JP7236458B2 - トリアゾロキナゾリノンの合成 - Google Patents
トリアゾロキナゾリノンの合成 Download PDFInfo
- Publication number
- JP7236458B2 JP7236458B2 JP2020554957A JP2020554957A JP7236458B2 JP 7236458 B2 JP7236458 B2 JP 7236458B2 JP 2020554957 A JP2020554957 A JP 2020554957A JP 2020554957 A JP2020554957 A JP 2020554957A JP 7236458 B2 JP7236458 B2 JP 7236458B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- under conditions
- conditions sufficient
- reacting
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 230000015572 biosynthetic process Effects 0.000 title description 9
- 238000003786 synthesis reaction Methods 0.000 title description 6
- 150000001875 compounds Chemical class 0.000 claims description 259
- 238000000034 method Methods 0.000 claims description 70
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 68
- 230000008569 process Effects 0.000 claims description 45
- 150000003839 salts Chemical class 0.000 claims description 36
- -1 tri-tert-butylphosphonium tetrafluoroborate Chemical compound 0.000 claims description 26
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 25
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 24
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 24
- 239000003153 chemical reaction reagent Substances 0.000 claims description 23
- 229910052725 zinc Inorganic materials 0.000 claims description 23
- 239000011701 zinc Substances 0.000 claims description 23
- 125000004432 carbon atom Chemical group C* 0.000 claims description 21
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 20
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 16
- 238000006243 chemical reaction Methods 0.000 claims description 16
- 125000000217 alkyl group Chemical group 0.000 claims description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 claims description 12
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 12
- AHVORQFVDUDBGE-UHFFFAOYSA-N chembl1644833 Chemical compound C1=CC(C)=CC=C1N1C(=O)N2C(=O)NC3=CC=C(CC=4C=CC=CC=4)C=C3C2=N1 AHVORQFVDUDBGE-UHFFFAOYSA-N 0.000 claims description 11
- 229910052736 halogen Inorganic materials 0.000 claims description 11
- 150000002367 halogens Chemical class 0.000 claims description 11
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 claims description 11
- 229910000041 hydrogen chloride Inorganic materials 0.000 claims description 11
- 238000004519 manufacturing process Methods 0.000 claims description 11
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 10
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 claims description 9
- 229910021595 Copper(I) iodide Inorganic materials 0.000 claims description 9
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 9
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 9
- 239000012458 free base Substances 0.000 claims description 9
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 7
- 239000001257 hydrogen Substances 0.000 claims description 7
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 claims description 7
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 claims description 7
- 239000004202 carbamide Substances 0.000 claims description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 claims description 6
- 235000017557 sodium bicarbonate Nutrition 0.000 claims description 6
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 claims description 5
- 229940073608 benzyl chloride Drugs 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 125000004181 carboxyalkyl group Chemical group 0.000 claims description 4
- 125000003884 phenylalkyl group Chemical group 0.000 claims description 4
- 229910052717 sulfur Inorganic materials 0.000 claims description 4
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 claims description 3
- 125000000304 alkynyl group Chemical group 0.000 claims description 3
- 150000002431 hydrogen Chemical class 0.000 claims description 3
- 229910052760 oxygen Inorganic materials 0.000 claims description 3
- 239000001301 oxygen Substances 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 125000004434 sulfur atom Chemical group 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 239000000243 solution Substances 0.000 description 34
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 24
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 23
- 229940125797 compound 12 Drugs 0.000 description 23
- 239000000725 suspension Substances 0.000 description 23
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- 239000000203 mixture Substances 0.000 description 19
- 239000000126 substance Substances 0.000 description 17
- 238000007792 addition Methods 0.000 description 16
- 238000002360 preparation method Methods 0.000 description 15
- 102000005962 receptors Human genes 0.000 description 14
- 108020003175 receptors Proteins 0.000 description 14
- 239000012065 filter cake Substances 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 12
- 239000008194 pharmaceutical composition Substances 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 229940049706 benzodiazepine Drugs 0.000 description 11
- 238000004128 high performance liquid chromatography Methods 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- 239000011521 glass Substances 0.000 description 10
- 230000000949 anxiolytic effect Effects 0.000 description 9
- LIQBAAKGMWRWPS-UHFFFAOYSA-M chlorozinc(1+);methanidylbenzene Chemical compound [Zn+]Cl.[CH2-]C1=CC=CC=C1 LIQBAAKGMWRWPS-UHFFFAOYSA-M 0.000 description 9
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 208000024891 symptom Diseases 0.000 description 9
- 239000002253 acid Substances 0.000 description 8
- 239000001961 anticonvulsive agent Substances 0.000 description 8
- 239000002249 anxiolytic agent Substances 0.000 description 8
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- 239000004480 active ingredient Substances 0.000 description 7
- 230000001773 anti-convulsant effect Effects 0.000 description 7
- 229960003965 antiepileptics Drugs 0.000 description 7
- 230000000338 anxiogenic effect Effects 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 210000002027 skeletal muscle Anatomy 0.000 description 7
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 238000006411 Negishi coupling reaction Methods 0.000 description 6
- 239000003610 charcoal Substances 0.000 description 6
- 125000001309 chloro group Chemical group Cl* 0.000 description 6
- 230000002920 convulsive effect Effects 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 239000005554 hypnotics and sedatives Substances 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000003158 myorelaxant agent Substances 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 239000012535 impurity Substances 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 4
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- 150000001557 benzodiazepines Chemical class 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 206010021118 Hypotonia Diseases 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000036640 muscle relaxation Effects 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108091008681 GABAA receptors Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 206010039897 Sedation Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical group [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- KBOSVOZLQUMJKM-UHFFFAOYSA-N [3-[[methyl-[4-(9-oxoxanthen-3-yl)oxybutyl]amino]methyl]phenyl] n-methylcarbamate Chemical compound CNC(=O)OC1=CC=CC(CN(C)CCCCOC=2C=C3C(C(C4=CC=CC=C4O3)=O)=CC=2)=C1 KBOSVOZLQUMJKM-UHFFFAOYSA-N 0.000 description 2
- UNKQVRFPUYCEJA-UHFFFAOYSA-N [Zn]CC1=CC=CC=C1 Chemical compound [Zn]CC1=CC=CC=C1 UNKQVRFPUYCEJA-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 229960004381 flumazenil Drugs 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 229940125425 inverse agonist Drugs 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 238000002390 rotary evaporation Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000036280 sedation Effects 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- AWNXKZVIZARMME-UHFFFAOYSA-N 1-[[5-[2-[(2-chloropyridin-4-yl)amino]pyrimidin-4-yl]-4-(cyclopropylmethyl)pyrimidin-2-yl]amino]-2-methylpropan-2-ol Chemical compound N=1C(NCC(C)(O)C)=NC=C(C=2N=C(NC=3C=C(Cl)N=CC=3)N=CC=2)C=1CC1CC1 AWNXKZVIZARMME-UHFFFAOYSA-N 0.000 description 1
- JYZVAJRSNNFELK-UHFFFAOYSA-N 1-amino-3-(ethylamino)urea Chemical compound CCNNC(=O)NN JYZVAJRSNNFELK-UHFFFAOYSA-N 0.000 description 1
- UDHAWRUAECEBHC-UHFFFAOYSA-N 1-iodo-4-methylbenzene Chemical compound CC1=CC=C(I)C=C1 UDHAWRUAECEBHC-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- SDQJTWBNWQABLE-UHFFFAOYSA-N 1h-quinazoline-2,4-dione Chemical compound C1=CC=C2C(=O)NC(=O)NC2=C1 SDQJTWBNWQABLE-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- PKRSYEPBQPFNRB-UHFFFAOYSA-N 2-phenoxybenzoic acid Chemical compound OC(=O)C1=CC=CC=C1OC1=CC=CC=C1 PKRSYEPBQPFNRB-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- BDTRIDKONHOQQN-UHFFFAOYSA-N 4h-pyrimidin-5-one Chemical class O=C1CN=CN=C1 BDTRIDKONHOQQN-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- 102000004300 GABA-A Receptors Human genes 0.000 description 1
- 108090000839 GABA-A Receptors Proteins 0.000 description 1
- 102000027484 GABAA receptors Human genes 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000002067 Protein Subunits Human genes 0.000 description 1
- 108010001267 Protein Subunits Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- VYUXIEKEACQAEO-UHFFFAOYSA-M [Cl-].[Zn+]CC1=CC=CC=C1 Chemical compound [Cl-].[Zn+]CC1=CC=CC=C1 VYUXIEKEACQAEO-UHFFFAOYSA-M 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 208000004793 anterograde amnesia Diseases 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 229940005530 anxiolytics Drugs 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000000712 assembly Effects 0.000 description 1
- 238000000429 assembly Methods 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- CFIHCJJFWPTAOV-UHFFFAOYSA-N benzyl(tributyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)CC1=CC=CC=C1 CFIHCJJFWPTAOV-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 239000003179 convulsant agent Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 150000002148 esters Chemical group 0.000 description 1
- QEPVFVNIQKEMFJ-UHFFFAOYSA-N ethyl N-[(6-benzyl-2-chloroquinazolin-4-yl)amino]-N-(4-methylphenyl)carbamate Chemical compound C(C1=CC=CC=C1)C=1C=C2C(=NC(=NC2=CC=1)Cl)NN(C(=O)OCC)C1=CC=C(C=C1)C QEPVFVNIQKEMFJ-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 238000001095 inductively coupled plasma mass spectrometry Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- WMFOQBRAJBCJND-UHFFFAOYSA-M lithium hydroxide Inorganic materials [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 229940035363 muscle relaxants Drugs 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000004172 nitrogen cycle Methods 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004344 phenylpropyl group Chemical group 0.000 description 1
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 1
- 150000003009 phosphonic acids Chemical class 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M potassium hydroxide Inorganic materials [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K potassium phosphate Substances [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 1
- 239000012041 precatalyst Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000004799 sedative–hypnotic effect Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 208000020685 sleep-wake disease Diseases 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- FFSJPOPLSWBGQY-UHFFFAOYSA-N triazol-4-one Chemical compound O=C1C=NN=N1 FFSJPOPLSWBGQY-UHFFFAOYSA-N 0.000 description 1
- JDPSKZFCBMHFKL-UHFFFAOYSA-N triazolo[4,5-h]quinazolin-4-one Chemical compound O=C1C=C2C=NC=NC2=C2C1=NN=N2 JDPSKZFCBMHFKL-UHFFFAOYSA-N 0.000 description 1
- 150000003628 tricarboxylic acids Chemical class 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- AIFRHYZBTHREPW-UHFFFAOYSA-N β-carboline Chemical class N1=CC=C2C3=CC=CC=C3NC2=C1 AIFRHYZBTHREPW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Anesthesiology (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
本出願は、2017年12月19日に出願され、参照により本明細書に組み込まれる米国特許出願第62/607,353号明細書の優先権を主張する。


R1、R2、及びR3は、本明細書に規定される。本発明はさらに、本発明の方法によって調製される式(I)の化合物及びその薬学的に許容される塩に関する。

本発明は、本発明の方法によって調製される9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオンにも関する。

R1は、ハロゲン、1~2個の炭素原子を有するアルキル、1~3個の炭素原子を有するカルボキシアルキル、アルキニル鎖に2~3個の炭素原子を有するフェニル-アルキニル、アルケニル鎖に1~3個の炭素原子を有するフェニル-アルケニル、アルキル鎖に1~3個の炭素原子を有するフェニル-アルキルであって、フェニル部分がさらに、任意の位置で酸素又は硫黄原子によって置換され得、アルキル鎖の1~2個の炭素原子を有するピリジル-アルキル及びトリフルオロメチルから成る群から選択される。
R2は、水素及びハロゲンから成る群から選択され、並びに
R3は、水素、ハロゲン及び1~2個の炭素原子を有するアルキルから成る群から選択される。



表2:最後の産物の化合物12の分離された回収物

表3:化合物12の選択された金属の含有量(ICP-MS)

本発明の一態様を以下に示すが、本発明はそれに限定されない。
[発明1]
式(I)の化合物及びその薬学的に許容される塩を調製するプロセスであって、
[化1]

R 1 は、ハロゲン、1~2個の炭素原子を有するアルキル、1~3個の炭素原子を有するカルボキシアルキル、アルキニル鎖に2~3個の炭素原子を有するフェニル-アルキニル、アルケニル鎖に1~3個の炭素原子を有するフェニル-アルケニル、アルキル鎖に1~3個の炭素原子を有するフェニル-アルキルであって、フェニル部分がさらに、任意の位置で酸素又は硫黄原子によって置換され得る、フェニル-アルキル、アルキル鎖に1~2個の炭素原子を有するピリジル-アルキル及びトリフルオロメチルから成る群から選択され、
R 2 は、水素及びハロゲンから成る群から選択され、並びに
R 3 は、水素、ハロゲン及び1~2個の炭素原子を有するアルキルから成る群から選択され、
工程1において、式(IV)の化合物を形成するのに充分な条件下で、式(II)の化合物を式(III)の化合物と反応させ、
[化2]

工程2において、式(VI)の化合物を形成するのに充分な条件下で、式(V)の化合物を反応させ、
[化3]

工程3において、式(VII)の化合物を形成するのに充分な条件下で、式(VI)の化合物を式(XI)の亜鉛試薬と反応させ、
[化4]

工程4において、式(VIII)の化合物を形成するのに充分な条件下で、式(VII)の化合物を反応させ、
[化5]

工程5において、式(IX)の化合物を形成するのに充分な条件下で、式(VIII)の化合物を式(IV)の化合物と反応させ、
[化6]

工程6において、式(X)の化合物を形成するのに充分な条件下で、式(IX)の化合物を反応させ、
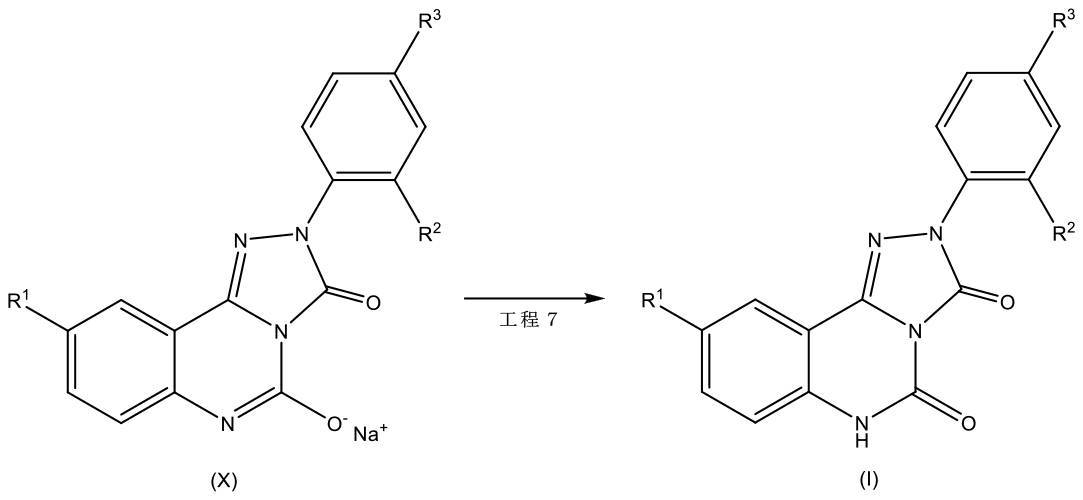
[化7]

並びに
工程7において、式(I)の化合物を形成するのに充分な条件下で、式(X)の化合物を反応させる、式(I)の化合物及びその薬学的に許容される塩を調製するプロセス。
[化8]
[発明2]
工程1において、ジメチルスルホキシド、前記式(III)の化合物、炭酸セシウム、前記式(II)の化合物、ヨウ化銅(I)が、式(IV)の粗化合物を形成するのに充分な条件下で反応され、
前記式(IV)の粗化合物は、前記式(IV)の化合物の塩酸塩を形成するのに充分な条件下で、塩化水素溶液と反応され、
前記式(IV)の化合物の塩化水素塩及び炭酸水素ナトリウムは、前記式(IV)の化合物の遊離塩基を形成するのに充分な条件下で反応される、発明1に記載のプロセス。
[発明3]
前記ジメチルスルホキシド、前記式(III)の化合物、前記炭酸セシウム、前記式(II)の化合物、及び前記ヨウ化銅(I)は、前記式(IV)の粗化合物を形成するためにこの順序で反応される、発明2に記載のプロセス。
[発明4]
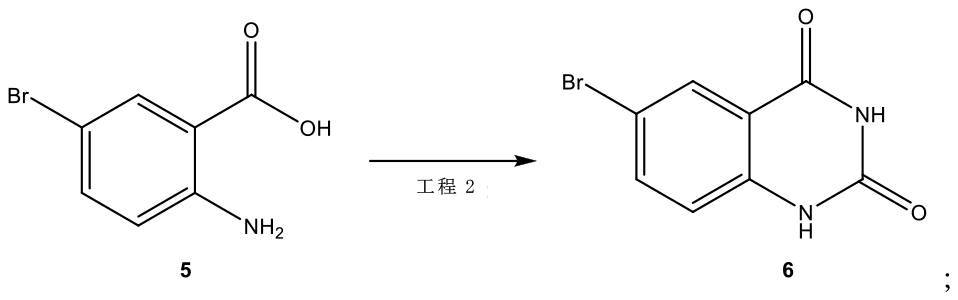
工程2において、前記式(V)の化合物及び尿素が前記式(VI)の化合物を形成するのに充分な条件下で反応される、発明1~3の何れか1項に記載のプロセス。
[発明5]
工程3において、前記式(VI)の化合物、前記式(XI)の亜鉛試薬、パラジウム(II)酢酸塩、及び、トリ-tert-ブチルホスホニウムテトラフルオロホウ酸塩が、前記式(VII)の化合物を形成するのに充分な条件下で反応される、発明1~4の何れか1項に記載のプロセス。
[発明6]
式R 1 -Clの化合物が、前記式(XI)の亜鉛試薬を形成するのに充分な条件下で反応される、発明1~5の何れか1項に記載のプロセス。
[発明7]
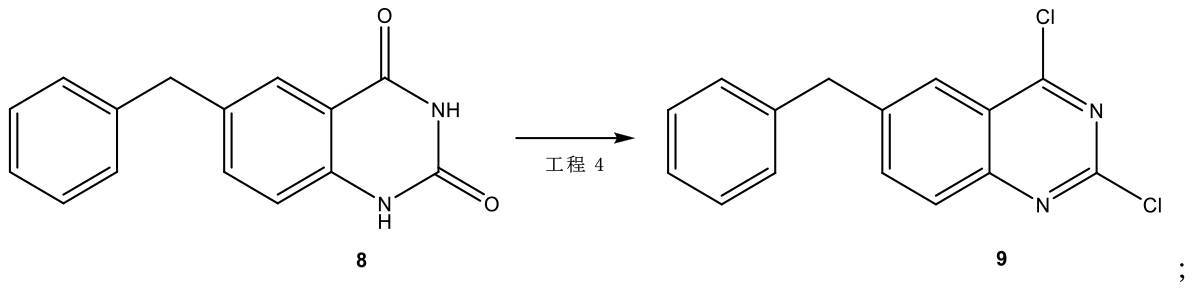
工程4において、前記式(VII)の化合物、オキシ塩化リン及びジイソプロピルエチルアミンが、前記式(VIII)の化合物を形成するのに充分な条件下で反応される、発明1~6の何れか1項に記載のプロセス。
[発明8]
工程5において、前記式(VIII)の化合物、前記式(IV)の化合物、及びN,N-ジイソプロピルエチルアミンが、前記式(IX)の化合物を形成するのに充分な条件下で反応される、発明1~7の何れか1項に記載のプロセス。
[発明9]
工程6において、前記式(IX)の化合物及び水酸化ナトリウムが前記式(X)の化合物を形成するのに充分な条件下で反応される、発明1~8の何れか1項に記載のプロセス。
[発明10]
工程7において、前記式(X)の化合物及び酢酸が前記式(I)の化合物を形成するのに充分な条件下で反応される、発明1~9の何れか1項に記載のプロセス。
[発明11]
前記式(I)の化合物が9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオンである、発明1~10の何れか1項に記載のプロセス。
[発明12]
発明1~10の何れか1項に記載のプロセスによって調製される、式(I)の化合物又はその薬学的に許容される塩。
[発明13]
前記式(I)の化合物が9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオンである、発明12に記載の化合物。
[発明14]
本明細書に記載のスキーム2に記載されるプロセスによって調製される式(I)の化合物又はその薬学的に許容される塩。
[発明15]
前記式(I)の化合物が9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオンである、発明14に記載の化合物。
[発明16]
以下の構造を有する式12の化合物を調製するプロセスであって、
[化9]

工程1において、式1及び式2の前記化合物を前記式4の化合物を形成するのに充分な条件下で反応させ、
[化10]

工程2において、前記式5の化合物を前記式6の化合物を形成するのに充分な条件下で反応させ、
[化11]

工程3において、前記式8の化合物を形成するのに充分な条件下で、前記式6の化合物を前記式13の亜鉛試薬と反応させ、
[化12]

工程4において、前記式8の化合物を前記式9の化合物を形成するのに充分な条件下で反応させ、
[化13]
工程5において、前記式9の化合物を前記式10の化合物を形成するのに充分な条件下で前記式4の化合物と反応させ、
[化14]

工程6において、前記式10の化合物を前記式11の化合物を形成するのに充分な条件下で反応させ、
[化15]

並びに
工程7において、前記式11の化合物を前記式12の化合物を形成するのに充分な条件下で反応させることを含む、式12の化合物を調製するプロセス。
[化16]

[発明17]
塩化ベンジルが前記式13の亜鉛試薬を形成するのに充分な条件下で反応される、発明16に記載の方法。
[発明18]
工程1において、ジメチルスルホキシド、前記式2の化合物、炭酸セシウム、前記式1の化合物及びヨウ化銅(I)が、式4の粗化合物を形成するのに充分な条件下で反応され、
前記式4の粗化合物が、前記式4の化合物の塩酸塩を形成するのに充分な条件下で塩化水素溶液と反応され、
前記式4の化合物の前記塩化水素塩及び炭酸水素ナトリウムが前記式4の化合物の遊離塩基を形成するのに充分な条件下で反応される、発明16又は17に記載のプロセス。
[発明19]
工程2において、前記式5の化合物及び尿素が、前記式6の化合物を形成するのに充分な条件下で反応される、発明16~18の何れか1項に記載のプロセス。
[発明20]
工程3において、前記式6の化合物、前記式13の亜鉛試薬、パラジウム(II)酢酸塩、及び、トリ-tert-ブチルホスホニウムテトラフルオロホウ酸塩が、前記式8の化合物を形成するのに充分な条件下で反応される、発明16~19の何れか1項に記載のプロセス。
[発明21]
工程4において、前記式8の化合物、オキシ塩化リン及びジイソプロピルエチルアミンが、前記式9の化合物を形成するのに充分な条件下で反応される、発明16~20の何れか1項に記載のプロセス。
[発明22]
工程5において、前記式9の化合物、前記式4の化合物、及びN,N-ジイソプロピルエチルアミンが、前記式10の化合物を形成するのに充分な条件下で反応される、発明16~21の何れか1項に記載のプロセス。
[発明23]
工程6において、前記式10の化合物及び水酸化ナトリウムが前記式11の化合物を形成するのに充分な条件下で反応される、発明16~22の何れか1項に記載のプロセス。
[発明24]
工程7において、前記式11の化合物及び酢酸が前記式12の化合物を形成するのに充分な条件下で反応される、発明16~23の何れか1項に記載のプロセス。
[発明25]
発明16~24の何れか1項に記載のプロセスによって調製される9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオン。
[発明26]
本明細書に記載のスキーム1に記載のプロセスによって調製される9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオン。
[発明27]
発明1~11又は16~24の何れか1項に記載のプロセスによって調製される式(I)の化合物又はその薬学的に許容される塩を含む医薬組成物、並びに薬学的に許容される担体又は賦形剤。
[発明28]
前記式(I)の化合物が9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオンである、発明27に記載の医薬組成物。
[発明29]
発明12~15、25又は26の何れか1項に記載の化合物及び薬学的に許容される担体又は賦形剤を含む医薬組成物。
[発明30]
発明1~11又は16~24の何れか1項に記載のプロセスによって調製される治療有効量の式(I)の少なくとも1つの化合物又はその薬学的に許容される塩を投与することによって、哺乳動物の抗不安、抗痙攣、催眠鎮静、骨格筋弛緩、不安惹起、睡眠障害(somnolytic)及び痙攣症状を治療する方法。
[発明31]
前記式(I)の化合物が9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオンである、発明29に記載の方法。
[発明32]
発明27~29の何れか1項に記載の少なくとも1つの医薬組成物の治療有効量を投与することによって、哺乳動物の抗不安、抗痙攣、催眠鎮静、骨格筋弛緩、不安惹起、睡眠障害(somnolytic)及び痙攣症状を治療する方法。
Claims (20)
- 式(I)の化合物及びその薬学的に許容される塩を調製するプロセスであって、

R1は、ハロゲン、1~2個の炭素原子を有するアルキル、1~3個の炭素原子を有するカルボキシアルキル、アルキニル鎖に2~3個の炭素原子を有するフェニル-アルキニル、アルケニル鎖に1~3個の炭素原子を有するフェニル-アルケニル、アルキル鎖に1~3個の炭素原子を有するフェニル-アルキルであって、フェニル部分がさらに、任意の位置で酸素又は硫黄原子によって置換され得る、フェニル-アルキル、アルキル鎖に1~2個の炭素原子を有するピリジル-アルキル及びトリフルオロメチルから成る群から選択され、
R2は、水素及びハロゲンから成る群から選択され、並びに
R3は、水素、ハロゲン及び1~2個の炭素原子を有するアルキルから成る群から選択され、
工程1において、式(IV)の化合物を形成するのに充分な条件下で、式(II)の化合物を式(III)の化合物と反応させ、

工程2において、式(VI)の化合物を形成するのに充分な条件下で、式(V)の化合物を反応させ、

工程3において、式(VII)の化合物を形成するのに充分な条件下で、式(VI)の化合物を式(XI)の亜鉛試薬と反応させ、

工程4において、式(VIII)の化合物を形成するのに充分な条件下で、式(VII)の化合物を反応させ、

工程5において、式(IX)の化合物を形成するのに充分な条件下で、式(VIII)の化合物を式(IV)の化合物と反応させ、

工程6において、式(X)の化合物を形成するのに充分な条件下で、式(IX)の化合物を反応させ、

並びに
工程7において、式(I)の化合物を形成するのに充分な条件下で、式(X)の化合物を反応させる、式(I)の化合物及びその薬学的に許容される塩を調製するプロセス。

- 工程1において、ジメチルスルホキシド、前記式(III)の化合物、炭酸セシウム、前記式(II)の化合物、ヨウ化銅(I)が、式(IV)の粗化合物を形成するのに充分な条件下で反応され、
前記式(IV)の粗化合物は、前記式(IV)の化合物の塩酸塩を形成するのに充分な条件下で、塩化水素溶液と反応され、
前記式(IV)の化合物の塩化水素塩及び炭酸水素ナトリウムは、前記式(IV)の化合物の遊離塩基を形成するのに充分な条件下で反応される、請求項1に記載のプロセス。 - 前記ジメチルスルホキシド、前記式(III)の化合物、前記炭酸セシウム、前記式(II)の化合物、及び前記ヨウ化銅(I)は、前記式(IV)の粗化合物を形成するためにこの順序で反応される、請求項2に記載のプロセス。
- 工程2において、前記式(V)の化合物及び尿素が前記式(VI)の化合物を形成するのに充分な条件下で反応される、請求項1~3の何れか1項に記載のプロセス。
- 工程3において、前記式(VI)の化合物、前記式(XI)の亜鉛試薬、パラジウム(II)酢酸塩、及び、トリ-tert-ブチルホスホニウムテトラフルオロホウ酸塩が、前記式(VII)の化合物を形成するのに充分な条件下で反応される、請求項1~4の何れか1項に記載のプロセス。
- 式R1-Clの化合物を亜鉛と反応させることにより前記式(XI)の亜鉛試薬が形成される、請求項1~5の何れか1項に記載のプロセス。
- 工程4において、前記式(VII)の化合物、オキシ塩化リン及びジイソプロピルエチルアミンが、前記式(VIII)の化合物を形成するのに充分な条件下で反応される、請求項1~6の何れか1項に記載のプロセス。
- 工程5において、前記式(VIII)の化合物、前記式(IV)の化合物、及びN,N-ジイソプロピルエチルアミンが、前記式(IX)の化合物を形成するのに充分な条件下で反応される、請求項1~7の何れか1項に記載のプロセス。
- 工程6において、前記式(IX)の化合物及び水酸化ナトリウムが前記式(X)の化合物を形成するのに充分な条件下で反応される、請求項1~8の何れか1項に記載のプロセス。
- 工程7において、前記式(X)の化合物及び酢酸が前記式(I)の化合物を形成するのに充分な条件下で反応される、請求項1~9の何れか1項に記載のプロセス。
- 前記式(I)の化合物が9-ベンジル-2-(4-メチルフェニル)-2,6-ジヒドロ[1,2,4]トリアゾロ[4,3-c]キナゾリン-3,5-ジオンである、請求項1~10の何れか1項に記載のプロセス。
- 以下の構造を有する式12の化合物を調製するプロセスであって、

工程1において、式1及び式2の前記化合物を前記式4の化合物を形成するのに充分な条件下で反応させ、

工程2において、前記式5の化合物を前記式6の化合物を形成するのに充分な条件下で反応させ、
工程3において、前記式8の化合物を形成するのに充分な条件下で、前記式6の化合物を前記式13の亜鉛試薬と反応させ、

工程4において、前記式8の化合物を前記式9の化合物を形成するのに充分な条件下で反応させ、

工程5において、前記式9の化合物を前記式10の化合物を形成するのに充分な条件下で前記式4の化合物と反応させ、

工程6において、前記式10の化合物を前記式11の化合物を形成するのに充分な条件下で反応させ、

並びに
工程7において、前記式11の化合物を前記式12の化合物を形成するのに充分な条件下で反応させることを含む、式12の化合物を調製するプロセス。

- 塩化ベンジルを亜鉛と反応させることにより前記式13の亜鉛試薬が形成される、請求項12に記載の方法。
- 工程1において、ジメチルスルホキシド、前記式2の化合物、炭酸セシウム、前記式1の化合物及びヨウ化銅(I)が、式4の粗化合物を形成するのに充分な条件下で反応され、
前記式4の粗化合物が、前記式4の化合物の塩酸塩を形成するのに充分な条件下で塩化水素溶液と反応され、
前記式4の化合物の前記塩化水素塩及び炭酸水素ナトリウムが前記式4の化合物の遊離塩基を形成するのに充分な条件下で反応される、請求項12又は13に記載のプロセス。 - 工程2において、前記式5の化合物及び尿素が、前記式6の化合物を形成するのに充分な条件下で反応される、請求項12~14の何れか1項に記載のプロセス。
- 工程3において、前記式6の化合物、前記式13の亜鉛試薬、パラジウム(II)酢酸塩、及び、トリ-tert-ブチルホスホニウムテトラフルオロホウ酸塩が、前記式8の化合物を形成するのに充分な条件下で反応される、請求項12~15の何れか1項に記載のプロセス。
- 工程4において、前記式8の化合物、オキシ塩化リン及びジイソプロピルエチルアミンが、前記式9の化合物を形成するのに充分な条件下で反応される、請求項12~16の何れか1項に記載のプロセス。
- 工程5において、前記式9の化合物、前記式4の化合物、及びN,N-ジイソプロピルエチルアミンが、前記式10の化合物を形成するのに充分な条件下で反応される、請求項12~17の何れか1項に記載のプロセス。
- 工程6において、前記式10の化合物及び水酸化ナトリウムが前記式11の化合物を形成するのに充分な条件下で反応される、請求項12~18の何れか1項に記載のプロセス。
- 工程7において、前記式11の化合物及び酢酸が前記式12の化合物を形成するのに充分な条件下で反応される、請求項12~19の何れか1項に記載のプロセス。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762607353P | 2017-12-19 | 2017-12-19 | |
| US62/607,353 | 2017-12-19 | ||
| PCT/IB2018/001573 WO2019123011A1 (en) | 2017-12-19 | 2018-12-19 | Triazoloquinazolinone synthesis |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021508342A JP2021508342A (ja) | 2021-03-04 |
| JP7236458B2 true JP7236458B2 (ja) | 2023-03-09 |
Family
ID=65494446
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020554957A Active JP7236458B2 (ja) | 2017-12-19 | 2018-12-19 | トリアゾロキナゾリノンの合成 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US11434243B2 (ja) |
| EP (1) | EP3752507B1 (ja) |
| JP (1) | JP7236458B2 (ja) |
| DK (1) | DK3752507T3 (ja) |
| ES (1) | ES2934885T3 (ja) |
| FI (1) | FI3752507T3 (ja) |
| PL (1) | PL3752507T3 (ja) |
| WO (1) | WO2019123011A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022074457A1 (en) * | 2020-10-06 | 2022-04-14 | Gabather Ab | Lipid formulations of triazoloquinazolinone compounds |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009123537A1 (en) | 2008-04-04 | 2009-10-08 | Forskarpatent I Syd Ab | GABAa RECEPTOR MODULATORS |
| WO2015164308A1 (en) | 2014-04-22 | 2015-10-29 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102179763B1 (ko) * | 2014-04-23 | 2020-11-17 | 덕산네오룩스 주식회사 | 유기전기 소자용 화합물, 이를 이용한 유기전기소자 및 그 전자 장치 |
-
2018
- 2018-12-19 FI FIEP18849402.5T patent/FI3752507T3/fi active
- 2018-12-19 JP JP2020554957A patent/JP7236458B2/ja active Active
- 2018-12-19 PL PL18849402.5T patent/PL3752507T3/pl unknown
- 2018-12-19 WO PCT/IB2018/001573 patent/WO2019123011A1/en not_active Ceased
- 2018-12-19 DK DK18849402.5T patent/DK3752507T3/da active
- 2018-12-19 EP EP18849402.5A patent/EP3752507B1/en active Active
- 2018-12-19 ES ES18849402T patent/ES2934885T3/es active Active
- 2018-12-19 US US16/954,795 patent/US11434243B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009123537A1 (en) | 2008-04-04 | 2009-10-08 | Forskarpatent I Syd Ab | GABAa RECEPTOR MODULATORS |
| WO2015164308A1 (en) | 2014-04-22 | 2015-10-29 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
Non-Patent Citations (3)
| Title |
|---|
| Bioorg. Med. Chem.,2011年,19,111-121 |
| Chem Bio Drug Des,2013年,81,583-590 |
| J Enzyme Inhib Med Chem,2016年,31(2),195-204 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019123011A1 (en) | 2019-06-27 |
| EP3752507B1 (en) | 2022-11-16 |
| US20210087193A1 (en) | 2021-03-25 |
| FI3752507T3 (fi) | 2023-02-16 |
| JP2021508342A (ja) | 2021-03-04 |
| PL3752507T3 (pl) | 2023-03-13 |
| DK3752507T3 (da) | 2023-01-09 |
| US11434243B2 (en) | 2022-09-06 |
| EP3752507A1 (en) | 2020-12-23 |
| ES2934885T3 (es) | 2023-02-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3950686A1 (en) | N-heteroaromatic amide derivatives for treatment of cancer | |
| CN102639536B (zh) | 吡啶衍生物的无水晶型 | |
| JP2006508092A (ja) | γ−セクレターゼの阻害のための環状スルファミド | |
| BG62619B1 (bg) | Кристални форми на 2-метил-тиено-бензодиазепин и метод заполучаването им | |
| EP3705478B1 (en) | Triazine compound and pharmaceutically acceptable salt thereof | |
| US6534505B2 (en) | Therapeutic polymorphs of a GABA-A alpha-5 inverse agonist and pamoate formulations of the same | |
| AU745005B2 (en) | Therapeutically active 1,2,4-triazolo(4.,3-b) pyridazine derivatives as ligands for gaba receptors | |
| JP2017061578A (ja) | 有機化合物 | |
| JP2022532141A (ja) | キナーゼ阻害剤として使用される化合物およびその応用 | |
| JP7682923B2 (ja) | ゲポチダシンの結晶形態 | |
| CN112351982A (zh) | 制备p300和/或cbp调节剂的方法 | |
| TW201016713A (en) | Method for purification of adefovir dipivoxil | |
| KR20110002090A (ko) | 오르베피탄트 말레에이트의 무수 결정 형태 | |
| JP7236458B2 (ja) | トリアゾロキナゾリノンの合成 | |
| CN102256975A (zh) | 氮杂二环-三氟甲基苯甲酰胺衍生物的新的多晶型形式 | |
| EP3004102A1 (en) | Crystalline form of n,n-dicyclopropyl-4-(1,5-dimethyl-1 h-pyrazol-3-ylamino)-6-ethyl-1 -methyl-1,6-dihydroimidazo[4,5- d]fy rrolo[2,3-b]pyridine-7-carboxamide for the treatment of myeloproliferative disorders | |
| EP3004103A1 (en) | Crystalline form of n,n-dicyclopropyl-4-(1,5-dimethyl-1 h-pyrazol-3-ylamino)-6-ethyl-1 -methyl-1,6-dihydroimidazo[4,5 d]fy rrolo[2,3-b]pyridine-7-carboxamide for the treatment of myeloproliferative disorders | |
| JPH07503727A (ja) | ベンゾジアゼピン受容体結合剤として有用な4−オキソ−および4H−イミダゾ(5,1−c)(1,4)ベンゾオキサジン類 | |
| EP3004104A1 (en) | Crystalline form of n,n-dicyclopropyl-4-(1,5-dimethyl-1 h-pyrazol-3-ylamino)-6-ethyl-1 -methyl-1,6-dihyrdroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide for the treatment of myeloproliferative disorders | |
| EP3004101A1 (en) | Crystalline form of n,n-dicyclopropyl-4-(1,5-dimethyl-1 h-pyrazol-3-ylamino)-6-ethyl-1 -methyl-1,6-dihydroimidazo[4,5- d]fy rrolo[2,3-b]pyridine-7-carboxamide for the treatment of myeloproliferative disorders | |
| JP2015516440A (ja) | 7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−メチル−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジンの2型多形 | |
| KR100545785B1 (ko) | 도파민 수용체에 활성을 지닌 신규 피페라지닐알킬트리아졸 화합물 | |
| US20060094764A1 (en) | Cyanothiophenes, their preparation and their use in pharmaceutical compositions | |
| JP3985117B2 (ja) | ジヒドロキノリン誘導体 | |
| US7714129B2 (en) | Methods of preparing anhydrous aripiprazole form II |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20211019 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20220922 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20221020 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230119 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230202 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230227 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7236458 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |