JP2017523169A - 呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2−d]ピリミジン、フロ[3,2−d]ピリミジン、およびピロロ[3,2−d]ピリミジン - Google Patents
呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2−d]ピリミジン、フロ[3,2−d]ピリミジン、およびピロロ[3,2−d]ピリミジン Download PDFInfo
- Publication number
- JP2017523169A JP2017523169A JP2017502192A JP2017502192A JP2017523169A JP 2017523169 A JP2017523169 A JP 2017523169A JP 2017502192 A JP2017502192 A JP 2017502192A JP 2017502192 A JP2017502192 A JP 2017502192A JP 2017523169 A JP2017523169 A JP 2017523169A
- Authority
- JP
- Japan
- Prior art keywords
- group
- pharmaceutically acceptable
- formula
- acceptable salt
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 C[C@@]([C@]1O2)O[C@](*)(C*)*1OC2=O Chemical compound C[C@@]([C@]1O2)O[C@](*)(C*)*1OC2=O 0.000 description 13
- JUVIBCKUPVAKRX-YELDHKJBSA-N Nc1ncnc2c1[s]cc2C(C1)O[C@@](CO)(CCl)[C@H]1O Chemical compound Nc1ncnc2c1[s]cc2C(C1)O[C@@](CO)(CCl)[C@H]1O JUVIBCKUPVAKRX-YELDHKJBSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H11/00—Compounds containing saccharide radicals esterified by inorganic acids; Metal salts thereof
- C07H11/04—Phosphates; Phosphites; Polyphosphates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/06—Heterocyclic radicals
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Virology (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description
特に呼吸器合胞体ウイルス感染を含めた、Pneumovirinaeウイルス感染を処置するための、置換チエノ[3,2−d]ピリミジン、フロ[3,2−d]ピリミジンおよびピロロ[3,2−d]ピリミジン化合物、方法および医薬製剤、ならびにこの化合物を調製するために有用な方法および中間体が、本明細書において提供される。
Pneumovirinaeウイルスは、流行している多くのヒトおよび動物疾患の原因となる、一本鎖マイナスセンスRNAウイルスである。このウイルスのPneumovirinaeサブファミリーは、Paramyxoviridaeファミリーの一部であり、ヒト呼吸器合胞体ウイルス(HRSV)を含む。ほとんどすべての児童が、2回目の誕生日までに、HRSV感染を有することになる。HRSVは、幼児および児童期における下気道感染症の主な原因であり、感染者の0.5%〜2%が入院を必要とする。慢性の心臓、肺疾患のある老人および成人、または免疫抑制されている老人および成人もやはり、重度のHRSV疾患を発症するリスクが高い(http://www.cdc.gov/rsv/index.html)。HRSV感染を予防するワクチンは、現在、利用可能ではない。モノクローナル抗体であるパリビズマブは、免疫学的予防に利用可能であるが、その使用は、高いリスクのある幼児、例えば、未熟児、または心臓もしくは肺の先天性疾患のどちらかを有する幼児に限定されており、一般使用の場合の費用は高く設定されていることが多い。さらに、ヌクレオシドアナログであるリバビリンは、HRSV感染を処置する唯一の抗ウイルス剤として承認されたが、有効性は限られている。したがって、抗Pneumovirinae治療薬が必要とされている。
ヒト呼吸器合胞体ウイルスによって引き起こされる感染の処置を含めた、Pneumovirinaeウイルスファミリーにより引き起こされる感染を処置するための化合物、方法および医薬製剤が提供される。
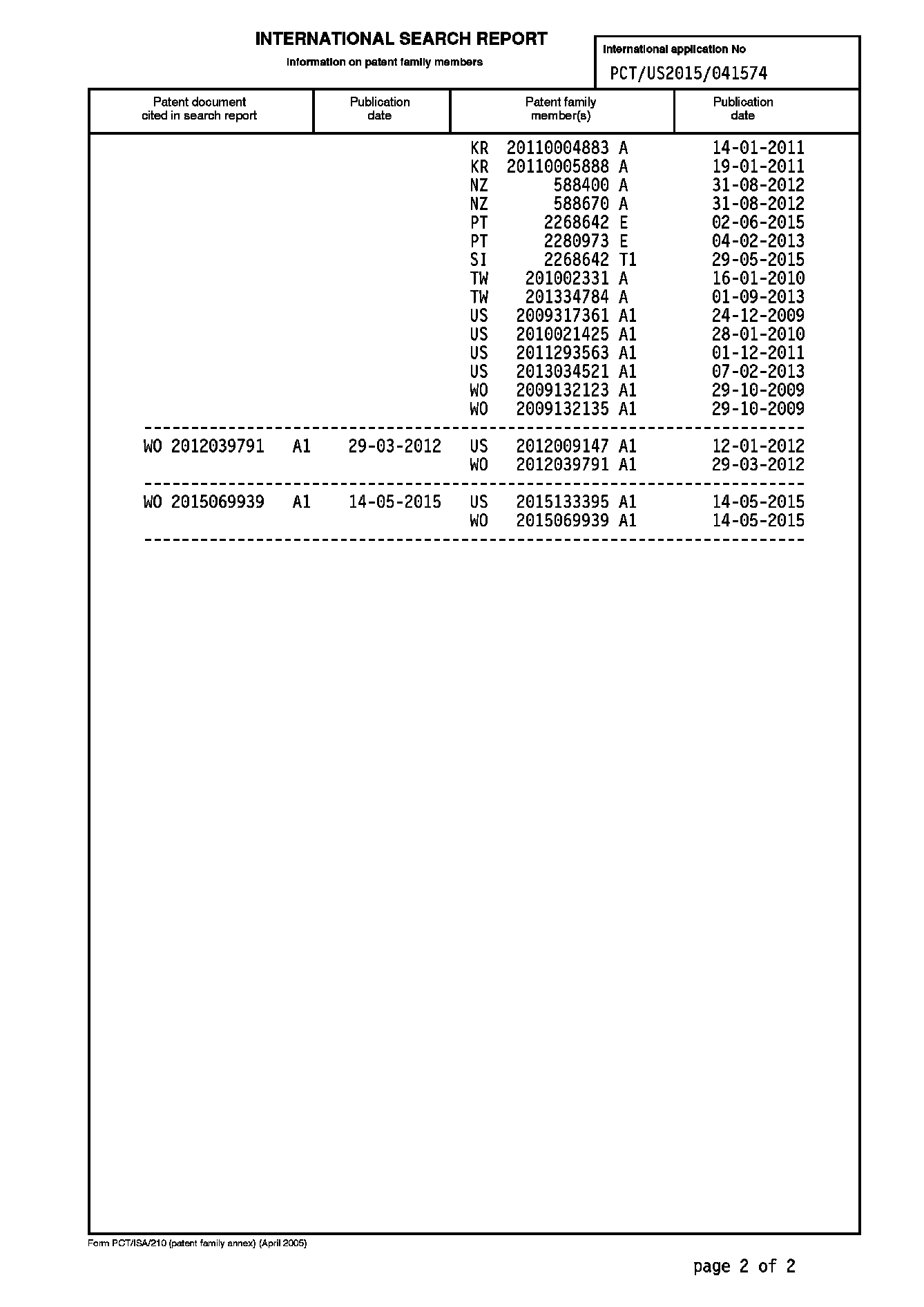
Xは、O、S、NHまたはN(C1〜C6アルキル)の群から選択され、
R1は、H、CH3、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、ORa、NHRa、CNおよびN3の群から選択され、
R3は、CN、ORa、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、−CH2−O−C1〜C6アルキル、−CH2−S−C1〜C6アルキル、C3〜C4シクロアルキル、アジド、ハロゲン、C1〜C3ハロアルキル、SRa、−CH2−C3〜C4シクロアルキル、−O−C3〜Cの群から選択され、R3は、CN、ORa、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、−CH2−O−C1〜C6アルキル、−CH2−S−C1〜C6アルキル、C3〜C4シクロアルキル、アジド、ハロゲン、C1〜C3ハロアルキル、SRa、−CH2−C3〜C4シクロアルキル、−O−C3〜C4シクロアルキル、および−O−C1〜C3ハロアルキルの群から選択されるか、
または
R2がORaである場合、2’位および3’位における2つのORa基は、それらが結合しているフラニル環と一緒になって、
R4は、H、−C(=O)R6、−C(=O)OR6および−C(=O)NR6R7の群から選択されるか、
または
a)R4は、式:
Yはそれぞれ、O、S、NR、+N(O)(R)、N(OR)、+N(O)(OR)もしくはN−NR2であり、
W1およびW2は、一緒になった場合、−Y3(C(Ry)2)3Y3−であるか、
もしくは、W1もしくはW2の一方は、3’ヒドロキシ基と一緒になって、−Y3−であり、W1もしくはW2のもう一方は、式Ia
であるか、
もしくは、W1およびW2はそれぞれ、独立して、式Ia:
ここで、
Y1はそれぞれ、独立して、O、S、NR、+N(O)(R)、N(OR)、+N(O)(OR)もしくはN−NR2であり、
Y2はそれぞれ、独立して、結合、O、CR2、−O−CR2−、NR、+N(O)(R)、N(OR)、+N(O)(OR)、N−NR2、S、S−S、S(O)もしくはS(O)2であり、
Y3はそれぞれ、独立して、O、SもしくはNRであり、
M1は、0、1、2もしくは3であり、
Rxはそれぞれ、独立して、Ryもしくは式:
ここで、
M2a、M2bおよびM2cはそれぞれ、独立して、0もしくは1であり、
M2dは、0、1、2、3、4、5、6、7、8、9、10、11もしくは12であり、
Ryはそれぞれ、独立して、H、F、Cl、Br、I、OH、R、−C(=Y1)R、−C(=Y1)OR、−C(=Y1)N(R)2、−N(R)2、−+N(R)3、−SR、−S(O)R、−S(O)2R、−S(O)(OR)、−S(O)2(OR)、−OC(=Y1)R、−OC(=Y1)OR、−OC(=Y1)(N(R)2)、−SC(=Y1)R、−SC(=Y1)OR、−SC(=Y1)(N(R)2)、−N(R)C(=Y1)R、−N(R)C(=Y1)OR、−N(R)C(=Y1)N(R)2、−SO2NR2、−CN、−N3、−NO2、−OR、またはW3であるか、
もしくは、同一炭素原子上の2つのRyは、一緒になった場合、3個、4個、5個、6個もしくは7個の炭素環原子を有する炭素環式環を形成するか、
もしくは、同一炭素原子上の2つのRyは、一緒になった場合、炭素原子と一緒になって、3個、4個、5個、6個もしくは7個の環原子を有する複素環を形成し、ここで、1個の環原子はOもしくはNから選択され、他の環原子はすべて炭素であり、
Rはそれぞれ、独立して、H、(C1〜C8)アルキル、(C1〜C8)置換アルキル、(C2〜C8)アルケニル、(C2〜C8)置換アルケニル、(C2〜C8)アルキニル、(C2〜C8)置換アルキニル、C6〜C10アリール、C6〜C10置換アリール、3〜10員の複素環、3〜10員の置換複素環、5〜12員のヘテロアリール、5〜12員の置換ヘテロアリール、アリールアルキル、置換アリールアルキル、ヘテロアリールアルキルもしくは置換ヘテロアリールアルキルであり、
W3は、W4もしくはW5であり、
W4は、R、−C(Y1)Ry、−C(Y1)W5、−SO2Ryもしくは−SO2W5であり、
W5は、フェニル、ナフチル、C3〜C8炭素環もしくは3〜10員の複素環から選択され、W5は、0、1つ、2つ、3つ、4つ、5つもしくは6つのRy基により独立して置換されており、
R6およびR7はそれぞれ、独立して、H、(C1〜C8)アルキル、(C2〜C8)アルケニル、(C2〜C8)アルキニル、(C4〜C8)カルボシクリルアルキル、C6〜C10アリール、C6〜C10置換アリール、5〜10員のヘテロアリール、5〜10員の置換ヘテロアリール、−C(=O)(C1〜C8)アルキル、−S(O)n(C1〜C8)アルキルもしくはアリール(C1〜C8)アルキルであるか、
もしくは、R6およびR7は、それらの両方が結合している窒素と一緒になって、3〜7員の複素環を形成し、ここで、前記複素環の任意の1個の環炭素原子が、−O−、−S−もしくは−NRa−で任意選択で置き換えられ得、
各R6もしくはR7の(C1〜C8)アルキル、(C2〜C8)アルケニル、(C2〜C8)アルキニルもしくはアリール(C1〜C8)アルキルはそれぞれ、ハロ、ヒドロキシ、CN、N3、N(Ra)2もしくはORaから選択される1つ、2つ、3つもしくは4つの置換基で任意選択で独立して置換されており、ここで、それぞれの前記(C1〜C8)アルキルの非末端炭素原子の1個、2個もしくは3個が、−O−、−S−もしくは−NRa−で任意選択で置き換えられていてもよいか、または
b)R4は、
R8は、フェニル、1−ナフチル、2−ナフチル、
R9は、HおよびCH3から選択され、
R10は、HもしくはC1〜C6アルキルから選択され、
R11は、H、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択されるか、または
c)R4および3’ヒドロキシ基が一緒になって、
XがSであり、Ra、R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、Y、Y1、Y2、Y3、W1、W2、W3、W4、W5、M1、M2a、M2b、M2c、M2d、RxおよびRyが、式(I)について上で定義されている通りである、式(I)の化合物または薬学的に許容されるその塩が提供される。
R1が、H、CH3、F、ClおよびNH2の群から選択され、
R2が、OH、F、Cl、N3、NH2およびCNの群から選択され、
R3が、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
X、Ra、R4、R5、R6、R7、R8、R9、R10、R11、Y、Y1、Y2、Y3、W1、W2、W3、W4、W5、M1、M2a、M2b、M2c、M2d、RxおよびRyが、式(I)について上で定義されている通りである、
式(I)の化合物または薬学的に許容されるその塩が提供される。
XがSであり、
R1が、H、CH3、F、ClおよびNH2の群から選択され、
R2が、OH、F、Cl、N3、NH2およびCNの群から選択され、
R3が、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
Ra、R4、R5、R6、R7、R8、R9、R10、R11、Y、Y1、Y2、Y3、W1、W2、W3、W4、W5、M1、M2a、M2b、M2c、M2d、RxおよびRyが、式(I)について上で定義されている通りである、
式(I)の化合物または薬学的に許容されるその塩が提供される。
XがOであり、
R1が、H、CH3、F、ClおよびNH2の群から選択され、
R2が、OH、F、Cl、N3、NH2およびCNの群から選択され、
R3が、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
Ra、R4、R5、R6、R7、R8、R9、R10、R11、Y、Y1、Y2、Y3、W1、W2、W3、W4、W5、M1、M2a、M2b、M2c、M2d、RxおよびRyが、式(I)について上で定義されている通りである、
式(I)の化合物または薬学的に許容されるその塩が提供される。
XがNHであり、
R1が、H、CH3、F、ClおよびNH2の群から選択され、
R2が、OH、F、Cl、N3、NH2およびCNの群から選択され、
R3が、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
Ra、R4、R5、R6、R7、R8、R9、R10、R11、Y、Y1、Y2、Y3、W1、W2、W3、W4、W5、M1、M2a、M2b、M2c、M2d、RxおよびRyが、式(I)について上で定義されている通りである、
式(I)の化合物または薬学的に許容されるその塩が提供される。
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
Ra、R4、R5、R6、R7、R8、R9、R10、R11、Y、Y1、Y2、Y3、W1、W2、W3、W4、W5、M1、M2a、M2b、M2c、M2d、RxおよびRyは、式(I)について上で定義されている通りである]。
R2は、F、Cl、OH、NH2、CNおよびN3であり、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
R4は、H、もしくは式:
W1およびW2はそれぞれ、独立して、OHもしくは式Ia:
Yはそれぞれ、独立して結合もしくはOであり、
mは、0、1、2もしくは3であり、
Rxはそれぞれ、H、ハロゲンもしくはOHであるか、または
R4は、Hおよび
n’は、1、2、3および4の群から選択され、
R7は、C1〜C8アルキル、−O−C1〜C8アルキル、ベンジル、−O−ベンジル、−CH2−C3〜C6シクロアルキル、−O−CH2−C3〜C6シクロアルキルおよびCF3の群から選択され、
R8は、フェニル、1−ナフチル、2−ナフチル、
R9は、HおよびCH3から選択され、
R10は、HもしくはC1〜C6アルキルから選択され、
R11は、H、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択される]。
n’は、1、2、3および4から選択され、
R7は、C1〜C8アルキル、−O−C1〜C8アルキル、ベンジル、−O−ベンジル、−CH2−C3〜C6シクロアルキル、−O−CH2−C3〜C6シクロアルキルおよびCF3から選択され、
R8は、フェニル、1−ナフチル、2−ナフチル
R9は、HおよびCH3から選択され、
R10は、HまたはC1〜C6アルキルから選択され、
R11は、H、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択される]から選択される、さらなる実施形態が存在する。
R7は、C1〜C8アルキル、−O−C1〜C8アルキル、ベンジルおよび−CH2−C3〜C6シクロアルキルから選択され、
R11は、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択される]から選択される、さらなる実施形態も存在する。
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2およびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
R11は、H、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択される]または薬学的に許容されるその塩が提供される。
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2およびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
R9は、HおよびCH3から選択され、
R10は、HまたはC1〜C6アルキルから選択され、
R11は、H、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択される]または薬学的に許容されるその塩を含む。
Xは、O、SおよびNHの群から選択され、
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択される]または薬学的に許容されるその塩が提供される。
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択される]または薬学的に許容されるその塩が提供される。
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択される]または薬学的に許容されるその塩が提供される。
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択される]または薬学的に許容されるその塩が提供される。
同様に、薬学的に有効な量の式(I)の化合物または薬学的に許容されるその塩、溶媒和物および/もしくはエステル、ならびに薬学的に許容される担体または添加剤を含む医薬製剤が本明細書において提供される。同様に、別々の医薬製剤であって、その各々が、薬学的に有効な量の式(II)、式(III)、式(IV)、式(V)、式(VI)、式(VII)、式(VIII)の化合物、および本明細書における実施例の具体的な化合物、または薬学的に許容されるその塩、溶媒和物および/もしくはエステル、ならびに薬学的に許容される担体または添加剤を含む、医薬製剤が提供される。
化合物(本明細書では、活性成分と呼ばれる)の一つまたは複数は、処置される状態に適した任意の経路により投与される。適切な経路には、経口、直腸、鼻腔、肺、局所(口腔および舌下を含む)、膣および非経口(皮下、筋肉内、静脈内、皮内、髄膜内および硬膜外を含む)などが含まれる。好ましい経路は、例えば、レシピエントの状態に応じて様々であり得ることが理解される。本明細書における化合物の優位性は、それらが経口で生体利用可能である点、および経口投薬され得る点である。
組成物はまた、他の活性成分と組み合わせて使用される。Pneumovirinaeウイルス感染の処置の場合、好ましくは、他の活性治療剤は、Pneumovirinaeウイルス感染、特に呼吸器合胞体ウイルス感染に対して活性である。こうした他の活性治療剤の非限定例は、リバビリン、パリビズマブ、モタビズマブ、RSV−IGIV(RespiGam(登録商標))、MEDI−557、A−60444(RSV604としても公知)、MDT−637、BMS−433771、ALN−RSV0、ALX−0171、およびそれらの混合物である。
同様に、本明細書において記載されている化合物のin vivoでの代謝産物は、当該産物が新規であり、かつ従来技術を超えた自明なものではない限り、本明細書の範囲内に収まる。当該産物は、例えば、主に酵素過程による、投与された化合物の酸化、還元、加水分解、アミド化、エステル化などに由来し得る。したがって、化合物をその代謝産物が生じるのに十分な時間、哺乳動物に接触させるステップを含むプロセスにより産生される、新規かつ非自明な化合物が含まれる。当該産物は、通常、放射標識されている(例えば、14Cまたは3H)化合物の調製、化合物の、ラット、マウス、モルモット、サルなどの動物またはヒトへの検出可能な用量(例えば、約0.5mg/kgを超える)での非経口投与、代謝が起こるのを可能にする十分な時間(通常、約30秒から30時間)、および尿、血液または他の生物試料からの化合物の変換産物の単離によって同定される。これらの産物は、標識されているので(他のものは、代謝物中で生き残ったエピトープに結合することが可能な抗体を使用することによって単離される)、容易に単離される。代謝物の構造は、従来の手法、例えば、MSまたはNMR分析によって決定される。一般に、代謝物の分析は、当業者に周知の従来の薬物代謝研究と同じ方法で行われる。変換産物は、別の方法でin vivoで見出されない限り、それら自体がHSV抗ウイルス活性を有していない場合でさえも、化合物を治療的に投薬した場合の診断的アッセイにおいて有用である。
スキーム1は、適切な核酸塩基S1b(X1=ハロゲン、−O−アルキル、−O−アリール、−S−アルキル、−S−アリール、NH2、NHRa、NRa 2;X2=Br、I)とのリチウム−ハロゲン交換(例えば、n−BuLi)反応、その後に続くラクトンS1aへの付加で始まる、本発明の化合物の一般合成を示している。ルイス酸性条件(例えば、BF3・Et2O、Et3SiH)下、側基である1’ヒドロキシル基の還元により、中間体S1cが生成する。ヒドロキシル保護基の標準的な変更により、適切に保護されている中間体S1dが得られる。次に、S1dの5’ヒドロキシル基は、対応するヨウ化物(例えば、I2、PPh3)に変換され、これは次に、塩基性条件(例えば、DBU)により処理すると脱離反応が起こり、中間体S1eが生成する。オレフィンS1eの酸化およびアジド付加(例えば、ICl、NaN3)により、中間体S1fが得られる。ヨウ化物S1fを酸素求核剤(例えば、MCPBA、MCBA)で置き換えると、S1gが生成し、X1基を−NH2に変換(例えば、NH4OH)すると、タイプS1hの最終化合物が生じる。
スキーム2は、酸化的条件下(例えば、EDCI)、5’ヒドロキシル基S1dのアルデヒドS2aへの変換から始まる、本発明の中間体の一般合成を示している。対応するエノレートをホルムアルデヒド(例えば、CH2O、NaOH)と縮合し、還元(例えば、NaBH4)すると中間体S2bが生じる。オルトゴナル保護基(orthogonal protecting group)によるヒドロキシル部分の逐次的な選択的保護により、中間体S2cが得られる。酸化的条件(例えば、EDCI)下でのヒドロキシル基S2cのアルデヒドへの変換により、中間体S2dが生成する。アルデヒドS2dのニトリルS2eへの処理は、オキシム形成(例えば、NH2OH)およびオキシムアルコールの脱離(例えば、CDI)を介して行うことができる。X1基の−NH2への変換(例えば、NH4OH)により、タイプS2fの最終化合物が生じる。
スキーム3は、アルコールS2cの処理による本発明の中間体の一般合成を示している。アルコールS2cの脱酸素化(例えば、PPh3、I2、次に、Bu3SnH、AIBN)、およびX1基の−NH2への変換(例えば、NH4OH)により、タイプS3aの最終化合物が生じる。アルコールS2cのメチル化(例えば、MeI)、およびX1基の−NH2への変換(例えば、NH4OH)により、タイプS3bの最終化合物が生じる。アルコールS2cの塩素化(例えば、POCl3)、およびX1基の−NH2への変換(例えば、NH4OH)により、タイプS3cの最終化合物が生じる。アルコールS2cのフッ素化(例えば、DAST)、およびX1基の−NH2への変換(例えば、NH4OH)により、タイプS3dの最終化合物が生じる。
スキーム4は、アルデヒドS2dの処理による本発明の中間体の一般合成を示している。アルデヒドS2dのオレフィン化(例えば、Ph3PCH2)、およびX1基の−NH2への変換(例えば、NH4OH)により、タイプS4aの最終化合物が生じる。オレフィンの還元(例えば、H2、Pd/C)により、タイプS4bの最終化合物が生じる。アルデヒドS2dの二フッ素化(例えば、DAST)、およびX1基の−NH2への変換(例えば、NH4OH)により、タイプS4cの最終化合物が生じる。オレフィンS4aのシクロプロパン化(例えば、CH2N2)、およびX1基の−NH2への変換(例えば、NH4OH)により、タイプS4dの最終化合物が生じる。
スキーム5は、トリフレートS5a(WO2013138236A1、WO2012037038A1、WO2012012776A1に従って調製した)の処理による本発明の中間体の一般合成を示している。トリフレートS5aのフッ素求核剤(例えばCsF)による置換、メトキシアセタールの加水分解(例えば、TFA、H2O)、および酸化(例えば、PDC)により、核酸塩基とのカップリングに適したタイプS5bの化合物が生じる。トリフレートS5aの塩素求核剤(例えば、LiCl)による置換、メトキシアセタールの加水分解(例えば、TFA、H2O)、および酸化(例えば、PDC)により、核酸塩基とのカップリングに適したタイプS5cの化合物が生じる。トリフレートS5aのアジド求核剤(例えば、NaN3)による置換、メトキシアセタールの加水分解(例えば、TFA、H2O)、および酸化(例えば、PDC)により、核酸塩基とのカップリングに適したタイプS5dの化合物が生じる。トリフレートS5aのシアン化物求核剤(例えば、NaCN)による置換、メトキシアセタールの加水分解(例えば、TFA、H2O)、および酸化(例えば、PDC)により、核酸塩基とのカップリングに適したタイプS5eの化合物が生じる。
スキーム6は、塩基S6a(WO2008073785A2に従って調製した)の処理による本発明の中間体の一般合成を示している。S6a塩基のアミノ化(例えば、NH3)によりS6bが生じ、これをアシル化(例えばAc2O)すると、S6cが生成する。求核性フッ素(例えば、CsF)の付加によりS6dが得られ、アシル基の除去(例えば、NH3)により、ラクトンへのカップリングに適した、タイプS6eの最終化合物が生じる。
スキーム7は、タイプS7bのリン酸化類似体の合成を含む、本発明の化合物の一般合成を示している。
スキーム8は、タイプS7bのリン酸化類似体の合成を含む、本発明の化合物の一般合成を示している。
スキーム9は、タイプS9cのリン酸化類似体の合成を含む、本発明の化合物の一般合成を示している。
MeCN(50mL)中の7−ブロモ−4−クロロチエノ[3,2−d]ピリミジン(1a、Pharmablock Inc.から購入、5.00g、20.0mmol)、フェノール(1.90g、20.2mmol)およびCs2CO3(7.8g、24.0mmol)の混合物を40℃で16時間、撹拌した。反応混合物をAcOH(1.8mL)によりクエンチし、減圧下で濃縮した。残留物をH2O(25mL)により処理し、次に、得られた固体沈殿物を真空濾過により回収した。固体をH2O、続いてヘキサン−酢酸エチル(4:1)により洗浄して乾燥すると、中間体1b(6.16g、88%)が白色固体として得られた。
1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 7.97 (s, 1H), 7.51-7.45 (m, 2H), 7.36-7.30 (m, 1H), 7.27-7.25 (m, 2H). MS m/z=307.1、309.1[M+1]
7−ブロモ−4−フェノキシチエノ[3,2−d]ピリミジン(1b、5.1g、16.6mmol)および(3R,4R,5R)−4−(ベンジルオキシ)−5−((ベンジルオキシ)メチル)−3−フルオロジヒドロフラン−2(3H)−オン(1c、WO2012012776A1、5.5g、16.6mmol)のTHF(300mL)溶液に、−78℃でnBuLi(ヘキサン中、2.5M、8.0mL、20mmol)を30分間かけて滴下添加した。反応混合物を−78℃で30分間、撹拌し、AcOH(1.5mL)によりクエンチした。得られた混合物を室温まで加温した。反応混合物を酢酸エチル(300mL)により希釈し、水により洗浄した。有機相を無水Na2SO4により乾燥させ、減圧下で濃縮した。ヘキサン−酢酸エチル(6:1〜3:1)により溶出するシリカゲルクロマトグラフィーによって残留物を精製すると、異性体混合物1d(5.5g、59%)が薄黄色油状物として得られた。
MS m/z=558.9[M+1]。
中間体1d(5.5g、10mmol)のCH2Cl2(50mL)溶液に、室温でEt3SiH(4.6g、39mmol)、次にBF3・Et2O(4.2g、30mmol)を加えた。混合物を室温で7日間、撹拌した。氷水浴を用いて反応混合物を冷却し、NaHCO3(20g)のH2O(100ml)溶液によりゆっくりとクエンチした。有機相を分離してNa2SO4で乾燥させ、ヘキサン−酢酸エチル(6:1〜4:1)により溶出するシリカゲルクロマトグラフィーによって精製すると、中間体1e(3g、56%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 8.15 (s, 1H), 7.5-7.45 (m, 2H), 7.4-7.2 (m, 13H), 5.71 (d, J = 24.8 Hz, 1H), 5.28 (dd, J = 55, 3.6 Hz, 1H), 4.75-4.45 (m, 4H), 4.38-4.25 (m, 2H), 3.98 (dd, J = 10.8, 2 Hz, 1H), 3.72 (dd, J = 10.8, 2 Hz, 1H). 19F NMR (376 MHz, CDCl3) δ -195.25〜-195.51(m). MS m/z=543.1[M+1]。
密封したフラスコ中で、中間体1e(3g、5.53mmol)、NH4OH(28%、50mL)およびMeCN(50mL)の混合物を65℃で64時間、撹拌した。HPLC分析により約40%の変換であることが示された。次に、追加のNH4OH(28%、50mL)およびMeCN(50mL)を加えた。反応混合物を70℃でさらに36時間、撹拌した。HPLC分析により約75%の変換であることが示された。次に、反応混合物を濃縮し、酢酸エチル(25〜100%)−ヘキサンにより溶出するシリカゲルクロマトグラフィーによって精製すると、中間体1f(1.3g)が得られた。残留出発物質(中間体1e、0.82g)を回収し、70℃で2Me−THF(25mL)およびEtOH(25mL)中、NH4OH(28%、50mL)により再処理した。64時間後、得られた混合物を減圧下で濃縮し、酢酸エチル(25〜100%)−ヘキサンにより溶出するシリカゲルカラムによって精製すると、中間体1fがさらに0.75g得られた。合計で、中間体1fが2.05g(80%)得られた。
1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 7.99 (d, J = 1.2 Hz, 1H), 7.38-7.25 (m, 10H), 5.75 (br s, 2H), 5.65 (d, J = 24.8 Hz, 1H), 5.25 (dd, J = 55, 3.6 Hz, 1H), 4.75-4.45 (m, 4H), 4.38-4.2 (m, 2H), 3.98 (dd, J = 10.8, 2 Hz, 1H), 3.72 (dd, J = 10.8, 2 Hz, 1H). 19F NMR (376 MHz, CDCl3) δ -195.12〜-195.39 (m). MS m/z=466.1[M+1]。
中間体1f(2.05g、4.4mmol)のピリジン(15mL)溶液に、0℃で塩化ベンゾイル(1.86g、13.2mmol)を加えた。反応混合物を室温で1時間、撹拌した。反応混合物を氷水浴で冷却し、MeOH(5mL)によりクエンチした。得た反応混合物を45℃で16時間、撹拌した。HPLCおよびLC−MSにより、6−NBz2が6−NHBzに変換されたことが示された。反応混合物を減圧下で濃縮した。残留物を酢酸エチルおよび水により処理した。有機相を分離してNa2SO4で乾燥させ、酢酸エチル(20〜50%)/ヘキサンにより溶出するシリカゲルクロマトグラフィーによって精製すると、中間体1g(2.1g、85%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.81 (s, 1H), 8.24 (s, 1H), 8.06 (d, J = 7.2 Hz, 2H), 7.66 (t, J = 7.2 Hz, 1H), 7.56 (app-t, J = 7.6 Hz, 2H), 7.37-7.25 (m, 10H), 5.73 (d, J = 24.8 Hz, 1H), 5.22 (dd, J = 54.8, 3.6 Hz, 1H), 4.71-4.45 (m, 4H), 4.4-4.20 (m, 2H), 3.98 (dd, J = 10.8, 2 Hz, 1H), 3.72 (dd, J = 10.8, 3.2 Hz, 1H). 19F NMR (376 MHz, CDCl3) δ -189.77〜-190.04 (m). MS m/z=570.1[M+1]。
中間体1g(2.1g、3.69mmol)をトルエン(2×6mL)と共蒸発させた。残留物を酢酸エチル(2mL)に溶解し、0℃でメタンスルホン酸(4mL)を加えた。3時間後、追加のメタンスルホン酸(1mL)を加えた。反応混合物を室温でさらに4時間、撹拌し、この時点で、反応混合物を酢酸エチル(50mL)により希釈した。得られた混合物を0℃まで冷却し、固体NaHCO3(12.0g)は4回に分けて加えた。得た混合物を0℃で1時間、次に、室温で16時間、撹拌した。反応混合物に、水(25mL)をゆっくりと加えた。混合物を0.5時間、撹拌し、次に濾過して残留固体を除去した。有機相をブラインにより洗浄し、Na2SO4で乾燥させて減圧下で濃縮した。メタノール(0〜10%)/ジクロロメタン(dicholomethane)で溶出するシリカゲルカラムによって残留物を精製すると、中間体1h(0.79g、55%)が得られた。
1H NMR (400 MHz, CD3OD) δ 8.87 (s, 1H), 8.37 (d, J = 0.8 Hz, 1H), 8.05 (d, J = 7.2 Hz, 2H), 7.66 (t, J = 7.6 Hz, 1H), 7.56 (app-t, J = 7.6 Hz, 2H), 5.57 (app dd, J = 24, 1.6 Hz, 1H), 5.12 (ddd, J = 54.8, 4, 1.6 Hz, 1H), 4.31 (ddd, J = 19.6, 8, 4 Hz, 1H), 4.07-4.02 (m, 1H), 4.0 (dd, J = 12.4, 2.4 Hz, 1H), 3.79 (dd, J = 12.4, 4 Hz, 1H). 19F NMR (376 MHz, CD3OD) δ -202.76〜-203.03 (m). MS m/z=390.1[M+1]。
中間体1h(0.79g、2.03mmol)、Ph3P(1.20g、4.58mmol)およびイミダゾール(0.277g、4.07mmol)のTHF(15mL)溶液に、室温でヨウ素(0.96g、3.78mmol)を加えた。4時間後、反応混合物にNaHCO3(固体、500mg)、次いで水(200μL)を加え反応をクエンチした。反応混合物を濃縮し、残留物を酢酸エチル−ヘキサン(1:1)により溶出するシリカゲルカラムにより精製すると、中間体1i(0.85g、84%)が固体として得られた。
1H NMR (400 MHz, CDCl3) δ 9.05 (br s, 1H), 8.84 (s, 1H), 8.29 (d, J = 1.2 Hz, 1H), 8.03 (d, J = 7.6 Hz, 2H), 7.68-7.63 (m, 1H), 7.56 (app-t, J = 7.6 Hz, 2H), 5.67 (app d, J = 27.2 Hz, 1H), 5.27 (ddd, J = 55.2, 4.4, 0.8 Hz, 1H), 4.17 (ddd, J = 20.8, 8.4, 4.4 Hz, 1H), 3.79-3.75 (m, 1H), 3.72 (dd, J = 10.8, 3.6 Hz, 1H), 3.53 (dd, J = 10.8, 4.8 Hz, 1H). 19F NMR 376 MHz, CDCl3) δ -193.33〜-193.61 (m). MS m/z=500.0[M+1]。
中間体1i(0.84g、1.68mmol)のTHF(5mL)溶液に、DBU(0.68g、4.47mmol)を加えた。反応混合物を室温で16時間、撹拌し、次に、45℃に加熱した。8時間後、この反応混合物を室温まで冷却し、反応混合物を減圧下で濃縮した。残留物をCH2Cl2に溶解し、酢酸エチル(50〜100%)−ヘキサンにより溶出するシリカゲルカラムに充填すると、中間体1j(0.45g、72%)が固体として得られた。
1H NMR (400 MHz, DMSO-d6) δ 11.65 (br s, 1H), 8.98 (s, 1H), 8.22 (d, J = 1.2 Hz, 1H), 8.08 (d, J = 7.6 Hz, 2H), 7.66 (t, J = 7.6 Hz, 1H), 7.55 (t, J = 7.6 Hz, 2H), 5.82 (d, J = 21.6 Hz, 1H), 5.81 (d, J = 8 Hz, 1H), 5.24 (dd, J = 54.4, 4 Hz, 1H), 4.85-4.72 (m, 1H), 4.43 (br s, 1H), 4.17 (t, J = 1.6 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ -201.19〜-201.46 (m). MS m/z=371.9[M+1]。
DMF(10mL)中のNaN3(400mg、6.15mmol)の懸濁液に、0℃(氷水浴)でICl(400mg、2.46mmol)を加えた。得られた混合物を0℃で10分間、撹拌し、20分間かけて室温まで加温した。反応混合物を氷−アセトン浴を用いて冷却し、中間体1j(400mg、1.08mmol)のDMF(2mL)溶液を加えた。得られた反応混合物を0℃で1時間、撹拌し、この時点で、反応物を、Na2S2O3水溶液(1M、3mL)によりクエンチした。反応混合物を減圧下で濃縮し、CH3CNと共蒸発させた。残留物をCH2Cl2で処理し、濾過した。濾液をヘキサン−酢酸エチル(1:1)により溶出するシリカゲルカラムに充填すると、中間体1k(410mg、70%)が固体として得られた。NMR分析により、この中間体は4’−位における45:55のアノマー混合物であることが示された。
1H NMR (400 MHz, DMSO-d6) δ 11.64 (br s, 1H), 8.98 (s, 1H), 8.56 (s, 0.45H), 8.43 (s, 0.55H), 8.08 (d, J = 8 Hz, 2H), 7.66 (t, J = 8 Hz, 1H), 7.55 (t, J = 8 Hz, 2H), 6.47 (d, J = 6 Hz, 0.45H), 6.26 (d, J = 7.2 Hz, 0.55H), 5.9-5.25 (m, 2H), 4.75-4.45 (m, 1H), 3.74 (s, 1H), 3.62-3.52 (m, 1H). 19F NMR (376 MHz, DMSO- d6) δ -197.24〜-197.51 (m) (主異性体), -208.52〜-208.73 (m) 副異性体). MS m/z=540.9[M+1]。
CH2Cl2(50mL)およびH2O(10mL)中の、中間体1k(400mg、0.74mmol)、3−クロロ安息香酸(300mg、1.92mmol)、硫酸水素テトラブチルアンモニウム(275mg、0.81mmol)およびリン酸水素二カリウム(3・H2O、830mg、3.64mmol)の溶液に、3−クロロ過安息香酸(77%、750mg、3.35mmol)を加えた。得た反応混合物を室温で4時間、撹拌した。反応混合物を氷水浴により冷却し、Na2S2O3水溶液(1M、5mL)でクエンチした。得られた混合物を減圧下で濃縮し、残留物を酢酸エチルに溶解した。有機相を飽和NaHCO3水溶液で洗浄して、無水硫酸ナトリウムにより乾燥させ、減圧下で濃縮した。残留物をヘキサン/酢酸エチル(2:1)により溶出するシリカゲルカラムによって精製すると、2つの異性体が得られた。最初に溶出する所望の異性体中間体1l(135mg、32%、シリカゲルおよびC−18 HPLCの両方で、異性体Bより速く溶出した)、および2番目に溶出した望ましくない異性体(70mg、17%、シリカゲルおよびC−18 HPLCの両方で、異性体Aより遅く溶出した)。
(2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−アジド−4−フルオロ−2−(ヒドロキシメチル)−テトラヒドロフラン−3−オール
中間体1l(135mg、0.237mmol)およびNH4OH(28%、3mL)のMeOH(3mL)溶液を45℃で16時間、撹拌した。この反応混合物を減圧下で濃縮した。この残留物を分取HPLCにより精製すると、実施例1(54mg、70%)が得られた。
1H NMR (400 MHz, DMSO-d6) δ 8.40 (s, 1H), 8.24 (s, 1H), 7.67 (br s, 2H), 5.85 (br s, 1H), 5.62 (d, J = 23.6 Hz, 1H), 5.14 (ddd, J = 55.2, 4.8, 2 Hz, 1H), 4.46 (dd, J = 24.0, 4.8 Hz, 1H), 3.70 (d, J = 12 Hz, 1H), 3.56 (d, J = 12 Hz, 1H). 19F NMR (376 MHz, DMSO-d6) δ -197.67〜-197.94 (m). MS m/z=327.0[M+1]。
中間体1h(2.0g、5.14mmol)のピリジン(20mL)溶液に、室温でDMTrCl(2.52g、7.45mmol)を1度に加えた。得られた混合物を室温で40分間、撹拌し、この時点でメタノール(5mL)を加えた。混合物を真空中で濃縮し、シリカゲルカラムクロマトグラフィー(ヘキサン中の0〜70%酢酸エチル)によって精製すると、中間体2a(2.5g、70%)が白色泡状物として得られた。MS m/z=692[M+1]
中間体2a(2.5g、3.61mmol)のDMF(20mL)溶液に、室温でイミダゾール(738mg、10.8mmol)およびTBSCl(817mg、5.42mmol)を加えた。混合物を7時間、撹拌し、MeOH(5mL)を加えた。5分間の撹拌後、混合物をEtOAcにより希釈して水および炭酸水素ナトリウム溶液により洗浄した。有機層を硫酸ナトリウムで乾燥し、真空中で濃縮した。残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜70%酢酸エチル)によって精製すると、中間体2b(3.0g、86%、84%純度)が白色固体として得られた。MS m/z=806[M+1]
DCM(20mL)およびMeOH(10mL)中の中間体2b(3.0g、3.7mmol)の溶液に、室温でTFA(1.50mL、19.6mmol)を滴下添加した。混合物を1時間、撹拌し、この時点で炭酸水素ナトリウム溶液(5mL)により処理し、ジクロロメタンにより希釈した。相を分離して、有機相を炭酸水素ナトリウム溶液により洗浄して、硫酸ナトリウムで乾燥して減圧下で濃縮した。残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜90%酢酸エチル)によって精製すると、中間体2c(1.6g、85%)が白色固体として得られた。 MS m/z=504[M+1]
中間体2c(1.6g、3.18mmol)のDMSO−トルエン(10:5mL)溶液に、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドHCl(1.83g、9.53mmol)、ピリジン(0.26mL、3.12mmol)およびTFA(0.15mL、1.91mL)を加えた。得られた混合物を室温で2時間、撹拌し、メタノール(5mL)を加えた。得られた混合物をジクロロメタンにより希釈して水およびブラインにより洗浄し、硫酸ナトリウムで乾燥して真空中で濃縮すると、粗製中間体2d(1.4g)が得られ、これを次の反応に直接使用した。
粗製中間体2d(1.4g、2.79mmol)をTHF(20mL)に溶解し、37重量%のホルムアルデヒド(1.70mL、22.8mmol)および2N NaOH水溶液(2.80mL、5.58mmol)の両方を加えた。得られた混合物を室温で2時間、撹拌した。次に、追加のホルムアルデヒド(2mL)および2N NaOH水溶液(2mL)を加えた。30分間後に、混合物をAcOHで中和し、EtOAcにより希釈し、炭酸水素ナトリウム溶液およびブラインにより洗浄し、硫酸ナトリウムで乾燥して真空中で濃縮すると、中間体2e(1.4g)が黄色泡状物として得られ、これを次のステップに直接使用した。
次に、中間体2e(1.4g、2.63mmol)をエタノール(15mL)に溶解し、0℃で水素化ホウ素ナトリウム(110mg、2.90mmol)を少量ずつ5分間かけて加えた。氷水浴中で15分間、撹拌した後、反応混合物をAcOHにより中和し、EtOAcにより希釈して飽和NaHCO3およびブラインにより洗浄し、硫酸ナトリウムで脱水した。残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜100%酢酸エチル)によって精製すると、中間体2f(1.0g、3ステップの通算で59%)が白色固体として得られた。
1H NMR (400 MHz, CDCl3) δ 9.40 (s, 1H), 8.84 (s, 1H), 8.13 - 7.97 (m, 3H), 7.73 - 7.61 (m, 1H), 7.6 - 7.47 (m, 2H), 5.55 (dd, J = 7.9, 5.5 Hz, 0.5H), 5.49 - 5.30 (m, 1.5H), 4.76 (dd, J = 5.4, 2.6 Hz, 1H), 3.94 (d, J = 12.1 Hz, 1H), 3.85 (d, J = 12.0 Hz, 1H), 3.70 (d, J = 7.6 Hz, 1H), 3.67 (d, J = 7.6 Hz, 1H), 0.96 (s, 9H), 0.19 - 0.14 (m, 6H). 19F NMR (376 MHz, CDCl3) δ -202.47 (dd, J = 53.2, 14.9 Hz). MS m/z=534[M+1]。
中間体2f(1.0g、1.94mmol)およびトリエチルアミン(0.7mL)のジクロロメタン(40mL)溶液に、シリンジポンプを使用し、0℃でDMTrCl(984mg、2.90mmol)を1時間かけて、ゆっくりと加えた。添加の完了時に、メタノール(2mL)を加えることにより反応をクエンチし、混合物をジクロロメタンにより希釈し、水および飽和炭酸水素ナトリウム水溶液により洗浄し、硫酸ナトリウムで乾燥して、減圧下で濃縮した。残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜70%酢酸エチル)によって精製すると、中間体2g(660mg)および回収された出発物質である中間体2f(340mg)が得られた。回収された出発中間体2f(340mg)に同じ反応条件を再度供し、さらなる中間体2gを白色固体として単離した(合計で1.0g、64%)。
MS m/z=836[M+1]。
中間体2g(1.0g、1.20mmol)をDMF(10mL)に溶解した。イミダゾール(244mg、3.59mmol)およびTBSCl(270mg、1.79mmol)を加えた。得られた混合物を室温で2時間、撹拌し、メタノール(2mL)を加えた。得られた混合物をEtOAcにより希釈し、水および炭酸水素ナトリウム溶液により洗浄し、硫酸ナトリウムで乾燥して真空中で濃縮した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜70%酢酸エチル)によって精製すると、完全に保護されている中間体2h(1.0g、88%)が白色固体として得られた。
MS m/z=950[M+1]。
中間体2h(1.0g、1.05mmol)をジクロロメタン(15mL)に溶解し、0℃に冷却した。PTSA(200mg、1.05mmol)のMeOH(5mL)溶液を5分間かけてゆっくりと加え、次に、激しく撹拌しながら、炭酸水素ナトリウム溶液(5mL)を加えた。ジクロロメタンにより希釈した後、混合物をブラインにより洗浄し、硫酸ナトリウムで乾燥して、真空中で濃縮した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜60%酢酸エチル)によって精製すると、中間体2i(570mg、84%)が白色固体として得られた。
MS m/z=648[M+1]。
トルエン−DMSO(2:4mL)中の、中間体2i(570mg、0.88mmol)とEDCI(506mg、2.64mmol)との懸濁液に、室温でピリジン(0.1mL、1.24mmol)およびTFA(0.05mL、0.65mmol)を順に加えた。得られた混合物を室温で1時間、撹拌し、酢酸エチルにより希釈し、炭酸水素ナトリウム水溶液およびブラインで洗浄して硫酸ナトリウムで乾燥させ、真空中で濃縮すると、中間体2j(570mg)が得られ、これを次のステップに直接使用した。
MS m/z=646[M+1]。
中間体2j(570mg、粗製)のピリジン(5mL)溶液に、室温でヒドロキシルアミン塩酸塩(92mg、1.32mmol)を加えた。得られた混合物を室温で1時間、撹拌し、酢酸エチルにより希釈してブラインにより洗浄し、硫酸ナトリウムで乾燥して真空中で濃縮した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜80%酢酸エチル)によって精製すると、中間体2k(500mg、85%)が白色固体として得られた。
MS m/z=661[M+1]。
中間体2k(500mg、0.76mmol)のMeCN−THF(10:5mL)溶液に、室温でCDI(430mg、2.65mmol)を加えた。得られた混合物を室温で4時間、撹拌し、酢酸エチルにより希釈してブラインにより洗浄し、硫酸ナトリウムで乾燥して真空中で濃縮した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜80%酢酸エチル)によって精製すると、中間体2l(480mg、99%)がガラス状固体として得られた。
1H NMR (400 MHz, CDCl3) δ 9.40 (s, 1H), 8.81 (s, 1H), 8.22 (d, J = 1.0 Hz, 1H), 8.09 - 8.03 (m, 2H), 7.71 - 7.63 (m, 1H), 7.57 (dd, J = 8.5, 7.0 Hz, 2H), 5.80 (dt, J = 24.4, 1.4 Hz, 1H), 5.20 (ddd, J = 54.4, 4.4, 1.8 Hz, 1H), 4.72 (dd, J = 20.4, 4.4 Hz, 1H), 4.16 (d, J = 11.3 Hz, 1H), 3.93 (d, J = 11.2 Hz, 1H), 0.96 (s, 9H), 0.90 (s, 9H), 0.12 (m, 12H). 19F NMR (376 MHz, CDCl3) δ -191.15 - -191.55 (m). MS m/z=643[M+1]。
中間体2l(480mg、0.75mmol)のTHF(10mL)溶液に、室温で酢酸(0.047mL、0.82mL)、次いで、THF中の1M TBAF(0.82mL、0.82mmol)を加えた。得られた混合物を室温で15時間、撹拌し、真空中で濃縮した。得られた残留物をシリカゲルカラムクロマトグラフィー(DCM中の0〜7%MeOH)によって精製すると、中間体2m(300mg、97%)がシロップ状物として得られた。
MS m/z=415[M+1]。
(2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−4−フルオロ−3−ヒドロキシ−2−(ヒドロキシメチル)テトラヒドロフラン−2−カルボニトリル
中間体2m(300mg、0.72mmol)を7Mメタノール性アンモニア(30mL)に溶解し、室温で撹拌した。24時間後、反応混合物を真空中で濃縮し、得られた残留物をシリカゲルカラムクロマトグラフィー(DCM中の0〜50%MeOH)により精製すると、実施例2(90mg、40%)が白色固体として得られた。出発中間体2m(120mg)も回収された。
1H NMR (400 MHz, CD3OD) δ 8.37 (s, 1H), 8.14 (d, J = 0.9 Hz, 1H), 5.67 (ddd, J = 23.6, 2.5, 1.0 Hz, 1H), 5.23 (ddd, J = 54.4, 4.5, 2.5 Hz, 1H), 4.65 (dd, J = 19.8, 4.5 Hz, 1H), 4.04 (d, J = 12.2 Hz, 1H), 3.89 (d, J = 12.2 Hz, 1H). 19F NMR (376 MHz, CD3OD) δ -195.68 (ddd, J = 54.4, 23.7, 19.8 Hz). MS m/z=311[M+1]。
中間体3a(Carbosynthから購入、6.8g、16.27mmol)および中間体1b(5g、16.3mmol)のTHF(100mL)溶液に、−60℃未満の内部温度を維持しながらヘキサン中の2.5M n−ブチルリチウム(7.2mL、17.9mmol)をこの反応混合物に滴下添加した。2時間後、追加のヘキサン中の2.5M n−ブチルリチウム(1mL)を滴下添加した。さらに2時間後、酢酸(2mL、35.8mmol)を滴下添加し、pH=3にした。冷却浴を取り去り、10分間、撹拌した。酢酸エチル(100mL)により希釈し、飽和NaHCO3(水性)(50mL)、次に、飽和NaCl(水性)(50mL)により洗浄した。有機物を無水Na2SO4で乾燥させ、減圧下で濃縮した。シリカゲルカラム(ヘキサン中の0〜40%酢酸エチル)により精製すると、粗製中間体3b(9g、85%)が得られた。
MS m/z=647.0[M+1]。
粗製中間体3b(9g、13.9mmol)を90mL無水DCMに溶解し、アルゴン下、反応混合物を氷浴中、0℃で撹拌した。トリエチルシラン(5.6mL、34.8mmol)、次いで三フッ化ホウ素ジエチルエーテル錯体(2.6mL、20.9mmol)を滴下添加した。反応混合物を0℃で60分間、撹拌し、反応混合物を室温まで加温した。16時間後、追加のトリエチルシラン(1.2mL)および三フッ化ホウ素ジエチルエーテル錯体(870uL)を加えた。20時間後、反応混合物を氷浴中で0℃まで冷却し、TEA(6.8mL、48.7mmol)を滴下添加した。次に、反応混合物を減圧下で濃縮した。残留物を酢酸エチル(200mL)に溶解し、飽和NaHCO3(水性)(50mL)、次に飽和NaCl(水性)(50mL)により洗浄した。有機相を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルクロマトグラフィー(ヘキサン中の0〜20〜30%酢酸エチル)により精製すると、中間体3c(2.98g、34%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 8.11 (s, 1H), 7.55 - 7.41 (m, 4H), 7.41 - 7.19 (m, 16H), 5.70 (s, 1H), 4.95 (s, 2H), 4.67 - 4.51 (m, 3H), 4.50 - 4.41 (m, 2H), 4.28 (m, 1H), 4.23 (m, 1H), 3.98 (dd, J = 10.8, 2.8 Hz, 1H), 3.73 (dd, J = 10.8, 3.2 Hz, 1H). MS m/z=631.2[M+1]。
密封容器中で、中間体3c(2.98g、4.7mmol)をアセトニトリル25mLに溶解して30%水酸化アンモニウム溶液25mLと混合し、反応混合物を80℃まで加熱した。16時間後、30%水酸化アンモニウム溶液(15mL)を加え、得られた混合物を90℃で24時間、撹拌した。追加のアセトニトリル(15mL)を加え、得られた混合物を90℃で4日間、撹拌した。反応混合物を室温まで冷却し、EtOAcにより希釈して飽和NaCl水溶液(3×)で洗浄した。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルカラム(ヘキサン中の0〜50%酢酸エチル)により精製すると、中間体3d(1.4g、56%)が得られた。
1H NMR (400 MHz, CDCl3) δ 7.95 (s, 1H), 7.50 - 7.16 (m, 16H), 5.62 (s, 1H), 4.93 (s, 2H), 4.66 - 4.51 (m, 2H), 4.47 (m, 1H), 4.41 (m, 1H), 4.27 (m, 1H), 4.17 (s, 1H), 4.14 - 4.08 (m, 1H), 3.97 (dd, J = 10.8, 2.7 Hz, 1H), 3.71 (dd, J = 10.7, 3.0 Hz, 1H). MS m/z=554.2[M+1]。
アルゴン雰囲気下、中間体3d(1.4g、2.5mmol)を無水DCM 3mLに溶解し、−78℃で撹拌した。DCM中の1M三塩化ホウ素(8.8mL、8.8mmol)を滴下添加した。反応混合物を2時間、撹拌し、追加のDCM中の1M三塩化ホウ素(1.25mL)を加えた。1時間後、反応混合物に1M炭酸水素トリエチルアンモニウム溶液(40mL)を1度に加え、得られた混合物をアセトニトリル(50mL)により希釈した。反応混合物を室温までゆっくりと加温し、減圧下で濃縮すると、粗製固体が得られた。固体を酢酸エチルに懸濁し、得られた混合物を30分間、撹拌した。溶媒をデカンテーションし、固体を真空下で乾燥すると、中間体3e(714mg)が得られ、これをさらに精製することなく次の反応に直接、使用した。
MS m/z=284.1[M+1]。
中間体3e(714mg、2.52mmol)をアセトン50mLに溶解し、室温で撹拌した。2,2−ジメトキシプロパン(619uL、5.04mmol)を加え、続いてメタンスルホン酸(245uL、3.78mmol)を滴下添加した。2時間後、追加の2,2−ジメトキシプロパン(620uL)およびメタンスルホン酸(163uL)を加えた。20時間後、追加の2,2−ジメトキシプロパン(1.2mL)およびメタンスルホン酸(163uL)を加えた。24時間後、追加の2,2−ジメトキシプロパン(1.2mL)およびメタンスルホン酸(163uL)を加えた。24時間後、追加の2,2−ジメトキシプロパン(1.2mL)を加えた。24時間後、反応混合物を酢酸エチル(100mL)により希釈し、飽和炭酸水素ナトリウム水溶液を加えてpH=8にした。有機抽出物をブラインにより洗浄し、無水硫酸ナトリウムで乾燥させて減圧下で濃縮した。得られたゲル状残留物をヘキサン中に懸濁し、2時間、撹拌した。固体を回収し、ヘキサンにより洗浄すると、中間体に3f(720mg、88%)が得られた。
1H NMR (400 MHz, CD3OD) δ 8.52 (s, 1H), 8.30 (s, 1H), 5.17 (d, J = 5.9 Hz, 1H), 4.95 - 4.91 (m, 1H), 4.80 (t, J = 6.1 Hz, 1H), 4.36 (q, J = 2.5 Hz, 1H), 3.88 (dd, J = 2.6, 1.0 Hz, 2H), 1.62 (s, 3H), 1.37 (s, 3H). MS m/z=324.1[M+1]。
中間体3f(720mg、2.23mmol)を無水DMF 10mLに溶解した。イミダゾール(395mg、5.8mmol)および塩化t−ブチルジメチルシリル(436mg、2.89mmol)を加えた。3時間後、追加のイミダゾール(395mg)および塩化t−ブチルジメチルシリル(436mg)を加えた。16時間後、反応混合物を酢酸エチル(75mL)により希釈し、飽和NaHCO3(水性)、次にブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルクロマトグラフィー(ヘキサン中の0〜50%酢酸エチル)により精製すると、中間体3g(1g、99%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.91 (s, 1H), 5.57 - 5.44 (m, 3H), 4.97 (m, 1H), 4.80 (m, 1H), 4.26 (m, 1H), 3.98 - 3.74 (m, 2H), 1.67 (s, 3H), 1.41 (s, 3H), 0.92 (s, 9H), 0.10 (s, 6H). MS m/z=438.1[M+1]。
中間体3g(976mg、2.23mmol)をTHF 12mLに溶解した。次に、TEA(311uL、2.23mmol)およびジ−tert−ブチルジカーボネート(583mg、2.68mmol)、次いでDMAP(136mg、1.12mmol)を加えた。1時間後、追加のジ−tert−ブチルジカーボネート(583mg)およびDMAP(136mg)を加えた。さらに2時間後、反応混合物を酢酸エチル(75mL)により希釈し、5%水性クエン酸、次にブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をMeOHに溶解し、1N NaOH(水性)を加えてpH=12に到達させ、16時間後に、追加の1N NaOH(水性)を加えてpH=13に到達させた。さらに14時間後、反応混合物を酢酸エチル(75mL)により希釈し、ブライン(3×)により洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製中間体3h(1g、83%)をさらに精製することなく、次の反応に直接使用した。
MS m/z=538.0[M+1]、536.3[M−1]。
中間体3h(1g、1.86mmol)をTHF 15mLに溶解した。次に、フッ化テトラブチルアンモニウム三水和物(880mg、2.79mmol)を1度に加えた。2時間後、追加のフッ化テトラブチルアンモニウム三水和物(293mg、0.5当量)を加えた。16時間後、反応混合物を酢酸エチル(75mL)により希釈し、飽和NaHCO3(水性)、次にブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルカラム(ヘキサン中の0〜50%酢酸エチル)により精製すると、中間体3i(648mg、82%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.81 (s, 1H), 8.17 (s, 1H), 8.01 (s, 1H), 5.10 (m, 3H), 4.50 (s, 1H), 3.99 (dt, J = 12.3, 1.7 Hz, 1H), 3.83 (dt, J = 12.3, 1.8 Hz, 1H), 1.68 (s, 3H), 1.65 (s, 9H), 1.40 (s, 3H). MS m/z=424.0[M+1]、422.2[M−1]
窒素雰囲気下、中間体3i(551mg、1.3mmol)を無水DMSO 7mLに溶解した。次に、EDCI(374mg、1.95mmol)を1度に、続いて、ピリジントリフルオロアセテート(126mg、0.65mmol)を加えた。1時間後、追加のEDCI(374mg、1.95mmol)を加えた。1時間後、追加のEDCI(374mg、1.95mmol)を加えた。さらに1時間後、反応混合物を酢酸エチル(75mL)により希釈し、飽和NaHCO3(水性)、次にブラインにより洗浄した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮した。粗製残留物をジオキサン10mLおよび水1mLに溶解した。37%ホルムアルデヒド水溶液(774uL、10.4mmol)、次いで、NaOH(水性)溶液(62mg、水500uL中の1.56mmol)を加えた。1時間後、反応混合物を酢酸エチル(75mL)により希釈し、ブライン(3×)により洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。次に、粗製残留物をMeOH(50mL)に溶解し、氷浴中で撹拌した。次に、水素化ホウ素ナトリウム(98mg、2.6mmol)を1度に加えた。30分後、追加の水素化ホウ素ナトリウム((98mg、2.6mmol)を加えた。さらに30分後、反応混合物を酢酸エチル(75mL)により希釈し、飽和NaHCO3(水性)およびブラインにより洗浄した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルクロマトグラフィー(ヘキサン中の0〜50%酢酸エチル)により精製すると、中間体3j(360mg、61%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.07 (s, 1H), 7.99 (d, J = 3.0 Hz, 1H), 5.21 (m, 2H), 5.15 (m, 1H), 3.93 (d, J = 11.9 Hz, 2H), 3.81 (m, 2H), 1.69 (s, 3H), 1.61 (s, 9H), 1.41 (s, 3H). MS m/z=454.0[M+1]、452.2[M−1]。
窒素雰囲気下、中間体3j(360mg、0.79mmol)を無水DCM 10mLに溶解し、氷浴中で撹拌した。TEA(220uL、1.58mmol)、次いでDMTrCl(401mg、1.19mmol)を加えた。90分後、追加のTEA(110uL)およびDMTrCl(134mg)を加えた。2時間後、反応混合物を酢酸エチル(50mL)により希釈し、飽和NaHCO3(水性)およびブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルクロマトグラフィー(ヘキサン中の0〜30%酢酸エチル)により精製すると、中間体3k(453mg、76%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.77 (d, J = 3.6 Hz, 1H), 7.97 (s, 1H), 7.78 (s, 1H), 7.58 - 7.49 (m, 1H), 7.41 (m, 2H), 7.37 - 7.26 (m, 3H), 7.26 - 7.14 (m, 2H), 6.91 - 6.77 (m, 4H), 5.25 - 5.19 (m, 1H), 5.19 - 5.12 (m, 2H), 4.96 - 4.85 (m, 1H), 4.07 - 3.88 (m, 2H), 3.88 - 3.72 (m, 6H), 3.66 - 3.56 (m, 1H), 3.19 (d, J = 9.4 Hz, 1H), 1.60 (m, 12H), 1.40 (d, J = 3.3 Hz, 3H). MS m/z=778.1[M+Na]、754.2[M−1]。
中間体3k(453mg、0.6mmol)を無水DMF 5mLに溶解した。次に、イミダゾール(123mg、1.8mmol)およびTBSCl(136mg、0.9mmol)を加えた。2時間後、追加のイミダゾール(123mg、1.8mmol)およびTBSCl(136mg、0.9mmol)を加えた。2時間後、反応混合物を酢酸エチル(50mL)により希釈し、飽和NaHCO3(水性)およびブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルクロマトグラフィー(ヘキサン中の0〜20%酢酸エチル)により精製すると、中間体3l(519mg、99%)が得られた。
MS m/z=892.1[M+Na]、868.3[M−1]。
窒素雰囲気下、0℃で中間体3l(519mg、0.60mmol)をDCM 6mLに溶解し、氷浴中で撹拌した。反応物に、PTSA(125mg、0.66mmol)のMeOH 6mL溶液を滴下添加した。1時間後、反応混合物を酢酸エチル(50mL)により希釈し、飽和NaHCO3(水性)およびブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルクロマトグラフィー(ヘキサン中の0〜50%酢酸エチル)により精製すると、中間体3m(254mg、75%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.86 (dd, J = 4.7, 1.6 Hz, 1H), 8.16 - 8.08 (m, 1H), 8.01 (s, 1H), 5.57 (d, J = 4.4 Hz, 1H), 5.07 (dd, J = 6.4, 4.1 Hz, 1H), 4.87 (t, J = 6.2 Hz, 1H), 4.00 - 3.78 (m, 4H), 1.71 (d, J = 5.4 Hz, 3H), 1.61 (d, J = 5.8 Hz, 9H), 1.42 (d, J = 5.4 Hz, 3H), 0.98 - 0.87 (m, 9H), 0.10 (m, 6H). MS m/z=568.0[M+1]、566.2[M−1]。
窒素雰囲気下、中間体3m(100mg、0.176mmol)を無水DMSO 3mLに溶解し、撹拌した。EDCI(51mg、0.26mmol)を1度に、続いて、ピリジントリフルオロアセテート(17mg、0.088mmol)を加えた。45分後、追加のEDCI(51mg、0.26mmol)を加えた。30分後、追加のEDCI(75mg)を加えた。30分後、追加のEDCI(75mg)を加えた。30分後、反応物を酢酸エチル(50mL)により希釈し、飽和NaHCO3(水性)およびブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物を無水ピリジン5mLに溶解した。次に、ヒドロキシルアミン塩酸塩(18mg、0.264mmol)を加えた。90分後、反応混合物を減圧下で濃縮した。残留物を酢酸エチル(50mL)に溶解し、得られた混合物を飽和NaHCO3(水性)およびブラインにより洗浄した。有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。次に、粗製残留物をアセトニトリル5mLに溶解した。次に、CDI(43mg、0.264mmol)を加え、反応物を30分間、撹拌した。次に、追加のCDI(45mg)を加え、反応混合物を30分間、撹拌した。次に、追加のCDI(45mg)を加え、反応混合物を30分間、撹拌した。次に、反応混合物を酢酸エチル(50mL)により希釈し、飽和NaHCO3(水性)ブラインにより洗浄した。次に、有機層を無水Na2SO4により乾燥させ、減圧下で濃縮した。粗製残留物をシリカゲルクロマトグラフィー(ヘキサン中の0〜30%酢酸エチル)により精製すると、中間体3n(88mg、89%)が得られた。
1H NMR (400 MHz, CDCl3) δ 8.81 (dd, J = 4.1, 1.6 Hz, 1H), 8.14 - 7.93 (m, 1H), 5.68 (dt, J = 3.4, 2.1 Hz, 1H), 5.16 (m, 1H), 5.09 - 4.99 (m, 1H), 4.09 - 3.96 (m, 2H), 1.87 - 1.77 (m, 3H), 1.67 - 1.54 (m, 9H), 1.47 - 1.36 (m, 3H), 1.01 - 0.88 (m, 9H), 0.20 - 0.07 (m, 6H). MS m/z=563.0[M+1]、561.2[M−1]。
(2R,3S,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−3,4−ジヒドロキシ−2−(ヒドロキシメチル)テトラヒドロフラン−2−カルボニトリル
中間体3n(88mg、0.156mmol)をTFA/H2O(1:1)溶液5mLに溶解した。20時間後、反応混合物を減圧下で濃縮した。粗製残留物を20mM炭酸水素トリエチルアンモニウム溶液に溶解し、分取HPLC(水中の2〜70%アセトニトリル)により精製すると、実施例3(39mg、81%)が得られた。
1H NMR (400 MHz, CD3OD) δ 8.41 (s, 1H), 8.14 (s, 1H), 5.27 (d, J = 7.2 Hz, 1H), 4.47 (dd, J = 7.2, 5.4 Hz, 1H), 4.37 (d, J = 5.4 Hz, 1H), 4.04 -3.85 (m, 2H). MS m/z=309.1[M+1]、307.1[M−1]。
(2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−アジド−4−クロロ−2−(ヒドロキシメチル)テトラヒドロフラン−3−オール
氷浴中で、塩化メチレン(30mL)およびピリジン(15mL)中の中間体4a(Combi−Blocksから購入、1.4g、0.07mmol)の溶液に、DCM中の1Mトリフルオロメタンスルホン酸無水物(12.2mL)を15分間かけて加えた。氷浴下で混合物を30分間、撹拌し、塩化メチレンにより希釈してNaHCO3水溶液により洗浄し、硫酸ナトリウムで乾燥させてトルエンと共蒸発させた。粗製残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜40%EtOAc)によって精製すると、化合物4b(1.2g、62%)がシロップ状物として得られた。
HMPA(12mL)中の化合物4b(1.9g、4.00mmol)とLiCl(845mg、20.00mmol)との混合物を室温で1時間、撹拌し、EtOAcにより希釈してブラインにより洗浄し(4x)、硫酸ナトリウムで乾燥して真空中で濃縮し、次に残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜30%EtOAc)により精製すると、化合物4cがアノマー混合物として(より速く移動した方の異性体は800mg、より遅く移動した方の異性体は300mg)、シロップ状物として(76%)得られた。より遅く移動した方のアノマーの場合。
1H NMR (400 MHz, CDCl3) δ 7.47 - 7.07 (m, 10H), 5.02 (d, J = 4.3 Hz, 1H), 4.80 (d, J = 12.6 Hz, 1H), 4.57 (d, J = 12.6 Hz, 1H), 4.50 (d, J = 12.1 Hz, 1H), 4.42 (d, J = 12.1 Hz, 1H), 4.24 (q, J = 3.7 Hz, 1H), 4.18 (dd, J = 6.7, 4.3 Hz, 1H), 3.96 (dd, J = 6.7, 3.9 Hz, 1H), 3.51 (s, 4H), 3.36 (dd, J = 10.7, 3.7 Hz, 1H).
化合物4c(3.0g、8.27mmol)をTFA(10mL)−水(10mL)に溶解した。得られた混合物を室温で16時間、撹拌し、50℃で4時間、加熱し、真空中で濃縮した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜50%EtOAc)によって精製すると、ラクトール中間体(2.1g、72%)がシロップ状物として得られた。次に、ラクトール中間体(2.1g、6.02mmol)を塩化メチレン(40mL)に溶解し、4A MS(5g)および二クロム酸ピリジニウム(6.80g、18.06mmol)により処理した。得られた混合物を室温で4時間、撹拌し、次に、セライトのパットにより濾過して減圧下で濃縮した。残留物にシリカゲルクロマトグラフィー(ヘキサン中の0〜30%EtOAc)を供すると、化合物4d(1.7g、81%)がシロップ状物として得られた。
1H NMR (400 MHz, CDCl3) δ 7.42 - 7.23 (m, 10H), 4.76 (d, J = 11.7 Hz, 1H), 4.70 (dd, J = 5.9, 1.1 Hz, 1H), 4.63 - 4.51 (m, 3H), 4.48 (d, J = 11.9 Hz, 1H), 4.32 (dd, J = 5.8, 3.7 Hz, 1H), 3.76 (dd, J = 11.3, 2.4 Hz, 1H), 3.62 (dd, J = 11.3, 2.6 Hz, 1H).
−78℃のTHF(40mL)中の化合物4d(1.7g、4.90mmol)および化合物1b(2.08g、5.88mmol)の溶液に、1M BuLi(2.35mL、5.88mmol)を30分間、滴下添加した。得られた混合物を−78℃で30分間、撹拌した。次に、AcOH(2mL)を加えた。次に、反応混合物をEtOAcにより希釈し、ブラインにより洗浄して真空中で濃縮した。残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜60%EtOAc)によって精製すると、化合物4e(2.82g、35%)が得られた。
MS m/z 575[M+1]。
化合物4e(2.0g、1.74mmol)をDCM(20mL)に懸濁し、氷浴下でTES(5.56mL、34.78mmol)、次にBF3エーテル錯体(1.12mL、8.70mmol)を加えた。得られた混合物を室温で6時間、撹拌し、氷水下、反応混合物を炭酸水素ナトリウム溶液で中和した。次に、混合物をDCMにより希釈し、水により洗浄して硫酸ナトリウムで乾燥して真空中で濃縮した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン中の0〜50%EtOAc)によって精製すると、化合物4f(460mg、22%)が白色泡状物として得られた。
MS m/z 559[M+1]。
化合物4f(460mg、0.53mmol、64%純度)のDCM(5mL)溶液に、氷浴中、メタンスルホン酸(0.9mL、13.16mmol)を滴下添加した。得られた混合物を室温で15時間、撹拌し、TEAで中和して真空中で濃縮し、残留物をシリカゲルカラムクロマトグラフィー(DCM中の0〜5%MeOH)によって精製すると、化合物4g(77mg、39%)が灰色固体として得られた。
1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H), 7.99 (s, 1H), 7.48 (t, J = 7.9 Hz, 2H), 7.39 - 7.30 (m, 1H), 7.25 (dd, J = 8.7, 1.3 Hz, 2H), 5.23 (d, J = 10.0 Hz, 1H), 4.97 (dd, J = 10.0, 4.4 Hz, 1H), 4.52 (d, J = 4.4 Hz, 1H), 4.38 (s, 1H), 3.99 (dd, J = 12.7, 1.9 Hz, 1H), 3.79 (dd, J = 12.7, 1.4 Hz, 1H). MS m/z 379[M+1]。
THF(4mL)中の化合物4g(100mg、0.264mmol)、Ph3P(104mg、0.396mmol)およびイミダゾール(27mg、0.396mmol)の混合物に、室温でヨウ化物(101mg、0.396mmol)を加えた。反応混合物を室温で5時間、撹拌し、NaHCO3水溶液(2mL)を加えた。次に、混合物を真空中で濃縮し、シリカゲルカラムクロマトグラフィー(ヘキサン中の0〜60%EtOAc)によって精製すると、化合物4h(97mg、75%)が白色固体として得られた。
MS m/z 489[M+1]。
化合物4h(90mg、0.184mmol)およびDBU(0.165mL、1.11mmol)を室温で15時間、撹拌した。次に、反応混合物を、EtOAcにより希釈して水により洗浄し、硫酸ナトリウム下で乾燥して真空中で濃縮すると、粗製残留物4iが得られ、これを高真空下で乾燥し、次の反応に直接使用した。
MS m/z 530[M+1]。
1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.04 (d, J = 0.8 Hz, 1H), 7.97 (t, J = 1.8 Hz, 1H), 7.90 (dt, J = 7.9, 1.3 Hz, 1H), 7.54 (ddd, J = 8.0, 2.1, 1.1 Hz, 1H), 7.52 - 7.44 (m, 2H), 7.41 - 7.30 (m, 2H), 7.28 - 7.20 (m, 2H), 5.80 (d, J = 3.4 Hz, 1H), 5.16 (dd, J = 6.7, 3.4 Hz, 1H), 5.00 (dd, J = 11.1, 6.8 Hz, 1H), 4.77 (d, J = 11.9 Hz, 1H), 4.67 (d, J = 11.9 Hz, 1H). MS m/z 558[M+1]。
(2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−アジド−4−クロロ−2−(ヒドロキシメチル)テトラヒドロフラン−3−オール
密封したフラスコ中、化合物4k(7mg、0.013mmol)、濃水性水酸化アンモニウム(0.5mL)およびアセトニトリル(0.5mL)の混合物を48時間、70℃で加熱した。次に、得られた混合物を真空中で濃縮し、メタノール(1mL)に溶解して、HPLC(20分間で、水中のアセトニトリルを0から30%)を用いて精製すると、化合物4(2.7mg、63%)がオフホワイトの固体として得られた。
1H NMR (400 MHz, メタノール-d4) δ 8.38 (s, 1H), 8.21 (s, 1H), 5.49 (d, J = 9.4 Hz, 1H), 4.95 (dd, J = 9.4, 4.8 Hz, 1H), 4.45 (d, J = 4.8 Hz, 1H), 3.73 (d, J = 12.1 Hz, 1H), 3.54 (d, J = 12.1 Hz, 1H). MS m/z=343(M+1)。
((2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−アジド−4−フルオロ−3−ヒドロキシテトラヒドロフラン−2−イル)メチル四水素トリホスフェート
実施例1(15mg、0.046mmol)およびNaHCO3(10mg、0.119mmol)のリン酸トリメチル(0.6mL)溶液に、0℃でPOCl3(50mg、0.326mmol)を加えた。反応混合物を0℃で6時間、撹拌した。イオン交換HPLCにより、約65%変換であることが示された。ピロリン酸トリブチルアミン塩(250mg、0.688mmol)のMeCN(0.6mL)溶液、次いでトリブチルアミン(121mg、0.65mmol)を加えた。反応混合物を0℃で0.5時間、撹拌した。反応を炭酸水素トリエチルアンモニウム緩衝水溶液(1M、6mL)によりクエンチした。反応混合物を室温で0.5時間、撹拌し、次に、濃縮して、水により2回、共蒸発させた。残留物をH2O(5mL)に溶解し、イオン交換カラムに充填して、H2O、次に、10〜35%の炭酸水素トリエチルアンモニウム緩衝液(1M)−H2Oにより溶出させた。生成物フラクションを合わせて濃縮し、H2Oにより共蒸発させると、約20mgの物質が得られた。物質をH2O(1mL)に溶解し、NaOH水溶液(1N、0.12mL)により処理して、約0.5mLまで濃縮し、H2Oにより溶出させたC−18カラムを用いて精製した。生成物フラクションを合わせて濃縮すると、所望のトリホスフェートTP1が四ナトリウム塩(14mg、47%)として得られた。
1H NMR (400 MHz, D2O) δ 8.19 (s, 1H), 8.06 (s, 1H), 5.73 (d, J = 24 Hz, 1H), 5.06 (dd, J = 55.6, 4.4 Hz, 1H), 4.63 (dd, J = 28.0, 4.4 Hz, 1H), 4.19 (br s, 2H). 19F NMR (376 MHz, D2O) δ -195.32〜-195.60 (m). 31P NMR (162 MHz, D2O) δ -8.16 (d, J = 48 Hz, 1P), -14.10 (d, J = 48 Hz, 1P), -23.9 (t, J = 48 Hz, 1P). MS m/z=566.97[M+1]。
((2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−シアノ−4−フルオロ−3−ヒドロキシテトラヒドロフラン−2−イル)メチル四水素トリホスフェート
実施例TP2(6mg、53%)は、出発物質として実施例2を使用し、実施例TP1と同様の方法で四ナトリウム塩として調製した。
1H NMR (400 MHz, D2O) δ 8.26 (s, 1H), 8.07 (s, 1H), 5.77 (d, J = 24.8 Hz, 1H), 5.16 (dd, J = 54, 4 Hz, 1H), 4.75 (dd, J = 26.0, 4.0 Hz, 1H), 4.4 (d, J = 4.8 Hz, 2H). 19F NMR (376 MHz, D2O) δ -193.49〜-193.77 (m). 31P NMR (162 MHz, D2O) δ -8.22 (d, J = 48 Hz, 1P), -14.45 (d, J = 48 Hz, 1P), -24.0 (t, J = 48 Hz, 1P). MS m/z=550.89[M+1]。
((2R,3S,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−シアノ−3,4−ジヒドロキシテトラヒドロフラン−2−イル)メチル四水素トリホスフェート
実施例TP3(3mg、26%)は、出発物質として実施例3を使用して、実施例TP1と同様の方法で四ナトリウム塩として調製した。
1H NMR (400 MHz, D2O) δ 8.22 (s, 1H), 8.08 (s, 1H), 5.4 (d, J = 4.4 Hz, 1H), 4.55 (d, J = 4.4 Hz, 1H), 4.37 (dd, J = 4.4, 4.4 Hz, 1H), 4.18-4.3 (m, 2H). 31P NMR (162 MHz, D2O) δ -4.27 (d, J = 48 Hz, 1P), -10.44 (d, J = 48 Hz, 1P), -20.1 (t, J = 48 Hz, 1P). MS m/z=548.95[M+1]。
((2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−アジド−4−クロロ−3−ヒドロキシテトラヒドロフラン−2−イル)メチル四水素トリホスフェート
1H NMR (400 MHz, D2O) δ 8.28 (s, 1H), 8.24 (s, 1H), 5.76 (d, J = 4.4 Hz, 1H), 4.77 (d, J = 4.4 Hz, 1H), 4.70 (dd, J = 4.8, 4.8 Hz, 1H), 4.20-4.10 (m, 2H). 31P NMR (162 MHz, D2O) δ -4.85 (d, J = 48.4 Hz, 1P), -10.32 (d, J = 48.4 Hz, 1P), -20.44 (t, J = 48.4 Hz, 1P). MS m/z=582.88[M+1]。
(2S)−エチル2−(((((2R,3R,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2−アジド−4−フルオロ−3−ヒドロキシテトラヒドロフラン−2−イル)メトキシ)(フェノキシ)ホスホリル)アミノ)プロパノエート
実施例1(5.00mg、15.3μmol)をNMP(0.2mL)に溶解した。アルゴン雰囲気下、室温で、THF(0.1mL)、次いで、塩化tert−ブチルマグネシウム(THF中の1.0M溶液、0.024mL、23μmol)を加えた。20分後、中間体PD1a(US20120009147A1に従って調製した、12.1mg、30.7μmol)のTHF(0.1mL)溶液を加え、反応混合物を50℃まで加温した。23時間後、追加の中間体PD1a(12.1mg、30.7μmol)および塩化tert−ブチルマグネシウム(THF中の1.0M溶液、0.024mL、23μmol)を加えた。5時間後、得られた混合物を分取HPLC(Phenominex Synergi 4u Hydro−RR 80Å 150x30mmカラム、40〜100%アセトニトリル/水のグラジエント)により直接、精製した。主要ジアステレオマーを単離すると、実施例PD1(1.0mg、20%)が薄黄色固体として得られた。
1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.94 (s, 1H), 7.38 - 7.12 (m, 5H), 5.81 (d, J = 25.1 Hz, 1H), 5.76 (br s, 1H), 5.25 (dd, J = 54.9, 4.8 Hz, 1H), 4.59 (br d, J = 24.4 Hz, 1H), 4.45 (dd, J = 11.2, 7.2 Hz, 1H), 4.34 (dd, J = 11.2, 7.1 Hz, 1H), 4.21 - 4.08 (m, 2H), 4.02 (td, J = 8.9, 6.8 Hz, 1H), 3.83 - 3.72 (m, 1H), 1.36 (d, J = 7.1 Hz, 3H), 1.23 (t, J = 7.1 Hz, 3H). 19F NMR (376 MHz, D2O) δ -193.64 (dt, J = 54.4, 24.4 Hz). 31P NMR (162 MHz, CDCl3) δ 2.57 (s). MS m/z=581.90[M+1]。
a) (2R,3R,4R,5S)−5−(4−アミノ−2−フルオロチエノ[3,2−d]ピリミジン−7−イル)−2−アジド−4−フルオロ−2−(ヒドロキシメチル)テトラヒドロフラン−3−オール:
c) (2R,3S,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−2,4−ジアジド−2−(ヒドロキシメチル)テトラヒドロフラン−3−オール:
j) (2R,3S,4R,5S)−5−(4−アミノチエノ[3,2−d]ピリミジン−7−イル)−4−アジド−2−(クロロメチル)−2−(ヒドロキシメチル)テトラヒドロフラン−3−オール:
本発明の別の態様は、ウイルス感染を阻害する方法であって、こうした阻害が必要と疑われる試料または被験体を本発明の組成物により処置するステップを含む方法に関する。
抗RSV活性
RSVに対する抗ウイルス活性は、HEp−2細胞において、感染性の細胞変性細胞保護アッセイを使用して決定される。このアッセイでは、ウイルス感染および/または複製を阻害する化合物は、細胞生存試薬を使用して定量することができるウイルス誘発性の細胞死滅に対する、細胞保護効果をもたらす。ここで使用される技法は、公開されている文献(Chapmanら、Antimicrob Agents Chemother. 2007年、51巻(9号):3346〜53頁)において記載されている方法を新規に適応したものである。
試験化合物の細胞毒性は、以前に他の細胞タイプに関して記載されている手法と同様の手法で細胞生存試薬を使用して、抗ウイルス活性と並行して、感染していないHEp−2細胞において決定する(Cihlarら、Antimicrob Agents Chemother. 2008年、52巻(2号):655〜65頁)。細胞がRSVに感染していない点を除いて、抗ウイルス活性の決定の場合と同じプロトコールを、化合物の細胞毒性の測定に使用する。代わりに、同じ密度の感染していない細胞混合物を、やはり100nl/試料に予め希釈した化合物を含有しているプレートに、20ul/ウェルで加える。次に、アッセイプレートを4日間、インキュベートし、続いて、同じCellTiter Glo試薬の添加を使用する細胞生存試験、および発光の読取り測定を行う。未処理細胞、および2uMのピューロマイシン(Sigma、St.Louis、MO)で処理された細胞は、それぞれ100%および0%細胞生存率の対照として働く。細胞生存率の百分率を0%および100%対照に対する、各試験化合物濃度について算出し、CC50値を細胞生存率が50%低下する化合物濃度として、非線形回帰により決定する。
MT−4細胞系は、NIH AIDS Research and Reference Reagent Program(Germantown、MD)から入手し、10%FBS、ペニシリン100単位/mL、ストレプトマイシン100単位/mLおよび2mM L−グルタミンを補充した、RPMI−1640培地(Irvine Scientific、Santa Ana、CA、カタログ番号9160)において培養した。MT−4細胞を1週あたり2回、継代し、0.6×106細胞/mL未満の細胞密度を維持した。26nM〜530μMの範囲の3倍連続希釈化合物の100×濃度を含有する完全RPMI−1640培地を、黒色384ウェルプレートに四連でスタンプした(stamped in)。化合物を添加した後、2×103個のMT−4細胞を、MicroFlo液体分注器(BioTek、Winooski、VT)を使用して各ウェルに加え、細胞を5%CO2のインキュベータ中、37℃で5日間、培養した。インキュベート後、細胞を25℃で平衡化し、細胞生存率をCell−Titer Glo生存試薬25μLを加えることにより決定した。混合物を25℃で10分間、インキュベートし、発光シグナルをVictor Luminescenceプレートリーダーで定量した。CC50値は、Cell−Titer Gloのシグナルにより決定される場合、細胞生存率を50%低下させる化合物の濃度として定義する。データは、Pipeline Pilot Plate Data Analytics Collectionソフトウェア(Version 7.0、Accelrys、San Diego、CA)を使用して解析した。CC50値は、4−パラメータS字用量−応答式:Y=Bottom+(Top−Bottom)/(1+10^[(LogCC50−X)*HillSlope])(式中、TopおよびBottomは、それぞれ100%および0%の細胞生存率に固定した)を使用して、非線形回帰分析から算出した。CC50値は、三つの独立した実験の平均±標準偏差として算出した。
RSVリボ核タンパク質(RNP)複合体は、Masonら(1)のものを改変した方法から調製した。HEp−2細胞は、MEM+10%ウシ胎児血清(FBS)中、7.1×104個の細胞/cm2の密度でプレート培養し、37℃(5%CO2)で一晩、付着させた。付着後、細胞を35mL MEM+2%FBSにおいて、RSV A2(MOI=5)に感染させた。感染から20時間後に、培地をアクチノマイシンD2μg/mLを補充したMEM+2%FBSに置き換え、1時間、37℃に戻した。次に、細胞をPBSにより1回洗浄し、35mLのPBS+リソ−レシチン250μg/mLにより1分間処理し、この後、すべての液体を吸引した。細胞を1.2mLの緩衝液A[50mM TRIS酢酸塩(pH8.0)、100mM酢酸カリウム、1mM DTTおよびアクチノマイシンD2μg/mL]にスクラップすることにより収集し、18ゲージのニードルにより継代を繰り返すことにより溶解させた(10回)。細胞溶解物を10分間、氷中に置き、次に、2400gで、4℃で10分間遠心分離機にかけた。上澄み液(S1)を除去し、ペレット(P1)を1%Triton X−100を補充した緩衝液B[10mM TRIS酢酸塩(pH8.0)、10mM酢酸カリウムおよび1.5mM MgCl2]600uL中で、18ゲージのニードルにより継代を繰り返す(10回)ことによって粉砕した。再懸濁させたペレットを10分間、氷中に置き、次に、2400gで、4℃で10分間、遠心分離機にかけた。上澄み液(S2)を除去し、ペレット(P2)を、0.5%デオキシコレートおよび0.1%Tween40を補充した緩衝液B600μL中で粉砕した。再懸濁させたペレットを10分間、氷中に置き、次に、2400gで、4℃で10分間、遠心分離機にかけた。RSV RNP複合体を豊富に含有している上澄み液(S3)フラクションを収集し、タンパク質濃度を280nmにおけるUV吸光度によって決定した。一定分量のRSV RNP S3フラクションを−80℃で保管した。
転写反応は、30μLの反応緩衝液[50mM TRIS酢酸塩(pH8.0)、120mM酢酸カリウム、5%グリセロール、4.5mM MgCl2、3mM DTT、2mM エチレングリコール−ビス(2−アミノエチルエーテル)−四酢酸(EGTA)、50μg/mL BSA、2.5U RNasin(Promega)、ATP、GTP、UTP、CTPおよび1.5uCi[α−32P]NTP(3000Ci/mmol)]中で、粗製RSV RNP複合体25μgを含有していた。転写アッセイにおいて使用される放射標識ヌクレオチドは、RSV RNP転写の阻害を評価されるヌクレオチドアナログに適合するよう選択した。冷却した競合NTPを、そのKmの半分の最終濃度で加えた(ATP=20μM、GTP=12.5μM、UTP=6μMおよびCTP=2μM)。残りの3種のヌクレオチドは、最終濃度100μMで加えた。
Claims (28)
- 式(I):
Xは、O、S、NHまたはN(C1〜C6アルキル)の群から選択され、
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、ORa、NHRa、CNおよびN3の群から選択され、
R3は、CN、ORa、C1〜C6アルキル、C2〜C6アルケニル、C2〜C6アルキニル、−CH2−O−C1〜C6アルキル、−CH2−S−C1〜C6アルキル、C3〜C4シクロアルキル、アジド、ハロゲン、C1〜C3ハロアルキル、SRa、−CH2−C3〜C4シクロアルキル、−O−C3〜C4シクロアルキル、および−O−C1〜C3ハロアルキルの群から選択されるか、
または
R2がORaである場合、2’位および3’位における2つのORa基は、それらが結合しているフラニル環と一緒になって、
R4は、H、−C(=O)R6、−C(=O)OR6および−C(=O)NR6R7の群から選択されるか、
または
b)R4は、式:
Yはそれぞれ、O、S、NR、+N(O)(R)、N(OR)、+N(O)(OR)もしくはN−NR2であり、
W1およびW2は、一緒になった場合、−Y3(C(Ry)2)3Y3−であるか、
もしくは、W1もしくはW2の一方は、3’ヒドロキシ基と一緒になって、−Y3−であり、W1もしくはW2のもう一方は、式Ia
であるか、
もしくは、W1およびW2はそれぞれ、独立して、式Ia:
ここで、
Y1はそれぞれ、独立して、O、S、NR、+N(O)(R)、N(OR)、+N(O)(OR)もしくはN−NR2であり、
Y2はそれぞれ、独立して、結合、O、CR2、−O−CR2−、NR、+N(O)(R)、N(OR)、+N(O)(OR)、N−NR2、S、S−S、S(O)もしくはS(O)2であり、
Y3はそれぞれ、独立して、O、SもしくはNRであり、
M1は、0、1、2もしくは3であり、
Rxはそれぞれ、独立して、Ryもしくは式:
ここで、
M2a、M2bおよびM2cはそれぞれ、独立して、0もしくは1であり、
M2dは、0、1、2、3、4、5、6、7、8、9、10、11もしくは12であり、
Ryはそれぞれ、独立して、H、F、Cl、Br、I、OH、R、−C(=Y1)R、−C(=Y1)OR、−C(=Y1)N(R)2、−N(R)2、−+N(R)3、−SR、−S(O)R、−S(O)2R、−S(O)(OR)、−S(O)2(OR)、−OC(=Y1)R、−OC(=Y1)OR、−OC(=Y1)(N(R)2)、−SC(=Y1)R、−SC(=Y1)OR、−SC(=Y1)(N(R)2)、−N(R)C(=Y1)R、−N(R)C(=Y1)OR、−N(R)C(=Y1)N(R)2、−SO2NR2、−CN、−N3、−NO2、−OR、またはW3であるか、
もしくは、同一炭素原子上の2つのRyは、一緒になった場合、3個、4個、5個、6個もしくは7個の炭素環原子を有する炭素環式環を形成するか、
もしくは、同一炭素原子上の2つのRyは、一緒になった場合、前記炭素原子と一緒になって、3個、4個、5個、6個もしくは7個の環原子を有する複素環を形成し、ここで、1個の環原子はOもしくはNから選択され、他の環原子はすべて炭素であり、
Rはそれぞれ、独立して、H、(C1〜C8)アルキル、(C1〜C8)置換アルキル、(C2〜C8)アルケニル、(C2〜C8)置換アルケニル、(C2〜C8)アルキニル、(C2〜C8)置換アルキニル、C6〜C10アリール、C6〜C10置換アリール、3〜10員の複素環、3〜10員の置換複素環、5〜12員のヘテロアリール、5〜12員の置換ヘテロアリール、アリールアルキル、置換アリールアルキル、ヘテロアリールアルキルもしくは置換ヘテロアリールアルキルであり、
W3は、W4もしくはW5であり、
W4は、R、−C(Y1)Ry、−C(Y1)W5、−SO2Ryもしくは−SO2W5であり、
W5は、フェニル、ナフチル、C3〜C8炭素環もしくは3〜10員の複素環から選択され、W5は、0、1つ、2つ、3つ、4つ、5つもしくは6つのRy基により独立して置換されており、
R6およびR7はそれぞれ、独立して、H、(C1〜C8)アルキル、(C2〜C8)アルケニル、(C2〜C8)アルキニル、(C4〜C8)カルボシクリルアルキル、C6〜C10アリール、C6〜C10置換アリール、5〜10員のヘテロアリール、5〜10員の置換ヘテロアリール、−C(=O)(C1〜C8)アルキル、−S(O)n(C1〜C8)アルキルもしくはアリール(C1〜C8)アルキルであるか、
もしくは、R6およびR7は、それらの両方が結合している窒素と一緒になって、3〜7員の複素環を形成し、ここで、前記複素環の任意の1個の環炭素原子が、−O−、−S−もしくは−NRa−で任意選択で置き換えられ得、
各R6もしくはR7の(C1〜C8)アルキル、(C2〜C8)アルケニル、(C2〜C8)アルキニルもしくはアリール(C1〜C8)アルキルはそれぞれ、ハロ、ヒドロキシ、CN、N3、N(Ra)2もしくはORaから選択される1つ、2つ、3つもしくは4つの置換基で任意選択で独立して置換されており、ここで、それぞれの前記(C1〜C8)アルキルの非末端炭素原子の1個、2個もしくは3個が、−O−、−S−もしくは−NRa−で任意選択で置き換えられていてもよいか、または
b)R4は、
R8は、フェニル、1−ナフチル、2−ナフチル、
R9は、HおよびCH3から選択され、
R10は、HもしくはC1〜C6アルキルから選択され、
R11は、H、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択されるか、または
d)R4および3’ヒドロキシ基が一緒になって、
の化合物または薬学的に許容されるその塩。 - XがSである、請求項1に記載の化合物、または薬学的に許容されるその塩。
- R3が、CN、ORa、C1〜C4アルキル、C2〜C4アルケニル、C2〜C4アルキニル、−CH2−O−C1〜C4アルキル、−CH2−S−C1〜C4アルキル、C3〜C4シクロアルキル、アジド、ハロゲン、C1〜C3クロロアルキル、C1〜C3ブロモアルキルおよびC1〜C3フルオロアルキルの群から選択される、請求項1および2のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R1が、H、CH3、F、ClおよびNH2の群から選択され、R2が、OH、F、Cl、N3、NH2およびCNの群から選択され、R3が、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeから選択される、請求項1、2および3のいずれかに記載の化合物、または薬学的に許容されるその塩。
- 式(II):
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
Ra、R4、R5、R6、R7、R8、R9、R10、R11、Y、Y1、Y2、Y3、W1、W2、W3、W4、W5、M1、M2a、M2b、M2c、M2d、RxおよびRyは、請求項1において定義されている通りである]
の、請求項1、2、3および4のいずれかに記載の化合物、または薬学的に許容されるその塩。 - 式(III):
R2は、F、Cl、OH、NH2、CNおよびN3であり、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択され、
R4は、H、もしくは式:
ここで、W1およびW2はそれぞれ、独立して、OHもしくは式Ia:
Yはそれぞれ、独立して結合もしくはOであり、
mは、0、1、2もしくは3であり、
Rxはそれぞれ、H、ハロゲンもしくはOHであるか、または
R4は、Hおよび
ここで、
n’は、1、2、3および4から選択され、
R7は、C1〜C8アルキル、−O−C1〜C8アルキル、ベンジル、−O−ベンジル、−CH2−C3〜C6シクロアルキル、−O−CH2−C3〜C6シクロアルキルおよびCF3から選択され、
R8は、フェニル、1−ナフチル、2−ナフチル、
R9は、HおよびCH3から選択され、
R10は、HもしくはC1〜C6アルキルから選択され、
R11は、H、C1〜C8アルキル、ベンジル、C3〜C6シクロアルキルおよび−CH2−C3〜C6シクロアルキルから選択される]
の、請求項1、2、3および4のいずれかに記載の化合物、または薬学的に許容されるその塩。 - 式(VI):
Xは、O、SおよびNHの群から選択され、
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択される]
の、請求項1、2、3および4のいずれかに記載の化合物、または薬学的に許容されるその塩。 - XがSである、請求項1、2、3、4、5、6および7のいずれかに記載の化合物、または薬学的に許容されるその塩。
- 式(IX):
R1は、H、CH3、F、ClおよびNH2の群から選択され、
R2は、F、Cl、OH、NH2、CNおよびN3の群から選択され、
R3は、CN、N3、メチル、エチル、プロピル、ビニル、プロペニル、エチニル、CH2F、CHF2、CH2Cl、CH2SMeおよびCH2OMeの群から選択される]
の、請求項1、2、3および4のいずれかに記載の化合物、または薬学的に許容されるその塩。 - R1がHである、請求項1、2、3、4、5、6、7および8のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R1がFである、請求項1、2、3、4、5、6、7および8のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R2がFである、請求項1、2、3、4、5、6、7、8、9、10および11のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R2がClである、請求項1、2、3、4、5、6、7、8、9、10および11のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R2がN3である、請求項1、2、3、4、5、6、7、8、9、10および11のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R3が、CNおよびN3の群から選択される、請求項1、2、3、4、5、6、7、8、9、10、11、12、13および14のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R3が、メチル、エチルおよびプロピルの群から選択される、請求項1、2、3、4、5、6、7、8、9、10、11、12、13および14のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R3が、CH2F、CHF2およびCH2Clの群から選択される、請求項1、2、3、4、5、6、7、8、9、10、11、12、13および14のいずれかに記載の化合物、または薬学的に許容されるその塩。
- R4が、
- R4が、
- R4が、
- R4が、
-
- ヒトにおけるPneumovirinaeウイルス感染を処置する方法であって、治療有効量の、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21および22のいずれかに記載の化合物、または薬学的に許容されるその塩を、前記処置を必要とする前記ヒトに投与するステップを含む方法。
- 前記Pneumovirinaeウイルス感染が、呼吸器合胞体ウイルス感染である、請求項23に記載の方法。
- 請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21および22のいずれかに記載の化合物または薬学的に許容されるその塩、ならびに薬学的に許容される担体または添加剤を含む医薬組成物。
- 請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21および22のいずれかに記載の化合物、または薬学的に許容されるその塩が使用されることを特徴とする、ヒトにおけるPneumovirinaeウイルス感染または呼吸器合胞体ウイルス感染を処置することが意図されている医薬を製造する方法。
- ヒトにおけるPneumovirinaeウイルス感染または呼吸器合胞体ウイルス感染の処置に使用するための、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21および22のいずれかに記載の化合物、または薬学的に許容されるその塩。
- ヒトにおけるPneumovirinaeウイルス感染または呼吸器合胞体ウイルス感染を処置するのに有用な医薬の調製における、請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21および22のいずれかに記載の化合物、または薬学的に許容されるその塩の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462029896P | 2014-07-28 | 2014-07-28 | |
| US62/029,896 | 2014-07-28 | ||
| PCT/US2015/041574 WO2016018697A1 (en) | 2014-07-28 | 2015-07-22 | Thieno [3,2-d] pyrimidine, furo [3,2,d] pyrimidine, and pyrrolo [3,2-d] pyrimidines useful for treating respiratory syncitial virus infections |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019212344A Division JP6991188B2 (ja) | 2014-07-28 | 2019-11-25 | 呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2-d]ピリミジン、フロ[3,2-d]ピリミジン、およびピロロ[3,2-d]ピリミジン |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017523169A true JP2017523169A (ja) | 2017-08-17 |
| JP2017523169A5 JP2017523169A5 (ja) | 2018-08-30 |
Family
ID=53765598
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017502192A Withdrawn JP2017523169A (ja) | 2014-07-28 | 2015-07-22 | 呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2−d]ピリミジン、フロ[3,2−d]ピリミジン、およびピロロ[3,2−d]ピリミジン |
| JP2019212344A Active JP6991188B2 (ja) | 2014-07-28 | 2019-11-25 | 呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2-d]ピリミジン、フロ[3,2-d]ピリミジン、およびピロロ[3,2-d]ピリミジン |
| JP2021181870A Pending JP2022010149A (ja) | 2014-07-28 | 2021-11-08 | 呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2-d]ピリミジン、フロ[3,2-d]ピリミジン、およびピロロ[3,2-d]ピリミジン |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019212344A Active JP6991188B2 (ja) | 2014-07-28 | 2019-11-25 | 呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2-d]ピリミジン、フロ[3,2-d]ピリミジン、およびピロロ[3,2-d]ピリミジン |
| JP2021181870A Pending JP2022010149A (ja) | 2014-07-28 | 2021-11-08 | 呼吸器合胞体ウイルス感染の処置のために有用な、チエノ[3,2-d]ピリミジン、フロ[3,2-d]ピリミジン、およびピロロ[3,2-d]ピリミジン |
Country Status (20)
| Country | Link |
|---|---|
| US (5) | US9828388B2 (ja) |
| EP (3) | EP4023650A1 (ja) |
| JP (3) | JP2017523169A (ja) |
| KR (1) | KR102460181B1 (ja) |
| CN (1) | CN106573939B (ja) |
| AR (1) | AR101280A1 (ja) |
| AU (1) | AU2015298207C1 (ja) |
| BR (1) | BR112017001565A2 (ja) |
| CA (1) | CA2956571C (ja) |
| EA (1) | EA032490B1 (ja) |
| ES (2) | ES2802175T3 (ja) |
| IL (1) | IL249706A0 (ja) |
| MX (1) | MX2017001284A (ja) |
| NZ (1) | NZ727996A (ja) |
| PL (2) | PL3693367T3 (ja) |
| PT (2) | PT3693367T (ja) |
| SG (1) | SG11201700213SA (ja) |
| SI (2) | SI3174870T1 (ja) |
| TW (1) | TWI678369B (ja) |
| WO (1) | WO2016018697A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020536069A (ja) * | 2017-09-29 | 2020-12-10 | エナンタ ファーマシューティカルズ インコーポレイテッド | Rsv阻害剤としての組み合わせ医薬剤 |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR122020020745B8 (pt) | 2010-07-22 | 2023-10-31 | Gilead Sciences Inc | Composto antiviral para o tratamento de infecções por paramyxoviridae e composição farmacêutica que o compreende |
| TWI678369B (zh) * | 2014-07-28 | 2019-12-01 | 美商基利科學股份有限公司 | 用於治療呼吸道合胞病毒感染之噻吩並[3,2-d]嘧啶、呋喃並[3,2-d]嘧啶及吡咯並[3,2-d]嘧啶化合物類 |
| DK3236972T3 (en) | 2014-12-26 | 2021-10-04 | Univ Emory | Antivirale N4-hydroxycytidin-derivativer |
| MY197231A (en) | 2015-07-22 | 2023-06-06 | Enanta Pharm Inc | Benzodiazepine derivatives as rsv inhibitors |
| US20190077797A1 (en) * | 2015-10-30 | 2019-03-14 | Ozkan SARI | Processes for preparing 2-dihalo ribolactones |
| CN110573518A (zh) * | 2017-01-26 | 2019-12-13 | 尤拉·S·赞特里佐斯 | 被取代的双环嘧啶基化合物及其组合物和用途 |
| JP6804790B1 (ja) | 2017-12-07 | 2020-12-23 | エモリー ユニバーシティー | N4−ヒドロキシシチジンおよび誘導体ならびにそれらに関連する抗ウイルス用途 |
| KR102926658B1 (ko) | 2019-03-18 | 2026-02-11 | 이난타 파마슈티칼스, 인코포레이티드 | Rsv 억제제로서의 벤조디아제핀 유도체 |
| US11505558B1 (en) | 2019-10-04 | 2022-11-22 | Enanta Pharmaceuticals, Inc. | Antiviral heterocyclic compounds |
| TWI794742B (zh) | 2020-02-18 | 2023-03-01 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| TWI775313B (zh) | 2020-02-18 | 2022-08-21 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| AU2021224588B2 (en) | 2020-02-18 | 2024-07-18 | Gilead Sciences, Inc. | Antiviral compounds |
| CN111548384B (zh) * | 2020-03-29 | 2021-04-27 | 常州安蒂卫生物科技有限公司 | 用于抗病毒治疗的被取代的n4-羟基胞苷衍生物及其前药 |
| US11945824B2 (en) | 2020-10-19 | 2024-04-02 | Enanta Pharmaceuticals, Inc. | Heterocyclic compounds as anti-viral agents |
| EP4323362B1 (en) | 2021-04-16 | 2025-05-07 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
| AU2022328698B2 (en) | 2021-08-18 | 2025-02-20 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| IL315102A (en) | 2022-03-02 | 2024-10-01 | Gilead Sciences Inc | COMPOSITIONS AND METHODS FOR THE TREATMENT OF VIRAL INFECTIONS |
| AR129003A1 (es) | 2022-04-07 | 2024-07-03 | Enanta Pharm Inc | Compuestos heterocíclicos antivirales |
| US12162857B2 (en) | 2022-04-27 | 2024-12-10 | Enanta Pharmaceuticals, Inc. | Antiviral compounds |
| US20250109157A1 (en) * | 2023-09-28 | 2025-04-03 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010533659A (ja) * | 2007-07-16 | 2010-10-28 | 鄭州大学 | 2’−フルオロ−4’−置換−ヌクレオシド類似体、その製造方法及び使用 |
| JP2011513195A (ja) * | 2007-05-10 | 2011-04-28 | バイオクライスト ファーマシューティカルズ, インコーポレイテッド | ウイルス感染およびがんの処置に使用するためのテトラヒドロフロ[3,4−d]ジオキソラン化合物 |
| JP2011521903A (ja) * | 2008-04-23 | 2011-07-28 | ギリアード サイエンシーズ, インコーポレイテッド | 抗ウイルス処置のための1’−置換カルバ−ヌクレオシドアナログ |
| WO2012040124A1 (en) * | 2010-09-22 | 2012-03-29 | Alios Biopharma, Inc. | Azido nucleosides and nucleotide analogs |
| WO2013096679A1 (en) * | 2011-12-22 | 2013-06-27 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| EP2615101A1 (en) * | 2010-09-07 | 2013-07-17 | High & New Technology Research Center, Henan Academy of Sciences | Nucleoside derivatives, synthesis methods and uses thereof for preparing anti-tumor and anti-virus medicaments |
| JP2013535453A (ja) * | 2010-07-22 | 2013-09-12 | ギリード・サイエンシズ・インコーポレーテッド | パラミクソウイルス科ウイルス感染症を治療するための方法及び化合物 |
| WO2013142525A1 (en) * | 2012-03-21 | 2013-09-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| JP2013538230A (ja) * | 2010-09-20 | 2013-10-10 | ギリアード サイエンシーズ, インコーポレイテッド | 抗ウイルス治療用の2’−フルオロ置換カルバヌクレオシド類似体 |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5458A (en) | 1848-02-22 | Bench-vise | ||
| US135A (en) | 1837-03-03 | Jesse j | ||
| US3361306A (en) | 1966-03-31 | 1968-01-02 | Merck & Co Inc | Aerosol unit dispensing uniform amounts of a medically active ingredient |
| US3565070A (en) | 1969-02-28 | 1971-02-23 | Riker Laboratories Inc | Inhalation actuable aerosol dispenser |
| FR2224175B1 (ja) | 1973-04-04 | 1978-04-14 | Isf Spa | |
| US4069819A (en) | 1973-04-13 | 1978-01-24 | Societa Farmaceutici S.P.A. | Inhalation device |
| IT1017153B (it) | 1974-07-15 | 1977-07-20 | Isf Spa | Apparecchio per inalazioni |
| SE438261B (sv) | 1981-07-08 | 1985-04-15 | Draco Ab | Anvendning i dosinhalator av ett perforerat membran |
| US4584369A (en) * | 1981-07-31 | 1986-04-22 | Sloan-Kettering Institute For Cancer Research | Anti-leukemic beta-glycosyl C-nucleosides |
| US4805811A (en) | 1985-03-29 | 1989-02-21 | Aktiebolaget Draco | Dosage device |
| SE448277B (sv) | 1985-04-12 | 1987-02-09 | Draco Ab | Indikeringsanordning vid en doseringsanordning for lekemedel |
| IT1228459B (it) | 1989-02-23 | 1991-06-19 | Phidea S R L | Inalatore con svuotamento regolare e completo della capsula. |
| US4955371A (en) | 1989-05-08 | 1990-09-11 | Transtech Scientific, Inc. | Disposable inhalation activated, aerosol device for pulmonary medicine |
| WO1993000951A1 (en) | 1991-07-02 | 1993-01-21 | Inhale, Inc. | Method and device for delivering aerosolized medicaments |
| US5261538A (en) | 1992-04-21 | 1993-11-16 | Glaxo Inc. | Aerosol testing method |
| US5785049A (en) | 1994-09-21 | 1998-07-28 | Inhale Therapeutic Systems | Method and apparatus for dispersion of dry powder medicaments |
| US5388572A (en) | 1993-10-26 | 1995-02-14 | Tenax Corporation (A Connecticut Corp.) | Dry powder medicament inhalator having an inhalation-activated piston to aerosolize dose and deliver same |
| US5522385A (en) | 1994-09-27 | 1996-06-04 | Aradigm Corporation | Dynamic particle size control for aerosolized drug delivery |
| US5544647A (en) | 1994-11-29 | 1996-08-13 | Iep Group, Inc. | Metered dose inhalator |
| US5622163A (en) | 1994-11-29 | 1997-04-22 | Iep Group, Inc. | Counter for fluid dispensers |
| US6116234A (en) | 1999-02-01 | 2000-09-12 | Iep Pharmaceutical Devices Inc. | Metered dose inhaler agitator |
| CN1968605A (zh) * | 2004-06-15 | 2007-05-23 | 默克公司 | 作为rna依赖性rna病毒聚合酶抑制剂的c-嘌呤核苷类似物 |
| JP5089395B2 (ja) | 2004-10-29 | 2012-12-05 | バイオクライスト ファーマシューティカルズ, インコーポレイテッド | 治療用フロピリミジンおよびチエノピリミジン |
| ES2571028T3 (es) | 2006-12-07 | 2016-05-23 | Genentech Inc | Compuestos inhibidores de fosfoinositida 3-cinasa y métodos de uso |
| SI2114980T1 (sl) | 2007-01-12 | 2012-11-30 | Biocryst Pharm Inc | Protivirusni nukleozidni analogi |
| EP2313102A2 (en) | 2008-07-03 | 2011-04-27 | Biota Scientific Management | Bycyclic nucleosides and nucleotides as therapeutic agents |
| TWI483950B (zh) | 2009-09-21 | 2015-05-11 | Gilead Sciences Inc | 用於製備1’-取代碳核苷類似物之方法及中間物 |
| PE20120995A1 (es) | 2009-09-21 | 2012-08-01 | Gilead Sciences Inc | Analogos de carba-nucleosidos sustituidos con 2'-fluoro para tratamiento antiviral |
| US8455451B2 (en) | 2009-09-21 | 2013-06-04 | Gilead Sciences, Inc. | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment |
| TW201201815A (en) | 2010-05-28 | 2012-01-16 | Gilead Sciences Inc | 1'-substituted-carba-nucleoside prodrugs for antiviral treatment |
| KR101995598B1 (ko) | 2010-07-19 | 2019-07-02 | 길리애드 사이언시즈, 인코포레이티드 | 부분입체 이성질성으로 순수한 포스포라미데이트 전구약물의 제조 방법 |
| US20120077814A1 (en) | 2010-09-10 | 2012-03-29 | Zhong Wang | Sulfonamide, sulfamate, and sulfamothioate derivatives |
| TW201305185A (zh) | 2010-09-13 | 2013-02-01 | 吉李德科學股份有限公司 | 用於抗病毒治療之2’-氟取代之碳-核苷類似物 |
| PL2898885T3 (pl) | 2010-10-15 | 2018-04-30 | Biocryst Pharmaceuticals, Inc. | Pochodne pirolopirymidynowe do zastosowania w leczeniu zakażeń wirusowych |
| AU2011349844B2 (en) | 2010-12-20 | 2017-06-01 | Gilead Sciences, Inc. | Combinations for treating HCV |
| WO2012142075A1 (en) | 2011-04-13 | 2012-10-18 | Merck Sharp & Dohme Corp. | 2'-azido substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| CN103748098B (zh) * | 2011-10-11 | 2016-06-29 | 弗·哈夫曼-拉罗切有限公司 | 用于治疗或预防呼吸道合胞病毒疾病的化合物 |
| CN106749272B (zh) | 2012-03-13 | 2019-08-30 | 吉利德科学公司 | 用于抗病毒治疗的2’-取代的卡巴-核苷类似物 |
| UA119050C2 (uk) * | 2013-11-11 | 2019-04-25 | Ґілеад Саєнсиз, Інк. | ПІРОЛО[1.2-f][1.2.4]ТРИАЗИНИ, ЯКІ ВИКОРИСТОВУЮТЬСЯ ДЛЯ ЛІКУВАННЯ РЕСПІРАТОРНО-СИНЦИТІАЛЬНИХ ВІРУСНИХ ІНФЕКЦІЙ |
| TWI678369B (zh) * | 2014-07-28 | 2019-12-01 | 美商基利科學股份有限公司 | 用於治療呼吸道合胞病毒感染之噻吩並[3,2-d]嘧啶、呋喃並[3,2-d]嘧啶及吡咯並[3,2-d]嘧啶化合物類 |
-
2015
- 2015-07-08 TW TW104122181A patent/TWI678369B/zh active
- 2015-07-22 EP EP22154464.6A patent/EP4023650A1/en active Pending
- 2015-07-22 SI SI201531250T patent/SI3174870T1/sl unknown
- 2015-07-22 WO PCT/US2015/041574 patent/WO2016018697A1/en not_active Ceased
- 2015-07-22 BR BR112017001565A patent/BR112017001565A2/pt not_active IP Right Cessation
- 2015-07-22 KR KR1020177005142A patent/KR102460181B1/ko active Active
- 2015-07-22 NZ NZ727996A patent/NZ727996A/en unknown
- 2015-07-22 PT PT201664380T patent/PT3693367T/pt unknown
- 2015-07-22 EP EP15745083.4A patent/EP3174870B1/en active Active
- 2015-07-22 MX MX2017001284A patent/MX2017001284A/es unknown
- 2015-07-22 US US14/806,227 patent/US9828388B2/en active Active
- 2015-07-22 AU AU2015298207A patent/AU2015298207C1/en active Active
- 2015-07-22 JP JP2017502192A patent/JP2017523169A/ja not_active Withdrawn
- 2015-07-22 PL PL20166438T patent/PL3693367T3/pl unknown
- 2015-07-22 CA CA2956571A patent/CA2956571C/en active Active
- 2015-07-22 AR ARP150102330A patent/AR101280A1/es unknown
- 2015-07-22 ES ES15745083T patent/ES2802175T3/es active Active
- 2015-07-22 SI SI201531826T patent/SI3693367T1/sl unknown
- 2015-07-22 EA EA201790146A patent/EA032490B1/ru not_active IP Right Cessation
- 2015-07-22 CN CN201580041342.0A patent/CN106573939B/zh active Active
- 2015-07-22 EP EP20166438.0A patent/EP3693367B1/en active Active
- 2015-07-22 PT PT157450834T patent/PT3174870T/pt unknown
- 2015-07-22 ES ES20166438T patent/ES2910382T3/es active Active
- 2015-07-22 SG SG11201700213SA patent/SG11201700213SA/en unknown
- 2015-07-22 PL PL15745083T patent/PL3174870T3/pl unknown
-
2016
- 2016-12-21 IL IL249706A patent/IL249706A0/en unknown
-
2017
- 2017-09-27 US US15/717,493 patent/US10179794B2/en active Active
-
2018
- 2018-11-28 US US16/203,112 patent/US10501476B2/en active Active
-
2019
- 2019-11-25 JP JP2019212344A patent/JP6991188B2/ja active Active
-
2021
- 2021-11-08 JP JP2021181870A patent/JP2022010149A/ja active Pending
-
2024
- 2024-03-05 US US18/596,486 patent/US20250042913A1/en not_active Abandoned
- 2024-10-16 US US18/917,217 patent/US20250270229A1/en active Pending
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011513195A (ja) * | 2007-05-10 | 2011-04-28 | バイオクライスト ファーマシューティカルズ, インコーポレイテッド | ウイルス感染およびがんの処置に使用するためのテトラヒドロフロ[3,4−d]ジオキソラン化合物 |
| JP2010533659A (ja) * | 2007-07-16 | 2010-10-28 | 鄭州大学 | 2’−フルオロ−4’−置換−ヌクレオシド類似体、その製造方法及び使用 |
| JP2011521903A (ja) * | 2008-04-23 | 2011-07-28 | ギリアード サイエンシーズ, インコーポレイテッド | 抗ウイルス処置のための1’−置換カルバ−ヌクレオシドアナログ |
| JP2013535453A (ja) * | 2010-07-22 | 2013-09-12 | ギリード・サイエンシズ・インコーポレーテッド | パラミクソウイルス科ウイルス感染症を治療するための方法及び化合物 |
| EP2615101A1 (en) * | 2010-09-07 | 2013-07-17 | High & New Technology Research Center, Henan Academy of Sciences | Nucleoside derivatives, synthesis methods and uses thereof for preparing anti-tumor and anti-virus medicaments |
| JP2013538230A (ja) * | 2010-09-20 | 2013-10-10 | ギリアード サイエンシーズ, インコーポレイテッド | 抗ウイルス治療用の2’−フルオロ置換カルバヌクレオシド類似体 |
| WO2012040124A1 (en) * | 2010-09-22 | 2012-03-29 | Alios Biopharma, Inc. | Azido nucleosides and nucleotide analogs |
| WO2013096679A1 (en) * | 2011-12-22 | 2013-06-27 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| WO2013142525A1 (en) * | 2012-03-21 | 2013-09-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020536069A (ja) * | 2017-09-29 | 2020-12-10 | エナンタ ファーマシューティカルズ インコーポレイテッド | Rsv阻害剤としての組み合わせ医薬剤 |
| JP7198811B2 (ja) | 2017-09-29 | 2023-01-04 | エナンタ ファーマシューティカルズ インコーポレイテッド | Rsv阻害剤としての組み合わせ医薬剤 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10501476B2 (en) | Thieno[3,2-d]pyrimidine, furo[3,2-d]pyrimidine, and pyrrolo[3,2-D]pyrimidines useful for treating respiratory syncitial virus infections | |
| JP2020111608A (ja) | 2’−クロロアミノピリミジノンおよびピリミジンジオンヌクレオシド | |
| HK40074916A (en) | 4'-substituted-furo[3,2-d]pyrimidine and -pyrrolo[3,2- d]pyrimidine derivatives useful for treating respiratory syncitial virus infections | |
| HK40031992A (en) | Thieno [3,2-d]pyrimidines useful for treating respiratory syncitial virus infections | |
| HK40031992B (en) | Thieno [3,2-d]pyrimidines useful for treating respiratory syncitial virus infections | |
| HK1237766A1 (en) | Thieno [3,2-d]pyrimidines, furo [3,2,d]pyrimidines and pyrrolo [3,2-d]pyrimidines useful for treating respiratory syncitial virus infections | |
| HK1237766B (en) | Thieno [3,2-d]pyrimidines, furo [3,2,d]pyrimidines and pyrrolo [3,2-d]pyrimidines useful for treating respiratory syncitial virus infections | |
| HK1232857B (zh) | 用於治療呼吸道合胞病毒感染的噻吩並[3,2-d]嘧啶、呋喃並[3,2-d]嘧啶和吡咯並[3,2-d]嘧啶 | |
| HK1232857A1 (en) | Thieno [3,2-d] pyrimidine, furo [3,2,d] pyrimidine, and pyrrolo [3,2-d] pyrimidines useful for treating respiratory syncitial virus infections |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180717 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180717 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190311 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20190314 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190603 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20191024 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20191209 |