JP2017522016A - 培養哺乳動物輪部幹細胞、その産生方法及びその使用 - Google Patents
培養哺乳動物輪部幹細胞、その産生方法及びその使用 Download PDFInfo
- Publication number
- JP2017522016A JP2017522016A JP2016575401A JP2016575401A JP2017522016A JP 2017522016 A JP2017522016 A JP 2017522016A JP 2016575401 A JP2016575401 A JP 2016575401A JP 2016575401 A JP2016575401 A JP 2016575401A JP 2017522016 A JP2017522016 A JP 2017522016A
- Authority
- JP
- Japan
- Prior art keywords
- lsc
- cells
- sesc
- population
- pax6
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
- C12N5/0621—Eye cells, e.g. cornea, iris pigmented cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/30—Nerves; Brain; Eyes; Corneal cells; Cerebrospinal fluid; Neuronal stem cells; Neuronal precursor cells; Glial cells; Oligodendrocytes; Schwann cells; Astroglia; Astrocytes; Choroid plexus; Spinal cord tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/36—Skin; Hair; Nails; Sebaceous glands; Cerumen; Epidermis; Epithelial cells; Keratinocytes; Langerhans cells; Ectodermal cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/10—Ophthalmic agents for accommodation disorders, e.g. myopia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0625—Epidermal cells, skin cells; Cells of the oral mucosa
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/11—Epidermal growth factor [EGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/235—Leukemia inhibitory factor [LIF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/33—Insulin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases [EC 2.]
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/998—Proteins not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/03—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from non-embryonic pluripotent stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2509/00—Methods for the dissociation of cells, e.g. specific use of enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2513/00—3D culture
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/90—Substrates of biological origin, e.g. extracellular matrix, decellularised tissue
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/106—Pharmacogenomics, i.e. genetic variability in individual responses to drugs and drug metabolism
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Ophthalmology & Optometry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Developmental Biology & Embryology (AREA)
- General Engineering & Computer Science (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Pathology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
Abstract
Description
本発明の分野は、眼科的障害、疾患及び傷害を処置するための方法及び組成物に関する。詳細には、本発明の分野は、角膜及び眼表面の障害、疾患、欠損及び傷害を処置するための方法、キット及び組成物に関する。本開示は、角膜輪部組織に由来する、培養哺乳動物輪部幹細胞の調製に関する。好ましい実施形態では、輪部幹細胞株は、自己再生性であり、角膜上皮組織に分化する能力がある。輪部幹細胞株を培養する方法、並びにそれらの使用の方法及び組成物も開示する。
用語「培養培地」、「細胞培養培地」又は「細胞培地」は、細胞、例えば幹細胞、前駆細胞又は分化細胞を増殖させる細胞増殖培地を記述するために使用される。培養培地は当技術分野において公知であり、少なくとも最小必須培地に加えて任意選択の薬剤、例えば、増殖因子(例えば、線維芽細胞増殖因子、好ましくは塩基性線維芽細胞増殖因子(bFGF)、及び上皮増殖因子(EGF)を含む)、サイトカイン(例えば、白血病抑制因子(LIF))、ホルモン(例えば、グルココルチコイド(例えばヒドロコルチゾン)及び甲状腺ホルモン(例えば、3,3',5-トリヨード-L-チロニン)を含む)、グルコース、非必須アミノ酸、グルタミン、インスリン、トランスフェリン、ベータメルカプトエタノール、ROCK阻害剤、コレラ毒素、及び当技術分野において周知の他の薬剤を含む。そのような培地は、L-グルタミン、ノックアウト血清代替物(KSR)、ウシ胎仔血清(FBS)、非必須アミノ酸、白血病抑制因子(LIF)、上皮増殖因子(EGF)、ベータ-メルカプトエタノール、塩基性線維芽細胞増殖因子(bFGF)、ヒドロコルチゾン、3,3',5-トリヨード-L-チロニン、ROCK阻害剤、抗生物質、B27培地サプリメント及び/又は他の培地サプリメントのいずれか1つ以上が補足されていてもよい、DMEM/F12(1:1)などの市販の培地を含む。本発明に有用な細胞培地は市販されており、非常に多数の商業的供給源の中でもInvitrogen Corp.(GIBCO)及びBiological Industries、Beth HaEmek、Israelから入手できる市販の成分をそのような細胞培地に補足することができる。
本発明は、染色体に組み込まれた、あるいは組み込まれていない染色体外遺伝物質のままのPAX6をコードする、化学合成、組換え、又は単離核酸を含む、単離された輪部幹若しくは前駆細胞(LSC)集団又はLSC様集団であって、前記単離されたLSC集団は非LSC細胞を実質的に含まず、又は前記LSC様集団は非LSC様細胞を実質的に含まず、又は前記単離されたLSC又はLSC様集団は、非LSC細胞及び非LSC様細胞を実質的に含まない、上記単離されたLSC集団又はLSC様集団を提供する。LSC集団又はLSC様集団は、ヒトなどの哺乳動物からのものであってもよい。LSC集団又はLSC様集団は、遺伝子改変されていてもよい。さらに、LSC集団又はLSC様集団は、LSC又はLSC様細胞運命のままであることもあり、又はLSC又はLSC様細胞運命を維持することもある。
本発明の一実施形態では、本開示は、哺乳動物輪部幹細胞(LSC)の培養に関する。輪部幹細胞は、ヒトドナーからの角強膜又は角膜輪部組織に由来する。詳細には、本開示は、自己再生輪部幹細胞を有する系であり、LSC、例えば、少なくとも約70%、少なくとも約80%、又は少なくとも約90%輪部幹細胞の大集団を含むことができる。角膜輪部組織を単離するための典型的な手技は、ドナーの眼の角膜表面の上又は耳側四分円(temporal quadrant)からの、0.8〜3mm2の輪部組織からなる小さい検体材料の、外科的除去である。角膜輪部からそのような検体材料を例えば表層角膜切除術によって得るための手技は、当業者には公知である。輪部幹細胞を産生するために使用される輪部組織検体材料のドナーは、組織系移植片、インプラント又はグラフトのレシピエントであることもある(すなわち自家組織系)。あるいは、輪部組織検体材料のドナーがレシピエントでない場合、ドナーは、一例として、生体適合性ドナー、例えば移植片若しくはグラフトのレシピエントの近親者であり、又は生体適合性(例えば組織適合性)の死体であることもある(すなわち同種組織系)。移植される細胞又は組織は、組織拒絶反応に関する問題を回避するためにその移植片のレシピエントと遺伝学的に適合性又は同一であることが一般には望ましい。
本発明は、輪部幹細胞又は角膜上皮細胞の機能不全に関連した疾患を有する対象を処置する方法を提供する。一実施形態では、この方法は、(a)本発明の、又は本発明の方法によって生産された、LSC、LSC様、CEC又はCEC様細胞を対象の罹患した眼に移植するステップを含み、移植された細胞が前記対象の罹患した眼の角膜又は輪部に定着し、正常な角膜透明性及び透明度を回復させ、それによって、輪部幹若しくは前駆細胞又は角膜上皮細胞の機能不全に関連した疾患を有する対象を処置する。
本発明は、眼疾患又は状態を有する対象を処置する方法であって、本明細書に記載の輪部幹細胞の治療量を含む組成物を前記対象への送達に適している治療製剤で前記対象に投与することを含む方法を企図している。角膜損傷は、病態生理に関与することに関係づけられているいずれの傷害、状態又は疾患であってもよい。非限定的な例を下で述べる。
角膜創傷は、眼表面の傷害であり、熱的創傷(すなわち熱傷)、化学的(すなわち酸)創傷、物理的創傷(すなわち擦過創)若しくは手術(すなわち角膜移植)創傷又はこれらの組合せであることができる。本発明の組成物及び方法は、角膜創傷の処置に有効でありうる。
眼の傷害全体の約25%は、角膜表面の異物に関わる。傷害が角膜上皮しか侵さない場合は瘢痕化は起こらないであろうが、ボーマンゾーンも侵す場合には瘢痕化が起こりうる。異物の除去後、眼をスルホンアミド又は抗生物質で処置し、毛様体うっ血及び羞明がある場合又は異物の除去が難しい場合には眼を約5%ホマトロピンなどの毛様体調節薬で処置する。場合によっては、本発明の治療用組成物は、異物に起因する傷害の治癒を促進するように、感染を予防するように、及び臨床成績を向上させるように設計される。
化学熱傷は、眼に液体を流すことによって化学薬品を先ず希釈し、その後、局所抗生物質の使用によって感染を予防することにより処置される。
角膜の裂傷後、損傷部を閉鎖する虹彩脱出が生ずる。すべての眼傷害の場合と同様、感染症のリスクがある。裂傷は強膜に及ぶこともあり、これは、よりいっそう重篤な傷害である。そのような場合、脱出したぶどう膜組織を傷害領域から除去するために手術が必要であり、強膜を縫合で閉じる。本発明の治療用組成物は、裂傷の治癒を促進するように及び感染症を予防するように設計される。
角膜炎は、角膜の炎症を指す。原因としては、アメーバ、細菌、真菌若しくはウイルス感染、光線角膜炎、被曝(眼瞼機能障害)、化学的傷害、外傷、手術(LASIK、PRK、白内障、角膜移植、翼状片手術)、又は先天的原因、例えば円錐角膜、フックスジストロフィー、若しくは乾性角結膜炎が挙げられるが、これらに限定されない。眼の他の炎症媒介状態としては、ぶどう膜炎、黄斑浮腫、加齢性黄斑変性、網膜剥離、眼腫瘍、多巣性脈絡膜炎、糖尿病性ぶどう膜炎、増殖性硝子体網膜症(PVR)、交感神経性眼炎、フォークト・小柳・原田(VKH)症候群、ヒストプラスマ症、及びぶどう膜滲出が挙げられるが、これらに限定されない。
本発明によるマイクロスフェアの組成物は、眼の手術に関連した炎症を改善するためにも使用されることがあり、これに関連して、予防的様式ではもちろん、上で詳述したような治癒の促進及び瘢痕化の低減にも特に有用である。
前眼状態は、前眼(すなわち眼の前方)領域又は部位、例えば、眼周囲の筋肉、眼瞼若しくは眼球組織、又は水晶体嚢の前壁から後壁に位置する流体、又は毛様体筋を侵す又はそれらに関わる疾患、病気又は状態である。したがって、前眼状態は、結膜、角膜、前眼房、虹彩、後眼房(虹彩の後ろ、しかし水晶体嚢の後壁の前)、水晶体又は水晶体嚢、並びに前眼領域又は部位に血管形成する又は神経支配する血管及び神経を侵す、又はそれらに関わる。
輪部幹細胞を含む組成物を対象に投与して、様々な細胞又は組織機能をもたらし、例えば、外傷、手術、遺伝的特徴、疾患などに起因する眼科障害を処置することができる。本明細書で使用する場合、「対象」は、ヒトを意味することもあり、非ヒト動物を意味することもある。
本発明は、最小必須培地、増殖因子、ホルモン、可溶性因子及び血清又は代用血清を含む、LSC、LSC様、SESC又はSESC様の短期インビトロ培養のためのフィーダーフリー細胞培養培地も提供する。一実施形態では、短期インビトロ培養を使用して、約0〜約第4継代のLSC、LSC様、SESC又はSESC様細胞を継代する。
以下の実施例は、本発明の方法及び組成物の製造及び使用方法の完全な開示及び説明を当業者に与えるために提示するものであり、本発明者らが自らの発明と考える範囲を限定することを意図したものではない。使用する数値(例えば、量、温度など)に関して正確を期すように努めたが、多少の実験的誤差及び偏差を考慮すべきである。別段の指示がない限り、部は重量部であり、分子量は平均分子量であり、温度は摂氏度であり、圧力は大気圧又はほぼ大気圧である。
材料及び方法
ヒト病理サンプル
角膜上皮扁平上皮化生及び他のすべての組織は、非特定化された手術標本として入手し、免疫蛍光研究のために5%ホルマリンで固定し、パラフィンに包埋し、薄片にし、染色した。
死後ヒト眼球をアイバンクから入手し、輪部領域を採取し、100IUペニシリン及び100μg/mlストレプトマイシンを含有する冷PBSで洗浄し、小片に切断した。37℃で2時間の0.2%コラゲナーゼIV消化によって細胞クラスターを得、37℃で15分間の0.25%トリプシン-EDTAでのさらなる消化によって単一細胞を得た。2%増殖因子低減Matrigel(354230、BD Biosciences, Inc.)で被覆されたプラスチックプレートに初代細胞を播種した。GFP標識ラット及びウサギからの輪部幹細胞を単離し、ヒトLSCについての方法と同じ方法を使用して培養した。
3D分化を24ウェルプレート又は8ウェルチャンバーで行った。簡単に言うと、解離させた単個幹細胞をMatrigel(登録商標)に2×104細胞/50μlゲルで包埋した。分化培地CnT-30(輪部幹細胞分化)又はCnT-02(皮膚表皮幹細胞分化)(CellnTec, Inc.)で14〜18日培養後、3D構造が形成された。
培養細胞中のタンパク質の局在を検出するために、細胞を4%パラホルムアルデヒドで20分間固定し、次いで0.3%Trion X-100-PBSで5分間、2回、透過処理し、5%ウシ血清アルブミン及び0.3%Triton X-100を含有するPBSでブロックし、その後、一次抗体中、4℃で一晩インキュベートした。PBSでの3回の洗浄後、細胞を二次抗体とともにインキュベートした。細胞核をDAPIで対比染色した。パラフィン包埋組織切片の免疫蛍光のために、脱パラフィンを行い、その後、上で説明したのと同じ免疫蛍光プロトコルを行った。
RNeasyキット(Qiagen, Inc.)を使用してRNAを単離し、オンカラムDNアーゼ消化に付した。Superscript III逆転写酵素キットをその製造業者(Invitrogen, Inc.)の説示に従って使用してcDNA合成を行った。7500リアルタイムPCRシステム(Applied Biosystems, Inc.)で遺伝子特異的プライマー(表1)及びPower SYBR Green PCR Master Mixを使用して40サイクル増幅によって定量的PCRを行った。測定を3回ずつ行い、内因性GAPDHレベルに正規化した。ΔΔCT法(CT値<30)を使用して発現の相対倍数変化を計算した。データを3回反復に基づいて平均±SDとして示す。

3D分化アッセイから、全RNAをLSC、SESC及び分化CECから単離した。Illuminaヒトゲノムマイクロアレイシステムを使用して、サンプルごとに生物学的反復実験で(群あたりn=2、Human HT-12 v4 Expression BeadChip、Illumina、San Diego、CA)、遺伝子発現マイクロアレイ解析を行った。生データを寄託番号GSE32145でGEOデータベースに寄託した。Illumina BeadStudioバージョン3.4.0によって発現レベルデータを生成し、四分位数正規化を使用して正規化した。少なくとも1つのサンプルで発現レベルが64の閾値を超えるプローブが検出されると考えた。閾値は、log2発現レベルの分布プロットから調査によって見つけた。検出されたプローブを、プローブが有意とみなされる最小偽陽性率(FDR)であるそれらのp値に従って選別した。マイクロアレイの有意性分析及び公式統計パッケージsam19でのその実装を用いてFDRを評価した。偽小分散に起因する偽陽性コールを回避するために、正則化t統計量において交換可能性因子s0に使用する標準偏差値の百分位数を50に設定した。本発明者らは、LESC及びCECサンプルを組み合わせて4サンプルの1群にし、この群とSESCサンプル間で差次的に発現される遺伝子を探した。この比較における上位100有意遺伝子を図4に提示する。この図中のすべての遺伝子は、0.01以下のFDRレベルで有意である。研究所内階層的クラスタリングソフトウェアを使用してヒートマップを生成した。色は倍数変化に定性的に対応する。
全RNAをPicropure RNA単離キット(Life Technology)によって精製した。RNA-seqは、以前に記載された20ように行った。簡単に言うと、600ngの全RNAを、superscript IIIファーストストランド合成キットとプライマーBiotin-B-Tによって、先ずcDNAに変換した。そのcDNAをNucleoSpin Gel and PCR Clean-Up Kitカラム(Clontech)によって精製して、遊離プライマー及び酵素を除去した。次いで、末端トランスフェラーゼ(NEB)を利用してcDNA 3'末端の端部をブロックした。ストレプトアビジン被覆磁気ビーズ(Life Technology)をさらに利用してcDNAを単離した。水酸化ナトリウムによるRNA消化後、第二鎖cDNAをプライマーA-N8でのランダムプライミングによって合成した。その第二鎖cDNAを熱変性によってビーズから溶離した。次いで、そのcDNAをテンプレートとして使用して、バーコードプライマー及びプライマーPBでの増幅によりライブラリーを構築した。シークエンシングは、Hiseq2000システムで行った。
PAX6、WNT7A及びFZD5遺伝子を標的にするレンチウイルスshRNAをAge IとEcoR I間のpLKO.1プラスミドにクローニングした、又はSigmaから直接購入した。遺伝子特異的ノックダウンのためのshRNA標的化配列は次の通りであった:PAX6、CGTCCATCTTTGCTTGGGAAA及びAGTTTGAGAGAACCCATTATC;WNT7A、CGTGCTCAAGGACAAGTACAA及びGCGTTCACCTACGCCATCATT;FZD5、CGCGAGCCCTTCGTGCCCATT及びTCCTAAGGTTGGCGTTGTAAT。本発明者らは、いずれの種からのいずれの既知遺伝子も標的にしないshRNAをコードするレンチウイルスpLKO.1-puro Non-Target shRNA対照プラスミド(Sigma, Inc.)を、すべての遺伝子ノックダウン実験において陰性対照として使用した。
ウェスタンブロッティングのために、細胞をPBSで1回洗浄し、その後、細胞溶解緩衝液(50mM Tris-HCl、pH6.8;2%SDS;10%グリセロール;100mM DTT)に回収した。タンパク質濃度をNanodropによって定量し、ブロモフェノールブルーを0.1%の最終濃度まで添加し、次いで25μgの全溶解物を4-12%NUPAGEゲル(Life Technology, Inc)で分画した。タンパク質を100Vで1時間、ニトロセルロース膜に転写した。その膜を5%ミルクでブロックし、関連抗体及びマウス抗β-アクチンモノクローナル抗体(A5316、Sigma, Inc.)でプローブした。
すべての動物研究は、眼科及び視覚研究における動物使用についてのARVO声明(ARVO statement for the Use of Animals in Ophthalmic and Vision Research)を完全に順守して行い、施設内実験動物管理委員会の承認を得た。
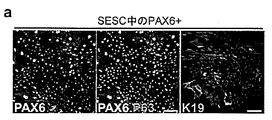
角膜及び皮膚上皮は、重層上皮の典型的な形態、並びに輪部及び表皮の基底細胞層のケラチン5/ケラチン14+(K5/K14)におけるp63によるそれらの幹細胞の維持を含む、多くの類似点を共有する4〜8(図1a、1b、2a及び2b)。しかし、それらには顕著な違いがある。皮膚上皮幹細胞(SESC)は、分化中に深部から基底上層へと上方に垂直に移動し9、10、そこでK5及びK14は皮膚特異的K1/K10によって置き換えられる(参考文献11、図2c及び2d)。対照的に、LSC(輪部でK19によって規定される12、図1a及び2eを参照されたい)は、分化を受けている間に中心角膜の方に求心的に数ミリメートル遊走し、K5/K14は角膜特異的K3及びK12によって置き換えられる(参考文献13及び4、図1c及び2f)。
参考文献
材料及び方法
動物
ROSAmT/mGマウスは以前に記載されており(PMID:17868096;28)、それをホモ接合体マウスとして飼育した。Pax6 P0エンハンサーの制御下でEGFP-Creリコンビナーゼ融合タンパク質を発現するP0-3.9-GFPCreマウスをFVBバックグラウンドで飼育し、記載されている(29)ようにPCR遺伝子型判定を行った。
ホモ接合体GFPレポーターマウス(ROSAmT/mG)と、Cre発現がマウスPax6外胚葉エンハンサーの制御下にある水晶体特異的Creトランスジェニックマウス(P0-3.9-GFPCre)とを交配させることによって、系統追跡実験を行った。生後第1日(P1)及びP60に眼を切開し、4%ホルムアルデヒドで一晩固定した。その後、組織を10%スクロース中でインキュベートし、凍結切片作成のために最適切断温度培地に包埋した。凍結切片をPBSで洗浄し、Zeiss Axio Imager蛍光顕微鏡で撮像した。
死後ヒト眼球は、アイバンクから入手し、ヒト表皮は、眼瞼のドナー皮膚生検から得た。輪部領域を切除し、100IU/mlペニシリン及び100μg/mlストレプトマイシンを含有する冷PBSで洗浄した。輪部領域を小片に切断した後、37℃で2時間の0.2%コラゲナーゼIV消化によって細胞クラスターを得た。この後、37℃で15分間、0.25%トリプシン/EDTAでさらに消化して単一細胞を得た。2%増殖因子低減Matrigel(登録商標)(354230、BD Biosciences, Inc.)で被覆されたプラスチックプレートに初代細胞を播種した。同じ処置を使用して毛包間表皮から初代ヒトSESCを単離した。
RNeasyキット(Qiagen, Inc.)を使用してRNAを単離した。オンカラムDNアーゼ消化を行った。Superscript III逆転写酵素キット(Invitrogen, Inc.)をその製造業者の説示に従ってcDNA合成に使用した。リアルタイムPCRシステム(Applied Biosystems, Inc.)を使用して定量的PCRを行った。遺伝子特異的プライマー(表3)及びPower SYBR Green PCR Master Mixを使用して40サイクルの増幅を行った。測定を3回ずつ行い、内因性GAPDHレベルに正規化した。ΔΔCT法(CT値<30)を使用して発現の相対倍数変化を計算した。データを3回反復に基づいて平均±S.D.として示す。
pLKO.1プラスミドのAge IとEcoR Iの間にクローニングしたレンチウイルスshRNAを使用してPAX6遺伝子を標的化した。遺伝子特異的ノックダウンのためのshRNA標的化配列は、5'-CGTCCATCTTTGCTTGGGAAA-3'及び5'-AGTTTGAGAGAACCCATTATC-3'であった。すべての遺伝子ノックダウン実験において、本発明者らは、いずれの種からのいずれの既知遺伝子も標的にしないshRNAをコードするレンチウイルスpLKO.1-puro non-target shRNA対照プラスミド(Sigma, Inc.)を陰性対照として使用した。レンチウイルスshRNA粒子を調製するために、複製能力のないレンチウイルス粒子を、shRNA構築物とパッケージング混合物(比率9:1でpCMV-dR8.2及びpCMV-VSVG)の共トランスフェクションによって、293T細胞にパッケージングした。ウイルスを2回、トランスフェクションの48時間及び72時間後に回収した。個々のウイルス及びポリブレンを8μg/mlの最終濃度で含有する新鮮培地を用いて、16〜20時間、細胞をレンチウイルスに感染させた。感染細胞をさらに2μg/mlピューロマイシンによって48時間選択した。
細胞を4%パラホルムアルデヒドで15分間、室温で固定し、0.3%Triton X-100を含有するリン酸緩衝食塩水(PBS)で10分間透過処理し、5%ウシ血清アルブミンと0.3%Triton X-100とを含有するPBSでブロックした。それらの細胞を一次抗体とともに18時間、4℃でインキュベートし、PBSで3回洗浄し、二次抗体とともに1時間インキュベートした。細胞核をDAPIで対比染色した。パラフィン包埋組織切片の免疫蛍光を、標準的な脱パラフィン、続いての上で説明したのと同じ免疫蛍光プロトコルによって遂行した。
本発明者らの以前の研究(15)の場合と同様に全RNAをLSC及びSESCから単離した。生データを寄託番号GSE32145でGene Expression Omnibus(GEO)データベースに寄託した。この研究で生成したマイクロアレイベースの遺伝子発現データを全サンプルにわたって正規化し、Cluster3.0/Tree Viewソフトウェアパッケージを使用する平均連結階層的クラスタリング(16)に付した。Wnt及びNotch経路に属する遺伝子の選択を以前の研究に基づいて行った。Reactomeネットワーク用のGeneSet解析ツール(17)を使用し、Cytoscape3用のReactome Functional Interaction(FI)プラグイン(18)を使用して生成したネットワークに発現値をオーバーレイした。
マウスにおける眼発生中のPAX6及びp63発現
角膜発生中のPAX6及びp63の機能を解明するために、本発明者らは、胎生12.5日(E12.5)〜E18.5のマウス胚におけるそれらの発現プロファイルを先ず研究した。E12.5で、強いPAX6発現が眼領域、特に初期角膜上皮において検出されたが、p63発現は陰性であった(図15A)。対照的に、p63陽性細胞は、眼瞼の発生及び融合後、E14.5で、眼表面に出現し、後に輪部及び角膜に拡大した(図15B)。PAX6発現は、眼発生中は眼領域に限定された(図15A〜D)。加えて、本発明者らは、GFPレポーターを駆動するPax6プロモーターを使用して系統追跡実験を行った。本発明者らは、ROSAmT/mGの角膜上皮において、P1及びP60のPAX6-GFPCreマウスにおいて強いGFP発現を観察した(図15E)。これらの結果は、初期発生期から成人期の輪部幹細胞及び角膜上皮におけるPAX6の中心的役割を示唆している。
本発明者らは、扁平上皮細胞発生の主要レギュレーターが、輪部と皮膚表皮両方の基底層において主として発現されることを観察した。これは、これら2つの上皮細胞タイプの類似性を示唆している。しかし、PAX6は角膜の上皮層において高度に発現されたが、成人の皮膚表皮では検出不能であった(図16)。皮膚表皮上皮及び角膜上皮のさらなる特徴づけのために、本発明者らは、組織特異的ケラチンの免疫染色を行った。LSCは、K3及びK12を角膜特異的マーカーとして、分化すると中心角膜に遊走する(図16A)が、皮膚表皮特異的ケラチン、K1及びK10、は、表皮基底上層で発現され(図16B)、PAX6及びp63は、角膜輪部に共局在した(図17A)。本発明者らは、さらに、LSC及び皮膚表皮幹細胞(SESC)を単離し、インビトロで培養した。LSCを拡大させ、PAX6及びp63発現によって同定することができ(図17B)、SESCをp63及びK5発現によって同定することができた(図17C)。
角膜細胞運命を決定する上でのPAX6の役割を調査するために、本発明者らは、ヒトLSCにおけるレンチウイルス媒介PAX6ノックダウンを使用した。本発明者らは、ピューロマイシン選択によってPAX6 shRNA LSCを精製した。RNAを幹細胞と分化細胞両方から抽出し、関連遺伝子の発現レベルを定量的PCRによって比較した。PAX6に2つの異なるshRNAを使用し、それらは同様の結果を示した。LSCでのPAX6の4.5倍ノックダウンは、活性Ki67発現に伴って増殖異常を生じさせなかった(図18A)が、角膜特異的マーカーK3及びK12は、分化時に、対照と比較してそれぞれ17.7倍及び14.5倍(p<0.05)、有意にダウンレギュレートされた。対照的に、皮膚特異的K1及びK10発現は、それぞれ4.1倍及び4.4倍(p<0.05)アップレギュレートされた(図18B)。これらの結果は、LSCにおけるPAX6の喪失が皮膚様分化につながることを示す。
LSC及び角膜上皮細胞(CEC)におけるPAX6発現の臨床的関連性を判定するために、本発明者らは、角膜実質に血管新生及び無秩序な細胞を有する皮膚表皮病態(図19A)を示す、ヒト角膜輪部デルモイドを研究した。本発明者らは、PAX6発現が角膜デルモイド領域に完全に存在しないことを見出した(図19B)。さらに、本発明者らは、基底層におけるp63及びK5の局在発現(図19B)、並びに基底上層における皮膚特異的ケラチンK1及びK10の局在発現(図19B)を観察した。これらの結果は、発生中の患者組織における角膜上皮細胞の皮膚様上皮細胞への転換を示唆しており、角膜上皮細胞運命決定におけるPAX6の本質的役割を強く裏づける。
角膜細胞運命決定を制御する機能的特徴をさらに決定するために、本発明者らは、LSC及びSESCにおいて差次的に活性化されうるシグナル伝達経路を同定しようとした。本発明者らは、マイクロアレイを使用してLSC及びSESCでのRNA発現解析を行い、続いて、DAVIDを使用する遺伝子オントロジー及び経路解析(19、20)を行った。本発明者らは、LSCとSESC間で少なくとも2倍の発現差を示す遺伝子のサブセットを同定し、その結果、合計1185の遺伝子を得た。この解析によって、非常に多くのGO期と、多くの細胞及び代謝プロセスに影響を与えるシグナル経路とを同定した。しかし、特に、Notch、Wnt及びTGF-ベータ経路がこの解析から重要な経路として浮上した。これは、上皮を含む様々な組織からの幹細胞の自己再生及び分化系統決定におけるそれらの重要な役割(21〜23)と一致する。これらの経路の注目すべきメンバーについての発現変化のグラフィック表示を図20に提供する。
澄んだ透明な角膜は、LSCの自己再生及びCECへのそれらの分化によって維持される(24、25)。これらの2つのプロセスは、角膜上皮の完全性及び恒常性を維持するために高度に組織化されている。LSCの病的変化は、角膜の透明度の喪失をもたらすこともあり、半盲目又は全盲の原因となることもある(2、15)。この研究において、本発明者らは、PAX6が、LSCの特徴の維持及びCEC系統へのそれらのさらなる分化系統決定に不可欠であることを見出した。注目すべきこととして、p63は、一般的扁平上皮、例えば、角膜、表皮及び前立腺の扁平上皮の自己再生及び分化の主要遺伝子として文書で十分に裏づけられている(4〜6)が、本発明者らは、p63がPAX6後に発現することを観察した。これは、角膜細胞運命制御における中心事象としてのPAX6発現を示唆する。
参考文献
ドナー角膜修復材料の調製方法
1.材料
輪部幹細胞培養培地又は輪部幹細胞維持培地:次のものを補足したDMEM/栄養素混合物F-12(比率3:1での体積:体積のDMEM:F-12)基礎培地:10%ウシ胎仔血清、0.4μg/mlヒドロコルチゾン、10-10Mコレラ毒素、5μg/mlトランスフェリン、2×10-9M3,3',5-トリヨード-L-チロニン、5μg/mlインスリン、10ng/ml上皮増殖因子(EGF)、100U/mlペニシリン及び100μg/mlストレプトマイシン。細胞第4継代後、ROCK阻害剤を輪部幹細胞培養培地に添加して、例えば、1μM Y-27632の添加によって、LSCを増殖状態で維持する。上記成分、DMEM、F12培地及びウシ胎仔血清は、GIBCO(登録商標)(Life Technologies)から購入し、残りの成分は、Sigma, USAから購入する。
眼球:ドナーから。
輪部幹細胞単離及び拡大:輪部の標本を眼球から単離し、PBS中の100IU/mlペニシリン及び100μg/mlストレプトマイシンで洗浄し、小片(組織サイズ約2×2mm2)に刻み、37℃で1時間、0.2%コラゲナーゼIVで消化した。次いで、37℃で15分間の0.25%トリプシン及び1mMエチレンジアミン四酢酸(EDTA)でのさらなる処置によって単一細胞を得る。2%増殖因子低減(GFR)Matrigel(登録商標)マトリックス(BD Biosciencesカタログ番号354230)で被覆されたプラスチック皿に初代細胞を播種した。フィーダー細胞を含有しない(フィーダーフリー)輪部幹細胞培養培地で細胞を培養し、70〜90%の集密度から継代後に15〜20%の集密度で継代する。
ドナー角膜修復材料の調製方法
1.材料
実施例3の場合と同じ。
輪部幹細胞単離及び拡大:輪部の標本を眼球から単離し、PBS中の100IU/mlのペニシリン及び100μg/mlストレプトマイシンで洗浄し、小片(組織サイズ約2×2mm2)に刻み、37℃で2時間、0.2%コラゲナーゼIVで消化した。次いで、37℃で15分間の0.25%トリプシン及び1mM EDTAでのさらなる処置によって単一細胞を得る。2%増殖因子低減Matrigel(登録商標)マトリックス(BD Biosciencesカタログ番号354230)で被覆されたプラスチック皿に初代細胞を播種した。フィーダー細胞を含有しない(フィーダーフリー)輪部幹細胞培養培地で細胞を培養し、70〜90%の集密度から継代後に15〜20%の集密度で継代する。
ドナー角膜修復材料の調製方法
1.材料
実施例3の場合と同じ。
輪部幹細胞単離及び拡大:輪部組織を眼球から切除し、PBS中の100IU/mlのペニシリン及び100μg/mlストレプトマイシンで洗浄する。輪部組織を2mm×2mm片に切断し、0.2%コラゲナーゼIVとともに37℃で2.5時間インキュベートして消化し、その後、37℃で20分間、0.25%トリプシン及び1mM EDTAで処置して単一細胞浮遊液を得る。その後、これらの消化された初代細胞をMatrigel(登録商標)被覆皿(増殖因子低減Matrigel(登録商標)、BD Biosciencesカタログ番号354230)に播種する。フィーダー細胞を含有しない(フィーダーフリー)輪部幹細胞培養培地で細胞を培養してLSCを拡大させ、70〜90%の集密度から継代後に15〜20%の集密度で継代する。
ドナー角膜修復材料の調製方法
1.材料
実施例3の場合と同じ。
輪部幹細胞単離及び拡大:輪部の標本を眼球から単離し、PBS中の100IU/mlのペニシリン及び100μg/mlストレプトマイシンで洗浄し、小片(約2mm×2mm)に刻み、37℃で3.5時間、0.2%コラゲナーゼIVで消化した。次いで、細胞及び細胞塊を37℃で10分間、0.25%トリプシン及び1mM EDTAでさらに処置して単一細胞懸濁液を得る。その後、その初代単一細胞を、2%増殖因子低減Matrigel(登録商標)、BD Biosciencesカタログ番号354230)で被覆された皿で培養する。フィーダー細胞を含有しない(フィーダーフリー)輪部幹細胞培養培地で細胞を培養してLSCを拡大させ、70〜90%の集密度から継代後に15〜20%の集密度で継代する。典型的に、ROCK阻害剤、Y-27632をP4後にLSC培養培地に添加して、LSC増殖を維持する。
LSC細胞の播種前に、プラスチックプレートを30分間、37℃で2%増殖因子低減Matrigel(登録商標)(354230、BD Biosciences, Inc.)で被覆することによって処理する。LSC培養培地又は維持培地は、1/100 Pen-Strep、10%ウシ胎仔血清、10ng/ml EGF、5μg/mlインスリン、0.4μg/mlヒドロコルチゾン、10-10Mコレラ毒素及び2×10-9M 3,3',5-トリヨード-L-チロニンを含有する、DMEM/F12及びDMEM(1:1)であり、LSC細胞がゆっくり増殖している場合にのみそれに1μM Y-27632を添加する。細胞を70〜90%の集密度で継代し、増殖因子低減Matrigel(登録商標)被覆皿での連続培養についての継代直後の細胞は約15〜20%の集密度である。インビトロでLSCを分化させるために、LSCを解離させて単一細胞浮遊液を得、個々のLSCをMatrigel(登録商標)に2×104細胞/50μlゲルで包埋する。分化培地CnT-30(CellnTec Advanced Cell Systems AG、Bern、Switzerland)又はLSCのCECへの分化を支持する等価の培地で14〜18日培養後、3D構造が形成された。
Claims (196)
- 染色体に組み込まれた、あるいは組み込まれていない染色体外遺伝物質のままのPAX6をコードする、化学合成、組換え、又は単離核酸を含む、単離された輪部幹若しくは前駆細胞(LSC)集団又はLSC様集団であって、単離されたLSC集団は非LSC細胞を実質的に含まず、又はLSC様集団は非LSC様細胞を実質的に含まず、又は単離されたLSC又はLSC様集団は非LSC細胞及び非LSC様細胞を実質的に含まない、単離されたLSC集団又はLSC様集団。
- 化学合成、組換え、又は単離核酸が、PAX6又はその断片を発現することができ、PAX6又はその断片が、LSC又はLSC様状態を維持することができ、又は幹細胞又は前駆細胞をLSC又はLSC様状態へと方向づけることができ、LSC又はLSC様状態が、結果として角膜上皮細胞(CEC)に至る分化経路に細胞集団を制限する、請求項1に記載の単離されたLSC集団又はLSC様集団。
- 化学合成、組換え、又は単離核酸が、PAX6又はその断片を発現し、PAX6又はその断片が、LSC又はLSC様状態を維持し、又は幹細胞又は前駆細胞をLSC又はLSC様状態へと方向づけ、LSC又はLSC様状態が、結果として角膜上皮細胞に至る分化経路に細胞集団を制限する、請求項2に記載の単離されたLSC集団又はLSC様集団。
- 染色体に組み込まれた、あるいは組み込まれていない染色体外遺伝物質のままのPAX6をコードする、化学合成、組換え、又は単離核酸を含む、単離された皮膚上皮幹細胞(SESC)集団又はSESC様集団であって、単離されたSESC集団は非SESC細胞を実質的に含まず、又はSESC様集団は非SESC様細胞を実質的に含まず、又は単離されたSESC又はSESC様集団は非SESC細胞及び非SESC様細胞を実質的に含まず、又は単離されたSESC又はSESC様集団は非SESC、非SESC様、非LSC及び非LSC様細胞を実質的に含まない、単離されたSESC集団又はSESC様集団。
- 化学合成、組換え、又は単離核酸が、PAX6又はその断片を発現することができ、PAX6又はその断片が、LSC又はLSC様状態を維持することができ、又は幹細胞又は前駆細胞をLSC又はLSC様状態へと方向づけることができ、LSC又はLSC様状態が、結果として角膜上皮細胞に至る分化経路に細胞集団を制限する、請求項4に記載の単離されたSESC集団又はSESC様集団。
- 化学合成、組換え、又は単離核酸が、PAX6又はその断片を発現し、PAX6又はその断片が、SESC又はSESC様細胞をLSC又はLSC様状態へと方向づけ、LSC又はLSC様状態が、結果として角膜上皮細胞に至る分化経路に細胞集団を制限する、請求項5に記載の単離されたSESC集団又はSESC様集団。
- 請求項1に記載のLSC集団又はLSC様集団及び適する担体を含む、医薬組成物。
- 請求項4に記載のSESC集団又はSESC様集団及び適する担体を含む、医薬組成物。
- CECに分化することなく少なくとも17継代にわたって培養することができる、請求項1に記載のLSC集団又はLSC様集団。
- 第3継代でのPAX6及びp63を発現する細胞の割合が、第17継代での割合と同じである、請求項1に記載のLSC集団又はLSC様集団。
- 第3継代でK19及びKi67を発現する細胞の割合が、第17継代以降での割合よりわずかに大きい、請求項1に記載のLSC集団又はLSC様集団。
- CECに分化することなく40〜60世代にわたって安定的に繁殖できる細胞を含む、請求項1に記載のLSC集団又はLSC様集団。
- 角膜上皮細胞集団に分化する、請求項1に記載のLSC集団又はLSC様集団。
- 皮膚表皮細胞又は角膜上皮細胞に分化することなく少なくとも17継代にわたって培養することができる、請求項4に記載のSESC集団又はSESC様集団。
- 皮膚表皮細胞又は角膜上皮細胞に分化することなく40〜60世代にわたって安定的に繁殖できる細胞を含む、請求項4に記載のSESC集団又はSESC様集団。
- 細胞運命がLSC又はLSC様細胞運命に切り替えられたSESC又はSESC様細胞を含む、請求項4に記載のSESC集団又はSESC様集団。
- LSC又はLSC様細胞運命を採用したSESC集団又はSESC様集団であって、SESC又はSESC様細胞運命が存在しない、請求項16に記載のSESC集団又はSESC様集団。
- 角膜上皮細胞に分化する、請求項4、16又は17に記載のSESC集団又はSESC様集団。
- 実質的に皮膚表皮細胞を含まない、角膜上皮細胞に分化する、請求項18に記載のSESC集団又はSESC様集団。
- LSC集団又はLSC様集団の90〜95%が、p63、PAX6、K19及びKi67を発現する、請求項1に記載のLSC集団又はLSC様集団。
- LSC集団の5%未満が、K5及びK14を発現する、請求項1に記載のLSC集団又はLSC様集団。
- LSC集団の95%より多くが、WNT7A及びFZD5を発現する、請求項1に記載のLSC集団。
- LSC様集団の5%未満が、WNT7Aを発現する、請求項1に記載のLSC様集団。
- 細胞集団の90〜95%が、SESC又はSESC様細胞運命のままで、p63、K5及びKi67を発現する、請求項4に記載のSESC集団又はSESC様集団。
- K3又はK12発現が、SESC又はSESC様細胞運命のままである細胞では検出されない、請求項4に記載のSESC集団又はSESC様集団。
- WNT7Aが、SESC又はSESC様細胞運命のままである細胞において、LSC細胞におけるレベルより約4倍〜5倍低いレベルで発現される、請求項4に記載のSESC集団。
- PAX6が、SESC又はSESC様細胞運命のままである細胞において発現されないか、又はLSC細胞におけるレベルの約8分の1未満のレベルで発現される、請求項4に記載のSESC集団又はSESC様集団。
- WNT7Aが、SESC様運命のままである細胞の70%より多くで発現される、請求項4に記載のSESC様集団。
- LSC又はLSC様細胞運命に切り替えられた集団内の細胞の90〜95%が、p63、PAX6、K19及びKi67を発現する、請求項16に記載のSESC集団又はSESC様集団。
- 角膜上皮細胞がPAX6並びに角膜上皮マーカー、K3及びK12を発現する、請求項2又は18に記載のLSC集団又はLSC様集団。
- 皮膚表皮細胞が、皮膚表皮分化マーカー、K1及びK10を発現する、請求項19に記載のSESC集団又はSESC様集団。
- ヒトである、請求項1に記載のLSC集団又はLSC様集団。
- ヒトである、請求項4に記載のSESC集団又はSESC様集団。
- 遺伝子改変されている、請求項1に記載のLSC集団又はLSC様集団。
- 遺伝子改変されている、請求項4に記載のSESC集団又はSESC様集団。
- LSC又はLSC様細胞運命のままであるか、又はLSC又はLSC様細胞運命を維持する、請求項1に記載のLSC集団又はLSC様集団。
- SESC又はSESC様細胞運命からLSC又はLSC様細胞運命に切り替えられる、請求項4に記載のSESC集団又はSESC様集団。
- 複数の請求項1又は4に記載の細胞を含む被定義細胞集団。
- 均一である、請求項38に記載の被定義細胞集団。
- 不均一である、請求項38に記載の被定義細胞集団。
- クローン性であるか、又は単一細胞に由来する、請求項38に記載の被定義細胞集団。
- 角膜上皮細胞に発達するように決定された、請求項38に記載の幹細胞の子孫細胞。
- 請求項38に記載の細胞を含む組織。
- 対象において組織を形成する方法であって、対象中又は対象上に、請求項42に記載の子孫細胞を、前記対象の角膜上皮細胞の形成に十分な量で導入することを含む方法。
- 対象において組織を再生又は修復する方法であって、対象中又は対象上に、請求項1又は4に記載の細胞を、組織の再生又は修復に十分な量で導入することを含む方法。
- 再生又は修復される組織が、輪部幹又は前駆細胞(LSC)と角膜上皮細胞とを含む角膜上皮細胞系統の組織を含む、請求項45に記載の方法。
- 対象の皮膚上皮幹細胞(SESC)から輪部幹細胞又は前駆細胞(LSC)様細胞を得る方法であって、SESCをLSC様細胞に転換するのに十分なレベルにSESC中のPAX6タンパク質を増加させることによって対象のSESCからLSC様細胞を得るための、SESCへのPAX6遺伝子の導入又はSESCにおけるPAX6遺伝子発現のアップレギュレーションを含む方法。
- 対象の皮膚上皮幹細胞(SESC)から輪部幹又は前駆細胞(LSC)様細胞を得るためのSESCへのPAX6遺伝子の導入又はSESCにおけるPAX6遺伝子発現のアップレギュレーションが、
(a)SESCを対象から得るステップ、
(b)SESCをインビトロ又はエクスビボでのフィーダーフリー細胞培養で培養するステップ、
(c)SESCに少なくとも1つのPAX6遺伝子を導入して又はSESCにおけるPAX6遺伝子発現をアップレギュレートさせて、SESCを輪部幹細胞又は前駆細胞(LSC)様細胞に転換するのに十分なレベルにSESC中のPAX6タンパク質を増加させ、それによって対象からの皮膚上皮幹細胞(SESC)から哺乳動物輪部幹細胞又は前駆細胞(LSC)様細胞を得るステップ
を含む、請求項47に記載の方法。 - インビトロの方法であるか、エクスビボ方法であるか、又は生体内原位置(in situ)で若しくは直接対象に適用される、請求項47に記載の方法。
- PAX6タンパク質をコードする核酸を導入するか、PAX6遺伝子発現をアップレギュレートするか、又はPAX6活性を増加させる薬剤で、対象が処置される、請求項47に記載の方法。
- 薬剤が、遺伝子治療ベクター、ウイルス粒子、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、組換え核酸、組換えタンパク質、PAX6タンパク質、PAX6発現の小分子レギュレーター、PAX6発現の負のレギュレーターの阻害剤、PAX活性の負のレギュレーターの小分子阻害剤、PAX6活性の小分子エンハンサー、又はこれらの組合せを含む、請求項47に記載の方法。
- PAX6遺伝子が、PAX6a遺伝子、PAX6b遺伝子、操作されたPAX6a遺伝子、操作されたPAXb遺伝子、PAX6遺伝子ファミリーの任意のメンバー、PAX6aタンパク質のすべて又は一部をコードする核酸、PAX6bタンパク質のすべて又は一部をコードする核酸、及びPAX6又はPAX6様活性を有するタンパク質をコードする任意の核酸のセットから選択される、請求項47又は48に記載の方法。
- PAX6タンパク質が、PAX6aタンパク質、PAX6bタンパク質、タンパク質のPAX6ファミリーの任意のメンバー、及びPAX6又はPAX6様活性を有する任意のタンパク質のセットから選択される、請求項47又は48に記載の方法。
- PAX6又はPAX6様活性を有するタンパク質が、内因性K19の発現増加を生じさせることができる任意のタンパク質であり、K19がアップレギュレートされたSECSが、K3及びK12遺伝子発現増加並びにK1及びK10遺伝子発現減少を有する角膜上皮細胞(CEC)又はCEC様細胞に分化しうる、請求項52又は53に記載の方法。
- 請求項48に記載のLSC様細胞から角膜上皮細胞(CEC)様細胞を得る方法であって、フィーダーフリーLSC分化培地で(c)の細胞を分化させてLSC様細胞をCEC様細胞に転換するステップをさらに含む方法。
- フィーダーフリーLSC分化培地が既知組成である、請求項55に記載の方法。
- 培地がゼノフリーであるか、又は培地に培養細胞と同じ種に由来する成分以外の成分を含まない、請求項56に記載の方法。
- 培地が無血清である、請求項56に記載の方法。
- 培地が、一切の動物又はヒト産物を有さない、請求項56に記載の方法。
- 皮膚上皮幹細胞(SESC)から輪部幹又は前駆細胞(LSC)様細胞を得る方法であって、
(a)SESCを対象から得るステップ、
(b)SESCをインビトロでのフィーダーフリー細胞培養で培養するステップ、
(c)SESCにおけるPAX6遺伝子発現をアップレギュレートさせる薬剤とSESCを接触させて、SESCを輪部幹細胞又は前駆細胞(LSC)様細胞に転換するの十分なレベルにSESC中のPAX6タンパク質を増加させ、それによって対象からの皮膚上皮幹細胞(SESC)から哺乳動物輪部幹細胞又は前駆細胞(LSC)様細胞を得るステップ
を含む、方法。 - 薬剤が、PAX6遺伝子を含む核酸、遺伝子治療ベクター、ウイルス粒子、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、組換えタンパク質、PAX6タンパク質、PAX6発現の小分子レギュレーター、PAX6発現の負のレギュレーターの阻害剤、PAX活性の負のレギュレーターの小分子阻害剤、PAX6活性の小分子エンハンサー、及びこれらの組合せからなる群から選択される、請求項60に記載の方法。
- PAX6遺伝子が、PAX6a遺伝子、PAX6b遺伝子、操作されたPAX6a遺伝子、操作されたPAXb遺伝子、PAX6遺伝子ファミリーの任意のメンバー、PAX6aタンパク質のすべて又は一部をコードする核酸、PAX6bタンパク質のすべて又は一部をコードする核酸、及びPAX6又はPAX6様活性を有するタンパク質をコードする任意の核酸のセットから選択される、請求項61に記載の方法。
- PAX6タンパク質が、PAX6aタンパク質、PAX6bタンパク質、タンパク質のPAX6ファミリーの任意のメンバー、並びにPAX6又はPAX6様活性を有する任意のタンパク質及びその断片のセットから選択される、請求項61に記載の方法。
- 対象から哺乳動物LSCを得るためのインビトロの方法であって、
(a)対象の眼の輪部領域から組織のサンプルを採取するステップ、
(b)組織を解離させて単一細胞を得るステップ、及び
(c)LSCの増殖を可能にするようにフィーダーフリー細胞培養培地で(b)の単一細胞を培養するステップであって、ここで増殖したLSCは角膜上皮細胞(CEC)に分化する潜在力を有し、それによって対象からインビトロで哺乳動物LSCを得る、ステップ
を含む、方法。 - 輪部領域が眼の角膜輪部を含む、請求項64に記載の方法。
- (b)における組織の解離が、解離剤での処置を含み、解離剤が、組織をより小さい塊及び単一細胞に機械的に解離させるための機器若しくはツール、酵素、プロテアーゼ、化学薬品、金属キレート剤、レーザー又はこれらの組合せである、請求項64に記載の方法。
- フィーダーフリー細胞培養培地が、rho関連プロテインキナーゼ(ROCK)阻害剤若しくは白血病抑制因子(LIF)又は両方をさらに含む、請求項60又は64に記載の方法。
- ROCK阻害剤が、Y-27632(4-[(1R)-1-アミノエチル]-N-4-ピリジニル-トランス-シクロヘキサンカルボキサミド二塩酸塩)である、請求項67に記載の方法。
- LSC又はLSC様細胞を、マトリックス又は細胞外マトリックス上で培養することによって、CEC様細胞に転換するステップをさらに含む、請求項60又は64に記載の方法。
- 対象から哺乳動物輪部幹又は前駆細胞(LSC)を得てフィーダーフリーLSC培養培地においてインビトロで拡大させる方法であって、
(a)対象の眼の輪部領域から組織のサンプルを採取するステップ、
(b)組織を解離させて単一細胞を得るステップ、及び
(c)LSCの増殖を可能にするようにフィーダーフリー細胞培養培地で(b)の単一細胞を培養するステップであって、ここで増殖したLSCは角膜上皮細胞(CEC)に分化する潜在力を有し、それによって対象から哺乳動物輪部幹細胞を得てインビトロで拡大させる、ステップ
を含む、方法。 - (c)において、単一細胞がマトリックス又は細胞外マトリックス上で培養される、請求項48又は70に記載の方法。
- マトリックス又は細胞外マトリックスが、Matrigel(登録商標)又はその等価物、増殖因子低減Matrigel(登録商標)又はその等価物、コラーゲン、コラーゲンIV、コラーゲンIVシート、哺乳動物羊膜、ヒト羊膜、フィブリノーゲン、トロンビン、パールカン、ラミニン、フィブロネクチン、組換えフィブロネクチン、プロテオグリカン、プロコラーゲン、ヒアルロン酸、エンタクチン、ヘパラン硫酸、テネイシン、ポリ-L-リシン、ゼラチン、ポリ-L-オルニチン、細胞外マトリックスタンパク質、トロンビンシート、フィブリノーゲンとトロンビンシート、及びこれらの任意の組合せからなる群から選択される、請求項71に記載の方法。
- 増殖したLSC又はLSC様細胞を継代前に70〜90%の集密度及び継代後に15〜20%の集密度で継代するステップ(d)をさらに含む、請求項70に記載の方法。
- 増殖したLSC又はLSC様細胞をLSC又はLSC様細胞として安定的に継代することができる回数が、17継代以上である、請求項73に記載の方法。
- LSCが、約16〜20時間の世代時間で増殖する、請求項73に記載の方法。
- LSC又はLSC様細胞が、CECに分化することなく40〜60世代にわたって安定的に繁殖する、請求項73に記載の方法。
- フィーダーフリーLSC培養培地が1日おきに交換される、請求項70に記載の方法。
- 輪部領域が、眼の角膜輪部、角膜と結膜の間の周縁部、角膜と強膜の境界、角強膜輪部、柵間乳頭間突起を含む領域、又はVogt柵を含む領域を含む、請求項70に記載の方法。
- (b)における組織の解離が、解離剤での処置を含み、解離剤が、組織をより小さい塊及び単一細胞に機械的に解離させるための機器若しくはツール、酵素、プロテアーゼ、化学薬品、金属キレート剤、レーザー又はこれらの組合せである、請求項70に記載の方法。
- プロテアーゼが、トリプシン、コラゲナーゼIV、又はこれらの組合せを含む、請求項79に記載の方法。
- 金属キレート剤が、EDTA、EGTA、又はこれらの組合せを含む、請求項79に記載の方法。
- フィーダーフリー細胞培養培地が、最小必須培地、増殖因子、ホルモン及び可溶性因子を含む、請求項70に記載の方法。
- フィーダーフリー細胞培養培地が、好ましくは、LSCを得て拡大させる種からの血清、又は代用血清をさらに含む、請求項82に記載の方法。
- フィーダーフリー細胞培養培地が、rho関連プロテインキナーゼ(ROCK)阻害剤をさらに含む、請求項82に記載の方法。
- ROCK阻害剤が、(R)-(+)-トランス-4-(1-アミノエチル)-N-(4-ピリジル)シクロヘキサンカルボキサミド二塩酸塩一水和物(Y-27632)、5-(1,4-ジアゼパン-1-イルスルホニル)イソキノリン(ファスジル又はHA1077)、H-1152、H-1152P、(S)-(+)-2-メチル-1-[(4-メチル-5-イソキノリニル)スルホニル]ホモピペラジン二塩酸塩、ジメチルファスジル(diMF、H-1152P)、N-(4-ピリジル)-N'-(2,4,6-トリクロロフェニル)尿素、Y-39983、Wf-536、SNJ-1656、及び(S)-(+)-2-メチル-1-[(4-メチル-5-イソキノリニル)スルホニル]-ヘキサヒドロ-1H-1,4-ジアゼピン二塩酸塩(H-1152)、イミダゾール含有ベンゾジアゼピン、イミダゾピリジン誘導体、インダゾールコア、2-アミノピリジン/ピリミジンコア、9-デアザグアニン誘導体、ベンズアミド又はアミノフラザンを含む化合物、並びにこれらの誘導体及び類似体、並びにそれらの組合せからなる群から選択される、請求項84に記載の方法。
- 対象からのLSCの単離後、LSCの第4継代後にフィーダーフリー細胞培養培地にROCK阻害剤が添加される、請求項84に記載の方法。
- フィーダーフリー細胞培養培地が、白血病抑制因子(LIF)をさらに含む、請求項82に記載の方法。
- 対象からのLSCの単離後、LSCの第4継代後にフィーダーフリー細胞培養培地にLIFが添加される、請求項87に記載の方法。
- フィーダーフリー細胞培養培地が、DMEM/F12培地、DMEM、ペニシリン-ストレプトマイシン、血清、EGF、インスリン、ヒドロコルチゾン、コレラ毒素、3,3',5-トリヨード-L-チロニン、又はこれらの組合せを含み、血清が、ウシ胎仔血清、しかし好ましくは培養されるLSCと同じ種からの血清、又は代用血清でありうる、請求項70に記載の方法。
- フィーダーフリー細胞培養培地が、DMEM/F12培地、DMEM、ペニシリン-ストレプトマイシン、血清、EGF、インスリン、ヒドロコルチゾン、コレラ毒素、及び3,3',5-トリヨード-L-チロニンを含み、血清が、ウシ胎仔血清、しかし好ましくは培養されるLSCと同じ種からの血清、又は代用血清でありうる、請求項70に記載の方法。
- フィーダーフリー細胞培養培地が、ROCK阻害剤、Y-27632をさらに含む、請求項89又は90に記載の方法。
- ROCK阻害剤、Y-27632が、細胞第4継代後にフィーダーフリー細胞培養培地に添加される、請求項91に記載の方法。
- フィーダーフリー細胞培養培地が、白血病抑制因子(LIF)をさらに含む、請求項89又は90に記載の方法。
- 対象からのLSCの単離後、LSCの第4継代後にフィーダーフリー細胞培養培地にLIFが添加される、請求項93に記載の方法。
- LSCが、WNT7A、FZD5、PAX6、p63、ケラチン5(K5)、ケラチン14(K14)、ケラチン19(K19)及びKi67を含むマーカーのセットを発現する、請求項70に記載の方法。
- LSCの90〜95%が、p63、PAX6、K19及びKi67を発現する、請求項70に記載の方法。
- LSCの5%未満が、K5及びK14を発現する、請求項70に記載の方法。
- LSCの95%より多くが、WNT7A及びFZD5を発現する、請求項70に記載の方法。
- CECが、WNT7A、FZD5、PAX6、ケラチン3(K3)及びケラチン12(K12)を含むマーカーのセットを発現し、ここでK3及びK12発現はLSCと比較して統計的に有意に高く、K19発現はLSCと比較して統計的に有意に低い、請求項70に記載の方法。
- CECが、p63を発現しないか、又はLSCより有意に低いレベルのp63を発現し、ケラチン1(K1)及びケラチン10(K10)を発現しないか、又は皮膚又は表皮細胞より有意に低いレベルのケラチン1(K1)及びケラチン10(K10)を発現する、請求項70に記載の方法。
- 単離されたLSCをインビトロで角膜上皮細胞(CEC)に分化させる方法であって、
(a)元々対象に由来した、好ましくは単一細胞状態に解離されている、事前に培養されたLSCを得るステップ、
(b)単離されたLSCを、分化に適している3次元細胞培養物を形成する又は形成を可能にするように、マトリックス又は細胞外マトリックス中及び/又は上に配置するステップ、及び
(c)単離されたLSCのCECへのインビトロでの分化を可能にするようにLSC分化培地でLSCを培養することによって、単離されたLSCをインビトロで角膜上皮細胞に分化させるステップ
を含む方法。 - マトリックス又は細胞外マトリックスが、Matrigel(登録商標)又はその等価物、増殖因子低減Matrigel(登録商標)又はその等価物、コラーゲン、コラーゲンIV、コラーゲンIVシート、哺乳動物羊膜、ヒト羊膜、フィブリノーゲン、トロンビン、パールカン、ラミニン、フィブロネクチン、組換えフィブロネクチン、プロテオグリカン、プロコラーゲン、ヒアルロン酸、エンタクチン、ヘパラン硫酸、テネイシン、ポリ-L-リシン、ゼラチン、ポリ-L-オルニチン、細胞外マトリックスタンパク質(Fischer又はLife Tech)、トロンビンシート(Fibrin Sealant、Reliseal(商標)、Reliance Life Sciences)、フィブリノーゲンとトロンビンのシート(Reliance Life)、及びこれらの任意の組合せからなる群から選択される、請求項101に記載の方法。
- マトリックス又は細胞外マトリックスが、増殖因子低減Matrigel(登録商標)若しくはその等価物、又はコラーゲンを含む、請求項101に記載の方法。
- 輪部幹細胞分化培地が、フィーダーフリー及び既知組成培地である、請求項101に記載の方法。
- フィーダーフリー及び既知組成培地が、CnT-30培地(Cellntec Advanced Cell Systems AG、Bern、Switzerland)、CnT-02(Cellntec)、CnT-02-3DP5(Cellntec)又は機能的等価物を含み、前記培地がLSCのCECへの分化を促進する、請求項104に記載の方法。
- CECが、WNT7A、FZD5、PAX6、ケラチン3(K3)及びケラチン12(K12)を含むマーカーのセットを発現し、ここでK3及びK12発現はLSCと比較して統計的に有意に高く、K19発現はLSCと比較して統計的に有意に低い、請求項101に記載の方法。
- CECが、p63を発現しないか、又はLSCより有意に低いレベルのp63を発現し、ケラチン1(K1)及びケラチン10(K10)を発現しないか、又は皮膚又は表皮細胞より有意に低いレベルのケラチン1(K1)及びケラチン10(K10)を発現する、請求項106に記載の方法。
- LSC分化培地が1日おきに交換される、請求項101に記載の方法。
- 請求項60又は64に記載の方法由来の単離された輪部幹細胞(LSC)又はLSC様細胞の集団。
- LSCが、WNT7A、FZD5、PAX6、p63、ケラチン5(K5)、ケラチン14(K14)、ケラチン19(K19)、及びKi67を含むマーカーのセットを発現する、請求項109に記載の細胞。
- 請求項101に記載の方法由来の単離された角膜上皮細胞(CEC)の集団。
- CECが、(i)WNT7A、FZD5、PAX6、ケラチン3(K3)及びケラチン12(K12)を含むマーカーのセットを発現し、ここでK3及びK12発現はLSCと比較して統計的に有意に高く、K19発現はLSCと比較して統計的に有意に低く、(ii)p63を発現しないか、又はLSCより有意に低いレベルのp63を発現し、ケラチン1(K1)及びケラチン10(K10)を発現しないか、又は皮膚又は表皮細胞より有意に低いレベルのケラチン1(K1)及びケラチン10(K10)を発現する、請求項111に記載の細胞。
- 請求項60、64又は101に記載の方法によって生産されたLSC、LSC様細胞若しくはCEC、又はこれらの細胞の組合せ、包装材料及び使用説明書を含む、角膜組織修復用キット。
- 医薬的に許容される担体をさらに含む、請求項113に記載のキット。
- 医薬的に許容される担体が、ヒト又は動物の眼の湾曲部のような湾曲部での細胞付着又は増殖を支持するために使用されるコンタクトレンズ又はその等価物である、請求項114に記載のキット。
- ヒト羊膜又は動物羊膜をさらに含む、請求項115に記載のキット。
- 輪部幹細胞又は角膜上皮細胞の機能不全に関連した疾患を有する対象を処置する方法であって、
a.請求項1、6、7又は8に記載のLSC、LSC様、CEC又はCEC様細胞を対象の罹患した眼に移植するステップを含み、移植された細胞が対象の罹患した眼の角膜又は輪部に定着し、正常な角膜透明性及び透明度を回復させ、
それによって、輪部幹若しくは前駆細胞又は角膜上皮細胞の機能不全に関連した疾患を有する対象を処置する方法。 - 輪部幹細胞又は角膜上皮細胞の機能不全に関連した疾患を有する対象を処置する方法であって、
(a)請求項47、48、55、60、64、70又は101に記載の方法によって生産されたLSC、LSC様、CEC又はCEC様細胞を対象の罹患した眼に移植するステップを含み、移植された細胞が対象の罹患した眼の角膜又は輪部に定着し、正常な角膜透明性及び透明度を回復させ、
それによって、輪部幹若しくは前駆細胞又は角膜上皮細胞の機能不全に関連した疾患を有する対象を処置する方法。 - 疾患又は状態が、輪部幹若しくは前駆細胞の欠損、角膜上皮細胞の欠損、角膜輪部の損傷、眼の角膜の損傷、輪部幹細胞の損傷、角膜上皮細胞の損傷、角膜の発生若しくは機能に影響を与える先天性欠損、角膜の発生若しくは機能に影響を与える後天性欠損、角膜が皮膚系統に切り替えられる細胞運命決定に影響を与える先天性欠損、角膜が皮膚系統に切り替えられる細胞運命決定に影響を与える後天性欠損、異常表皮分化、スティーブンス・ジョンソン症候群、無虹彩、再発性翼状片、角膜疾患、角膜上皮扁平上皮化生、炎症性角膜症、外傷、化学熱傷、アルカリ熱傷、半盲目又は全盲を含む、請求項117又は118に記載の方法。
- 疾患又は状態が、半盲目、全盲、角膜表面疾患、角膜疾患、角膜上皮扁平上皮化生、炎症性角膜症、外傷又はアルカリ熱傷である、請求項117又は118に記載の方法。
- 半盲目、全盲、角膜表面疾患、角膜疾患、角膜上皮扁平上皮化生、炎症性角膜症、外傷又はアルカリ熱傷を有する対象の正常な角膜透明性及び透明度を回復させる方法であって、
(a)請求項1、6、7又は8に記載のLSC、LSC様、CEC又はCEC様細胞を対象の罹患した眼に移植するステップを含み、移植された細胞が対象の罹患した眼の角膜又は輪部に定着し、正常な角膜透明性及び透明度を回復させ、
それによって、半盲目、全盲、角膜表面疾患、角膜疾患、角膜上皮扁平上皮化生、炎症性角膜症、外傷又はアルカリ熱傷を有する対象の正常な角膜透明性及び透明度を回復させる方法。 - 半盲目、全盲、角膜表面疾患、角膜疾患、角膜上皮扁平上皮化生、炎症性角膜症、外傷又はアルカリ熱傷を有する対象の正常な角膜透明性及び透明度を回復させる方法であって、
(a)請求項47、48、55、60、64、70又は101に記載の方法によって生産されたLSC、LSC様、CEC又はCEC様細胞を対象の罹患した眼に移植するステップを含み、移植された細胞が対象の罹患した眼の角膜又は輪部に定着し、正常な角膜透明性及び透明度を回復させ、
それによって、半盲目、全盲、角膜表面疾患、角膜疾患、角膜上皮扁平上皮化生、炎症性角膜症、外傷又はアルカリ熱傷を有する対象の正常な角膜透明性及び透明度を回復させる方法。 - 対象の角膜組織を再生又は修復する方法であって、請求項1、6、7又は8に記載の単離されたLSC、LSC様、CEC又はCEC様細胞集団を角膜組織の再生又は修復に十分な量で対象に導入することを含む方法。
- 対象の角膜組織を再生又は修復する方法であって、請求項47、48、55、60、64、70又は101に記載の方法によって生産された単離されたLSC、LSC様、CEC又はCEC様細胞集団を角膜組織の再生又は修復に十分な量で対象に導入することを含む方法。
- 細胞が、対象以外の個体からのものである、請求項121、122、123又は124に記載の方法。
- 請求項121、122、123又は124に記載の方法であって、細胞が、前記方法によって生産された細胞で処置される対象からのものである、方法。
- 対象が哺乳動物である、請求項121、122、123又は124に記載の方法。
- 哺乳動物が、ヒト、ラット、イヌ、ネコ、ブタ、ウマ、ウサギ、ウシ、サル又はマウスである、請求項127に記載の方法。
- 眼化生を有する患者がPAX6遺伝子又は遺伝子産物での処置の利益を受け得るかどうかを判定する方法であって、化生領域におけるWNT7A、PAX6、K3及び/又はK12の遺伝子発現又はタンパク質レベルを評価することを含み、化生領域におけるWNT7A、PAX6、K3及び/又はK12の非存在が、眼化生を有する患者が、PAX6遺伝子若しくは遺伝子産物又は請求項1若しくは4に記載の細胞での処置の利益を受け得ることを示す方法。
- 眼化生を有する対象がWNT7A遺伝子又は遺伝子産物での処置の利益を受ける可能性があるかどうかを判定する方法であって、化生領域におけるWNT7A、PAX6、K3及び/又はK12の遺伝子発現又はタンパク質レベルを評価することを含み、化生領域におけるWNT7A、PAX6、K3及び/又はK12の非存在が、眼化生を有する患者が、WNT7A遺伝子又は遺伝子産物での処置の利益を受け得ることを示す方法。
- 眼化生を有する対象がWNT7A及びPAX6両方の遺伝子又は遺伝子産物での処置の利益を受け得るかどうかを判定する方法であって、化生領域におけるWNT7A、PAX6、K3及び/又はK12の遺伝子発現又はタンパク質レベルを評価することを含み、化生領域におけるWNT7A、PAX6、K3及び/又はK12の非存在が、眼化生を有する患者が、WNT7A及びPAX6両方の遺伝子又は遺伝子産物での処置の利益を受け得ることを示す方法。
- 眼化生を有する対象がPAX6遺伝子若しくは遺伝子産物又は請求項47、48、55、60、64、70若しくは101に記載の方法によって生産された細胞での処置の利益を受け得るかどうかを判定する方法であって、化生領域におけるWNT7A、PAX6、K3及び/又はK12の遺伝子発現又はタンパク質レベルを評価することを含み、化生領域におけるWNT7A、PAX6、K3及び/又はK12の非存在が、眼化生を有する患者が、PAX6遺伝子若しくは遺伝子産物又は請求項47、48、55、60、64、70若しくは101に記載の方法によって生産された細胞での処置の利益を受け得ることを示す方法。
- 対象の角膜組織を再生又は修復する方法であって、請求項1、64又は70に記載の単離されたLSC集団を角膜組織の再生又は修復に十分な量で対象に導入することを含む方法。
- 対象が哺乳動物である、請求項133に記載の方法。
- 哺乳動物が、ヒト、ラット、イヌ、ネコ、ブタ、ウマ、ウサギ、ウシ、サル又はマウスである、請求項134に記載の方法。
- 請求項47、48、55、60、64、70若しくは101に記載の方法によって生産されたLSC、LSC様、CEC若しくはCEC様細胞又はこれらの細胞の組合せの有効量及び適する医薬担体を含む、医薬組成物。
- 眼疾患を処置する方法であって、請求項1に記載の単離されたLSC若しくはLSC様集団又は請求項4に記載のSESC若しくはSESC様集団を、請求項1又は4に記載の単離されたLSC又はLSC様集団が、CEC又はCEC様細胞への分化を可能にするLSC又はLSC様状態を生じさせる又は維持するのに十分な量のPAX6を生産又は過剰発現するのに十分な量で、対象に投与することによって、眼疾患を処置することを含む方法。
- 眼疾患が、ヒト角膜疾患、角膜上皮扁平上皮化生、炎症性角膜症、外傷及びアルカリ熱傷である、請求項137に記載の方法。
- 眼疾患が、ヒト眼疾患である、請求項137に記載の方法。
- 対象の角膜組織を再生又は修復する方法であって、対象の角膜組織、角膜組織の推定位置又は眼の前面を角膜組織の再生又は修復に十分な量のPAX6と接触させることを含む方法。
- 対象の角膜組織を再生又は修復する方法であって、請求項47、48、55、60、64、70若しくは101に記載の方法によって生産されたLSC、LSC様、CEC若しくはCEC様細胞又はこれらの細胞の組合せを角膜組織の再生又は修復に十分な量で角膜又は眼の前面に投与することを含む方法。
- 対象の角膜組織を再生又は修復する方法であって、角膜組織、角膜組織の推定位置又は眼の前面に角膜組織の再生又は修復に十分な量のPAX6タンパク質を投与することを含む方法。
- 請求項1又は6に記載のLSC又はLSC様集団を増殖及び維持するためのキットであって、LSC集団を維持するための細胞培養培地を含み、前記培地が本質的にフィーダーフリーである、キット。
- 請求項4に記載のSESC又はSESC様集団のLSC又はLSC様集団を増殖及び維持するためのキットであって、LSC集団を維持するための細胞培養培地を含み、前記培地が本質的にフィーダーフリーである、キット。
- 培地が、本質的にゼノフリーである、請求項143又は144に記載のキット。
- LSC又はLSC様集団をCEC又はCEC様集団に分化させるためのLSC分化培地をさらに含む、請求項143又は144に記載のキット。
- 培地が、本質的に無血清である、請求項143又は144に記載のキット。
- 培地に本質的に動物産物を含まない、請求項143又は144に記載のキット。
- 培地に本質的にヒト産物を含まない、請求項143又は144に記載のキット。
- 培地が、既知組成である、請求項143又は144に記載のキット。
- 請求項47、48、55、60、64、70若しくは101に記載の方法によって生産されたLSC、LSC様、CEC若しくはCEC様細胞又はこれらの細胞の組合せ、包装材料及び使用説明書を含む、角膜組織修復用のキット。
- 医薬的に許容される担体をさらに含む、請求項151に記載のキット。
- 医薬的に許容される担体が、ヒト又は動物の眼の湾曲部のような湾曲部での細胞付着又は増殖を支持するために使用されるコンタクトレンズ又はその等価物である、請求項152に記載のキット。
- ヒト羊膜又は動物羊膜をさらに含む、請求項153に記載のキット。
- 対象の角膜又は角膜機能に影響を与える眼疾患を発症するリスクを評価する方法であって、(a)LSC又は角膜上皮細胞におけるWNTZ7A、FZD5及びPAX6又はこれらの組合せの活性を評価するステップを含み、WNTZ7A、FZD5及びPAX6又はこれらの組合せの活性がより低い又はないことが、対象の角膜又は角膜機能に影響を与える眼疾患を発症するリスクがより高いことを示し、それによって対象の角膜又は角膜機能に影響を与える眼疾患を発症するリスクを評価する方法。
- 請求項47、48、55、60、64、70若しくは101に記載の方法によって生産されたLSC、LSC様、CEC若しくはCEC様細胞又はこれらの細胞の組合せでの細胞移植、あるいはPAX6タンパク質又はPAX6をコードする核酸での処置に適している患者集団であって、角膜の異常な皮膚表皮様細胞及び随伴角質化、並びにWNTZ7A、FZD5及びPAX6遺伝子又は遺伝子の組合せの発現喪失又は減少を有する患者集団を同定する方法。
- 輪部幹細胞が欠損している対象の輪部にLSC又はLSC様細胞を再定着させる方法であって、請求項47、48、64又は70に記載の方法によって生産されたLSC又はLSC様細胞を対象の眼の前面に投与するステップ、及び前記細胞のLSCニッチへの遊走を可能にするステップであって、それによって対象の輪部にLSC又はLSC様細胞を再定着させる、ステップを含む、方法。
- LSC又はLSC様細胞を投与するステップが、LSC又はLSC様細胞のシートを対象の眼表面にグラフトするステップ、あるいは、LSC又はLSC様細胞を含む細胞若しくは組織浮遊液を投与するステップを含む、請求項157に記載の方法。
- 対象の眼へのLSC又はLSC様細胞の投与後、LSC又はLSC様細胞が羊膜、好ましくはヒト羊膜で覆われる、請求項158に記載の方法。
- 輪部幹細胞が欠損している対象の輪部にLSC又はLSC様細胞を再定着させる方法であって、結膜、推定角膜位置、眼又は眼瞼の皮膚上皮幹細胞(SESC)をLSC又はLSC様細胞に転換するために、対象の結膜、推定角膜位置、眼又は眼瞼の領域にPAX6活性又は発現を増加させる薬剤を投与することを含み、輪部への移動時に、対象の輪部にLSC又はLSC様細胞を再定着させ、それによって対象の輪部にLSC又はLSC様細胞を再定着させる方法。
- 最小必須培地、増殖因子、ホルモン、可溶性因子及び血清又は代用血清を含む、LSC、LSC様、SESC又はSESC様の短期インビトロ培養のためのフィーダーフリー細胞培養培地。
- 短期インビトロ培養が、LSC、LSC様、SESC又はSESC様細胞の第0〜約第4継代である、請求項161に記載のフィーダーフリー細胞培養培地。
- DMEM/F12培地、DMEM、ペニシリン-ストレプトマイシン、ウシ胎仔血清、EGF、インスリン、ヒドロコルチゾン、コレラ毒素及び3,3',5-トリヨード-L-チロニンを含み、前記ウシ胎仔血清はヒト血清又は代用血清に代替されてもよく、EGF及びインスリンが、それぞれ、組換えEGF及び/又は組換えインスリン、好ましくは組換えヒトEGF及び組換えヒトインスリンであってもよく、ここでLSC、LSC様、SESC又はSESC様細胞が増殖してCEC又は皮膚表皮細胞に分化しない限り、成分の各々が機能的に等価の成分に置換されてもよい、請求項161に記載のフィーダーフリー細胞培養培地。
- DMEM/F12及びDMEM(1:1)とともに10〜20%の範囲のウシ胎仔血清又は10〜20%の範囲の血清の代用血清、10〜20ng/mlの範囲のEGF、5〜10μg/mlの範囲のインスリン、0.2〜0.8μg/mlの範囲のヒドロコルチゾン、5×10-11〜5×10-10Mの範囲のコレラ毒素及び10-9M〜4×10-9Mの範囲の3,3',5-トリヨード-L-チロニンを含み、EGF及びインスリンが、それぞれ、組換えEGF及び/又は組換えインスリン、好ましくは組換えヒトEGF及び組換えヒトインスリンであってもよく、ここでLSC、LSC様、SESC又はSESC様細胞が増殖してCEC又は皮膚表皮細胞に分化しない限り、成分の各々が機能的に等価の成分に置換されてもよい、請求項161に記載のフィーダーフリー細胞培養培地。
- DMEM/F12及びDMEM(1:1)とともに100U/mlペニシリン、100μg/mlストレプトマイシン、10%ウシ胎仔血清、10ng/ml EGF、5μg/mlインスリン、0.4μg/mlヒドロコルチゾン、10-10Mコレラ毒素及び2×10-9M 3,3',5-トリヨード-L-チロニンを含み、ウシ胎仔血清はヒト血清又は代用血清に代替されてもよく、EGF及びインスリンが、それぞれ、組換えEGF及び/又は組換えインスリン、好ましくは組換えヒトEGF及び組換えヒトインスリンであってもよく、ここでLSC、LSC様、SESC又はSESC様細胞が増殖してCEC又は皮膚表皮細胞に分化しない限り、成分の各々が機能的に等価の成分に置換されてもよい、請求項161に記載のフィーダーフリー細胞培養培地。
- 最小必須培地、増殖因子、ホルモン、可溶性因子、血清又は代用血清、及びrho関連プロテインキナーゼ(ROCK)阻害剤を含む、LSC、LSC様、SESC又はSESC様細胞の長期インビトロ培養のためのフィーダーフリー細胞培養培地。
- 長期インビトロ培養が、LSC、LSC様、SESC又はSESC様細胞の17継代以上の継代である、請求項166に記載のフィーダーフリー細胞培養培地。
- DMEM/F12培地、DMEM、ペニシリン-ストレプトマイシン、ウシ胎仔血清、EGF、インスリン、ヒドロコルチゾン、コレラ毒素及び3,3',5-トリヨード-L-チロニン、及びY-27632を含み、ウシ胎仔血清はヒト血清又は代用血清に代替されてもよく、EGF及びインスリンが、それぞれ、組換えEGF及び/又は組換えインスリン、好ましくは組換えヒトEGF及び組換えヒトインスリンであってもよく、Y-27632は別のROCK阻害剤に置換されてもよく、LSC、LSC様、SESC又はSESC様細胞が増殖してCEC又は皮膚表皮細胞に分化しない限り、成分の各々の代わりに機能的に等価の成分を代用してもよい、請求項166に記載のフィーダーフリー細胞培養培地。
- DMEM/F12及びDMEM(1:1)とともに10〜20%の範囲のウシ胎仔血清又は10〜20%の範囲の血清の代用血清、10〜20ng/mlの範囲のEGF、5〜10μg/mlの範囲のインスリン、0.2〜0.8μg/mlの範囲のヒドロコルチゾン、5×10-11〜5×10-10Mの範囲のコレラ毒素、10-9M〜4×10-9Mの範囲の3,3',5-トリヨード-L-チロニン及び1〜10μMの範囲のY-27632を含み、EGF及びインスリンが、それぞれ、組換えEGF及び/又は組換えインスリン、好ましくは組換えヒトEGF及び組換えヒトインスリンであってもよく、Y-27632は別のROCK阻害剤に置換されてもよく、ここでLSC、LSC様、SESC又はSESC様細胞が増殖してCEC又は皮膚表皮細胞に分化しない限り、成分の各々が機能的等価物に置換されてもよい、請求項166に記載のフィーダーフリー細胞培養培地。
- DMEM/F12及びDMEM(1:1)とともに100U/mlペニシリン、100μg/mlストレプトマイシン、10%ウシ胎仔血清、10ng/ml EGF、5μg/mlインスリン、0.4μg/mlヒドロコルチゾン、10-10Mコレラ毒素、2×10-9M 3,3',5-トリヨード-L-チロニン及び1μM Y-27632を含み、ウシ胎仔血清はヒト血清又は代用血清に代替されてもよく、EGF及びインスリンが、それぞれ、組換えEGF及び/又は組換えインスリン、好ましくは組換えヒトEGF及び組換えヒトインスリンであってもよく、Y-27632は別のROCK阻害剤に置換されてもよく、ここでLSC、LSC様、SESC又はSESC様細胞が増殖してCEC又は皮膚表皮細胞に分化しない限り、成分の各々が機能的等価物に置換されてもよい、請求項166に記載のフィーダーフリー細胞培養培地。
- 最小必須培地、増殖因子、ホルモン、可溶性因子、血清又は代用血清、rho関連プロテインキナーゼ(ROCK)阻害剤及び白血病抑制因子(LIF)を含む、LSC又はLSC様細胞の長期インビトロ培養のためのフィーダーフリー細胞培養培地。
- 長期インビトロ培養が、LSC又はLSC様細胞の17継代以上の継代である、請求項171に記載のフィーダーフリー細胞培養培地。
- DMEM/F12培地、DMEM、ペニシリン-ストレプトマイシン、ウシ胎仔血清、EGF、インスリン、ヒドロコルチゾン、コレラ毒素及び3,3',5-トリヨード-L-チロニン、Y-27632及びLIFを含み、ウシ胎仔血清はヒト血清又は代用血清に代替されてもよく、EGF、インスリン及びLIFが、それぞれ、組換えEGF、組換えインスリン及び/又は組換えLIF、好ましくは組換えヒトEGF、組換えヒトインスリン及び組換えヒトLIFであってもよく、Y-27は別のROCK阻害剤に置換されてもよく、ここでLSC又はLSC様細胞が増殖してCECに分化しない限り、成分の各々の代わりに機能的に等価の成分を代用してもよい、請求項171に記載のフィーダーフリー細胞培養培地。
- DMEM/F12及びDMEM(1:1)とともに10〜20%の範囲のウシ胎仔血清又は10〜20%の範囲の血清の代用血清、10〜20ng/mlの範囲のEGF、5〜10μg/mlの範囲のインスリン、0.2〜0.8μg/mlの範囲のヒドロコルチゾン、5×10-11〜5×10-10Mの範囲のコレラ毒素、10-9M〜4×10-9Mの範囲の3,3',5-トリヨード-L-チロニン、1〜10μMの範囲のY-27632及び5〜20ng/ml LIFを含み、EGF、インスリン及びLIFが、それぞれ、組換えEGF、組換えインスリン及び/又は組換えLIF、好ましくは組換えヒトEGF、組換えヒトインスリン及び組換えヒトLIFであってもよく、Y-27632は別のROCK阻害剤に置換されてもよく、ここでLSC、LSC様、SESC又はSESC様細胞が増殖してCECに分化しない限り、成分の各々が機能的等価物に置換されてもよい、請求項161に記載のフィーダーフリー細胞培養培地。
- 培地が、DMEM/F12及びDMEM(1:1)とともに100U/mlペニシリン、100μg/mlストレプトマイシン、10%ウシ胎仔血清、10ng/ml EGF、5μg/mlインスリン、0.4μg/mlヒドロコルチゾン、10-10Mコレラ毒素、2×10-9M 3,3',5-トリヨード-L-チロニン、1μM Y-27632及び10ng/ml LIFを含み、ウシ胎仔血清の代わりにヒト血清又は代用血清を使用してもよく、EGF、インスリン及びLIFが、それぞれ、組換えEGF、組換えインスリン及び/又は組換えLIF、好ましくは組換えヒトEGF、組換えヒトインスリン及び組換えヒトLIFであってもよく、Y-27632は別のROCK阻害剤に置換されてもよく、ここでLSC又はLSC様細胞が増殖してCECに分化しない限り、成分の各々が機能的等価物に置換されてもよい、請求項161に記載のフィーダーフリー細胞培養培地。
- ROCK阻害剤が、(R)-(+)-トランス-4-(1-アミノエチル)-N-(4-ピリジル)シクロヘキサンカルボキサミド二塩酸塩一水和物(Y-27632、Sigma-Aldrich)、5-(1,4-ジアゼパン-1-イルスルホニル)イソキノリン(ファスジル又はHA1077、Cayman Chemical)、H-1152、H-1152P、(S)-(+)-2-メチル-1-[(4-メチル-5-イソキノリニル)スルホニル]ホモピペラジン二塩酸塩、ジメチルファスジル(diMF、H-1152P)、N-(4-ピリジル)-N'-(2,4,6-トリクロロフェニル)尿素、Y-39983、Wf-536、SNJ-1656、及び(S)-(+)-2-メチル-1-[(4-メチル-5-イソキノリニル)スルホニル]-ヘキサヒドロ-1H-1,4-ジアゼピン二塩酸塩、イミダゾール含有ベンゾジアゼピン、イミダゾピリジン誘導体、インダゾールコア、2-アミノピリジン/ピリミジンコア、9-デアザグアニン誘導体、ベンズアミド又はアミノフラザンを含む化合物、並びにこれらの誘導体及び類似体、並びにそれらの組合せからなる群から選択される、請求項166又は171に記載のフィーダーフリー細胞培養培地。
- ウシ胎仔血清がヒト血清又は代用血清に代替される、請求項163、164、165、168、169、170、173、174又は175に記載のフィーダーフリー細胞培養培地。
- EGF、インスリン又はLIFが、それぞれ、組換えEGF、組換えインスリン又は組換えLIFである、請求項163、164、165、168、169、170、173、174又は175に記載のフィーダーフリー細胞培養培地。
- EGFが、組換えヒトEGFである、請求項178に記載のフィーダーフリー細胞培養培地。
- インスリンが、組換えヒトインスリンである、請求項178に記載のフィーダーフリー細胞培養培地。
- LIFが、組換えヒトLIFである、請求項178に記載のフィーダーフリー細胞培養培地。
- SESCを得てインビトロでフィーダーフリー細胞培養培地において拡大させる方法であって、単離されたSESC皮膚を請求項161又は166に記載のフィーダーフリー細胞培養培地で培養することを含む方法。
- SESCが、毛包間表皮から単離される、請求項102に記載の方法。
- SESCが、ヒト又は動物のSESCを有する任意のSESCニッチから単離される、請求項102に記載の方法。
- 輪部幹細胞(LSC)から上皮幹細胞(SESC)又はSESC様細胞を得る方法であって、(a)細胞運命をLSCからSESC運命に変えるために十分にWNT7A又はPAX6遺伝子又はタンパク質の発現又は活性をノックダウンするステップを含み、それによってLSC細胞からSESC又はSESC様細胞を得る方法。
- LSCを、WNT7A若しくはPAX6に指向されたshRNAと接触させて、あるいは、LSCが、WNT7A若しくはPAX6遺伝子に指向されたshRNA、RNAi若しくはアンチセンスRNAを生産する導入遺伝子を発現して、LSCをSESC又はSESC様細胞に転換するために十分にWNT7A又はPAX6の発現を低減させる、請求項185に記載の方法。
- 対象から皮膚上皮幹細胞(SESC)を得て、フィーダーフリー細胞培養培地においてインビトロで拡大させる方法であって、
(a)対象の毛包間表皮から又は対象のSESCを有する任意のSESC幹細胞ニッチから組織サンプルを採取するステップ、
(b)組織を解離させて単一細胞を得るステップ、及び
(c)SESCの増殖を可能にするようにフィーダーフリー細胞培養培地で(b)の単一細胞を培養するステップであって、ここで増殖したSESCは皮膚表皮細胞に分化する潜在力を有し、それによって対象から皮膚上皮幹細胞を得てインビトロで拡大させる、ステップ
を含む、方法。 - SESCが、請求項161、163、164、165、166、168、169又は170に記載のフィーダーフリー細胞培養培地においてインビトロで培養される、請求項185に記載の方法。
- インビトロでSESC又はSESC様細胞から皮膚表皮細胞又は皮膚表皮様細胞を得る方法であって、SESC又はSESC様細胞の皮膚表皮細胞又は皮膚表皮様細胞への分化を支持する既知組成分化培地で単離されたSESC又はSESC様細胞を培養することによって、インビトロでSESC又はSESC様細胞から皮膚表皮細胞又は皮膚表皮様細胞を得ることを含む方法。
- 既知組成分化培地が、CnT-02(CellnTec)又は等価の培地である、請求項189に記載の方法。
- 新たな皮膚を必要とする対象を処置する方法であって、請求項185に記載の方法によって得た又は請求項182、187若しくは189に記載の方法によって得て拡大させたSESC細胞、SESC様細胞、皮膚表皮細胞又は表皮上皮様細胞を対象に投与して、例えば、対象のSESC細胞集団又は皮膚表皮細胞集団を再定着させ、対象に新たな皮膚をもたらすことによって、新たな皮膚を必要とする対象を処置することを含む方法。
- 皮膚を必要とする対象が、皮膚ジストロフィー、皮膚疾患、皮膚感染症、熱傷傷害、皮膚潰瘍、擦過創、黒色腫、癌腫、創傷、加齢、皮膚の遺伝障害、皮膚生検、手術、美容上の欠陥、若しくは皮膚に影響を与える再建術を患う、又は皮膚を必要とする対象が、皮膚置換術を必要とする対象、又は皮膚に影響を与える美容整形若しくは再建術を受ける対象である、請求項191に記載の方法。
- 輪部幹若しくは前駆細胞(LSC)及び/又はその子孫をSESC又はSESC様細胞に変化させる方法であって、LSC及び/又はその子孫をSESC又はSESC様細胞に変化させるために十分にLSCにおけるWNT7A又はPAX6遺伝子の発現をダウンレギュレートさせることによって、LSC及び/又はその子孫をSESC又はSESC様細胞に変化させることを含む方法。
- LSC様細胞及び/又はその子孫をSESC又はSESC様細胞に変化させる方法であって、LSC様細胞及び/又はその子孫をSESC又はSESC様細胞に変化させるために十分にLSC様細胞におけるWNT7A又はPAX6遺伝子の発現をダウンレギュレートさせることによって、LSC様細胞及び/又はその子孫をSESC又はSESC様細胞に変化させることを含む方法。
- 皮膚上皮幹細胞(SESC)及び/又はその子孫をLSC又はLSC様細胞に変化させる方法であって、SESC細胞及び/又はその子孫をLSC又はLSC様細胞に変化させるために十分にSESCにおいてPAX6又はWNT7A遺伝子をアップレギュレート又は過剰発現させることによって、SESC及び/又はその子孫をLSC又はLSC様細胞に変化させることを含む方法。
- SESC様細胞及び/又はその子孫をLSC又はLSC様細胞に変化させる方法であって、SESC様細胞及び/又はその子孫をLSC又はLSC様細胞に変化させるために十分にSESC様細胞においてPAX6又はWNT7A遺伝子をアップレギュレート又は過剰発現させることによって、SESC様及び/又はその子孫をLSC又はLSC様細胞に変化させることを含む方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462018396P | 2014-06-27 | 2014-06-27 | |
| US62/018,396 | 2014-06-27 | ||
| PCT/US2015/038384 WO2015200920A1 (en) | 2014-06-27 | 2015-06-29 | Cultured mammalian limbal stem cells, methods for generating the same, and uses thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017522016A true JP2017522016A (ja) | 2017-08-10 |
| JP2017522016A5 JP2017522016A5 (ja) | 2018-08-09 |
Family
ID=54938873
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016575401A Pending JP2017522016A (ja) | 2014-06-27 | 2015-06-29 | 培養哺乳動物輪部幹細胞、その産生方法及びその使用 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20170233698A1 (ja) |
| EP (1) | EP3161127A4 (ja) |
| JP (1) | JP2017522016A (ja) |
| KR (1) | KR20170020527A (ja) |
| CN (1) | CN107075469A (ja) |
| CA (1) | CA2953524A1 (ja) |
| IL (1) | IL249828A0 (ja) |
| MX (1) | MX2017000181A (ja) |
| SG (2) | SG10201806498RA (ja) |
| WO (1) | WO2015200920A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023182856A1 (ko) * | 2022-03-24 | 2023-09-28 | 서울대학교산학협력단 | 로-키나제 억제제를 포함하는 켈로이드 등 피부 섬유화 질환의 예방 또는 치료용 약학적 조성물 및 그의 용도 |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2905558T3 (es) * | 2015-11-13 | 2022-04-11 | Avellino Lab Usa Inc | Procedimientos para el tratamiento de las distrofias corneales |
| EA201891338A1 (ru) | 2015-12-04 | 2018-12-28 | Новартис Аг | Композиции и способы для иммуноонкологии |
| KR101645901B1 (ko) * | 2015-12-31 | 2016-08-04 | 가톨릭대학교 산학협력단 | 양막 슬라이드 지지체를 이용한 윤부줄기세포 배양방법 |
| CN106011065B (zh) * | 2016-07-15 | 2019-08-02 | 江苏大学 | 人眼球结膜胬肉干细胞细胞株 |
| CN106591220A (zh) * | 2016-12-29 | 2017-04-26 | 深圳市永生原代细胞生物科技有限公司 | 一种人正常皮肤上皮细胞及其用途 |
| CA3048388A1 (en) * | 2017-01-13 | 2018-07-19 | Osaka University | Method for producing a corneal epithelial cell population |
| WO2018175636A2 (en) | 2017-03-22 | 2018-09-27 | Novartis Ag | Compositions and methods for immunooncology |
| EP3823635A4 (en) * | 2018-07-19 | 2022-08-24 | Texas Tech University System | CORNEAL EPITHELIAL CELLS AND PRODUCTS THEREOF FOR THE TREATMENT OF CORNEAL DISEASES |
| CN109321599B (zh) * | 2018-09-27 | 2023-08-22 | 同济大学 | 人多能干细胞中的谱系示踪系统的构建与应用 |
| UY38427A (es) * | 2018-10-26 | 2020-05-29 | Novartis Ag | Métodos y composiciones para terapia con células oculares |
| JP7641902B2 (ja) * | 2019-01-29 | 2025-03-07 | ザ リージェンツ オブ ザ ユニヴァーシティ オブ カリフォルニア | 緑内障の新規治療法 |
| CN109897823A (zh) * | 2019-01-31 | 2019-06-18 | 华中科技大学 | 一种基于Matrigel制备组织工程角膜缘的方法 |
| EP3947651A1 (en) * | 2019-04-01 | 2022-02-09 | Stemsight Oy | Method for obtaining or maintaining abcg2-positive corneal limbal stem cells |
| WO2021009778A2 (en) * | 2019-07-18 | 2021-01-21 | Pandorum Technologies Private Limited | Methods for culturing mesenchymal stem cells, products thereof, and applications thereof |
| EP4025206A1 (en) * | 2019-09-02 | 2022-07-13 | Institut National de la Santé et de la Recherche Médicale (INSERM) | Methods and compositions for treating pax6- deficiency related disease |
| WO2021092564A1 (en) * | 2019-11-07 | 2021-05-14 | Massachusetts Eye And Ear Infirmary | Cultivated autologous limbal epithelial cell (calec) transplantation |
| CN111500526B (zh) * | 2020-03-31 | 2022-02-18 | 厦门大学 | 一种用于原代小鼠角膜上皮细胞体外培养的组合物及其用途 |
| CN115667504A (zh) * | 2020-04-27 | 2023-01-31 | 诺华股份有限公司 | 用于眼细胞疗法的方法和组合物 |
| CN113736735B (zh) * | 2020-05-27 | 2024-02-20 | 深圳华大生命科学研究院 | 体外诱导类角膜缘干细胞的方法及试剂盒 |
| MY208025A (en) * | 2020-12-23 | 2025-04-08 | Univ Kebangsaan Malaysia | A corneal-healing device and method for producing the same |
| CN112626019A (zh) * | 2020-12-28 | 2021-04-09 | 武汉爱尔眼科医院有限公司 | 一种眼角膜及角膜缘单细胞悬液的制备方法 |
| CN115068504A (zh) * | 2021-03-12 | 2022-09-20 | 广州康睿生物医药科技股份有限公司 | 一种角膜修复材料及其制备方法和用途 |
| CN115094038A (zh) * | 2022-07-15 | 2022-09-23 | 中国医学科学院北京协和医院 | 一种角膜基质细胞原代培养的方法 |
| PL442250A1 (pl) * | 2022-09-12 | 2024-03-18 | Acellmed Spółka Z Ograniczoną Odpowiedzialnością | Opatrunek fibrynowy zwierający komórki nabłonka rogówki i sposób wytwarzania opatrunku |
| CN116042510B (zh) * | 2023-01-31 | 2025-07-25 | 浙江大学 | 湖羊瘤胃上皮类器官培养基、上皮类器官的体外构建方法及应用 |
| CN117018165A (zh) * | 2023-09-13 | 2023-11-10 | 中国人民解放军总医院第三医学中心 | Reelin蛋白或Reln基因在角膜损伤治疗药物中的应用 |
| CN117925825B (zh) * | 2024-01-31 | 2024-09-13 | 中国医学科学院北京协和医院 | 包含ncoa7的标志物组合在诊断圆锥角膜中的应用 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003030959A1 (en) * | 2001-10-06 | 2003-04-17 | Btg International Limited | Corneal repair device |
| JP2007524411A (ja) * | 2004-01-27 | 2007-08-30 | リライアンス ライフ サイエンシーズ ピーヴィーティー. リミテッド | 角膜輪部由来の未分化幹細胞を有する組織系 |
| JP2012516685A (ja) * | 2009-02-03 | 2012-07-26 | コーニンクレッカ ネザーランド アカデミー ヴァン ウェテンシャッペン | 上皮幹細胞および該幹細胞を含むオルガノイドのための培養培地 |
| WO2012168930A2 (en) * | 2011-06-10 | 2012-12-13 | Koninklijke Nederlandse Akademie Van Wetenschappen (Knaw) | Culture media for stem cells |
| JP2013000121A (ja) * | 2011-06-16 | 2013-01-07 | Tokyo Women's Medical College | 上皮細胞培養用培地、それを用いた上皮細胞培養方法及びそれより得られた上皮細胞 |
| WO2013012087A1 (ja) * | 2011-07-15 | 2013-01-24 | 国立大学法人大阪大学 | 角膜内皮細胞の調製方法 |
| WO2013061608A1 (ja) * | 2011-10-27 | 2013-05-02 | 国立大学法人東京医科歯科大学 | 大腸上皮幹細胞の単離・培養技術と、これを用いた大腸上皮移植技術 |
| WO2013136372A1 (ja) * | 2012-03-16 | 2013-09-19 | 株式会社日立製作所 | 細胞シート、細胞培養方法および細胞培養装置 |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5057578A (en) | 1990-04-10 | 1991-10-15 | E. I. Du Pont De Nemours And Company | Silicone-containing block copolymers and macromonomers |
| US5314960A (en) | 1990-04-10 | 1994-05-24 | Permeable Technologies, Inc. | Silicone-containing polymers, oxygen permeable hydrophilic contact lenses and methods for making these lenses and treating patients with visual impairment |
| US5371147A (en) | 1990-10-11 | 1994-12-06 | Permeable Technologies, Inc. | Silicone-containing acrylic star polymers, block copolymers and macromonomers |
| US6011029A (en) | 1996-02-26 | 2000-01-04 | Bristol-Myers Squibb Company | Inhibitors of farnesyl protein transferase |
| US5989835A (en) | 1997-02-27 | 1999-11-23 | Cellomics, Inc. | System for cell-based screening |
| NZ513800A (en) | 1996-08-12 | 2001-09-28 | Welfide Corp | Treatment of diseases using Rho kinase inhibitors |
| US6152142A (en) | 1997-02-28 | 2000-11-28 | Tseng; Scheffer C. G. | Grafts made from amniotic membrane; methods of separating, preserving, and using such grafts in surgeries |
| US7217722B2 (en) | 2000-02-01 | 2007-05-15 | Kirin Beer Kabushiki Kaisha | Nitrogen-containing compounds having kinase inhibitory activity and drugs containing the same |
| US20030125344A1 (en) | 2001-03-23 | 2003-07-03 | Bayer Corporation | Rho-kinase inhibitors |
| US20030096813A1 (en) | 2001-04-20 | 2003-05-22 | Jingrong Cao | Compositions useful as inhibitors of GSK-3 |
| JPWO2002100833A1 (ja) | 2001-06-12 | 2004-09-24 | 住友製薬株式会社 | Rhoキナーゼ阻害剤 |
| US6642263B2 (en) | 2001-11-19 | 2003-11-04 | Iconix Pharmaceuticals Inc. | Modulators of Rho C activity |
| DE60320933D1 (de) | 2002-01-10 | 2008-06-26 | Bayer Healthcare Ag | Rho-kinase inhibitoren |
| JP4469179B2 (ja) | 2002-01-23 | 2010-05-26 | バイエル ファーマセチカル コーポレーション | Rhoキナーゼ阻害剤としてのピリミジン誘導体 |
| MXPA04007196A (es) | 2002-01-23 | 2005-06-08 | Bayer Pharmaceuticals Corp | Inhibidores de rho-quinasa. |
| TW200306819A (en) | 2002-01-25 | 2003-12-01 | Vertex Pharma | Indazole compounds useful as protein kinase inhibitors |
| US7645878B2 (en) | 2002-03-22 | 2010-01-12 | Bayer Healthcare Llc | Process for preparing quinazoline Rho-kinase inhibitors and intermediates thereof |
| GB0206860D0 (en) | 2002-03-22 | 2002-05-01 | Glaxo Group Ltd | Compounds |
| EP1500643A4 (en) | 2002-04-03 | 2007-03-28 | Dainippon Sumitomo Pharma Co | benzamidine |
| ES2273047T3 (es) | 2002-10-28 | 2007-05-01 | Bayer Healthcare Ag | Fenilaminopirimidinas sustituidas con heteroariloxi como inhibidores de rho-cinasa. |
| JP4869068B2 (ja) | 2003-06-19 | 2012-02-01 | スミスクライン ビーチャム コーポレーション | 化合物 |
| CA2530389A1 (en) | 2003-07-02 | 2005-01-13 | Biofocus Discovery Limited | Pyrazine and pyridine derivatives as rho kinase inhibitors |
| CN1289156C (zh) * | 2003-07-25 | 2006-12-13 | 吕伟光 | 组织工程自体角膜上皮及其制备方法 |
| CN1590541A (zh) * | 2004-05-27 | 2005-03-09 | 天津医科大学眼科中心 | 角膜缘干细胞组织工程复合体及其制备方法 |
| US20080124276A1 (en) * | 2006-07-24 | 2008-05-29 | Lifeline Cell Technology | Synthetic cornea from retinal stem cells |
| CN101121926A (zh) * | 2007-07-02 | 2008-02-13 | 西北农林科技大学 | 一种角膜缘干细胞无血清培养基 |
| US20110171180A1 (en) | 2009-03-19 | 2011-07-14 | Worcester Polytechnic Institute | Bioengineered skin substitutes |
| GB0908927D0 (en) * | 2009-05-22 | 2009-07-01 | Univ Reading The | Synthetic graft |
| EP2638149B1 (en) | 2010-11-12 | 2019-05-15 | Georgetown University | Immortalization of epithelial cells and methods of use |
| CN103492555A (zh) | 2011-04-20 | 2014-01-01 | 国立大学法人大阪大学 | 角膜上皮分化取向性iPS细胞 |
| CN102952779B (zh) * | 2012-11-30 | 2014-07-16 | 山东大学 | 诱导人胚胎干细胞定向分化为角膜缘干细胞的培养方法 |
| CN103053511A (zh) * | 2012-12-10 | 2013-04-24 | 山东省眼科研究所 | 一种角膜中期保存液及其制备和使用方法 |
-
2015
- 2015-06-29 CA CA2953524A patent/CA2953524A1/en not_active Abandoned
- 2015-06-29 WO PCT/US2015/038384 patent/WO2015200920A1/en not_active Ceased
- 2015-06-29 EP EP15812593.0A patent/EP3161127A4/en not_active Withdrawn
- 2015-06-29 SG SG10201806498RA patent/SG10201806498RA/en unknown
- 2015-06-29 SG SG11201610857TA patent/SG11201610857TA/en unknown
- 2015-06-29 JP JP2016575401A patent/JP2017522016A/ja active Pending
- 2015-06-29 US US15/322,423 patent/US20170233698A1/en not_active Abandoned
- 2015-06-29 CN CN201580046185.2A patent/CN107075469A/zh active Pending
- 2015-06-29 KR KR1020177002448A patent/KR20170020527A/ko not_active Withdrawn
- 2015-06-29 MX MX2017000181A patent/MX2017000181A/es unknown
-
2016
- 2016-12-25 IL IL249828A patent/IL249828A0/en unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003030959A1 (en) * | 2001-10-06 | 2003-04-17 | Btg International Limited | Corneal repair device |
| JP2007524411A (ja) * | 2004-01-27 | 2007-08-30 | リライアンス ライフ サイエンシーズ ピーヴィーティー. リミテッド | 角膜輪部由来の未分化幹細胞を有する組織系 |
| JP2012516685A (ja) * | 2009-02-03 | 2012-07-26 | コーニンクレッカ ネザーランド アカデミー ヴァン ウェテンシャッペン | 上皮幹細胞および該幹細胞を含むオルガノイドのための培養培地 |
| WO2012168930A2 (en) * | 2011-06-10 | 2012-12-13 | Koninklijke Nederlandse Akademie Van Wetenschappen (Knaw) | Culture media for stem cells |
| JP2013000121A (ja) * | 2011-06-16 | 2013-01-07 | Tokyo Women's Medical College | 上皮細胞培養用培地、それを用いた上皮細胞培養方法及びそれより得られた上皮細胞 |
| WO2013012087A1 (ja) * | 2011-07-15 | 2013-01-24 | 国立大学法人大阪大学 | 角膜内皮細胞の調製方法 |
| WO2013061608A1 (ja) * | 2011-10-27 | 2013-05-02 | 国立大学法人東京医科歯科大学 | 大腸上皮幹細胞の単離・培養技術と、これを用いた大腸上皮移植技術 |
| WO2013136372A1 (ja) * | 2012-03-16 | 2013-09-19 | 株式会社日立製作所 | 細胞シート、細胞培養方法および細胞培養装置 |
Non-Patent Citations (2)
| Title |
|---|
| MICROSCOPY RESEARCH AND TECHNIQUE, JPN6019034439, 2010, pages 1045 - 1052, ISSN: 0004474864 * |
| STEM CELLS, vol. 25, JPN6019034443, 2007, pages 1145 - 1155, ISSN: 0004474865 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023182856A1 (ko) * | 2022-03-24 | 2023-09-28 | 서울대학교산학협력단 | 로-키나제 억제제를 포함하는 켈로이드 등 피부 섬유화 질환의 예방 또는 치료용 약학적 조성물 및 그의 용도 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3161127A1 (en) | 2017-05-03 |
| WO2015200920A1 (en) | 2015-12-30 |
| US20170233698A1 (en) | 2017-08-17 |
| IL249828A0 (en) | 2017-02-28 |
| EP3161127A4 (en) | 2018-06-06 |
| KR20170020527A (ko) | 2017-02-22 |
| SG10201806498RA (en) | 2018-08-30 |
| MX2017000181A (es) | 2017-05-01 |
| CA2953524A1 (en) | 2015-12-30 |
| CN107075469A (zh) | 2017-08-18 |
| WO2015200920A8 (en) | 2017-04-06 |
| SG11201610857TA (en) | 2017-01-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2017522016A (ja) | 培養哺乳動物輪部幹細胞、その産生方法及びその使用 | |
| JP7162221B2 (ja) | 網膜組織を含む細胞凝集体及びその製造方法 | |
| ES2688019T3 (es) | Métodos y composiciones para la producción acelerada de células epiteliales pigmentadas retinianas a partir de células pluripotentes | |
| Shatos et al. | Isolation and characterization of progenitor cells in uninjured, adult rat lacrimal gland | |
| ES2716200T3 (es) | Método de diferenciación dirigida que produce células endoteliales corneales | |
| JP4950660B2 (ja) | 分娩後由来細胞を使用する眼組織の修復および再生 | |
| TW201632620A (zh) | 使用前驅細胞治療視網膜變性 | |
| Limnios et al. | Efficient differentiation of human embryonic stem cells to retinal pigment epithelium under defined conditions | |
| TW201908478A (zh) | 調節米勒神經膠質細胞之方法 | |
| US12553027B2 (en) | Generation and cryopreservation of pluripotent stem cell-derived clinical grade corneal endothelial cells | |
| CN112538458B (zh) | 用于重编程细胞的方法 | |
| TW201636032A (zh) | 使用前驅細胞治療眼睛病況 | |
| CN118302517A (zh) | 片状视网膜组织的制造方法 | |
| Higa et al. | Human corneal organoid has a limbal function that supplies epithelium to the cornea with limbal deficiency | |
| Higa et al. | Effects of Amniotic Membrane–Derived Fibroblast Supernatant on Corneal Epithelium | |
| WO2022239868A1 (ja) | 網膜組織の製造方法 | |
| US20230392116A1 (en) | Method for differentiating corneal endothelial cell-like cells from pluripotent stem cells | |
| TW201729819A (zh) | 使用前驅細胞治療視網膜變性 | |
| CN112334574A (zh) | 用于产生视网膜色素上皮细胞的方法和组合物 | |
| Ho et al. | Corneal Endothelium Regeneration: Future Prospects | |
| Higa et al. | Regenerative Therapy | |
| Ladan Espandar et al. | Adult corneal stem cells and alternative sources for regenerative therapy for the cornea: an update | |
| Suresh et al. | Standardization of human corneal endothelial cell isolation and the use of denuded human amniotic membrane as a scaffold for human corneal endothelial cells | |
| Suresh et al. | Standardization of Human Corneal Endothelial Cell Isolation and the Use of | |
| VATTULAINEN | Human Pluripotent Stem Cells for Corneal Applications |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170510 |
|
| RD01 | Notification of change of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7426 Effective date: 20170823 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20170823 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180628 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180628 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190910 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20191203 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200310 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200901 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20210330 |