (v/v); Gradiente: del 35% al 50% de B durante 8,5 minutos y después mantenido en 100% de B durante 2 minutos; Caudal: 27 ml/minuto. Temperatura: 25 °C; Detección: DAD 215 nm, 254 nm; intervalo de EM (+) de 150-2000 daltons; Volumen de inyección: hasta 5 ml. Instrumento: 305 RP Waters FractionLynx LCMS.
Procedimiento P: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 25 % al 100 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento Q: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 20 % al 100 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento R: Phenomenex Luna C18, 150 x 21,2 mm, columna de 5 pm; Fase móvil A: ácido acético al 0,02% en agua (v/v); Fase móvil B: ácido acético al 0,02% en acetonitrilo (v/v); Gradiente: del 35% al 50% de B durante 8,5 minutos y después mantenido en 100% de B durante 2 minutos; Caudal: 27 ml/minuto. Temperatura: 25 °C; Detección: DAD 215 nm, 254 nm; intervalo de EM (+) de 150-2000 daltons; Volumen de inyección: hasta 5 ml. Instrumento: 305 RP Waters FractionLynx LCMS.
Procedimiento S: Phenomenex Luna C18, 100 x 30 mm, columna de 5 pm; Fase móvil A: ácido trifluoroacético al
0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: del 20 % al 90 % de B durante 20 minutos; Caudal: 20 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml. Instrumento: Gilson 215.
Procedimiento T: Phenomenex Synergi Max C18, 250 x 50 mm, columna de 10 um; Fase móvil A: ácido fórmico al 0,2 % en agua (v/v); Fase móvil B: acetonitrilo; Gradiente: del 35,0 % al 65 % de B en 30 minutos, aumentando después hasta 100% de B en 2 minutos, mantenido durante 5 minutos; Caudal: 30 ml/minuto. Temperatura: no controlada; Detección: DAD 220 nm.
Procedimiento U: Isco MPLC, columna Gold C18 RediSep de 43 g; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente: 20-100% de B en 20 minutos; Caudal: 40 ml/min; Temperatura: no controlada; DAD 214, 254 nm.
Procedimiento V: Isco MPLC, columna Gold C18 RediSep de 43 g; Fase móvil A: ácido trifluoroacético al 0,02% en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,02% en acetonitrilo (v/v); Gradiente 10-80% de B en 20 minutos; Caudal: 40 ml/min; Temperatura: no controlada; DAD 214, 254 nm.
Procedimiento W. Phenomenex Synergi Max-RP 250 x 50 mm, columna de 10 pm; Fase móvil A: ácido trifluoroacético al 0,1 % en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,1 % en acetonitrilo (v/v); Gradiente: del 24 % al 54 % de B durante 30 minutos; Caudal: 30 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml.
Procedimiento X: Luna C18 150 x 25, columna de 5 pm; Fase móvil A: ácido trifluoroacético al 0,2 % en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,2 % en acetonitrilo (v/v); Gradiente: del 30 % al 50% de B durante 11 minutos; Caudal: 35 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml.
Procedimiento Y: Phenomenex Synergi Max-RP 250 x 50 mm, 100 x 30 mm, columna de 10 pm; Fase móvil A: ácido trifluoroacético al 0,1 % en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,1 % en acetonitrilo (v/v); Gradiente: del 30% al 60% de B durante 22 minutos; Caudal: 80 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml.
Procedimiento Z: Phenomenex Synergi Max-RP 250 x 50 mm, columna de 10 pm; Fase móvil A: ácido trifluoroacético al 0,1 % en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,1 % en acetonitrilo (v/v); Gradiente: del 30 % al 60 % de B durante 21,5 minutos; Caudal: 80 ml/minuto. Temperatura: no controlada; Detección: DAD 210, 254 nm; Volumen de inyección: hasta 5 ml.
Procedimiento AA: Phenomenex Synergi Max-RP 250 x 80, columna de 10 pm; Fase móvil A: ácido trifluoroacético al 0,1 % en agua (v/v); Fase móvil B: ácido trifluoroacético al 0,1 % en acetonitrilo (v/v); Gradiente: del 40 % al 70 % de B durante 30 minutos; Caudal: 30 ml/minuto. Temperatura: no controlada; Detección: DAD 220 nm; Volumen de inyección: hasta 5 ml.
Ejemplo 1: Preparación de N~2~-Benzoil-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amin o}propil]p irrolidin-1-il}- 2-oxoetil)-5,5,10-trimetN-3,6,9-trioxo-8-(propan-2-M)-2,13-dioxa-4,7,10-triazatetradec-1-N]fenM}-N~5~- carbamoil-L-ornitinamida (6)

Etapa 1: Síntesis de N~2~-benzoil-N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (2). A una solución de N~2~-benzoil-N~6~-(terc-butoxicarbonil)-L-lisina (100 mg, 0,285 mmol) y L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-omitinamida (1,108 mg, 0,285 mmol) en 1 ml de N, N-dimetilformamida se le añadió hexafluorofosfato de 0 -(7-azabenzotriazol-1-il)-A/,W,W,W-tetrametiluronio (HATU, 111 mg, 0,285 mmol) seguido de N,N-diisopropiletilamina (201 ul, 149 mg, 1,14 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 16 horas a temperatura ambiente. Después, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento A). Las fracciones que contenían el producto se liofilizaron para proporcionar 127 mg (63%) del producto deseado. CL-EM m/z 712,4 [M+H+]; 710,4 [M-H+]; tiempo de retención = 1,30 minutos.
Etapa 2: Síntesis de N~2~-benzoil-N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (3). Una solución de N~2~-benzoil-N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (2, 125 mg, 0,352 mmol) en 1 ml de N,N-dimetilformamida se trató con carbonato de p-nitrofenilo (107 mg, 0,352 mmol) y trietilamina (101 ul, 73,4 mg, 0,704 mmol). La mezcla se agitó durante dos horas a temperatura ambiente y después se concentró casi a sequedad y se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 5% al 20% en diclorometano para dar 129 mg (83,6%) del producto deseado. CL-EM m/z 877,4 [M+H+]; 899,3 [M+Na+]; tiempo de retención = 1,73 minutos.
Etapa 3: Síntesis de N~2~-benzoil-N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (5). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)- 1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-5-metil-1 -oxoheptan-4-il]-N-metil-L-valinamida (4 , 20 mg, 0,027 mmol) N~2~-benzoil-N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (3 , 28 mg, 0,032 mmol) en 0,2 ml de N,N-dimetilformamida se le añadieron 2,6-lutidina (5,8 mg, 0,032 mmol), N,N-diisopropiletilamina (9 ul, 7 mg, 0,054 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (HOAt, 4,4 mg, 0,032 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 16 horas a temperatura ambiente. Después, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento B). Las fracciones que contenían el producto se liofilizaron para proporcionar 30 mg (75 %) del producto deseado. CL-EM m/z 1481,8 [M+H+]; 1478,8 [M-H+]; tiempo de retención = 1,91 minutos.
Etapa 4: Síntesis de N~2~-benzoil-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1 -(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (6). A una solución de N~2~-benzoil-N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9
trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (5, 30 mg, 0,02 mmol) en 2,5 ml de diclorometano se le añadieron 2,5 ml de una solución al 10% de ácido trifluoroacético en diclorometano. La mezcla se controló por CL-EM y se dejó en agitación durante 2,25 horas a temperatura ambiente. Después, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento C). Las fracciones que contenían el producto se liofilizaron para proporcionar 22 mg (74 %) del producto deseado. CL-EM m/z 1381,7 [M+H+]; tiempo de retención = 1,54 minutos. tiempo de retención de HPLC analítica = 6,00 minutos.
Ejemplo 2: Preparación de
N~2~-(2,2-Dimetilpropanoil)-L-lisil-L-valil-N-£4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-om itinamida (12)

Etapa 1: Síntesis de N-(ferc-butoxicarbonil)-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,l3-dioxa-4, 7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (8). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida (4, 80 mg, 0,11 mmol) y N-(fercbutoxicarbonil)-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (7, 76,7 mg, 0,119 mmol) en 0,5 ml de N,N-dimetilformamida se le añadieron 2,6-lutidina (46,3 mg, 0,432 mmol), N,N-diisopropiletilamina (67,7 ul, 56,4 mg, 0,432 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (HOAt, 17,7 mg, 0,130 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 16 horas a 45 °C. Después, la reacción se concentró y se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 5% al 20% en diclorometano para proporcionar 90 mg (67%) del producto deseado. CL-EM m/z 1248,7 [M+H+]; tiempo de retención = 1,84 minutos.
Etapa 2: Síntesis de L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il] fenil}-N~5~-carbamoil-L-ornitinamida (9). A una solución de N-(ferc-butoxicarbonil)-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (8 , 91 mg, 0,072 mmol) en 5 ml de diclorometano se le añadieron 2 ml de una solución al 20% de ácido trifluoroacético en diclorometano. La mezcla se controló por CL-EM y se dejó en agitación durante 2,75 horas a temperatura ambiente. Después, la reacción se concentró para proporcionar 91 mg (100%) del producto deseado que se usó directamente en la siguiente etapa sin purificación. CL-EM m/z 1148,7 [M+H+]; tiempo de retención = 1,43 minutos.
Etapa 3: Síntesis de N~6~-(ferc-butoxicarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]- 12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (10). A una solución de N~6~-(ferc-butoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisina (50,6 mg, 0,108 mmol) en 0,5 ml de N,N-dimetilformamida se le añadieron hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N,,N'-tetrametiluronio (HATU, 41,6 mg, 0,108 mmol) seguido de N,N-diisopropiletilamina (51 ul, 37,6 mg, 0,288 mmol) y la mezcla se agitó durante 15 minutos a temperatura ambiente. Después, se añadió una solución de L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxo etil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec1-il] fenil}-N~5~-carbamoil-L-omitinamida (9, 91 mg, 0,072 mmol) en 1 ml de N,N-dimetilformamida. La mezcla se controló por CL-EM y se dejó en agitación durante 2 horas a temperatura ambiente antes de que se añadiera lentamente dietilamina (2 ml, 1,41 g, 19,3 mmol). La reacción se dejó en agitación durante 16 horas y después se acidificó mediante la adición de ácido trifluoroacético, se concentró y se purificó por HPLC de fase inversa (Procedimiento D). Las fracciones que contenían el producto se liofilizaron para proporcionar 99 mg (92 %) del producto deseado. CL-EM m/z 1376,8 [M+H+]; 1374,8 [M-H+]; tiempo de retención = 1,40 minutos.
Etapa 4: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-(2,2-dimetilpropanoil)-L-lisil-L-valil-N-{4-[(8S,11S, 12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil -3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (11). A una solución de trifluoracetato de N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-((2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-([(1S)-2-feniM-(1,3-tiazol-2-il)etil] amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida trifluoracetato (10, 21,5 mg, 0,015 mmol) y anhídrido piválico (5,6 mg, 0,030 mmol) en 0,2 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (26 ul, 19,4 mg, 0,15 mmol) y la mezcla se agitó a temperatura ambiente durante 45 minutos. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento E). Las fracciones que contenían el producto se liofilizaron para proporcionar 21,9 mg (100 %) del producto deseado. CL-EM m/z 1460,7 [M+H+]; 1458,5 [M-H+]; tiempo de retención = 1,93 minutos.
Etapa 5: Síntesis de N~2~-(2,2-dimetilpropanoil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (12). A una solución de N~6~-(terc-butoxicarbonil)-N~2~-(2,2-dimetilpropanoil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (11, 21,9 mg, 0,015 mmol) en 1 ml de diclorometano se le añadieron 0,33 ml de ácido trifluoroacético. La mezcla se controló por CL-EM y se dejó en agitación durante 1 hora a temperatura ambiente. Después, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento F). Las fracciones que contenían el producto se liofilizaron para proporcionar 8 mg (36 %) del producto deseado. CL-EM m/z 1360,7 [M+H+]; tiempo de retención = 1,45 minutos. tiempo de retención de HPLC analítica = 6,01 minutos.
Ejemplo 3: Preparación de N~2~-(Femlacetil)-L-lisil-L-valil-N-£4-[(8S,11S,12R)-1H(2S)-butan-2-il]- 12-(2-{(2S)-2- [(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-femM-(1,3-tiazol-2-il)etil]ammo}propil]pirrolidm-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]feml}-N~5~-carbamoil-L-ornitinamida (14)

Etapa 1: Síntesis de N—6—-(terc-butoxicarbonil)-N-2—-(fenilacetil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1s)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (13). A una solución de ácido fenilacético (15 mg, 0,11 mmol) en 0,2 ml de N,N-dimetilformamida se le añadió hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU, 43,1 mg, 0,11 mmol) seguido de N,N-diisopropiletilamina (20 ul, 14,5 mg, 0,11 mmol) y la mezcla se agitó a temperatura ambiente durante 45 minutos. Se añadió una solución de trifluoroacetato de N—6—-(terc-butoxicarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil] amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (10, 20 mg, 0,013 mmol) en 0,2 ml de N,N-dimetilformamida y la mezcla se agitó a temperatura ambiente durante 16 horas. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento G). Las fracciones que contenían el producto se liofilizaron para proporcionar 10 mg (52 %) del producto deseado. CL-EM m/z 1495,3 [M+H+]; 1493,1 [M-H+]; tiempo de retención = 1,93 minutos.
Etapa 2: Síntesis de N~2~-(fenilacetil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino} propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (14). A una solución de N~6~-(terc-butoxicarbonil)-N~2~-(fenilacetil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trio xo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (13, 10 mg, 0,007 mmol) en 1 ml de diclorometano se le añadió 1 ml de una solución al 20% de ácido trifluoroacético en diclorometano. La mezcla se controló por CL-EM y se dejó en agitación durante 2 horas a temperatura ambiente. Después, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento G). Las fracciones que contenían el producto se liofilizaron para proporcionar 10 mg (100%) del producto deseado. CL-EM m/z 1394,7 [M+H+]; tiempo de retención = 1,52 minutos. tiempo de retención de HPLc analítica = 5,39 minutos.
Ejemplo 4: Preparación de N—2—-(1H-imidazol-5-NcarbonN)-L-NsN-L-valN-N-{4-[(8S,11S,l2R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metM-3-oxo-3-{[(1S)-2-fenN-1-(1,3-tiazol-2-M)etM]ammo}propN]pirroMdin-1-M}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5— carbamoil-L-ornitinamida (16)

Etapa 1: Síntesis de N~6~-(terc-butoxicarbonN)-N~2~-(1H-imidazol-5-Ncarbonil)-L-NsN-L-valN-N-(4-[(8S,11S,12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-{[(1S) -2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (15). A una solución de trifluoroacetato de N~6~-(terc-butoxicarbonil)-L-Nsil-L-valN-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-([(1S)-2-fenil-1-(1,3-tiazol-2-il)etil] amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (10, 20,5 mg, 0,014 mmol) y ácido 1H-imidazol-5-carboxílico (15,7 mg, 0,14 mmol) en 0,2 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (25 ul, 18,3 mg, 0,14 mmol) y la mezcla se agitó a temperatura ambiente durante 5 minutos. Se añadió hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU, 54,3 mg, 0,14 mmol) y la mezcla se agitó durante una hora más a temperatura ambiente. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento G). Las fracciones que contenían el producto se liofilizaron para proporcionar 13 mg (59 %) del producto deseado. CL-EM m/z 1471,6 [M+H+]; 1469,0 [M-H+]; tiempo de retención = 1,70 minutos.
Etapa 2: Síntesis de N~2~-(1H-imidazol-5-ilcarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2- [(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (16). A una solución de N~6~-(terc-butoxicarbonil)-N~2~-(1H-imidazol-5-ilcarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (15, 13 mg, 0,008 mmol) en 5 ml de diclorometano se le añadieron 2 ml de una solución al 20% de ácido trifluoroacético en diclorometano. La mezcla se controló por CL-EM y se dejó en agitación durante 15 minutos a temperatura ambiente. Después, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento D). Las fracciones que contenían el producto se liofilizaron para proporcionar 6 mg (50 %) del producto deseado. CL-EM m/z 1370,7 [M+H+]; 1368,6 [M-H+]; tiempo de retención = 1,39 minutos. tiempo de retención de HPLC analítica = 5,30 minutos.
Ejemplo 5: Preparación de N~2~(1H-Imidazol-5-NacetM)-L-MsM-L-vam-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-M]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1 -(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (18)

Etapa 1: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-(1H-imidazol-5-ilacetil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (17). A una solución de trifluoroacetato de N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (10, 20 mg, 0,013 mmol) y ácido 2-(1H-imidazol-5-il)acético (30,0 mg, 0,20 mmol) en 0,2 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (23 ul, 17 mg, 0,13 mmol) y la mezcla se agitó a temperatura ambiente durante 5 minutos. Se añadió hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU, 50,4 mg, 0,13 mmol) y la mezcla se agitó durante una hora más a temperatura ambiente. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento G). Las fracciones que contenían el producto se liofilizaron para proporcionar 13,6 mg (65 %) del producto deseado. CL-EM m/z 1485,6 [M+H+]; 1483,5 [M-H+]; tiempo de retención = 1,44 minutos.
Etapa 2: Síntesis de N~2~-(1H-imidazol-5-ilacetil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (18). A una solución de N~6~-(terc-butoxicarbonil)-N~2~-(1H-imidazol-5-ilacetil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (17, 13,6 mg, 0,009 mmol) en 5 ml de diclorometano se le añadieron 2 ml de una solución al 20% de ácido trifluoroacético en diclorometano. La mezcla se controló por CL-EM y se dejó en agitación durante 15 minutos a temperatura ambiente. Después, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento D). Las fracciones que contenían el producto se liofilizaron para proporcionar 13 mg (96 %) del producto deseado. CL-EM m/z 1384,9 [M+H+]; 1382,8 [M-H+]; tiempo de retención = 1,26 minutos. tiempo de retención de HPLC analítica = 5,21 minutos.
Ejemplo 6: Preparación de N~2~-(3-MetNbutanoM)-L-NsN-L-vam-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-M]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (19)

Una solución de acido 3-metilbutanoico (10,2 mg, 0,100 mmol) en 0,4 ml de N,N-dimetilformamida se trato con N,N-diisopropiletilamina (35 ul, 26,1 mg, 0,200 mmol) y hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N’,N'-tetrametiluronio (HATU, 77,6 mg, 0,200 mmol) y la mezcla se agitó a temperatura ambiente durante cinco minutos. Se añadió una solución de trifluoroacetato de N~6~-(terc-butoxicarbonil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (10, 30 mg, 0,020 mmol) en 0,4 ml de N,N-dimetilformamida y la mezcla se dejó en agitación a temperatura ambiente durante 1 hora. Después, la solución se concentró casi a sequedad, después se disolvió en 2 ml de diclorometano y se trató con 0,5 ml de ácido trifluoroacético. Después de agitar a temperatura ambiente durante 30 minutos más, la reacción se concentró y se purificó por HPLC de fase inversa (Procedimiento H). Las fracciones que contenían el producto se liofilizaron para proporcionar 13 mg (44 %) del producto deseado. CL-EM m/z 1360,8 [M+H+]; 1358,4 [M-H+]; tiempo de retención = 1,49 minutos. tiempo de retención de HPLC analítica = 5,98 minutos.
Ejemplo 7: Preparación de N~2~-(2-Metilpropanoil)-L-lisil-L-valil-N-£4-[(85,115,12R)-11-[(25)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino)propil]pirrolidin-1-il)-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (25)

Etapa 1: Síntesis de N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (20). A una solución de L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (1, 6,5 g, 17,1 mmol) y N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisina (8,03 g, 17,1 mmol) en 200 ml de N,N-dimetilformamida se le añadieron N,N-diisopropiletilamina (3,32 g, 25,7 mmol) y hexafluorofosfato de 0-(7-azabenzotriazoM-il)-A/,W,W',W-tetrametiluronio (HATU, 7,8 g, 20,56 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 12 horas, después se concentró y se purificó por cromatografía sobre sílice usando un gradiente de elución de metanol del 5% al 17% en diclorometano para dar 4,00 g (28,2%) del producto deseado en forma de un sólido de color blanco.
Etapa 2: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (21). Una mezcla de N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (20, 4,8 g, 5,78 mmol) en 96 ml de ácido clorhídrico acuoso 4 M se agitó a temperatura ambiente durante 12 horas. La mezcla de reacción se ajustó a pH 7,5 mediante la adición lenta de K2CO3 acuoso saturado y después la suspensión resultante se filtró. El sólido se recogió y se secó al vacío para dar 4,00 g (94,9%) del producto deseado en forma de un sólido de color amarillo.
Etapa 3: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(2-metilpropanoil)-L-lisil-L-valil-N-5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (22). A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (21, 500 mg, 0,685 mmol) y 1-[(2 metilpropanoil)oxi]pirrolidin-2,5-diona (190 mg, 1,03 mmol) en 20 ml de N,N-dimetilformamida se le añadió N-N-diisopropiletilamina (221 mg, 1,7 mmol). Después de agitar durante 2 horas a temperatura ambiente, la mezcla se concentró y se purificó por HPLC de fase inversa (Procedimiento I) para dar 100 mg (18,2%) del producto deseado en forma de un sólido de color blanco.
Etapa 4: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(2-metilpropanoil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (23). A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(2-metilpropanoil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (22, 100 mg, 0,125 mmol) y carbonato de p-nitrofenilo (42 mg, 0,138 mmol) en 5 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (32 mg, 0,25 mmol) y la mezcla se agitó a temperatura ambiente durante 12 horas. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento J) para dar 110 mg (91,7%) del
producto deseado en forma de un sólido de color blanco. EMAR m/z para C50H60N8O12: 987,4036 (M+Na)+. 1H RMN (400 MHz, DMSO): □ 10,12 (s, 1H), 8,30 (m, 2H), 8,18 (m, 1H), 7,88 (m, 3H), 7,64 (m, 6H), 7,27 (m, 6H), 6,00 (s, 1H), 5,44 (s, 2H), 5,23 (s, 2H), 4,21 (m, 6H), 2,95 (m, 5H), 1,99 (m, 1H), 1,24 (m, 12H), 0,99 (d, 6H), 0,85 (d, 6H);
Etapa 4: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(2-metilpropanoil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5 ~-carbamoil-L-ornitinamida (24). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida (4 , 27,5 mg, 0,032 mmol) y N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(2-metilpropanoil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (23, 36,7 mg, 0,038 mmol) en 1 ml de N,N-dimetilacetamida se le añadieron 2,6-lutidina (13,7 mg, 0,128 mmol), N,N-diisopropiletilamina (23 ul, 16,7 mg, 0,128 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (HOAt, 5,2 mg, 0,038 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 16 horas a temperatura ambiente. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento K). Las fracciones que contenían el producto se liofilizaron para proporcionar 30 mg (60 %) del producto deseado.
Etapa 5: Síntesis de N~2~-(2-metilpropanoil)-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2 -{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (25). Una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(2-metilpropanoil)-L-lisil-L-valil-N-{4 -[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3 -{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (24, 30 mg, 0,019 mmol) en 1 ml de diclorometano se trató con 1 ml de dietilamina. La solución se agitó a temperatura ambiente durante cinco horas, después se concentró y se purificó por HPLC de fase inversa (Procedimiento K). Las fracciones que contenían el producto se liofilizaron para proporcionar 6,0 mg (21 %) del producto deseado. CL-EM m/z 1346,9 [M+H+]; tiempo de retención = 1,47 minutos. tiempo de retención de HPLC analítica = 5,87 minutos.
Procedimiento para los Ejemplos 8-18 y 49-71: Conjugación, purificación de ADC anti-Trop2 específicos del sitio
Para la conjugación de anticuerpos anti-Trop2 con las cargas útiles de enlazador (LP) de los Ejemplos 1 a 7, se ajustó la cantidad indicada de anticuerpo a 5 mg/ml en tampón que contenía Tris-HCl 25 mM, pH 8,0-8,5. La cantidad indicada de LP se añadió en un exceso molar de 5-50 veces sobre el anticuerpo y se inició la reacción enzimática mediante la adición de transglutaminasa bacteriana al 2% (p/v) (Ajinomoto Activa TI, Japón) y se incubó con agitación suave a 37 °C durante 16-24 horas. El ADC se purificó usando Butil Sepharose™ High Performance (Buti1HP, GE Healthcare Biosciences) ajustando la mezcla de reacción para obtener una composición tampón de sulfato de amonio 0,75 M, fosfato potásico 25 mM, pH 7 (Tampón A). El material se aplicó a un Butil HP, se lavó con 5 VC de Tampón A, y se eluyó con un gradiente lineal de 20 VC en fosfato potásico 25 mM, pH 7. Las fracciones que contenían el ADC se reunieron, se dializaron contra PBS, se concentraron usando una unidad de filtro centrífuga Amicon Ultra de 10 kDa (Millipore Corporation) y se esterilizaron por filtración a través de un filtro de 0,2 ^m. Como alternativa, el ADC se purificó usando resina de proteína A MabSelect (GE Healthcare Biosciences) siguiendo protocolos convencionales, y el tampón se intercambió en PBS.
Los siguientes ADC (unidos en varias posiciones y en varias etiquetas en el anticuerpo Trop2, como se indica en las Tablas 2 a 7) se fabricaron usando la técnica descrita anteriormente (la porción entre corchetes indica un residuo de glutamina, y las líneas onduladas muestran puntos de unión al resto del anticuerpo Anti-Trop2):
Ejemplo comparativo 8: C6vcMMAD:


Ejemplo 14:
Ċ 

Se obtuvieron muestras de plasma de ratones en puntos de tiempo que variaban de 1 a 10 días después de una dosis única de 9 mg/kg de ADC. Las muestras se diluyeron con un volumen igual de 1x PBS, y los ADC se aislaron usando cuentas MabSelect o antígeno Trop2 acoplado a Sepharose activada con CNBr(GE Healthcare) usando protocolos convencionales. Las relaciones fármaco-anticuerpo se evaluaron antes y después de la exposición in vivo usando procedimientos HIC y espectrometría de masas.
Ejemplo 20: Ensayo de estabilidad in vivo - Rata
Se obtuvieron muestras de plasma de ratas en puntos de tiempo que variaban de 1 a 10 días después de una dosis única de 9 mg/kg de ADC. Las muestras se diluyeron con un volumen igual de 1x PBS, y los ADC se aislaron usando antígeno Trop2 acoplado a Sepharose activada con CNBr(GE Healthcare) usando protocolos convencionales. Las relaciones fármaco-anticuerpo se evaluaron antes y después de la exposición in vivo usando procedimientos HIC y espectrometría de masas.
Ejemplo 21A: Ensayo de estabilidad in vitro - Plasma de ratón
Se incubaron 100-200 ug de ADC en plasma de ratón (proporcionado por Bioreclamation) suplementado con 1x PBS a la concentración final de ADC de 0,125 mg/ ml. Después de la incubación durante 3-7 días, las muestras se diluyeron con un volumen igual de 1x PBS, y los ADC se aislaron usando antígeno Trop2 acoplado a Sepharose activada con CNBr(GE Healthcare) usando protocolos convencionales. Las relaciones fármaco-anticuerpo se evaluaron antes y después de la exposición in vitro usando procedimientos HIC y espectrometría de masas.
Ejemplo 21B: Identificación de la enzima plasmática de ratón responsable de la escisión del enlazador
Para clasificar la enzima plasmática de ratón, se realizaron ensayos de estabilidad in vitro en presencia de diversos inhibidores de la proteasa usando conjugado anti-Trop2 L11B C6 vc0101 como sustrato. Los resultados mostraron que la enzima plasmática de ratón es probablemente una serina hidrolasa inhibida por Pefabloc 1 mM (Roche) y BNPP 100 uM (Sigma). Para identificar la enzima, se enriqueció la actividad de hidrólisis del enlazador anti-Trop2 L11B C6 vc0101 fraccionando el suero de ratón Balb/c usando una secuencia de etapas. Primero, El suero Balb/c (BioreclamationIVT) se agotó de IgG y albúmina usando las resinas de Proteína A y Proteína G de MabSelect disponibles en el mercado (GE Healthcare), seguido del kit de agotamiento de albúmina murina Qproteome (Qiagen). Después, el suero agotado se sometió a precipitación secuencial con sulfato de amonio. Las fracciones con la mayor actividad de hidrolasa enlazadora se aplicaron después a una columna HiTrap SP de intercambio catiónico (GE Healthcare) usando eluciones por gradiente escalonado con cloruro sódico. Después de esto, las fracciones SP con la mayor actividad se aplicaron a una columna HiTrap Q de intercambio aniónico (GE Healthcare) usando eluciones por gradiente escalonado con cloruro sódico. Después, las fracciones con la mayor actividad de hidrolasa más alta se combinaron y fraccionaron por tamaño usando una columna Superdex 200 (GE Healthcare). El análisis proteómico de las fracciones Superdex 200 agrupadas con la mayor actividad reveló que la Carboxilesterasa 1C es capaz de hidrolizar enlaces amida, y puede inhibirse con Pefabloc y BNPP. Para verificar la identificación correcta de Carboxilesterasa 1C de ratón como la enzima plasmática responsable de la escisión del enlazador, la proteína recombinante se expresó en células Expi 293 y se purificó usando una etiqueta HIS siguiendo protocolos convencionales. Se confirmó que la proteína purificada escinde múltiples conjugados con enlazadores basados en VC con la misma actividad relativa que la mostrada para el plasma de ratón.
Ejemplo 22: Ensayo de estabilidad in vitro - Plasma de rata
Se incubaron 100-200 ug de ADC en plasma de rata (proporcionado por Bioreclamation) suplementado con 1x PBS a la concentración final de ADC de 0,125 mg/ ml. Después de la incubación durante 3-7 días, las muestras se diluyeron con un volumen igual de 1x PBS, y los ADC se aislaron usando antígeno Trop2 acoplado a Sepharose activada con CNBr(GE Healthcare) usando protocolos convencionales. Las relaciones fármaco-anticuerpo se evaluaron antes y después de la exposición in vitro usando procedimientos HIC y espectrometría de masas.
Ejemplo 23: Ensayo de estabilidad in vitro - Plasma de Cynomolgus
Se incubaron 100-200 ug de ADC en plasma de cynomolgus (proporcionado por Bioreclamation) suplementado con 1x PBS a la concentración final de ADC de 0,125 mg/ ml. Después de la incubación durante 3-7 días, las muestras se diluyeron con un volumen igual de 1x PBS, y los ADC se aislaron usando antígeno Trop2 acoplado a Sepharose activada con CNBr(GE Healthcare) usando protocolos convencionales. Las relaciones fármaco-anticuerpo se evaluaron antes y después de la exposición in vitro usando procedimientos HIC y espectrometría de masas.
Ejemplo 24: Ensayo de citotoxicidad in vitro
Como se muestra en Fig. 1 (A a I), se realizaron estudios de citotoxicidad in vitro de anticuerpos quiméricos anti-Trop2 conjugados en una variedad de sitios con la carga útil citotóxica aminocaproílo (C6) vc Aur0101 con células BxPC3 que expresan la diana. Los compuestos no tratados (línea continua) y sus metabolitos aislados después de 4,5 días de tratamiento en plasma de ratón (línea discontinua) se ensayaron uno al lado del otro para determinar cualquier cambio en la citotoxicidad. BxPc3 es una línea celular de cáncer con altos niveles de expresión de diana (Trop-2 ++). Las células se sembraron en placas de fondo transparente de paredes blancas a 2000 células por pocillo durante 24 horas antes del tratamiento. Las células se trataron con conjugados anticuerpo-fármaco diluidos en serie 4 veces por triplicado. La viabilidad celular se determinó mediante el ensayo de viabilidad celular luminiscente CellTiter-Glo® 96 (Promega, Madison, WI) 96 horas después del tratamiento. La viabilidad celular relativa se determinó como porcentaje de control no tratado.
También, como se muestra en la Fig. 1 (J a O), se realizaron estudios de citotoxicidad in vitro de anticuerpos quiméricos anti-Trop2 conjugados en los sitios LCQ04 o L11B con una de las tres cargas útiles enlazadoras (cargas útiles enlazadoras de los Ejemplos 2, 3 y 5) con células BxPC3 que expresan la diana. Los compuestos no tratados (línea continua) y sus metabolitos aislados después de 4,5 días de tratamiento en plasma de ratón (línea discontinua) se ensayaron uno al lado del otro para determinar cualquier cambio en la citotoxicidad. BxPc3 es una línea celular de cáncer con altos niveles de expresión de diana (Trop-2 ++). Las células se sembraron en placas de fondo transparente de paredes blancas a 2000 células por pocillo durante 24 horas antes del tratamiento. Las células se trataron con conjugados anticuerpo-fármaco diluidos en serie 4 veces por triplicado. La viabilidad celular se determinó mediante el ensayo de viabilidad celular luminiscente CellTiter-Glo® 96 (Promega, Madison, WI) 96 horas después del tratamiento. La viabilidad celular relativa se determinó como porcentaje de control no tratado.
Ejemplo 25: Preparación de N~2~-(hidroxiacetN)-L-NsNvam-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-ornitinamida (30)

Etapa 1: Síntesis de N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (26). A una solución de N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-omitinamida (20, 998 mg, 1,20 mmol) y carbonato de p-nitrofenilo (748 mg, 2,41 mmol)) en 7 ml de N,N-dimetilformamida y en 7 ml de diclorometano se le añadió N,N-diisopropiletilamina (452 ul, 0,328 g, 2,41 mmol) y la mezcla se agitó a temperatura ambiente durante 3 horas. El residuo se diluyó con acetato de etilo y éter dietílico y la suspensión resultante se agitó durante 30 minutos. El sólido se aisló por filtración y se lavó con éter varias veces para dar, después del secado al aire, el producto deseado en forma de un sólido de color amarillo (1319 mg) que se usó tal cual sin purificación adicional. CL-EM m/z 995,5 [M+H+]; tiempo de retención = 1,02 minutos. tiempo de retención de HPLC analítica = 8,181 minutos.
Etapa 2: Síntesis de N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (27). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida (4, 371 mg, 0,499 mmol) y N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (26, 802 mg, 0,64 mmol) en 2,5 ml de N,N-dimetilacetamida se le añadieron 2,6-lutidina (216 ul, 200 mg, 1,9 mmol), N,N-diisopropiletilamina (326 ul, 240 mg, 1,9 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (HOAt, 17,7 mg, 0,130 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 90 minutos a 50 °C y después a temperatura ambiente durante 16 horas. Después, la reacción se purificó por cromatografía de fase inversa (Procedimiento U) para proporcionar, después de la liofilización, 339 mg (42%, 2 etapas) del producto deseado en forma de un sólido de color blanco. CL-EM m/z 1599,9 [M+H+]; tiempo de retención = 1,09 minutos. tiempo de retención de HPLC analítica = 8,163 minutos.
Etapa 3: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~- carbamoil-L-ornitinamida (28). A una suspensión de N~2~-(terc-butoxicarbonil)-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S, 12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (27, 439 mg, 0,275 mmol) en 2 ml de diclorometano se le añadió 1 ml de ácido trifluoroacético. La mezcla se controló por CL-EM y se dejó en agitación durante 20 minutos a temperatura
ambiente. Después, la reacción se concentró y se purificó por cromatografía de fase inversa (Procedimiento V) para proporcionar, después de la liofilización, 410 mg (96%). CL-Em m/z 1499,8 [M+H+]; tiempo de retención = 0,86 minutos. tiempo de retención de HPLC analítica = 6,778 minutos.
Etapa 4: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(hidroxiacetil)-L-lisilvalil-N-{4-[(8S, 11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (29). A una solución de ácido hidroxiacético (14,7 mg, 0,193 mmol) en 0,3 ml de N,N-dimetilformamida se le añadió clorhidrato de W-(3-dimetilaminopropil)-W'-etilcarbodiimida (EDCI, 7,99 mg, 0,0417 mmol), (hidroxiimino)cianoacetato de etilo (Oxyma, 20 mg, 0,14 mmol) y A/-metilmorfolina (40 ul, 37 mg, 0,36 mmol). La solución de color amarillo resultante se agitó durante 20 minutos y después se añadió una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 50 mg, 0,032 mmol) en 0,5 ml de N,N-dimetilformamida. La mezcla se controló por CL-EM y se dejó en agitación durante una hora a temperatura ambiente, y después se concentró y se purificó por HPLC de fase inversa (Procedimiento F). Las fracciones que contenían el producto se liofilizaron para proporcionar, después de la liofilización, 12,2 mg (24%). CL-EM m/z 1557,8 [M+H+]; tiempo de retención = 0,98 minutos. tiempo de retención de HPLC analítica = 7,371 minutos.
Etapa 5: Síntesis de N~2~-(hidroxiacetil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (30) A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(hidroxiacetil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (29 , 12,2 mg, 0,00784 mmol) en 0,3 ml de N,N-dimetilformamida se le añadió piperidina (0,1 ml, 90 mg, 1 mmol) y la mezcla se agitó a temperatura ambiente durante una hora. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento E). Las fracciones que contenían el producto se liofilizaron para proporcionar 8 mg (70 %) del producto deseado. CL-EM m/z 1334,8 [M+H+]; tiempo de retención = 0,76 minutos. tiempo de retención de HPLC analítica = 5,788 minutos.
Ejemplo de referencia 26: Preparación de N~2~-(metilcarbamoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2- N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metM-3-oxo-3-{[(1S)-2-femM-(1,3-tiazol-2-N)etN]amino}propN]pirroNdm-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (33)

Etapa 1: Síntesis de N-metiMH-imidazoM-carboxairiida (31). Una mezcla de 1,1'-carbonildiimidazol (1,76 g, 10,7 mmol) y clorhidrato de metilamina (661 mg, 9,7 mmol) se disolvió en 1,8 ml de N,N-dimetilformamida y 5,5 ml de acetonitrilo. La mezcla se controló por CL-EM y se dejó en agitación durante 3,25 horas a temperatura ambiente. Después, la reacción se concentró y se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 0% al 10% en diclorometano para proporcionar 696 mg (57%) del producto deseado. CL-EM m/z 126,0 [M+H+]; tiempo de retención = 0,17 minutos. 1H RMN (400 MHz, DMSO-d6) 8 = 8,22 (s, 1H), 7,65 (s, 1H), 7,03 (s, 1H), 2,83 (d, J = 4,3 Hz, 3H).
Etapa 2: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(metilcarbamoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-([(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (32). A una mezcla de N-metil-1H-imidazol-1-carboxamida (40,1 mg, 0,321 mmol) y N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 50 mg, 0,032 mmol) en 0,5 ml de N,N-dimetilformamida se le añadió trietilamina (44,7 ul, 32,5 mg, 0,321 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 1,5 a temperatura ambiente, y después se diluyó con dimetilsulfóxido y se purificó por HPLC de fase inversa (Procedimiento l) para proporcionar, después de la liofilización, 8,8 mg (18%) del producto deseado. CL-EM m/z 1556,8 [M+H+]; tiempo de retención = 0,99 minutos. tiempo de retención de HPLC analítica = 7,521 minutos.
Etapa 3: Síntesis de N~2~-(metilcarbamoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (33) A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-(metilcarbamoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5.5.10- trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,1 0-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (32, 8,8 mg, 0,0057 mmol) en 0,3 ml de N,N-dimetilformamida se le añadió piperidina (0,1 ml, 90 mg, 1 mmol) y la mezcla se agitó a temperatura ambiente durante 1,5 hora. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento E y después M). Las fracciones que contenían el producto se liofilizaron para proporcionar 4,6 mg (58 %) del producto deseado. CL-EM m/z 1333,7 [M+H+]; tiempo de retención = 0,76 minutos. tiempo de retención de HPLC analítica = 5,825 minutos.
Ejemplo 27: Preparación de N-metilglicil-L-lisilvalil-N-{4-[(8S,11S,12R)-1H(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil] pirrolidin-1 -il}-2-oxoetil)-5.5.10- trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]feml}-N~5~-carbamoil-L-ornitinamida (34)

A una solución de N-[(9H-fluoren-9-ilmetoxi)carbonil]-N-metilgliCinato de 2,5-dioxopirrolidin-1 -ilo (11,9 mg, 0,0416 mmol) en 0,1 ml de N,N-dimetilformamida se le añadieron N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-N)etN]amino}propN]pirroNdin-1-N}-2-oxoetN)-5,5,10-trimetN-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-N]fenN}-N~5~-carbamoil-L-ornitinamida (28, 50 mg, 0,032 mmol) y N,N-diisopropiletNamina (17 ul, 13 mg, 0,096 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante una hora a temperatura ambiente (CL-EM m/z 1670,71 [M+H+]; tiempo de retención = 1,03 minutos). La mezcla se concentró con un fuerte flujo de nitrógeno y el residuo se disolvió de nuevo en metanol y se pasó a través de una columna SCX (prelavada con 2 volúmenes de columna de metanol). El cartucho se lavó con un volumen de columna de metanol y el producto se dejó reposar sobre la columna durante una noche. La columna se lavó abundantemente con una solución 7 M de amoniaco en metanol y después de la concentración el residuo se purificó por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 4,2 mg (8 %) del producto deseado. CL-EM m/z 1347,51 [M+H+]; tiempo de retención = 0,68 minutos. tiempo de retención de HPLC analítica = 5,486 minutos.
Ejemplo 28: Preparación de N,N-dimetilglicil-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil] p irrolidin-1-il}- 2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-ornitinamida (35)


A una solución de N,N-dimetilglicina (4,5 mg, 0,044 mmol) en 0,1 ml de N,N-dimetilformamida se le añadieron 0,1 ml de una solución madre de clorhidrato de W-(3-dimetilaminopropil)-W'-etilcarbodiimida (EDCI, 79,9 mg, 0,417 mmol), (hidroxiimino)cianoacetato de etilo (Oxyma, 10 mg, 0,07 mmol) y W-metilmorfolina (10 ul, 9 mg, 0,09 mmol) en 1 ml de N,N-dimetilformamida. La solución de color amarillo resultante se agitó durante 15 minutos, después se añadió una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 50 mg, 0,032 mmol) en 0,5 ml de N,N-dimetilformamida. La mezcla se controló por CL-EM, se dejó en agitación durante 40 minutos a temperatura ambiente y se añadió más cantidad N-metil morfolina (50 ul, 45 mg, 0,45 mmol). La mezcla se agitó durante 16 horas, se concentró y se purificó por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 5,4 mg (11 %) del producto deseado. CL-EM m/z 1361,8 [M+H+]; tiempo de retención = 0,71 minutos. tiempo de retención de HPLC analítica = 5,481 minutos.
Ejemplo 29: Preparación de glicil-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-N)-2,13-dioxa-4,7,10-triazatetradec-1-N]feml}-N~5~-carbamoN-L-omitmamida (37)

Etapa 1: Síntesis de N-[(9H-fluoren-9-ilmetoxi)carbonil]glicil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)- 2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N-5--carbamoil-L-ornitinamida (36) A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2
il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 65,0 mg, 0,043 mmol)) en 0,650 ml de N,N-dimetilformamida se le añadieron N-[(9H-fluoren-9-ilmetoxi)carbonil]glicinato de 2,5-dioxopirrolidin-1-ilo (68,4 mg, 0,173 mmol) y N,N-diisopropiletilamina (23,1 mg, 0,173 mmol, 30,8 ul). La mezcla se controló por CL-EM y se dejó en agitación durante 2 horas a temperatura ambiente y se purificó directamente por HPLC de fase inversa (Procedimiento N). Las fracciones que contenían el producto se liofilizaron para proporcionar 22,3 mg (29 %) del producto deseado. c L-EM m/z 1777,2 [M+H+]; tiempo de retención = 1,10 minutos. tiempo de retención de HPLC analítica = 8,544 minutos.
Etapa 2: Síntesis de glicil-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (37) A una solución de N-[(9H-fluoren-9-ilmetoxi)carbonil]glicil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (36, 20 mg, 0,011 mmol) en 0,6 ml de N,N-dimetilacetamida se le añadió piperidina (0,3 ml, 270 mg, 3 mmol) y la mezcla se agitó a temperatura ambiente durante 15 minutos. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento L y después M). Las fracciones que contenían el producto se liofilizaron para proporcionar 14,4 mg (90 %) del producto deseado. CL-EM m/z 1333,8 [M+H+]; tiempo de retención = 0,70 minutos. tiempo de retención de HPLC analítica = 5,825 minutos.
Ejemplo 30: Preparación de N~2~-[(3-metiloxetan-3-il)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2- N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-{[(1S)-2-femM-(1,3-tiazol-2-N)etN]ammo}propN]pirroNdm-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (39)

Etapa 1: Síntesis de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-[(3-metiloxetan-3-il)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-feniM-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec1- il]fenil}-N-5--carbamoil-L-ornitinamida (38) En un baño de agua enfriada con hielo, a una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2- il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (28, 50,0 mg, 0,031 mmol) en 0,5 ml de N, N-dimetilacetamida se le añadieron ácido 3-metiloxetano-3-carboxílico (22 mg, 0,19 mmol), hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’-tetrametiluronio (HATU, 65 mg, 0,17 mmol) y N,N-diisopropiletilamina (35 ul, 26 mg, O, 20 mmol). La mezcla se agitó durante 40 minutos más en el baño de agua enfriada con hielo. Después, la solución se purificó directamente por HPLC de fase inversa (Procedimiento P). Las fracciones que contenían el producto se liofilizaron para proporcionar 37,4 mg (55 %) del producto deseado. CL-EM m/z 1597,8 [M+H+]; tiempo de retención = 1,00 minutos. tiempo de retención de HPLC analítica = 7,567 minutos.
Etapa 2: Síntesis de N~2~-[(3-metiloxetan-3-il)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5.5.10- trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (39) A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-N~2~-[(3-metiloxetan-3-il)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (38, 27,4 mg, 0,0172 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadió una solución acuosa de hidróxido de litio (411 ul, 41,1 mg, 0,0343 mmol) y la mezcla se agitó a temperatura ambiente durante una hora. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 9,3 mg (36 %) del producto deseado. CL-EM m/z 1374,4 [M+H+]; tiempo de retención = 0,77 minutos. tiempo de retención de HPLC analítica = 5,841 minutos.
Ejemplo 31: Preparación de 2-metilalanil-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil] pirrolidin-1-il}- 2-oxoetil)-5.5.10- trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-om itinam ida (41)

Etapa 1: Síntesis de N-[(9H-fluoren-9-ilmetoxi)carbonil]-2-metilalanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-thazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (40) En un baño de agua enfriada con hielo, a una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (28, 50,0 mg, 0,031 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron N-[(9H-fluoren-9-ilmetoxi)carbonil]-2-metilalanina (17,5 mg, 0,0538 mmol), hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N’,N’-tetrametiluronio (HATU, 25,9 mg, 0,0681 mmol) y N,N-diisopropiletilamina (25 ul, 19 mg, 0,14 mmol). La mezcla se agitó durante 40 minutos más en el baño de agua enfriada con hielo. Después, la solución se purificó directamente por HPLC de fase inversa (Procedimiento Q). Las fracciones que contenían el producto se liofilizaron para proporcionar 56 mg (100%) del producto deseado. CL-EM m/z 1806,9 [M+H+]; tiempo de retención = 1,12 minutos. tiempo de retención de HPLC analítica = 8,817 minutos.
Etapa 2: Síntesis de 2-metilalanil-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (40) A una solución de N-[(9H-fluoren-9-ilmetoxi)carbonil]-2-metilalanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-thazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (40, 56 mg, 0,031 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadió una solución acuosa de hidróxido de litio (100 ul, 10 mg, 0,4 mmol) y la mezcla se agitó a temperatura ambiente durante una hora. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 7,6 mg (15 %) del producto deseado. CL-EM m/z 1384,2 [M+Na+]; tiempo de retención = 0,71 minutos. tiempo de retención de HPLC analítica = 5,467 minutos.
Ejemplo 32: Preparación de N~2~-(metoxicarboml)-L-MsNvalN-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-M]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinam ida (43)

Etapa 1: Síntesis de N-(metoxicarbonil)-2-metilalanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S, 12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (42) A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 30,0 mg, 0,019 mmol)) en 0,3 ml de N,N-dimetilacetamida and 0,2 ml de
diclorometano se le añadió cloroformiato de metilo (29 ul, 35,2 mg, 0,372 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 2 horas a temperatura ambiente. Después de 2 h, se añadió N,N-diisopropiletilamina (10,0 ul, 7,5 mg, 0,056 mmol) y la mezcla se agitó durante 16 horas más. El análisis por CL-EM aún mostraba que la reacción no se había completado así que se añadió cloroformiato de metilo (60 ul, 73 mg, 0,78 mmol) seguido de N,N-diisopropiletilamina (10 ul, 7,5 mg, 0,056 mmol). Después de agitar a temperatura ambiente durante 4 horas más, la mezcla se purificó directamente por HPLC de fase inversa (Procedimiento Q). Las fracciones que contenían el producto se liofilizaron para proporcionar 6,4 mg (22 %) del producto deseado. CL-EM m/z 1557,9 [M+H+]; tiempo de retención = 1,05 minutos. tiempo de retención de HPLC analítica = 7,709 minutos.
Etapa 2: Síntesis de N~2~-(metoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (43) A una solución de N-(metoxicarbonil)-2-metilalanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (42, 6,4 mg, 0,0041 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadió dietilamina (0,3 ml, 200 mg, 3 mmol) y la mezcla se agitó a temperatura ambiente durante 3 horas. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 2,6 mg (44%) del producto deseado. CL-EM m/z 1334,8 [M+H+]; tiempo de retención = 0,83 minutos. tiempo de retención de HPLC analítica = 5,908 minutos.
Ejemplo de referencia 33: Preparación de L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-ornitinamida (44)

A una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 20,0 mg, 0,012 mmol)) en 0,2 ml de N,N-dimetilacetamida se le añadió piperidina (0,1 ml, 90 mg, 1 mmol) y la mezcla se agitó a temperatura ambiente durante 4 horas. Después, la solución se purificó directamente por HPLC de fase inversa (Procedimiento L y después R). Las fracciones que contenían el producto se liofilizaron para proporcionar 7,6 mg (44%) del producto deseado. CL-EM m/z 1276,7 [M+H+]; tiempo de retención = 0,70 minutos. tiempo de retención de HPLC analítica = 5,516 minutos.
Ejemplo 34: Preparación de L-alanil-L-lisilvalil-N-£4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (46)

Etapa 1: Síntesis de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-alanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -N}-2-oxoetN)-5,5,10-trimetN-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-thazatetradec-1- il]fenil}-N~5~-carbamoil-L-omitinamida (45) En un baño de agua enfriada con hielo, a una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2- il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 8,7 mg, 0,0054 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-alaninato de 2,5-dioxopirrolidin-1-ilo ((8,81 mg, 0,0216 mmol) y N,N-diisopropiletilamina (10,0 ul, 7,5 mg, 0,056 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante 16 horas a temperatura ambiente y se purificó directamente por HPLC de fase inversa (Procedimiento S). Las fracciones que contenían el producto se liofilizaron para proporcionar 5,3 mg (55 %) del producto deseado. CL-EM m/z 1791,7 [M+H+]; tiempo de retención = 1,11 minutos. tiempo de retención de HPLC analítica = 8,655 minutos.
Etapa 2: Síntesis de L-alanil-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (46) A una solución de N-[(9H-fluoren-9-ilmetoxi)carbonil]-L-alanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-ornitinamida (45, 5,3 mg, 0,0030 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadió una solución acuosa de hidróxido de litio (210 ul, 20 mg, 0,9 mmol) y la mezcla se agitó a temperatura ambiente durante una hora. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 3,4 mg (73 %) del producto deseado. CL-EM m/z 1347,7 [M+H]+; tiempo de retención = 0,69 minutos. tiempo de retención de HPLC analítica = 5,487 minutos.
Ejemplo 35: Preparación de D-alanil-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-N)-2,13-dioxa-4,7,10-triazatetradec-1-N]fenN}-N~5~-carbamoN-L-omitmamida (48)

Etapa 1: Síntesis de N-(terc-butoxicarbonil)-D-alanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-thazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (47) En un baño de agua enfriada con hielo, a una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2- il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 50 mg, 0,03 mmol) en 0,5 ml de N, N-dimetilacetamida se le añadieron N-(terc-butoxicarbonil)-D-alaninato de 2,5-dioxopirrolidin-1-ilo (15,7 mg, O, 0548 mmol) y N,N-diisopropiletilamina (22,0 ul, 16 mg, 0,12 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante 16 horas a temperatura ambiente y se purificó directamente por HPLC de fase inversa (Procedimiento S). Las fracciones que contenían el producto se liofilizaron para proporcionar 20,0 mg (40 %) del producto deseado. CL-EM m/z 1791,7 [M+H+]; tiempo de retención = 1,08 minutos. tiempo de retención de HPLC analítica = 8,024 minutos.
Etapa 2: Síntesis de D-alanil-L-lisilvalil-N-{4-[(8S,11S,12R)-1H(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida(48) A una solución de N-(tercbutoxicarbonil)-D-alanil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (47, 20,0 mg, 0,012 mmol) en 1 ml de acetonitrilo se le añadieron 0,5 ml de ácido trifluoroacético. La mezcla se agitó a temperatura ambiente durante 15 minutos y después se concentró. El residuo se disolvió en 0,5 ml de N,N-dimetilacetamida y se enfrió con un baño de agua enfriada con hielo. A esto se le añadió una solución acuosa de hidróxido de litio (50,3 ul, 5,3 mg, 0,12 mmol) y la mezcla se agitó a temperatura ambiente durante 30 minutos. Después, la solución se purificó directamente por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 8,9 mg (47%) del producto deseado. CL-EM m/z 1347,9 [M+H]+; tiempo de retención = 0,77 minutos. tiempo de retención de HPLC analítica = 5,429 minutos.
Ejemplo de referencia 36: Preparación de L-alfa-aspartM-L-MsM-L-valM-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-M]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-{[(1S)-2-femM-(1,3-tiazol-2-N)etN]ammo}propN]pirroNdm-1-N}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~S~-carbamoil-L-ornitinamida (50)

Etapa 1: Síntesis de (6S,9S,12S,15S)-1-amino-6-({4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1s)-2-fenil-1-(1,3-tiazol-2-il)etil]amino} propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,l3-dioxa-4,7,10-triazatetradec-1-il]fenil}carbamoil)-15-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-12-(4-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}butil)-1,8,11,14-tetraoxo-9-(propan-2-il)-2,7,10,13-tetraazaheptadecan-17-oato de terc-butilo (49) En un baño de agua enfriada con hielo, a una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 50 mg, 0,03 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron ácido (2S)-4-terc-butoxi-2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-4-oxobutanoico (20,9 mg, 0,0722 mmol), hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU, 28,0 mg, 0,074 mmol) y N,N-diisopropiletilamina (22,0 ul, 16 mg, 0,12 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante una hora y se purificó directamente por HPLC de fase inversa (Procedimiento Q). Las fracciones que contenían el producto se liofilizaron para proporcionar 41 mg (70 %) del producto deseado. CL-EM m/z 1771,2 [M+H+]; tiempo de retención = 1,16 minutos. tiempo de retención de HPLC analítica = 8,591 minutos.
Etapa 2: Síntesis de L-alfa-aspartil-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (50) A una solución de (6S,9S,12S,15S)-1-amino-6-({4-[(8S,11S,12R)-11-[(2S)-butan-2-il]- 12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}carbamoil)- 15-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}-12-(4-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}butil)-1,8,11,14-tetraoxo-9-(propan-2-il)-2,7,10,13-tetraazaheptadecan-17-oato de terc-butilo (49, 41,0 mg, 0,023 mmol) en 1 ml de acetonitrilo se le añadieron 0,5 ml de ácido trifluoroacético. La mezcla se agitó a temperatura ambiente durante 25 minutos (CL-EM m/z 1671,0 [M+H]+; tiempo de retención = 0,93 minutos) y después se concentró. El residuo se disolvió en 0,5 ml de N,N-dimetilacetamida y se enfrió con un baño de agua enfriada con hielo. A esto se le añadió una solución acuosa de hidróxido de litio (50,3 ul, 5,3 mg, 0,12 mmol) y la mezcla se agitó a temperatura ambiente durante 20 minutos. Después, la solución se purificó directamente por HPLC de fase inversa (Procedimiento Q). Las fracciones que contenían el producto se liofilizaron para proporcionar 1,1 mg (3 %) del producto deseado. CL-EM m/z 1392,1 [M+H]+; tiempo de retención = 0,80 minutos. tiempo de retención de Hp l C analítica = 5,309 minutos.
Ejemplo 37: Preparación de N-acetikjlicil-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2
[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino} propil]pirrolidm-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]feml}-N~5~-carbamoil-L-om itinam ida (52)

Etapa 1: Síntesis de N-acetilgMcil-N~6— [(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N^4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (51) En un baño de agua enfriada con hielo, a una solución de N~6~-[(9H-fluoren-9-ilmetoxi)carbonN]-L-lisilvalil-N-{4-[(8S,11S, 12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-{[(1S)-2-feniM-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (28, 30 mg, 0,02 mmol) en 0,3 ml de N,N-dimetilacetamida se le añadieron N-acetilglicinato de 2,5-dioxopirrolidin-1-ilo (13,7 mg, 0,0640 mmol) y N,N-diisopropiletilamina (22 ul, 16 mg, 0,12 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante 16 horas a temperatura ambiente y se purificó directamente por HPLC de fase inversa (Procedimiento Q). Las fracciones que contenían el producto se liofilizaron para proporcionar 17,4 mg (60 %) del producto deseado. CL-EM m/z 1598,9 [M+H+]; tiempo de retención = 1.04 minutos. tiempo de retención de HPLC analítica = 7,326 minutos.
Etapa 2: Síntesis de N-acetNgNcN-L-NsN-L-valN-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (52) A una solución de N-acetilglicil-N~6— [(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (51, 17.4 mg, 0,0109 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadió una solución acuosa de hidróxido de litio (100 ul, 10 mg, 0,2 mmol) y la mezcla se agitó a temperatura ambiente durante 20 minutos. Después, la solución se purificó directamente por HPLC de fase inversa (Procedimiento B). Las fracciones que contenían el producto se liofilizaron para proporcionar 9,4 mg (58%) del producto deseado. CL-EM m/z 1398,1 [M+Na+]; tiempo de retención = 0,84 minutos. tiempo de retención de HPLC analítica = 5,715 minutos.
Ejemplo 38: Preparación de L-seril-L-lisilvalil-N-£4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,l0-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (57)

Etapa 1: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N~5~- carbamoil-N-[4-(hidroximetil)fenil]-L-omitinamida (53). En un baño de agua enfriada con hielo, a una solución de N~6~-(tercbutoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisina (1090 mg, 2,32 mmol) en 15 ml de N,N-dimetilformamida se le añadió hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-W,W,W',W'-tetrametiluronio (HATU, 882 mg, 2,32 mmol). Después de 40 minutos, se añadió L-valil-N~5~-carbamoil-N-[4-(hidroximetil) fenil]-L-ornitinamida (1, 800 mg, 2,11 mmol) seguido de N,N-diisopropiletilamina (1,114 ml, 817 mg, 6,32 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 2 horas a temperatura ambiente. Después, la reacción se vertió en metil tercbutil éter (150 ml), se agitó durante 15 minutos, y después se filtró. El sólido resultante se lavó con una mezcla de diclorometano:metil terc-butil éter:metanol (10:10:2) para proporcionar el producto en bruto (1,3 g) en forma de un sólido de color gris. Este sólido se purificó por HPLC de fase inversa (Procedimiento T). Las fracciones que contenían el producto se liofilizaron para proporcionar 570 mg (33 %) del producto deseado. tiempo de retención de HPLC analítica = 4,55 minutos. 1H RMN (400 MHz, DMSO-d6) S = 9,97 (s a, 1H), 8,14 (d, J = 6,5 Hz, 1H), 7,90 (d, J = 7,5 Hz, 2H), 7,73 (s a, 3H), 7,54 (d, J = 8,5 Hz, 3H), 7,42 (t, J = 7,3 Hz, 2H), 7,33 (t, J = 7,3 Hz, 2H), 7,23 (d, J = 8,0 Hz, 2H), 6,78 (s a, 1H), 5,98 (s a, 1H), 5,43 (s a, 2H), 5,11 (s a, 1H), 4,45 - 4,34 (m, 3H), 4,34 - 4,15 (m, 4H), 4,02 (s a, 1H), 3,02 (d, J = 5,5 Hz, 1H), 2,89 (s a, 3H), 1,98 (d, J = 6,5 Hz, 1H), 1,61 (s a, 3H), 1,52 (s a, 1H), 1,42-1,30 (m, 13H), 1,24 (s a, 2H), 0,83 (d, J = 12,0 Hz, 3H), 0,85 (d, J = 11,5 Hz, 3H).
Etapa 2: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N~5~- carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (54). En un baño de agua enfriada con hielo, a una solución de N~6~-(terc-butoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (53, 570 mg, 0,687 mmol) y carbonato de p-nitrofenilo (418 mg, 1,37 mmol) en 10 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (257 ul, 178 mg, 1,37 mmol) y la mezcla se agitó a temperatura ambiente durante 6 horas. Después, la reacción se vertió en metil terc-butil éter (150 ml), se agitó durante
15 minutos, y después se filtró. El sólido resultante se dispersó en diclorometano (20 ml) durante 20 minutos y después se filtró para proporcionar 400 mg (58%) del producto deseado. CL-EM m/z 996,0 [M+H+]; tiempo de retención = 4,829 minutos. tiempo de retención de HPLC analítica = 5,43 minutos. 1H RMN (400 MHz, DMSO-da) 8 = 10,14 (s, 1H), 8,36 -8,29 (m, 2H), 8,18 (d, J = 7,5 Hz, 1H), 7,90 (d, J = 7,5 Hz, 2H), 7,74 (d, J = 7,0 Hz, 2H), 7,66 (d, J = 8,0 Hz, 2H), 7,62 - 7,50 (m, 2H), 7,42 (d, J = 7,0 Hz, 3H), 7,38 - 7,11 (m, 3H), 6,78 (s a, 1H), 6,00 (s a, 1H), 5,44 (s a, 2H), 5,26 (s, 2H), 4,39 (s a, 1H), 4,35 -4,16 (m, 4H), 4,04 (s a, 1H), 3,05 (d, J = 7,0 Hz, 1H), 2,90 (s a, 3H), 2,74 (s, 1H), 2,34 (s a, 1H), 2,10 (s, 1H), 1,99 (d, J = 6,0 Hz, 1H), 1,68 (s a, 1H), 1,62 (s a, 2H), 1,53 (s a, 2H), 1,38 (s, 11H), 1,42 - 1,20 (m, 1H), 1,42 - 1,20 (m, 1H), 0,85 (d, J = 12,5 Hz, 3H), 0,86 (d, J = 12,5 Hz, 3H).
Etapa 3: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (55). A una solución de 2-metilalanil-N-[(3R,4S,5S)- 3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida (4 , 440 mg, 0,59 mmol) y N~6~-(terc-butoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (54, 658,0 mg, 0,661 mmol) en 1,3 ml de N,N-dimetilacetamida se le añadieron 2,6-lutidina (137 ul, 130 mg, 1,2 mmol), N,N-diisopropiletilamina (207,0 ul, 160 mg, 1,2 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (HOAt, 92,1 mg, 0,677 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 2,5 horas a 50 °C y después a temperatura ambiente durante 16 horas. Después, la reacción se purificó por cromatografía de fase inversa (Procedimiento U) para proporcionar, después de la liofilización, 630 mg (67%) del producto deseado en forma de un sólido de color blanco. CL-EM m/z 1600,6 [M+H+]; tiempo de retención = 1,07 minutos. tiempo de retención de HPLC analítica = 8,213 minutos.
Etapa 4: Síntesis de N~6~-(terc-butoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (56) En un baño de agua enfriada con hielo, a una solución de N~6~-(terc-butoxicarbonil)-N~2~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil] pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (55, 630 mg, 0,394 mmol) en 5 ml de N,N-dimetilacetamida se le añadió una solución de hidróxido de litio (29,4 mg, 1,23 mmol) en 0,5 ml de agua y la mezcla se agitó a temperatura ambiente durante 30 minutos, después de lo cual se añadió hidróxido de litio (40,4 mg, 1,69 mmol). Después de un total de 3 horas, la solución se enfrió en un baño de agua enfriada con hielo, después se inactivó con ácido acético (0,2 ml) y se purificó por HPLC de fase inversa (Procedimiento 2). Las fracciones que contenían el producto se liofilizaron para proporcionar 350 mg (60%) del producto deseado. CL-EM m/z 1377,6 [M+H+]; tiempo de retención = 0,77 minutos. tiempo de retención de HPLC analítica = 6,181 minutos.
Etapa 5: Síntesis de L-seril-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2- il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (57). En un baño de agua enfriada con hielo, a una solución de N~6~-(terc-butoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~- carbamoil-L-ornitinamida (56, 50,0 mg, 0,0335 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron N-(terc-butoxicarbonil)-L-serina (6,88 mg, 0,0335 mmol) en 0,1 ml de N,N-dimetilacetamida, hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-A/,W,W',W-tetrametiluronio (HATU, 15,3 mg, 0,0402 mmol mmol) en 0,1 ml de N,N-dimetilacetamida y N,N-diisopropiletilamina (17,9 ul, 13,4 mg, 0,101 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante 45 minutos a temperatura ambiente y después se concentró en una corriente fuerte de nitrógeno para dar el producto en bruto (CL-EM m/z 1565,6 [M+H+], tiempo de retención = 0,96 minutos). El residuo amarillo se disolvió en 1,2 ml de metanol y se cargó en partes iguales en dos cartuchos SCX pre-eluidos con 3 volúmenes de columna de metanol. Las columnas se lavaron con 3 volúmenes de columna de metanol (sin trazas de producto en los lavados de acuerdo con el análisis por CL-EM). El producto se dejó reposar sobre la columna durante una noche. El producto deseado se eluyó con 2 volúmenes de columna de trietilamina 1 N en metanol. Después de la concentración a presión reducida, el residuo se purificó por cromatografía de fase inversa (Procedimiento V) para proporcionar, después de la liofilización, 8,3 mg (16%) del producto deseado. CL-EM m/z 1364,5 [M+H+], tiempo de retención = 0,68 minutos. tiempo de retención de HPLC analítica = 5,464 minutos.
Ejemplo 39: Preparación de D-seril-L-lisilvalil-N-£4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-£(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-omitinamida (58)

En un baño de agua enfriada con hielo, a una solución de N~6~-(terc-butoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-omitinamida (56, 50,0 mg, 0,0335 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron N-(terc-butoxicarbonil)-D-serina (6,88 mg, 0,0335 mmol) en 0,1 ml de N,N-dimetilacetamida, hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N’,N-tetrametiluronio (HATU, 15,3 mg, 0,0402 mmol mmol) en 0,1 ml de N,N-dimetilacetamida y N,N-diisopropiletilamina (17,9 ul, 13,4 mg, 0,101 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante 45 minutos a temperatura ambiente y después se concentró en una corriente fuerte de nitrógeno para dar el producto en bruto (CL-EM m/z 1565,6 [M+H+], tiempo de retención = 0,96 minutos). El residuo amarillo se disolvió en 1,2 ml de metanol y se cargó en partes iguales en dos cartuchos SCX pre-eluidos con 3 volúmenes de columna de metanol. Las columnas se lavaron con 3 volúmenes de columna de metanol (sin trazas de producto en los lavados de acuerdo con el análisis por CL-EM). El producto se dejó reposar sobre la columna durante una noche. El producto deseado se eluyó con 2 volúmenes de columna de trietilamina 1 N en metanol. Después de la concentración a presión reducida, el residuo se purificó por cromatografía de fase inversa (Procedimiento V) para proporcionar, después de la liofilización, 10,4 mg (19%) del producto deseado. CL-EM m/z 1364,5 [M+H+], tiempo de retención = 0,67 minutos. tiempo de retención de Hp LC analítica = 5,424 minutos.
Ejemplo 40: Preparación de N~2~-[(2S)-2-hidroxipropanoN]-L-NsNvalN-N-{4-[(85,11S,12R)-11-[(2S)-butan-2-N]-2-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1 -(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2- oxoetN)-5,5,10-trimetN-3,6,9-trioxo-8-(propan-2-N)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-ornitinamida (59)

En un baño de agua enfriada con hielo, a una solución de N~6~-(terc-butoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1 -(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (56, 50,0 mg, 0,0335 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron ácido (2S)-2-hidroxipropanoico (3,02 mg, 0,0335 mmol) en 0,1 ml de N,N-dimetilacetamida, hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-W,A/,W',W-tetrametiluronio (HATU, 15,3 mg, 0,0402 mmol mmol) en 0,1 ml de N,N-dimetilacetamida y N,N-diisopropiletilamina (17,9 ul, 13,4 mg, 0,101 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante 45 minutos a temperatura ambiente y después se concentró en una corriente fuerte de nitrógeno para dar el producto en bruto (CL-EM m/z 1449,5 [M+H+], tiempo de retención = 0,91 minutos). El residuo amarillo se disolvió en 1,2 ml de metanol y se cargó en partes iguales en dos cartuchos SCX pre-eluidos con 3 volúmenes de columna de metanol. Las columnas se lavaron con 3 volúmenes de columna de metanol (sin trazas de producto en los lavados de acuerdo con el análisis por CL-EM). El producto se dejó reposar sobre la columna durante una noche. El producto deseado se eluyó con 2 volúmenes de columna de trietilamina 1 N en metanol. Después de la concentración a presión reducida, el residuo se purificó por cromatografía de fase inversa (Procedimiento V) para proporcionar, después de la liofilización, 12,9 mg (26 %) del producto deseado. CL-EM m/z 1349,4 [M+H+], tiempo de retención = 0,74 minutos. tiempo de retención de HPLC analítica = 5,813 minutos.
Ejemplo 41: Preparación de N~2~-[(2R)-2-hidroxipropanoN]-L-NsNvalN-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-{[(1S)-2-femM-(1,3-tiazol-2-N)etN]ammo}propN]pirroNdm-1-N}-2- oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-ornitinamida (60)

En un baño de agua enfriada con hielo, a una solución de N~6~-(terc-butoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1 -(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (56, 50,0 mg, 0,0335 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron ácido (2R)-2-hidroxipropanoico (3,01 mg, 0,0334 mmol) en 0,1 ml de N,N-dimetilacetamida, hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-W,A/,W',W-tetrametiluronio (HATU, 15,3 mg, 0,0402 mmol mmol) en 0,1 ml de N,N-dimetilacetamida y N,N-diisopropiletilamina (17,9 ul, 13,4 mg, 0,101 mmol). La mezcla se controló por CL-EM, se dejó en agitación durante 45 minutos a temperatura ambiente y después se concentró en una corriente fuerte de nitrógeno para dar el producto en bruto (CL-EM m/z 1449,4 [M+H+], tiempo de retención = 0,90 minutos). Después, el residuo se purificó por cromatografía de fase inversa (Procedimiento V) para proporcionar, después de la liofilización,
29,6 mg (60%) del producto deseado. CL-EM m/z 1349,4 [M+H+], tiempo de retención = 0,75 minutos. tiempo de retención de HPLC analítica = 5,740 minutos.
Ejemplo 42: Preparación de N~2~-[(2S)-2,3-dihidroxipropanoN]-L-NsNvalN-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1 -(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -N}-2-oxoetN)-5,5,10-trimetN-3,6,9-trioxo-8-(propan-2-N)-2,13-dioxa-4,7,10-triazatetradec-1-N]fenN}-N~5~-carbamoil-L-ornitinamida (62)

Etapa 1: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-[(2S)-2,3-dihidroxipropanoil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoil-L-ornitinamida (61). En un baño de agua enfriada con hielo, a una solución de N~6~-(tercbutoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (56, 50,0 mg, 0,0335 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron ácido (2S)-2,3-dihidroxipropanoico (4,02 mg, 0,0379 mmol) en 0,1 ml de N,N-dimetilacetamida, hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N’,N-tetrametiluronio (HATU, 15,3 mg, 0,0402 mmol mmol) en 0,1 ml de N,N-dimetilacetamida y N,N-diisopropiletilamina (17,9 ul, 13,4 mg, 0,101 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 45 minutos a temperatura ambiente y después se concentró en una corriente fuerte de nitrógeno. Después, el residuo amarillo se purificó por cromatografía de fase inversa (Procedimiento U) para proporcionar, después de la liofilización, 10,8 mg (22%) del producto deseado. CL-EM m/z 1465,5 [M+H+], tiempo de retención = 0,89 minutos. tiempo de retención de HPLC analítica = 6,607 minutos.
Etapa 2: Síntesis de N~2~-[(2S)-2,3-dihidroxipropanoil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]- 12-(2-{(2S)-2- [(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (62). A una suspensión de N~6~-(terc-butoxicarbonil)-N~2~-[(2S)-2,3-dihidroxipropanoil]-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (61, 10,8 mg, 0,0074 mmol) en 1 ml de diclorometano se le añadieron 0,5 ml de ácido trifluoroacético. La mezcla se controló por CL-EM y se dejó en agitación durante 20 minutos a temperatura ambiente. Después, la reacción se concentró y se purificó por cromatografía de fase inversa (Procedimiento V) para
proporcionar, después de la liofilización, 3,6 mg (33%). CL-EM m/z 1365,4 [M+H+]; tiempo de retención = 0,74 minutos. tiempo de retención de HPLC analítica = 5,749 minutos.
Ejemplo 43: Preparación de N~2~-(3-hidroxipropanoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1 -(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoil-L-ornitinamida (64)

Etapa 1: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-(3-hidroxipropanoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,l0-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (63). En un baño de agua enfriada con hielo, a una solución de N~6~-(terc-butoxicarbonil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,l0-trimetil-3,6,9-trioxo-8-(propan-2-il)- 2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-om itinamida (56, 50,0 mg, 0,0335 mmol) en 0,5 ml de N,N-dimetilacetamida se le añadieron ácido 3-hidroxipropanoico (4,01 mg, 0,0445 mmol) en 0,1 ml de N,N-dimetilacetamida, hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N’,N-tetrametiluronio (HATU, 15,3 mg, 0,0402 mmol mmol) en 0,1 ml de N,N-dimetilacetamida y N,N-diisopropiletilamina (17,9 ul,d 13,4 mg, 0,101 mmol). La mezcla se controló por CL-EM y se dejó en agitación durante 45 minutos a temperatura ambiente y después se concentró en una corriente fuerte de nitrógeno. Después, el residuo amarillo se purificó por cromatografía de fase inversa (Procedimiento U) para proporcionar, después de la liofilización, 13,9 mg (29 %) del producto deseado. CL-EM m/z 1449,6 [M+H+], tiempo de retención = 0,91 minutos. tiempo de retención de HPLC analítica = 6,721 minutos.
Etapa 2: Síntesis de N~2~-(3-hidroxipropanoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (64). A una suspensión de N~6~-(terc-butoxicarbonil)-N~2~-(3-hidroxipropanoil)-L-lisilvalil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida (63, 13,9 mg, 0,0096 mmol) en 1 ml de diclorometano se le añadieron 0,5 ml de ácido trifluoroacético. La mezcla se controló por CL-EM y se dejó en agitación durante 20 minutos a temperatura ambiente. Después, la reacción se concentró y se purificó por cromatografía de fase inversa (Procedimiento V) para proporcionar, después de la liofilización, 4,1 mg (29%). CL-EM m/z 1349,4 [M+H+]; tiempo de retención = 0,75 minutos. tiempo de retención de HPLC analítica = 5,758 minutos.
Ejemplo de referencia 44: Preparación de N~2~-(metilsulfonil)lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-N]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metN-3-oxo-3-{[(1S)-2-femM-(1,3-tiazol-2-N)etN]ammo}propN]pirroNdm-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil)-N~5~-carbamoilornitinamida, sal trifluoroacética (70)

Etapa 1: Síntesis de N~6~-(ferc-butoxicarbonil)-N~2~-(metilsulfonil)-L-lisinato de metilo (65). A una solución enfriada con hielo de N~6~-(ferc-butoxicarbonil)-L-lisinato de metilo (2,0 g, 6,73 mmol) en tetrahidrofurano:agua (1:1,20 ml) se le añadió carbonato potásico (4,6 g, 33,7 mmol) seguido de la adición gota a gota de cloruro de mesilo (0,62 ml, 8,08 mmol). La mezcla se agitó a temperatura ambiente durante 3 horas y después la mezcla se filtró. El filtrado se extrajo tres veces con acetato de etilo. Los extractos combinados se lavaron con salmuera, se secaron sobre sulfato sódico y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 0% al 2% en diclorometano para proporcionar 2,0 g (87%) del producto deseado en forma de un aceite incoloro. 1H RMN (400 MHz, CLOROFORMO-d) 5 = 5,23 (d, J = 9,0 Hz, 1H), 4,62 (s a, 1H), 4,10 (dt, J = 5,0, 8,5 Hz, 1H), 3,78 (s, 3H), 3,16 - 3,04 (m, 2H), 2,95 (s, 3H), 1,88 - 1,78 (m, 2H), 1,77 - 1,64 (m, 1H), 1,59 - 1,47 (m, 3H), 1,45 (s, 9H).
Etapa 2: Síntesis de N~6~-(ferc-butoxicarbonil)-N~2~-(metilsulfonil)-L-lisina (66). A una solución agitada de N~6~-(ferc-butoxicarbonil)-N~2~-(metilsulfonil)-L-lisinato de metilo (65, 2,0 g, 5,90 mmol) en tetrahidrofurano:agua:metanol (1:1:1, 30 ml) se le añadió hidróxido de litio monohidrato (496 mg, 11,8 mmol). La mezcla se agitó a temperatura ambiente durante 3 horas. La mezcla se concentró a presión reducida. La capa acuosa restante se extrajo dos veces con acetato de etilo. La capa acuosa se acidificó a pH 3 con una solución acuosa 1 N de ácido clorhídrico y después se extrajo dos veces con alcohol isopropílico:acetato de etilo (1:5). Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre sulfato sódico y se concentraron a presión reducida para proporcionar 1,3 g (68%) del producto deseado en forma de un sólido de color blanco. EM m/z 323,1 [M-H+]; 1H RMN (400 MHz, CLOROFORMO-d) 5 = 6,40 (s a, 1H), 5,81 - 5,66 (m, 1H), 4,79 (s a, 1H), 3,23 - 3,06 (m, 2H), 2,99 (s, 3H), 1,96 -1,71
(m, 2H), 1,55 - 1,39 (m, 13H).
Etapa 3: Síntesis de N~6~-(terc-butox¡carbon¡l)-N~2~-(met¡lsulfon¡l)-L-l¡s¡l-L-val¡l-N~5~-carbamo¡l-N-[4-(h¡drox¡met¡l)fen¡l]orn¡t¡nam¡da (67). A una soluc¡ón enfriada con h¡elo de N~6~-(terc-butox¡carbon¡l)-N~2~-(met¡lsulfon¡l)-L-l¡s¡na (66, 1,0 g, 3,09 mmol) y L-val¡l-N~5~-carbamo¡l-N-[4-(h¡drox¡met¡l)fen¡l]-L-orn¡t¡nam¡da (1, 1,28 g, 3,40 mmol) en 20 ml N,N-d¡met¡lformam¡da se le añad¡eron N,N-d¡¡soprop¡let¡lam¡na (600 ul, 438 mg, 3,40 mmol), clorh¡drato de N-(3-d¡met¡lam¡noprop¡l)-N-et¡lcarbod¡¡m¡da (649 mg, 3,40 mmol) y 1-h¡drox¡benzotr¡azol h¡drato (459 mg, 3,40 mmol). La mezcla se dejó calentar a temperatura amb¡ente y se ag¡tó durante 16 horas. Después, la mezcla se añad¡ó gota a gota a 150 ml de terc-but¡l met¡l éter. El sól¡do se purificó por HPLC de fase ¡nversa. Las fracc¡ones que contenían el producto se l¡of¡l¡zaron para proporc¡onar 580 mg (28%) del producto deseado en forma de un sól¡do de color blanco.
Etapa 4: Síntes¡s de N~6~-(terc-butox¡carbon¡l)-N~2~-(met¡lsulfon¡l)-L-l¡s¡l-L-val¡l-N~5~-carbamo¡l-N-[4-({[(4-n¡trofenox¡)carbon¡l]ox¡}met¡l)fen¡l]om¡t¡nam¡da (68). A una soluc¡ón ag¡tada de N~6~-(terc-butox¡carbon¡l)-N~2~-(met¡lsulfon¡l)-L-l¡s¡l-L-val¡l-N~5~-carbamo¡l-N-[4-(h¡drox¡met¡l)fen¡l]orn¡t¡nam¡da (67, 580 mg, 0,85 mmol) y p-n¡trofen¡lo carbonato (770 mg, 2,56 mmol) en 10 ml de N,N-d¡met¡lformam¡da se le añad¡ó N,N-d¡¡soprop¡let¡lam¡na (451 ul, 330 mg, 2,56 mmol). La mezcla se ag¡tó a temperatura amb¡ente durante 16 horas. La mezcla se añad¡ó gota a gota a 150 ml de terc-but¡l met¡l éter. El sól¡do se purificó por HPLC de fase ¡nversa (ProcedimientoXXX). Las fracc¡ones que contenían el producto se l¡of¡l¡zaron para proporc¡onar 200 mg (28%) del producto deseado en forma de un sól¡do de color blanco. EM m/z 851,4 [M+H+].'H RMN (400 MHz, DMSO-d6) 5 = 10,16 (s, 1H), 8,32 (d, J = 9,5 Hz, 2H), 8,26 (d, J = 6,5 Hz, 1H), 7,95 (t, J = 7,4 Hz, 1H), 7,65 (d, J = 8,5 Hz, 2H), 7,58 (t, J = 5,9 Hz, 2H), 7,45 (d, J = 9,0 Hz, 1H), 7,42 (d, J = 8,5 Hz, 2H), 6,79 (t, J = 5,5 Hz, 1H), 6,00 (t, J = 5,8 Hz, 1H), 5,45 (s, 2H), 4,40 (d, J = 5,0 Hz, 1H), 4,33 -4,06 (m, 1H), 3,84 (d, J = 4,5 Hz, 1H), 3,30 (s a, 1H), 3,09 - 2,99 (m, 1H), 2,98 - 2,93 (m, 1H), 2,93 - 2,86 (m, 2H), 2,83 (s, 3H), 2,01 (d, J = 6,5 Hz, 1H), 1,59 (s a, 3H), 1,47 (d, J = 13,1 Hz, 2H), 1,38 (s, 13H), 0,89 (d, J = 7,0 Hz, 3H), 0,85 (d, J = 7,0 Hz, 3H).
Etapa 5: Síntes¡s de N~6~-(terc-butox¡carbon¡l)-N~2~-(met¡lsulfon¡l)-D-l¡s¡l-L-val¡l-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-¡l]-12-(2-{(2S)-2-[(1R,2R)-1-metox¡-2-met¡l-3-oxo-3-{[(1s)-2-fen¡l-1-(1,3-t¡azol-2-¡l)et¡l]am¡no}prop¡l]p¡rrol¡d¡n-1-¡l}-2-oxoet¡l)-5,5,10-tr¡met¡l-3,6,9-tr¡oxo-8-(propan-2-¡l)-2,13-d¡oxa-4,7,10-tr¡azatetradec-1-¡l]fen¡l}-N~5~-carbamo¡lorn¡t¡nam¡da (69). A una soluc¡ón de 2-met¡lalan¡l-N-[(3R,4S,5S)-3-metox¡-1-{(2S)-2-[(1R,2R)-1-metox¡-2-met¡l-3-oxo-3-{[(1S)-2-fen¡l-1-(1,3-t¡azol-2-¡l)et¡l]am¡no}prop¡l]p¡rrol¡d¡n-1-¡l}-5-met¡l-1-oxoheptan-4-¡l]-N-met¡l-L-val¡nam¡da (4, 52,0 mg, 0,070 mmol) y N~6~-(terc-butox¡carbon¡l)-N~2~-(met¡lsulfon¡l)-L-l¡s¡l-L-val¡l-N~5~-carbamo¡l-N-[4-({[(4-n¡trofenox¡)carbon¡l]ox¡}met¡l)fen¡l]orn¡t¡nam¡da (68, 60,6 mg, 0,0710 mmol) en 2 ml de N,N-d¡met¡lacetam¡da se le añad¡eron 2,6-lut¡d¡na (31 ul, 28,3 mg, 0,264 mmol), N,N-d¡¡soprop¡let¡lam¡na (46 ul, 34,1 mg, 0,264 mmol) y 3H-[1,2,3]tr¡azolo[4,5-b]p¡r¡d¡n-3-ol (9 mg, 0,066 mmol). La mezcla se dejó en ag¡tac¡ón durante 16 horas a 45 °C. Después, la reacc¡ón se purificó por HPLC de fase ¡nversa (Procedimiento S) para proporc¡onar, después de la l¡of¡l¡zac¡ón, 70 mg (69%) del producto deseado en forma de un sól¡do de color blanco. CL-EM m/z 1455,09 [M+H+], 1453,77 [M-H+]; t¡empo de retenc¡ón = 1,80 m¡nutos.
Etapa 6: Síntes¡s de N-2~-(met¡lsulfon¡l)l¡s¡l-L-val¡l-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-¡l]-12-(2-{(2S)-2-[(1R,2R)-1-metox¡-2-met¡l-3-oxo-3-{[(1S)-2-fen¡l-1-(1,3-t¡azol-2-¡l)et¡l]am¡no}prop¡l]p¡rrol¡d¡n-1-¡l}-2-oxoet¡l)-5,5,10-tr¡met¡l-3,6,9-tr¡oxo-8-(propan-2-¡l)-2,13-d¡oxa-4,7,10-tr¡azatetradec-1-¡l]fen¡l}-N~5~-carbamo¡lorn¡t¡nam¡da, sal tr¡fluoroacét¡ca (70). A una soluc¡ón de N~6~-(terc-butox¡carbon¡l)-N~2~-(met¡lsulfon¡l)-D-l¡s¡l-L-val¡l-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-¡l]-12-(2-{(2S)-2-[(1R,2R)-1-metox¡-2-met¡l-3-oxo-3-{[(1s)-2-fen¡l-1-(1,3-t¡azol-2-¡l)et¡l]am¡no}prop¡l]p¡rrol¡d¡n-1-¡l}-2-oxoet¡l)-5,5,10-tr¡met¡l-3,6,9-tr¡oxo-8-(propan-2-¡l)-2,13-d¡oxa-4,7,10-tr¡azatetradec-1-¡l]fen¡l}-N~5~-carbamo¡lorn¡t¡nam¡da (69, 30 mg, 0,021 mmol) en 0,8 ml de d¡clorometano se le añad¡eron 0,2 ml de ác¡do tr¡fluoroacét¡co. La mezcla se controló por CL-e M y se dejó en ag¡tac¡ón durante 30 m¡nutos a temperatura amb¡ente. Después, la soluc¡ón se purificó d¡rectamente por HPLC de fase ¡nversa (Procedimiento B). Las fracc¡ones que contenían el producto se l¡of¡l¡zaron para proporc¡onar, después de la l¡of¡l¡zac¡ón, 15 mg (48 %). CL-EM m/z 1355,4 [M+H+], 1353,3 [M-H+]; t¡empo de retenc¡ón = 1,42 m¡nutos.
Ejemplo 45: Preparación de N~2~-(fenilsulfonil)lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]- 12-(2-{(2S)-2-[(lR,2R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-feml-1-(1,3-tiazol-2-il)etil]ammo}propil]pirrolidm-1 -il}-2-oxoetil)-5,5,10-trimetN-3,6,9-trioxo-8-(propan-2-N)-2,13-dioxa-4,7,10-triazatetradec-1 -il]fenil}-N~5~-carbamoilornitinamida, sal trifluoroacética (75)

Etapa 1: Síntesis de N~6~-(ferc-butoxicarbonil)-N~2~-(fenilsulfonil)-L-lisina (71). A una solución enfriada con hielo de N~6~-(ferc-butoxicarbonil)-L-lisina (3,0 g, 12,2 mmol) en tetrahidrofurano:agua (1:1, 100 ml) se le añadió carbonato potásico (8,4 g, 61,0 mmol) seguido de la adición gota a gota de cloruro de bencenosulfonilo (1,86 ml, 14,6 mmol). La mezcla se agitó a temperatura ambiente durante 3 horas y después la mezcla se filtró. El filtrado se extrajo tres veces con 10:1 de acetato de etilo:alcohol isopropílico. Los extractos combinados se lavaron con salmuera, se secaron sobre sulfato sódico y se concentraron a presión reducida. Al residuo se le añadieron 100 ml de ferc-butil metil éter y la mezcla se agitó durante 30 minutos. El sólido se recogió por filtración y se purificó por HPLC de fase inversa. Las fracciones que contenían el producto se liofilizaron para proporcionar 538 mg (11 %) del producto deseado en forma de un sólido de color blanco. EM m/z 408,9 [M+Na+].1H RMN (400 MHz, DMSO-da) 8 = 7,76 (d, J = 7,5 Hz, 2H), 7,63 - 7,50 (m, 3H), 3,61 - 3,53 (m, 1H), 2,82 - 2,73 (m, 2H), 1,61 -1,40 (m, 2H), 1,35 (s, 9H), 1,28 - 1,06 (m, 4H).
Etapa 2: Síntesis de N~6~-(ferc-butoxicarbonil)-N~2~-(fenilsulfonil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]ornitinamida (72). A una solución enfriada con hielo de N~6~-(ferc-butoxicarbonil)-N~2~-(fenilsulfonil)-L-lisina (71, 1,2 g, 3,10 mmol) y L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (1, (1,30 g, 3,41 mmol) en 20 ml N,N-dimetilformamida se le añadieron W,A/-diisopropiletilamina (600 ul, 438 mg, 3,40 mmol), clorhidrato de W-(3-dimetilaminopropil)-W'-etilcarbodiimida (650 mg, 3,41 mmol) y 1-Hidroxibenzotriazol hidrato (460 mg, 3,41 mmol). La mezcla se dejó calentar a temperatura ambiente y se agitó durante 16 horas. La mezcla se añadió gota a gota a 150 ml de ferc-butil metil éter. El sólido se aisló por filtración y después se purificó por HPLC de fase inversa. Las fracciones que contenían el producto se liofilizaron para proporcionar 850 mg (37 %) del producto deseado en forma de un sólido de color blanco.
Etapa 3: Síntesis de N~6~-(ferc-butoxicarbonil)-N~2~-(fenilsulfonil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]ornitinamida (73). A una solución agitada de N~6~-(ferc-butoxicarbonil)-N~2~-(fenilsulfonil)-L-lisil-L-valil-N~5— carbamoil-N-[4-(hidroximetil)fenil]ornitinamida (72, 850 mg, 1,14 mmol) y p-nitrofenilo carbonato (853 mg, 2,85 mmol) en 15 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (501 ul, 365 mg, 2,85 mmol). La mezcla se agitó a temperatura ambiente durante 16 horas. La mezcla se añadió gota a gota a 150 ml de ferc-butil metil éter. El sólido se purificó por HPLC de fase inversa. Las fracciones que contenían el producto se liofilizaron para proporcionar 450 mg (43 %) del producto deseado en forma de un sólido de color blanco. EM m/z 935,1 [M+Na+]; 1H RMN (400 MHz, DMSO-d6) 8 = 10,11 (s, 1H), 8,35 -8,27 (m, 2H), 8,10 (d, J = 7,0 Hz, 1H), 7,94 (d, J = 8,5 Hz, 1H), 7,83 (d, J = 8,5 Hz, 1H), 7,76 (d, J = 7,5 Hz, 2H), 7,63 (d, J = 8,5 Hz, 2H), 7,60 - 7,48 (m, 5H), 7,40 (t, J = 5,8 Hz, 2H), 6,70 (s, J = 5,6, 5,6 Hz, 1H), 6,01 - 5,93 (m, 1H), 5,43 (s, 2H), 4,36 (d, J = 6,0 Hz, 1H), 4,01 (t, J = 7,5 Hz, 1H), 3,78 (d, J = 5,0 Hz, 1H), 3,01 (d, J = 6,0 Hz, 1H), 2,97 - 2,88 (m, 1H), 2,76 (c, J = 6,5 Hz, 2H), 1,86 (d, J = 6,5 Hz, 1H), 1,44 (d, J = 5,5 Hz, 4H), 1,41 - 1,40 (m, 1H), 1,36 (s, 9H), 1,31 - 1,13 (m, 3H), 1,05 (s a, 1H), 0,75 (d, J = 7,0 Hz, 3H), 0,73 (d, J = 7,0 Hz, 3H).
Etapa 4: Síntesis de N~6~-(terc-butoxicarbonil)-N~2~-(fenilsulfonil)lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,l3-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (74). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida (4 , 52,0 mg, 0,070 mmol) y N~6~-(terc-butoxicarbonil)-N~2~-(fenilsulfonil)-L-lisil-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]ornitinamida (73 , 60,4 mg, 0,066 mmol) en 2 ml de N,N-dimetilacetamida se le añadieron 2,6-lutidina (31 ul, 28,3 mg, 0,264 mmol), N,N-diisopropiletilamina (46 ul, 34,1 mg, 0,264 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (9 mg, 0,066 mmol). La mezcla se dejó en agitación durante 16 horas a 45 °C. Después, la reacción se purificó por HPLC de fase inversa (Procedimiento S) para proporcionar, después de la liofilización, 85 mg (85 %) del producto deseado en forma de un sólido de color blanco. CL-EM m/z 1518,4 [M+H+], 1516,3 [M-H+]; tiempo de retención = 1,93 minutos.
Etapa 5: Síntesis de N~2~-(fenilsulfonil)lisil-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida, sal trifluoroacética (75). A una solución de N~6~-(terc-butoxicarbonil)-N~2~-(fenilsulfonil)lisil-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (75, 82 mg, 0,054 mmol) en 0,8 ml de diclorometano se le añadieron 0,2 ml de ácido trifluoroacético. La mezcla se controló por CL-EM y se dejó en agitación durante 30 minutos a temperatura ambiente. Después, la solución se purificó directamente por HPLC de fase inversa (Procedimiento B). Las fracciones que contenían el producto se liofilizaron para proporcionar, después de la liofilización, 35 mg (42 %). CL-EM m/z 1416,87 [M+H+]; tiempo de retención = 1,35 minutos.
Ejemplo de referencia 46: Preparación de N-acetil-O-(2-ammoetil)-D-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-U(1S)-2-femM-(1,3-tiazol-2-il)etil]ammo}propil]pirrolidm-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilomitinamida, sal del ácido trifluoroacético (83)

Etapa 1: Síntesis de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil] amino}etil)-L-serinato de metilo (78). A una suspensión de (2-hidroxietil)carbamato de 9H-fluoren-9-ilmetilo (1000 mg, 3,53 mmol) y (2S)-1-acetilaziridin-2-carboxilato de metilo (documento US 20110021482, 758 mg, 5,29 mmol) en 20 ml de diclorometano a -30 °C se le añadió gota a gota dieterato de trifluoruro de boro (532 ul, 601 mg, 4,24 mmol). La mezcla se agitó a esa temperatura durante 2 horas y después se dejó calentar a temperatura ambiente. Después de otra hora, a la reacción se le añadió una solución saturada acuosa de bicarbonato sódico y se diluyó con diclorometano (80 ml). La fase orgánica se separó y se lavó con salmuera, se secó sobre sulfato sódico y se concentró a presión reducida. El residuo se purificó por HPLC de fase inversa (Procedimiento W). Las fracciones apropiadas se combinaron y los disolventes orgánicos se retiraron a presión reducida. La mezcla acuosa restante se extrajo con acetato de etilo (2 x 100 ml). Los extractos combinados se lavaron con salmuera, se secaron sobre sulfato sódico y se concentraron. El residuo se co-evaporó
con diclorometano (50 ml) once para dar el compuesto del título 160 mg (11%) en forma de una goma de color amarillo claro. EM m/z 448,9 [M+Na+]; 1H RMN (400 MHz, CLOROFORMO-d) 8 = 7,78 (d, J = 7,5 Hz, 2H), 7,60 (d, J = 7,5 Hz, 2H), 7,42 (t, J = 7,5 Hz, 2H), 7,37 - 7,31 (m, 2H), 6,66 (s a, 1H), 5,07 (s a, 1H), 4,89 - 4,72 (m, 1H), 4,45 (d, J = 6,5 Hz, 2H), 4,23 (d, J = 6,5 Hz, 1H), 3,88 (d, J = 6,5 Hz, 1H), 3,72 (s a, 4H), 3,54 (s a, 2H), 3,36 (s a, 2H), 2,09 (s, 3H).
Etapa 2: Síntesis de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)-L-serina (79). A una solución de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)-L-serinato de metilo (78, 160 mg, 0,375 mmol) en isopropanol:agua (7:3, 10 ml) se le añadió cloruro de calcio (833 g, 7,5 mmol). Cuando la mezcla se volvió transparente, se añadió hidróxido sódico (18 mg, 0,450 mmol). La mezcla resultante se agitó a temperatura ambiente durante 3 horas. Después, la mezcla se acidificó a pH~1-2 con una solución acuosa 3 M de ácido clorhídrico y se diluyó con agua (10 ml). La mezcla se extrajo con acetato de etilo (20 ml) y la capa orgánica se aisló y se concentró a presión reducida para retirar la mayor parte del isopropanol. La fase acuosa se extrajo con acetato de etilo (2 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato sódico y se concentraron para proporcionar 107 mg (96%) el compuesto deseado en forma de un sólido de color azul, que se usará tal cual en la siguiente etapa sin purificación adicional. EM m/z 434,8 [M+Na+]; 1H RMN (400 MHz, CLOROFORMO-d) 8 = 8,48 (d, J = 9,0 Hz, 1H), 7,78 (d, J = 7,5 Hz, 2H), 7,56 (d, J = 7,5 Hz, 2H), 7,47 -7,37 (m, 2H), 7,35 - 7,29 (m, 2H), 7,15 (t, J = 6,1 Hz, 1H), 5,03 (d, J = 9,0 Hz, 1H), 4,51 (dd, J = 6,8, 10,8 Hz, 1H), 4,41 (dd, J = 7,0, 10,5 Hz, 2H), 4,30 - 4,23 (m, 1H), 4,32 - 4,21 (m, 1H), 4,21 - 4,09 (m, 1H), 3,57 (t, J = 8,3 Hz, 3H), 3,53 - 3,45 (m, 2H), 3,40 (s a, 1H), 3,31 (d, J = 9,0 Hz, 1H), 2,00 (s, 3H).
Etapa 3: Síntesis de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)-L-serilvalil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (80). A una solución de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]aminoletil)-L-serina (79, 105 mg, 0,255 mmol) y L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (1, 96,6 mg, 0,255 mmol) en 3 ml de N,N-dimetilformamida se le añadió hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (106 mg, 0,280 mmol) seguido de N,N-diisopropiletilamina (98,7 mg, 0,764 mmol). La mezcla de color amarillo transparente se agitó a temperatura ambiente durante 2 horas. La mezcla se diluyó con terc-butil metil éter (60 ml) y se agitó durante 30 minutos. El sólido se aisló por filtración y después se purificó por HPLC de fase inversa (Procedimiento X). Las fracciones que contenían el producto se liofilizaron para proporcionar 84 mg (43 %) del producto deseado en forma de un sólido de color blanco. EM m/z 774,2 [M+H+].
Etapa 4: Síntesis de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil) serilvalil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (81). A una solución agitada de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)-L-serilvalil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (80, 200 mg, 0,258 mmol) y carbonato de p-nitrofenilo (853 mg, 2,85 mmol) en 5 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (100,8 ul, 73,5 mg, 0,569 mmol). La mezcla se agitó a temperatura ambiente durante 3 horas y después se añadió más cantidad de carbonato de p-nitrofenilo (78,6 mg, 0,258 mmol). Después de una hora más, la mezcla se añadió gota a gota a 100 ml de terc-butil metil éter. El sólido se aisló por filtración y se lavó con algo de tercbutil metil éter para proporcionar, después del secado al aire, 180 mg (74%) del producto deseado en forma de un sólido de color amarillo. EM m/z 939,2 [M+H+] 961,1 [M+Na+].
Etapa 5: Síntesis de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)-D-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoilornitinamida (82 ). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida (4 , 60,0 mg, 0,081 mmol) y N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)serilvalil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (81, 121 mg, 0,188 mmol) en 2,5 ml de N,N-dimetilacetamida se le añadieron 2,6-lutidina (37,4 ul, 34,6 mg, 0,323 mmol), N,N-diisopropiletilamina (42,2 ul, 31,3 mg, 0,242 mmol) y 3H-[1,2,3]triazolo[4,5-b] piridin-3-ol (11,0 mg, 0,081 mmol). La mezcla se dejó en agitación durante 6 horas a 45 °C. Después, la reacción se purificó por HPLC de fase inversa (Procedimiento G) para proporcionar, después de la liofilización, 62 mg (50%) del producto deseado en forma de un sólido de color blanco. CL-Em m/z 1543,3 [M+H+]; tiempo de retención = 2,25 minutos. tiempo de retención de HPLC analítica = 7,435 minutos. Etapa 6: Síntesis de N-acetil-O-(2-aminoetil)-D-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida, sal del ácido trifluoroacético (83) A una solución de N-acetil-O-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etil)-D-seril-L-valil-N-{4-[(8S,11S, 12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (82, 42 mg, 0,027 mmol) en 2,5 ml de N,N-dimetilformamida se añadió una solución acuosa de hidróxido de litio (3,26 mg, 0,136 mmol) y la mezcla se agitó a temperatura ambiente durante una hora. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento l). Las fracciones que contenían el producto se liofilizaron para proporcionar 29 mg (74 %) del producto deseado. CL-EM m/z 1321,2 [M+H+]; tiempo de retención = 1,30 minutos. tiempo de retención de HPLC analítica = 5,804 minutos.
Ejemplo 47: N-acetil-O-[2-(2-aminoetoxi)etil]-L-seril-L-valil-N-£4-[(8S,11S,12R)-11-[(2S)-butan-2-il]- 12-(2-{(2S)-2 - [( lR ,2 R)-1 -metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)5,5,10-trimetN-3,6,9-trioxo-8-(propan-2-N)-2,13-dioxa-4,7,10-triazatetradec-1-N]feml}-N~5— carbamoilornitinamida, sal del ácido trifluoroacético (90)

Etapa 1: Síntesis de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-serinato de metilo (85). A una solución de [2-(2-hidroxietoxi)etil]carbamato de 9H-fluoren-9-ilmetilo (84, 7,55 g, 23,1 mmol) y (2S)-1-acetilaziridin-2-carboxilato de metilo (758 mg, 5,29 mmol) en 130 ml de diclorometano se le añadió gota a gota dietileterato de trifluoruro de boro (3,94 ml, 4,46 g, 31,4 mmol). Después de una hora, a la reacción se le añadió una solución saturada acuosa de bicarbonato sódico. La fase orgánica se separó y se lavó con salmuera, se secó sobre sulfato sódico y se concentró a presión reducida. El residuo se purificó por HPLC de fase inversa (Procedimiento Y). Las fracciones apropiadas se combinaron y los disolventes orgánicos se retiraron a presión reducida. La mezcla acuosa restante se extrajo con diclorometano (2 x 100 ml). Los extractos combinados se lavaron con salmuera, se secaron sobre sulfato sódico y se concentraron a presión reducida para dar el compuesto del título 3,8 g (26%) en forma de un aceite incoloro. EM m/z 493,0 [M+Na+]; 1H RMN (400 MHz, CLOROFORMO-d) 5 = 7,77 (d, J = 7,5 Hz, 2H), 7,62 (d, J = 7,0 Hz, 2H), 7,44 - 7,37 (m, 2H), 7,35 - 7,29 (m, 2H), 6,69 (d, J = 6,5 Hz, 1H), 5,49 (s a, 1H), 4,78 (d, J = 8,0 Hz, 1H), 4,42 (d, J = 6,5 Hz, 2H), 4,25 (d, J = 7,0 Hz, 1H), 3,95 (d, J = 10,0 Hz, 1H), 3,75 (s, 3H), 3,66 - 3,50 (m, 6H), 3,41 (s a, 2H), 2,04 (s, 3H).
Etapa 2: Síntesis de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino} etoxi)etil]-L-serina (86). A una solución de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-serinato de metilo (85, 3200 mg, 6,8 mmol) en
90 ml de isopropanol y 40 ml de agua se le añadió cloruro de calcio (15,1 g, 136 mmol). Cuando la mezcla se volvió transparente, se añadió hidróxido sódico (326 mg, 8,16 mmol). Después de 2 horas, la mezcla se acidificó a pH~1-2 con una solución acuosa 3 M de ácido clorhídrico y se diluyó con agua (300 ml). La mezcla se extrajo con acetato de etilo (3 x 250 ml) y la capa orgánica se aisló y se concentró a presión reducida para proporcionar 3000 mg (97%) del compuesto deseado en forma de un sólido de color blanco, que se usó tal cual en la siguiente etapa sin purificación adicional. EM m/z 456,9 [M+H+]; 1H RMN (400 MHz, CLOROFORMO-d) 8 = 7,77 (d, J = 7,5 Hz, 2H), 7,63 - 7,53 (m, 2H), 7,44 -7,38 (m, 2H), 7,36 -7,30 (m, 2H), 6,65 (d, J = 6,0 Hz, 1H), 4,77 (s a, 1H), 4,51 (s a, 1H), 4,43 (d, J = 6,5 Hz, 1H), 4,26 (d, J = 6,0 Hz, 1H), 3,95 (s a, 1H), 3,76 (s a, 1H), 3,70 - 3,57 (m, 3H), 3,54 (d, J = 7,5 Hz, 2H), 3,41 (d, J = 13,1 Hz, 2H), 3,29 (s a, 1H), 2,05 (s a, 3H).
Etapa 3: Síntesis de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino} etoxi)etil]-L-serilvalil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (87). A una solución de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-serina (86 , 3000 mg, 6,57 mmol) en 45 ml de N,N-dimetilformamida se le añadió hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU, 3000 mg, 7,89 mmol). Después de 20 minutos, se añadió L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (1, 2490 mg, 6,57 mmol) seguido de N,N-diisopropiletilamina (2550 mg, 19,7 mmol). Después de 3 horas, la mezcla se vertió en una mezcla de tercbutil metil éter (500 ml) y éter de petróleo (200 ml) y se agitó durante 20 minutos. El sólido se aisló por filtración y después se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 1% al 20% en diclorometano para proporcionar 3100 mg (58%) del producto deseado en forma de un sólido de color blanco. EM m/z 818,1 [M+H+].
Etapa 4: Síntesis de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-serilvalil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (88). A una solución agitada y enfriada con hielo de N-acetil-0 - [2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-serilvalil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (87, 500 mg, 0,611 mmol) y carbonato de p-nitrofenilo (279 mg, 0,917 mmol) en 6 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (216,7 ul, 158 mg, 1,22 mmol). La mezcla se agitó a temperatura ambiente durante 5 horas y después se añadió más cantidad de carbonato de p-nitrofenilo (100 mg, 0,538 mmol). Después de 12 horas más, la mezcla se vertió en una mezcla de terc-butil metil éter (60 ml) y éter de petróleo (20 ml) y se agitó durante 15 minutos. El sólido se aisló por filtración y después se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 1% al 10% en diclorometano para proporcionar 300 mg (50%) del producto deseado en forma de un sólido de color blanco. EM m/z 983,5 [M+H+].
Etapa 5: Síntesis de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-seril-L-valil-N-{4-[(8S,11S, 12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1- il]fenil}-N~5~-carbamoilornitinamida (89). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-ox o-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxo heptan-4-il]-N-metil-L-valinamida (4, 60,0 mg, 0,081 mmol) y N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-serilvalil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (88 , 75,4 mg, 0,077 mmol) en 2,5 ml de N,N-dimetilacetamida se le añadieron 2,6-lutidina (37,4 ul, 34,6 mg, 0,323 mmol), N,N-diisopropiletilamina (42,2 ul, 31,3 mg, 0,242 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (HOAt, 11,0 mg, 0,081 mmol). La mezcla se dejó en agitación durante 16 horas a 45 °C. Después, la reacción se purificó por HPLC de fase inversa (Procedimiento G) para proporcionar, después de la liofilización, 76 mg (58 %) del producto deseado en forma de un sólido de color blanco. CL-EM m/z 1587,3 [M+H+]; tiempo de retención = 2,26 minutos. tiempo de retención de HPLC analítica = 7,459 minutos.
Etapa 6: Síntesis de N-acetil-O-[2-(2-aminoetoxi)etil]-L-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~- carbamoilornitinamida, sal del ácido trifluoroacético (90) A una solución de N-acetil-O-[2-(2-{[(9H-fluoren-9-ilmetoxi)carbonil]amino}etoxi)etil]-L-seril-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[( 1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1 -il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (89, 40 mg, 0,025 mmol) en 2,5 ml de N,N-dimetilformamida se le añadió una solución acuosa de hidróxido de litio (3,02 mg, 0,126 mmol) y la mezcla se agitó a temperatura ambiente durante 30 minutos. Después, la solución se purificó por HPLC de fase inversa (Procedimiento D). Las fracciones que contenían el producto se liofilizaron para proporcionar 27 mg (72 %) del producto deseado. CL-EM m/z 1365,2 [M+H+]; tiempo de retención = 1,26 minutos. tiempo de retención de HPLC analítica = 5,768 minutos.
Ejemplo 48: Preparación de N~2~-(3-hidroxipropanoN)-L-NsNvaMl-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-M]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoil-L-ornitinamida, sal del ácido trifluoroacético (97)

Etapa 1: Síntesis de N-acetil-O-[1-(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21-il]-L-serinato de metilo (92). A una solución de (l7-hidroxi-3,6,9,12,15-pentaoxaheptadec-1-il)carbamato de 9H-fluoren-9-ilmetilo (91, 7600 mg, 15,09 mmol) y (2S)-1-acetilaziridin-2-carboxilato de metilo (4320 mg, 30,2 mmol) en 150 ml de diclorometano se le añadió gota a gota dietileterato de trifluoruro de boro (2,84 ml, 3210 mg, 22,6 mmol). Después de 2,5 horas, la reacción se lavó con una solución saturada acuosa de bicarbonato sódico y después con salmuera, se secó sobre sulfato sódico y se concentró a presión reducida. El residuo se purificó por HPLC de fase inversa (Procedimiento Z). Las fracciones apropiadas se combinaron y los disolventes orgánicos se retiraron a presión reducida. La mezcla acuosa restante se extrajo con acetato de etilo (2 x 100 ml). Los extractos combinados se secaron sobre sulfato sódico y se concentraron para dar el compuesto del título 5 g (51%) en forma de un aceite. EM m/z 647,1 [M+H+]; 1H RMN (400 MHz, CLOROFORMO-d) 5 = 7,76 (d, J = 7,5 Hz, 2H), 7,61 (d, J = 7,5 Hz, 2H), 7,40 (t, J = 7,5 Hz, 2H), 7,31 (t, J = 7,4 Hz, 2H), 6,72 (d, J = 7,5 Hz, 1H), 5,47 (s a, 1H), 4,73 (td, J = 3,5, 8,0 Hz, 1H), 4,41 (d, J = 7,0 Hz, 2H), 4,26 - 4,19 (m, 1H), 3,94 (dd, J = 3,5, 10,0 Hz, 1H), 3,75 (s, 3H), 3,70 (dd, J = 3,5, 10,0 Hz, 1H), 3,67 - 3,56 (m, 22H), 3,40 (c, J = 5,2 Hz, 2H), 2,07 (s, 3H).
Etapa 2: Síntesis de N-acetil-O-[1-(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21-il]-L-serina (93). A una solución de N-acetil-O-[1-(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21-il]-L-serinato de metilo (92, 4200 mg, 6,49 mmol) en isopropanol:agua (7:3, 170 ml) se le añadió cloruro de calcio (14,4 g, 130 mmol). Cuando la mezcla se volvió transparente, se añadió hidróxido sódico (312 mg, 7,79 mmol). La mezcla
resultante se agitó a temperatura ambiente durante 3 horas. Después, la mezcla se acidificó a pH~1-2 con una solución acuosa 3 M de ácido clorhídrico y se diluyó con agua (100 ml). La mezcla se extrajo con acetato de etilo (100 ml) y la capa orgánica se aisló y se concentró a presión reducida para retirar la mayor parte del isopropanol. La fase acuosa se extrajo con acetato de etilo (2 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato sódico y se concentraron a presión reducida. Después, el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 0% al 15% en diclorometano para proporcionar 3,4 g (83%) el compuesto deseado en forma de un aceite de color amarillo. EM m/z 633,1 [M+H+].
Etapa 3: Síntesis de N-acetil-O-[1-(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21-il]-L-serilvalil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (94). A una solución enfriada con hielo de N-acetil-O-[1-(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21-il]-L-serina (93, 1,20 g, 1,90 mmol) y L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (1, 720 mg, 1,90 mmol) en 15 ml de N,N-dimetilformamida se le añadió hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (794 mg, 2,09 mmol) seguido de N,N diisopropiletilamina (736 mg, 5,69 mmol). La mezcla de color amarillo transparente se agitó a temperatura ambiente durante 2 horas. La mezcla se diluyó con acetato de etilo (3 x 30 ml), se lavó con agua (60 ml) y se secó sobre sulfato sódico y se concentró a presión reducida. Después, el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 0% al 15% en diclorometano para proporcionar 1,5 g (80%) del producto deseado en forma de un vidrio incoloro. EM m/z 994,8 [M+H+].
Etapa 4: Síntesis de N-acetil-O-[2-(2-aminoetoxi)etil]-L-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5.5.10- trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (95). En un baño de hielo, a una solución agitada de N-acetil-O-[1-(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21-il]-L-serilvalil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida (94, 1,5 g, 1,5 mmol) y pnitrofenilo carbonato (918 mg, 3,02 mmol) en 15 ml de N,N-dimetilformamida se le añadió N,N-diisopropiletilamina (402 ul, 293 mg, 2,26 mmol). La mezcla se agitó a temperatura ambiente durante 16 horas y después se añadió más cantidad de carbonato de p-nitrofenilo (918 mg, 3,02 mmol). Después de 3 horas más, la mezcla se diluyó con acetato de etilo (30 ml) y se lavó con agua (40 ml). La fase acuosa se extrajo con acetato de etilo (30 ml). Las capas orgánicas combinadas se concentraron a presión reducida, después se purificaron por cromatografía sobre gel de sílice usando un gradiente de elución de metanol del 0% al 15% en diclorometano y después se purificaron por HPLC de fase inversa (Procedimiento AA) para proporcionar, después de la liofilización, 944 mg (54%) del producto deseado en forma de un sólido de color blanco. EM m/z 1159,8 [M+H+].
Etapa 5: Síntesis de N-acetil-O-[1-(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21-il]-L-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)- 2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (96). A una solución de 2-metilalanil-N-[(3R,4S,5S)-3-metoxi-1-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-5-metil-1-oxoheptan-4-il]-N-metil-L-valinamida (4, 55,0 mg, 0,074 mmol) y N-acetil-O-[2-(2-aminoetoxi)etil]-L-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino} propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (95,
85,8 mg, 0,074 mmol) en 2,5 ml de N,N-dimetilacetamida se le añadieron 2,6-lutidina (34,3 ul, 31,7 mg, 0,296 mmol), N, N-diisopropiletilamina (38,7 ul, 28,7 mg, 0,222 mmol) y 3H-[1,2,3]triazolo[4,5-b]piridin-3-ol (10,1 mg, 0,074 mmol). La mezcla se dejó en agitación durante 6 horas a 45 °C. Después, la reacción se purificó por HPLC de fase inversa (Procedimiento G) para proporcionar, después de la liofilización, 90 mg (69%) del producto deseado en forma de un sólido de color blanco. CL-Em m/z 1764,4 [M+H+]; tiempo de retención = 1,99 minutos. tiempo de retención de HPLC analítica = 7,476 minutos.
Etapa 6: Síntesis de N-acetil-O-(17-amino-3,6,9,12,15-pentaoxaheptadec-1-il)-L-seril-L-valil-N-{4-[(8S,11S,12R)-11-[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5,5,10-trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida, sal del ácido trifluoroacético (97) A una solución de N-acetil-O-[1 -(9H-fluoren-9-il)-3-oxo-2,7,10,13,16,19-hexaoxa-4-azahenicosan-21 -il]-L-seril-L-valil-N-{4-[(8S,11S,12R)-11 -[(2S)-butan-2-il]-12-(2-{(2S)-2-[(1R,2R)-1-metoxi-2-metil-3-oxo-3-{[(1S)-2-fenil-1-(1,3-tiazol-2-il)etil]amino}propil]pirrolidin-1-il}-2-oxoetil)-5.5.10- trimetil-3,6,9-trioxo-8-(propan-2-il)-2,13-dioxa-4,7,10-triazatetradec-1-il]fenil}-N~5~-carbamoilornitinamida (96, 46 mg, 0,026 mmol) en 2,5 ml de N,N-dimetilformamida se le añadió una solución acuosa de hidróxido de litio (3,1 mg, O, 130 mmol) y la mezcla se agitó a temperatura ambiente durante 20 minutos. Después, la solución se concentró y se purificó por HPLC de fase inversa (Procedimiento D y después Procedimiento H). Las fracciones que contenían el producto se liofilizaron para proporcionar 15 mg (35 %) del producto deseado. CL-EM m/z 1542,2 [M+H+]; tiempo de retención = 1,41 minutos. tiempo de retención de HPLC analítica = 5,836 minutos. Los siguientes ADC adicionales (unidos en varias posiciones y en varias etiquetas en el anticuerpo Trop2, como se indica en las Tablas 2 a 7) se fabricaron usando la técnica de conjugación descrita anteriormente (la porción entre corchetes indica un residuo de glutamina, y las líneas onduladas muestran puntos de unión al resto del anticuerpo Anti-Trop2):
Ejemplo de referencia 49:


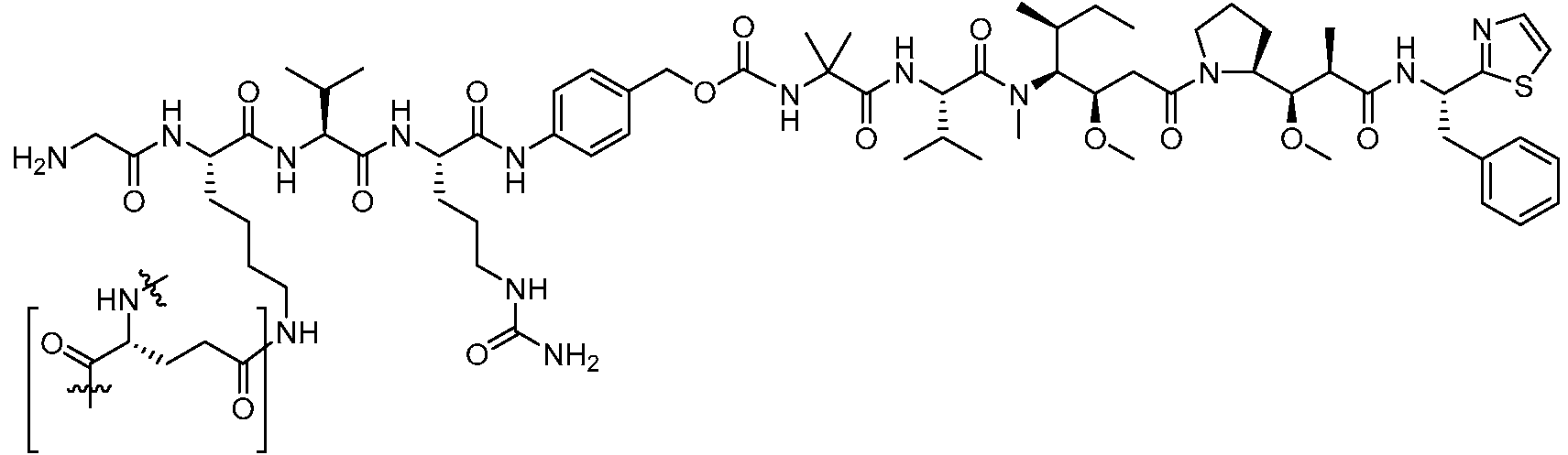
Ejemplo 58:

Ejemplo 59:

Ejemplo 60:

Ejemplo 61:
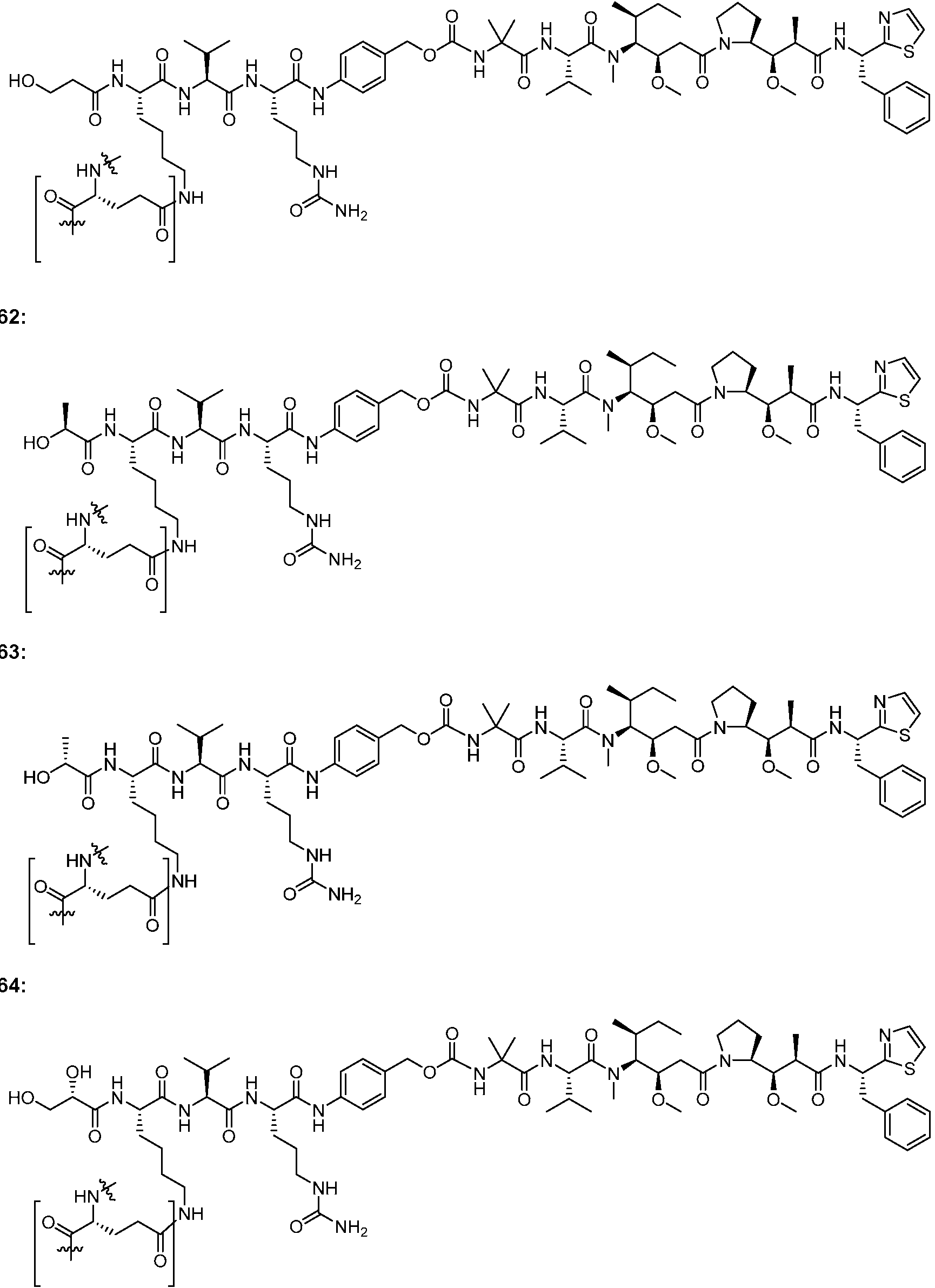

Ejemplo 66:

Ejemplo 67:

Ejemplo 68 :

Ejemplo de referencia 69:

Ejemplo 72 Confirmación de la carboxilesterasa 1C de ratón como la enzima responsable de la escisión del enlazador VC-PABC en el plasma
Se usaron muestras de plasma de la cepa de ratón defectiva C56/BL6 CeslC -/-, la cepa heterocigota C56/BL6 CeslC /- y la cepa de tipo salvaje C56/BL6 Ces1C /+ para evaluar la estabilidad del conjugado anti-Trop2 L11B-C6-VC-PABC-Aur0101. Se incubaron 56 |jg de CAF en el plasma en plasma al 35 % suplementado con 1x PBS hasta la concentración final de CAF de 0,125 mg/ml. Después de la incubación durante 18 horas, los CAF se aislaron usando antígeno M1S1 acoplado a Sepharose activada por CNBr (GE Healthcare) usando protocolos estándar. Los valores DAR se evaluaron antes y después de la incubación mediante análisis HIC. La estabilidad del conjugado (%) se calcula
como % de DAR restante después de la incubación. Véase la figura 3.
DATOS BIOLÓGICOS
Los datos de estabilidad generados en los procedimientos descritos en los Ejemplos 8 a 18 se resumen en las Tablas 2 a 7.
Tabla 2 - Estabilidad del conjugado in vivo en ratón

Estabilidad del conjugado in vivo en el ratón. Los valores de DAR para los conjugados purificados de animales a los que se administraron dosis de 9 mg/kg se compararon con los valores de DAR antes de la exposición in vivo. Los valores de estabilidad anteriores se expresan como el % de DAR restante después del período de exposición dado (mostrado entre paréntesis).
Tabla 3 - Estabilidad del conjugado in vivo en ratas

Estabilidad del conjugado in vivo en rata. Los valores de DAR para los conjugados purificados de animales a los que se administraron dosis de 9 mg/kg se compararon con los valores de DAR antes de la exposición in vivo. Los valores de estabilidad anteriores se expresan como el % de DAR restante después del período de exposición dado (mostrado entre paréntesis).
Tabla 4 - Estabilidad del conjugado in vitro en plasma de ratón

(continuación)

Estabilidad del conjugado in vitro en plasma de ratón. Los valores de DAR para conjugados purificados después de la incubación en plasma se compararon con los valores de DAR antes del tratamiento con plasma. Los valores de estabilidad anteriores se expresan como el % de DAR restante después del período de incubación dado (mostrado entre paréntesis).
Tabla 5 - Estabilidad del conjugado in vitro en plasma de rata

Estabilidad del conjugado in vitro en plasma de rata. Los valores de DAR para conjugados purificados después de la incubación en plasma se compararon con los valores de DAR antes del tratamiento con plasma. Los valores de estabilidad anteriores se expresan como el % de DAR restante después del período de incubación dado (mostrado entre paréntesis).
Tabla 6 - Estabilidad del conjugado in vitro en plasma de cyno

Estabilidad del conjugado in vitro en plasma de cyno. Los valores de DAR para conjugados purificados después de la incubación en plasma se compararon con los valores de DAR antes del tratamiento con plasma. Los valores de estabilidad anteriores se expresan como el % de DAR restante después del período de incubación dado (mostrado entre paréntesis).
Tabla 7A - Valores ex erimentales de DAR con el % de estabilidad calculada

Tabla 7B - Valores de DAR ex erimentales adicionales con el % de estabilidad calculada

Claims (16)
1. Un compuesto de Fórmula (I):

o una sal o solvato farmacéuticamente aceptable del mismo;
en la que:
M es un modulador de la estabilidad en el que M es -M1-M2;
en el que M1 es -NR1-C(O)-, -NR1-S(O)2-, o está ausente, y
M2 se selecciona entre el grupo que consiste en:

P es una secuencia de péptidos que incluye uno o más residuos de glutamina;
Q es un residuo de glutamina presente en P;
cada E se selecciona independientemente entre el grupo que consiste en:-C(R1)2-, -O-C(R1)2-C(R1)2- en el que cuando E es -O-C(R1)2-C(R1)2 r es al menos 2, y -C(R1)2-C(R1)2-O- en el que cuando E es -c (r 1)2-c (r 1)2-O- s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H y alquilo C1-C6 lineal o ramificado; en la que -Xm-Yn- es -X-Y- y en la que X-Y se selecciona entre el grupo que consiste en Gly, p-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg, Phe-N9-Nitro-Arg, Val-Ala y Ala-Ala-Asn;
cada Z es PABC (p-aminobencil-carbamoílo);
p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y
D es un agente citotóxico.
2. Un compuesto de Fórmula (II):

o una sal o solvato farmacéuticamente aceptable del mismo;
en la que:
M es un modulador de la estabilidad en el que M es -M1-M2;
en el que M1 es -NR1-C(O)-, -NR1-S(O)2-, o está ausente, y
M2 se selecciona entre el grupo que consiste en:


P es una secuencia de péptidos que incluye uno o más residuos de glutamina;
Q es uno de dichos residuos de glutamina presentes en P;
cada E se selecciona independientemente entre el grupo que consiste en: -C(R1)2-, -O-C(R1)2-C(R1)2- en el que cuando E es -O-C(R1)2-C(R1)2 r es al menos 2, y -C(R1)2-C(R1)2-O- en el que cuando E es -C(R1)2-C(R1)2-O- s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H y alquilo C1-C6 lineal o ramificado; en la que -Xm-Yn- es -X-Y- y en la que X-Y se selecciona entre el grupo que consiste en Gly, p-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg, Phe-N9-Nitro-Arg, Val-Ala y Ala-Ala-Asn;
cada Z es PABC (p-aminobencil-carbamoílo);
p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y
D es un agente citotóxico.
3. Un compuesto de Fórmula (III):

o una sal o solvato farmacéuticamente aceptable del mismo;
en la que:
M es un modulador de la estabilidad en el que M es -M1-M2;
en el que M1 es -NR1-C(O)-, -NR1-S(O)2-, o está ausente, y
M2 se selecciona entre el grupo que consiste en:

cada E se selecciona independientemente entre el grupo que consiste en: -C(R1)2-, -O-C(R1)2-C(R1)2- en el que cuando E es -O-C(R1)2-C(R1)2 r es al menos 2, y -C(R1)2-C(R1)2-O- en el que cuando E es -C(R1)2-C(R1)2-O- s es al menos 1;
cada R1 se selecciona independientemente entre el grupo que consiste en: H y alquilo C1-C6 lineal o ramificado; en la que -Xm-Yn- es -X-Y- y en la que X-Y se selecciona entre el grupo que consiste en Gly, p-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-tosil-Arg, Phe-N9-Nitro-Arg, Val-Ala y Ala-Ala-Asn;
cada Z es PABC (p-aminobencil-carbamoílo);
p es 0-2, q es 0-10, r es 0-2 y s es 0-2, en el que q+r+s=2 o más; y
D es un agente citotóxico.
4. El compuesto de cualquiera de las reivindicaciones 1-3, en el que el agente citotóxico D se selecciona entre el grupo que consiste en: una antraciclina, una auristatina, una spliceostatina, un dímero de CBI/CPI, una calicheamicina, una duocarmicina, un enodiino, una geldanamicina, una maitansina, una puromicina, un taxano, un alcaloide de la vinca, SN-38, una tubulisina, una hemiasterlina, una camptotecina, una combretastatina, una dolastatina, un dímero de indolino-benzodiazepina, un dímero de pirrolobenzodiazepina y una pladienolida.
5. El compuesto de cualquiera de las reivindicaciones 1-4, en el que el agente citotóxico D es una auristatina seleccionada entre grupo que consiste en dolestatina, MMAD, MMAE, MMAF, PF-06380101, PF-06463377 y PF-06456780.
6. El compuesto de la reivindicación 1 o la reivindicación 2, en el que P es un péptido que comprende una secuencia de aminoácidos seleccionada entre el grupo que consiste en: Q, Lq G, LLQg G (SEQ ID NO: 1), LLQG (SEQ ID NO: 2), LSLSQG (SEQ ID NO: 3), GGGLLQGG (SEQ ID NO: 4), GLLQG (SEQ ID NO: 5), LLQ, GSPLAQSHGG (SEQ ID NO: 6), GLLQGGG (SEQ ID NO: 7), GLLQGG (SEQ ID NO: 8), GLLQ (SEQ ID NO: 9), LLQLLQGA (SEQ ID NO: 10), LLQGA (SEQ ID NO: 11), LLQYQGA (SEQ ID NO: 12), LLQGSG (SEQ ID NO: 13), LLQYQG (SEQ ID NO: 14), LLQLLQG (SEQ ID NO: 15), SLLQG (SEQ ID NO: 16), LLQLQ (SEQ ID NO: 17), LLQLLQ (SEQ ID NO: 18), LLQGR (SEQ ID NO: 19), LLQGPP (SEQ ID NO: 20), LLQGPA (SEQ ID NO: 21), GGLLQGPP (SEQ ID NO: 22), GGLLQGA (SEQ ID NO: 23), LLQGA (SEQ ID NO: 24), LLQGPGK (SEQ ID NO: 25), LLQGPG (SEQ ID NO: 26), LLQGP (SEQ ID NO: 27), LLQP (SEQ ID NO: 28), LLQPGK (SEQ ID NO: 29), LLQAPGK (SEQ ID NO: 30), LLQGAPG (SEQ ID NO: 31), LLQGAP (SEQ ID NO: 32), LLQGPA (SEQ ID NO: 33), LLQGPP (SEQ ID NO: 34), GGLLQGPP (SEQ ID NO: 35), LLQLQG (SEQ ID NO: 36) y XX-QX (SEQ ID NO: 37).
7. Un compuesto de la reivindicación 3 seleccionado entre el grupo que consiste en:

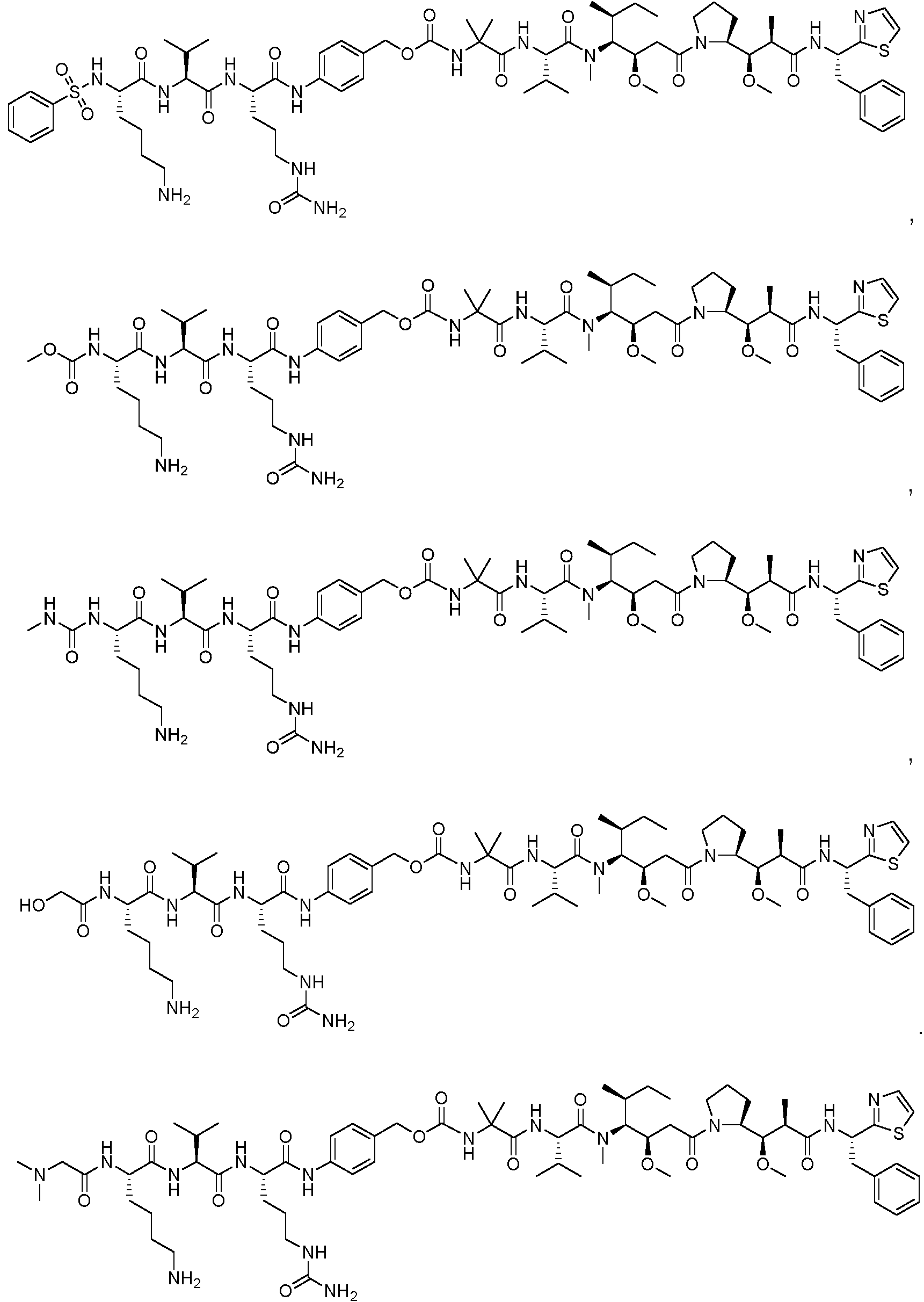


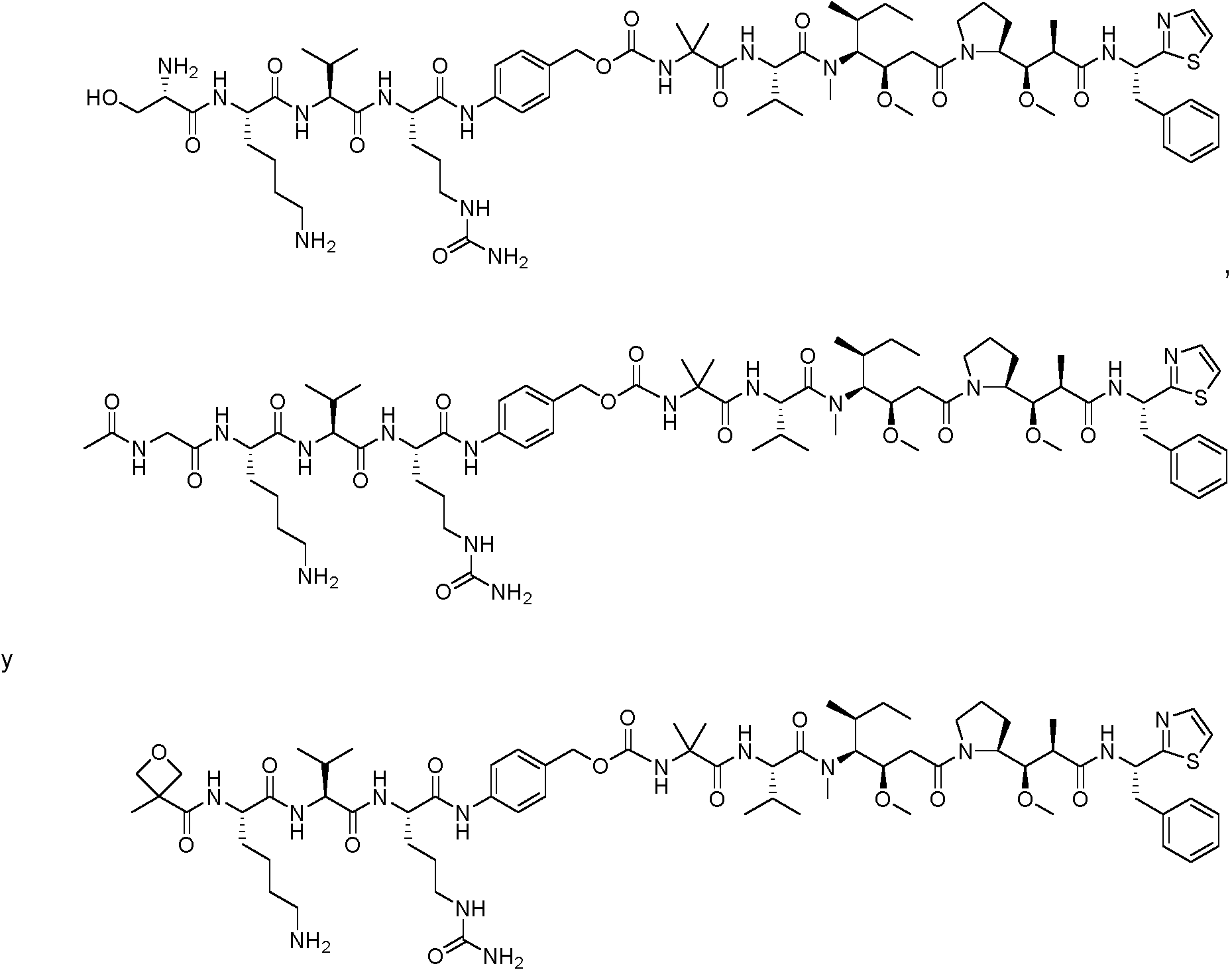
o una sal o solvato farmacéuticamente aceptable del mismo.
8. Un compuesto de una cualquiera de las reivindicaciones 1 o 2 seleccionado entre el grupo que consiste en:





o una sal o solvato farmacéuticamente aceptable del mismo, en el que la parte entre corchetes del compuesto representa un residuo de glutamina con una secuencia de péptidos.
9. El compuesto de la reivindicación 1 o la reivindicación 2, en el que P se selecciona entre el grupo que consiste en: un anticuerpo monoclonal, un anticuerpo policlonal, un anticuerpo humano, un anticuerpo humanizado, un anticuerpo quimérico, un anticuerpo biespecífico, un minicuerpo y un fragmento de anticuerpo.
10. El compuesto de la reivindicación 9, en el que dicho anticuerpo se selecciona entre el grupo que consiste en: trastuzumab, mutantes de trastuzumab, oregovomab, edrecolomab, cetuximab, un anticuerpo monoclonal humanizado para el receptor de vitronectina (avp3), alemtuzumab, anticuerpos anti-HLA-DR, 1311 Lym-1, anticuerpos anti-HLA-Dr10, anticuerpo anti-cd33, anticuerpo anti-cd22, labetuzumab, bevacizumab, ibritumomab tiuxetan, ofatumumab, panitumumab, rituximab, tositumomab, ipilimumab y gemtuzumab.
11. El compuesto de la reivindicación 1 o la reivindicación 2, en el que cada Q es independientemente un residuo de una glutamina endógena para dicha secuencia de péptidos P, o un residuo de una glutamina proporcionado en una secuencia etiqueta diseñada en dicha secuencia de péptidos P.
12. El compuesto de la reivindicación 1 o la reivindicación 2, en el que uno o más Q es un residuo de una glutamina endógena a dicha secuencia de péptidos P.
13. El compuesto de la reivindicación 1 o la reivindicación 2, en el que uno o más Q es un residuo de una glutamina proporcionada en una secuencia etiqueta diseñadas en dicha secuencia de péptidos P.
14. El compuesto de la reivindicación 13, en el que una o más etiquetas es el único aminoácido glutamina.
15. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1-14, o una sal o solvato farmacéuticamente aceptable del mismo, y un excipiente o vehículo farmacéuticamente aceptable.
16. Un compuesto de una cualquiera de las reivindicaciones 1-14, o una sal o solvato farmacéuticamente aceptable del mismo, o una composición farmacéutica de acuerdo con la reivindicación 15 para su uso en el tratamiento de cáncer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462042901P | 2014-08-28 | 2014-08-28 | |
| PCT/IB2015/056211 WO2016030791A1 (en) | 2014-08-28 | 2015-08-14 | Stability-modulating linkers for use with antibody drug conjugates |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2797754T3 true ES2797754T3 (es) | 2020-12-03 |
Family
ID=54266589
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15777735T Active ES2797754T3 (es) | 2014-08-28 | 2015-08-14 | Enlazadores moduladores de la estabilidad para su uso con conjugados de fármaco y anticuerpo |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US10967068B2 (es) |
| EP (1) | EP3185908B1 (es) |
| JP (2) | JP6832621B2 (es) |
| KR (1) | KR102051503B1 (es) |
| CN (1) | CN106795203B (es) |
| AU (1) | AU2015308179B2 (es) |
| CA (1) | CA2901684C (es) |
| DK (1) | DK3185908T3 (es) |
| ES (1) | ES2797754T3 (es) |
| HU (1) | HUE049325T2 (es) |
| IL (1) | IL250319B (es) |
| MX (2) | MX384379B (es) |
| PL (1) | PL3185908T3 (es) |
| PT (1) | PT3185908T (es) |
| RU (1) | RU2680238C2 (es) |
| SI (1) | SI3185908T1 (es) |
| WO (1) | WO2016030791A1 (es) |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11566082B2 (en) | 2014-11-17 | 2023-01-31 | Cytiva Bioprocess R&D Ab | Mutated immunoglobulin-binding polypeptides |
| WO2017147542A2 (en) | 2016-02-26 | 2017-08-31 | Regeneron Pharmaceuticals, Inc. | Optimized transglutaminase site-specific antibody conjugation |
| US10703774B2 (en) | 2016-09-30 | 2020-07-07 | Ge Healthcare Bioprocess R&D Ab | Separation method |
| US10889615B2 (en) | 2016-05-11 | 2021-01-12 | Cytiva Bioprocess R&D Ab | Mutated immunoglobulin-binding polypeptides |
| US10654887B2 (en) | 2016-05-11 | 2020-05-19 | Ge Healthcare Bio-Process R&D Ab | Separation matrix |
| US11708390B2 (en) | 2016-05-11 | 2023-07-25 | Cytiva Bioprocess R&D Ab | Method of storing a separation matrix |
| US10730908B2 (en) | 2016-05-11 | 2020-08-04 | Ge Healthcare Bioprocess R&D Ab | Separation method |
| JP7031934B2 (ja) | 2016-05-11 | 2022-03-08 | サイティバ・バイオプロセス・アールアンドディ・アクチボラグ | 分離マトリックス |
| JP6987424B2 (ja) | 2016-05-11 | 2022-01-05 | サイティバ・バイオプロセス・アールアンドディ・アクチボラグ | 分離マトリックスを洗浄および/または消毒する方法 |
| CN109071613A (zh) | 2016-05-11 | 2018-12-21 | 通用电气医疗集团生物工艺研发股份公司 | 分离基质 |
| WO2018025168A1 (en) * | 2016-08-03 | 2018-02-08 | Pfizer Inc. | Heteroaryl sulfone-based conjugation handles, methods for their preparation, and their use in synthesizing antibody drug conjugates |
| US12448411B2 (en) | 2016-09-30 | 2025-10-21 | Cytiva Bioprocess R&D Ab | Separation method |
| MX2019008773A (es) | 2017-01-24 | 2019-09-18 | Pfizer | Derivados de caliqueamicina y conjugados anticuerpo-farmaco de los mismos. |
| CN118652192A (zh) | 2017-05-24 | 2024-09-17 | 德克萨斯州大学系统董事会 | 用于抗体药物缀合物的接头 |
| WO2019057772A1 (en) | 2017-09-19 | 2019-03-28 | Paul Scherrer Institut | METHOD FOR CONJUGATING TRANSGLUTAMINASE AND BINDING SEQUENCE |
| CA3082271A1 (en) * | 2017-11-14 | 2019-05-23 | Debiopharm Research & Manufacturing S.A. | Ligand-drug-conjugates as substrates for selective cleavage by the exopeptidase activity of cathepsin b |
| JOP20200268A1 (ar) | 2018-06-01 | 2020-10-26 | Eisai R&D Man Co Ltd | تضفير مترافقات عقار جسم مضاد معدل وطرق استخدامها |
| CN110468175A (zh) * | 2019-08-22 | 2019-11-19 | 赵秋萍 | 一种复合植物肽粉及其制备方法和用途 |
| WO2021234593A1 (en) * | 2020-05-20 | 2021-11-25 | Janssen Biotech, Inc. | Methods for site specific conjugation of proteins containing glycosylated fc domains |
| CA3183689A1 (en) * | 2020-05-20 | 2021-11-25 | Janssen Biotech, Inc. | Site-specific conjugation of glycosylated monoclonal antibodies with transglutaminase |
| US20230331867A1 (en) | 2020-09-04 | 2023-10-19 | Novarock Biotherapeutics, Ltd. | Nectin-4 antibodies and uses thereof |
| CN116472064A (zh) * | 2020-10-12 | 2023-07-21 | 昆山新蕴达生物科技有限公司 | 抗体-药物偶联物及其应用 |
| CA3196218A1 (en) | 2020-10-25 | 2022-04-28 | Philipp SPYCHER | Means and methods for producing antibody-linker conjugates |
| AU2022286137A1 (en) | 2021-06-01 | 2023-12-14 | Ajinomoto Co., Inc. | Conjugate of antibody and functional substance or salt of said conjugate, and compound for use in production of said conjugate or salt of said compound |
| CN118159298A (zh) * | 2021-10-25 | 2024-06-07 | 阿拉里斯生物技术股份公司 | 生产抗体-接头偶联物的方法 |
| WO2025026338A1 (zh) * | 2023-07-31 | 2025-02-06 | 石药集团巨石生物制药有限公司 | 具有高药物负载的抗体-药物缀合物及其用途 |
| WO2026020031A2 (en) | 2024-07-18 | 2026-01-22 | Novarock Biotherapeutics, Ltd. | Cdh17 antibodies and uses thereof |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1245217A (en) * | 1981-12-10 | 1988-11-22 | Joshua S. Boger | Renin inhibitory peptides having phe su13 xx deletion |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| IL85035A0 (en) | 1987-01-08 | 1988-06-30 | Int Genetic Eng | Polynucleotide molecule,a chimeric antibody with specificity for human b cell surface antigen,a process for the preparation and methods utilizing the same |
| EP1400536A1 (en) | 1991-06-14 | 2004-03-24 | Genentech Inc. | Method for making humanized antibodies |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| US5663149A (en) | 1994-12-13 | 1997-09-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide heterocyclic and halophenyl amides |
| WO1998052976A1 (en) | 1997-05-21 | 1998-11-26 | Biovation Limited | Method for the production of non-immunogenic proteins |
| WO2000034317A2 (en) | 1998-12-08 | 2000-06-15 | Biovation Limited | Method for reducing immunogenicity of proteins |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| EP1718667B1 (en) | 2004-02-23 | 2013-01-09 | Genentech, Inc. | Heterocyclic self-immolative linkers and conjugates |
| RU2402548C2 (ru) * | 2004-05-19 | 2010-10-27 | Медарекс, Инк. | Химические линкеры и их конъюгаты |
| CN101837130B (zh) | 2009-03-20 | 2013-10-23 | 北京大学 | 聚乙二醇-二肽-抗肿瘤药物复合物及其用途 |
| US8829033B2 (en) | 2009-09-23 | 2014-09-09 | The University Of North Carolina At Chapel Hill | N-benzylamide substituted derivatives of 2-(acylamido)acetic acid and 2-(acylamido)propionic acids: potent neurological agents |
| US9676871B2 (en) | 2010-11-05 | 2017-06-13 | Pfizer Inc. | Engineered polypeptide conjugates and methods for making thereof using transglutaminase |
| AU2012335205A1 (en) | 2011-11-11 | 2014-05-29 | Rinat Neuroscience Corp. | Antibodies specific for Trop-2 and their uses |
| MY168297A (en) | 2011-11-17 | 2018-10-23 | Pfizer | Cytotoxic Peptides and Antibody Drug Conjugates Thereof |
| RU2015129800A (ru) * | 2012-12-21 | 2017-01-30 | Биоэллаенс К. В. | Гидрофильные саморазрушающиеся линкеры и их конъюгаты |
| EP3027224B1 (en) | 2013-07-31 | 2020-06-03 | Rinat Neuroscience Corp. | Engineered polypeptide conjugates using transglutaminase |
| PE20160671A1 (es) * | 2013-08-02 | 2016-07-09 | Pfizer | Anticuerpos anti-cxcr4 y conjugados de anticuerpo y farmaco |
| CN106459055B (zh) | 2014-01-27 | 2019-11-01 | 辉瑞大药厂 | 双功能细胞毒类药剂 |
| SI3134127T1 (sl) * | 2014-04-25 | 2020-06-30 | Rinat Neuroscience Corp. | Konjugati zdravil s protitelesi z visoko stopnjo zdravila |
| KR102451080B1 (ko) * | 2014-06-12 | 2022-10-06 | 씨에스피씨 메가리스 바이오파마슈티칼 씨오., 엘티디. | 효소적 방법을 통한 균일한 항체 약물 접합체 |
-
2015
- 2015-08-14 ES ES15777735T patent/ES2797754T3/es active Active
- 2015-08-14 CN CN201580053977.2A patent/CN106795203B/zh active Active
- 2015-08-14 US US15/504,801 patent/US10967068B2/en active Active
- 2015-08-14 PL PL15777735T patent/PL3185908T3/pl unknown
- 2015-08-14 WO PCT/IB2015/056211 patent/WO2016030791A1/en not_active Ceased
- 2015-08-14 HU HUE15777735A patent/HUE049325T2/hu unknown
- 2015-08-14 AU AU2015308179A patent/AU2015308179B2/en active Active
- 2015-08-14 RU RU2017104511A patent/RU2680238C2/ru active
- 2015-08-14 MX MX2017002600A patent/MX384379B/es unknown
- 2015-08-14 PT PT157777350T patent/PT3185908T/pt unknown
- 2015-08-14 DK DK15777735.0T patent/DK3185908T3/da active
- 2015-08-14 EP EP15777735.0A patent/EP3185908B1/en active Active
- 2015-08-14 SI SI201531244T patent/SI3185908T1/sl unknown
- 2015-08-14 KR KR1020177005202A patent/KR102051503B1/ko active Active
- 2015-08-25 CA CA2901684A patent/CA2901684C/en active Active
- 2015-08-26 JP JP2015166643A patent/JP6832621B2/ja active Active
-
2017
- 2017-01-26 IL IL250319A patent/IL250319B/en active IP Right Grant
- 2017-02-27 MX MX2021008334A patent/MX2021008334A/es unknown
-
2021
- 2021-02-02 JP JP2021014690A patent/JP2021073274A/ja active Pending
- 2021-02-23 US US17/183,265 patent/US20210205463A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| RU2017104511A3 (es) | 2018-09-28 |
| CA2901684A1 (en) | 2016-02-28 |
| US20210205463A1 (en) | 2021-07-08 |
| RU2680238C2 (ru) | 2019-02-18 |
| JP2016050204A (ja) | 2016-04-11 |
| CA2901684C (en) | 2020-06-09 |
| JP6832621B2 (ja) | 2021-02-24 |
| KR102051503B1 (ko) | 2019-12-03 |
| WO2016030791A1 (en) | 2016-03-03 |
| CN106795203B (zh) | 2021-01-26 |
| PT3185908T (pt) | 2020-06-18 |
| PL3185908T3 (pl) | 2020-09-07 |
| MX384379B (es) | 2025-03-14 |
| RU2017104511A (ru) | 2018-09-28 |
| DK3185908T3 (da) | 2020-06-15 |
| MX2017002600A (es) | 2017-05-19 |
| IL250319A0 (en) | 2017-03-30 |
| HUE049325T2 (hu) | 2020-09-28 |
| IL250319B (en) | 2020-04-30 |
| BR112017002912A2 (pt) | 2018-01-30 |
| SI3185908T1 (sl) | 2020-08-31 |
| EP3185908A1 (en) | 2017-07-05 |
| US20180140714A1 (en) | 2018-05-24 |
| US10967068B2 (en) | 2021-04-06 |
| KR20170036048A (ko) | 2017-03-31 |
| EP3185908B1 (en) | 2020-04-15 |
| CN106795203A (zh) | 2017-05-31 |
| AU2015308179A1 (en) | 2017-02-02 |
| MX2021008334A (es) | 2021-08-05 |
| AU2015308179B2 (en) | 2020-07-02 |
| JP2021073274A (ja) | 2021-05-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2797754T3 (es) | Enlazadores moduladores de la estabilidad para su uso con conjugados de fármaco y anticuerpo | |
| ES2783376T3 (es) | Conjugados de anticuerpo-fármaco con alta carga de fármaco | |
| ES2804614T3 (es) | Conjugados de polipéptidos manipulados usando transglutaminasa | |
| JP7390315B2 (ja) | スプライシング調節薬抗体-薬物コンジュゲート及びその使用方法 | |
| KR102345175B1 (ko) | 결합 링커, 그러한 결합 링커를 함유하는 세포 결합 분자-약물 결합체, 링커를 갖는 그러한 결합체의 제조 및 사용 | |
| CN113195487A (zh) | 2,3-二氨基琥珀酰基偶联物连接子 | |
| EP3856258B1 (en) | Sulfomaleimide-based linkers and corresponding conjugates | |
| KR20170139110A (ko) | 칼리케아마이신 구성체 및 사용 방법 | |
| BR112017002912B1 (pt) | Compostos ligantes de modulação de estabilidade, composição farmacêutica e uso dos mesmos | |
| HK1237350A1 (en) | Stability-modulating linkers for use with antibody drug conjugates | |
| HK1237350B (zh) | 用於抗体药物缀合物的稳定性调节接头 | |
| BR112016024370B1 (pt) | Conjugados de anticorpo-fármaco e composição farmacêutica que os compreende |