ES2819374T3 - Métodos de síntesis del hidrocloruro de (1R,2R,5R)-5-amino-2-metilciclohexanol e intermedios útiles en los mismos - Google Patents
Métodos de síntesis del hidrocloruro de (1R,2R,5R)-5-amino-2-metilciclohexanol e intermedios útiles en los mismos Download PDFInfo
- Publication number
- ES2819374T3 ES2819374T3 ES16831114T ES16831114T ES2819374T3 ES 2819374 T3 ES2819374 T3 ES 2819374T3 ES 16831114 T ES16831114 T ES 16831114T ES 16831114 T ES16831114 T ES 16831114T ES 2819374 T3 ES2819374 T3 ES 2819374T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- solvent
- contacting
- base
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000000543 intermediate Substances 0.000 title description 13
- HKWIETGWMFZMPJ-RYLOHDEPSA-N (1r,2r,5r)-5-amino-2-methylcyclohexan-1-ol;hydrochloride Chemical compound Cl.C[C@@H]1CC[C@@H](N)C[C@H]1O HKWIETGWMFZMPJ-RYLOHDEPSA-N 0.000 title description 8
- 238000001308 synthesis method Methods 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 100
- 238000000034 method Methods 0.000 claims abstract description 47
- 239000002904 solvent Substances 0.000 claims abstract description 39
- 239000002585 base Substances 0.000 claims abstract description 24
- 239000000243 solution Substances 0.000 claims abstract description 24
- 239000000203 mixture Substances 0.000 claims abstract description 17
- 239000002841 Lewis acid Substances 0.000 claims abstract description 16
- 150000007517 lewis acids Chemical class 0.000 claims abstract description 16
- 239000003960 organic solvent Substances 0.000 claims abstract description 15
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims abstract description 12
- DXDIHODZARUBLA-IODNYQNNSA-N (2s,3s)-2,3-dibenzoyloxybutanedioic acid;hydrate Chemical compound O.O([C@H](C(=O)O)[C@H](OC(=O)C=1C=CC=CC=1)C(O)=O)C(=O)C1=CC=CC=C1 DXDIHODZARUBLA-IODNYQNNSA-N 0.000 claims abstract description 8
- 239000012320 chlorinating reagent Substances 0.000 claims abstract description 8
- 239000007800 oxidant agent Substances 0.000 claims abstract description 8
- 230000001590 oxidative effect Effects 0.000 claims abstract description 8
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 7
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims abstract description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims abstract description 5
- 239000012458 free base Substances 0.000 claims abstract description 5
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical class CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 claims abstract description 4
- 239000007864 aqueous solution Substances 0.000 claims abstract description 4
- 229910019093 NaOCl Inorganic materials 0.000 claims abstract description 3
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 claims abstract description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims abstract 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 45
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 9
- GRWFGVWFFZKLTI-IUCAKERBSA-N (-)-α-pinene Chemical compound CC1=CC[C@@H]2C(C)(C)[C@H]1C2 GRWFGVWFFZKLTI-IUCAKERBSA-N 0.000 claims description 5
- GRWFGVWFFZKLTI-UHFFFAOYSA-N rac-alpha-Pinene Natural products CC1=CCC2C(C)(C)C1C2 GRWFGVWFFZKLTI-UHFFFAOYSA-N 0.000 claims description 5
- 239000012279 sodium borohydride Substances 0.000 claims description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 claims description 4
- MVNCAPSFBDBCGF-UHFFFAOYSA-N alpha-pinene Natural products CC1=CCC23C1CC2C3(C)C MVNCAPSFBDBCGF-UHFFFAOYSA-N 0.000 claims description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 45
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 37
- 208000035475 disorder Diseases 0.000 description 25
- 238000004128 high performance liquid chromatography Methods 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 17
- 238000003756 stirring Methods 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- 239000000047 product Substances 0.000 description 13
- 201000010099 disease Diseases 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 11
- 108010055717 JNK Mitogen-Activated Protein Kinases Proteins 0.000 description 10
- 102000019145 JUN kinase activity proteins Human genes 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 230000003176 fibrotic effect Effects 0.000 description 10
- 239000011541 reaction mixture Substances 0.000 description 10
- GJIKAJOLWXMKEG-ZETCQYMHSA-N (1r)-4-methylcyclohex-3-en-1-amine Chemical compound CC1=CC[C@H](N)CC1 GJIKAJOLWXMKEG-ZETCQYMHSA-N 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 210000004185 liver Anatomy 0.000 description 9
- 230000037361 pathway Effects 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 238000004821 distillation Methods 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 208000024891 symptom Diseases 0.000 description 7
- GJIKAJOLWXMKEG-UHFFFAOYSA-N 4-methylcyclohex-3-en-1-amine Chemical compound CC1=CCC(N)CC1 GJIKAJOLWXMKEG-UHFFFAOYSA-N 0.000 description 6
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 6
- 102000001253 Protein Kinase Human genes 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 108060006633 protein kinase Proteins 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 208000001145 Metabolic Syndrome Diseases 0.000 description 5
- 108091000080 Phosphotransferase Proteins 0.000 description 5
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 102000020233 phosphotransferase Human genes 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- CBZNOKAFOMSSLW-OPRDCNLKSA-N tert-butyl n-[(1r,3r,4r)-3-hydroxy-4-methylcyclohexyl]carbamate Chemical compound C[C@@H]1CC[C@@H](NC(=O)OC(C)(C)C)C[C@H]1O CBZNOKAFOMSSLW-OPRDCNLKSA-N 0.000 description 5
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- OYOQOLNBTPTFEM-UHFFFAOYSA-N 4-methylcyclohex-3-ene-1-carboxylic acid Chemical compound CC1=CCC(C(O)=O)CC1 OYOQOLNBTPTFEM-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 206010063209 Chronic allograft nephropathy Diseases 0.000 description 4
- 201000001200 Crouzon syndrome-acanthosis nigricans syndrome Diseases 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- 206010039710 Scleroderma Diseases 0.000 description 4
- 201000009594 Systemic Scleroderma Diseases 0.000 description 4
- 206010042953 Systemic sclerosis Diseases 0.000 description 4
- 206010052779 Transplant rejections Diseases 0.000 description 4
- 238000005356 chiral GC Methods 0.000 description 4
- 238000004296 chiral HPLC Methods 0.000 description 4
- 235000019439 ethyl acetate Nutrition 0.000 description 4
- 206010025135 lupus erythematosus Diseases 0.000 description 4
- 231100000240 steatosis hepatitis Toxicity 0.000 description 4
- QBBRJRLJWXRSHQ-CKYFFXLPSA-N 2-(tert-butylamino)-4-[[(1r,3r,4r)-3-hydroxy-4-methylcyclohexyl]amino]pyrimidine-5-carboxamide Chemical compound C1[C@@H](O)[C@H](C)CC[C@H]1NC1=NC(NC(C)(C)C)=NC=C1C(N)=O QBBRJRLJWXRSHQ-CKYFFXLPSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 101000950695 Homo sapiens Mitogen-activated protein kinase 8 Proteins 0.000 description 3
- 101000950669 Homo sapiens Mitogen-activated protein kinase 9 Proteins 0.000 description 3
- 208000029523 Interstitial Lung disease Diseases 0.000 description 3
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 3
- 108091054455 MAP kinase family Proteins 0.000 description 3
- 102000043136 MAP kinase family Human genes 0.000 description 3
- 102100037808 Mitogen-activated protein kinase 8 Human genes 0.000 description 3
- 102100037809 Mitogen-activated protein kinase 9 Human genes 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical group CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 208000019425 cirrhosis of liver Diseases 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- YOUXMVZSALAVJO-UHFFFAOYSA-N 4-methylcyclohex-3-ene-1-carboxamide Chemical compound CC1=CCC(C(N)=O)CC1 YOUXMVZSALAVJO-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- 229910015900 BF3 Inorganic materials 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- 208000004930 Fatty Liver Diseases 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- 206010019708 Hepatic steatosis Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical group [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 241000286209 Phasianidae Species 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 2
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 description 2
- 230000007882 cirrhosis Effects 0.000 description 2
- 208000010706 fatty liver disease Diseases 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 208000006454 hepatitis Diseases 0.000 description 2
- 231100000283 hepatitis Toxicity 0.000 description 2
- WQYVRQLZKVEZGA-UHFFFAOYSA-N hypochlorite Chemical compound Cl[O-] WQYVRQLZKVEZGA-UHFFFAOYSA-N 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 2
- 208000019423 liver disease Diseases 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 description 2
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical group ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000003909 protein kinase inhibitor Substances 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- 230000007863 steatosis Effects 0.000 description 2
- 238000004808 supercritical fluid chromatography Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- PCOYCFJCVTWMFV-JTQLQIEISA-N tert-butyl n-[(1r)-4-methylcyclohex-3-en-1-yl]carbamate Chemical compound CC1=CC[C@H](NC(=O)OC(C)(C)C)CC1 PCOYCFJCVTWMFV-JTQLQIEISA-N 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 229930006720 (-)-alpha-pinene Natural products 0.000 description 1
- YONLFQNRGZXBBF-ZIAGYGMSSA-N (2r,3r)-2,3-dibenzoyloxybutanedioic acid Chemical compound O([C@@H](C(=O)O)[C@@H](OC(=O)C=1C=CC=CC=1)C(O)=O)C(=O)C1=CC=CC=C1 YONLFQNRGZXBBF-ZIAGYGMSSA-N 0.000 description 1
- -1 (R) - (4-methylcyclohex-3-en-1-yl) tert-butyl Chemical group 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- QPQNOKYVGHJYNX-UHFFFAOYSA-N 1-(methylamino)cyclohexan-1-ol Chemical class CNC1(O)CCCCC1 QPQNOKYVGHJYNX-UHFFFAOYSA-N 0.000 description 1
- MVODTGURFNTEKX-UHFFFAOYSA-N 2-bromo-n-(2-bromoethyl)-n-(thiophen-2-ylmethyl)ethanamine;hydrobromide Chemical compound Br.BrCCN(CCBr)CC1=CC=CS1 MVODTGURFNTEKX-UHFFFAOYSA-N 0.000 description 1
- 102000051485 Bcl-2 family Human genes 0.000 description 1
- 108700038897 Bcl-2 family Proteins 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 208000008964 Chemical and Drug Induced Liver Injury Diseases 0.000 description 1
- 206010008609 Cholangitis sclerosing Diseases 0.000 description 1
- 206010072268 Drug-induced liver injury Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 206010019728 Hepatitis alcoholic Diseases 0.000 description 1
- 101000628949 Homo sapiens Mitogen-activated protein kinase 10 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102100025087 Insulin receptor substrate 1 Human genes 0.000 description 1
- 101710201824 Insulin receptor substrate 1 Proteins 0.000 description 1
- 206010023126 Jaundice Diseases 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102000007999 Nuclear Proteins Human genes 0.000 description 1
- 108010089610 Nuclear Proteins Proteins 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- WTARULDDTDQWMU-UHFFFAOYSA-N Pseudopinene Natural products C1C2C(C)(C)C1CCC2=C WTARULDDTDQWMU-UHFFFAOYSA-N 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 208000002353 alcoholic hepatitis Diseases 0.000 description 1
- 125000003447 alpha-pinene group Chemical group 0.000 description 1
- 239000012223 aqueous fraction Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 235000013330 chicken meat Nutrition 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- AKPUJVVHYUHGKY-UHFFFAOYSA-N hydron;propan-2-ol;chloride Chemical compound Cl.CC(C)O AKPUJVVHYUHGKY-UHFFFAOYSA-N 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000000865 phosphorylative effect Effects 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 201000000742 primary sclerosing cholangitis Diseases 0.000 description 1
- 230000009822 protein phosphorylation Effects 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 208000010157 sclerosing cholangitis Diseases 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/54—Preparation of compounds containing amino groups bound to a carbon skeleton by rearrangement reactions
- C07C209/58—Preparation of compounds containing amino groups bound to a carbon skeleton by rearrangement reactions from or via amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/82—Purification; Separation; Stabilisation; Use of additives
- C07C209/86—Separation
- C07C209/88—Separation of optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/33—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C211/39—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of an unsaturated carbon skeleton
- C07C211/40—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of an unsaturated carbon skeleton containing only non-condensed rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/04—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups from amines with formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/06—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups by reactions not involving the formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C67/347—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by addition to unsaturated carbon-to-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/16—Systems containing only non-condensed rings with a six-membered ring the ring being unsaturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Un método para preparar un compuesto de fórmula (A), **(Ver fórmula)** comprendiendo método comprende las etapas de: (a) poner en contacto un compuesto de fórmula (1), **(Ver fórmula)** con un compuesto de fórmula (2), **(Ver fórmula)** en presencia de un ácido de Lewis en un disolvente para proporcionar un compuesto de fórmula (3), **(Ver fórmula)** (b) poner en contacto el compuesto de fórmula (3) de la etapa (a) con una base acuosa para proporcionar un compuesto de fórmula (4), **(Ver fórmula)** (c) poner en contacto el compuesto de fórmula (4) de la etapa (b) con DMF y un agente clorante en un disolvente orgánico, seguido del tratamiento del derivado de cloruro de ácido resultante con amoniaco acuoso para proporcionar un compuesto de fórmula (5), **(Ver fórmula)** (d) poner en contacto el compuesto de fórmula (5) de la etapa (c) con una disolución acuosa de NaOH y NaOCl para proporcionar un compuesto de fórmula (6), **(Ver fórmula)** (e) poner en contacto el compuesto de fórmula (6) de la etapa (d) con monohidrato del ácido (+)-dibenzoil-D-tartárico en un disolvente para proporcionar un compuesto de fórmula (7), (f) poner en contacto el compuesto de fórmula (7) de la etapa (e) con una base acuosa, seguido del tratamiento de la base libre resultante con BoC2O en un disolvente orgánico para proporcionar un compuesto de fórmula (8), (g) poner en contacto el compuesto de fórmula (8) de la etapa (f) con una mezcla de un agente reductor, un auxiliar quiral y un ácido de Lewis en un disolvente, seguido de un tratamiento con un oxidante en presencia de una base para proporcionar un compuesto de fórmula (9), y (h) poner en contacto el compuesto de fórmula (9) de la etapa (g) con una disolución de ácido clorhídrico en un disolvente para proporcionar el compuesto de fórmula (A).
Description
DESCRIPCIÓN
Métodos de síntesis del hidrocloruro de (1 R,2R,5R)-5-amino-2-metilciclohexanol e intermedios útiles en los mismos
Esta solicitud reivindica el beneficio de prioridad de la Solicitud Provisional de los Estados Unidos con No de Serie 62/196.363, presentada el 24 de julio de 2015.
Campo
En la presente memoria se proporcionan métodos e intermedios para preparar el hidrocloruro de (1 R,2R,5R)-5-amino-2-metilciclohexanol, que son útiles para la preparación de compuestos útiles para el tratamiento de una enfermedad, trastorno o afección asociada con la ruta de JNK.
Antecedentes
La conexión entre la fosforilación anormal de proteínas y la causa o consecuencia de enfermedades se conoce desde hace más de 20 años. Por consiguiente, las proteínas quinasas se han convertido en un grupo muy importante de dianas farmacológicas. [Véase, Cohen, Nature, 1:309-315 (2002), Gaestel et al. Curr.Med.Chem.14: 2214-223 (2007); Grimminger et al. Nat. Rev. Drug Disc. 9(12):956-970 (2010)]. Se han utilizado diversos inhibidores de proteínas quinasas a nivel clínico en el tratamiento de una amplia variedad de enfermedades, tales como cáncer y enfermedades inflamatorias crónicas, incluyendo artritis reumatoide y psoriasis (Véase Cohen, Eur. J. Biochem., 268:5001-5010 (2001); Protein Kinase Inhibitors for the Treatment of Disease: The Promise and the Problems, Handbook of Experimental Pharmacology, Springer Berlin Heidelberg, 167 (2005)).
JNK es una serina/treonina quinasa expresada de forma ubicua perteneciente, junto con ERK (quinasa regulada extracelular) y p38, a la familia de las proteínas quinasas activadas por mitógenos (MAPK). (Kyriakis JM, Sci. STKE (48):pe1 (2000); Whitmarsh AJ, et al. Sci. STKE (1):pe1 (1999); Schramek H, News Physiol. Sci.17:62-7 (2002); Ichijo H, Oncogene 18(45):6087-93 (1999)). Las MAPK son mediadores importantes de la transducción de señales desde la superficie celular al núcleo, que usan cascadas de fosforilación para generar una respuesta coordinada de una célula a un estímulo externo mediante la fosforilación de proteínas intracelulares seleccionadas, incluyendo factores de transcripción. Además, JNK también fosforila proteínas no nucleares, por ejemplo, IRS-1 y miembros de la familia Bcl-2. (Davis RJ, Trends Biochem. Sci. 9(11):470-473 (1994); Seger R et al., FASEB J.; 9(9):726-35 (1995); Fanger GR et al., Curr. Opin. Genet. Dev.; 7(1):67-74 (1997)).
La elucidación de la complejidad de las rutas de las proteínas quinasas y la complejidad de la relación e interacción entre las diversas proteínas quinasas y las rutas de las quinasas resalta la importancia de desarrollar agentes farmacéuticos que puedan actuar como moduladores, reguladores o inhibidores de proteínas quinasas que tengan actividad beneficiosa sobre múltiples quinasas o múltiples rutas de quinasas. El compuesto denominado químicamente 2-(terf-butilamino)-4-((1 R,3R,4R)-3-hidroxi-4-metilciclohexilamino)-pirimidina-5-carboxamida (denominado alternativamente 2-[(1,1-dimetiletil)amino]-4-[[(1 R,3R,4R)-3-hidroxi-4-metilciclohexil]amino]-5-pirimidinacarboxamida; y referido en la presente memoria como "Compuesto I"), un inhibidor de la ruta de JNK, se describe en la Publicación de la Solicitud de Patente de EE. UU. No. 2013/0029987, publicada el 31 de enero de 2013, Pub. Internacional No. WO 2012/145569 y Solicitud de Patente de EE. UU. No. 14/608.314, presentada el 29 de enero de 2015. WO 2010/027500 describe la preparación de metilamino ciclohexanoles ópticamente resueltos. Por consiguiente, sigue existiendo la necesidad de nuevos procesos para la preparación del Compuesto I.
La mención o identificación de cualquier referencia en la Sección 2 de esta solicitud no debe interpretarse como una admisión de que la referencia sea la técnica anterior a la presente solicitud.
Resumen
En la presente memoria se proporcionan procesos e intermedios útiles para la preparación del Compuesto I:

que tiene el nombre 2-(terc-butilamino)-4-((1 R,3R,4R)-3-hidroxi-4-metilciclohexilamino)-pirimidin-5-carboxamida, que es útil para el tratamiento de una enfermedad, trastorno o afección asociada con la ruta de JNK.
En particular, en la presente memoria se proporcionan procesos para preparar un compuesto de fórmula (A):

que tiene el nombre hidrocloruro de (1R,2R,5R)-5-amino-2-metilciclohexanol.
Se proporciona un método para preparar un compuesto de fórmula (A),

comprende las etapas de:
(a) poner en contacto un compuesto de fórmula (1),

con un compuesto de fórmula (2),

en presencia de un ácido de Lewis en un disolvente para proporcionar un compuesto de fórmula (3),

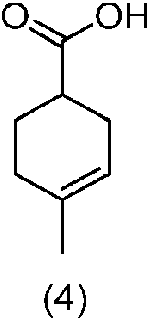
(b) poner en contacto el compuesto de fórmula (3) de la etapa (a) con una base acuosa para proporcionar un compuesto de fórmula (4),

(c) poner en contacto el compuesto de fórmula (4) de la etapa (b) con DMF y un agente clorante en un disolvente orgánico, seguido del tratamiento del derivado de cloruro de ácido resultante con amoniaco acuoso para proporcionar un compuesto de fórmula (5),

(d) poner en contacto el compuesto de fórmula (5) de la etapa (c) con una disolución acuosa de NaOH y NaOCI para proporcionar un compuesto de fórmula (6),

(e) poner en contacto el compuesto de fórmula (6) de la etapa (d) con monohidrato del ácido (+)-dibenzoil-D-tartárico en un disolvente para proporcionar un compuesto de fórmula (7),

(f) poner en contacto el compuesto de fórmula (7) de la etapa (e) con una base acuosa, seguido del tratamiento de la base libre resultante con Boc2O en un disolvente orgánico para proporcionar un compuesto de fórmula (8),

( 8 )
(g) poner en contacto el compuesto de fórmula (8) de la etapa (f) con una mezcla de un agente reductor, un auxiliar quiral y un ácido de Lewis en un disolvente, seguido de un tratamiento con un oxidante en presencia de una base para proporcionar un compuesto de fórmula (9),

y
(h) poner en contacto el compuesto de fórmula (9) de la etapa (g) con una disolución de ácido clorhídrico en un disolvente para proporcionar el compuesto de fórmula (A). En una realización, el ácido de Lewis de la etapa (a) es ALCl3. En una realización adicional, el disolvente de la etapa (a) es DCM. En una realización, la base de la etapa (b) es NaOH. En una realización, el agente clorante de la etapa (c) es SOCl2. En una realización adicional, el disolvente orgánico de la etapa (c) es DCM. En una realización, el disolvente de la etapa (e) es MeOH. En una realización, la base de la etapa (f) es NaOH. En una realización adicional, el disolvente orgánico de la etapa (f) es DCM. En una realización, el agente reductor de la etapa (g) es NaBH4. En una realización adicional, el auxiliar quiral de la etapa (g) es a-pineno. En realizaciones particulares, el ácido de Lewis de la etapa (g) es BF3^Et2O. En otra realización particular, el disolvente de la etapa (g) es THF. En todavía otra realización, el oxidante de la etapa (g) es H2O2. En una realización específica, la base de la etapa (g) es NaOH. En una realización, el disolvente de la etapa (h) es IPA.
En ciertos aspectos, el Compuesto I es útil para inhibir una quinasa en una célula que expresa dicha quinasa, por ejemplo, JNK1 o JNK2. En otros aspectos, el Compuesto I es útil para tratar o prevenir una afección tratable o prevenible mediante la inhibición de una ruta de JNK, como se describe en la presente memoria. En otro aspecto, el Compuesto I es útil para tratar o prevenir uno o más trastornos seleccionados entre fibrosis pulmonar intersticial, esclerosis sistémica, esclerodermia, nefropatía crónica por aloinjerto, rechazo mediado por anticuerpos o lupus. En otro aspecto más, el Compuesto I es útil para tratar o prevenir trastornos fibróticos hepáticos, o diabetes y/o síndrome metabólico que conduce a trastornos fibróticos hepáticos, como se describe en la presente memoria.
Las presentes realizaciones pueden comprenderse de manera más completa por referencia a la descripción detallada y los ejemplos, que pretenden ejemplificar realizaciones no limitativas.
Breve descripción de los dibujos
La FIG. 1 representa un espectro de1H RMN del Compuesto (A) en D2O.
La FIG. 2 representa un espectro expandido (~0,2-3,6 ppm) de 1H RMN del Compuesto (A) en D2O.
La FIG. 3 representa un cromatograma de HPLC del Compuesto (A).
La FIG. 4 representa un cromatograma de GC quiral del Compuesto (A).
Descripción detallada
Definiciones
Tal como se usa en la presente memoria, y en la memoria descriptiva y en las reivindicaciones adjuntas, los artículos indefinidos “un”, “una” y el artículo definido “el/la” incluyen referentes en plural, así como en singular, a no ser que el contexto indique claramente otra cosa.
Tal como se usa en la presente memoria, y a no ser que se especifique otra cosa, los términos "aproximadamente" y "alrededor de", cuando se usan en relación con cantidades o porcentajes en peso de ingredientes de un proceso, significan una cantidad o porcentaje en peso que es reconocido por un experto en la técnica para proporcionar un efecto equivalente al obtenido a partir de la cantidad especificada, o porcentaje en peso. En ciertas realizaciones, los términos "aproximadamente" y "alrededor de", cuando se usan en este contexto, contemplan una cantidad o porcentaje en peso dentro del 30 %, dentro del 20 %, dentro del 15 %, dentro del 10 %, o dentro del 5 %, de la cantidad o porcentaje en peso especificado.
"JNK" significa una proteína o una isoforma de la misma expresada por un gen JNK1, JNK2 o JNK3 (Gupta, S., Barrett, T., Whitmarsh, AJ, Cavanagh, J., Sluss, HK, Derijard, B. y Davis, RJ The EMBO J. 15:2760-2770 (1996)).
"Tratar", tal y como se usa en la presente memoria, significa un alivio, total o parcial, de un trastorno, enfermedad o afección, o uno o más de los síntomas asociados con un trastorno, enfermedad o afección, o ralentización o parada de una mayor progresión o empeoramiento de esos síntomas, o un alivio o erradicación de la o las causas del trastorno, enfermedad o afección en sí mismo. En una realización, el trastorno es una afección que se puede tratar o prevenir mediante la inhibición de una ruta de JNK, como se describe en la presente memoria. En otra realización, el trastorno se selecciona de fibrosis pulmonar intersticial, esclerosis sistémica, esclerodermia, nefropatía crónica por aloinjerto, rechazo mediado por anticuerpos o lupus. En otra realización más, el trastorno es un trastorno fibrótico hepático, o diabetes y/o síndrome metabólico que conduce a trastornos fibróticos hepáticos, como se describe en la presente memoria. En algunas realizaciones, el trastorno es un trastorno fibrótico hepático, tal como esteatohepatitis no alcohólica, esteatosis (es decir, hígado graso), cirrosis, colangitis esclerosante primaria, cirrosis biliar primaria, hepatitis, carcinoma hepatocelular o fibrosis hepática coincidente con la ingesta de alcohol crónica o repetida (hepatitis alcohólica), con infección (p. ej ., infección viral tal como VHC), con trasplante de hígado o con lesión hepática inducida por fármacos (p. ej ., toxicidad por acetaminofén). En algunas realizaciones, "tratar" significa un alivio, total o parcial, de un trastorno, enfermedad o afección, o síntomas asociados con la diabetes o el síndrome metabólico que conduce a trastornos fibróticos hepáticos, tales como esteatohepatitis no alcohólica, esteatosis (es decir, hígado graso), hepatitis o cirrosis, o una ralentización o parada de una mayor progresión o empeoramiento de esos síntomas. En una realización, el síntoma es ictericia.
"Prevenir", tal y como se usa la presente memoria, significa un método para retrasar y/o impedir el inicio, recurrencia o diseminación, total o parcial, de un trastorno, enfermedad o afección; impedir que un sujeto adquiera un trastorno, enfermedad o afección; o reducir el riesgo de un sujeto de adquirir un trastorno, enfermedad o afección. En una realización, el trastorno es una afección que se puede tratar o prevenir mediante la inhibición de una ruta de JNK, como se describe en la presente memoria. En otra realización, el trastorno se selecciona de fibrosis pulmonar intersticial, esclerosis sistémica, esclerodermia, nefropatía crónica por aloinjerto, rechazo mediado por anticuerpos o lupus. En una realización, el trastorno es un trastorno fibrótico hepático, o diabetes o síndrome metabólico que conduce a trastornos fibróticos hepáticos, como se describe en la presente memoria, o síntomas de los mismos.
"Paciente" o “sujeto” se define en la presente memoria para incluir animales, tales como mamíferos, que incluyen, pero no están limitados a, primates (p. ej., seres humanos), vacas, ovejas, cabras, caballos, perros, gatos, conejos, ratas, ratones, monos, pollos, pavos, perdices, o cobayas y similares, en una realización un mamífero, en otra realización un ser humano. En una realización, un sujeto es un ser humano que tiene o está en riesgo de tener fibrosis pulmonar intersticial, esclerosis sistémica, esclerodermia, nefropatía crónica por aloinjerto, rechazo mediado por anticuerpos o lupus. En otra, un sujeto es un ser humano que tiene o está en riesgo de tener trastornos fibróticos hepáticos o diabetes o síndrome metabólico que conduce a trastornos fibróticos hepáticos, o una afección, tratable o prevenible mediante la inhibición de una ruta de JNK, o un síntoma de los mismos.
Compuesto (A)
Como se describe en la Publicación de Solicitud de Patente de EE. UU. No. 2013/0029987, publicada el 31 de enero de 2013, Pub. Internacional No. WO 2012/145569 y Solicitud de Patente de EE. UU. No. 14/608.314, presentada el 29 de enero de 2015, los compuestos de fórmula I se pueden preparar como se muestra en el Esquema A.

Los procesos proporcionados en la presente memoria se refieren a métodos para preparar un compuesto de fórmula (A):

que tiene el nombre hidrocloruro de (1 R,2R,5R)-5-amino-2-metilciclohexanol, e intermedios útiles en dichos procesos.
Debe indicarse que, de haber una discrepancia entre una estructura representada y un nombre otorgado a esa estructura, prevalecerá la estructura representada. Además, si la estereoquímica de una estructura o porción de una estructura no se indica, por ejemplo, en negrita o con líneas punteadas, se interpretará que la estructura o porción de la estructura engloba todos los estereoisómeros de la misma.
Métodos para preparar el compuesto (A)
A modo de ejemplo y no de limitación, el compuesto de fórmula (A) se puede preparar como se describe en el Esquema 1 mostrado a continuación, así como en los ejemplos mostrados en la presente memoria.


NaBH+.Brj- E!í O
aifa pirene, HF fPA-HCI
nísOH HjD?


Esquema
En un aspecto, en la presente memoria se proporcionan métodos para preparar un compuesto de fórmula (A):

comprendiendo los métodos poner en contacto un compuesto de fórmula (9):

con ácido clorhídrico en un disolvente.
En algunas realizaciones, el disolvente es metanol, 2-propanol (IPA), éter o dioxano. En una realización, el disolvente es 2-propanol (IPA).
En algunas realizaciones, los métodos comprenden además preparar un compuesto de fórmula (9):

comprendiendo los métodos poner en contacto un compuesto de fórmula (8):

con una mezcla de un agente reductor, un auxiliar quiral y un ácido de Lewis en un disolvente, seguido de tratamiento con un oxidante en presencia de una base.
En una realización, el agente reductor es NaBH4. En otra realización, el auxiliar quiral es a-pineno. En otra realización, el ácido de Lewis es BF3^Et2O. En una realización, el disolvente es THF o EtOH. En otra realización, el disolvente es THF. En una realización, el oxidante es H2O2 u oxona. En otra, el oxidante es H2O2. En una realización, la base es NaOH.
En algunas realizaciones, los métodos comprenden además preparar un compuesto de fórmula (8):

comprendiendo los métodos poner en contacto un compuesto de fórmula (7):
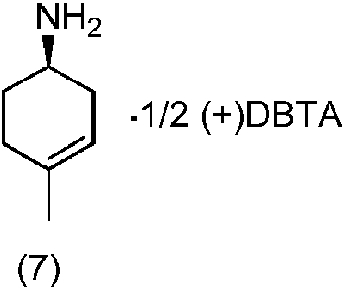
con una base acuosa, seguido de tratamiento de la base libre resultante con Boc2O en un disolvente orgánico, opcionalmente en presencia de una segunda base.
En una realización, la base acuosa es NaOH acuoso. En una realización, el disolvente orgánico es DCM o éter. En otra realización, el disolvente orgánico es DCM. En una realización, la segunda base es trietilamina.
En algunas realizaciones, los métodos comprenden además preparar un compuesto de fórmula (7):

comprendiendo los métodos poner en contacto un compuesto de fórmula (6):

con monohidrato del ácido (+)-dibenzoil-D-tartárico en un disolvente.
En una realización, el disolvente es metanol.
En algunas realizaciones, los métodos comprenden además preparar un compuesto de fórmula (6):

comprendiendo los métodos poner en contacto un compuesto de fórmula (5):

con una disolución acuosa de NaOH y NaOCl.
En algunas realizaciones, los métodos comprenden además preparar un compuesto de fórmula (5):

comprendiendo los métodos poner en contacto un compuesto de fórmula (4):

con DMF y un agente clorante en un disolvente orgánico, seguido del tratamiento del derivado de cloruro de ácido resultante con amoniaco acuoso.
En una realización, el agente clorante es cloruro de oxalilo o SOCl2. En una realización, el agente clorante es SOCl2. En una realización, el disolvente orgánico es DCM.
En algunas realizaciones, los métodos comprenden además preparar un compuesto de fórmula (4):
comprendiendo los métodos poner en contacto un compuesto de fórmula (3)

con una base acuosa.
En una realización, la base es LiOH o NaOH. En otra realización, la base es NaOH.
En algunas realizaciones, los métodos comprenden además preparar un compuesto de fórmula (3):

comprendiendo los métodos poner en contacto un compuesto de fórmula (1):

con un compuesto de fórmula (2):

en un disolvente, en presencia de un ácido de Lewis.
En una realización, el ácido de Lewis es AlCta. En una realización, el disolvente es DCM.
Los intermedios útiles en los procesos proporcionados en la presente memoria incluyen:

Utilidad del Compuesto I
El Compuesto I tiene utilidad como un producto farmacéutico para tratar, prevenir o mejorar afecciones en animales o seres humanos. En particular, el Compuesto I es activo frente a proteínas quinasas, particularmente JNK1 y/o JNK2. Los usos del Compuesto I se describen en la Publicación de Patente de EE.UU. 2013/0029987, publicada el 31 de enero de 2013.
Abreviaturas
Las siguientes abreviaturas se usan en las descripciones y en los ejemplos:
ACN: Acetonitrilo
Boc: terc-Butoxicarbonilo
n-BuOH: n-Butanol
DBTA: Ácido dibenzoil-D-tartárico
DBU: 1,8-Diazabiciclo[5.4.0]undec-7-eno
DCM: Diclorometano
DIPEA: N,N-Diisopropiletilamina
DMAc: N,N-Dimetilacetamida
DMF: N, N-Dimetilformida
DMSO: Dimetilsulfóxido
EtOAc: Acetato de etilo
EtOH: Etanol
GC: Cromatografía de gases
IPA: 2-propanol
IPAc: Acetato de isopropilo
LC: Cromatografía líquida
MeOH: Metanol
2-MeTHF: 2-Metil tetrahidrofurano
MS: Espectrometría de masas
MTBE: terc-Butil metil éter
NMP: N-Metil-2-pirrolidona
RMN: Resonancia magnética nuclear
OR: Rotación óptica
SFC: Cromatografía de fluidos supercríticos
Tf: Triflato o trifluorometanosulfonilo
TFE: 2,2,2-Trifluoroetanol
THF: Tetrahidrofurano
Ejemplos sintéticos
Los siguientes ejemplos sintéticos, presentados a modo de ilustración y no de limitación, muestran métodos para la preparación del Compuesto (A). Se usó ACD/NAME (Advanced Chemistry Development, Inc., Ontario, Canadá) para generar nombres para las estructuras químicas y Chemdraw (Cambridgesoft, Perkin Elmer, Waltham, MA) para dibujar las estructuras químicas. En ciertos casos, se usó Chemdraw para generar nombres para estructuras químicas. Ejemplo 1: Síntesis de hidrocloruro de (1 R,2R,5R)-5-amino-2-metilciclohexanol

4-Metilciclohex-3-enocarboxilato de metilo (3): A un reactor se añadió DCM (2,5 L) y acrilato de metilo (1) (500,0 g) a 20-25 °C bajo una atmósfera de nitrógeno. Después de agitar durante 5 min., el lote se enfrió hasta 0 °C, y se añadió isopreno (2) (593,4 g) durante 5-10 min. Después de agitar durante 5 min. a 25 °C, se añadió AlCÍ3 anhidro (116,2 g) durante 60-90 min. mientras se mantenía la temperatura entre 0-10 °C. Después de agitar a 0-10 °C durante 30 min., el lote se calentó gradualmente hasta 25 °C y se agitó (> 3 h) a esa temperatura hasta que la HPLC indicó < 1 % de acrilato de metilo sin reaccionar (1). Una vez completada la reacción, según lo indicado por HPLC, el lote se enfrió hasta 0 °C y se inactivó con una disolución de HCl (250 mL HCl conc. y 1.750 ml de agua) durante un período de 30 60 min. mientras se mantenía la temperatura por debajo de 10 °C durante el período de inactivación. Se dejó que el lote se calentara hasta 25 °C y se filtró a través de Hyflo para eliminar el sólido no disuelto mientras se enjuagaba el residuo con DCM (500 mL). El filtrado se extrajo con DCM (1 L) y las capas orgánicas combinadas se lavaron sucesivamente con disolución acuosa al 5 % de NaHCÜ3 (1 L) y salmuera (1 L). El DCM se eliminó por destilación de la fracción orgánica a 40-50 °C bajo condiciones atmosféricas para suministrar 4-metilciclohex-3-enocarboxilato de metilo crudo (3) como un líquido marrón (-1.200 g, rendimiento cuantitativo, 85,54 % de pureza por HPLC), que se usó para la siguiente etapa sin purificación.
Ácido 4-metilciclohex-3-enocarboxílico (4): A una disolución de NaOH (290,4 g de NaOH en 1.800 mL de agua) en un reactor a 15-20 °C se añadió lentamente 4-metilciclohex-3-enocarboxilato (3) (1.200 g de material crudo de anteriormente; 895,63 g considerando un rendimiento del 100 %), mientras se mantenía la temperatura por debajo de 25 °C. El lote se calentó gradualmente hasta 35-40 °C y la disolución transparente resultante se agitó (> 2 h) a esa temperatura hasta que la HPLC indicó < 1 % de intermedio sin reaccionar (3). Una vez completada la reacción, como se indica por HPLC, el lote se llevó hasta 25 °C y se inactivó con agua (900 mL). La mezcla acuosa que contenía el producto se lavó con DCM (2 x 900 mL). La capa acuosa se enfrió hasta 0-10 °C y se acidificó con HCl conc. (630 mL) hasta pH 1 -2 mientras se mantenía la temperatura por debajo de 20 °C. Después de agitar la mezcla durante 10 min. a 20-25 °C, el producto se extrajo de la capa acuosa con DCM (2 x 900 mL). Las capas orgánicas combinadas se lavaron con agua (900 mL). El DCM se eliminó por destilación de la fracción orgánica a 40-45 °C y la masa sólida resultante se secó en vacío durante 1 h a 40-45 °C para suministrar, después de enfriar hasta temperatura ambiente, ácido 4-metilciclohex-3-enocarboxílico (4) (707,51 g, 86,90 % de rendimiento basado en HPLC, 85,28 % de pureza por HPLC). El producto así obtenido se disolvió en DCM (750 mL) y se usó para la siguiente etapa sin purificación.
4-Metilciclohex-3-enocarboxamida (5): A un reactor que contenía una disolución de ácido 4-metilciclohex-3-enocarboxílico (4) en DCM de anteriormente (~1.614 g, que contenían ~690 g de intermedio (4)) se añadió DMF (6,9 mL) a 25 °C bajo una atmósfera de nitrógeno. Después de agitar la reacción durante 5 min., se añadió cloruro de tionilo (673,44 g) durante un periodo de 30-60 min. mientras se mantenía la temperatura por debajo de 20 °C. Después de agitar durante 10 min. a 15-20 °C, la reacción se calentó hasta 25-30 °C y se agitó (> 2 h) a esa temperatura hasta que la TLC indicó < 2 % de intermedio sin reaccionar (4). Una vez completada la reacción, según lo indicado por TLC, los disolventes se eliminaron completamente por destilación en vacío. La mezcla resultante se secó en vacío durante 30 min. a 35-40 °C y luego se llevó a temperatura ambiente. La masa así obtenida se añadió lentamente durante un periodo de 30-60 min. a una disolución enfriada en hielo (0-5 °C) de amoniaco acuoso (2,76 L) en un reactor separado manteniendo la temperatura por debajo de 10 °C. Después de agitar la mezcla de reacción durante 30 min. a 0-10 °C, el residuo resultante se retiró por filtración, se lavó con agua y se secó en vacío sobre aire. El producto se secó
adicionalmente en un horno de aire a 45-50 °C y se llevó hasta temperatura ambiente para suministrar 4-metilciclohex-3- enocarboxamida (5) como un sólido blanquecino (604 g, 88,15 % de rendimiento basado en HPLC, 86,55 % de pureza por HPLC), que se usó para la siguiente etapa sin purificación.
4- Metilciclohex-3-enamina (6): A una disolución de NaOH (481,68 g) y agua (2,16 L) en un reactor a -5 °C a 5 °C se añadió lentamente una disolución de hipoclorito de sodio al 10,5 % p/p (4.587,4 g) bajo una atmósfera de nitrógeno. Después de agitar durante 10 min., se añadió gradualmente en porciones 4-metilciclohex-3-enocarboxamida (5) (600 g) a una temperatura de -5 °C a 5 °C. La reacción se agitó durante 6 h a una temperatura por debajo de 10 °C, se calentó gradualmente hasta 25 °C y se agitó (> 5 h) a esa temperatura hasta que la HPLC indicó < 5 % de intermedio sin reaccionar (5). Después de la compleción de la reacción, según se indica por HPLC, se añadió tolueno (1,2 L). La mezcla se enfrió hasta 0-5 °C y se acidificó con HCl conc. (1,5 L) hasta pH 1 -1,5 mientras se mantenía la temperatura por debajo de 20 °C. Después de agitar durante 5 min., la capa orgánica se separó, y la capa acuosa se lavó con tolueno (1,2 L). La capa acuosa se enfrió entonces hasta 0-5 °C, y se basificó con disolución acuosa de NaOH (2,0 kg de NaOH y 1.340 mL de H2O) hasta pH >13 mientras se mantenía la temperatura por debajo de 20 °C. El producto se extrajo con DCM (2 x 1,5 L), y las capas orgánicas combinadas se secaron sobre sulfato de sodio y se filtraron. El DCM se eliminó por destilación del filtrado a 40-60 °C bajo condiciones atmosféricas. El residuo resultante se enfrió hasta temperatura ambiente para suministrar 4-metilciclohex-3-enamina (6) (377,4 g, 78,74 % de rendimiento basado en HPLC, 85,74 % de pureza por HPLC), que se usó para la siguiente etapa sin purificación.
Hemi-dibenzoil-D(+)-tartarato de (R)-4-metilciclohex-3-enamina (7): Una disolución de monohidrato del ácido (+)-dibenzoil-D-tartárico (1.015,3 g) en metanol (3 L) se llevó gradualmente a reflujo. A esta disolución a reflujo se le añadió lentamente una disolución de 4-metilciclohex-3-enamina (6) (300 g) en metanol (300 mL) durante un período de 60-75 min. La mezcla de reacción se calentó a reflujo durante 2 h y luego se enfrió gradualmente hasta 25 °C durante 4-5 h. Después de agitar la mezcla de reacción durante 1 h adicional a 25 °C, el residuo resultante se filtró, se lavó con metanol, y se secó en vacío durante 30 min. La HPLC quiral del producto protegido con Boc (preparado convirtiendo una parte alícuota en el derivado Boc) indicó un 71,22 % del hemi-dibenzoil-D(+)-tartarato de (R)-4-metilciclohex-3-enamina deseado (7), y un 28,72 % del S-isómero correspondiente.
El producto crudo obtenido anteriormente (~645 g) se trató con monohidrato del ácido (+)-dibenzoil-D-tartárico (123,1 g) y metanol (3,8 L), y la mezcla resultante se puso a reflujo durante 2 h, y se enfrió entonces gradualmente hasta 25 °C durante 4-5 h. Después de agitar la mezcla de reacción durante 1 h adicional a 25 °C, el residuo resultante se filtró, se lavó con metanol, y se secó en vacío durante 30 min. La HPLC quiral de una parte alícuota del producto que se convirtió en el derivado BOC indicó un 82,11 % del hemi-dibenzoil-D(+)-tartarato de (R)-4-metilciclohex-3-enamina deseado (7), y un 17,82 % del S-isómero correspondiente.
El producto crudo obtenido anteriormente (~480 g) se trató con monohidrato del ácido (+)-dibenzoil-D-tartárico (93,3 g) y metanol (2,9 L), y la mezcla resultante se puso a reflujo durante 2 h, y se enfrió entonces gradualmente hasta 25 °C durante 4-5 h. Después de agitar la mezcla de reacción durante 1 h adicional a 25 °C, el residuo resultante se filtró, se lavó con metanol, y se secó en vacío durante 30 min. El producto así obtenido se secó adicionalmente en un horno de aire a 45-50 °C para suministrar hemi-dibenzoil-D(+)-tartarato de (R)-4-metilciclohex-3-enamina (7) (220,5 g, 28,5 % de rendimiento, 97,81 % de pureza por GC, rango de punto de fusión: 205,2-206,3 °C, OR: 110,0 ° (C=1 % en ácido acético a 25 °C)); la HPLC quiral indicó un 86,88 % del R-isómero deseado y un 13,12 % del S-isómero correspondiente.
(R)-(4-Metilciclohex-3-en-1 -il) carbamato de terc-butilo (8): A una disolución de NaOH (124 g) y agua (1.200 mL) a 10 20 °C se añadió lentamente hemi-dibenzoil-D(+)-tartarato de (R)-4-metilciclohex-3-enamina (7) (300 g) mientras se mantenía la temperatura por debajo de 25 °C. Después de agitar la mezcla de reacción durante 15 min, la base libre resultante se extrajo de la capa acuosa usando DCM (2 x 300 mL, 1 x 150 mL). Las capas orgánicas se combinaron, y la disolución resultante (~850 mL) se trató con anhídrido de Boc (236,8 g) a 0-5 °C. La mezcla de reacción se dejó calentar hasta 25 °C y se agitó
(> 2 h) a esa temperatura hasta que la HPLC indicó < 1 % de intermedio sin reaccionar (7). Después de la compleción de la reacción, según lo indicado por HPLC, se añadió agua (230 mL) y la mezcla se agitó durante 10 min. La capa orgánica se separó y se lavó con disolución acuosa al 2 % de ácido cítrico (230 mL) seguida de agua (230 mL). El DCM se eliminó por destilación a 30-40 °C en vacío, y la masa amarilla clara resultante se secó en vacío durante 30 min. a 40-45 °C para suministrar (R)-(4-metilciclohex-3-en-1-il)carbamato de terc-butilo (8) (216 g, 98,91 % de rendimiento basado en HPLC,
98,36 % de pureza por HPLC), que contenía un 14,85 % del S-isómero correspondiente según lo indicado por HPLC quiral. El producto así obtenido se recogió en THF (437 mL), se agitó durante 10 min. para obtener una disolución transparente y se almacenó en una atmósfera de nitrógeno para usarlo en la siguiente etapa.
((1 R,3R,4R)-3-hidroxi-4-metilciclohexil)carbamato de terc-butilo (9): A una suspensión de borohidruro de sodio (76,97 g) en THF (1.290 mL) a 25 °C se añadió lentamente (-)-a-pineno (582,07 g) durante un periodo de 15 min. bajo una atmósfera de nitrógeno. Después de enfriar la mezcla de reacción hasta 0-5 °C, se añadió lentamente eterato de trifluoruro de boro (57 %, 531,95 g) durante un periodo de 30-60 min. Se dejó que la reacción se calentara hasta 25 °C,
se agitó durante 8 h, y después se trató con la disolución de (R)-(4-metilciclohex-3-en-1-il)carbamato de terc-butilo (8) en THF preparado anteriormente
(623 g, que contenía 215 g de (8)). La mezcla de reacción resultante se agitó (> 3 h) a 25 °C hasta que e1HPLC indicó < 1 % de intermedio sin reaccionar (8). Después de enfriar hasta 0-5 °C, la reacción se inactivó lentamente por la adición de agua durante un periodo de 30-60 min., seguido de la adición posterior de NaOH acuoso (244,15 g de NaOH y 716 mL de agua) y una disolución de peróxido de hidrógeno al 48 % (432,36 g). La mezcla de reacción se calentó gradualmente hasta 25 °C y se agitó durante 3 h, después de lo cual se añadió una disolución de tiosulfato de sodio (75 g de tiosulfato de sodio y 75 mL de agua). Después de agitar durante 30 min., se añadió una disolución de ácido cítrico (254 g de ácido cítrico y 860 mL de agua) y la mezcla se agitó durante 30 min. adicionales, después de lo cual se añadió acetato de etilo (1.290 mL). Después de agitar durante 10 min., la capa orgánica se separó y la fracción acuosa se extrajo con acetato de etilo (2 x 430 mL). Las capas orgánicas se combinaron y el disolvente se eliminó por destilación a 40-50 °C en vacío. La masa resultante se secó en vacío durante 1 h para rendir un rendimiento cuantitativo de ((1 R,3R,4R)-3-hidroxi-4-metilciclohexil)carbamato de terc-butilo (9)
(858 g crudo), que se usó para la siguiente etapa sin purificación.
Hidrocloruro de (1R,2R,5R)-5-amino-2-metilciclohexanol (A): Una mezcla de ((1R,3R,4R)-3-hidroxi-4-metilciclohexil)carbamato de terc-butilo (9) de anteriormente (853 g, que contenían 233 g de (9)) e IPA-HCl (disolución al 14 % p/p; 699 mL) se agitó (> 2 h) a 25 °C hasta que el HPLC indicó <1 % de intermedio sin reaccionar (9). El disolvente se eliminó por destilación a 40-60 °C en vacío. Se añadió IPA reciente (233 mL) a 40-45 °C y el disolvente se eliminó de nuevo por destilación a 40-60 °C en vacío. Después de desgasificar durante 30 min. a 40-60 °C, la masa resultante se trató con IPA reciente (699 mL) y la mezcla se agitó bajo nitrógeno a 30-35 °C durante 30 min. y luego a 0-5 °C durante 30 min adicionales. El producto sólido se filtró a 0-5 °C y se lavó con IPA enfriado. El producto resultante se secó en vacío a 40-60 °C para rendir ~70 g de producto crudo, que contenía ~96,49 % del RRR-isómero deseado según lo indicado por GC quiral (estaban presentes otros isómeros como impurezas en la cantidad del 2,06 % (isómero SSS), 0,18 % (isómero SRR ), y 1,26 % (isómero RSS)). Se añadió IPA (3 L) y la suspensión de sólidos resultante se calentó a reflujo durante 30 min. La mezcla se enfrió gradualmente hasta 70-75 °C, y la impureza no disuelta se retiró por filtración y se lavó con IPA (140 mL). El disolvente se eliminó por destilación a 40-60 °C para rendir una masa blanca que se enfrió gradualmente hasta 25 °C y después se trató con agua (31,5 mL) y acetonitrilo (31,5 mL). La mezcla resultante se calentó a 75-80 °C durante 10 min. para obtener una disolución transparente, que se trató entonces lentamente con acetonitrilo (574 mL) a 75-80 °C durante un período de 1 h. Después de agitar durante 15 min. a 75-80 °C, la masa resultante se enfrió hasta 0-5 °C durante 2-3 h y se agitó a esa temperatura durante 30 min. El producto se filtró bajo nitrógeno a 0-5 °C y la torta sólida se lavó con acetonitrilo enfriado
(70 mL) y se secó en vacío para producir el isómero RRR deseado. El proceso anterior de precipitar el producto deseado fuera de una mezcla de agua y acetonitrilo por la adición de acetonitrilo a 75-80 °C se repitió hasta que la GC quiral indicó la presencia de no más del 0,5 % de cualquier otro isómero único (isómero SSS, SRR y RSS). El producto así obtenido se secó más en vacío a 40-60 °C para rendir hidrocloruro de (1 R,2R,5R)-5-amino-2-metilciclohexanol (A) como un sólido blanco
(63 g, 37,4 % de rendimiento, 100 % de pureza por HPLC, rango de punto de fusión: 244,0-245,5 °C, SOR: -31,2 ° (c =1 % en MeOH a 25 °C)); la GC quiral indicó el 99,83 % del RRR-isómero deseado y el 0,17 % del SSS-isómero correspondiente.
1H-RMN (D2O) (400 MHz): 53,18-3,08 (m, 2H), 2,15-2,12 (m, 1H), 1,86-1,83 (m, 1H), 1,72-1,68 (m, 1H), 1,32-1,16 (m, 3H), 1,02-0,91 (m, 1H), 0,86-0,85 (d, 3H, J = 6,4Hz).
Claims (14)
1. Un método para preparar un compuesto de fórmula (A),

(a) poner en contacto un compuesto de fórmula (1),

con un compuesto de fórmula (2),

(2 )
en presencia de un ácido de Lewis en un disolvente para proporcionar un compuesto de fórmula (3),

(b) poner en contacto el compuesto de fórmula (3) de la etapa (a) con una base acuosa para proporcionar un compuesto de fórmula (4),

(c) poner en contacto el compuesto de fórmula (4) de la etapa (b) con DMF y un agente clorante en un disolvente orgánico, seguido del tratamiento del derivado de cloruro de ácido resultante con amoniaco acuoso para proporcionar un compuesto de fórmula (5),

(d) poner en contacto el compuesto de fórmula (5) de la etapa (c) con una disolución acuosa de NaOH y NaOCl para proporcionar un compuesto de fórmula (6),

(6 )
(e) poner en contacto el compuesto de fórmula (6) de la etapa (d) con monohidrato del ácido (+)-dibenzoil-D-tartárico en un disolvente para proporcionar un compuesto de fórmula (7),

(f) poner en contacto el compuesto de fórmula (7) de la etapa (e) con una base acuosa, seguido del tratamiento de la base libre resultante con BoC2O en un disolvente orgánico para proporcionar un compuesto de fórmula (8),

(g) poner en contacto el compuesto de fórmula (8) de la etapa (f) con una mezcla de un agente reductor, un auxiliar quiral y un ácido de Lewis en un disolvente, seguido de un tratamiento con un oxidante en presencia de una base para proporcionar un compuesto de fórmula (9),

(h) poner en contacto el compuesto de fórmula (9) de la etapa (g) con una disolución de ácido clorhídrico en un disolvente para proporcionar el compuesto de fórmula (A).
2. El método de la reivindicación 1, en donde el ácido de Lewis de la etapa (a) es AlCh y/o en donde el disolvente de la etapa (a) es DCM.
3. El método de la reivindicación 1 o 2, en donde la base de la etapa (b) es NaOH.
4. El método de una cualquiera de las reivindicaciones 1 a 3, en donde el agente clorante de la etapa (c) es SOCl2 y/o en donde el disolvente orgánico de la etapa (c) es DCM.
5. El método de una cualquiera de las reivindicaciones 1 a 4, en donde el disolvente de la etapa (e) es MeOH.
6. El método de una cualquiera de las reivindicaciones 1 a 5, en donde la base de la etapa (f) es NaOH y/o en donde el disolvente orgánico de la etapa (f) es DCM.
7. El método de una cualquiera de las reivindicaciones 1 a 6, en donde el agente reductor de la etapa (g) es NaBH4, y/o en donde el auxiliar quiral de la etapa (g) es a-pineno, y/o en donde el ácido de Lewis de la etapa (g) es BF3^Et2O, y/o en donde el disolvente de la etapa (g) es THF, y/o en donde el oxidante de la etapa (g) es H2O2, y/o en donde la base de la etapa (g) es NaOH.
8. El método de una cualquiera de las reivindicaciones 1 a 7, en donde el disolvente de la etapa (h) es IPA.
9. Un método para preparar un compuesto de fórmula (3),

comprendiendo el método poner en contacto un compuesto de fórmula (1),

con un compuesto de fórmula (2),

en presencia de un ácido de Lewis en un disolvente.
10. El método de la reivindicación 9, en donde el ácido de Lewis es AlCh.
11. El método de la reivindicación 9 o 10, en donde el disolvente es DCM.
12. Un método para preparar un compuesto de fórmula (7),

comprendiendo el método poner en contacto un compuesto de fórmula (6),

con monohidrato del ácido (+)-dibenzoil-D-tartárico en un disolvente.
13. El método de la reivindicación 12, en donde el disolvente es MeOH.
14. Un compuesto de fórmula (7),
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562196363P | 2015-07-24 | 2015-07-24 | |
| PCT/US2016/043511 WO2017019487A1 (en) | 2015-07-24 | 2016-07-22 | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2819374T3 true ES2819374T3 (es) | 2021-04-15 |
Family
ID=57885143
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20193348T Active ES2994877T3 (en) | 2015-07-24 | 2016-07-22 | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| ES16831114T Active ES2819374T3 (es) | 2015-07-24 | 2016-07-22 | Métodos de síntesis del hidrocloruro de (1R,2R,5R)-5-amino-2-metilciclohexanol e intermedios útiles en los mismos |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20193348T Active ES2994877T3 (en) | 2015-07-24 | 2016-07-22 | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
Country Status (9)
| Country | Link |
|---|---|
| US (4) | US10252981B2 (es) |
| EP (2) | EP3325432B1 (es) |
| JP (2) | JP6805232B2 (es) |
| CN (1) | CN107922287B (es) |
| AU (2) | AU2016297784B2 (es) |
| CA (2) | CA3208587A1 (es) |
| ES (2) | ES2994877T3 (es) |
| MX (2) | MX2018001004A (es) |
| WO (1) | WO2017019487A1 (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016123291A1 (en) | 2015-01-29 | 2016-08-04 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| CA3208587A1 (en) * | 2015-07-24 | 2017-02-02 | Celgene Corporation | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| CN111848423B (zh) * | 2019-04-30 | 2022-10-14 | 尚科生物医药(上海)有限公司 | 3-氧代环丁基氨基甲酸叔丁酯的制备方法 |
Family Cites Families (80)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2859239A (en) * | 1956-08-17 | 1958-11-04 | Dow Chemical Co | Acrylic acid compounds |
| CH549339A (de) | 1971-05-12 | 1974-05-31 | Ciba Geigy Ag | Herbizides mittel. |
| US5763647A (en) * | 1990-03-30 | 1998-06-09 | Shionogi & Co., Ltd. | Preparation of optically active 1,4-bridged-cyclohexane carboxylic acid derivatives |
| EP0520419A3 (en) * | 1991-06-26 | 1993-06-16 | Union Carbide Chemicals & Plastics Technology Corporation | Production of unsaturated cycloaliphatic esters and derivatives thereof |
| AU1507199A (en) | 1997-12-15 | 1999-07-05 | Yamanouchi Pharmaceutical Co., Ltd. | Novel pyrimidine-5-carboxamide derivatives |
| ES2274634T3 (es) | 1998-08-29 | 2007-05-16 | Astrazeneca Ab | Compuestos de pirimidina. |
| AU5107900A (en) | 1999-06-09 | 2000-12-28 | Yamanouchi Pharmaceutical Co., Ltd. | Novel heterocyclic carboxamide derivatives |
| WO2000076980A1 (en) | 1999-06-10 | 2000-12-21 | Yamanouchi Pharmaceutical Co., Ltd. | Novel nitrogen-contaiing heterocyclic derivatives or salts thereof |
| AU5636900A (en) | 1999-06-30 | 2001-01-31 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| MXPA02001565A (es) | 1999-08-13 | 2005-07-14 | Vertex Pharma | Inhibidores de cinasas c-jun n-terminal (jnk) y de otras cinasas proteicas. |
| TWI329105B (en) | 2002-02-01 | 2010-08-21 | Rigel Pharmaceuticals Inc | 2,4-pyrimidinediamine compounds and their uses |
| GB0206215D0 (en) | 2002-03-15 | 2002-05-01 | Novartis Ag | Organic compounds |
| JPWO2003082855A1 (ja) | 2002-03-28 | 2005-08-04 | 協和醗酵工業株式会社 | 抗炎症剤 |
| AU2003244098A1 (en) | 2002-06-28 | 2004-01-19 | Yamanouchi Pharmaceutical Co., Ltd. | Diaminopyrimidinecarboxa mide derivative |
| ES2445208T3 (es) | 2002-07-29 | 2014-02-28 | Rigel Pharmaceuticals, Inc. | Compuestos de 2,4-pirimidindiamina para uso en métodos para tratar o prevenir enfermedades autoinmunitarias |
| JPWO2004054617A1 (ja) | 2002-12-13 | 2006-04-20 | 協和醗酵工業株式会社 | 中枢疾患の予防および/または治療剤 |
| US7358384B2 (en) * | 2003-01-16 | 2008-04-15 | Toray Fine Chemicals Co., Ltd. | Processes for the recovery of optically active diacyltartaric acids |
| EP1590341B1 (en) | 2003-01-17 | 2009-06-17 | Warner-Lambert Company LLC | 2-aminopyridine substituted heterocycles as inhibitors of cellular proliferation |
| ATE440087T1 (de) | 2003-01-30 | 2009-09-15 | Boehringer Ingelheim Pharma | 2,4-diaminopyrimidinderivate, die sich als inhibitoren von pkc-theta eignen |
| NZ585188A (en) | 2003-08-15 | 2011-09-30 | Novartis Ag | 2,4-Pyrimidinediamines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| JP2007506746A (ja) | 2003-09-24 | 2007-03-22 | ワイス・ホールディングズ・コーポレイション | 抗癌剤としての5−アリールピリミジン類 |
| WO2005095382A1 (ja) | 2004-03-30 | 2005-10-13 | Kyowa Hakko Kogyo Co., Ltd. | 抗腫瘍剤 |
| JP2008505910A (ja) | 2004-07-08 | 2008-02-28 | ベーリンガー インゲルハイム ファーマシューティカルズ インコーポレイテッド | Pkc−シータのインヒビターとして有用なピリミジン誘導体 |
| EP1796679A1 (en) | 2004-09-10 | 2007-06-20 | Altana Pharma AG | Ciclesonide and syk inhibitor combination and methods of use thereof |
| CA2579017A1 (en) | 2004-09-10 | 2006-03-16 | Altana Pharma Ag | Roflumilast and syk inhibitor combination and methods of use thereof |
| JP2006124387A (ja) | 2004-09-30 | 2006-05-18 | Taisho Pharmaceut Co Ltd | 新規なキノリン、テトラヒドロキナゾリン、及びピリミジン誘導体と、これらを使用することに関連した治療方法 |
| ATE542802T1 (de) | 2004-09-30 | 2012-02-15 | Tibotec Pharm Ltd | Hiv-inhibierende 5-substituierte pyrimidine |
| WO2006091737A1 (en) | 2005-02-24 | 2006-08-31 | Kemia, Inc. | Modulators of gsk-3 activity |
| KR20080004488A (ko) | 2005-03-10 | 2008-01-09 | 바이엘 파마슈티칼스 코포레이션 | 과다증식성 장애 치료용 피리미딘 유도체 |
| WO2007032445A1 (ja) | 2005-09-16 | 2007-03-22 | Kyowa Hakko Kogyo Co., Ltd. | タンパク質キナーゼ阻害剤 |
| MX2009000769A (es) | 2006-07-21 | 2009-01-28 | Novartis Ag | Compuestos de 2,4-di(arilaminio)-pirimidin-5-carboxamida como inhibidores de cinasas jak. |
| US8188113B2 (en) | 2006-09-14 | 2012-05-29 | Deciphera Pharmaceuticals, Inc. | Dihydropyridopyrimidinyl, dihydronaphthyidinyl and related compounds useful as kinase inhibitors for the treatment of proliferative diseases |
| US20080139531A1 (en) | 2006-12-04 | 2008-06-12 | Alcon Manufacturing Ltd. | Use of connective tissue mast cell stabilizers to facilitate ocular surface re-epithelization and wound repair |
| CL2007003693A1 (es) * | 2006-12-22 | 2008-06-27 | Actelion Pharmaceuticals Ltd | Compuestos derivados de pirido [3,2-b] [1,4] tiazina; composicion farmaceutica que contiene dichos compuestos; y su uso en el tratamiento de infecciones bacterianas. |
| BRPI0810411B8 (pt) | 2007-04-18 | 2021-05-25 | Pfizer Prod Inc | derivados de sulfonil amida para o tratamento de crescimento celular anormal, seu sos, bem como composição farmacêutica |
| WO2008132505A1 (en) | 2007-04-27 | 2008-11-06 | Astrazeneca Ab | N' - (phenyl) -n- (morpholin-4-yl-pyridin-2-yl) -pyrimidine-2, 4-diamine derivatives as ephb4 kinase inhibitors for the treatment of proliferative conditions |
| EP2183242A2 (en) | 2007-07-16 | 2010-05-12 | AstraZeneca AB | Pyrimidine derivatives 934 |
| CA2693594A1 (en) | 2007-07-17 | 2009-01-22 | Rigel Pharmaceuticals, Inc. | Cyclic amine substituted pyrimidinediamines as pkc inhibitors |
| JP5269444B2 (ja) * | 2008-03-11 | 2013-08-21 | 東ソー・ファインケム株式会社 | 固体ルイス酸触媒、それを用いたディールスアルダー付加物の製造方法 |
| SG2014015085A (en) | 2008-04-16 | 2014-06-27 | Portola Pharm Inc | 2,6-diamino-pyrimidin-5-yl-carboxamides as syk or jak kinases inhibitors |
| EA024109B1 (ru) | 2008-04-16 | 2016-08-31 | Портола Фармасьютиклз, Инк. | Ингибиторы протеинкиназ |
| NZ588830A (en) | 2008-04-22 | 2012-11-30 | Portola Pharm Inc | Inhibitors of protein kinases |
| WO2009132980A1 (en) | 2008-04-29 | 2009-11-05 | F. Hoffmann-La Roche Ag | Pyrimidinyl pyridone inhibitors of jnk. |
| PT2300013T (pt) | 2008-05-21 | 2017-10-31 | Ariad Pharma Inc | Derivados de fósforo como inibidores de cinases |
| UY31929A (es) | 2008-06-25 | 2010-01-05 | Irm Llc | Compuestos y composiciones como inhibidores de cinasa |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| TWI546290B (zh) | 2008-06-27 | 2016-08-21 | 賽基艾維洛米斯研究股份有限公司 | 雜芳基化合物及其用途 |
| JP5644499B2 (ja) | 2008-09-01 | 2014-12-24 | アステラス製薬株式会社 | 2,4−ジアミノピリミジン化合物 |
| EP2159220A1 (en) * | 2008-09-02 | 2010-03-03 | Nabriva Therapeutics AG | Organic compounds |
| TWI453207B (zh) * | 2008-09-08 | 2014-09-21 | Signal Pharm Llc | 胺基三唑并吡啶,其組合物及使用其之治療方法 |
| MX2011002825A (es) | 2008-09-18 | 2011-04-05 | Astellas Pharma Inc | Compuestos heterociclicos de carboxamida. |
| TW201016676A (en) | 2008-10-03 | 2010-05-01 | Astrazeneca Ab | Heterocyclic derivatives and methods of use thereof |
| PL2346988T3 (pl) | 2008-10-31 | 2017-10-31 | Janssen Biotech Inc | Różnicowanie ludzkich zarodkowych komórek macierzystych do linii endokrynnej trzustki |
| US8513242B2 (en) | 2008-12-12 | 2013-08-20 | Cystic Fibrosis Foundation Therapeutics, Inc. | Pyrimidine compounds and methods of making and using same |
| WO2010080864A1 (en) | 2009-01-12 | 2010-07-15 | Array Biopharma Inc. | Piperidine-containing compounds and use thereof |
| AU2010219097A1 (en) | 2009-01-13 | 2011-08-04 | Glaxo Group Limited | Pyrimidinecarboxamide derivatives as inhibitors of SYK kinase |
| CA2749837C (en) | 2009-01-21 | 2017-07-11 | Rigel Pharmaceuticals, Inc. | Derivatives of n2-(3-pyridil or phenyl)-n4-(4-piperidyl)-2,4-pyrimidinediamine useful in the treatment of inflammatory, autoimmune or proliferative diseases |
| AR076550A1 (es) | 2009-05-06 | 2011-06-22 | Portola Pharm Inc | Inhibidores de la janus tirosina kinasa (jak) |
| SI2428508T1 (sl) | 2009-05-08 | 2016-04-29 | Astellas Pharma Inc. | Diamino heterociklična karboksamidna spojina |
| JP2012148977A (ja) | 2009-05-20 | 2012-08-09 | Astellas Pharma Inc | アミノシクロヘキシルアルキル基を有する2,4−ジアミノピリミジン化合物 |
| WO2010144468A1 (en) | 2009-06-10 | 2010-12-16 | Abbott Laboratories | 2- ( lh-pyrazol-4 -ylamino ) -pyrimidine as kinase inhibitors |
| JP2012197231A (ja) | 2009-08-06 | 2012-10-18 | Oncotherapy Science Ltd | Ttk阻害作用を有するピリジンおよびピリミジン誘導体 |
| WO2011065800A2 (ko) | 2009-11-30 | 2011-06-03 | 주식회사 오스코텍 | 피리미딘 유도체, 이의 제조방법 및 이를 함유하는 약학적 조성물 |
| US20110130415A1 (en) | 2009-12-01 | 2011-06-02 | Rajinder Singh | Protein kinase c inhibitors and uses thereof |
| EP2582671A4 (en) * | 2010-06-17 | 2014-02-26 | Reddys Lab Ltd Dr | PROCESS FOR PREPARING A SINGLE ENANTIOMER FROM 3-AMINOPIPERIDIN DIHYDROCHLORIDE |
| EP2595982B1 (en) | 2010-07-21 | 2018-06-13 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| US8580805B2 (en) | 2010-08-31 | 2013-11-12 | Hubert Maehr | Pyrimidine carboxamide derivatives |
| WO2012044936A1 (en) | 2010-09-30 | 2012-04-05 | Portola Pharmaceuticals, Inc. | Combination therapy with 4-(3-(2h-1,2,3-triazol-2-yl)phenylamino)-2-((1r,2s)-2-aminocyclohexylamino)pyrimidine-5-carboxamide |
| WO2012045020A1 (en) | 2010-09-30 | 2012-04-05 | Portola Pharmaceuticals, Inc. | Combinations of 4-(cyclopropylamino)-2-(4-(4-(ethylsulfonyl)piperazin-1-yl)phenylamino)pyrimidine-5-carboxamide and fludarabine |
| SMT202400031T1 (it) * | 2011-04-22 | 2024-03-13 | Signal Pharm Llc | Diamminocarbossammide e diamminocarbonitrile pirimidine sostituite, loro composizioni e metodi di trattamento con esse |
| WO2014019166A1 (zh) * | 2012-08-01 | 2014-02-06 | 上海威智医药科技有限公司 | 高活性硼烷类化合物的工业化生产方法 |
| JP2014031327A (ja) * | 2012-08-02 | 2014-02-20 | Kaneka Corp | 光学活性シス−2−アミノ−シクロヘキサンカルボン酸誘導体およびその前駆体の製造法 |
| EP3082819B1 (en) | 2013-12-20 | 2020-06-17 | Signal Pharmaceuticals, LLC | Substituted diaminopyrimidyl compounds, compositions thereof, and methods of treatment therewith |
| NZ715903A (en) | 2014-01-30 | 2017-06-30 | Signal Pharm Llc | Solid forms of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| TWI523838B (zh) * | 2014-03-21 | 2016-03-01 | Far Eastern New Century Corp | Terephthalic acid and 4-methyl-3-cyclohexene-1-carboxylic acid Ester preparation method |
| MX388321B (es) | 2014-10-06 | 2025-03-19 | Signal Pharm Llc | Compuestos de aminopurina sustituida, composiciones del mismo, y metodos de tratamiento con los mismos. |
| EP3233808B1 (en) | 2014-12-16 | 2021-07-14 | Signal Pharmaceuticals, LLC | Medical uses comprising methods for measurement of inhibition of c-jun n-terminal kinase in skin |
| BR112017012795A2 (pt) | 2014-12-16 | 2018-01-02 | Signal Pharmaceuticals, Llc | Formulações de 2-(terc-butilamino)-4-((1r,3r,4r)-3- hidroxi-4-metilciclo-hexilamino)-pirimidina-5- carboxamida |
| WO2016123291A1 (en) | 2015-01-29 | 2016-08-04 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| CA3208587A1 (en) * | 2015-07-24 | 2017-02-02 | Celgene Corporation | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
-
2016
- 2016-07-22 CA CA3208587A patent/CA3208587A1/en active Pending
- 2016-07-22 WO PCT/US2016/043511 patent/WO2017019487A1/en not_active Ceased
- 2016-07-22 MX MX2018001004A patent/MX2018001004A/es active IP Right Grant
- 2016-07-22 JP JP2018503482A patent/JP6805232B2/ja active Active
- 2016-07-22 US US15/746,853 patent/US10252981B2/en active Active
- 2016-07-22 CA CA2993173A patent/CA2993173C/en active Active
- 2016-07-22 AU AU2016297784A patent/AU2016297784B2/en active Active
- 2016-07-22 ES ES20193348T patent/ES2994877T3/es active Active
- 2016-07-22 MX MX2020001720A patent/MX385379B/es unknown
- 2016-07-22 EP EP16831114.0A patent/EP3325432B1/en active Active
- 2016-07-22 EP EP20193348.8A patent/EP3795553B1/en active Active
- 2016-07-22 ES ES16831114T patent/ES2819374T3/es active Active
- 2016-07-22 CN CN201680043308.1A patent/CN107922287B/zh active Active
-
2019
- 2019-01-28 US US16/258,802 patent/US10774033B2/en active Active
-
2020
- 2020-08-13 US US16/992,219 patent/US11192847B2/en active Active
- 2020-12-03 JP JP2020200723A patent/JP7165178B2/ja active Active
-
2021
- 2021-03-09 AU AU2021201493A patent/AU2021201493B2/en active Active
- 2021-11-03 US US17/518,552 patent/US11780801B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP7165178B2 (ja) | 2022-11-02 |
| JP2021046426A (ja) | 2021-03-25 |
| EP3795553B1 (en) | 2024-05-15 |
| EP3795553A1 (en) | 2021-03-24 |
| AU2016297784B2 (en) | 2020-12-24 |
| JP2018521085A (ja) | 2018-08-02 |
| AU2021201493B2 (en) | 2022-07-14 |
| MX2020001720A (es) | 2021-08-13 |
| AU2021201493A1 (en) | 2021-03-25 |
| EP3325432B1 (en) | 2020-09-02 |
| AU2016297784A1 (en) | 2018-02-08 |
| JP6805232B2 (ja) | 2020-12-23 |
| CN107922287B (zh) | 2021-04-09 |
| MX385379B (es) | 2025-03-18 |
| CN107922287A (zh) | 2018-04-17 |
| US20200369600A1 (en) | 2020-11-26 |
| US20190152893A1 (en) | 2019-05-23 |
| US11192847B2 (en) | 2021-12-07 |
| US20220234990A1 (en) | 2022-07-28 |
| ES2994877T3 (en) | 2025-02-03 |
| HK1255553A1 (en) | 2019-08-23 |
| CA3208587A1 (en) | 2017-02-02 |
| EP3325432A1 (en) | 2018-05-30 |
| EP3325432A4 (en) | 2019-04-24 |
| US10252981B2 (en) | 2019-04-09 |
| US20180215700A1 (en) | 2018-08-02 |
| CA2993173A1 (en) | 2017-02-02 |
| MX2018001004A (es) | 2018-06-07 |
| US10774033B2 (en) | 2020-09-15 |
| WO2017019487A1 (en) | 2017-02-02 |
| US11780801B2 (en) | 2023-10-10 |
| CA2993173C (en) | 2023-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111039944B (zh) | Mst1激酶抑制剂及其用途 | |
| US11780801B2 (en) | Methods of synthesis of (1R,2R,5R)-5-amino-2-methyl-cyclohexanol hydrochloride and intermediates useful therein | |
| ES2927021T3 (es) | Compuestos heteroaromáticos como inhibidores de vanina | |
| CN104507946A (zh) | 氘代依鲁替尼 | |
| TWI906958B (zh) | 嘧啶胺類nuak抑制劑及其製備方法和用途 | |
| TWI905843B (zh) | 嘧啶胺類nuak抑制劑及其製備方法和用途 | |
| TWI902351B (zh) | 嘧啶胺類nuak抑制劑及其製備方法和用途 | |
| EP3339299B1 (en) | Novel catechol derivative and pharmaceutical composition comprising same | |
| HK40040730A (en) | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein | |
| HK40040730B (en) | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein | |
| HK1255553B (en) | Methods of synthesis of (1r,2r,5r)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein | |
| TWI920642B (zh) | 嘧啶胺類nuak抑制劑及其製備方法和用途 | |
| HK40122633A (zh) | Jak抑制剂类似物、制剂及其用途 | |
| JP2006232671A (ja) | 新規セレナゾリン誘導体 | |
| TW202502347A (zh) | 嘧啶胺類nuak抑制劑及其製備方法和用途 |