KR800000079B1 - Method for preparing N-[(1-piperidinyl) alkyl] carboxamide derivative - Google Patents
Method for preparing N-[(1-piperidinyl) alkyl] carboxamide derivative Download PDFInfo
- Publication number
- KR800000079B1 KR800000079B1 KR7601895A KR760001895A KR800000079B1 KR 800000079 B1 KR800000079 B1 KR 800000079B1 KR 7601895 A KR7601895 A KR 7601895A KR 760001895 A KR760001895 A KR 760001895A KR 800000079 B1 KR800000079 B1 KR 800000079B1
- Authority
- KR
- South Korea
- Prior art keywords
- parts
- piperidinyl
- ethyl
- general formula
- filtered
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- 125000000217 alkyl group Chemical group 0.000 title claims description 27
- 238000000034 method Methods 0.000 title description 39
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 title description 4
- 125000003917 carbamoyl group Chemical class [H]N([H])C(*)=O 0.000 title 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 70
- -1 1-methyl-2-pyrrolyl Chemical group 0.000 claims description 52
- 229910052739 hydrogen Inorganic materials 0.000 claims description 29
- 239000001257 hydrogen Substances 0.000 claims description 29
- 238000006243 chemical reaction Methods 0.000 claims description 21
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 14
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 12
- 150000002431 hydrogen Chemical class 0.000 claims description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 8
- 150000003053 piperidines Chemical class 0.000 claims description 7
- 235000013305 food Nutrition 0.000 claims description 6
- 239000003960 organic solvent Substances 0.000 claims description 6
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims description 4
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 claims description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 4
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 claims description 3
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 3
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000003545 alkoxy group Chemical group 0.000 claims description 2
- 125000003118 aryl group Chemical group 0.000 claims description 2
- 125000004185 ester group Chemical group 0.000 claims description 2
- 125000004076 pyridyl group Chemical group 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 5
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- UUEVFMOUBSLVJW-UHFFFAOYSA-N oxo-[[1-[2-[2-[2-[4-(oxoazaniumylmethylidene)pyridin-1-yl]ethoxy]ethoxy]ethyl]pyridin-4-ylidene]methyl]azanium;dibromide Chemical compound [Br-].[Br-].C1=CC(=C[NH+]=O)C=CN1CCOCCOCCN1C=CC(=C[NH+]=O)C=C1 UUEVFMOUBSLVJW-UHFFFAOYSA-N 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 114
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 78
- 239000000203 mixture Substances 0.000 description 74
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 71
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 63
- 150000001875 compounds Chemical class 0.000 description 51
- 238000002844 melting Methods 0.000 description 48
- 230000008018 melting Effects 0.000 description 48
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 46
- 239000000047 product Substances 0.000 description 45
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 42
- 239000000243 solution Substances 0.000 description 39
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 35
- 229960001701 chloroform Drugs 0.000 description 35
- 239000011541 reaction mixture Substances 0.000 description 27
- 239000000706 filtrate Substances 0.000 description 24
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 239000002253 acid Substances 0.000 description 21
- 239000003054 catalyst Substances 0.000 description 20
- 150000003839 salts Chemical class 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 15
- 239000003480 eluent Substances 0.000 description 15
- 239000000543 intermediate Substances 0.000 description 15
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 15
- 239000007858 starting material Substances 0.000 description 15
- 238000003756 stirring Methods 0.000 description 15
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 14
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 13
- 238000004440 column chromatography Methods 0.000 description 13
- 239000000741 silica gel Substances 0.000 description 12
- 229910002027 silica gel Inorganic materials 0.000 description 12
- 238000001816 cooling Methods 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 10
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 239000000284 extract Substances 0.000 description 10
- 125000005843 halogen group Chemical group 0.000 description 10
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000004480 active ingredient Substances 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 9
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 8
- 239000000908 ammonium hydroxide Substances 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 241000501667 Etroplus Species 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 6
- 239000003513 alkali Substances 0.000 description 6
- 150000008064 anhydrides Chemical class 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 150000003254 radicals Chemical class 0.000 description 6
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- 206010047700 Vomiting Diseases 0.000 description 5
- VMWNQDUVQKEIOC-CYBMUJFWSA-N apomorphine Chemical compound C([C@H]1N(C)CC2)C3=CC=C(O)C(O)=C3C3=C1C2=CC=C3 VMWNQDUVQKEIOC-CYBMUJFWSA-N 0.000 description 5
- 229960004046 apomorphine Drugs 0.000 description 5
- SXDBWCPKPHAZSM-UHFFFAOYSA-N bromic acid Chemical compound OBr(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-N 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 150000004820 halides Chemical class 0.000 description 5
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 5
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 230000008673 vomiting Effects 0.000 description 5
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 4
- HTQWGIHCFPWKAS-UHFFFAOYSA-N 1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one Chemical compound C1CNCCC11C(=O)NCN1C1=CC=CC=C1 HTQWGIHCFPWKAS-UHFFFAOYSA-N 0.000 description 4
- 125000005999 2-bromoethyl group Chemical group 0.000 description 4
- MHXACMFKKGZLNW-UHFFFAOYSA-N 2-n-(1-benzylpiperidin-4-yl)benzene-1,2-diamine Chemical compound NC1=CC=CC=C1NC1CCN(CC=2C=CC=CC=2)CC1 MHXACMFKKGZLNW-UHFFFAOYSA-N 0.000 description 4
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 4
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 229910000564 Raney nickel Inorganic materials 0.000 description 4
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 238000005917 acylation reaction Methods 0.000 description 4
- 239000012458 free base Substances 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- JFDLNTWOJUPVTD-UHFFFAOYSA-N n-(2-nitrophenyl)piperidin-4-amine Chemical compound [O-][N+](=O)C1=CC=CC=C1NC1CCNCC1 JFDLNTWOJUPVTD-UHFFFAOYSA-N 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- BYNBAMHAURJNTR-UHFFFAOYSA-N 3-piperidin-4-yl-1h-benzimidazol-2-one Chemical compound O=C1NC2=CC=CC=C2N1C1CCNCC1 BYNBAMHAURJNTR-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 230000003474 anti-emetic effect Effects 0.000 description 3
- 239000002111 antiemetic agent Substances 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 238000006114 decarboxylation reaction Methods 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- VNYZLKGRVHTMIQ-UHFFFAOYSA-N n-(2-bromoethyl)-2-nitrobenzamide Chemical compound [O-][N+](=O)C1=CC=CC=C1C(=O)NCCBr VNYZLKGRVHTMIQ-UHFFFAOYSA-N 0.000 description 3
- FWFRKOQCZMBNKS-UHFFFAOYSA-N n-(2-chloroethyl)furan-2-carboxamide Chemical compound ClCCNC(=O)C1=CC=CO1 FWFRKOQCZMBNKS-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 239000000052 vinegar Substances 0.000 description 3
- 235000021419 vinegar Nutrition 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- YUBDLZGUSSWQSS-UHFFFAOYSA-N 1-benzylpiperidin-4-amine Chemical compound C1CC(N)CCN1CC1=CC=CC=C1 YUBDLZGUSSWQSS-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 2
- ONRREFWJTRBDRA-UHFFFAOYSA-N 2-chloroethanamine;hydron;chloride Chemical compound [Cl-].[NH3+]CCCl ONRREFWJTRBDRA-UHFFFAOYSA-N 0.000 description 2
- YTMSMHJTXDYIKR-UHFFFAOYSA-N 2-nitrobenzamide;hydrochloride Chemical compound Cl.NC(=O)C1=CC=CC=C1[N+]([O-])=O YTMSMHJTXDYIKR-UHFFFAOYSA-N 0.000 description 2
- DOAYWDKFDPSTSV-UHFFFAOYSA-N 5-chloro-1,3-dihydro-1-(4-piperidinyl)-2h-benzimidazol-2-one Chemical compound O=C1NC2=CC(Cl)=CC=C2N1C1CCNCC1 DOAYWDKFDPSTSV-UHFFFAOYSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 238000005903 acid hydrolysis reaction Methods 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 230000003457 anti-vomiting effect Effects 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 125000006196 aroyl alkyl group Chemical group 0.000 description 2
- HGBCPYMIZWPKMI-UHFFFAOYSA-N aziridin-1-yl(phenyl)methanone Chemical compound C=1C=CC=CC=1C(=O)N1CC1 HGBCPYMIZWPKMI-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- WBYWAXJHAXSJNI-UHFFFAOYSA-N cinnamic acid Chemical compound OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- BESCFIAGVMCBAI-UHFFFAOYSA-N ethyl n-(2-bromoethyl)carbamate Chemical compound CCOC(=O)NCCBr BESCFIAGVMCBAI-UHFFFAOYSA-N 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical group 0.000 description 2
- 239000008240 homogeneous mixture Substances 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 2
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 230000003340 mental effect Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- WUSZVYUCESUCSQ-UHFFFAOYSA-N n-(2-chloroethyl)-1-methylpyrrole-2-carboxamide Chemical compound CN1C=CC=C1C(=O)NCCCl WUSZVYUCESUCSQ-UHFFFAOYSA-N 0.000 description 2
- PAEWUPLHSBIJMQ-UHFFFAOYSA-N n-(2-chloroethyl)pyridine-2-carboxamide Chemical compound ClCCNC(=O)C1=CC=CC=N1 PAEWUPLHSBIJMQ-UHFFFAOYSA-N 0.000 description 2
- GJYDUQKSXUDKCH-UHFFFAOYSA-N n-(2-hydroxyethyl)-1-methylpyrrole-2-carboxamide Chemical compound CN1C=CC=C1C(=O)NCCO GJYDUQKSXUDKCH-UHFFFAOYSA-N 0.000 description 2
- SIUMVFGIULAFNI-UHFFFAOYSA-N n-(2-hydroxyethyl)pyridine-2-carboxamide Chemical compound OCCNC(=O)C1=CC=CC=N1 SIUMVFGIULAFNI-UHFFFAOYSA-N 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 150000002828 nitro derivatives Chemical class 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 229940100692 oral suspension Drugs 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 208000020016 psychiatric disease Diseases 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 229960005137 succinic acid Drugs 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000013589 supplement Substances 0.000 description 2
- 230000009897 systematic effect Effects 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- SKYZYDSNJIOXRL-BTQNPOSSSA-N (6ar)-6-methyl-5,6,6a,7-tetrahydro-4h-dibenzo[de,g]quinoline-10,11-diol;hydrochloride Chemical compound Cl.C([C@H]1N(C)CC2)C3=CC=C(O)C(O)=C3C3=C1C2=CC=C3 SKYZYDSNJIOXRL-BTQNPOSSSA-N 0.000 description 1
- AXDCNQMJSHRGHN-UHFFFAOYSA-N (methoxycarbonylamino)methyl methanimidate Chemical compound COC(=O)NCOC=N AXDCNQMJSHRGHN-UHFFFAOYSA-N 0.000 description 1
- SILNNFMWIMZVEQ-UHFFFAOYSA-N 1,3-dihydrobenzimidazol-2-one Chemical compound C1=CC=C2NC(O)=NC2=C1 SILNNFMWIMZVEQ-UHFFFAOYSA-N 0.000 description 1
- HWSOUGJPWCIYGW-UHFFFAOYSA-N 1-(1-benzylpiperidin-4-yl)benzimidazol-2-amine Chemical compound NC1=NC2=CC=CC=C2N1C(CC1)CCN1CC1=CC=CC=C1 HWSOUGJPWCIYGW-UHFFFAOYSA-N 0.000 description 1
- OGFAWKRXZLGJSK-UHFFFAOYSA-N 1-(2,4-dihydroxyphenyl)-2-(4-nitrophenyl)ethanone Chemical compound OC1=CC(O)=CC=C1C(=O)CC1=CC=C([N+]([O-])=O)C=C1 OGFAWKRXZLGJSK-UHFFFAOYSA-N 0.000 description 1
- YZKQBUWNKRTZQC-UHFFFAOYSA-N 1-benzyl-n-(2-nitrophenyl)piperidin-4-amine Chemical compound [O-][N+](=O)C1=CC=CC=C1NC1CCN(CC=2C=CC=CC=2)CC1 YZKQBUWNKRTZQC-UHFFFAOYSA-N 0.000 description 1
- BFCFYVKQTRLZHA-UHFFFAOYSA-N 1-chloro-2-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1Cl BFCFYVKQTRLZHA-UHFFFAOYSA-N 0.000 description 1
- BZCOSCNPHJNQBP-UHFFFAOYSA-N 2,3-dihydroxybut-2-enedioic acid Chemical compound OC(=O)C(O)=C(O)C(O)=O BZCOSCNPHJNQBP-UHFFFAOYSA-N 0.000 description 1
- ZYHQGITXIJDDKC-UHFFFAOYSA-N 2-[2-(2-aminophenyl)ethyl]aniline Chemical group NC1=CC=CC=C1CCC1=CC=CC=C1N ZYHQGITXIJDDKC-UHFFFAOYSA-N 0.000 description 1
- WJAXXWSZNSFVNG-UHFFFAOYSA-N 2-bromoethanamine;hydron;bromide Chemical compound [Br-].[NH3+]CCBr WJAXXWSZNSFVNG-UHFFFAOYSA-N 0.000 description 1
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical group CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- OFTKFKYVSBNYEC-UHFFFAOYSA-N 2-furoyl chloride Chemical compound ClC(=O)C1=CC=CO1 OFTKFKYVSBNYEC-UHFFFAOYSA-N 0.000 description 1
- SOYVSEQEWSKZDK-UHFFFAOYSA-N 2-hydroxy-2-phenylacetic acid;methanesulfonic acid Chemical compound CS(O)(=O)=O.OC(=O)C(O)C1=CC=CC=C1 SOYVSEQEWSKZDK-UHFFFAOYSA-N 0.000 description 1
- LODHFNUFVRVKTH-ZHACJKMWSA-N 2-hydroxy-n'-[(e)-3-phenylprop-2-enoyl]benzohydrazide Chemical compound OC1=CC=CC=C1C(=O)NNC(=O)\C=C\C1=CC=CC=C1 LODHFNUFVRVKTH-ZHACJKMWSA-N 0.000 description 1
- UWYVPFMHMJIBHE-UHFFFAOYSA-N 2-hydroxybut-2-enedioic acid Chemical compound OC(=O)C=C(O)C(O)=O UWYVPFMHMJIBHE-UHFFFAOYSA-N 0.000 description 1
- FRPNIZFUGKDSSS-UHFFFAOYSA-N 2-n-piperidin-4-ylbenzene-1,2-diamine Chemical compound NC1=CC=CC=C1NC1CCNCC1 FRPNIZFUGKDSSS-UHFFFAOYSA-N 0.000 description 1
- KLGQWSOYKYFBTR-UHFFFAOYSA-N 2-nitrobenzamide Chemical compound NC(=O)C1=CC=CC=C1[N+]([O-])=O KLGQWSOYKYFBTR-UHFFFAOYSA-N 0.000 description 1
- AGQILSDICOUDEO-UHFFFAOYSA-N 3-ethyl-2-nitrobenzamide Chemical compound C(C)C=1C(=C(C(=O)N)C=CC1)[N+](=O)[O-] AGQILSDICOUDEO-UHFFFAOYSA-N 0.000 description 1
- PGALEJNMXOZBGY-UHFFFAOYSA-N 3-piperidin-4-yl-1,2-dihydrobenzimidazole Chemical compound C1NC2=CC=CC=C2N1C1CCNCC1 PGALEJNMXOZBGY-UHFFFAOYSA-N 0.000 description 1
- NCPDIORJCOFBTD-UHFFFAOYSA-N 4-(4-chlorophenyl)-3-methylpiperidin-4-ol Chemical compound CC1CNCCC1(O)C1=CC=C(Cl)C=C1 NCPDIORJCOFBTD-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- WRFYIYOXJWKONR-UHFFFAOYSA-N 4-bromo-2-methoxyaniline Chemical compound COC1=CC(Br)=CC=C1N WRFYIYOXJWKONR-UHFFFAOYSA-N 0.000 description 1
- YLUCXHMYRQUERW-UHFFFAOYSA-N 4-fluoro-2-nitrobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C=C1[N+]([O-])=O YLUCXHMYRQUERW-UHFFFAOYSA-N 0.000 description 1
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 1
- LVQFHDAKZHGEAJ-UHFFFAOYSA-M 4-methylbenzenesulfonate Chemical compound [CH2]C1=CC=C(S([O-])(=O)=O)C=C1 LVQFHDAKZHGEAJ-UHFFFAOYSA-M 0.000 description 1
- XUPXMIAWKPTZLZ-UHFFFAOYSA-N 4-methylhexan-2-one Chemical compound CCC(C)CC(C)=O XUPXMIAWKPTZLZ-UHFFFAOYSA-N 0.000 description 1
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- YRQDTSXAKBGGJC-UTCJRWHESA-O C/[NH+]=C\C1[IH]C1 Chemical compound C/[NH+]=C\C1[IH]C1 YRQDTSXAKBGGJC-UTCJRWHESA-O 0.000 description 1
- PCBZRNYXXCIELG-WYFCWLEVSA-N COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 Chemical compound COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 PCBZRNYXXCIELG-WYFCWLEVSA-N 0.000 description 1
- XZMCDFZZKTWFGF-UHFFFAOYSA-N Cyanamide Chemical compound NC#N XZMCDFZZKTWFGF-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WZELXJBMMZFDDU-UHFFFAOYSA-N Imidazol-2-one Chemical compound O=C1N=CC=N1 WZELXJBMMZFDDU-UHFFFAOYSA-N 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- NMMIHXMBOZYNET-UHFFFAOYSA-N Methyl picolinate Chemical compound COC(=O)C1=CC=CC=N1 NMMIHXMBOZYNET-UHFFFAOYSA-N 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 238000007126 N-alkylation reaction Methods 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000108664 Nitrobacteria Species 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-N Salicylic acid Natural products OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 1
- 206010039897 Sedation Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- SOIFLUNRINLCBN-UHFFFAOYSA-N ammonium thiocyanate Chemical compound [NH4+].[S-]C#N SOIFLUNRINLCBN-UHFFFAOYSA-N 0.000 description 1
- 230000003354 anti-apomorphinic effect Effects 0.000 description 1
- 230000001062 anti-nausea Effects 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 229960003990 apomorphine hydrochloride Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- REWFUSHVOGNMCQ-UHFFFAOYSA-N azanium;iron;chloride Chemical compound [NH4+].[Cl-].[Fe] REWFUSHVOGNMCQ-UHFFFAOYSA-N 0.000 description 1
- ACPCIEDHXWSQIW-UHFFFAOYSA-N aziridin-1-yl(thiophen-2-yl)methanone Chemical group C=1C=CSC=1C(=O)N1CC1 ACPCIEDHXWSQIW-UHFFFAOYSA-N 0.000 description 1
- WOKMXDQPPPOWBG-UHFFFAOYSA-N aziridin-1-yl-(2,5-dichlorophenyl)methanone Chemical compound ClC1=CC=C(Cl)C(C(=O)N2CC2)=C1 WOKMXDQPPPOWBG-UHFFFAOYSA-N 0.000 description 1
- FAYTWMURNZSRFC-UHFFFAOYSA-N aziridin-1-yl-(2-hydroxyphenyl)methanone Chemical group OC1=CC=CC=C1C(=O)N1CC1 FAYTWMURNZSRFC-UHFFFAOYSA-N 0.000 description 1
- WJMOWGFGRONZPA-UHFFFAOYSA-N aziridin-1-yl-(4-fluorophenyl)methanone Chemical compound C1=CC(F)=CC=C1C(=O)N1CC1 WJMOWGFGRONZPA-UHFFFAOYSA-N 0.000 description 1
- IAVXPMUEHXYCGG-UHFFFAOYSA-N aziridin-1-yl-(5-chloro-2-methoxy-4-nitrophenyl)methanone Chemical compound COC1=CC([N+]([O-])=O)=C(Cl)C=C1C(=O)N1CC1 IAVXPMUEHXYCGG-UHFFFAOYSA-N 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- QDKFWHFXUNNUGZ-UHFFFAOYSA-N benzamide;hydron;bromide Chemical compound Br.NC(=O)C1=CC=CC=C1 QDKFWHFXUNNUGZ-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- MYONAGGJKCJOBT-UHFFFAOYSA-N benzimidazol-2-one Chemical compound C1=CC=CC2=NC(=O)N=C21 MYONAGGJKCJOBT-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- SNIABFMMCKVXSY-UHFFFAOYSA-N benzoylazanium;chloride Chemical compound Cl.NC(=O)C1=CC=CC=C1 SNIABFMMCKVXSY-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 230000001055 chewing effect Effects 0.000 description 1
- 239000000084 colloidal system Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- MKJDUHZPLQYUCB-UHFFFAOYSA-N decan-4-one Chemical compound CCCCCCC(=O)CCC MKJDUHZPLQYUCB-UHFFFAOYSA-N 0.000 description 1
- DIOQZVSQGTUSAI-NJFSPNSNSA-N decane Chemical compound CCCCCCCCC[14CH3] DIOQZVSQGTUSAI-NJFSPNSNSA-N 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 239000000686 essence Substances 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- HCPOCMMGKBZWSJ-UHFFFAOYSA-N ethyl 3-hydrazinyl-3-oxopropanoate Chemical compound CCOC(=O)CC(=O)NN HCPOCMMGKBZWSJ-UHFFFAOYSA-N 0.000 description 1
- DCBDUYKOEGKBBZ-UHFFFAOYSA-N ethyl n-[2-[4-(2-oxo-3h-benzimidazol-1-yl)piperidin-1-yl]ethyl]carbamate Chemical compound C1CN(CCNC(=O)OCC)CCC1N1C(=O)NC2=CC=CC=C21 DCBDUYKOEGKBBZ-UHFFFAOYSA-N 0.000 description 1
- NUMIBNFCMMUFSV-UHFFFAOYSA-N ethyl n-[2-[4-(5-chloro-2-oxo-3h-benzimidazol-1-yl)piperidin-1-yl]ethyl]carbamate Chemical compound C1CN(CCNC(=O)OCC)CCC1N1C(=O)NC2=CC(Cl)=CC=C21 NUMIBNFCMMUFSV-UHFFFAOYSA-N 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 150000002338 glycosides Chemical class 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000002650 habitual effect Effects 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- QMEZUZOCLYUADC-UHFFFAOYSA-N hydrate;dihydrochloride Chemical compound O.Cl.Cl QMEZUZOCLYUADC-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 150000004694 iodide salts Chemical group 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000012263 liquid product Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical group O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- APHVGKYWHWFAQV-UHFFFAOYSA-N methyl 1-methylpyrrole-2-carboxylate Chemical compound COC(=O)C1=CC=CN1C APHVGKYWHWFAQV-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- TTWJBBZEZQICBI-UHFFFAOYSA-N metoclopramide Chemical compound CCN(CC)CCNC(=O)C1=CC(Cl)=C(N)C=C1OC TTWJBBZEZQICBI-UHFFFAOYSA-N 0.000 description 1
- 229960004503 metoclopramide Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- LOCPURVUTOUVKC-UHFFFAOYSA-N n-(2-chloroethyl)pyridine-2-carboxamide;hydrochloride Chemical compound Cl.ClCCNC(=O)C1=CC=CC=N1 LOCPURVUTOUVKC-UHFFFAOYSA-N 0.000 description 1
- WKGCTLNRIMYYPG-UHFFFAOYSA-N n-(2-chloroethyl)pyridine-3-carboxamide;hydrochloride Chemical compound Cl.ClCCNC(=O)C1=CC=CN=C1 WKGCTLNRIMYYPG-UHFFFAOYSA-N 0.000 description 1
- JHBTVPLZRMXZKD-UHFFFAOYSA-N n-(pyridin-2-ylmethyl)benzamide Chemical compound C=1C=CC=CC=1C(=O)NCC1=CC=CC=N1 JHBTVPLZRMXZKD-UHFFFAOYSA-N 0.000 description 1
- RXGGJJGJPVTQDB-UHFFFAOYSA-N n-[2-[4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl]ethyl]-4-fluoro-2-nitrobenzamide Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCNC(=O)C1=CC=C(F)C=C1[N+]([O-])=O RXGGJJGJPVTQDB-UHFFFAOYSA-N 0.000 description 1
- DIOQZVSQGTUSAI-UHFFFAOYSA-N n-butylhexane Natural products CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 1
- SDIDYFBTIZOPLA-UHFFFAOYSA-N n-ethylbenzamide Chemical compound CCNC(=O)C1=CC=CC=C1 SDIDYFBTIZOPLA-UHFFFAOYSA-N 0.000 description 1
- LKPFBGKZCCBZDK-UHFFFAOYSA-N n-hydroxypiperidine Chemical compound ON1CCCCC1 LKPFBGKZCCBZDK-UHFFFAOYSA-N 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000004526 pharmaceutical effect Effects 0.000 description 1
- SCWKRWCUMCMVPW-UHFFFAOYSA-N phenyl n-methylcarbamate Chemical compound CNC(=O)OC1=CC=CC=C1 SCWKRWCUMCMVPW-UHFFFAOYSA-N 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- IBBMAWULFFBRKK-UHFFFAOYSA-N picolinamide Chemical compound NC(=O)C1=CC=CC=N1 IBBMAWULFFBRKK-UHFFFAOYSA-N 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- NJBNJRILTKLNQZ-UHFFFAOYSA-N propyl 4-hydroxybenzoate Chemical compound CCCOC(=O)C1=CC=C(O)C=C1.CCCOC(=O)C1=CC=C(O)C=C1 NJBNJRILTKLNQZ-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 230000036280 sedation Effects 0.000 description 1
- 238000010956 selective crystallization Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- AYGJDUHQRFKLBG-UHFFFAOYSA-M sodium;1,1-dioxo-1,2-benzothiazol-3-olate;dihydrate Chemical compound O.O.[Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 AYGJDUHQRFKLBG-UHFFFAOYSA-M 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- BGRJTUBHPOOWDU-UHFFFAOYSA-N sulpiride Chemical compound CCN1CCCC1CNC(=O)C1=CC(S(N)(=O)=O)=CC=C1OC BGRJTUBHPOOWDU-UHFFFAOYSA-N 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 125000004360 trifluorophenyl group Chemical group 0.000 description 1
- UJMBCXLDXJUMFB-UHFFFAOYSA-K trisodium;5-oxo-1-(4-sulfonatophenyl)-4-[(4-sulfonatophenyl)diazenyl]-4h-pyrazole-3-carboxylate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)C1=NN(C=2C=CC(=CC=2)S([O-])(=O)=O)C(=O)C1N=NC1=CC=C(S([O-])(=O)=O)C=C1 UJMBCXLDXJUMFB-UHFFFAOYSA-K 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/52—Oxygen atoms attached in position 4 having an aryl radical as the second substituent in position 4
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
내용 없음.No content.
Description
본 발명은 N-[(1-피페리디닐)알킬/아릴카르복스아마이드 유도체에 관한 것이다.The present invention relates to N-[(1-piperidinyl) alkyl / arylcarboxamide derivatives.
N-[(1-피페리디닐)알킬]벤즈아마이드를 포함한 어떤 N-[(디알킬아미노)알킬]벤즈아마이드와 약학적 성질을 갖인 어떤 N-[(2-피로디닐)메틸]벤즈아마이드가 문헌에 수록되였다. 이러한 류의 공지된 화합물의 예를들면 통상 메토크로프라미드라 칭하고 구토 억제제로서 유용한 4-아미노-5-크로로-N-[2-디에틸아미노)에틸]-2-메톡시벤즈아마이드와 통상 술피리드로 칭하는 구토억제 및 신경 작용제로서 유용한 5-아미노술포닐-N-[(1-에틸-2-피로리디닐)메틸]-2-메톡시벤즈아마이드가 있다.Any N-[(2-pyridinyl) methyl] benzamide having pharmaceutical properties with any N-[(dialkylamino) alkyl] benzamide, including N-[(1-piperidinyl) alkyl] benzamide In the literature. Examples of known compounds of this class are, for example, 4-amino-5-chloro-N- [2-diethylamino) ethyl] -2-methoxybenzamide, commonly referred to as metoclopramide and useful as antiemetic agents. 5-aminosulfonyl-N-[(1-ethyl-2-pyrrolidinyl) methyl] -2-methoxybenzamide is useful as an antiemetic and neurological agent called sulfide.
본 발명의 화합물들은 알킬-측쇄에 부착된 치환 피페리딘 핵의 성질에 의하여 종래의 화합물들과 상이하다. 종래의 다양한 화합물들은 하기 문헌에서 찾을 수 있다.The compounds of the present invention differ from conventional compounds by the nature of the substituted piperidine nucleus attached to the alkyl-side chain. Various conventional compounds can be found in the literature.
C.A., 59,11358C ;C.A., 59,11358 C;
미국 특허 3,342,826호U.S. Patent 3,342,826
C.A., 71, P811 413C.C.A., 71, P811 413C.
본 발명의 신규 N-[(1-피페리디닐)알킬]아릴카르복스아마이드 유도체는 하기 구조식으로 나타낼 수 있다.The novel N-[(1-piperidinyl) alkyl] arylcarboxamide derivatives of the present invention can be represented by the following structural formula.
상기 식중에서In the above formula
Ar는 페닐, 치환된 페닐, 2-티에닐, 2-푸라닐, 피리디닐 및 1-메틸-2-피로릴로 부터 선택한 아릴기인데 이중 치환된 페닐은 할로, 저급알킬, 저급알킬옥시, 트리플루오로메틸, 니트로, 하이드록시, 아미노, 저급알킬카르보닐옥시 및 저급알킬카르보닐아미노로 부터 독립적으로 선택한 1-3개의 치환기를 갖인 페닐이고 상기 치환기가 하나 이상일때 이들의 단 하나는 하이드록시, 아미노, 저급 알킬카르보닐옥시 및 저급알킬카르보닐아미노로부터 선택한 것임.Ar is an aryl group selected from phenyl, substituted phenyl, 2-thienyl, 2-furanyl, pyridinyl and 1-methyl-2-pyrrolyl, wherein the substituted phenyl is halo, lower alkyl, lower alkyloxy, trifluoro Phenyl having 1-3 substituents independently selected from rommethyl, nitro, hydroxy, amino, lower alkylcarbonyloxy and lower alkylcarbonylamino and when one or more of these substituents is hydroxy, amino , Lower alkylcarbonyloxy and lower alkylcarbonylamino.
R는 수로와 저급알킬로부터 선택한 것이며R is selected from channel and lower alkyl
n는 2-3의 정수이고n is an integer from 2-3
![]()
![]()
a) 하기 일반식을 갖인 라디칼a) a radical having the general formula

식 중에서In the formula
R1와 R2는 수로, 할로, 저급알킬 및 트리플루오로메틸로 부터 독립적으로 선택한 것임.R 1 and R 2 are independently selected from channel, halo, lower alkyl and trifluoromethyl.
b) 하기 일반식을 갖인 라디칼b) radicals having the general formula

식중에서In the food
R3과 R4는 수소, 할로, 저급알킬 및 트리플루오로메틸로 부터 각각 선택한 것이고; M는 수소, 저급알킬, 저급알킬카르보닐 및 2-시아노에틸로 부터 선택한 것이며; Y는 O,S 및 저급알킬카르보닐이마노로 부터 선택한 것이고 점선은 피페리딘 핵의 3-및 4-탄소원자 사이의 이중 결합이 임의적 임을 나타내며 만일 3-및 4-탄소원자간의 이중 결합이 존재할때 상기 Y는 0이고 M는 수소임.R 3 and R 4 are each selected from hydrogen, halo, lower alkyl and trifluoromethyl; M is selected from hydrogen, lower alkyl, lower alkylcarbonyl and 2-cyanoethyl; Y is selected from O, S and lower alkylcarbonylimano, and the dashed line indicates that the double bond between the 3- and 4-carbon atoms of the piperidine nucleus is optional and if the double bond between the 3- and 4-carbon atoms When present, Y is 0 and M is hydrogen.
c) 하기 일반식을 갖인 라디칼c) radicals having the general formula
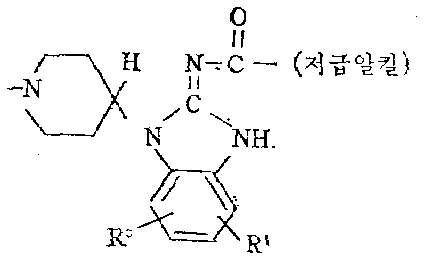
식중에서In the food
R3및 R4는 수소, 할로, 저급알킬 및 트리풀루오로메틸로 부터 각각 독립적으로 선택한 것임R 3 and R 4 are each independently selected from hydrogen, halo, lower alkyl, and trifluomethyl
d) 하기 일반식을 갖인 라디칼d) radicals having the general formula

식중에서In the food
R5는 수소와 메틸로 부터 선택한 것이고;R 5 is selected from hydrogen and methyl;
R6는 수소와 할로로 부터 선택한 것이며R 6 is selected from hydrogen and halo
R7은 수소, 할로, 저급알킬 및 트리풀루오로메틸임.R 7 is hydrogen, halo, lower alkyl and trifluomethyl.
본 명세서에 사용된 "저급알킬"은 직쇄나 분지쇄이고 1-5개의 탄소 원자를 갖이며 예컨데 메틸, 에틸, 프로필, 1-메틸에틸, 부틸, 펜틸이다. "할로"란 용어는 127 이하의 원자량을 갖인 할로겐으로서 예컨데 풀루오로, 크로로, 브로모 및 요오도 등이 있다.As used herein, "lower alkyl" is straight or branched and has 1-5 carbon atoms, for example methyl, ethyl, propyl, 1-methylethyl, butyl, pentyl. The term "halo" refers to halogens having an atomic weight of 127 or less, such as pulluro, chromo, bromo and iodo.
Ar와![]()
![]()
![]()
![]()
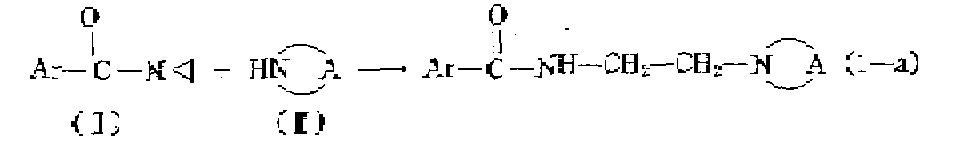
상기 반응은 적당한 반응-불활성 유기 용매 예컨데 메탄올, 에탄올, 프로판올, 부탄올 등과 같은 알콜; 메틸벤젠, 디메틸벤젠 등과 같은 방향족 탄화수소; 4-메틸-2-펜타논 같은 케톤, 1,4-디옥산, 1,1'-옥시비 스에탄과 같은 에테르; N,N-디메틸포름아마이드; 니트로벤젠 및 이들 용매의 혼합물 내에서 실시한다. 반응 속도를 증가시키기 위하여 상승온도가 적당하며 특히 반응혼합물의 환류 온도에서 실시하는 것이 좋다. 다음 공정에서 반응 생성물은 매체로 부터 분리하며 필요에 따라 당해 분야의 공지된 방법을 적용하여 더 정제한다.The reaction may be carried out with a suitable reaction-inert organic solvent such as methanol, ethanol, propanol, butanol and the like; Aromatic hydrocarbons such as methylbenzene, dimethylbenzene and the like; Ketones such as 4-methyl-2-pentanone, ethers such as 1,4-dioxane, 1,1'-oxybisethane; N, N-dimethylformamide; It is carried out in a mixture of nitrobenzene and these solvents. In order to increase the reaction rate, an elevated temperature is suitable, particularly at the reflux temperature of the reaction mixture. In the next process the reaction product is separated from the medium and further purified by application of methods known in the art as required.
식중 R가 저급알킬이고 n가 3인 일반식(1)의 화합물들은 또한 식중 Ar, R 및 n가 상술한 바와같고 X가 예컨데 할로, 메탄술포닐, 4-메틸벤젠술포닐 등과 같은 해당하는 알콜로 부터 유도된 적당한 반응성 에스테르기인 일반식(Ⅳ)의 적당한 반응성 에스테르와 일반식(Ⅲ)의 적당한 피페리딘 유도체와 반응 시킴에 의하여 제조한다.The compounds of formula (1) wherein R is lower alkyl and n is 3 are also the corresponding alcohols, wherein Ar, R and n are as described above and X is for example halo, methanesulfonyl, 4-methylbenzenesulfonyl and the like. It is prepared by reacting a suitable reactive ester of formula (IV) which is a suitable reactive ester group derived from and a suitable piperidine derivative of formula (III).
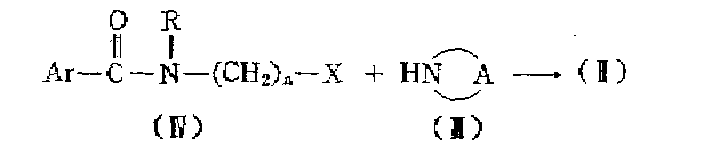
상기 반응은 예컨데 N,N-디메틸포름 아마이드, N,N-디메틸아세트아마이드, 4-메틸-2-펜타논, 2-프로판올, 메탄올, 에탄올, 2-프로판올등과 같은 적당한 유기 용매내에서 실시한다. 적당한 염기 예컨데 알카리 금속이나 토금속의 탄산화물이나 중탄산화물의 첨가는 반응 중 유리되는 산을 제거하는데 이용된다. 반응속도를 증가시키기 위하여 다소 상승된 온도가 적당하여 특히 반응은 환류 온도에서 실시한다.The reaction is carried out in a suitable organic solvent such as, for example, N, N-dimethylformamide, N, N-dimethylacetamide, 4-methyl-2-pentanone, 2-propanol, methanol, ethanol, 2-propanol and the like. . The addition of suitable bases such as alkali or earth metal carbonates or bicarbonates is used to remove the free acid during the reaction. A slightly elevated temperature is suitable to increase the reaction rate, in particular the reaction is carried out at reflux temperature.
일반식(Ⅰ)의 화합물을 제조하는 또 다른 방법은 아릴할라이드와 아민으로 부터 출발하여 아마이드를 제조하는 공지된 방법에 따라 일반식(Ⅴ)의 적당히 치환된 아로일할라이드를 일반식(Ⅵ)의 적당한 아민과 반응시키는 것이다.Another method for preparing a compound of formula (I) is to form an appropriately substituted aroyl halide of formula (V) according to the known method for preparing amides starting from aryl halides and amines. To react with a suitable amine.

상기 반응은 적당한 반응-불활성 유기 용매 예컨데 메탄올, 에탄올, 프로판올, 부탄올 등과 같은 저급 알칸올; 벤젠, 메틸벤젠, 디메틸벤젠 등과 같은 방향족 탄화수소; 4- 메틸-2-펜타논과 같은 케톤; 1,4-디옥산, 1,1'-옥시비스에탄등과 같은 에테르; N,N-디메틸포름아마이드; 니트로벤젠 또는 이들의 혼합물내에서 반응물들을 함께 환류 시킴에 의하여 실시한다. 반응 속도를 증가 시키기 위하여 상승온도가 적당하며 특히 반응 혼합물의 환류온도에서 실시한다.The reaction may be carried out with a suitable reaction-inert organic solvent such as lower alkanols such as methanol, ethanol, propanol, butanol and the like; Aromatic hydrocarbons such as benzene, methylbenzene, dimethylbenzene and the like; Ketones such as 4-methyl-2-pentanone; Ethers such as 1,4-dioxane, 1,1'-oxybisethane and the like; N, N-dimethylformamide; By reacting the reactants together in nitrobenzene or mixtures thereof. In order to increase the reaction rate, an elevated temperature is suitable, especially at the reflux temperature of the reaction mixture.
식중 Ar가 2 내에 아미노균만을 또는 다른 치환기들과 함께 아미노군을 갖인 페닐군인 일반식(Ⅰ)의 화합물들은 해당하는 니트로-치환된 유사물을 공지된 공정에 따라 예컨데 라네이-닉켈 활탄상의 팔라듐이나 이산화 백금 촉매를 사용한 촉매적 수소화 또는 니트로 화합물을 철-염화암모니움이나 아연-식초산으로 처리함에 의하여 니트로를 아민으로 환원 사킴에 의하여 제조한다.The compounds of formula (I) wherein Ar is a group of phenyls having amino groups alone or with other substituents within 2, can be used to prepare the corresponding nitro-substituted analogues according to known processes, for example, palladium on ranei-nickel glide Or nitro is reduced to amine by treating catalytic hydrogenation or nitro compounds with a platinum dioxide catalyst or iron-ammonium chloride or zinc-vinegar acid.
식중에 Ar가 2 내에 저급 알킬카르보닐아미노 또는 저급 알킬카르보닐옥시를 갖인 페닐군인 일반식(1)의 화합물들은 해당하는 아미노나 하이드록시 치환된 유도체를 예컨데 적당한 저급 알킬 카르복실산으로부터 유도된 할라이드 또는 무수물 같은 적당한 아실화제로 아실화 시킴에 의하여 용이하게 유도된다. 아실화 반응은 물내에서 적당한 저급 알킬카르복실산을 사용하여 실시한다.Wherein compounds of formula (1), wherein Ar is a group of phenyls having lower alkylcarbonylamino or lower alkylcarbonyloxy in 2, are used for the halides derived from the appropriate lower alkyl carboxylic acids, for example with the corresponding amino or hydroxy substituted derivatives. Or by acylation with a suitable acylating agent such as anhydride. The acylation reaction is carried out using a suitable lower alkylcarboxylic acid in water.
하기 일반식(1-b)로 표시되는 일반식(1)의 화합물들은 하기 일반식(1-c)의 해당하는 치환되지 않은 화합물을 적당한 저급 알킬 카르복실산으로 부터 유도된 적당한 아실화제 예컨데 아실할라이드나 무수물로 아실화 시킴에 의하여 제조할 수 있다.Compounds of the general formula (1) represented by the following general formula (1-b) may be prepared by appropriate acylating agents derived from suitable lower alkyl carboxylic acids, such as acyl, with the corresponding unsubstituted compounds of the general formula (1-c) It can be prepared by acylating with a halide or anhydride.

상기 아실화 반응은 예컨데 벤젠, 메틸벤젠, 디메틸벤젠등과 같은 적당한 유기 용매내에서 적당한 무수물을 사용하여 실시한다.The acylation reaction is carried out using a suitable anhydride in a suitable organic solvent such as benzene, methylbenzene, dimethylbenzene and the like.
하기 일반식(1-d)로 표시되는 일반식(1)화합물들은 하기 일반식(Ⅶ)의 적당한 다아민을 예컨데 이의 유화 탄소, 티오우레아, 카르보노티오디크로라이드, 암모니움 티오시아네이트 등과 같은 유황-함유 환상화상제로서 고리 폐쇄 시킴에 의하여 제조한다.The compounds of the general formula (1) represented by the following general formula (1-d) include suitable polyamines of the following general formula (i), for example, emulsified carbons, thiourea, carbonothiodichromide, ammonium thiocyanate, and the like. Prepared by ring closure as the same sulfur-containing cyclic burner.

하기 일반식(1-e)로 표시되는 일반식(1)의 화합물들은 일반식(1-d)의 해당하는 화합물을 표준 S-알킬화 공정에 따라 S-알킬화 시킴에 의하여 예를들면(1-d)를 적당한 할로 저급알칸이나 적당한 디(저급알킬) 설페이트와 반응 시킴에 의하여 제조한다.Compounds of the general formula (1) represented by the following general formula (1-e) are exemplified by S-alkylation of the corresponding compound of the general formula (1-d) according to a standard S-alkylation process. d) is prepared by reacting with a suitable halo lower alkane or a suitable di (lower alkyl) sulfate.

상기 제조에 사용된 시발 물질들은 후술한 하기 공정에 따라 얻는다.Starting materials used in the preparation are obtained according to the following process described below.
공지된 화합물인 일반식(Ⅱ)의 아로일아지리딘 중간체는 문헌에 기술된 공지된 공정을 적용시킴에 의하여 예를들면 반응중 유리되는 산을 중화 시키기에 의하여 적당한 염기의 존재하에 일반식(Ⅴ)의 아로일할라이드를 아지리딘(Ⅷ)과 반응 시킴에 의하여 쉽게 제조한다. 반응은 예컨데 물과 트리크로토메탄의 혼합물 같은 적당한 용매계 내에서 실시하며 상기 반응은 하기 반응식으로 설명된다.Aroyl aziridine intermediates of the general formula (II), which are known compounds, are formulated in the presence of a suitable formula (V) in the presence of a suitable base, for example by neutralizing the acid liberated during the reaction by applying known processes described in the literature. Aroyl halides of) are readily prepared by reacting with aziridine. The reaction is carried out in a suitable solvent system such as, for example, a mixture of water and tricrotomethane, which reaction is illustrated by the following scheme.

약간은 문헌에 기술된 일반식(Ⅳ)의 중간체는 예컨데 표준 알로일화 공정에 따라 일반식(Ⅸ)의 적당한 아미노알칸올을 적당한 알로일 할라이드(Ⅴ)와 또는 일반식(Ⅹ)의 적당한 저급 알킬아릴카르복시레이트와 N-알로일화시킨 다음 수득한 (XI)의 알킬 측쇄상의 수산기를 공지된 공정에 따라 반응성 에스테르로 전환시킴에 의하여 제조한다. 할라이드의 제조에서 예컨데 카르보니크 디크로라이드, 술피닐 크로라이드, 술퍼릴크로라이드, 5염화인, 5 브롬화인, 포스포릴크로라이드 같은 통상 할로겐화제를 사용한다. 반응성 에스테르가 요오다이드일때 할로겐을 요오딘으로 대치시킴에 의하여 해당하는 크로라이드나 요오다이드로부터 제조한다. 메탄술포네이트와 4-메틸벤젠술포네이트 같은 기타 반응성 에스테르들은 알콜을 예컨데 메탄술포닐크로라이드 및 4-메틸벤젠술포닐크로라이드와 각각 반응시킴에 의하여 수득한다. 상기 반응들은 하기 반응식으로 설명될 수 있다.Some intermediates of general formula (IV) described in the literature can be prepared by, for example, the appropriate aminoalkanols of general formula (A) with the appropriate alloyl halide (V) or with the appropriate lower alkyl of general formula (III) N-alloylated with arylcarboxylate, followed by conversion of the hydroxyl groups on the alkyl side chains obtained (XI) to reactive esters according to known processes. In the preparation of halides, conventional halogenating agents are used, for example carbonic dichloride, sulfinyl chromide, sulphryl chloride, phosphorus pentachloride, phosphorus pentabromide, phosphoryl chloride. It is prepared from the corresponding chromide or iodide by replacing halogen with iodine when the reactive ester is iodide. Other reactive esters such as methanesulfonate and 4-methylbenzenesulfonate are obtained by reacting alcohols with, for example, methanesulfonyl chloride and 4-methylbenzenesulfonyl chloride. The reactions can be described by the following schemes.

Ⅹ가 할로인 일반식(Ⅳ)의 화합물(Ⅳ-a)은 예컨데 N,N-디메틸포름아마이드(DMF)와 N,N-디메틸아세트아마이드 각은 적당한 용매 내에서 N,N-디에틸에탄아민 같은 적당한 염기의 존재하에 적당한 아로일 할라이드(Ⅴ)를 적당한 할로-알칸아민(XⅡ)과 반응 시킴에 의하여 한 단계에서 얻는다.Compound (IV-a) of Formula (IV), wherein the valent is halo, is, for example, N, N-dimethylformamide (DMF) and N, N-dimethylacetamide angles in N, N-diethylethanamine in a suitable solvent. A suitable aroyl halide (V) is obtained in one step by reaction with a suitable halo-alkanamine (XII) in the presence of the same suitable base.

상기 반응을 알카리 메채내에서 실시하고 식중 R가 수소이며 n가 2인 일반식(XⅡ)의 중간체를 사용함에 의하여 일반식(Ⅱ)의 해당하는 마지리딘 수득한다.The reaction is carried out in an alkali medium and the corresponding marziridine of formula (II) is obtained by using an intermediate of formula (XII) wherein R is hydrogen and n is 2.
일반식(Ⅵ)의 중간체들은 하기와 같이 제조한다.Intermediates of formula (VI) are prepared as follows.
일반식(XⅢ)의 적당한 저급 알킬 N-(할로알킬) 카르바메이트를 일반식(Ⅲ)의 적당한 피페리딘 유도체와 통상의 N-알킬화 공정에 따라 반응시킴에 의하여 예컨데 반응물들을 함께 적당한 반응 불활성 유기용매 즉 메탄올, 에탄올 등과 같은 저급 알칸올내에서 가열함에 의하여 반응시켜 일반식(XIV)의 카르바메이트를 수득한 다음 이것을 산이나 알카리 가수분해 시키면 탈카르복실화가 일어나 원하는 일반식(Ⅵ)의 중간체를 수득한다.By reacting a suitable lower alkyl N- (haloalkyl) carbamate of the general formula (XIII) with a suitable piperidine derivative of the general formula (III) according to a conventional N-alkylation process, for example, the reactants may be reacted together appropriately. Reaction by heating in an organic solvent, i.e., lower alkanol such as methanol or ethanol, to give a carbamate of the general formula (XIV), and then hydrolyzing the acid or alkali to decarboxylation, resulting in the desired intermediate of the general formula (VI). To obtain.

알카리 가수분해를 실시하는데 있어서 수산화 나트륨이나 칼륨 같은 금속 염기가 사용된다. 산 가수분해에 사용될 수 있는 산은 염산, 브롬산, 황산, 인산등과 같은 강한 무기산을 포함한다.In carrying out alkali hydrolysis, metal bases such as sodium hydroxide and potassium are used. Acids that can be used for acid hydrolysis include strong inorganic acids such as hydrochloric acid, bromic acid, sulfuric acid, phosphoric acid and the like.
중간체(Ⅵ) 내![]()
![]()
일반식(Ⅵ)의 중간체들은 신규이며 일반식(1)의 화합물을 제조하는데 유용한 중간체로서 본 발명의 부가적인 형테이다.Intermediates of formula (VI) are additional forms of the invention as novel and useful intermediates for preparing compounds of formula (1).
일반식(Ⅶ)의 중간체는 일반식(XV)의 N-(2-니트로페닐)-4-피페리딘아민을 일반식(Ⅱ)의 아지리딘 또는 일반식(Ⅳ)의 반응성 에스테르와 반응시켜 아로일 알킬을 일반식(XV) N-(2-니트로페닐)-4-피페리딘아민내에 도입시킨 다음 수득한 (XVI)의 니트로균을 상술한 니트로를 아민으로 환원하는 표준공정에 따라 환원의 팔라듐 같은 적당한 촉매를 사용하여 촉매적 수소화에 의하여 실신한다.The intermediate of formula (VII) reacts N- (2-nitrophenyl) -4-piperidineamine of formula (XV) with aziridine of formula (II) or a reactive ester of formula (IV) Aroyl alkyl is introduced into the general formula (XV) N- (2-nitrophenyl) -4-piperidineamine, and then the nitro bacteria of (XVI) obtained are reduced according to the standard procedure of reducing the above-mentioned nitro to amine. Faint by catalytic hydrogenation using a suitable catalyst such as palladium.

일반식(Ⅵ)의 중간체들은 신규이며 일반식(1)의 화합물을 제조하는데 유용한 중간체로서 본 발명의 부가적인 형테이다.Intermediates of formula (VI) are additional forms of the invention as novel and useful intermediates for preparing compounds of formula (1).
일반식(Ⅶ)의 중간체는 일반식(XV)의 N-(2-니트로페닐)-4-피페리딘아민을 일반식(Ⅱ)의 아지리딘 또는 일반식(Ⅳ)의 반응성 에스테르와 반응시켜 아로일 알킬을 일반식(XV) N-(2-니트로페닐)-4-피페리딘아민내에 도입시킨 다음 수득한 (XVI)의 니트로군을 상술한 니트로를 아민으로 환원하는 표준 공정에 따라 환원를 아민으로 환원하는 표준 공정에 따라 환원시킴에 의하여 제조한다.The intermediate of formula (VII) reacts N- (2-nitrophenyl) -4-piperidineamine of formula (XV) with aziridine of formula (II) or a reactive ester of formula (IV) Reduction was carried out according to the standard procedure of introducing aroyl alkyl into the general formula (XV) N- (2-nitrophenyl) -4-piperidineamine and then reducing the nitro group of (XVI) obtained to the above-mentioned nitro to amine. Prepared by reduction according to standard processes of reducing to amines.

하기 일반식으로 표시되는 일반식(Ⅲ)의 시발물질과 이들의 제조 방법들은 하기 문헌에 수록되였다.The starting materials of the general formula (III) represented by the following general formula and their preparation methods are listed in the following documents.

a) 미국 특허 3,238,216호a) US Patent 3,238,216
b) 미국 특허 3,161,645호b) US Patent 3,161,645
벨지움 특허 830,403호Belgium Patent 830,403
c) 미국 특허 3,518,276호c) US Patent 3,518,276
미국 특허 3,575,990호U.S. Patent 3,575,990
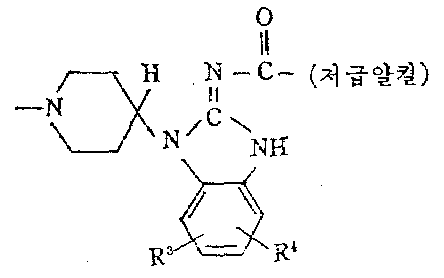
하기 일반식(Ⅲ-d)와 (Ⅲ-e)로 표시되는 일반식(Ⅲ)의 시발물질들은 일반적으로 하기 일반식(XVⅡ)의 적당한 N-(2-아미노페닐)-4-피페리딘아민으로 부터 출발하여 제조한다.The starting materials of general formula (III) represented by the following general formulas (III-d) and (III-e) are generally suitable N- (2-aminophenyl) -4-piperidine of general formula (XVII) Prepared starting from amines.

시발물질(Ⅲ-d)는 (XVⅡ)을 이유화탄소 같은 적당한 환상 화제로 환상화시킨 다음 수득한 (XVⅢ)의 저급알킬옥시카르보닐을 알카리 가수분해에 의하여 제거함에 의하여 제조한다.The starting material (III-d) is prepared by cyclizing (XVII) with a suitable cyclizing agent such as carbon wed and then removing the lower alkyloxycarbonyl of (XVIII) obtained by alkali hydrolysis.
시발물질(Ⅲ-e)는 (1-d)로 부터 출발하여 (1-e)의 제조에 대하여 기술한 바와같이 (XVⅢ)를 S-알킬화시킨 다음 수득한 (XIX)의 저급알킬옥시카르보닐군을 제거함에 의하여 제조한다.The starting material (III-e) is a lower alkyloxycarbo of (XIX) obtained after S-alkylation of (XVIII) as described for the preparation of (1-e) starting from (1-d) Produced by removing the Neal group.
공지된 화합물인 일반식(XVⅡ)의 N-(2-아미노페닐)-4-피페리딘아민은 미국 특허 3,910,930호 및 벨지움 특허 830,403호에 기술된 공정에 따라 제조한다.N- (2-aminophenyl) -4-piperidineamine of the general formula (XVII), a known compound, is prepared according to the processes described in US Pat. No. 3,910,930 and Belgian Patent 830,403.
상기 공정은 하기 반응식으로 설명된다.The process is illustrated by the following scheme.

하기 일반식(Ⅲ-f)로 표시되는 일반식(Ⅲ)의 중간체들은 다음과 같이 제조한다.The intermediates of the general formula (III) represented by the following general formula (III-f) are prepared as follows.

일반식(XX)의 적당한 N-(2-아미노페닐)-1-(페닐메틸)-4-피페리딘아민을 공지된 바와 같이 시안아마이드 같은 적당한 환상화제로 환상화 시킴에 의하여 해당하는 1-1-(페닐메틸)-4-피페리디닐-1H-벤즈이미다졸-2-아민(XXI)으로 전환시키고 수득한 (XXI)을 적당한 저급 앝킬카르복실산으로 부터 유도된 아실화제 예컨테 할라이드나 무수물로 아실화시켜 일반식(XXⅡ)의 중간체를 수득한다. 그리고 원하는 (Ⅲ-f)는 활탄상의 팔라듐 같은 적당한 촉매를 사용한 촉매적 수소화에 의하여 (XXⅡ)의 페닐메틸군을 제거함에 의하여 얻는다.The corresponding N- (2-aminophenyl) -1- (phenylmethyl) -4-piperidinamine of the general formula (XX) is subjected to the corresponding 1- by cyclizing it with a suitable cyclic agent such as cyanamide, as is known. (XXI) obtained by converting to 1- (phenylmethyl) -4-piperidinyl-1H-benzimidazol-2-amine (XXI) and acylating agent derived from a suitable lower hydroxyalkyl acid such as halide Acylation with anhydride affords the intermediate of formula (XXII). And the desired (III-f) is obtained by removing the phenylmethyl group of (XXII) by catalytic hydrogenation with a suitable catalyst, such as palladium on charcoal.
중간체(XXI)은 또한 (XX)를 1,2-벤젠디아민으로 부터 출발하여 2-벤즈이미다졸 카르바메이트를 제조하는데 공지된 바와 같은 적당한 환상화제 예컨데 일반식(XXⅢ)의 저급 알킬(이미노메톡시메틸) 카르바메이트로 환상화 시킴에 의하여 저급 알킬 1,3-디하이드로-1-1-(페닐메틸)-4-피페리디닐-2H-벤즈이미다졸-2-이리덴 카르바메이트(XXIV)로 전환시킨 다음 이것을 산 가수분해에 의하여 탈카르복실화 시킴에 의하여 제조한다.Intermediates (XXI) are also suitable cyclic agents such as those known to prepare 2-benzimidazole carbamates starting from 1,2-benzenediamine (XX) such as lower alkyl (iminome) of general formula (XXIII). Lower alkyl 1,3-dihydro-1-1- (phenylmethyl) -4-piperidinyl-2H-benzimidazole-2-iriden carbamate (by cyclizing to methoxymethyl) carbamate XXIV), which is then prepared by decarboxylation by acid hydrolysis.
상기 반응은 하기 반응식으로 설명된다.The reaction is illustrated by the following scheme.

일반식(XX)의 시발물질은 예컨대 1-(페닐메틸)-4-피페리딘아민과 일반식(XXV)의 적당한 2-할로-니트로 벤젠을 축합시킨 다음 수득한 (XXVI)의 니트로군을 라네이-닉켈과 같은 적당한 촉매를 사용하여 촉매적 수소화에 의하여 환원 시킴에 의한 것과 같은 공지된 방법을 사용하여 수득한다.The starting material of the general formula (XX) is, for example, a condensation of 1- (phenylmethyl) -4-piperidineamine with a suitable 2-halo-nitro benzene of general formula (XXV), and then the nitro group of (XXVI) obtained Obtained using known methods such as by reduction by catalytic hydrogenation using a suitable catalyst such as ranei-nickel.

상기 공정들에서 대개는 공지되였고 공지된 방법에 따라 제조할 수 있다.In the above processes are usually known and can be prepared according to known methods.
식중![]()
![]()
피페리딘 핵의 평면에 대하여 메틸과 수산기의 상대적 위치에 따라 화합물들은 시스 또는 트란스 배열을 갖이며 이들 형태들의 각각은 2개의 광학적 이성체를 갖는다. 본 발명의 범위내에 속하는 이들 화합물의 입체 화학적 및 광학적 이성체들은 당해 분야에 공지된 방법에 따라 제조한다. 수득한 화합물 및 이들 선구물의 시스와 한다. 수득한 화합물 및 이들 선구물의 시스와 트란스 이성체들은 선택적 결정화 같은 물리적 방법에 의하여 따로따로 얻을 수 있다. 그들의 실제 입체 화학적 배열에 관계없이 먼저 분리된 형테를 A-형태라 칭하고 남은것을 B-형태라 칭하기로 하다. 포함된 화합물들이 염기성이므로 라세미크시스와 트란스의 분해에 광학적으로 활성인 산을 사용하여 이들의 광학적 이성체를 얻는다.Depending on the relative position of methyl and hydroxyl groups with respect to the plane of the piperidine nucleus, the compounds have a cis or trans configuration and each of these forms has two optical isomers. Stereochemical and optical isomers of these compounds that fall within the scope of the present invention are prepared according to methods known in the art. It is with the cis obtained compound and these precursors. The obtained compounds and cis and trans isomers of these precursors can be obtained separately by physical methods such as selective crystallization. Regardless of their actual stereochemical configuration, the first separated form is called the A-form and the remainder is called the B-form. Since the compounds involved are basic, their optical isomers are obtained using optically active acids for the resolution of racemic and trans.
본 발명의 화합물들은 적당한 산 예컨데 염산, 브롬산과 같은 할로겐화산, 황산, 질산, 인산 등과 같은 무기산과 식초산-프로파노산, 2-하이드록시아세트산, 2-하이드록시프로파노산, 2-옥소프로파노산, 프로판디오산, 부탄디오산, (Z)-2-부탄디오산, (E)-2-부텐디오산, 2-하이드록시부텐디오산, 2,3-디하이드록시부텐디오산, 2-하이드록시-1,2,3-프로판트리카르복실산, 벤조산, 3-페닐-2-프로페노산, α-하이드록시벤젠아세트산 메탄술폰산, 에탄술폰산, 벤젠 술폰산, 4-메틸벤젠술폰산, 싸이크로헥산술팜산, 2-하이드록시벤조산, 4-아미노-2-하이드록시 벤조산과 같은 유기산으로 처리함에 의하여 이들의 약학적으로 유용한 산 부가염으로 전환시킬 수 있고 반대로 염 형태는 알카리로 처리함에 의하여 유리 염기 형태로 전환시킬 수 있다.The compounds of the present invention may be selected from suitable acids such as hydrochloric acid, halogenated acids such as bromic acid, sulfuric acid, nitric acid, phosphoric acid and the like and vinegar-propanoic acid, 2-hydroxyacetic acid, 2-hydroxypropanoic acid, 2-oxopro Panoic acid, propanedioic acid, butanedioic acid, (Z) -2-butanedioic acid, (E) -2-butenedioic acid, 2-hydroxybutenedioic acid, 2,3-dihydroxybutenedioic acid, 2-hydroxy-1,2,3-propanetricarboxylic acid, benzoic acid, 3-phenyl-2-propenoic acid, α-hydroxybenzeneacetic acid methanesulfonic acid, ethanesulfonic acid, benzene sulfonic acid, 4-methylbenzenesulfonic acid, Treatment with organic acids such as cyclohexanesulfamic acid, 2-hydroxybenzoic acid, 4-amino-2-hydroxy benzoic acid can be converted to their pharmaceutically useful acid addition salts and conversely the salt forms are treated with alkali. By the free base form.
일반식(1)의 화합물 및 이들의 약학적으로 활성인 산 부가염은 개에서 구토를 유발시키는 아포몰핀을 억제하는 이들의 작용에 의하여 확실한 바와같이 강력한 구토 억제 작용을 갖는다. 사용한 방법은 P.A.J. 잔센 및 C.J.E. 니에메기에스에 의하여 Arzneim. -Forsch(drug Res),9 765-767(1959)에 기술되였다.Compounds of formula (1) and their pharmaceutically active acid addition salts have potent vomiting inhibitory effects, as is evident by their action of inhibiting apomorphine, which causes vomiting in dogs. The method used was P.A.J. Xansen and C.J.E. Arzneim by Niemegies. Forsch (drug Res), 9 765-767 (1959).
하기에 수록한 화합물들은 상이한 투약량으로 비글개에게 피하 주사를 하고 1시간후 0.31㎎/㎏의 아포몰핀의 표준 투약량을 피하 주사한다.The compounds listed below are injected subcutaneously into Beagle dogs at different dosages and a standard dose of apomorphine at 0.31 mg / kg after 1 hour.
하기 표는 많은 화합물에 대한 ED50값으로 주어졌다. 본 명세서에 사용한 ED50값은 구토로 부터 50%의 동물을 보호한 투약량을 표시한다.The table below gives the ED 50 values for many compounds. As used herein, the ED 50 value indicates the dose that protected 50% of the animal from vomiting.
하기 표에 나타낸 화합물은 본 발명을 한정할 목적으로 수록한 것이 아니고 일반식(1)의 범위내의 모든 화합물의 구토 방지적 성질을 설명하기 위한 것이다.The compounds shown in the following table are not listed for the purpose of limiting the present invention but are intended to explain the anti-emetic properties of all compounds within the range of the general formula (1).
[표 Ⅰ]TABLE I



[표 Ⅱ]TABLE II


[표 Ⅲ]TABLE III

[표 Ⅳ]Table IV

일반식(1)의 화합물은 강력한 신경안정제이므로 예컨데 정신적인 비정상 및 정신 분열증 같은 정신적 비정상의 처리에 유용하다. 본 화합물(1)의 신경 안정작용은 증추 신경계에 대한 진정작용을 나타내는 쥐에서 아포몰핀 시험에서 얻은 시험 데이타에 의하여 입증되였다. 시험은 후술한 공정에 따라 실시하고 수득한 시험 데이타는 표 4에 요약하였는데 여기에서 화합물들은 표 1-Ⅳ내의 해당하는 구조식을 참조로 하였다.Compounds of formula (1) are potent neurostabilizers and are therefore useful for the treatment of mental abnormalities such as mental abnormalities and schizophrenia. Neural stabilization of the compound (1) was demonstrated by test data obtained in the apomorphine test in mice exhibiting sedation against the central nervous system. The test was carried out according to the process described below and the test data obtained were summarized in Table 4, wherein the compounds were referenced to the corresponding structural formulas in Tables 1-IV.
표 Ⅴ에 수록된 화합물들은 본 발명을 한정할 목적으로 주어진 것이 아니고 일반식(1)의 범위내의 모든 화합물의 유용한 신경 안정 작용을 예를들기 위한 것이다.The compounds listed in Table V are not given for the purpose of limiting the invention and are intended to illustrate the useful neurostable action of all compounds within the scope of formula (1).
[쥐에 대한 아포몰핀 시험]Apomorphine Test in Rats
본 시험에 사용한 실험동물을 성숙한 숫놈의 위스타 쥐(체중 240±10g)이다. 1야간 단식시킨 후 동물을 화합물의 수용액(1㎖/100g)으로 피하 주사하고 분리된 관찰할 수 있는 상자에 넣고 30분후 1.25㎎/㎏의 아포몰핀 하이드로크로라이드를 정맥 주사하고 쥐를 아포몰핀이 유발하는 현상 즉 경동 및 상습적인 씹음의 존재 또는 부재에 대하여 1시간에 걸쳐 관찰한다. 표 Ⅴ는 최저의 효과적인 투약량(LED) 즉 투약량 준위로 주어졌으며 이들의 효과는 해당하는 처리되지 않은 비교 동물에서 관찰된 것과 통계적으로 확실히 상이하다(피쉬 확율시험).The test animals used in this study were mature male Wistar rats (240 ± 10 g body weight). After overnight fasting, animals were injected subcutaneously with an aqueous solution of the compound (1 mL / 100 g), placed in a separate observable box, and 30 minutes later, intravenously injected with 1.25 mg / kg of apomorphine hydrochloride, and the rats were apomorphine-containing. Observe over 1 hour for the phenomena causing, ie, the presence or absence of tilting and habitual chewing. Table V is given as the lowest effective dose (LED), or dose level, and their effects are statistically different from those observed in the corresponding untreated comparative animals (fish probability test).
[표 Ⅳ] 쥐에 대한 화합물(1)의 항-아포몰핀 작용TABLE IV Anti-apomorphine action of Compound (1) on rats


이들의 유용한 구토억제 및 정신 안정 작용의 견지에서 본 화합물들은 급여 목적에 따라 여러가지 제약적 형태로 만들수 있다. 본 발명의 제약적 조성물을 제조하기 위하여 작용 성분으로서 염기 또는 산-부가염 형태로된 특정화합물의 효과적인 구토억제 또는 정신 안정제적 양을 약학적 허용담체와 혼합시키며 담체는 원하는 급여에 적당한 형태에 따라 광범위 형태를 취할수 있다. 이 제약적 조성물들은 경구적 또는 비경구적 주사에 의한 급여에 적당한 단위투약량 형태가 바람직하다. 예를들면 경구적 투약량 형태로된 조성물을 제조하는데 있어서는 현탁액, 시럽, 에끼스 및 용액같은 경구적 액체제품의 경우 예컨데 물, 글리골, 오일, 알콜같은 통상의 제약적 매체와 분말, 환약, 캡슐 및 정제의 경우 전분, 설탕, 활석, 윤활제, 결합제 분해제같은 고체담체가 사용된다. 급여의 용이성 때문에 정제와 캡슐이 가장 좋은 경구적 투약형태이며 이런 경우 고체의 제약적 담체가 사용된다. 비경구적 조성물의 경우 담체의 용해도를 증가시키기 위한 다른 성분과 함께 대부분이 소독된 물로 되였다. 주사용액은 담체가 식염수, 포도당 용액 또는 식염과 포도당의 혼합물로 되였고 주사용 현탁액은 적당한 액체담체, 현탁화제 등을 사용하여 제조한다. 해당하는 염기 형태보다 물에 대한 용해도가 큰 일반식(1)의 산부가 염이 수용성 조성물의 제조에 보다 적당하다.In view of their useful anti-nausea and psychostable effects, the compounds can be formulated into various pharmaceutical forms depending on the purpose of the supplement. To prepare a pharmaceutical composition of the present invention, an effective nausea suppressor or psychostabilizing amount of a specific compound in the form of a base or acid-addition salt as a working ingredient is mixed with a pharmaceutically acceptable carrier and the carrier can be used in a wide range depending on the form suitable for the desired diet. Can take the form. These pharmaceutical compositions are preferably in unit dosage form suitable for feeding by oral or parenteral injection. For example, in the preparation of compositions in oral dosage form, oral liquid products such as suspensions, syrups, essences and solutions, for example, conventional pharmaceutical media such as water, glycol, oils, alcohols and powders, pills, capsules and In the case of tablets, solid carriers such as starch, sugar, talc, lubricants and binder disintegrators are used. Because of ease of feeding, tablets and capsules are the best oral dosage forms, in which case solid pharmaceutical carriers are used. In the case of parenteral compositions, most of it was disinfected water along with other ingredients to increase the solubility of the carrier. Injectable solutions consist of a carrier in saline solution, glucose solution or a mixture of saline and glucose, and injectable suspensions are prepared using suitable liquid carriers, suspending agents and the like. Acid addition salts of the general formula (1) having a higher solubility in water than the corresponding base forms are more suitable for the preparation of the water-soluble composition.
상술한 제약적 조성물을 급여가 용이하고 투약량의 균일성을 제공할 수 있는 단위 투약량 형태로 만드는 것이 특히 적당하다.It is particularly suitable to make the above-mentioned pharmaceutical compositions in unit dosage form which is easy to feed and can provide uniformity of dosage.
본 명세서 및 청구범위에 사용한 단위 투약량 형태는 단위 투약에 적당한 물리적으로 분리된 단위로서 각 단위는 원하는 제약적 담체와 함께 원하는 약학적 효과를 얻도록 계산된 사전 결정된 양의 작용성분을 포함한다. 그러한 단위 투약량 형태의 예를들면 정제, 캡슐, 환약, 분말포장, 주사용액이나 현탁액, 차한숫갈, 식숫갈 가득히 등이 있다.Unit dosage forms as used herein and in the claims are physically discrete units suited for unit dosage, each unit comprising a predetermined amount of active ingredient calculated to achieve the desired pharmaceutical effect with the desired pharmaceutical carrier. Examples of such unit dosage forms include tablets, capsules, pills, powder packaging, injectable solutions or suspensions, chilled water, filled with sake.
구로방지 또는 신경안정 목적을 위한 단위 투약량당 작용성분의 양은 1-200㎎ 특히 약 5-100㎎이다The amount of active ingredient per unit dose for prophylactic or neurostable purposes is 1-200 mg, especially about 5-100 mg.
하기 형태들은 전통적인 구토억제 및 신경 안정적 제약 조성물을 동물 및 인간에게 계통적으로 급여하기에 적당한 단위 투약량 형태로 제조하는 예를든 것이다.The following forms are examples of preparing traditional anti-emetic and neuro-stable pharmaceutical compositions in unit dosage forms suitable for systematic feeding to animals and humans.
[경구적 드롤(drop)][Oral drop]
하기형태는 ㎖당 유효 성분으로서 5㎖의 N{1-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-일피페리디닐]에틸}-4-플루오로벤즈아마이드를 포함한 10ℓ의 경구적 드롭용액을 제조한다.The following form is 5 ml of N {1- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-ylpipet as an active ingredient per ml. A 10 L oral drop solution containing ridinyl] ethyl} -4-fluorobenzamide is prepared.

피로겐 유리 물을 첨가하여 10ℓ로 만듬10 l with pyrogen-free water
메틸과 프로필 4-하이드록시 벤조에이트를 5ℓ의 비등하는 피로겐 부재의 물에 용해시키고 약 50℃로 냉각한 후 교반하면서 2-하이드록시 프로파노산을 첨가한 다음 유효성분을 첨가한다. 용액을 실온으로 냉각하고 피로겐 부재의 물을 일정용량까지 보충시킨 다음 용액을 여과(U.S.P. XVⅡ p.811)에 의하여 소독하고 소독된 용기내에 채운다.Methyl and propyl 4-hydroxy benzoate are dissolved in 5 L of boiling pyrogen-free water, cooled to about 50 ° C., and 2-hydroxy propanoic acid is added with stirring followed by the addition of the active ingredient. The solution is cooled to room temperature and the pyrogen-free water is replenished to a certain volume, then the solution is sterilized by filtration (U.S.P. XVII p.811) and filled into sterilized containers.
[주사용액][Injection amount]
상술한 경구적 드롭용액은 주사용액으로 사용할 수도 있다The oral drop solution described above may be used as an injection solution.
[캡슐][capsule]
각 캡슐이 유효성분(A, I)로서 20㎎의 N{1-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸}-4-플루오로벤즈아마이드를 포함한 10,000개의 단단한 제라틴캡슐을 하기 조성물로부터 제조한다.Each capsule contains 20 mg of N {1- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1 as the active ingredient (A, I). 10,000 hard gelatin capsules containing -piperidinyl] ethyl} -4-fluorobenzamide are prepared from the following composition.

유효성분 및 보충성분의 균일 혼합물을 제조하여 2-쪽으로된 단단한 제라틴 캡슐에 채운다.A homogeneous mixture of active ingredients and supplements is prepared and filled in a two-sided hard gelatin capsule.
[정제][refine]
각 정제가 유효성분으로서 25㎎의 N{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-일피페리디닐]에틸}-4-플루오로벤즈아마이드를 포함한 5,000개 의 압측정제를 하기 조성물로부터 제조한다.Each tablet is 25 mg of N {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-ylpiperidinyl as an active ingredient. 5,000 pressure gauges, including] ethyl} -4-fluorobenzamide, are prepared from the following compositions.

미세분말 성분들을 잘 혼합하여 10%의 전분풀과 함께 과립으로 만들어 과립을 건조하여 정제로 압축시킨다.The fine powder components are mixed well into granules with 10% starch paste, the granules are dried and compressed into tablets.
[경구적 현탁액][Oral suspension]
하기형태로 유형성분(A, I)로서 차숫갈(5-㎖)당 15㎎의 N{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸}-4-풀루오로벤즈아마이드를 포함한 5ℓ의 경구 현탁액을 제조한다.15 mg of N {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimi per 15 mg (5-ml) as tangible ingredient (A, I) in the following form A 5 L oral suspension is prepared comprising dazol-1-yl) -1-piperidinyl] ethyl} -4-pululobenzamide.


파라벤을 폴리에티렌 글리곧내에 용해시키고 이 용액을 절반의 물내의 쏘디움 싸이크라메이트, 쏘디움 사카린 및 설탕의 용액에 첨가한다. 벤토나이트를 뜨거운(약 85℃) 물에 현탁시키고 60분간 교반한 다음 상기 용액에 첨가한다. 술포 썩시네이트를 약간의 물에 용해시킨 다음 수득한 용액내에 작용성분을 현탁시키고 최소량의 물로서 로우쉰 형태까지 희석시킨 거품방지 A.F.에멀존을 첨가한 후 잘 혼합시킨다. 수득한 유효성분의 현탁액을 상기 용액에 첨가하고 잘 혼합한후 소량의 물에 용해시킨 FD C 황색 5를 첨가하고 오랜지 향료를 첨가한 다음 물을 일정 용량까지 채우고 균일 혼합물이 될때까지 교반한다. 이 혼합물을 코로이드 밑을 통과시킨후 적당한 용기에 채운다.Parabens are dissolved in polystyrene glycoside and this solution is added to a solution of sodium cyclomate, sodium saccharin and sugar in half water. Bentonite is suspended in hot (about 85 ° C.) water, stirred for 60 minutes and then added to the solution. The sulfo rosinate is dissolved in a little water, and then the active ingredient is suspended in the obtained solution and mixed well after addition of an antifoam A.F.emulsion diluted to a Roesch form with a minimum amount of water. The suspension of the obtained active ingredient is added to the above solution, mixed well, and then FD C yellow 5 dissolved in a small amount of water is added, orange fragrance is added, the water is filled to a certain volume and stirred until a homogeneous mixture is obtained. The mixture is passed under a colloid and filled into a suitable container.
본 화합물의 구토 억제작용의 견지에서 본 발명은 효과적인 구토 억제적 양의 일반식(1)의 화합물 및 이들의 약학적 허용 산부가염을 약학적 담체와 혼합하여 계통적 급여에 의하여 구토에 의하여 고생하는 온혈 동물에게 구토를 억제시키는 방법을 제공한다. 또한 본 화합물의 신경안정제적 작용의 견지에서 본 발명은 약학적 담체와 배합된 일반식(1)의 화합물 및 이들의 약학적 허용산 부가염의 효과적인 정신질환 억제적 양을 계동적으로 급여함에 의하여 정신적 비정상인 온혈동물을 치료하는 방법을 제공한다.In view of the anti-vomiting effect of the present invention, the present invention provides an effective anti-vomiting amount of the compound of formula (1) and their pharmaceutically acceptable acid addition salts with a pharmaceutical carrier, and warm blood suffering from vomiting by systematic feeding. An animal is provided with a method of inhibiting vomiting. In addition, in view of the neurostabilizing action of the compound, the present invention provides a method for treating mentally psychiatric disorders by systematically feeding an effective mental disease inhibitory amount of the compound of formula (1) and their pharmaceutically acceptable acid addition salts in combination with a pharmaceutical carrier. Provided are methods for treating abnormal warm blooded animals.
하기 실시예들은 본 발명의 범위를 한정할려는 것이 아니고 설명하기 위한 것이며 다른 설명이 없는한 모든 부는 중량에 의한 것이다.The following examples are intended to illustrate and not limit the scope of the invention and all parts are by weight unless otherwise indicated.
[실시예 1]Example 1
76부의 4-플루오로-2-니트로벤조산과 225부의 벤젠의 교반 혼합물에 71부의 술피닐 크로라이드를 방울방울 첨가한 다음 환류 온도에서 5시간 그리고 실온에서 1야간 교반시킨다. 반응혼합물을 증발시키고 잔유물을 벤젠내에 넣고 벤젠을 증발시키는 것을 2회 반복하면 잔유물로서 85부의 4-플루오로-2-니트로벤벤조일 크로라이드를 수득한다.To a stirring mixture of 76 parts of 4-fluoro-2-nitrobenzoic acid and 225 parts of benzene were added dropwise 71 parts of sulfinyl chloride, followed by stirring at reflux for 5 hours and at room temperature overnight. Evaporating the reaction mixture, putting the residue into benzene and repeating the evaporation of benzene twice gives 85 parts of 4-fluoro-2-nitrobenbenzoyl chromide as a residue.
[실시예 2]Example 2
물내의 231부의 아리자린 0.95m용액에 0℃에서 교반하면서 16부의 중탄산나르륨을 첨가한 다음 0℃를 유지하면서 150부의 트리크로로메탄내 36부의 4-플루오로-2-메톡시 벤조일 크로라이드 용액을 45분간에 걸쳐 방울방울 첨가한 다음 냉각하지 않고 30-45분간 교반시킨다. 반응혼합물을 묽은 수산화나트륨용액을 사용하여 pH8로 조절하고 생성물을 트리크로메탄으로 3회 추출한후 합친 추출액을 물로 3회 세척 건조 및 여과하여 증발시키면 잔유물로서 40.5부의 1-(4-플루오로-2-메톡시벤조일) 아지리딘을 수득한다.16 parts of sodium bicarbonate were added to 231 parts of arizine 0.95 m solution in water with stirring at 0 ° C., followed by 36 parts of 4-fluoro-2-methoxy benzoyl chromide in 150 parts of trichloromethane while maintaining at 0 ° C. The solution is added dropwise over 45 minutes and then stirred for 30-45 minutes without cooling. The reaction mixture was adjusted to pH 8 with dilute sodium hydroxide solution, the product was extracted three times with trichromemethane, and the combined extracts were washed three times with water, dried, filtered and evaporated to give 40.5 parts of 1- (4-fluoro-2) as a residue. -Methoxybenzoyl) aziridine is obtained.
[실시예 3]Example 3
일반식 2의 공정을 따르고 사용된 4-플루오로-2-메톡시벤조일 크로라이드 대신에 당량의 적당한 아릴카르보닐 크로라이드를 사용하여 하기 1-(아닐 카르보닐) 아지리딘을 제조한다.The following 1- (anyl carbonyl) aziridine is prepared by following the process of formula 2 and using the equivalent arylcarbonyl fluoride equivalents in place of the 4-fluoro-2-methoxybenzoyl fluoride used.
1-(2-하이드록시벤조일) 아지리딘:잔유물1- (2-hydroxybenzoyl) aziridine: residue
1-(4-풀루오로-2-니트로벤조일) 아지리딘:잔유물1- (4-Pluoro-2-nitrobenzoyl) aziridine: residue
1-(2-티에닐카르보닐) 아지리딘:잔유물1- (2-thienylcarbonyl) aziridine: residue
1-(5-크로로-2-메톡시-4-니트로벤조일) 아지리딘1- (5-Chloro-2-methoxy-4-nitrobenzoyl) aziridine
[실시예 4]Example 4
12.94부의 에틸(2-브로모에틸) 카르바메이트, 13.51부의 1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원, 10.1부의 중탄산나트륨 및 160부의 에탄올의 혼합물을 1야간 교반 및 환류시킨다. 반응혼합물을 냉각하여 여과하고 여과액을 증발시킨다. 잔유물을 용출액으로서 트리크로로메탄, 10% 메탄올 및 1방울의 수산화암모니움의 혼합물을 사용하여 시리카겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키면 잔유물로서 4.4부의 에틸 2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸카르바메이트를 수득한다.12.94 parts ethyl (2-bromoethyl) carbamate, 13.51 parts 1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole-2-one, 10.1 parts sodium bicarbonate and 160 parts The mixture of ethanol is stirred and refluxed overnight. The reaction mixture is cooled, filtered and the filtrate is evaporated. The residue is purified by column-chromatography on silica gel using a mixture of trichloromethane, 10% methanol and 1 drop of ammonium hydroxide as eluent. Collecting pure fractions and evaporating the eluate gave 4.4 parts of ethyl 2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethylcarb as a residue. Obtain a barmate.
0.333부의 에틸 2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸카르바메이트, 1.65부의 48% 브롬산 및 0.11부의 물의 혼합물을 2시간 교반 및 환류시킨다. 브롬산과 물을 증발시키고 잔유물을 벤젠에 넣고 증발시키는 것을 3회 반복하고 오일상의 잔유물을 2-프로판올로부터 결정화시킨다. 생성물을 여과하고 소량의 에탄올로부터 재결정시키면 용융점이 +300℃(분해)인 0.27부의 1-[1-(2-아미노에틸)-4-피페리디닐]-1,3-디하이드로-2H-벤즈이미다졸-2-원 디하이드로 브로마이드를 얻는다.0.333 parts of ethyl 2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethylcarbamate, 1.65 parts of 48% bromic acid and 0.11 The mixture of portions of water is stirred and refluxed for 2 hours. Bromic acid and water are evaporated, the residue is placed in benzene and evaporated three times and the oily residue is crystallized from 2-propanol. The product was filtered and recrystallized from a small amount of ethanol and 0.27 parts 1- [1- (2-aminoethyl) -4-piperidinyl] -1,3-dihydro-2H-benz having a melting point of + 300 ° C (decomposition) Obtain imidazole-2-one dihydro bromide.
[실시예 5]Example 5
87부의 2-크로로에탄아민 하이드로크로라이드와 300부의 트리크로로메탄의 교반된 냉각(얼음-중탕)혼합물에 224.2부의 N,N-디에틸에탄아민을 첨가하고 다시 300부의 트리크로로메탄을 첨가한후 5℃이하의 온도에서 300부의 트리크로로메탄내의 96부의 2-푸란카르보닐 크로라이드용액을 1.50시간에 걸쳐 방울방울 첨가한 다음 실온에서 1야간 교반한다. 반응혼합물을 물에 쏟고 층을 분리하여 수용액층을 트리크로로메탄으로 추출한후 합친 유기상을 물로, 묽은 염산 용액으로 그리고 다시 물로 2회 세척, 건조 및 여과하여 증발시킨다. 잔유물을 용출액으로서 트리크로로메탄과 2%의 메탄올의 혼합물을 사용하여 시리카겔 상에서 컬럼-크로마토그라피에 의하여 정제한 다음 순수한 분율을 수집하여 용출액을 증발시키면 잔유물로서 95부의 N-(2-크로로에틸)-2-푸란카르복스아마이드를 수득한다.To a stirred cooling (ice-bath) mixture of 87 parts 2-chloroethaneamine hydrochloride and 300 parts trichloromethane, 224.2 parts N, N-diethylethanamine is added and 300 parts trichloromethane is added again. After the addition, 96 parts of 2-furancarbonyl chloride solution in 300 parts of trichloromethane was added dropwise over 1.50 hours at a temperature of 5 ° C. or lower, followed by stirring overnight at room temperature. The reaction mixture is poured into water, the layers are separated, the aqueous layer is extracted with trichloromethane, and the combined organic phases are washed with water, diluted hydrochloric acid solution and then twice with water, dried and filtered to evaporate. The residue was purified by column-chromatography on silica gel using a mixture of trichloromethane and 2% methanol as eluent and then pure fractions collected to evaporate the eluent to give 95 parts of N- (2-chloro) as a residue. Ethyl) -2-furancarboxamide is obtained.
[실시예 6]Example 6
150부의 물내의 32부의 2-브로모에탄아민 하이드로브로마이드의 교반 및 냉각혼합물에 54부의 벤젠내 23.5부의 4-풀루오로-2-니트로벤조일 크로마이드 용액을 첨가하고 0-5℃를 유지하고 교반하면서 200부의 물내의 13부의 수산화나트륨 용액을 방울방울 첨가한후 0-10℃에서 2시간 교반한다. 침전된 생성물을 여과하여 물로 세척한후 건조하면 30부(93.6%)의 N-(2-브로모에틸)-4-풀루오로-2-니트로벤즈아마이드를 얻는다.To a stirring and cooling mixture of 32 parts 2-bromoethanamine hydrobromide in 150 parts water, a solution of 23.5 parts 4-fluluoro-2-nitrobenzoyl chromide in 54 parts benzene was added and maintained at 0-5 ° C. and stirred 13 parts of sodium hydroxide solution in 200 parts of water was added dropwise, followed by stirring at 0-10 ° C. for 2 hours. The precipitated product was filtered off, washed with water and dried to afford 30 parts (93.6%) of N- (2-bromoethyl) -4- pulluro-2-nitrobenzamide.
[실시예 7]Example 7
75부의 물내의 21부의 수산화나트륨의 교반용액에 75부의 물내의 29부의 2-크로로에탄아민 하이드로크로라이드 용액을 첨가한 다음) 전체를 90℃에서 10분간 교반한다. 0℃로 냉각한후 19부의 중탄산나트륨을 첨가하고 세차게 교반하면서 0℃이하의 온도에서 75부의 트리크로로메탄내 49부의 2.5-디크로로벤조일 크로라이드 용액을 45분간에 걸쳐 방울방을 첨가한 다음 냉각하지 않고 30분간 교반시킨다. 반응혼합물을 25℃로 가온하고 묽은 수산화나트륨을 사용하여 pH8로 조절한 다음 생성물을 트리크로로메탄으로 3회 추출하여 합친 추출액을 물로 3회 세척, 건조 및 여과하여 증발시키면 잔유물로서 62부의 1-(2,5-디크로로벤조일) 아지리딘을 수득한다.To a stirred solution of 21 parts sodium hydroxide in 75 parts water is added 29 parts 2-chloroethaneamine hydrochloride solution in 75 parts water) and the whole is stirred at 90 ° C. for 10 minutes. After cooling to 0 ° C., 19 parts of sodium bicarbonate was added, and 49 parts of 2.5-dichlorobenzoyl fluoride solution in 75 parts of trichloromethane were added dropwise over 45 minutes at a temperature of 0 ° C. under vigorous stirring. It is then stirred for 30 minutes without cooling. The reaction mixture was warmed to 25 ° C., adjusted to pH 8 using dilute sodium hydroxide, the product was extracted three times with trichloromethane, and the combined extracts were washed three times with water, dried and filtered to evaporate. Obtain (2,5-dichlorobenzoyl) aziridine.
[실시예 8]Example 8
72부의 메틸 2-피리딘카르복시레이트와 32부의 2-아미노에탄올의 혼합물을 2시간 교반 및 환류시키고 반응 혼합물을 냉각하여 물에 쏟는다. 생성물을 트리크로로메탄으로 3회 추출하고 합친 추출액을 건조여과하여 증발시키면 잔유물로서 49부의 N-(2하이드록시에틸)-2-피리딘카르복스아마이드를 얻는다.A mixture of 72 parts methyl 2-pyridinecarboxylate and 32 parts 2-aminoethanol is stirred and refluxed for 2 hours and the reaction mixture is cooled and poured into water. The product is extracted three times with trichloromethane and the combined extracts are filtered dry and evaporated to yield 49 parts of N- (2hydroxyethyl) -2-pyridinecarboxamide as a residue.
49부의 N-(2-하이드록시에틸)-2-피리딘카르복스아마이드에 교반하면서 80부의 술피닐 크로라이드를 방울방울 첨가한 다음 먼저 환류온도에서 3시간 그리고 실온에서 2시간 교반시킨다. 잉여의 술피닐 크로라이드를 증발시키고 잔유물을 뜨거운 메탄올에 쏟은 다음 냉각하고 침전된 생성물을 여과하여(여과액은 보관) 2,2'-옥시비스 프로판으로 충분히 세척하면 14부의 N-(2-크로로에틸)-2-피리딘카르복스아마이드 하이드로크로라이드의 첫번째 분율을 수득한다. 보관된 상기 여과액을 2,2'-옥시비스 프로판과 교반하고 침전된 생성물을 여과하여 건조하면 30부의 N-(2-크로로에틸)-2-피리딘카르복스아마이드하이드로크토라이드의 제2분율을 수득한다.80 parts of sulfinyl chloride are added dropwise to 49 parts of N- (2-hydroxyethyl) -2-pyridinecarboxamide while stirring, followed by stirring for 3 hours at reflux and 2 hours at room temperature. Excess sulfinyl chloride was evaporated, the residue was poured into hot methanol, cooled and the precipitated product was filtered (keeping filtrate) and washed thoroughly with 2,2'-oxybispropane to 14 parts N- (2-chloro Obtain the first fraction of roethyl) -2-pyridinecarboxamide hydrochloride. The filtrate was stirred with 2,2'-oxybis propane and the precipitated product was filtered off and dried to obtain a second fraction of 30 parts of N- (2-chloroethyl) -2-pyridinecarboxamide hydrocoxide. To obtain.
[실시예 9]Example 9
5.5부의 메틸 1-메틸-1H-피롤-2-카르복시레이트와 2.4부의 2-아미노에탄의 혼합물을 2시간 교반 및 환류시킨다. 반응혼합물을 건조하도록 증발시키고 잔유물에 첨가한후 증발시키는 것을 2회 반복한 다음 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 95:5)의 혼합물을 사용하여 시리카켈 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 석유에테르내에 넣고 긁어주면 생성물이 고체화한다. 이것을 여과하여 건조하면 용융점이 78.1℃인 1.9부의 N-(2-하이드록시에틸)-1-메틸-1H-피롤-2-카르복스아마이드를 얻는다.A mixture of 5.5 parts methyl 1-methyl-1H-pyrrole-2-carboxylate and 2.4 parts 2-aminoethane is stirred and refluxed for 2 hours. The reaction mixture was evaporated to dryness, added to the residue and then evaporated twice, then the residue was subjected to column-chromatography on silica using a mixture of trichloromethane and methanol (95: 5 in volume) as eluent. To purify. Pure fractions are collected, the eluate is evaporated and the residue is placed in petroleum ether and scraped to solidify the product. This is filtered and dried to obtain 1.9 parts of N- (2-hydroxyethyl) -1-methyl-1H-pyrrole-2-carboxamide having a melting point of 78.1 占 폚.
450부의 트리크로로메탄내의 40부의 N-(2-하이드록시에틸)-1-메틸-1H-피롤-2-카르복스아마이드의 교반용액에 1방울의 피리딘을 첨가하고 28.3부의 술피닐 크로라이드(약간 발열반응:온도가 15℃로부터 25℃로 상승)를 30분간에 걸쳐 첨가한 다음 실온에서 1야간 교반한다. 반응혼합물을 증발시키고 잔유물을 용출제로서 트리크로로메탄과 메탄올(용량으로 96:4)의 혼합물을 사용하여 시리카겔 상에서 컬럼-크로마토그라피에 의하여 정제한다.One drop of pyridine is added to a stirring solution of 40 parts of N- (2-hydroxyethyl) -1-methyl-1H-pyrrole-2-carboxamide in 450 parts of trichloromethane, and 28.3 parts of sulfinyl chloride ( Slightly exothermic: temperature rises from 15 ° C. to 25 ° C.) over 30 minutes and then stirred at room temperature overnight. The reaction mixture is evaporated and the residue is purified by column-chromatography on silica gel using a mixture of trichloromethane and methanol (96: 4 in volume) as eluent.
순수한 분율을 수집하여 용출제를 증발시키고 잔유물을 2,2'-옥시비스프로판내에서 교반하고 생성물을 여과하여 건조시키면 15부의 N-(2-크로로에틸)-1-메틸-1H-피롤-2-카르복스아마이드를 수득한다Pure fractions were collected to evaporate the eluent and the residue was stirred in 2,2′-oxybispropane and the product was filtered and dried to give 15 parts of N- (2-chloroethyl) -1-methyl-1H-pyrrole- Obtain 2-carboxamide
[실시예 10]Example 10
12.94부의 에틸(2-브로모에틸) 카르바메이트, 15.68부의 5-크로로-1,3-디하이드로-1-(4-피페리피닐)-2H-벤즈이미다졸-2-원, 10.08부의 중탄산나트륨 및 160부의 에탄올의 혼합물을 1야간 교반 및 환류시킨다. 형성된 침전을 여과하여 트리크로로 메탄으로 세척하고 여과액으로부터 층을 분리하여 유기상을 건조, 여과후 증발시킨다. 잔유물을 2-프로판올내에서 교반하고 반응하지 않은 시발물질을 여과하고 여과액을 용출액으로서 트리크로로메탄과 메탄올(용량으로 90:10)의 혼합물을 사용하여 시리카겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 70% 에탄올로부터 결정화시킨후 생성물을 여과하여 건조하면 용융점이 187.7℃인 6.5부의 에틸 {2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸}카르바메이트를 수득한다.12.94 parts ethyl (2-bromoethyl) carbamate, 15.68 parts 5-chloro-1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole-2-one, 10.08 parts A mixture of sodium bicarbonate and 160 parts of ethanol is stirred and refluxed overnight. The precipitate formed is filtered off, washed with trichloromethane, the layers are separated from the filtrate, the organic phase is dried, filtered and evaporated. The residue was stirred in 2-propanol and the unreacted starting material was filtered off and the filtrate was purified by column-chromatography on silica gel using a mixture of trichloromethane and methanol (90:10 in volume) as eluent. do. Pure fractions were collected to evaporate the eluate, the residue was crystallized from 70% ethanol and the product was filtered and dried to give 6.5 parts of ethyl {2- [4- (5-chloro-2,3-dihydro with a melting point of 187.7 ° C. 2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} carbamate.
6.6부의 에틸 {2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페 리디닐]에틸}카르바메이트, 60부의 48% 브롬산과 4부의 물의 혼합물을 2.50시간 교반 및 환류시킨다음 냉각하고 침전된 생성물을 여과하여 물로부터 결정화시키면 용융점이 300℃이상인 5.5부의 1-[1-(2-아미노에틸)-4-피페리디닐]-5-크로로-1,3-디하이드로-2H-벤즈이미다졸-2-원디하이드로 브로마이드를 수득한다.6.6 parts of ethyl {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} carbamate, 60 After stirring and refluxing a mixture of 48% bromic acid and 4 parts of water for 2.50 hours, cooling, and the precipitated product was filtered and crystallized from water. 5.5 parts of 1- [1- (2-aminoethyl) -4- Piperidinyl] -5-chloro-1,3-dihydro-2H-benzimidazole-2-onedihydro bromide is obtained.
[실시예 11]Example 11
38부의 1-(페닐메틸)-4-피페리딘 아민, 40부의 2-크로로니트로벤젠, 32부의 탄산나트륨, 320부의 싸이크로헥산을 내 몇개의 요오드화칼륨 결정의 혼합물을 22시간 교반 및 환류시킨후 냉각하여 300부의 물을 첨가하고 유기층을 분리하여 160부의 벤젠으로 희석한 다음 전체를 150부의 물로 3회 세척한다. 유기상을 황산마그네슘상에서 건조시켜 여과하여 증발시킨 다음 잔유물을 40부의 2,2'-옥시비스프로판과 160부의 헥산의 혼합물에 용해시킨다. -15°C로 냉각한 후 침전을 여과하여 여과액은 보관한다. 침전을 160부의 2,2'-옥시비스프로판으로부터 재결정화시켜 여과하면 용융점이 93.4-94.6°C인 18.5부의 N-(2니트로페닐)-1-(페닐메틸)-4-피페리딘아민을 얻는다.A mixture of several potassium iodide crystals in 38 parts of 1- (phenylmethyl) -4-piperidine amine, 40 parts of 2-chloronitrobenzene, 32 parts of sodium carbonate, and 320 parts of cyclohexane was stirred and refluxed for 22 hours. After cooling, 300 parts of water was added, the organic layer was separated, diluted with 160 parts of benzene, and the whole was washed three times with 150 parts of water. The organic phase is dried over magnesium sulfate, filtered and evaporated and the residue is dissolved in a mixture of 40 parts 2,2'-oxybispropane and 160 parts hexane. After cooling to -15 ° C, the precipitate is filtered and the filtrate is stored. The precipitate was recrystallized from 160 parts of 2,2'-oxybispropane and filtered to give 18.5 parts of N- (2nitrophenyl) -1- (phenylmethyl) -4-piperidinamine having a melting point of 93.4-94.6 ° C. Get
합친 모액을 2,2'-옥시비스프로판으로 희석하여 건조한 염화수소기체를 도입시킨 후 침전된 하이드로크로라이드염을 여과한 다음 120부의 물로 세척하고 불용성부분을 건조하면 용융점이 206-220°C(분해)인 18부의 N-(2-니트로페닐)-1-(페닐메틸)-4-피페리디아민 하이드로크로라이드를 얻는다.The combined mother liquor was diluted with 2,2'-oxybispropane to introduce a dry hydrogen chloride gas, the precipitated hydrochloride salt was filtered off, washed with 120 parts of water, and the insoluble portion was dried, and the melting point was 206-220 ° C. 18 parts N- (2-nitrophenyl) -1- (phenylmethyl) -4-piperidiamine hydrochloride is obtained.
160부의 테트라하이드로푸란내 31부의 N-(2-니트로페닐)-1-(페닐메틸)-4-피페리디아민 용액을 보통 압력 및 40°C의 온도에서 20부의 라네이-닉켈 촉매로 수소화한다. 계산된 양의 수소를 취한후(3물)수소화를 정지하고 촉매를 여과한 다음 여과액으로부터 용매를 증발시킨다. 고체 잔유물을 160부의 2,2'-옥시비스프로판으로 세척하면 용융점이 112-113°C인 22부의 N-[1-(페닐메틸)-4-피페리디닐]-1.2-벤젠디아민을 수득한다. 여과액을 이것의 용량의 약![]()
![]()
5부의 N-[1-(페닐메틸)-4-피페리디닐]-1,2-벤젠디아민 2, 35부의 메틸(이미노메톡시메틸) 카르바메이트, 5부의 식초산과 75부의 트리크로로메탄의 혼합물을 48시간 교반 및 환류시킨다. 반응혼합물을 증발시키고 잔유물을 물에 넣은 다음 용액을 수산화암모니움 용액으로 중화시킨다. 침전된 생성물을 용출액으로서 트리크로로 메탄과 메탄올(용량으로 95:5)의 혼합물로 시리카겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키면 용융점이 169.7°C인 1.75부의 메틸{1,3-디하이드로-1-[1-(페닐메틸)-4-피페리디닐]-2H-벤즈이미다졸-2-이리덴} 카르바메이트를 얻는다.5 parts N- [1- (phenylmethyl) -4-piperidinyl] -1,2-benzenediamine 2, 35 parts methyl (iminomethoxymethyl) carbamate, 5 parts vinegar and 75 parts trichloromethane The mixture is stirred and refluxed for 48 hours. The reaction mixture is evaporated, the residue is placed in water and the solution is neutralized with ammonium hydroxide solution. The precipitated product is purified by column-chromatography on silica gel with a mixture of methane and methanol (95: 5 in volume) as trichloro as eluent. Collecting pure fractions and evaporating the eluate gave 1.75 parts of methyl {1,3-dihydro-1- [1- (phenylmethyl) -4-piperidinyl] -2H-benzimidazole-2 with a melting point of 169.7 ° C. -Iriden} carbamate.
3.64부의 메틸{2,3-디하이드로-1-[1-(페닐메틸)-4-피페리디닐]-1H-벤즈이미다졸-2-이리덴} 카르바메이트, 24부의 염산용액 및 40부의 에탄 올의 혼합물을 1야간 교반 및 환류 시킨다. 반응 혼합물을 증발시키고 잔유물을 물에 용해 시킨 다음 이 용액을 진한 수산화암모니움 용액으로 알카리화시킨다. 침전된 생성물을 여과하여 물로 세척한후 트리크로로메탄내에 용해 시키고 용액을 건조, 여과하여 증발시키면 잔유물로서 2.1부의 1-[1-(페닐메틸)-4-피페리디닐]-1H-벤즈이미다졸-2-아민을 얻는다.3.64 parts of methyl {2,3-dihydro-1- [1- (phenylmethyl) -4-piperidinyl] -1H-benzimidazole-2-yriden} carbamate, 24 parts hydrochloric acid solution and 40 parts The mixture of ethanol is stirred and refluxed overnight. The reaction mixture is evaporated and the residue is dissolved in water and then the solution is alkaline with concentrated ammonium hydroxide solution. The precipitated product was filtered, washed with water and dissolved in trichloromethane. The solution was dried, filtered and evaporated. As a residue, 2.1 parts of 1- [1- (phenylmethyl) -4-piperidinyl] -1H-benzimi Obtain dazol-2-amine.
2부의 1-[1-(페닐메틸)-4-피페리디닐]-1H-벤즈이미다졸-2-아민, 3부의 식초산 무수물 및 45부의 메틸벤젠의 혼합물을 3시간 교반 및 환류 시킨다. 반응 혼합물을 증발시키고 잔유물에 물을 첨가하고 전체를 진한 수산화암모니움 용액으로 알카리화 시킨다. 침전된 생성물을 여과하여 물로 세척하고 트리크로로메탄내에 용해 시킨다. 용액을 건조하여 여과후 증발시키고 잔유물을 2,2-옥시비스프로판으로 부터 결정화 시킨 다음 건조하면 용용점이 189.2°C인 0.8부의 N-{1,3-디하이드로-1-[1-(페닐메틸)-4-피페리디닐]-2H-벤즈이미다졸-2-이리덴} 아세트아마이드를 수득한다.A mixture of 2 parts 1- [1- (phenylmethyl) -4-piperidinyl] -1H-benzimidazol-2-amine, 3 parts vinegar anhydride and 45 parts methylbenzene is stirred and refluxed for 3 hours. The reaction mixture is evaporated and water is added to the residue and the whole is alkaline with concentrated ammonium hydroxide solution. The precipitated product is filtered off, washed with water and dissolved in trichloromethane. The solution was dried, filtered and evaporated. The residue was crystallized from 2,2-oxybispropane and dried. 0.8 parts of N- {1,3-dihydro-1- [1- (phenylmethyl) having a melting point of 189.2 ° C ) -4-piperidinyl] -2H-benzimidazole-2-iridene} acetamide.
12부의 N-{1,3-디하이드로-1-[1-(페닐메틸)-4-피페리디닐]-2H-벤즈이미다졸-2-이리덴} 아세트 아마이드와 120부의 메탄올의 혼합물은 보통 압력 및 실온에서 5부의 10% 팔라듐-활탄 촉매로서 수소화 한다. 계산된 양의 수소를 취한후 촉매를 여과해 내고 여과액을 증발시킨다. 잔유물을 2,2'-옥시비스프로판과 2-프로판올의 혼합물로 부터 결정화 시키고 생성물을 여과하여 에탄올과 2,2'-옥시비스프로판의 혼합물로 부터 재결정화시키면 용융점이 164.5°C인 2.6부의 N-[1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-이리덴] 아세트아마이드를 얻는다.A mixture of 12 parts N- {1,3-dihydro-1- [1- (phenylmethyl) -4-piperidinyl] -2H-benzimidazole-2-iriden} acetamide and 120 parts methanol is usually Hydrogenate as 5 parts 10% palladium-activated carbon catalyst at pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated. The residue was crystallized from a mixture of 2,2'-oxybispropane and 2-propanol, and the product was filtered to recrystallize from a mixture of ethanol and 2,2'-oxybispropane. 2.6 parts of N having a melting point of 164.5 ° C. -[1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole-2-iridene] acetamide is obtained.
[실시예 12]Example 12
1.68부의 1-(4-풀루오로벤조일) 아지리딘, 2.5부의 1-(4-풀루오로페닐)-1,3,8-트리아자스피로[4,5]-데칸-4-원, 10.8부의 벤젠 및 1.6부의 메탄올의 혼합물을 1.50시간 교반 및 환류 시킨다. 반응 혼합물을 냉각시키고 사전에 염화 수소기체로 포화시킨 2-프로판올로 산성화 시킨다. 형성된 하이드로크로라이드 염을여과하고 에탄올과 물(용량으로 8:2)의 혼합물로 부터 결정화 시킨 다음 건조하면 용융점이 279.6℃인 2.4부의 4-플루오로-N[2-[1-4-푸루오로페닐)-4-옥소-1,3,8-트리아자스피로[4,5]데크-8-일에틸} 벤즈 아마이드 하이드로크로라이드를 수득한다.1.68 parts 1- (4-fluorofluoroyl) aziridine, 2.5 parts 1- (4-fluorofluoro) -1,3,8-triazaspiro [4,5] -decane-4-one, 10.8 A mixture of parts benzene and 1.6 parts methanol is stirred and refluxed for 1.50 hours. The reaction mixture is cooled and acidified with 2-propanol previously saturated with hydrogen chloride gas. The hydrochloride salt formed was filtered and crystallized from a mixture of ethanol and water (8: 2 by volume), followed by drying to find 2.4 parts of 4-fluoro-N [2- [1-4-furuo with a melting point of 279.6 ° C. Rophenyl) -4-oxo-1,3,8-triazaspiro [4,5] dec-8-ylethyl} benzamide hydrochloride is obtained.
[실시예 13]Example 13
0.74부의 1-(벤조일) 아지리딘, 1.16부의 1-페닐-1,3,8-트리아자스피로 [4,5]데칸-4-원, 7.2부의 벤젠 및 0.8 부의 메탄올의 혼합물을 2시간 환류 및 교반한다. 반응 혼합물을 냉각하여 1,1'-옥시비스에탄을 첨가한후 전제를 에틸 아세테이트내에서 비등 시킨다. 냉각한후 침전된 생성물을 여과하여 70% 에탄올로 부터 결정화시킨다음 80℃하의진공에서 건조 시키면 용융점이 198.4℃인 0.67부의 N-[2-(4-옥소-1-페닐1,3,8-트리아자스피로[4,5]데크-8-일)에틸]벤즈아마이-드를 얻는다.A mixture of 0.74 parts 1- (benzoyl) aziridine, 1.16 parts 1-phenyl-1,3,8-triazaspiro [4,5] decane-4-one, 7.2 parts benzene and 0.8 parts methanol was refluxed for 2 hours and Stir. The reaction mixture is cooled to add 1,1'-oxybisethane and the whole is boiled in ethyl acetate. After cooling, the precipitated product was filtered and crystallized from 70% ethanol and dried in vacuo at 80 ° C. to 0.67 parts of N- [2- (4-oxo-1-phenyl1,3,8- Triazaspiro [4,5] dec-8-yl) ethyl] benzamide-de is obtained.
[실시예 14]Example 14
실시예 13의 공정을 따르고 당량의 적당한 시발물질을 사용하여 하기 화합물들을 염기 형태로 또는 염기를 적당한 산과 반응 시킴에 의하여 산 부가 염의 형태로 제조한다.The following compounds are prepared in the form of a base, or in the form of an acid addition salt by reacting the base with a suitable acid, using the process of Example 13 and using an equivalent equivalent starting material.



[실시예 15]Example 15
1.2부의 N-(2-브로모에틸)-2-니트로벤즈아마이드, 1.15부의 1-페닐-1,3,8-트리아자스피로[4,5]데칸-4-원과 45부의 N,N-디메틸포름아마이드의 혼합물을 3시간 교반 및 환류 시킨다. 실온에서 1시간 교반하고 80℃에서 진공하에 N,N-디메틸포름아마이드를 증발시킨 다음 잔유물을 2-프로판올과 물의 혼합물내에서 비등시키고 반응하지 않은 시발 물질들을 여과해 낸다. 여과액을 이것의 절반이 되도록 농축하고 농축액에 2-프로판올을 첨가한다. 침전된 생성물을 여과하여 건조하면 용융점이 259.4℃인7부의 2-니트로-N-[2-(4-옥소-1-페닐-1,3,8-트리아자스피로 4,5데크-8-일) 에틸]벤즈아마이드 하이드로 브로마이드를 수득한다.1.2 parts N- (2-bromoethyl) -2-nitrobenzamide, 1.15 parts 1-phenyl-1,3,8-triazaspiro [4,5] decane-4-membered and 45 parts N, N- The mixture of dimethylformamide is stirred and refluxed for 3 hours. After stirring for 1 hour at room temperature and evaporating N, N-dimethylformamide under vacuum at 80 ° C., the residue is boiled in a mixture of 2-propanol and water and the unreacted starting materials are filtered off. The filtrate is concentrated to half of this and 2-propanol is added to the concentrate. The precipitated product was filtered off and dried to 7 parts 2-nitro-N- [2- (4-oxo-1-phenyl-1,3,8-triazaspiro 4,5 deck-8-yl having a melting point of 259.4 ° C. ) Ethyl] benzamide hydrobromide is obtained.
[실시예 16]Example 16
6.9부의 N-(2-크로로에틸)-2-푸란카르복스아마이드, 0.2부의 1-페닐-1,3,8-트리아자스피로[4,5] 데칸-4-원, 6.6부의 요오드화 칼륨 및 135부의 N,N-디메틸포름아마이드의 혼합물을 먼저 환류 온도에서 3시간 그리고 실온에서 1야간 교반 시킨다. 반응 혼합물을 증발시키고 잔유물을 물에 넣은 다음 전체를 알카리화 시킨후 생성물을 4-메틸--2-펜타논으로 추출한다. 추출액을 건조하여 여과하고 증발시킨 다음 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 96:4)의 혼합물을 사용하여 시리카겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을수집하여 용출액을 증발시키고 고체잔유물을 2-프로파논으로 부터 결정화 시킨다. 생성물을 여과하여 건조하면 용융점이 186.3℃인 1.9 부의 N-[2-(4-옥소-1-페닐 1,3,8-트리아자스피로[4,5] 데크-8-일) 에틸]-2-푸란카르복스아마이드를 수득한다.6.9 parts N- (2-chloroethyl) -2-furancarboxamide, 0.2 parts 1-phenyl-1,3,8-triazaspiro [4,5] decane-4-one, 6.6 parts potassium iodide and A mixture of 135 parts of N, N-dimethylformamide is first stirred at reflux for 3 hours and at room temperature overnight. The reaction mixture is evaporated, the residue is taken up in water and the whole is alkaline, and then the product is extracted with 4-methyl-2-pentanone. The extract is dried, filtered and evaporated and the residue is purified by column-chromatography on silica gel using a mixture of trichloromethane and methanol (96: 4 in volume) as eluent. Collect pure fractions to evaporate the eluate and crystallize the solid residue from 2-propanone. The product was filtered off and dried to give 1.9 parts of N- [2- (4-oxo-1-phenyl 1,3,8-triazaspiro [4,5] deck-8-yl) ethyl] with a melting point of 186.3 ° C. -Furancarboxamide is obtained.
[실시예 17]Example 17
실시예 XVI의 공정을 따르고 N-(2-크로로에틸)-2-푸란카르복스아마이드를 1-(4-풀루오로페닐)-1,3,8-트리아자스피로 [4,5] 데칸-4-원과 반응시킴에 의하여 용융점이 254.2℃인 N-{2-[1-(4-풀루오로페닐)-4-옥소-1,3,8-트리아자스피로 [4,5] 데크-8-일] 에틸}-2-푸란카르복스아마이드하이드로그로라이드를 제조한다.Follow the process of Example XVI and replace N- (2-chloroethyl) -2-furancarboxamide with 1- (4- pullophenyl) -1,3,8-triazaspiro [4,5] decane. N- {2- [1- (4- pullophenyl) -4-oxo-1,3,8-triazaspiro [4,5] deck having a melting point of 254.2 ° C. by reaction with a 4-membered -8-yl] ethyl} -2-furancarboxamide hydrochloride is prepared.
[실시예 18]Example 18
4.4부의N-(2-크로로에틸)-2-피리딘카르복스 아마이드, 13.8부의 1-페닐-1,3,8-트리아자스피로[4,5] 데칸-4-원, 3,3부의 요오드화 칼륨 및 200부의 4-메틸-2-펜타논의 혼합물을 24시간 교반 및 환류 시킨다. 반응 혼합물을 증발시키고 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 98:2)의 혼합물을 사용하여 시리카 겔 상에서 컬럼-크로마토그라피에 의하여 정제한다.4.4 parts N- (2-chloroethyl) -2-pyridinecarboxamide, 13.8 parts 1-phenyl-1,3,8-triazaspiro [4,5] decane-4-one, 3,3 parts iodide A mixture of potassium and 200 parts of 4-methyl-2-pentanone is stirred and refluxed for 24 hours. The reaction mixture is evaporated and the residue is purified by column-chromatography on a silica gel using a mixture of trichloromethane and methanol (98: 2 in volume) as eluent.
순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 2-프로판올내에서 하이드로크로라이드 염으로 전환시키고 염을 여과하여 에탄올로 부터 결정화 시키면 용융점이 250.5℃인 1,1'부의 N-[2-(4-옥소-1-페닐-1,3,8-트리아자스피로 [4,5] 데크-8일) 에틸]-2-피리딘카르복스아마이드 디하이드로크로라이드 하이드레이트를 얻는다.Pure fractions were collected to evaporate the eluate, convert the residue to a hydrochloride salt in 2-propanol, and the salt was filtered and crystallized from ethanol to yield 1,1'part of N- [2- (4- Obtain oxo-1-phenyl-1,3,8-triazaspiro [4,5] deck-8 yl) ethyl] -2-pyridinecarboxamide dihydrochloride hydrate.
[실시예 19]Example 19
3.75부의 N-(2-크로로에틸)-1-메틸-1H-피롤-2-카르복스아마이드, 5부의 1-(4-플루오로페닐)-1,3,8-트리아자스피로 4,5 데칸-4-원, 1.7부의 중탄산나트륨, 0.1부의 요오드화 칼륨 및 160부의 4-메틸-2-펜타논의 혼합물을 48시간 교반 및 환류 시킨다. 반응 혼합물을 냉각하고 용매를 증발시킨다. 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 95:5)의 혼합물을 사용하여 시리카 겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 2-프로판올과 2,2'-옥시비스프로판 내에서 하이드로크로라이드 염으로 전환 시킨다. 염을 여과하여 메탄올로 부터 결정 화시키면 용융점이 273.4℃인 0.8부의 N-2-[1-(4-풀루오로페닐)-4-옥소-1,3,8-트리아자스피로[4,5] 데크-8-일] 에틸}-1-메틸-1-피롤-카르복스아마이드 하이드로크로 라이드를 수득한다.3.75 parts N- (2-chloroethyl) -1-methyl-1H-pyrrole-2-carboxamide, 5 parts 1- (4-fluorophenyl) -1,3,8-triazaspiro 4,5 A mixture of decan-4-one, 1.7 parts sodium bicarbonate, 0.1 parts potassium iodide and 160 parts 4-methyl-2-pentanone is stirred and refluxed for 48 hours. Cool the reaction mixture and evaporate the solvent. The residue is purified by column-chromatography on silica gel using a mixture of trichloromethane and methanol (95: 5 in volume) as eluent. Pure fractions are collected to evaporate the eluate and the residue is converted to the hydrochloride salt in 2-propanol and 2,2'-oxybispropane. The salt was filtered and crystallized from methanol. 0.8 parts of N-2- [1- (4-pulophenyl) -4-oxo-1,3,8-triazaspiro [4,5 having a melting point of 273.4 ° C. Deck-8-yl] ethyl} -1-methyl-1-pyrrole-carboxamide hydrochloride is obtained.
[실시예 20]Example 20
8부의 4-플루오로-2-니트로-N-[2-(4-옥소-1-페닐-1,3,8-트리아자스피로 [4,5] 데크-8-일) 에틸] 벤즈아마이드, 40부의 메탄올 및 90부의 테트라하이드로푸란의 혼합물을 보통 압력 및 실온에서 3부의 라네이-닉켈 촉매로 수소화 한다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨다. 잔유물을 2-프로판올내에서 하이드트크로라이드 염으로 전환시키고 염을 여과하여 에탄올로 부터 재결정 시키면 용융점이 167.3℃인 1부의 2-아미노-4-풀루오로-N-[2-(4-옥소-1-페닐-1,3,8-트리아자스피로 [4,5] 데크-8-일) 에틸] 벤즈아마이드 하이드로크로라이드를 얻는다.8 parts 4-fluoro-2-nitro-N- [2- (4-oxo-1-phenyl-1,3,8-triazaspiro [4,5] deck-8-yl) ethyl] benzamide, A mixture of 40 parts methanol and 90 parts tetrahydrofuran is hydrogenated with 3 parts Lanei-Nickel catalyst at normal pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated. The residue was converted to the hydrochloride salt in 2-propanol and the salt was filtered off and recrystallized from ethanol to give 1 part 2-amino-4- pullouro-N- [2- (4-oxo having a melting point of 167.3 ° C. -1-phenyl-1,3,8-triazaspiro [4,5] dec-8-yl) ethyl] benzamide hydrochloride is obtained.
[실시예 21]Example 21
8부의4-풀루오로-N-{2-[1-(4-풀루오로 페닐)-4-옥소-1,3,8-트리아자스로 [4,5] 데크-8-일] 에틸}-2-니트로벤즈아마이드 하이드로크로라이드, 120부의 메탄올 및 25부의 물의 혼합물을 보통압력 및 실온에서 5부의 라네이-닉켈 촉매로서 수소화한다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨다. 잔유물을 메탄올로부터 결정화 시켜 생성물을 여과하고 에탄올로부터 재결정시키면 용융점이 223.5℃인 1부의 2-아미노一4-풀루오로-N-{2-[1-(4-풀루오로페닐)-4-옥소-1,3-8-트리아자스피로 [4,5] 테크-8-일] 에틸} 벤즈아마이드 하이드로크로라이드 하이드레이트를 수득한다.8 parts 4-Pluoro-N- {2- [1- (4-Pluorophenyl) -4-oxo-1,3,8-triazathro [4,5] dec-8-yl] ethyl } A mixture of 2-nitrobenzamide hydrochloride, 120 parts methanol and 25 parts water is hydrogenated as 5 parts Lanei-Nickel catalyst at normal pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated. The residue was crystallized from methanol, the product was filtered off and recrystallized from ethanol to give 1 part 2-aminol4-pullouro-N- {2- [1- (4-pululophenyl) -4- with a melting point of 223.5 ° C. Oxo-1,3-8-triazaspiro [4,5] tech-8-yl] ethyl} benzamide hydrochloride hydrate is obtained.
[실시예 22]Example 22
6부의 5-크로로-2-메톡시-4-니트로-N-[2-(4-옥소-1-페닐-1,3,8-트리아자스피 로 [4,5] 데크-8-일) 에틸 벤즈아마이드와, 50부의 식초산의 혼합물을 보통압력 및 실온에서 1부의 라네이-닉켈 촉매로 수소화 한다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨다. 잔유물을물에 넣고 전체를 알카리화 시킨 다음 침전된 생정물을 여과하여 에탄올로 부터 2회 결정화 시키면 용융점이 247.4℃인 1.7부의 4-아미노-5-크로로-2-메톡시-N-[2-(4-옥소-1-페닐-1,3,8-트리아자스피로 [4,5] 테크-8-일) 에틸] 벤즈아마이드를 얻는다.6 parts 5-chloro-2-methoxy-4-nitro-N- [2- (4-oxo-1-phenyl-1,3,8-triazaspiro [4,5] deck-8-yl ) A mixture of ethyl benzamide and 50 parts of vinegar acid is hydrogenated with 1 part of Raney-Nickel catalyst at normal pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated. The residue was poured into water, the whole was alkalized, and the precipitated crude was crystallized twice from ethanol. 1.7 parts of 4-amino-5-chloro-2-methoxy-N- [2 having a melting point of 247.4 ° C. were obtained. -(4-oxo-1-phenyl-1,3,8-triazaspiro [4,5] tech-8-yl) ethyl] to obtain a benzamide.
[실시예 23]Example 23
실시예 22의 공정을 따르고 시발물질로서 5-크로로-N-{2-[1-(4-풀루오로페닐)-4-옥소-1,3,8-트리아자스피로 [4,5] 데크-8-일] 에틸} 2-메톡시-4-니트로벤즈아마이드를 사용하여 수득한 생성물을 하이드로크로라이드 염으로 전환시켜 용융점이 239.8℃인 4-아미노-5-크로로-N-{2-[1-(4-풀루오로페닐)-4-옥소-1,3,8-트리아자스피로 4,5 데크-8-일] 에틸}-2-메톡시벤즈아마이드 하이드로크로라이드를 수득한다.Following the process of Example 22 and as starting material 5-chloro-N- {2- [1- (4-fluorofluoro) -4-oxo-1,3,8-triazaspiro [4,5] Deck-8-yl] ethyl} 2-methoxy-4-nitrobenzamide is used to convert the product obtained to a hydrochloride salt to 4-amino-5-chloro-N- {2 having a melting point of 239.8 ° C. Obtain [-(1- (4-Pluorophenyl) -4-oxo-1,3,8-triazaspiro 4,5 deck-8-yl] ethyl} -2-methoxybenzamide hydrochloride .
[실시예 24]Example 24
9부의 N-{2-[1-(4-풀루오로페닐)-4-옥소-1,3,8-트리아자스피로 [4,5] 데크-8-일] 에틸-2-니트로벤즈아마이드 하이드로크로라이드와 150부의 식초산의 혼합물을 보통 압력 및 실온에서 2부의 10% 팔라듐-활탄 촉매로 수소화 한다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨다. 잔유물을 물에 넣고 전제를 수산화 암모니움으로 알카리화한 다음 생성물을 트리크로로메탄으로 2회 추출한다. 합친 추출액을 물로 2회 세척, 건조 및 여과하여 증발시킨다. 고체 잔유물을 2-프로판 올내에서 비등 시키고 여과한 다음 여과액으로 부터 결정화 하도록 방치한다. 생성물을 여과하여 진공에서 건조 시키면 용융점이 194.9℃인 5부의 2-아미노-N-{2-[1-(4-풀루오로페닐)-4-옥소-1,3,8-트리아자스피로 [4,5] 테크-8-일] 에틸} 벤즈아마이드를 수득한다.9 parts N- {2- [1- (4-fluorofluorophenyl) -4-oxo-1,3,8-triazaspiro [4,5] deck-8-yl] ethyl-2-nitrobenzamide The mixture of hydrochloride and 150 parts of vinegar acid is hydrogenated with 2 parts of 10% palladium-activated carbon catalyst at normal pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated. The residue is taken up in water and the whole is alkaline with ammonium hydroxide and the product is extracted twice with trichloromethane. The combined extracts are washed twice with water, dried and filtered to evaporate. The solid residue is boiled in 2-propanol, filtered and left to crystallize from the filtrate. The product was filtered and dried in vacuo to give 5 parts 2-amino-N- {2- [1- (4- pullophenyl) -4-oxo-1,3,8-triazaspiro [1] with a melting point of 194.9 ° C. 4,5] tech-8-yl] ethyl} benzamide.
[실시예 25]Example 25
9부의 2-아미노-4풀로오로-N-{2-[1-(4풀로오로페닐)-4-옥소-1,3,8-트라아자스피로[4,5]테크-8-일] 에틸}-벤즈아마이드, 8.5부의 식초산 무수물 및 85부의 물의 혼합물을 80℃의 물 중탕내에서 30분간 교반한다. 반응 혼합물을 냉각하여 수산화 암모니움으로 알카리화 시키고 생성물을 트리크로로메탄으로 추출한다. 수용액 상을 분리하여 트리크로로메탄으로 추출하고 합친 유기상을 물로 3회 세척, 건조 및 여과하여 증발시킨다. 잔유물을 2-프로판올로 부터 결정화시켜 생성물을 여과후 건조시키면 용융점이 195.1℃인 5.2부의 2-아미노-4풀로오로-N-{2-[1-(4풀로오로페닐)-4-옥소-1,3,8-트라아자스피로 [4,5]데크-8-일] 에틸} 벤즈아마이드를 얻는다.9 parts 2-amino-4 pullo-N- {2- [1- (4 pullophenyl) -4-oxo-1,3,8-traazaspiro [4,5] tech-8-yl] ethyl } -Benzamide, a mixture of 8.5 parts vinegar anhydride and 85 parts water is stirred for 30 minutes in a water bath at 80 ° C. The reaction mixture is cooled to alkaline with ammonium hydroxide and the product is extracted with trichloromethane. The aqueous phase is separated and extracted with trichloromethane and the combined organic phases are washed three times with water, dried and filtered and evaporated. The residue was crystallized from 2-propanol and the product was filtered and dried. 5.2 parts of 2-amino-4 pullolo-N- {2- [1- (4 pullophenyl) -4-oxo-1 having a melting point of 195.1 ° C. were obtained. , 3,8-traazaspiro [4,5] dec-8-yl] ethyl} benzamide.
[실시예 26]Example 26
1.68부의 1-(4-풀루오로벤조일) 아지리딘, 2.5부의 5-크로로-1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원, 10.8부의 벤젠 및 1.6부의 메탄올의 혼합물을 1.50시간 동안 교반 및 환류 시킨다. 침전된 생성물을 여과하여 2-프로파논으로 세척하여 2-프로판올로 부터 결정화 시키면 용융점이 239.6℃인 2.47부의 N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드를 얻는다.1.68 parts 1- (4-Pluorobenzoyl) aziridine, 2.5 parts 5-chloro-1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole-2-one, 10.8 A mixture of parts benzene and 1.6 parts methanol is stirred and refluxed for 1.50 hours. The precipitated product was filtered off, washed with 2-propanone and crystallized from 2-propanol to give 2.47 parts of N- {2- [4- (5-chloro-2,3-dihydro-2- with a melting point of 239.6 ° C. Oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4- pullobenzamide.
[실시예 XXVⅡ]Example XXVII
실시예 XXVI의 공정을 따르고 당량의 적당한 시발물질을 사용하여 하기 화합물을 유리 염기 형태 또는 유리 염기를 적당한 산과 반응시킴에 의하여 산부가 염 형태로 수득한다.The acid is added in the form of an acid addition salt by following the process of Example XXVI and using the equivalent starting material to react the following compounds with free base or free base with a suitable acid.
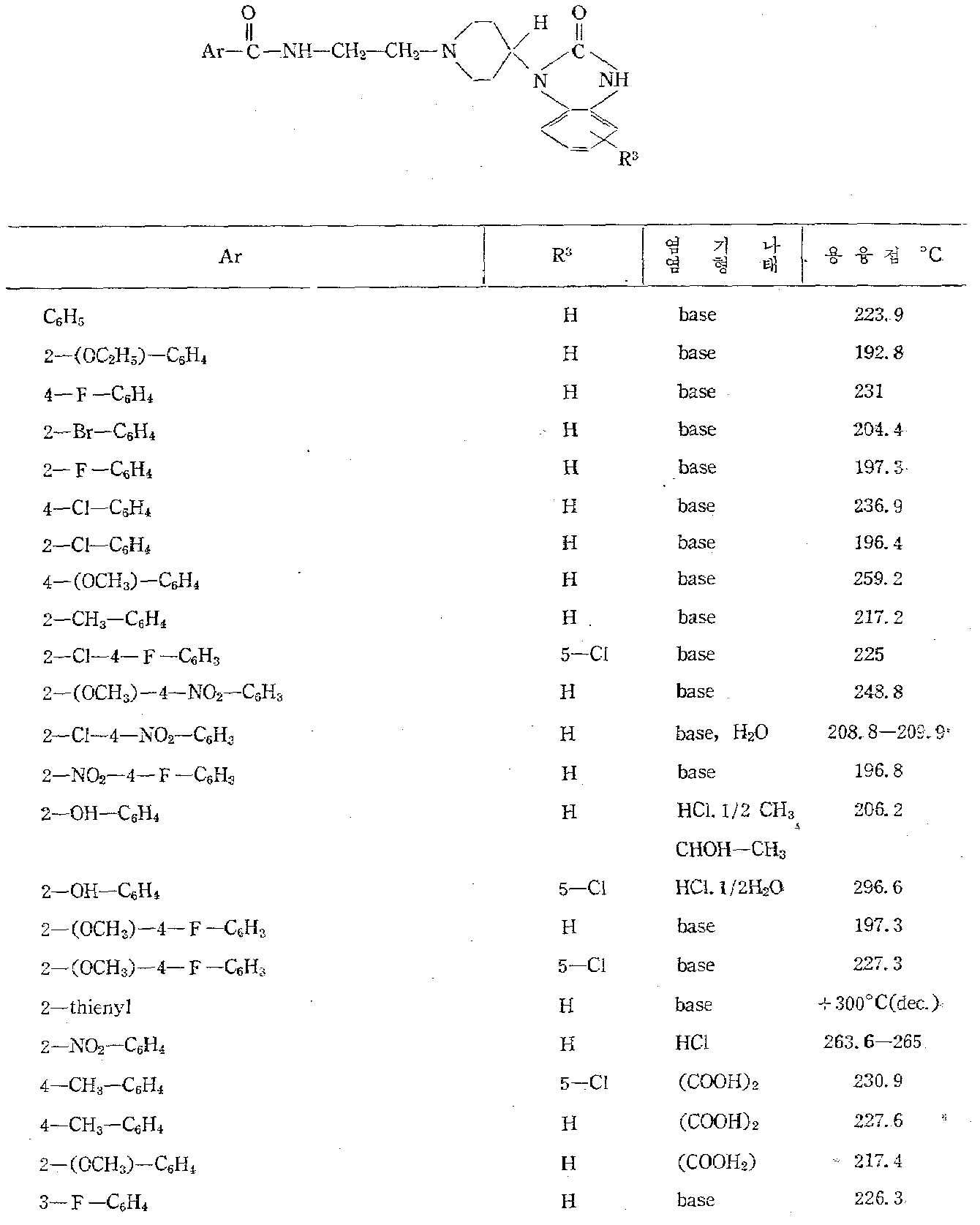

[실시예 XXVⅢ]Example XXVIII
실시예 XXVI의 공정을 따라 (1-아지리디닐) (4-풀루오로페닐) 메타논을 N-[1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-이리덴] 아세트아미드와 반응 시킴에 의하여 용융점이 216.9℃인 N-[2-{4(아세틸아미노)-2,3-디하이드로-1H-벤즈이미다조릴-1-일]-1-피페리디닐} 에틸]-4-풀루오로벤즈아마이드를 제조한다.Following the process of Example XXVI, (1-aziridinyl) (4- pullophenyl) methanone was replaced with N- [1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole N- [2- {4 (acetylamino) -2,3-dihydro-1H-benzimidazolyl-1-yl] -1 having a melting point of 216.9 ° C. by reacting with 2-iridene] acetamide -Piperidinyl} ethyl] -4- pullobenzamide is prepared.
[실시예 XXIX]Example XXIX
1.5부의 N-(2-브로모에틸)-2-니트로-벤즈아마이드, 1.55부의 5-크로로-1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원 및 27부의 N, N-디메틸포름아마이드의 혼합물을 5시간 교반 및 환류 시킨다. 반응 혼합물을 증발 시키고 오일상의 잔유물을 에탄올로부터 결정화시켜 생성물을 여과한 다음 건조 시키면 용융점이 273.2℃인 2.2부 (671)의 N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-2-니트로벤즈아마이드 하이드로 브로마이드 헤미하이드레이트를 수득한다.1.5 parts N- (2-bromoethyl) -2-nitro-benzamide, 1.55 parts 5-chloro-1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole-2 -A mixture of the original and 27 parts of N, N-dimethylformamide is stirred and refluxed for 5 hours. The reaction mixture was evaporated and the oily residue was crystallized from ethanol to filter the product and dried to give 2.2 parts 671 of N- {2- [4- (5-chloro-2,3-di having a melting point of 273.2 ° C. Hydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -2-nitrobenzamide hydrobromide hemihydrate is obtained.
[실시예 XXX][Example XXX]
8.7부의 4-풀루오로-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피폐리디닐] 에틸}-2-니트로벤즈아마이드와 120부의 메탄올의 혼합물을 보통 압력 및 실온에서 2부의 라네이-닉켈 촉매로 수소화 한다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨 다음 잔유물을 메탄올과 2-프로판올의 혼합물로부터 결정화 시킨다. 생성물을 여과하여 건조하면 용융점이 229.3℃인 4.5부의 2-아미노-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드를 수득한다.8.7 parts 4-Pluoro-N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -2- A mixture of nitrobenzamide and 120 parts of methanol is hydrogenated with 2 parts of Raney-Nickel catalyst at normal pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated and the residue is crystallized from a mixture of methanol and 2-propanol. The product was filtered off and dried to give 4.5 parts 2-amino-N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1- with a melting point of 229.3 ° C. Piperidinyl] ethyl} -4- pullobenzamide is obtained.
[실시예 XXXI][Example XXXI]
실시예 XXX의 공정에 따라 N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸]-4-풀루오로-2-니트로벤즈아마이드를 수소화한 다음 수득한 생성물을 염산으로서 하이드로 크로라이드 염으로 전환 시킴에 의하여 용융점이 250℃인 에틸}-4-풀루오로벤즈아마이드 하이드르 크로라이드를 수득한다.N- {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl according to the process of Example XXX ] -4-Pluoro-2-nitrobenzamide is hydrogenated and the resulting product is converted to hydrochloride salt as hydrochloric acid to yield ethyl} -4- pullobenzamide hydrochrom with a melting point of 250 ° C. Obtain a ride.
[실시예 XXXⅡ]Example XXXII
90부의 테트라하이드로푸란내 16부의 N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-2-니트로벤즈아마이드와 40부의 메탄올의 혼합물을 보통압력 및 실온에서 3부의 라네이-닉켈 촉매로 수소화한다. 계산된 양의 수소를 취한후 사전에 암모니아 기체로 포화시킨 메탄올 용액을 첨가하고 촉매를 여과한 다음 여과액을 증발시킨다. 잔유물을 산성화 시킨 물에 넣고 전체를 트리크로로메탄으로 2회 세척한다. 수용액상을 분리하여 묽은 수산화나트륨 용액으로 알카리화 하고 생성물을 트리크로로메탄으로 2회 추출하여 합친 추출액을 물로 3회 세척, 건조 및 여과후 증발시킨다. 잔유물을 2,2-옥시비스프로판내에서 분쇄하고 생성물을 여과후 에탄올과 소량의 물의 혼합물로부터 결정화시키면 용융점이 226.8℃인 2부의 2-아미노-N{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일(-1-피페리디닐]에틸} 벤즈아마이드를 수득한다.16 parts N- {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl in 90 parts of tetrahydrofuran] A mixture of ethyl} -2-nitrobenzamide and 40 parts of methanol is hydrogenated with 3 parts of Raney-Nickel catalyst at normal pressure and room temperature. After taking the calculated amount of hydrogen, a methanol solution previously saturated with ammonia gas is added, the catalyst is filtered off and the filtrate is evaporated. The residue is taken up in acidified water and washed twice with trichloromethane. The aqueous phase is separated, alkalined with dilute sodium hydroxide solution, the product is extracted twice with trichloromethane, and the combined extracts are washed three times with water, dried and filtered and then evaporated. The residue was triturated in 2,2-oxybispropane and the product was filtered and crystallized from a mixture of ethanol and a small amount of water to give 2 parts 2-amino-N {2- [4- (5-chloro- with a melting point of 226.8 ° C. 2,3-dihydro-2-oxo-1H-benzimidazol-1-yl (-1-piperidinyl] ethyl} benzamide is obtained.
[실시예 XXXⅢ]Example XXXIII
실시예 XXXⅡ의 공정에 따라 하기 화합물을 해당하는 니트로 화합물로 부터 제조한다.The following compounds were prepared from the corresponding nitro compounds according to the procedure of Example XXXII.
4-아미노-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤조이미다졸-1-일)-1-피페리디닐] 에틸}-2-메톡시벤즈아마이드 하이드레이트:용융점 220.6℃ 2-아미노-N- {2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸} 벤즈아마이드 헤미하드레이트:용융점 219.2℃4-amino-N- {2- [4- (2,3-dihydro-2-oxo-1H-benzoimidazol-1-yl) -1-piperidinyl] ethyl} -2-methoxybenzamide Hydrate: melting point 220.6 ° C. 2-amino-N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} benzamide Hemihydrate: Melting point 219.2 ℃
[실시예 XXXIV]Example XXXIV
4.2부의 5-크로로-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-2-메톡시-4-니트로 벤즈아마이드와 150부의 식초산의 혼합물을 보통 압력 및 실온에서 1부의 라네이-닉켈 촉매로 수소화 한다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨 다음 잔유물에 물을 첨가하고 전체를 묽은 수산화나트륨 용액으로 알카리화 한다. 침전된 생성물을 여과하여(여과액은 보관함) 메탄올로부터 결정화 시킨 다음 건조 시키면 용융점이 230.1℃인 0.8부의 4-아미노-5-크로로-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-2-메톡시벤즈아마이드의 첫번째 분율을 얻는다.4.2 parts 5-chloro-N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -2-meth A mixture of oxy-4-nitrobenzamide and 150 parts of vinegar acid is hydrogenated with 1 part of Raney-Nickel catalyst at normal pressure and room temperature. After taking up the calculated amount of hydrogen, the catalyst is filtered off, the filtrate is evaporated, water is added to the residue and the whole is alkaline with dilute sodium hydroxide solution. The precipitated product was filtrated (the filtrate was stored), crystallized from methanol and then dried to give 0.8 parts of 4-amino-5-chloro-N- {2- [4- (2,3-dihydro) having a melting point of 230.1 ° C. -2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -2-methoxybenzamide.
보관된 여과액을 농축하여 여과하면 용융점이 232.5℃인 1.6부의 4-아미노-5-크로로-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-2-메톡시벤즈아마이드를 수득한다.The stored filtrate was concentrated and filtered to 1.6 parts 4-amino-5-chloro-N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimidazole having a melting point of 232.5 ° C. -1-yl) -1-piperidinyl] ethyl} -2-methoxybenzamide.
[실시예 XXXV]Example XXXV
1.68부의 1-(4-풀루오로벤조일)아지리딘, 2.16부의 1.3-디하이드로-1-(3,6-디하이드로-1-(2H)피리디닐)-2H-벤즈이미다졸-2-원, 10.8부의 벤젠 및 1.6부의 메탄올을 혼합물을 1.50시간 교반 및 환류 시킨다. 냉각하여 침전된 생성물을 여과하고 2-프로파논으로 세척하여 에탄올로부터 결정화 시키면 용융점이 202.7℃인 1.76부의 N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-3,6-디하이드로-1-(2H)-피리디닐]에틸}-4-풀루오로벤즈아마이드를 얻는다.1.68 parts 1- (4-fluorobenzoyl) aziridine, 2.16 parts 1.3-dihydro-1- (3,6-dihydro-1- (2H) pyridinyl) -2H-benzimidazole-2-one , 10.8 parts benzene and 1.6 parts methanol were stirred and refluxed for 1.50 hours. After cooling, the precipitated product was filtered off, washed with 2-propanone and crystallized from ethanol. 1.76 parts of N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimimi having a melting point of 202.7 占 폚 was obtained. Dazol-1-yl) -3,6-dihydro-1- (2H) -pyridinyl] ethyl} -4- pullobenzamide is obtained.
[실시예 XXXVI]Example XXXVI
1.68부의 1-(4-풀루오로벤조일) 아지리딘, 2.07부의 2,3-디하이드로-2-옥소-3-(4-피페리디닐)-1H-벤즈이미다졸-1-프로판니트릴 하이드로크로라이드, 1.65부의 N, N-디에틸에탄아민, 10.8부의 벤젠 및 1.6부의 메탄올의 혼합물을 1.50 시간 교반 및 환류시킨다. 반응 혼합물을 냉각하여 물에 쏟고 생성물을 트리크로로메탄으로 추출한후 추출액을 건조, 여과한 다음 증발시킨다. 잔유물을 2-프로판올로부터 결정화하여 생성물을 여과하고 2-프로판올로부터 재결정시키면 용융점이 172.2℃인 1부의 N-[2-{4-[3-(2-시아노에틸)-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드를 수득한다.1.68 parts 1- (4-Pluorobenzoyl) aziridine, 2.07 parts 2,3-dihydro-2-oxo-3- (4-piperidinyl) -1H-benzimidazole-1-propanenitrile hydrochrome Ride, a mixture of 1.65 parts N, N-diethylethanamine, 10.8 parts benzene and 1.6 parts methanol is stirred and refluxed for 1.50 hours. The reaction mixture is cooled, poured into water, the product is extracted with trichloromethane, the extract is dried, filtered and evaporated. The residue was crystallized from 2-propanol to filter the product and recrystallized from 2-propanol to give 1 part N- [2- {4- [3- (2-cyanoethyl) -2,3-dihydro with a melting point of 172.2 ° C. -2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4- pullobenzamide.
[실시예 XXXVⅡ]Example XXXVII
5.6부의 N-(2-크로로에틸)-1-메틸-1H-피롤-2-카르복스아마이드, 6.52부의 1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원, 2.52부의 중탄산나트륨, 0.1부의 요오드화 칼륨 및 240부의 4-에틸-2-펜타논의 혼합물을 62시간 교반 및 환류 시킨다.5.6 parts N- (2-chloroethyl) -1-methyl-1H-pyrrole-2-carboxamide, 6.52 parts 1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole A mixture of 2-membered, 2.52 parts sodium bicarbonate, 0.1 parts potassium iodide and 240 parts 4-ethyl-2-pentanone is stirred and refluxed for 62 hours.
반응 혼합물을 냉각하여 용매를 증발시키고 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 95:5)의 혼합물을 사용하여 시리카 겔 상에서 컬럼 크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 에탄올, 2-프로판올 및 2,2'-옥시비스프로판내에서 하이드로르크로라이드 염으로 전환시킨다. 염을 여과하여 에탄올과 2,2'-옥시비스프로판의 혼합물로부터 결정화 시키면 용융점이 237.7℃인 0.6부의 N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-1-메틸 -1H-피롤-2-카르복스아마이드 하이드로크로라이드 하이드레이트를 수득한다.The reaction mixture is cooled to evaporate the solvent and the residue is purified by column chromatography on silica gel using a mixture of trichloromethane and methanol (95: 5 in volume) as eluent. Pure fractions are collected to evaporate the eluate and the residue is converted to hydrochloride salt in ethanol, 2-propanol and 2,2'-oxybispropane. The salts were filtered to crystallize from a mixture of ethanol and 2,2'-oxybispropane, and 0.6 parts of N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimimi having a melting point of 237.7 ° C was obtained. Dazol-1-yl) -1-piperidinyl] ethyl} -1-methyl-1H-pyrrole-2-carboxamide hydrochloride hydrate is obtained.
[실시예 XXXVⅢ]Example XXXVIII
실시예 XXXVⅡ의 공정에 따라 하기 화합물을 제조한다.The following compound was prepared according to the process of Example XXXVII.
N-(2-크로로에틸)-3-피리딘카르복스아마이드 하이드로크로라이드를 1,3-디하이 드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원과 반응시킴에 의하여 용융점이 214.1℃인 N-{2-[4-(2,3-디하이드로-2-1-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-3-피리딘카르복스아마이드 디하이드로크로라이드 하이드레이트:Reacts N- (2-chloroethyl) -3-pyridinecarboxamide hydrochloride with 1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole-2-one N- {2- [4- (2,3-dihydro-2-1-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -3 having a melting point of 214.1 ° C. Pyridinecarboxamide dihydrocride hydrate:
N-(2-브로모에틸)-4-풀루오로-2-니트로벤즈아마이드를 5-크로로-1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원과 반응시킴에 의하여 N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로-2-니트로벤즈아마이드N- (2-bromoethyl) -4-fuluro-2-nitrobenzamide to 5-chloro-1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole- N- {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] by reaction with a 2-membered Ethyl} -4-Pluoro-2-nitrobenzamide
[실시예 XXXIX]Example XXXIX
16부의 N-(2-브로모에틸)-4-풀루오로-2-니트로벤즈아마이드, 12.6부의 5-크로로-1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원과 216부의 N,N-디메틸포름아마이드의 혼합물을 3시간 교반 및 환류시킨다. N,N-디메틸포름아마이드를 증발시키고 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 95:5)의 혼합물을 사용하여 시리카 겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 2-프로파논과 2-프로판올 내에서 하이드로크로라이드 염으로 전환시킨다. 염을 여과하여 메탄올로부터 결정화시키면 용융점이 276.8℃인 1.5부의 N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸} -4-풀루오로-2-니트로 벤즈아마이드 하이드로크로라이드를 얻는다.16 parts N- (2-bromoethyl) -4-fuluro-2-nitrobenzamide, 12.6 parts 5-chloro-1,3-dihydro-1- (4-piperidinyl) -2H- A mixture of benzimidazole-2-one and 216 parts N, N-dimethylformamide is stirred and refluxed for 3 hours. N, N-dimethylformamide is evaporated and the residue is purified by column-chromatography on a silica gel using a mixture of trichloromethane and methanol (95: 5 in volume) as eluent. Pure fractions are collected to evaporate the eluate and convert the residue to a hydrochloride salt in 2-propanone and 2-propanol. The salt was filtered and crystallized from methanol, and 1.5 parts of N- {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) having a melting point of 276.8 ° C. was obtained. -1-piperidinyl] ethyl} -4- pullouro-2-nitro benzamide hydrochloride.
[실시예 XL]Example XL
2.2부의 N-(2-크로로에틸)-2-피리딘카르복스아마이드 하이드로크로라이드, 6.6부의 1,3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원, 1.66부의 요오드화칼륨, 및 120부의 4-메틸-2-펜다논의 혼합물을 1야간 교반 및 환류시킨다. 냉각후 침전된 생성물을 여과하여 물에 용해시키고 용액을 탄산나트륨으로 알카리화한 후 생성물을 4-메틸-2-펜타논으로 3회 추출한다. 합친 추출액을 물로 세척건조 및 여과하여 증발시킨후 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 95:5)의 혼합물을 사용하여 시리카 겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 2-프로파논과 2-프로판올내에서 하이드로 크로라이드 염으로 전환시킨 다음 염을 여과하여 에탄올과 2,2′-옥시비스프로판(용량으로 1:1)의 혼합물로부터 결정화시키면 용융점이 165.7℃인 2부의 N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸}-2-피리딘카르복스아마이드 디하이드로 크로라이드 디하이드레이트를 얻는다.2.2 parts N- (2-chloroethyl) -2-pyridinecarboxamide hydrochloride, 6.6 parts 1,3-dihydro-1- (4-piperidinyl) -2H-benzimidazole-2-one , A mixture of 1.66 parts of potassium iodide, and 120 parts of 4-methyl-2-pentanone are stirred and refluxed overnight. After cooling, the precipitated product is filtered, dissolved in water, the solution is alkaline with sodium carbonate, and the product is extracted three times with 4-methyl-2-pentanone. The combined extracts were washed, dried over water, filtered and evaporated, and the residue was purified by column-chromatography on silica gel using a mixture of trichloromethane and methanol (95: 5 in volume) as eluent. Pure fractions were collected to evaporate the eluate and the residue was converted to hydrochloride salts in 2-propanone and 2-propanol and the salts filtered to ethanol and 2,2′-oxybispropane (1: 1 by volume). Crystallization from a mixture of 2 parts N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl having a melting point of 165.7 ° C. } -2-pyridinecarboxamide dihydro chromide dihydrate is obtained.
[실시예 XLI]Example XLI
실시예 XL의 공정을 따르고 용매로서 N, N-디메틸포름아마이드를 사용하여 N-(2-크로로에틸)-2-푸란카르복스아마이드를 1.3-디하이드로-1-(4-피페리디닐)-2H-벤즈이미다졸-2-원과 반응시킴에 의하여 용융점이 231.7℃인 N-{2-[4-(2,3-드하이드로-2-옥소-1H-벤즈이디다졸-1-일)--피페리디닐]에틸}-2-푸란카르복스아마이드를 제조한다.Following the procedure of Example XL and using N, N-dimethylformamide as the solvent, N- (2-chloroethyl) -2-furancarboxamide was added 1.3-dihydro-1- (4-piperidinyl) N- {2- [4- (2,3-dehydro-2-oxo-1H-benzidazol-1-yl) having a melting point of 231.7 ° C by reacting with -2H-benzimidazole-2-one Prepare piperidinyl] ethyl} -2-furancarboxamide.
[실시예 XLⅡ]Example XLII
1부의 2-아미노-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸}-4-풀로오로벤즈아마이드, 1부의 무수식초산 및 15부의 물의 혼합물을 80-90℃의 물 중탕내에서 30분간 교반한다. 반응 혼합물을 냉각하여 수산화 암모니움 용액으로 알칼리화하고 생성물을 트리크로로메탄으로 추출한다. 추출액을 건조 및 여과하여 증발시킨 다음 잔유물을 4-메틸-2-펜타논으로부터 결정화시킨후 생성물을 여과하여 건조시키면 용융점이 210℃인 0.5부의 2-(아세틸아미노)-N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀르오로 벤즈아마이드를 수득한다.1 part 2-amino-N- {2- [4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4-pulolo A mixture of benzamide, 1 part acetic anhydride and 15 parts water is stirred for 30 minutes in a water bath at 80-90 ° C. The reaction mixture is cooled to alkalinize with ammonium hydroxide solution and the product is extracted with trichloromethane. The extract was dried, filtered and evaporated, the residue was crystallized from 4-methyl-2-pentanone and the product was filtered and dried to yield 0.5 parts of 2- (acetylamino) -N- {2- [4 having a melting point of 210 ° C. -(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4-fuluro to obtain benzamide.
[실시예 XLⅢ]Example XLIII
실시예 XLⅡ의 공정에 따라 2-아미노-N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐]에틸}-4-풀루오로벤즈아마이드로부터 출발하여 용융점이 196.5℃인 2-(아세틸아미노)-N-{2-[4-5-크로로-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드를 제조한다.2-amino-N- {2- [4- (5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-pipe according to the process of Example XLII 2- (acetylamino) -N- {2- [4-5-chloro- (2,3-dihydro-2) having a melting point of 196.5 ° C. starting from ridinyl] ethyl} -4-fulurobenzamide -Oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4- pullobenzamide is prepared.
[실시예 XLIV]EXAMPLE XLIV
1.68부의 4-풀루오로벤조일 크로라이드, 4.22부의 1-[1-(2-아미노 에틸)-4-피페리디닐-1,3-디하이드로-2H-벤즈이미다졸-2-원 디하이드로 브로마이드, 4.83부의 탄산칼륨 및 18부의 N, N-디메틸포름아마이드의 혼합물을 90℃에서 1야간 교반한다. 반응 혼합물을 냉각하여 여과하고 여과액을 소량의 N,N-디메틸포름아마이드로 세척한 후 N,N-디메틸포름아마이드를 진공에서 제거한 다음 잔유물에 물을 첨가한다. 생성물을 트리크로로메탄으로 추출하여 추출액을 건조 및 여과한 다음 증발시킨 후 잔유물을 2-프로판올로부터 결정화시키면 용융점이 231℃인 2.5부의 N-{2-[4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드를 얻는다.1.68 parts 4-Pluorobenzoyl Chloride, 4.22 parts 1- [1- (2-amino ethyl) -4-piperidinyl-1,3-dihydro-2H-benzimidazole-2-one dihydro bromide , 4.83 parts of potassium carbonate and 18 parts of N, N-dimethylformamide are stirred at 90 DEG C overnight. The reaction mixture is cooled and filtered, the filtrate is washed with a small amount of N, N-dimethylformamide, the N, N-dimethylformamide is removed in vacuo and water is added to the residue. The product was extracted with trichloromethane, the extract was dried, filtered and evaporated and the residue was crystallized from 2-propanol. 2.5 parts of N- {2- [4- (2,3-dihydro- with a melting point of 231 ° C. were obtained. 2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4- pullobenzamide is obtained.
[실시예 XLV]Example XLV
0.74부의 1-(벤조일)아지리딘, 1.06부의 4-(4-크로로페닐)-4-피페리디올, 5.4부의 벤젠 및 0.4부의 메탄올의 혼합물을 2시간 교반 및 환류시킨다. 반응 혼합물을 냉각하고 1,1'-옥시비스 에탄을 첨가하면 생성물이 침전된다. 15분간 교반한 후 생성물을 여과하여 건조하면 용융점이 169℃인, 1,1'부의 N-{2-[4-(4-크로로페닐)-4-하이드록시-1-피페리디닐] 에틸} 벤즈아마이드를 수득한다.A mixture of 0.74 parts 1- (benzoyl) aziridine, 1.06 parts 4- (4-chlorophenyl) -4-piperidiol, 5.4 parts benzene and 0.4 parts methanol is stirred and refluxed for 2 hours. Cool the reaction mixture and add 1,1'-oxybis ethane to precipitate the product. After stirring for 15 minutes, the product was filtered and dried to give 1,1 'part N- {2- [4- (4-chlorophenyl) -4-hydroxy-1-piperidinyl] ethyl having a melting point of 169 ° C. } Obtain benzamide.
[실시예 XLVI]Example XLVI
실시예 XLV의 공정을 따르고 당량의 정당한 시발물질을 사용하여 하기 화합물을 유리 염기 형태 또는 염기를 적당한 산과 반응시켜 산 부가 염의 형태로 제조한다.The following compound is prepared in the form of an acid addition salt by following the process of Example XLV and using an equivalent equivalent starting material by reacting the free base form or base with a suitable acid.

[실시예 XLVⅡ]Example XLVII
5.8부의 2-아미노-N-{2-[4-(4-크로로페닐)-4-하이드록시-1-피페리디닐] 에틸} -4-풀루오로벤즈아마이드, 5.5부의 무수식초산 및 55부의 물의 혼합물을 80℃의 물 중탕내에서 30분간 교반시킨다. 반응 혼합물을 냉각하여 수산화암모니움으로 알카리화하고 생성물을 트리크로로메탄으로 추출한다. 수용액 상을 분리하여 트리크로로메탄으로 추출하고 합친 유기상을 물로 3회 세척, 건조 및 여과하여 증발시킨다. 오일상의 잔유물을 4-메틸-2-펜타논으로 부터 결정화시켜 생성물을 여과한 후 건조시키면 용융점이 175.1℃인 4.5부의 2-(아세틸아미노)-N-{2-[4-(4-크로로페닐)-4-하이드록시-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드를 얻는다.5.8 parts 2-amino-N- {2- [4- (4-chlorophenyl) -4-hydroxy-1-piperidinyl] ethyl} -4- pullobenzamide, 5.5 parts of acetic anhydride and The mixture of 55 parts of water is stirred for 30 minutes in a water bath at 80 ° C. The reaction mixture is cooled to alkaline with ammonium hydroxide and the product is extracted with trichloromethane. The aqueous phase is separated and extracted with trichloromethane and the combined organic phases are washed three times with water, dried and filtered and evaporated. The oily residue was crystallized from 4-methyl-2-pentanone and the product was filtered and dried. 4.5 parts of 2- (acetylamino) -N- {2- [4- (4-chloro with a melting point of 175.1 占 폚 were obtained. Phenyl) -4-hydroxy-1-piperidinyl] ethyl} -4- pullobenzamide.
[실시예 XLVⅢ]Example XLVIII
10.6부의 N-(2-브로모에틸)-2-니트로벤즈아마이드, 9.2부의 A(±)-4-(4-크로로페닐)-3-메틸-4-피페리딘올 및 270부의 N,N-디메틸포름아마이드의 혼합물을 4시간 교반 및 환류시킨다. 반응혼합물을 건조하도록 증발시키고 잔유물을 용출액으로서 트리크로로메탄과 메탄올(용량으로 95:5)의 혼합물을 사용하여 시리카 겔 상에서 컬럼-크로마토그라피에 의하여 정제한다. 순수한 분율을 수집하여 용출액을 증발시키고 잔유물을 2-프로판올내에서 하이드로크로라이드 염으로 전환시킨 후 염을 여과하여 에탄올과 2,2'-옥시비스프로판(용량으로 1:1)의 혼합물로부터 결정화 시킨다. 생성물을 여과하여 60℃에서 1야간 건조시키면 용융점이 228℃인 4.5부의 A-(±)-N-{2-[4-(4-크로로페닐)-4-하이드록시-3-메틸-1-피페 리디닐] 에틸}-2-니트로벤즈아마이드 하이드로크로라이드를 수득한다.10.6 parts N- (2-bromoethyl) -2-nitrobenzamide, 9.2 parts A (±) -4- (4-chlorophenyl) -3-methyl-4-piperidinol and 270 parts N, N The mixture of dimethylformamide is stirred and refluxed for 4 hours. The reaction mixture is evaporated to dryness and the residue is purified by column-chromatography on silica gel using a mixture of trichloromethane and methanol (95: 5 in volume) as eluent. Pure fractions were collected to evaporate the eluate and the residue was converted to a hydrochloride salt in 2-propanol and the salts filtered to crystallize from a mixture of ethanol and 2,2'-oxybispropane (1: 1 in volume). . The product was filtered and dried overnight at 60 DEG C., 4.5 parts of A- (±) -N- {2- [4- (4-chlorophenyl) -4-hydroxy-3-methyl-1 having a melting point of 228 DEG C. -Piperidinyl] ethyl} -2-nitrobenzamide hydrochloride is obtained.
[실시예 IL]Example IL
실시예 XLVⅢ의 공정을 따라서 N-(2-브르모에틸)-4-풀루오로-2-니트로벤즈아마이드를 A-(±)-4-(4-크로로페닐)-3-메틸-4-피페리딘올과 반응시킴에 의하여 A-(±)-N-{2-[4-(4-크로로페닐)-4-하이드록시-3-메틸-1-피페리디닐] 에틸}-4-풀루오로-2-니 트로벤즈아마이드 하이드로크로라이드를 제조한다.Example N- (2-Bromoethyl) -4- pullouro-2-nitrobenzamide was prepared according to the process of Example XLVIII in A- (±) -4- (4-chlorophenyl) -3-methyl-4 -A- (±) -N- {2- [4- (4-chlorophenyl) -4-hydroxy-3-methyl-1-piperidinyl] ethyl} -4 by reaction with piperidinol -Pluoro-2-nitrobenzamide hydrochloride is prepared.
[실시예 L]Example L
2.8부의 A-(±)-N-{2-[4-(4-크로로페닐)-4-하이드록시-3-메틸-1-피페리디닐] 에틸}-4-풀루오로-2-니트로벤즈아마이드 하이드로 크로라이드와 160부의 메탄올의 혼합물을 보통 압력 및 실온에서 2부의 라네이-닉겔 촉매로 수소화시킨다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨 다음 잔유물을 2-프로파논과 2-프로판올내에서 하이드로크로라이드 염으로 전환시킨다. 염을 여과하여 2-프로판올로부터 결정화시키면 용융점이 l85℃인 1부의 A-(±)-2-아미노-N-{2-[4-(4크로로페닐)-4-하이드록시-3-메틸-1-피페리디닐]에틸}-4-풀루오로벤즈아마이드 디하이드로크로라이드 2-프로파노레이트를 얻는다.2.8 parts A- (±) -N- {2- [4- (4-chlorophenyl) -4-hydroxy-3-methyl-1-piperidinyl] ethyl} -4-pulluoro-2- A mixture of nitrobenzamide hydrochloride and 160 parts of methanol is hydrogenated with 2 parts of rani-nickel catalyst at normal pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated and the residue is converted to a hydrochloride salt in 2-propanone and 2-propanol. The salt was filtered to crystallize from 2-propanol to give 1 part A- (±) -2-amino-N- {2- [4- (4-chlorophenyl) -4-hydroxy-3-methyl with a melting point of 185 ° C. -1-piperidinyl] ethyl} -4-pulorobebenzamide dihydrochromide 2-propanoate is obtained.
[실시예 LI]Example LI
실시예 L의 공정에 따라 A-(±)-N-{2-[4-(4-크로로페닐)-4-하이드록시-3-메틸-1-피페리디닐] 에틸}-2-니트로벤즈아마이드를 수소화함에 의하여 용융점이 190.5℃인 A-(±)-2-아미노-N-{2-[4-(4-크로로페닐)-4-하이드록시-3-메틸-1-피페리디닐] 에틸} 벤즈아마이드 디하이드로 크로라이드를 제조한다.A- (±) -N- {2- [4- (4-chlorophenyl) -4-hydroxy-3-methyl-1-piperidinyl] ethyl} -2-nitro according to the process of example L A- (±) -2-amino-N- {2- [4- (4-chlorophenyl) -4-hydroxy-3-methyl-1-piperidi having a melting point of 190.5 ° C. by hydrogenating benzamide Nil] ethyl} benzamide dihydro chromide.
[실시예 LⅡ]Example LII
3.5부의 N-{2-[4-(4-크로로페닐)-4-하이드록시-1-피페리디닐] 에틸}-2-니트로벤즈아마이드, 하이드로크로라이드, 90부의 테트라하이드르푸란 및 40부의 메탄올의 혼합물을 보통 압력 및 실온에서 0.2부의 산화백금으로 수소화한다. 계산된 양의 수소를 취한 후 촉매를 여과하여 여과액을 증발시킨다. 잔유물을 2-프로판올과 2,2'-옥시비스프로판내에서 하이드로크로라이드 염으로 전환시키고 염을 여과하여 건조하면 용융점이 195.7℃인 2.6부의 2-아미노-N-{2-[4-(4-크로로페닐)-4-하이드록시-1-피페리디닐] 에틸} 벤즈아마이드 디하이드로크로라이드 헤미하이드레이트를 얻는다.3.5 parts N- {2- [4- (4-chlorophenyl) -4-hydroxy-1-piperidinyl] ethyl} -2-nitrobenzamide, hydrochloride, 90 parts tetrahydrofuran and 40 The mixture of parts methanol is usually hydrogenated with 0.2 parts of platinum oxide at pressure and room temperature. Take the calculated amount of hydrogen and then filter the catalyst to evaporate the filtrate. The residue was converted to a hydrochloride salt in 2-propanol and 2,2'-oxybispropane and the salt filtered off and dried to give 2.6 parts 2-amino-N- {2- [4- (4 having a melting point of 195.7 ° C. -Chrophenyl) -4-hydroxy-1-piperidinyl] ethyl} benzamide dihydrochromide hemihydrate is obtained.
[실시예 LⅢ]Example LIII
4.5부의 N-{2-[4-(4-크로로페닐)-4-하이드록시-1-피페리디닐] 에틸}-4-플루오로-2-니트로벤즈아마이드, 40부의 메탄올 및 90부의 테트라 하이드로푸란의 혼합물을 보통 압력 및 실온에서 2부의 라네이-닉겔 촉매로 수소화 한다. 계산된 양의 수소를 취한후 촉매를 여과하고 여과액을 증발시킨후 잔유물을 2-프로판올내에서 하이드로크로라이드 염으로 전환시킨다. 염을 여과하여 건조하면 용융점이 210.7℃인 2부의 2-아미노-N- {2-[4-(4-크로로페닐)-4-하이드록시-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드 디하이드로크로라이드를 수득한다.4.5 parts N- {2- [4- (4-chlorophenyl) -4-hydroxy-1-piperidinyl] ethyl} -4-fluoro-2-nitrobenzamide, 40 parts methanol and 90 parts tetra The mixture of hydrofuran is usually hydrogenated with two parts Lanei-Nigel catalyst at pressure and room temperature. After taking the calculated amount of hydrogen the catalyst is filtered off and the filtrate is evaporated and the residue is converted to the hydrochloride salt in 2-propanol. The salt was filtered off and dried to give 2 parts 2-amino-N- {2- [4- (4-chlorophenyl) -4-hydroxy-1-piperidinyl] ethyl} -4-pool having a melting point of 210.7 ° C. Luobenzamide dihydrochloride is obtained.
[실시예 LIV]EXAMPLE LIV
실시예 XIV의 공정을 따르고 당량의 적당한 시발물질을 사용하여 하기 화합물들을 하이드로크로라이드 염으로 수득한다The following compounds are obtained as hydrochloride salts following the process of Example XIV and using an equivalent equivalent starting material
N-{2-[1-(4-크로로페닐)-4-옥소-1,3,8-트리아자스피로[4,5] 데크-8-일] 에틸} -4-풀루오로벤즈아마이드:N- {2- [1- (4-Chlorophenyl) -4-oxo-1,3,8-triazaspiro [4,5] deck-8-yl] ethyl} -4- pullobenzamide :
N-{2-[1-(4-크로로페닐)-4-옥소-1,3,8-트리아자스피로 4,5 데크-8-일] 에틸} 벤즈아마이드:N- {2- [1- (4-Chlorophenyl) -4-oxo-1,3,8-triazaspiro 4,5 deck-8-yl] ethyl} benzamide:
N-{2-[1-(4-브로모페닐)-4-옥소-1,3,8-트리아자스피로 [4,5] 데크-8-일] 에틸}-4-풀루오로벤즈아마이드: :N- {2- [1- (4-bromophenyl) -4-oxo-1,3,8-triazaspiro [4,5] deck-8-yl] ethyl} -4- pullobenzamide ::
2-크로로-N-{2-[1-(4-크로로페닐)-4-옥소-1,3,8-트리아자스피로 [4,5]-데크-8-일] 에틸}-4-풀루오로벤즈아마이드:2-Chloro-N- {2- [1- (4-chlorophenyl) -4-oxo-1,3,8-triazaspiro [4,5] -deck-8-yl] ethyl} -4 -Fluorobenzamide:
2-아미노-N-{2-[1-(4-크로로페닐)-4-옥소-1,3,8-트리아자스피로[4,5]-데크-8-일] 에틸}-4-풀루오로벤즈아마이드:2-amino-N- {2- [1- (4-chlorophenyl) -4-oxo-1,3,8-triazaspiro [4,5] -deck-8-yl] ethyl} -4- Pulluorobesamide:
2-아미노-N-2-1-(4-부로모페닐)-4-옥소-1,3,8-트리아자스피로 [4,5] 데크-8-일]에 틸} -4-풀루오로벤즈아마이드2-amino-N-2-1- (4-bromophenyl) -4-oxo-1,3,8-triazaspiro [4,5] dec-8-yl] ethyl} -4-pulluo Robbenzamide
[실시예 LV]Example LV
실시예 XXVI의 공정을 따르고 당량의 적당한 시발물질을 사용하여 일반식(1)의 하기 화합물을 제조한다.The following compounds of formula (1) are prepared following the process of Example XXVI and using an equivalent equivalent starting material.
N-{2-[4-{5-브로모-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀로로벤즈아마이드:N- {2- [4- {5-bromo-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4- pullobenz Amide:
N-{2-[4-(2,3-디하이드로-5-메틸-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드:N- {2- [4- (2,3-dihydro-5-methyl-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4- pullorobenz Amide:
N-{2-[4-(4,5,6-디크로로-2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-피페리디닐 에틸-4-풀루오로벤즈아마이드:N- {2- [4- (4,5,6-dichloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl ethyl-4- Pulluorobesamide:
2-크로로-N-{2-[4-(5,6-디크로로-2,3-디하이드로-2-옥소-1H-벤즈아미다졸-1-일)-1-피페리디닐] 에틸]-4-풀루오로벤즈아마이드 :2-Chloro-N- {2- [4- (5,6-dichloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] Ethyl] -4-Pluorobenzamide:
2-아미노-{N-[2-4-(5-브로모-2,3-디하이드로-2-옥소-1H-벤즈아미다졸-1-일)-1-피페리디닐] 에틸}-4-풀루오로벤즈아미이드:2-amino- {N- [2-4- (5-bromo-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1-piperidinyl] ethyl} -4 -Fluorobenzamide:
N-{2-[3,5-디하이드로-4-(2,3-디하이드로-2-옥소-1H-벤즈이미다졸-1-일)-1-(2H)-피-페리디닐] 에틸}-벤즈아미드 :N- {2- [3,5-Dihydro-4- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1- (2H) -piperidinyl] ethyl } -Benzamide:
2-아미노-N-{2-[3,6-디하이드로-(2, 3-디하이드로-2-옥소-1H-벤즈아미다졸-1-일)-1-(2H) 피리디닐] 에틸}-4-크로로벤즈아마이드2-amino-N- {2- [3,6-dihydro- (2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1- (2H) pyridinyl] ethyl} 4-chlorobenzamide
2-크로로-N-{2-[3,6-디하이드로-4-(2, 3-디하이드로-2-옥소-1H-벤즈아미다졸-1-일)-1-(2H)-피리디닐] 에틸}-4-풀루오로벤즈아마이드:2-Chloro-N- {2- [3,6-dihydro-4- (2, 3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1- (2H) -pyri Diyl] ethyl} -4- pullorobebenzamide:
N-{2-[4-(5-크로로-2,3-디하이드로-2-옥소-1H-벤즈아미다졸-1-일)-1-(2H)-피 리디닐] 에틸}-4-풀루오로벤즈아미드:N- {2- [4- (5-Chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl) -1- (2H) -pyridinyl] ethyl} -4 -Pluorobenzamide:
[실시예 LVI]Example LVI
실시예 XLV공정을 따르고 당량의 적당한 시발물질을 사용한 하기 화합물을 제조한다.EXAMPLES The following compounds were prepared following the XLV process and using an equivalent amount of suitable starting material.
N-[2-{4-[4-크로로-3-(트리풀루오로메틸) 페닐]-4-하이드록시-1-피페리디닐} 에틸]-4-풀루오로벤즈아마이드N- [2- {4- [4-Chloro-3- (trifuluromethyl) phenyl] -4-hydroxy-1-piperidinyl} ethyl] -4-fulurobenzamide
N-아미노[2-{4-[4-크로로-3-(트리풀루오로메틸) 페닐]-4-하이드록시-1-피페리디닐} 메틸]-4-풀루오로벤즈아마이드N-amino [2- {4- [4-chloro-3- (trifuluromethyl) phenyl] -4-hydroxy-1-piperidinyl} methyl] -4-fulurobenzamide
2-크로로-[2-{4-[4-크로로-3-(트리풀루오로메틸) 페닐}-4-하이드록시-1-피페 리디닐}에틸]-4-풀루오로벤즈아미드2-Chloro- [2- {4- [4-Chloro-3- (tripulouromethyl) phenyl} -4-hydroxy-1-piperidinyl} ethyl] -4-pululobenzamide
N-[2-{4-(4-브로모페닐)-4-하이드록시-1-피페리디닐] 에틸}-4-풀루오로벤즈아마이드N- [2- {4- (4-bromophenyl) -4-hydroxy-1-piperidinyl] ethyl} -4- pullobenzamide
2-아미노-N-[2-{4-(4-브로모페닐)-4-하이드록시-1-피페리디닐}-에틸}-4-풀루오로벤즈아마이드2-amino-N- [2- {4- (4-bromophenyl) -4-hydroxy-1-piperidinyl} -ethyl} -4- pullobenzamide
Claims (1)




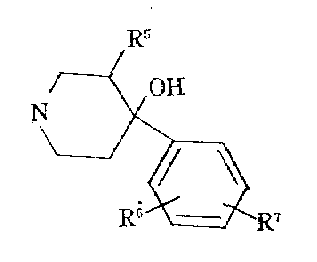
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR7601895A KR800000079B1 (en) | 1976-08-05 | 1976-08-05 | Method for preparing N-[(1-piperidinyl) alkyl] carboxamide derivative |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR7601895A KR800000079B1 (en) | 1976-08-05 | 1976-08-05 | Method for preparing N-[(1-piperidinyl) alkyl] carboxamide derivative |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR800000079B1 true KR800000079B1 (en) | 1980-01-30 |
Family
ID=19202549
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR7601895A Expired KR800000079B1 (en) | 1976-08-05 | 1976-08-05 | Method for preparing N-[(1-piperidinyl) alkyl] carboxamide derivative |
Country Status (1)
| Country | Link |
|---|---|
| KR (1) | KR800000079B1 (en) |
-
1976
- 1976-08-05 KR KR7601895A patent/KR800000079B1/en not_active Expired
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4031226A (en) | N-[(1-piperidinyl)alkyl]arylcarboxamide derivatives | |
| KR0159099B1 (en) | 2-amino pyrimidinone derivatives | |
| KR850000630B1 (en) | Process for preparing piperidine derivatives | |
| KR870001512B1 (en) | Process for preparing benzoxazol and benzothiazolamine derivatives | |
| KR101386679B1 (en) | Novel arylpiperazine-containing imidazole 4-carboxamide derivatives and pharmaceutical composition comprising same | |
| BG64447B1 (en) | Indazole derivatives, pharmaceutical compositions and application thereof | |
| CA2565065A1 (en) | Therapeutic compounds: pyridine as scaffold | |
| CS234044B2 (en) | Method of new 1-(4-arylcyclohexyl) piperidine derivatives preparation | |
| EP0244018A1 (en) | Amino-thiazole and oxazole derivatives | |
| KR100238562B1 (en) | Piperidine Derivative Compound | |
| EP0034415A1 (en) | 1-(Cyclohexyl or cyclohexenyl)-4-aryl-4-piperidinecarboxylic acid derivatives | |
| DK168291B1 (en) | N-substituted diphenylpiperidines, process for their preparation, drug containing the compounds and use of the compounds and use of the compounds for the preparation of a drug for use in the prevention or treatment of obesity | |
| KR970011156B1 (en) | New substituted n-(3-hydroxy -4- piperidinyl) benzamides | |
| KR102582197B1 (en) | Preparation of 2-([1,2,3]triazol-2-yl)-benzoic acid derivatives | |
| US4197304A (en) | N-Aryl-N-(1-L-4-piperidinyl)-arylacetamides | |
| KR800000079B1 (en) | Method for preparing N-[(1-piperidinyl) alkyl] carboxamide derivative | |
| JPH0784471B2 (en) | Novel 3-aminochroman spiro compounds, a process for their preparation and pharmaceutical compositions containing them | |
| US4196210A (en) | N-Aryl-N-(1-L-4-piperidinyl)-arylacetamides | |
| KR840002022B1 (en) | Process for preparing 1-(cyclohexyl)-4-aryl-4-piperidine carboxylic acid derivatives | |
| SU867304A3 (en) | Method of preparing derivatives of n-phenyl-n-(4-piperidinyl)amide or their salts | |
| NZ297312A (en) | 1-[2-phenyl-4-(4-(3-hydroxythien-2-yl)piperidino)butanoyl]azepane derivatives | |
| US4151286A (en) | N-aryl-N-(1-L-4-piperidinyl)-arylacetamides | |
| US7777048B2 (en) | Processes for preparing biaryl ureas and analogs thereof | |
| DE69305330T2 (en) | Aminoalkylchomones, processes for their preparation and the pharmaceutical compositions containing them | |
| FR2702211A1 (en) | New derivatives of 2-phenoxyethylamine, their preparation and their therapeutic application. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0109 | Patent application |
Patent event code: PA01091R01D Comment text: Patent Application Patent event date: 19760805 |
|
| PE0902 | Notice of grounds for rejection |
Comment text: Notification of reason for refusal Patent event date: 19790823 Patent event code: PE09021S01D |
|
| PG1605 | Publication of application before grant of patent |
Comment text: Decision on Publication of Application Patent event code: PG16051S01I Patent event date: 19791219 |
|
| PE0701 | Decision of registration |
Patent event code: PE07011S01D Comment text: Decision to Grant Registration Patent event date: 19800408 |