IMPROVED MELAMINYLTHIOARSENITES
BACKGROUND OF THE INVENTION The compound represented by the formula

and certain of its derivatives such as the disodium salt are well known as trypanocidal and macrofilaricidal agents. The usefulness of the compounds is impaired by the limited stability of their solutions in water. As a result, it is necessary to package the products in the form of a dry powder which must be taken up in water just prior to use. This is a distinct disadvantage under field conditions in developing countries which are the principal sites of such infections. One such infection is commonly known as African Sleeping Sickness. It results from infections by T. gambiense and T. rhodesiense. Another is onchocerciasis, a non-fatal but disfiguring and blinding disease caused by the filarial worm Onchocerca volvulus. There is a definite need for anti-infective agents useful for the treatment of such infections, but not suffering the disadvantage of instability. More specifically, there is a need for trypanocidal and macrofilarxcidal agents which will remain stable in a liquid medium, preferably aqueous media.
THE INVENTION
It has now been discovered that novel melaminylthioar senites which are trypanocidal and macrofilaricidal agents can be prepared. They can be provided as the principal active agent in therapeutically useful compositions in association with pharmaceutically acceptable excipients. The compounds of this invention may be represented by the formula:
wherein Y is selected from the group concisting of
and n and m are both integers from 2 to
12 with the proviso that the value of n plus m is no more than 12. The invention also includes pharmaceutically acceptable acid addition salts of the free bases within its scope.
The compounds of the invention are obtained by reaction of melarsenoxide with an appropriate thiol. The reaction can be represented by the equation:
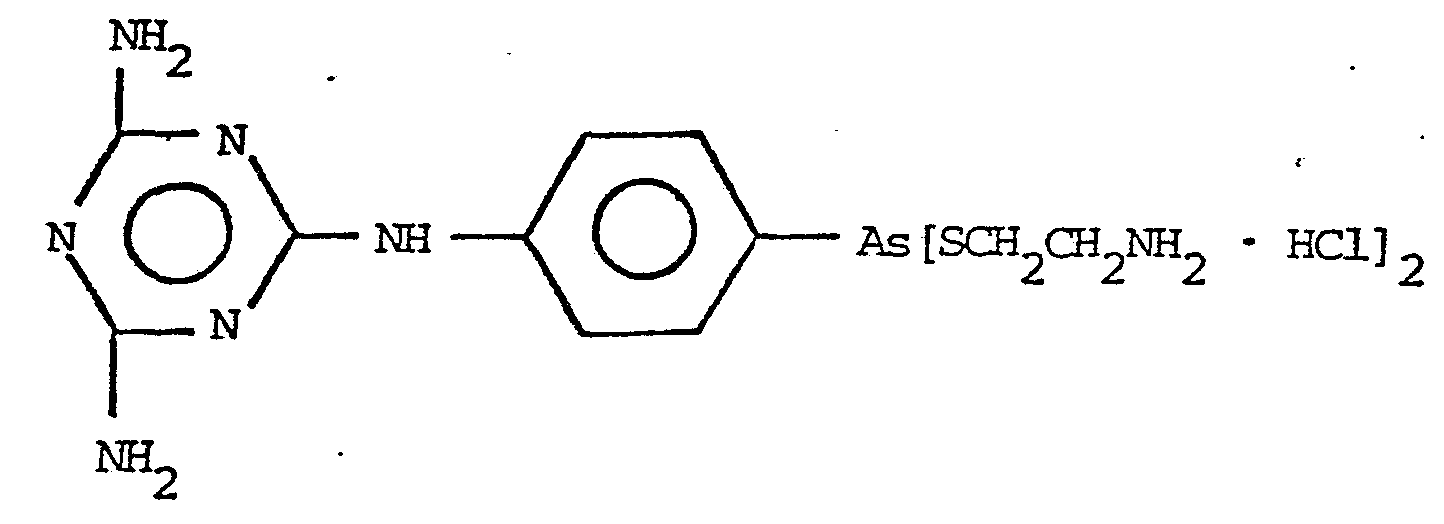
In the equation Y, n and m have the same meaning as above. The following compounds are the preferred thiols for use in this invention. They are preferred because they are readily available, known to be non-toxic and because they produce compounds with a high order of activity, a high therapeutic index and low toxicity. NH2CH2CH2SH · HCl V
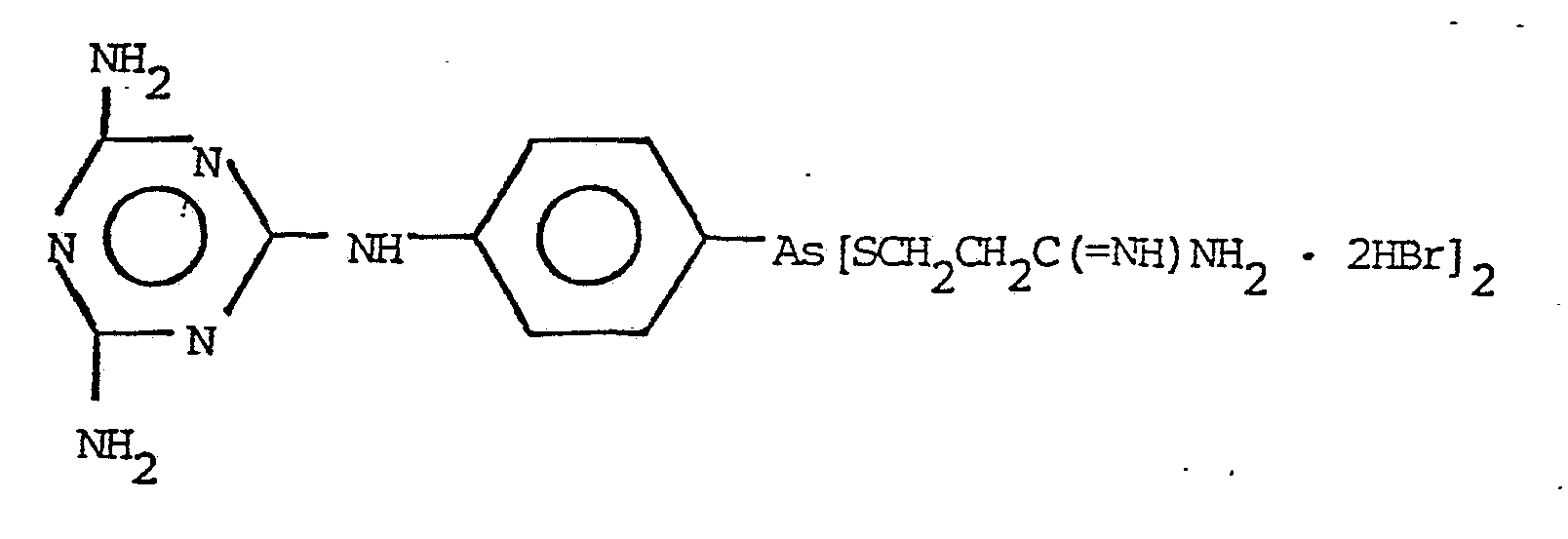
NH2C(=NH)CH2CH2SH · 2HBr VI
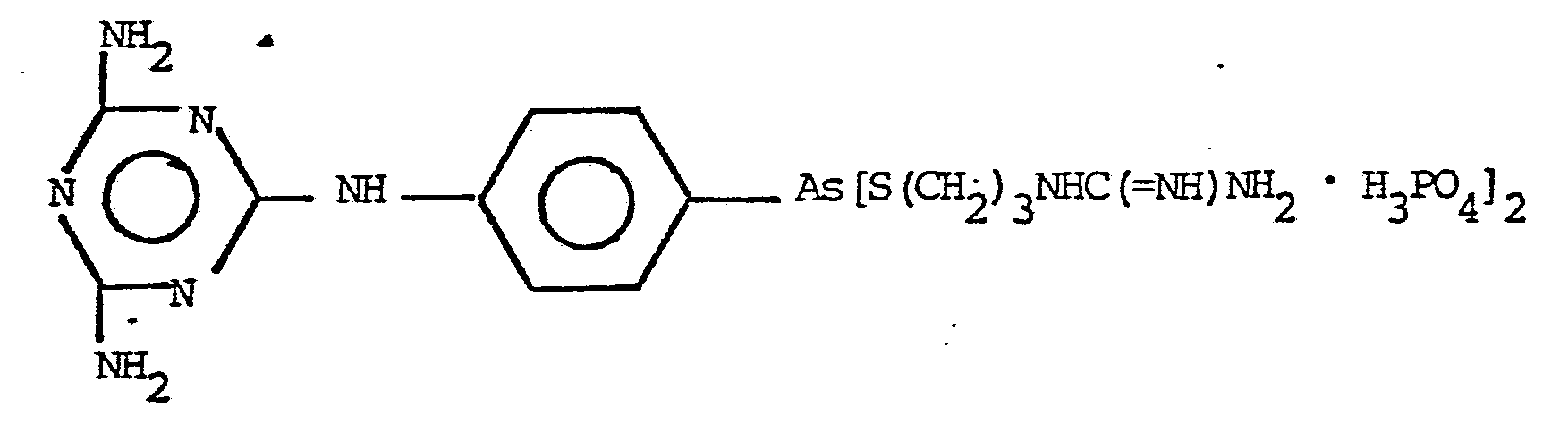
NH2C( NH)NH(CH2)3S H2PO3 VII
NH2(CH2)3NH(CH2)2 · S H2PO3 VIII
NH2(CH2)3NH(CH2)3 ·S H2PO3 IX It will be noted that the first two compounds are acid addition salts. The last three compounds are esters. In the course of the reaction, the esters rearrange so that the final products are
acid addition salts.
Pharmaceutically acceptable acid addition salts are those containing non-toxic anions and include, for example, hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, lactic, citric, tartaric, succinic, maleic, gluconic and saccharic acids. These acid addition salts are prepared by standard reactions either directly from the initial reaction or from the free base by reaction with the selected acid.
The general procedure for the preparation of the compounds of this invention is to react melarsenoxide with the selected thiol in an aqueous or lower alkanol medium at a temperature of from 40º to 100°C until all of the reactants have dissolved. The reaction products separate as a viscous bottom layer. The reaction solvent, which may be a mixture of water and a lower alkanol, preferably methanol or ethanol is separated, suitably by decantation. The product may be purified by repeated recrystallization together with trituration with a dehydrating solvent, preferably ethanol. The preferred recrystallization solvent is water which may contain a small amount of ethanol, e.g. up to 20% by weight. It is preferred to utilize a molar excess of thiol, i.e. more than two moles of thiol per mole of melarsenoxide. This insures as complete a reaction as possible of the toxic melarsenoxide and limits the amount which may be present as a contaminant in the final product. It is preferred to use at least 10% molar excess of thiol, and for most purposes an excess of from 10 to 20% is acceptable.
The thiols may be reacted either as free thiols, phosphothionites, or as acid salts such as the hydrochloride or hydrobromide. Accordingly, a product of the invention may be obtained as a free base or an acid addition salt.
The preferred products of the invention are those represented by the formulas:
From these salts the free cationic melaminylthioarsenites can be prepared according to conventional methods. The cationic thioarsenites of the present invention are white powders, soluble in water, dilute acids and alkali, insoluble in acetone, chloroform, and ether. They have no sharp melting point and decompose above
200°C. They are useful for the treatment of parasitic diseases such as trypanosomiasis and filariasis, and onchocerciasis.
The products of this invention may be administered alone but will generally be administered with pharmaceutically acceptable, non-toxic carriers, the proportions of which are determined by the suitability and chemical nature of the particular carrier, the chosen route of administration, and standard pharmaceutical practice. For example, in combatting various infections or in maintaining therapeutically effective levels in the blood or tissue thay may be administered orally in the form of tablets or capsules containing such excipients as starch, milk sugar, certain types of clay, etc. They may be enteric coated so as to be more resistant to the acid and digestive enzymes of the stomach. For intravenous and intramuscular administration they may be used in the form of a sterile solution containing other solutes, for example, enough saline or glucose to make the solution inotonic.
The compounds of this invention are orders of magnitude more stable than related prior art compounds which have been similarly employed. For example, if the disodium salt of Compound I, the first formula shown above, is dissolved in 1% water solution and held at 37°C, the solution becomes slightly cloudy after only 12 hours. A precipitate forms in 5 days. In contrast, Compounds
X and XIII of this invention in 1% aqueous solution have been held at 37°C for 5 days while still remaining clear. In fact, the solutions remain clear for as long as 3 months at 100°C.
The compounds of this invention have been screened for useful activity.
The test for trypanocidal activity is as described by Rane et al., Amer. J. Trop. Med. Hug. 25:394(1976).
This test system is patterned after the one developed and employed in testing of compounds for activity against Plasmodium berghei maleria in mice. The trypanosomiasis system is based on comparisons of responses to test compounds by ICR/HA Swiss mice infected with the Wellcome CT strain of T. rhodesiense as expressed in mean survival times compared with mean survival times of untreated controls. Using a standard inoculum, it is possible to produce a uniform disease fatal to 100% of untreated animals within 4 to 6 days with a mean survival time of 4.45 ± .25 days.
Test mice, six weeks of age, weighing 30-32 grams receive an intraperitoneal injection of 0.5 ml of a 1:50,000 dilution of heparinized heart blood drawn from donor mice infected 3 days earlier. Compounds are given as a single dose in peanut oil about two hours after parasite inoculation. Five mice per drug level, 20 infected untreated (negative) controls, and 10 infected positive controls are routinely used per test. Positive controls are mice infected and treated at 40 mg/kg with Stilbamidine Isethionate. Routinely compounds are first tested by the subcutaneous route of administration.
retested for confirmation at selected drug levels and if the activity is confirmed it is tested by the oral route of administration.
Deaths prior to the fourth day, when untreated controls begin to die, are regarded as non-parasitic and are scored as "toxic deaths". Treated animals are kept under observation for 30 days. Survivors at the end of this period of time are considered as "cured". An increase of 100% in mean survival time is considered the minimum effective ("active") response for a candidate compound. In calculating mean survival times, toxic deaths and 30-day survivors are not included.
Using this test it was found that with Compound XII the LD50 was of the order of 400 mg/kg of body weight and the DCur50 (dose curative - 50%) was of the order of 0.16 mg/kg of body weight. This corresponds to a therapeutic index of more than 2,000. The filaricidal activity of Compound XIII was tested in gerbils infected with L. carinii according to the standard method employed at the WHO screening center at the Justes Liebig University in Giessen, West Germany. The results, as a function of dose, are shown in the following table.
The following non-limiting examples are given by way of illustration only. All temperatures are in degrees Centigrade. Example 1 - Compound X
A solution of 3.6g III in 72 ml boiling EtOH is stirred into a solution of 3.0g of V in 30 ml boiling EtOH. A white precipitate is formed which, after cooling to 10°, is filtered off, washed with EtOH and dried in vacuo over -P2O5. X is reprecipitated by cooling out of boiling water. Yield 5.0g. White powder, soluble in water, insoluble in MeOH, EtOH, chloroform, ether, unchanged dissolved in boiling water for 1 month.
Example 2 - Compound XII 10g III are disolved in a solution of 20.2g VI in 200 ml boiling water. On cooling to 0° of the filtered reaction, mixture XII precipitates, is purified by 3 reprecipitations by cooling of the hot aqueous solution, filtered off, washed with
acetone and dried in vacuo. Yield: 7.1g. XII is a white hygroscopic powder, soluble in water, EtOH and MeOH, insoluble in acetone, ethylacetate, ether, chloroform. A 1% solution of XII in water remains unchanged for 4 weeks. Example 3 - Compound XII
XII 5g III are dissolved, with stirring, in a solution of 5.6g VI in 100 ml boiling MeOH. After cooling to 0°, the reaction mixture is filtered and added to 200 ml ether. The reaction product precipitates as a viscous colorless syrup, which is separated by decantation, washed with 3 portions of 50 ml ether and dried in vacuo. The resulting crusts are soluble in water, EtOH and MeOH, insoluble in ether, chloroform and ethylacetate.
Example 4 - Compound XII 13g III are dissolved with stirring in a solution of 15g VII in 150 ml water warmed to 80°. On cooling of the filtered reaction mixture to 0° , XII forms a viscous transparent bottom layer. The supernatant is decanted, the bottom layer washed with 3 portions of 50 ml ice water. After the last decantation the product is triturated with 200 ml hot EtOH, whereupon it becomes granular. It is filtered off, washed with EtOH and dried over P2O5 in vacuo. Yield 18.6g. XII is a white powder, soluble in water and dilute mineral acids. It is insoluble in EtOH, MeOH, acetone, ether and chloroform. A 1% solution of XII in water remains unchanged at 100 for 4 weeks. Example 5 - Compound XIII
12g VII are dissolved in 100 ml of water. 8g III are added with stirring, while the mixture is brought to a boil. The resulting
solution is filtered hot. On standing at 2º a colorless viscous bottom layer is formed. The supernatant is decanted. The bottom layer is dissolved in 50 ml boiling water and precipitated again by cooling. This procedure is repeated twice. After the last decantation the bottom layer has turned into a white powder, which is filtered off and dried in vacuo over P2O5. Yield 14.3g. XIII is a white powder, sparingly soluble in cold water, soluble in hot water, dilute mineral acids, glacial acetic acid, and insoluble in EtOH, MeOH, acetone, chloroform and ether. A 1% solution of XIII in water remains unchanged at 100° for 1 month.
Example 6 - Compound XIV 10g III are dissolved with stirring in a solution of 10.1g IX in 100 ml boiling water. The filtered solution, on standing at 0°, deposits a clear viscous bottom layer which is separated from the supernatant by decantation. On trituration with 100 ml EtOH, the bottom layer becomes solid, granular. It is filtered off and dried in vacuo. The yield is 18g. The reaction product is purified by dissolving in the minimal amount of boiling water, followed by cooling and treatment with EtOH as above. XIV is a white powder, soluble in water, insoluble in EtOH, MeOH, acetone, chloroform and ether. A 1% solution of XIV in water remains unchanged at 100° for 4 weeks.