JP2017505848A - 破砕抵抗性高吸水性樹脂およびその製造方法 - Google Patents
破砕抵抗性高吸水性樹脂およびその製造方法 Download PDFInfo
- Publication number
- JP2017505848A JP2017505848A JP2016551152A JP2016551152A JP2017505848A JP 2017505848 A JP2017505848 A JP 2017505848A JP 2016551152 A JP2016551152 A JP 2016551152A JP 2016551152 A JP2016551152 A JP 2016551152A JP 2017505848 A JP2017505848 A JP 2017505848A
- Authority
- JP
- Japan
- Prior art keywords
- superabsorbent resin
- water
- particles
- crush
- resistant
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images






Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
- B01J20/267—Cross-linked polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/22—Expanded, porous or hollow particles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28014—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their form
- B01J20/28047—Gels
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28054—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their surface properties or porosity
- B01J20/28057—Surface area, e.g. B.E.T specific surface area
- B01J20/28061—Surface area, e.g. B.E.T specific surface area being in the range 100-500 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28054—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their surface properties or porosity
- B01J20/28057—Surface area, e.g. B.E.T specific surface area
- B01J20/28064—Surface area, e.g. B.E.T specific surface area being in the range 500-1000 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28054—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their surface properties or porosity
- B01J20/28057—Surface area, e.g. B.E.T specific surface area
- B01J20/28066—Surface area, e.g. B.E.T specific surface area being more than 1000 m2/g
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/3021—Milling, crushing or grinding
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/3085—Chemical treatments not covered by groups B01J20/3007 - B01J20/3078
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/04—Polymerisation in solution
- C08F2/10—Aqueous solvent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/46—Polymerisation initiated by wave energy or particle radiation
- C08F2/48—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/02—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques
- C08J3/03—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques in aqueous media
- C08J3/075—Macromolecular gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
- C08J3/245—Differential crosslinking of one polymer with one crosslinking type, e.g. surface crosslinking
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/22—Expanded, porous or hollow particles
- C08K7/24—Expanded, porous or hollow particles inorganic
- C08K7/26—Silicon- containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/12—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity
- C08L101/14—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity the macromolecular compounds being water soluble or water swellable, e.g. aqueous gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/14—Water soluble or water swellable polymers, e.g. aqueous gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/04—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters
- C08J2333/06—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters of esters containing only carbon, hydrogen, and oxygen, the oxygen atom being present only as part of the carboxyl radical
- C08J2333/08—Homopolymers or copolymers of acrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/04—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters
- C08J2333/06—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters of esters containing only carbon, hydrogen, and oxygen, the oxygen atom being present only as part of the carboxyl radical
- C08J2333/10—Homopolymers or copolymers of methacrylic acid esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/04—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters
- C08J2333/14—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters of esters containing halogen, nitrogen, sulfur, or oxygen atoms in addition to the carboxy oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
- C08K2201/006—Additives being defined by their surface area
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Analytical Chemistry (AREA)
- Dispersion Chemistry (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Abstract
Description
高吸水性樹脂(A)、下記i)およびii)の特性を有する粒子(B)、および水(C)を含む破砕抵抗性高吸水性樹脂であって、
前記粒子(B)は、前記高吸水性樹脂(A)の100重量部に対して0.0001〜15重量部含まれ、水(C)は、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して0.1〜20.0重量部含まれることを特徴とする、破砕抵抗性高吸水性樹脂を提供する。
i)300〜1500m2/gのBET比表面積(specific surface area)、
ii)50%以上の孔隙率(porosity)
a)高吸水性樹脂(A)に、下記i)およびii)の特性を有する粒子(B)を、前記高吸水性樹脂(A)の100重量部に対して0.0001〜15重量部となるように添加して処理するステップと、
b)前記ステップa)で処理された高吸水性樹脂(A)および前記粒子(B)に、水(C)を、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して0.1〜20.0重量部となるように投入して、加水された高吸水性樹脂を製造するステップとを含む、破砕抵抗性高吸水性樹脂の製造方法を提供する。
i)300〜1500m2/gのBET比表面積(specific surface area)、
ii)50%以上の孔隙率(porosity)
高吸水性樹脂(A)、下記i)およびii)の特性を有する粒子(B)、および水(C)を含む破砕抵抗性高吸水性樹脂であって、前記粒子(B)は、前記高吸水性樹脂(A)の100重量部に対して0.0001〜15重量部含まれ、水(C)は、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して0.1〜20.0重量部含まれることを特徴とする。
i)300〜1500m2/gのBET比表面積(specific surface area)、
ii)50%以上の孔隙率(porosity)
a)水溶性エチレン系不飽和単量体および重合開始剤を含む単量体組成物を熱重合または光重合して、含水ゲル状重合体を用意するステップと、
b)前記含水ゲル状重合体を乾燥するステップと、
c)前記乾燥した含水ゲル状重合体を粉砕して高吸水性樹脂粒子を得るステップと、
d)前記高吸水性樹脂粒子に表面架橋剤を添加して表面架橋反応を進行させるステップとを含んで製造されることを特徴とする。
a)高吸水性樹脂(A)に、下記i)およびii)の特性を有する粒子(B)を、前記高吸水性樹脂(A)の100重量部に対して0.0001〜15重量部となるように添加して処理するステップと、
b)前記ステップa)で処理された高吸水性樹脂(A)および前記粒子(B)に、水(C)を、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して0.1〜20.0重量部となるように投入して、加水された高吸水性樹脂を製造するステップとを含む、破砕抵抗性高吸水性樹脂の製造方法を含む。
i)300〜1500m2/gのBET比表面積(specific surface area)、
ii)50%以上の孔隙率(porosity)
a)水溶性エチレン系不飽和単量体および重合開始剤を含む単量体組成物を熱重合または光重合して、含水ゲル状重合体を用意するステップと、
b)前記含水ゲル状重合体を乾燥するステップと、
c)前記乾燥した含水ゲル状重合体を粉砕して高吸水性樹脂粒子を得るステップと、
d)前記高吸水性樹脂粒子に表面架橋剤を添加して表面架橋反応を行うステップとを含んで製造されることを特徴とする。
上述した内容のほか、本発明の破砕抵抗性高吸水性樹脂を製造する方法に記載の高吸水性樹脂(A)、粒子(B)、水(C)に関する具体的な説明は、本明細書において破砕抵抗性高吸水性樹脂について説明した内容を援用する。
アクリル酸100g、架橋剤としてポリエチレングリコールジアクリレート0.3g、開始剤としてジフェニル(2,4,6−トリメチルベンゾイル)−ホスフィンオキシド0.033g、苛性ソーダ(NaOH)38.9g、および水103.9gの比率で混合して、単量体混合物を用意した。以後、前記単量体混合物を連続移動するコンベヤベルト上に投入し、紫外線を照射(照射量:2mW/cm2)して2分間UV重合を進行させて、含水ゲル重合体を得た。前記用意された含水ゲル状重合体を5*5mmの大きさに切断して、170℃の温度の熱風乾燥機で2時間乾燥し、ピンミル粉砕機で粉砕した後、篩(sieve)を用いて、粒径サイズが150〜850μmの高吸水性樹脂を得た。
高吸水性樹脂が最終的に製品に適用される時に現れる物性の低下程度を模写して確認すべく、ボールミリング(ball milling)を通した破砕抵抗性(attrition resistance)確認実験を行った。破砕抵抗性に水の及ぼす影響を確認すべく、高吸水性樹脂に600ppmの多孔性超疎水性微細粒子を処理した後、以下のように2.5、5.0、7.5重量%の水を加水した。多孔性超疎水性微細粒子のエアロゲル(JIOS社)を使用し、使用した多孔性超疎水性微細粒子のエアロゲルの粒度は5μmであり、720m2/gのBET比表面積を有し、水に対する接触角は144゜であり、孔隙率は95%であった。
[式1]
孔隙率(porosity、%)=(1−ρt/ρs)*100
前記製造例1による破砕抵抗性高吸水性樹脂の物性を評価するために、下記のような試験を行った。
前記製造例1で用意された高吸水性樹脂それぞれに対する保水能を測定した。
前記製造例1で用意された高吸水性樹脂それぞれに対する加圧吸水能を測定した。加圧吸水能の測定は、欧州不織布産業協会(European Disposables and Nonwovens Association、EDANA)規格EDANA WSP242.3(11)(IST242.2(02))に従って測定した。
本発明のSFC値は、米国特許第5669894号に開示されたSFC試験方法に従って、前記製造例1で用意された高吸水性樹脂それぞれに対する、荷重2.07kPaでの0.69重量%塩化ナトリウム水溶液の通液性を測定した。
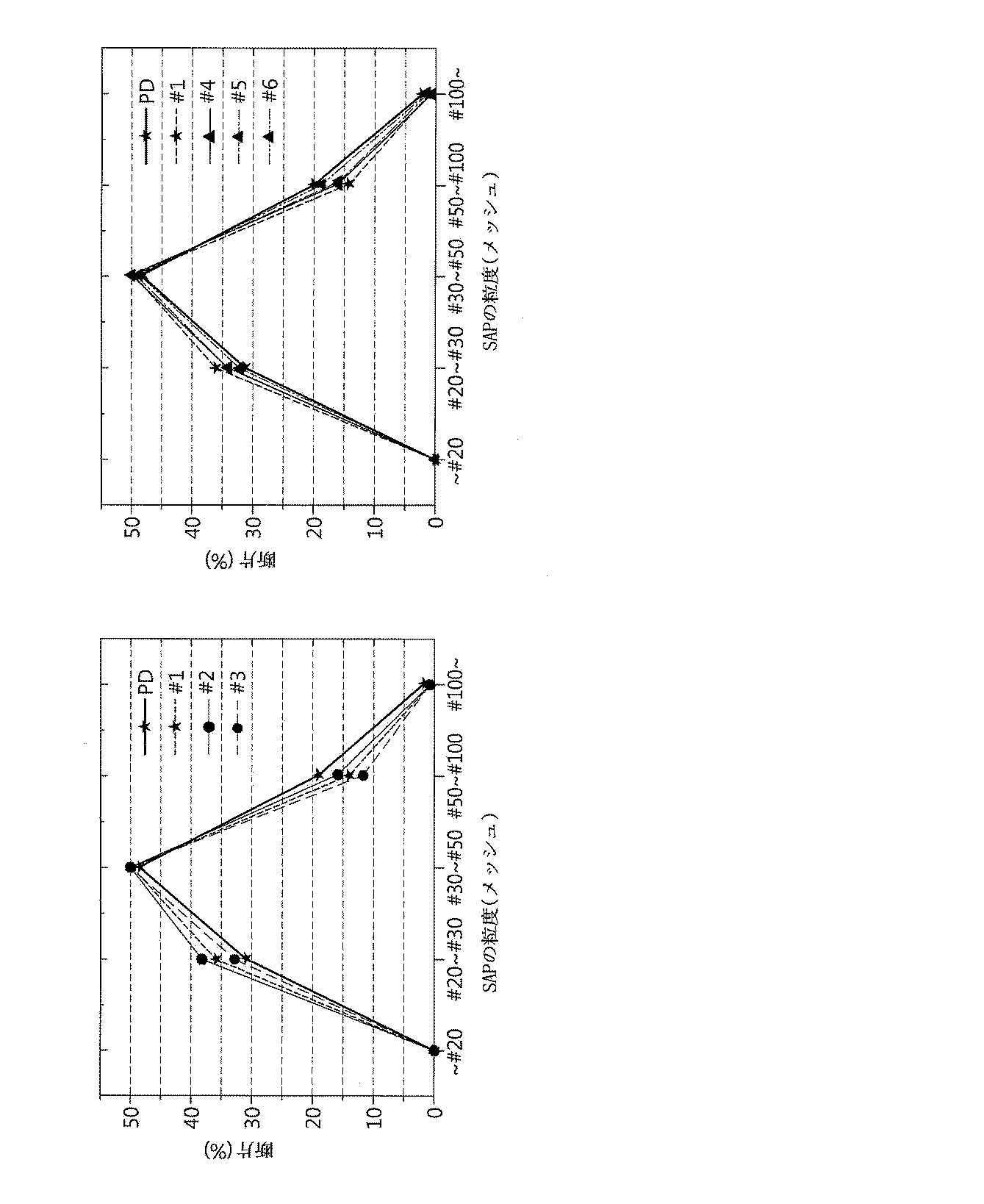
前記製造例1の結果に基づいてパイロット実験を行った。Batchあたり25kgの高吸水性樹脂を使用し、以下の表のように、高吸水性樹脂に多孔性超疎水性微細粒子を処理した後、水を投入して破砕抵抗性を確認した。高吸水性樹脂自体をP/D(比較例1)、高吸水性樹脂に何らかの処理を施さず撹拌だけを実施した試料を#1(比較例2)、多孔性超疎水性微細粒子(Aerogel)を処理し水を添加しなかった試料を#2(比較例3)と#4(比較例4)とし、多孔性超疎水性微細粒子(Aerogel)を処理し水の含有量に応じて分類した試料をそれぞれ#3(実施例1)、#5(実施例2)、#6(実施例3)として実験を行った。
ボールミリング後の粒度の変化をより確実に観察するために、前記製造例2の各試料を#30と#50の大きさの篩で分級した。以後、製造例1の方法でボールミリングを実施した。ボールミリングに破砕された高吸水性樹脂を再び#30と#50の大きさの篩で分級して、#30〜#50の区間の高吸水性樹脂の減少を確認した。これに対する結果は図3にグラフとして示した。図3において、高吸水性樹脂に何らかの処理を施さなかった試料(PD)の場合、ボールミリング後、#30〜#50の区間の高吸水性樹脂中の84.07%が残っていることが分かった。これは、約16%の高吸水性樹脂が破砕されたことを意味するのである。撹拌だけを施した試料#1(比較例2)の場合も、類似の結果が得られた。高吸水性樹脂にそれぞれ600ppmおよび1,000ppmの多孔性超疎水性微細粒子を導入した#3、#5の試料においても、類似する程度の破砕が起こることが分かる。しかし、#3、#5、#6の場合のように高吸水性樹脂に水が導入された場合、粒子の破砕程度が確実に減少することを確認することができた。前記結果から、高吸水性樹脂に水が投入されると、可塑剤の役割を果たして粒子の破砕を抑制する効果を示すことを確認することができた。
ボールミリング前/後の高吸水性樹脂に対する保水能の変化を観察するために、前記製造例2の各試料を#30と#50の大きさの篩で分級した。以後、製造例1の方法でボールミリングを実施した。ボールミリングで破砕された高吸水性樹脂を再び#30と#50の大きさの篩で分級して、#30〜#50の区間のそれぞれに対する保水能の変化を測定した。これに対する結果は図4にグラフとして示した。保水能の測定は、前記実験例1の方法を援用した。図4に示されているように、#3、#5、#6の場合、水を投入しただけのCRCの低下が起こることを確認することができた。ボールミリング前/後のCRCの増加幅は、水を投入しなかった場合より少なく上昇して、粒子の破砕によるCRC物性の変化が相対的に少ないことを、前記結果から間接的に確認することができた。
製造例3の方法で製造された破砕抵抗性高吸水性樹脂に対して、以後、前記実験例2の方法と同様に加圧吸水能(AUP、Absorption Under Pressure)を測定して物性の変化を確認し、その結果を図5Aに示した。高吸水性樹脂に何らかの処理を施さなかった試料対比、水が投入された試料の場合、ボールミリング後にAUPがさらに高いことを確認した。#2と#3の試料をみると、同量の多孔性超疎水性微細粒子を使用したにもかかわらず、水を投入した場合、ボールミリング後、AUPの低下が減少することを確認することができた。
前記製造例3の方法で製造された破砕抵抗性高吸水性樹脂に対して、以後、通液性(permeability)を測定して物性の変化を確認し、その結果を図5Bに示した。通液性も粒子の破砕と深い関連性があり、図5Bのグラフを通して確認することができた。高吸水性樹脂自体または高吸水性樹脂に多孔性超疎水性微細粒子のみを導入した場合は、ボールミリング後、すべて100%以上に通液性が悪くなる結果を現した。反面、高吸水性樹脂に水が投入された場合には、その半分またはそれ以下に相当する程度に通液性が悪くなることを確認することができた。この結果は、高吸水性樹脂に水が投入された場合には、外部の圧力および衝撃にもかかわらず、物性の低下が少なくなり、物理的破砕にも強いことを示すのである。したがって、本発明によれば、高吸水性樹脂に水を投入しても表面の粘度が増加せず粒度が維持され、したがって、加工性の減少がない破砕抵抗性高吸水性樹脂を提供することにより、製造工程での負荷の減少および粒度および物性の制御が容易になると予想することができる。
Claims (37)
- 高吸水性樹脂(A)、下記i)およびii)の特性を有する粒子(B)、および水(C)を含む破砕抵抗性高吸水性樹脂であって、
前記粒子(B)は、前記高吸水性樹脂(A)の100重量部に対して0.0001〜15重量部含まれ、水(C)は、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して0.1〜20.0重量部含まれることを特徴とする、破砕抵抗性高吸水性樹脂:
i)300〜1500m2/gのBET比表面積(specific surface area)、
ii)50%以上の孔隙率(porosity)。 - 前記粒子(B)は、前記高吸水性樹脂(A)の100重量部に対して0.001〜2.0重量部含まれることを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、前記高吸水性樹脂(A)の100重量部に対して0.05〜0.15重量部含まれることを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 水(C)は、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して1.0〜10.0重量部含まれることを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 水(C)は、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して2.5〜7.5重量部含まれることを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、2nm〜50μmの粒度を有することを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、水に対する接触角が125゜以上の超疎水性を有することを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、2nm〜50μmの粒度、および水に対する接触角が125゜以上の超疎水性を有することを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、500〜1500m2/gのBET比表面積(specific surface area)を有することを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、600〜1500m2/gのBET比表面積(specific surface area)を有することを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、水に対する接触角が135゜以上の超疎水性を有することを特徴とする、請求項7に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、水に対する接触角が140゜以上の超疎水性を有することを特徴とする、請求項7に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、90%以上の孔隙率(porosity)を有することを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記粒子(B)は、シリカ(SiO2)、アルミナ、炭素(Carbon)、およびチタニア(TiO2)からなる群より選択される1種以上であることを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- 前記破砕抵抗性高吸水性樹脂は、実含水率(Moisture content)が0.1重量%以上であることを特徴とする、請求項1に記載の破砕抵抗性高吸水性樹脂。
- a)高吸水性樹脂(A)に、下記i)およびii)の特性を有する粒子(B)を、前記高吸水性樹脂(A)の100重量部に対して0.0001〜15重量部となるように添加して処理するステップと、
b)前記ステップa)で処理された高吸水性樹脂(A)および前記粒子(B)に、水(C)を、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して0.1〜20.0重量部となるように投入して、加水された高吸水性樹脂を製造するステップとを含むことを特徴とする、破砕抵抗性高吸水性樹脂の製造方法:
i)300〜1500m2/gのBET比表面積(specific surface area)、
ii)50%以上の孔隙率(porosity)。 - 前記ステップb)の加水された高吸水性樹脂を製造するステップの後に、
c)前記加水された高吸水性樹脂を粉砕して破砕抵抗性を確認するステップを含むことを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。 - 前記粒子(B)は、前記高吸水性樹脂(A)の100重量部に対して0.001〜2.0重量部含まれることを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、前記高吸水性樹脂(A)の100重量部に対して0.05〜0.15重量部含まれることを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 水(C)は、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して1.0〜10.0重量部含まれることを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 水(C)は、高吸水性樹脂(A)および前記粒子(B)の100重量部に対して2.5〜7.5重量部含まれることを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、2nm〜50μmの粒度を有することを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、水に対する接触角が125゜以上の超疎水性を有することを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、2nm〜50μmの粒度、および水に対する接触角が125゜以上の超疎水性を有することを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、500〜1500m2/gのBET比表面積(specific surface area)を有することを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、600〜1500m2/gのBET比表面積(specific surface area)を有することを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、水に対する接触角が135゜以上の超疎水性を有することを特徴とする、請求項23に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、水に対する接触角が140゜以上の超疎水性を有することを特徴とする、請求項23に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、90%以上の孔隙率(porosity)を有することを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記粒子(B)は、シリカ(SiO2)、アルミナ、炭素(Carbon)、およびチタニア(TiO2)からなる群より選択される1種以上であることを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記高吸水性樹脂(A)は、
a)水溶性エチレン系不飽和単量体および重合開始剤を含む単量体組成物を熱重合または光重合して、含水ゲル状重合体を用意するステップと、
b)前記含水ゲル状重合体を乾燥するステップと、
c)前記乾燥した含水ゲル状重合体を粉砕して高吸水性樹脂粒子を得るステップと、
d)前記高吸水性樹脂粒子に表面架橋剤を添加して表面架橋反応を行うステップとを含んで製造されることを特徴とする、請求項16に記載の破砕抵抗性高吸水性樹脂の製造方法。 - 前記水溶性エチレン系不飽和単量体は、陰イオン性単量体とその塩、非イオン系親水性含有単量体、およびアミノ基含有不飽和単量体およびその4級化物からなる群より選択されるいずれか1つ以上であることを特徴とする、請求項31に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記熱重合のための重合開始剤は、過硫酸塩系開始剤、アゾ系開始剤、過酸化水素、およびアスコルビン酸からなる群より選択されるいずれか1つ以上であることを特徴とする、請求項31に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記光重合のための重合開始剤は、ベンゾインエーテル(benzoin ether)、ジアルキルアセトフェノン(dialkyl acetophenone)、ヒドロキシルアルキルケトン(hydroxyl alkylketone)、フェニルグリオキシレート(phenyl glyoxylate)、ベンジルジメチルケタール(Benzyl Dimethyl Ketal)、アシルホスフィン(acyl phosphine)、およびアルファ−アミノケトン(α−aminoketone)からなる群より選択されるいずれか1つ以上であることを特徴とする、請求項31に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記ステップb)の乾燥ステップは、150℃〜250℃の温度で行われることを特徴とする、請求項31に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記ステップc)の粉砕後に得られる高吸水性樹脂粒子の粒度範囲は、150〜850μmであることを特徴とする、請求項31に記載の破砕抵抗性高吸水性樹脂の製造方法。
- 前記表面架橋剤は、多価アルコール化合物;エポキシ化合物;ポリアミン化合物;ハロエポキシ化合物;ハロエポキシ化合物の縮合生成物;オキサゾリン化合物;モノ−、ジ−またはポリオキサゾリジノン化合物;環状ウレア化合物;多価金属塩;およびアルキレンカーボネート化合物からなる群より選択されるいずれか1つ以上であることを特徴とする、請求項31に記載の破砕抵抗性高吸水性樹脂の製造方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020140186684A KR101967807B1 (ko) | 2014-12-23 | 2014-12-23 | 파쇄 저항성 고흡수성 수지 및 그 제조 방법 |
| KR10-2014-0186684 | 2014-12-23 | ||
| PCT/KR2015/010865 WO2016104926A1 (ko) | 2014-12-23 | 2015-10-14 | 파쇄 저항성 고흡수성 수지 및 그 제조 방법 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017505848A true JP2017505848A (ja) | 2017-02-23 |
| JP6317462B2 JP6317462B2 (ja) | 2018-04-25 |
Family
ID=56150910
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016551152A Active JP6317462B2 (ja) | 2014-12-23 | 2015-10-14 | 破砕抵抗性高吸水性樹脂およびその製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US10307731B2 (ja) |
| EP (1) | EP3124529A4 (ja) |
| JP (1) | JP6317462B2 (ja) |
| KR (1) | KR101967807B1 (ja) |
| CN (1) | CN106062071B (ja) |
| BR (1) | BR112016018438B1 (ja) |
| TW (1) | TWI589629B (ja) |
| WO (1) | WO2016104926A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019216530A1 (ko) * | 2018-05-09 | 2019-11-14 | 주식회사 금빛 | 금속 나노입자가 포함된 항균 초흡수성 고분자 흡수재의 제조방법 |
| JP2024541167A (ja) * | 2022-10-21 | 2024-11-08 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20170070801A (ko) | 2015-12-14 | 2017-06-22 | 주식회사 엘지화학 | 내파쇄성 고흡수성 수지, 그의 제조 방법 및 제조용 조성물 |
| KR102025892B1 (ko) | 2016-02-17 | 2019-09-26 | 주식회사 엘지화학 | 내고화성이 향상된 고흡수성 수지의 제조 방법 |
| KR102665374B1 (ko) * | 2018-10-02 | 2024-05-09 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102578742B1 (ko) * | 2019-09-18 | 2023-09-14 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102671013B1 (ko) | 2019-11-05 | 2024-05-30 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| BR112022024465A2 (pt) | 2020-09-11 | 2023-04-04 | Lg Chemical Ltd | Polímero superabsorvente e método de preparação do mesmo |
| KR102888753B1 (ko) * | 2022-07-12 | 2025-11-21 | 주식회사 엘지화학 | 고흡수성 수지 복합체의 제조방법 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS644653A (en) * | 1987-06-26 | 1989-01-09 | Lion Corp | Polymeric absorbent composition |
| JPH08176338A (ja) * | 1994-12-22 | 1996-07-09 | Uni Charm Corp | 消臭性樹脂組成物およびその製法 |
| JPH08253597A (ja) * | 1995-03-15 | 1996-10-01 | Nippon Synthetic Chem Ind Co Ltd:The | 高吸水性樹脂の造粒法 |
| JP2000109714A (ja) * | 1998-08-07 | 2000-04-18 | Sanyo Chem Ind Ltd | 吸水剤およびその製造法 |
| JP2011178969A (ja) * | 2010-03-04 | 2011-09-15 | San-Dia Polymer Ltd | 吸収性樹脂粒子及びこの製造方法 |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2888852B2 (ja) * | 1989-03-13 | 1999-05-10 | 三菱化学株式会社 | 粉体高吸水性ポリマー組成物 |
| SE8903180D0 (sv) | 1989-09-27 | 1989-09-27 | Sca Development Ab | Saett att behandla fibrer av cellulosahaltigt material |
| DE4426008A1 (de) | 1994-07-22 | 1996-01-25 | Cassella Ag | Hydrophile, hochquellfähige Hydrogele |
| DE19524724A1 (de) | 1995-07-07 | 1997-01-09 | Hoechst Ag | Hydrophile, hochquellfähige Hydrogele |
| EP0797966A1 (en) | 1996-03-29 | 1997-10-01 | The Procter & Gamble Company | Collapsed superabsorbent foams |
| US6414214B1 (en) | 1999-10-04 | 2002-07-02 | Basf Aktiengesellschaft | Mechanically stable hydrogel-forming polymers |
| DE60143706D1 (de) * | 2000-07-18 | 2011-02-03 | Sanyo Chemical Ind Ltd | Absorbens und verfahren zu dessen herstellung, absorbierbare artikel und syntheseprodukte |
| DE10125599A1 (de) | 2001-05-25 | 2002-11-28 | Stockhausen Chem Fab Gmbh | Superabsorber, Verfahren zu ihrer Herstellung und ihre Verwendung |
| DE10161496A1 (de) | 2001-12-14 | 2003-06-26 | Stockhausen Chem Fab Gmbh | Kompaktierte absorbierende Polymere, deren Herstellung und Verwendung |
| US20040044321A1 (en) | 2002-08-27 | 2004-03-04 | Kainth Arvinder Pal Singh | Superabsorbent materials having controlled gel-bed friction angles and cohesion values and composites made from same |
| WO2005120594A1 (en) | 2004-06-07 | 2005-12-22 | Dow Global Technologies Inc. | Polymers with odor control properties and method for their preparation |
| CA2495473A1 (en) * | 2005-01-26 | 2006-07-26 | Stephane Chevigny | Polysaccharide-inorganic agglomerated particles as performance additives for superabsorbent polymers |
| JP5419712B2 (ja) | 2007-02-06 | 2014-02-19 | ビーエーエスエフ ソシエタス・ヨーロピア | モノマー溶液の液滴の重合による吸水性ポリマー粒子を製造する方法 |
| JP2009057496A (ja) * | 2007-08-31 | 2009-03-19 | San-Dia Polymer Ltd | 吸水性樹脂粒子、吸収体及び吸収性物品 |
| KR101887706B1 (ko) | 2009-09-30 | 2018-08-10 | 가부시키가이샤 닛폰 쇼쿠바이 | 입자상 흡수제 및 그 제조방법 |
| JP5485805B2 (ja) | 2010-06-15 | 2014-05-07 | 住友精化株式会社 | 吸水性樹脂 |
| JP2012052080A (ja) * | 2010-09-02 | 2012-03-15 | Ko Tamihiro | 吸湿又は吸放湿性樹脂ペレット |
| JP2012217599A (ja) * | 2011-04-08 | 2012-11-12 | San-Dia Polymer Ltd | 吸収性樹脂粒子、これを含む吸収体及び吸収性物品 |
| KR101559081B1 (ko) | 2012-11-15 | 2015-10-08 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 이로부터 제조되는 고흡수성 수지 |
| CN103044752A (zh) * | 2013-01-16 | 2013-04-17 | 合肥杰事杰新材料股份有限公司 | 一种用于再生塑料成型的吸水母粒及其制备方法 |
| KR101584719B1 (ko) * | 2013-04-22 | 2016-01-13 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR20140134219A (ko) * | 2013-05-13 | 2014-11-21 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| JP6700190B2 (ja) | 2013-12-03 | 2020-05-27 | エルジー・ケム・リミテッド | 高吸水性樹脂およびその製造方法 |
| KR20150064649A (ko) | 2013-12-03 | 2015-06-11 | 주식회사 엘지화학 | 고흡수성 수지의 제조방법 |
-
2014
- 2014-12-23 KR KR1020140186684A patent/KR101967807B1/ko active Active
-
2015
- 2015-10-14 JP JP2016551152A patent/JP6317462B2/ja active Active
- 2015-10-14 BR BR112016018438-6A patent/BR112016018438B1/pt active IP Right Grant
- 2015-10-14 WO PCT/KR2015/010865 patent/WO2016104926A1/ko not_active Ceased
- 2015-10-14 CN CN201580010885.6A patent/CN106062071B/zh active Active
- 2015-10-14 US US15/307,232 patent/US10307731B2/en active Active
- 2015-10-14 EP EP15873454.1A patent/EP3124529A4/en not_active Withdrawn
- 2015-11-25 TW TW104139171A patent/TWI589629B/zh active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS644653A (en) * | 1987-06-26 | 1989-01-09 | Lion Corp | Polymeric absorbent composition |
| JPH08176338A (ja) * | 1994-12-22 | 1996-07-09 | Uni Charm Corp | 消臭性樹脂組成物およびその製法 |
| JPH08253597A (ja) * | 1995-03-15 | 1996-10-01 | Nippon Synthetic Chem Ind Co Ltd:The | 高吸水性樹脂の造粒法 |
| JP2000109714A (ja) * | 1998-08-07 | 2000-04-18 | Sanyo Chem Ind Ltd | 吸水剤およびその製造法 |
| JP2011178969A (ja) * | 2010-03-04 | 2011-09-15 | San-Dia Polymer Ltd | 吸収性樹脂粒子及びこの製造方法 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019216530A1 (ko) * | 2018-05-09 | 2019-11-14 | 주식회사 금빛 | 금속 나노입자가 포함된 항균 초흡수성 고분자 흡수재의 제조방법 |
| JP2024541167A (ja) * | 2022-10-21 | 2024-11-08 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| JP7750619B2 (ja) | 2022-10-21 | 2025-10-07 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2016104926A1 (ko) | 2016-06-30 |
| EP3124529A4 (en) | 2017-05-24 |
| TWI589629B (zh) | 2017-07-01 |
| TW201638227A (zh) | 2016-11-01 |
| BR112016018438B1 (pt) | 2021-11-03 |
| US20170050171A1 (en) | 2017-02-23 |
| JP6317462B2 (ja) | 2018-04-25 |
| EP3124529A1 (en) | 2017-02-01 |
| US10307731B2 (en) | 2019-06-04 |
| KR101967807B1 (ko) | 2019-04-10 |
| KR20160076559A (ko) | 2016-07-01 |
| CN106062071A (zh) | 2016-10-26 |
| CN106062071B (zh) | 2019-04-16 |
| BR112016018438A2 (pt) | 2017-10-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6321822B2 (ja) | 耐固化性の向上した高吸水性樹脂およびその製造方法 | |
| JP6973870B2 (ja) | 高吸水性樹脂およびその製造方法 | |
| JP6317462B2 (ja) | 破砕抵抗性高吸水性樹脂およびその製造方法 | |
| JP6449902B2 (ja) | 高吸水性樹脂の製造方法 | |
| KR101507287B1 (ko) | 고흡수성 수지의 제조방법 | |
| JP6468538B2 (ja) | 高吸水性樹脂およびその製造方法 | |
| JP2016540106A5 (ja) | ||
| KR101960041B1 (ko) | 고흡수성 수지의 제조방법 | |
| CN106661235A (zh) | 用于制备超吸收性聚合物的方法和由此制备的超吸收性聚合物 | |
| CN106661299A (zh) | 用于制备超吸收性聚合物的方法 | |
| JP6466472B2 (ja) | 微細粒子を含む水分散液で処理された高吸水性樹脂の製造方法 | |
| US11111356B2 (en) | Attrition-resistant superabsorbent polymer, method for preparing the same and composition for preparing the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20160810 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170622 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170626 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170913 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180305 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20180329 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6317462 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |