WO1986005489A1 - Process for 4-amino quinolines - Google Patents
Process for 4-amino quinolines Download PDFInfo
- Publication number
- WO1986005489A1 WO1986005489A1 PCT/US1986/000484 US8600484W WO8605489A1 WO 1986005489 A1 WO1986005489 A1 WO 1986005489A1 US 8600484 W US8600484 W US 8600484W WO 8605489 A1 WO8605489 A1 WO 8605489A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mixture
- formula
- compound
- piperazine
- gal
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
- C07D215/42—Nitrogen atoms attached in position 4
- C07D215/44—Nitrogen atoms attached in position 4 with aryl radicals attached to said nitrogen atoms
Definitions
- the present invention provides a novel means for preparing a known pharmacological agent.
- the present invention provides a novel means of preparing 1[(4-fluorophenyl)sulfonyl]-4-[4-[[7(trifluoromethyl)-4-quinolinyl]amino]benzoyl]piperazine.
- This compound is disclosed and claimed in U.S. Patent 4,167,567. This compound is known to be useful as an antihypertensive agent, and is currently under development at The Upjohn Company. This compound has the USAN name "losulazine”.
- U.S. Patent 4,167,567 discloses a process for preparing 1[(4-fluorophenyl)sulfonyl]-4-[4-[[7(trifluoromethyl)-4-quinolinyl]amino]benzoyl]piperazine.
- the present invention particularly provides:
- a process for preparing a compound of the Formula C-7, or a pharmacologically acceptable acid addition salt thereof which comprises reacting a compound of the Formula C-5, or a pharmacologically acceptable acid addition salt thereof, with a compound of the Formula C-6 to yield the Formula C-7 compound.
- salts include the hydrochloride, hydrobromide, hydroiodide, sulfate, phosphate, acetate, propionate, lactate, maleate, malate, succinate, tartrate, and the like. These salts may be in hydrated form.
- the present invention provides a novel means to prepare losulazine.
- the prior art process employs the 4-chloro-7-trifluoromethyl quinoline, the most expensive compound used in the preparation of losulazine, in the first step of the process, as seen by Chart A.
- This reagent is very expensive, and while the reaction proceeds in high yield, when the remaining steps of the process are performed, the yield of the final product from this starting material becomes relatively low. In other words, this process is not efficient in terms of 7-trifluoro- methyl-4-chloroquinoline.
- Another means of preparing this compound, as disclosed in the prior art is that set forth in Chart B. Once again, the yield based on the expensive starting material is low.
- the present invention provides a novel means for employing 4-chloro-7-trifluoromethyl-quinoline in the last step, thereby obtaining a substantial yield of final product based on this reagent.
- Two means of obtaining the compound of the Formula II are set forth on Chart C. As can be seen, both of these procedures ultimately employ the process of the present invention as the last step.
- the Formula C-5 compound (1-(4-aminobenzoyl)-4-(4-fluorophenyl)sulfonyl piperazine) is prepared from the 4-aminobenzoyl piperazine compound (Formula C-3A) by treating it with p-fluorobenzene sulfonyl chloride.
- the Formula C-3 p-nitrobenzoyl piperazine is treated with p-fluorobenzene sulfonyl chloride to yield the Formula C-4 compound (4-[4-fluorobenzenesulfonyl]-1-[4-nitrobenzoyl]- piperazine) which is then hydrogenated by known means (e.g., using hydrogen gas with a platinum oxide or palladium-on-carbon catalyst to yield the Formula C-5 compound.
- known means e.g., using hydrogen gas with a platinum oxide or palladium-on-carbon catalyst
- the solution is basified by known means (e.g., using an aqueous wash of sodium hydroxide, tertiary organic amine bases like triethylamine, potassium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, or potassium bicarbonate).
- a co-solvent may be added (e.g., ethanol, methanol, n-propanol, and n-butanol).
- reaction will proceed in any of the following as the sole reaction solvent: chloroform, methylene chloride, 1,2-dichloroethane, the alcohols named above, dimethylformamide (DMF) or dimethylsulfoxide
- the preferred solvent for the reaction is a mixture of two solvents because, surprisingly, the product formed is more soluble in such solvent mixture than it is in a comparable volume of either solvent alone, thus the preferred solvent is a mixture of two solvents: one co-solvent selected from the chlorinated hydrocarbon solvents chloroform, methylene chloride and 1,2-dichloroethane; the other co-solvent selected from the group of alcohols methanol, ethanol, n-propanol and n-butanol.
- the ratio (volume/volume) of the chlorinated hydrocarbon solvent: alcohol solvent can vary from 11 to 10:1, preferably it is about 2:1 to 5:1.
- the chloroform-ethanol mixture is preferred because: (1) the chloroform is sufficiently high boiling to allow a beneficial reaction rate; and (2) the ethanol apparently reacts with the 4-chloro- 7-trifluoromethylquinoline reactant more slowly than e.g., methanol does.
- the product may be recrystall i zed from toluene, ethyl acetate, benzene , dioxane , diethyl ether or tetrahydrofuran, (after dissolving it in the original reaction solvents ) as described more fully in Example 2.
- Glacial acetic acid tank farm 40 gal
- the autoclave is charged with 18.0 g of Platinum oxide and the solution is stirred under 50 psig of hydrogen until the required 3 molar equivalents of hydrogen is absorbed. Whenever the solution reaches 35° C , the stirrer is stopped and the mixture is cooled to 25° C before resuming the agitation.
- the mixture is filtered through Solka Floe on a sparkler filter collecting the filtrate in a 50-gal glass-lined wash tank.
- the autoclave is rinsed and the mixture filtered with 2.0 gal of glacial acetic acid.
- Steps 1-3 are repeated with 4.77 kg of 4-[4-fluorobenzenesulfonyl)- 1-[4-nitrobenzoyl]piperazine combining the f iltrates and washes from both runs in the 50-gal wash tank.
- the mixture is chilled to 20° C with cold water.
- the distillate is diluted with 30 gal of absolute methanol and discarded.
- step 8 The crystals from step 8 are collected on a grounded stainless steel filter (the filtrate is saved) , the crystals are rinsed with 3.0 gal of ethyl acetate and dried on the filter.
- the crystals are assayed by TLC (Analtech GF, 6% methanol/methylene chloride-uv).
- the crystals contained two major components (Rf-0.43, 85%; Rf-0.31, 15%).
- the crystalline product is collected by filtration through a stainless steel filter.
- the filter cake is rinsed with 3.0 gal of ethyl acetate.
- the filtrate is concentrated to about 3 gal and 3 gal of ethyl acetate are blown into the hot solution.
- the mixture is cooled and the crystals are collected by filtration, and the crystals are rinsed with ethyl acetate. The filtrate is discarded. The crystals are dried in a vacuum oven at 50° C, wt. 450 g (5%).
- TLC (Analtech GF, 6% methanol/methylene chloride, uv) analysis shows the first crop >99.9% one spot; second crop >98% one spot.
- Example 1 1 -C ( 4-fluorophenyl)sulfonyl]-4-[4-[[7(trifluoromethyl )- 4-quinolinyl]amino]phenyl]benzoyl piperazine monohydrochloride hemi hydrate Refer to Chart C (conversion of C-5 to C-7) .
- Procedure 1 From a grounded drum, 256 liters of chloroform are drawn into the 100 gallon vessel , which is maintained under nitrogen.
- the sodium hydroxide solution is added to the chloroform suspension through the port all at once.
- the pH of the aqueous portion is monitored , and is basified via the addition of more sodium hydroxide if necessary .
- the layers are then separated.
- the chloroform (lower ) layer is separated into the 200 gal wash tank.
- the aqueous layer is discarded after pH is adj usted to 7.
- the organic layer is washed with 187 l of water .
- the organic (lower) layer is drawn into the second 100 gallon vessel which is clean and dry.
- the aqueous layer is monitored for the presence of residual starting material by TLC . When no significant starting material is present , the pH is adj usted to 6.5-8.5 and the material is discarded.
- the TLC assay is performed with Analtech GF plates , using 6% methanol/-methylene chloride , developed with ultraviolet light (uv) .
- anhydrous hydrogen chloride is prepared by bubbling gaseous hydrogen chloride into 900 ml of pre-weighed absolute ethanol until 58.4 g have been absorbed.
- the ethanolic hydrogen chloride solution is then drawn into the reaction mixture. Beware of corrosive hydrogen chloride fumes .
- the hot reaction mixture is assayed by treating a sample with a few drops of 10% sodium hydroxide solution.
- the organic portion is analyzed by TLC comparing with the starting material and the product , using Analtech GF plates , and 6% methanol/methyl ene chloride and developing with ultraviolet light .
- the reaction i is approximately 95% complete , the mixture is concentrated under reduced pressure to about 22 2, ( 6.0 gal) .
- the chloroform distillate is discarded.
- the mixture is chilled to 18° C with cold water cooling.
- the crystalline filter cake is dried in a vacuum oven at 50° C.
- TLC analysis Analtech GF, 6% methanol/methylene chloride, u.v. shows one spot with an Rf value of 0.51.
- reaction vessel is re-inerted with nitrogen 75 % of methanol is drawn into the vessel from a grounded drum through a Sethco filter.
- a Sethco (in-line) filter is used to remove any find articulate matter.
- the filtrate should have no appreciable quantity of desired product remaining therein.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Quinoline Compounds (AREA)
Abstract
Novel process for preparing known pharmacological agents useful in the treatment of hypertension of formula C-7.
Description
DESCRIPTION
Process for 4-Amino Quinolines
BACKGROUND OF THE INVENTION The present invention provides a novel means for preparing a known pharmacological agent. In particular, the present invention provides a novel means of preparing 1[(4-fluorophenyl)sulfonyl]-4-[4-[[7(trifluoromethyl)-4-quinolinyl]amino]benzoyl]piperazine. This compound is disclosed and claimed in U.S. Patent 4,167,567. This compound is known to be useful as an antihypertensive agent, and is currently under development at The Upjohn Company. This compound has the USAN name "losulazine". INFORMATION DISCLOSURE
U.S. Patent 4,167,567 discloses a process for preparing 1[(4-fluorophenyl)sulfonyl]-4-[4-[[7(trifluoromethyl)-4-quinolinyl]amino]benzoyl]piperazine. SUMMARY OF THE INVENTION
The present invention particularly provides:
A process for preparing a compound of the Formula C-7, or a pharmacologically acceptable acid addition salt thereof, which comprises reacting a compound of the Formula C-5, or a pharmacologically acceptable acid addition salt thereof, with a compound of the Formula C-6 to yield the Formula C-7 compound.
The term "pharmaceutically acceptable salts" include the hydrochloride, hydrobromide, hydroiodide, sulfate, phosphate, acetate, propionate, lactate, maleate, malate, succinate, tartrate, and the like. These salts may be in hydrated form.
The present invention provides a novel means to prepare losulazine. The prior art process employs the 4-chloro-7-trifluoromethyl quinoline, the most expensive compound used in the preparation of losulazine, in the first step of the process, as seen by Chart A. This reagent is very expensive, and while the reaction proceeds in high yield, when the remaining steps of the process are performed, the yield of the final product from this starting material becomes relatively low. In other words, this process is not efficient in terms of 7-trifluoro- methyl-4-chloroquinoline. Another means of preparing this compound,
as disclosed in the prior art, is that set forth in Chart B. Once again, the yield based on the expensive starting material is low.
In Chart A, 4-chloro-7-trifluoromethyl quinoline (Formula A-1) in alcohol in the presence of hydrochloric acid reacts with 4-aminobenzoic acid to yield p-[(7-trifluoro-methyl-4-quinolyl)amino]benzoic acid of the Formula A-3. This compound is treated with thionyl chloride to yield the corresponding chloride of the Formula A-4. This compound is then treated with anhydrous piperazine to yield 1-[p-[[7-(trifluoro-methyl)-4-quinyl]amine]benzoyl]piperazine of the Formula A-6. This compound is then treated with p-fluorobenzenesulfonyl chloride (Formula A-6) to yield the desired Formula A-8 product, 1 [ (4-fluorophenyl)sulfonyl] 4-[4-[[7-(trifluoromethyl)-4-quinolinyl]amino]benzoyl]piperazine. See, e.g., U.S. patent 4,167,567, Example 17.
In Chart B, piperazine hexahydrate (Formula A-1) is reacted with p-nitrobenzoyl chloride (see, U.S. patent 4,167,567, preparation 1, part (a)) to yield p-nitrobenzoylpiperazine (Formula B-3) . This compoun is then hydrogenated using a palladium-on-carbon catalyst (U.S. patent 4,167,567, preparation 1, part (b)) to yield the Formula B-4 p-aminobenzoylpiperazine. This compound is then treated with 4-chloro-7-trifluoromethylquinoline (Formula B-5) using the method described in U.S. patent 4,025,629, Example 9. This compound is converted to the desired product as described above (Chart A).
The present invention provides a novel means for employing 4-chloro-7-trifluoromethyl-quinoline in the last step, thereby obtaining a substantial yield of final product based on this reagent. Two means of obtaining the compound of the Formula II are set forth on Chart C. As can be seen, both of these procedures ultimately employ the process of the present invention as the last step.
In Chart C, the Formula C-3 and C-3A compounds are prepared as described above (as depicted in Chart B).
The Formula C-5 compound (1-(4-aminobenzoyl)-4-(4-fluorophenyl)sulfonyl piperazine) is prepared from the 4-aminobenzoyl piperazine compound (Formula C-3A) by treating it with p-fluorobenzene sulfonyl chloride. (See Preparation 1 below.) Alternatively, the Formula C-3 p-nitrobenzoyl piperazine is treated with p-fluorobenzene sulfonyl chloride to yield the Formula C-4 compound (4-[4-fluorobenzenesulfonyl]-1-[4-nitrobenzoyl]-
piperazine) which is then hydrogenated by known means (e.g., using hydrogen gas with a platinum oxide or palladium-on-carbon catalyst to yield the Formula C-5 compound. (See also Preparations 2, 3 and
4 below). The process of the instant invention (the conversion of C-5 to
C-7) proceeds as follows: 1-[4-fluorobenzenesulfonyl]-4-[4-aminobenzoyl]piperazine, or a pharmacologically acceptable acid addition salt thereof (e.g. the hydrochloride), is dissolved in an appropriate solvent
(e.g., chloroform, methylene chloride, or 1,2-dichloroethane, preferably chloroform). In the case of salt, the solution is basified by known means (e.g., using an aqueous wash of sodium hydroxide, tertiary organic amine bases like triethylamine, potassium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, or potassium bicarbonate).
The layers are separated and the organic layer is washed. A co-solvent may be added (e.g., ethanol, methanol, n-propanol, and n-butanol).
The reaction will proceed in any of the following as the sole reaction solvent: chloroform, methylene chloride, 1,2-dichloroethane, the alcohols named above, dimethylformamide (DMF) or dimethylsulfoxide
(DMSO). The preferred solvent for the reaction is a mixture of two solvents because, surprisingly, the product formed is more soluble in such solvent mixture than it is in a comparable volume of either solvent alone, thus the preferred solvent is a mixture of two solvents: one co-solvent selected from the chlorinated hydrocarbon solvents chloroform, methylene chloride and 1,2-dichloroethane; the other co-solvent selected from the group of alcohols methanol, ethanol, n-propanol and n-butanol.
The ratio (volume/volume) of the chlorinated hydrocarbon solvent: alcohol solvent can vary from 11 to 10:1, preferably it is about 2:1 to 5:1. The chloroform-ethanol mixture is preferred because: (1) the chloroform is sufficiently high boiling to allow a beneficial reaction rate; and (2) the ethanol apparently reacts with the 4-chloro- 7-trifluoromethylquinoline reactant more slowly than e.g., methanol does.
4-Chloro-7-trifluoromethylquinoline, dissolved in the same solvent as the 1-[4-fluorobenzenesulfonyl]-4-[4-aminobenzoyl]piperazine, is then added to the reaction vessel. Ethanolic hydrogen chloride can
be added at this point in the reaction. The temperature is maintained in a range from about 20° C to the reflux temperature of the mixture, preferably at the reflux temperature. The reaction proceeds for 15 to 35 hours , preferably 24 hours . The reaction mixture is concentrated under reduced pressure, and crystallized using e.g. , ethyl acetate , toluene, benzene , dioxane , diethyl ether , or tetrahydrof uran , preferably ethyl acetate. Optionally , the product may be recrystall i zed from toluene, ethyl acetate, benzene , dioxane , diethyl ether or tetrahydrofuran, (after dissolving it in the original reaction solvents ) as described more fully in Example 2.
The use of the compounds of this invention is disclosed in U.S. Patent 4, 167,567, which is expressly incorporated by reference herein. DESCRIPTION OF THE PREFERRED EMBODIMENTS
The present invention is seen more fully by the examples given below.
Preparation 1 1 , ( 4 -Aminobenzoyl )-4- ( 4-fluorophenyl )sulfonyl piperazine Refer to Chart C (conversion of C-3A to C-5) .
To a suspension of 4-aminobenzoyl piperazine (0.005 mole) and tri ethyl amine (0.005 mole) in 20 ml of methylene chloride is added dropwise to a solution of approximately 1 g p-fluorobenzene sulfonyl chloride in methylene chloride at room temperature. After complete addition, the resulting mixture is stirred for 30 minutes under nitrogen .
The above mixture is quenched into cold water and extracted twice with 50 ml portions of methylene chloride . The methylene chloride solution is then washed with water, dried over sodium sulfate and concentrated in vacuo to yield an oil , which on trituration with anhydrous ether gives 1 .4 g methylene chloride ( 80%) of white crystalline titled product . Preparation 2 1 - ( 4-N i troben zoyl )-4-( 4-fluorophenyl )sulfony piperazine Refer to Chart C , (conversion of C-3 to C-4) .
To a solution of 4-nitrobenzyl piperazine (0.01 mole) (prepared as described in preparation 1 ) ( 0.01 mole) in 25 ml of methylene chloride is added dropwise a solution of p-fluorobenzene sulfonyl chloride
( 0.01 mole) in methylene chloride. The reaction mixture is maintained
under nitrogen. After complete addition, the resulting mixture is stirred for 30 minutes. The mixture is then quenched into cold water and extracted twice with 50 ml portions of methylene chloride. The methylene chloride solution is washed with water, dried over sodium sulfate and concentrated in vacuo to yield an oil. Trituration with anhydrous ether gives 3.2 g (82%) of white crystalline titled product, mp 179-181°.
Preparation 3 4-[4-Fluorobenzenesulfonyl]-1-(4-amlnobenzoyl)- piperazine Refer to Chart C (conversion of C-4 to C-5).
A mixture of preparation 2 and 0.2 g of 5% palladium-on-carbon catalyst in 200 ml of absolute ethanol is hydrogenated for one hour. To the above mixture is theri added 200 ml of methylene chloride and the mixture is filtered. The filtrate is concentrated to an oil. This on trituration with anhydrous ether gives 1.5 g (83$) of white crystalline product, mp 208-210°.
Anal. Calcd. for C17H18FN3SO3 (MW 367.34): C, 56.19; H, 4.99; N, 11.57; F, 5.23; S, 8.81.
Found: C, 55.74; H, 4.76; N, 11.55; F. 4.99; S . Preparation 4 Preparation of 4-[4-Fluorobenzenesulfonyl]-1-[4 aminobenzoyl]piperazine hydrochloride Refer to Chart C (conversion of C-4 to C-5).
Materials 4-[4-Fluorobenzenesulfonyl]-1 [4-nitrobenzoyl]- piperazine (22.88 moles) 9.27 kg
Glacial acetic acid, tank farm 40 gal
Platinum oxide (Engelhardt 1638) 36.00 g
Methanol, absolute, 90 gal
Concentrated hydrochloric acid (25.2 moles) 2.10 l Ethyl acetate, C.P. 150 I
Solka Floe as needed
Methylene Chloride, C.P. 37.3 gal
Equipment 30-gallon Autoclave Sparkler filter
50-gallon glass-lined tank 100-gallon glass-lined reactor (GD-1006) 100-gallon glass-lined receiver (GR-1012) Stainless steel filter
Procedure
1. A nitrogen inerted 30-gal autoclave is charged with 4.5 kg of 4-[4-fluorobenzenesulfonyl]-1 -[ 4-nitrobenzoyl ]pi perazine and 67.5 l of glacial acetic acid. The mixture is warmed to 45° C to dissolve the solids and then the mixture is cooled to 25°
C.
2. The autoclave is charged with 18.0 g of Platinum oxide and the solution is stirred under 50 psig of hydrogen until the required 3 molar equivalents of hydrogen is absorbed. Whenever the solution reaches 35° C , the stirrer is stopped and the mixture is cooled to 25° C before resuming the agitation.
3. The mixture is filtered through Solka Floe on a sparkler filter collecting the filtrate in a 50-gal glass-lined wash tank. The autoclave is rinsed and the mixture filtered with 2.0 gal of glacial acetic acid.
4. Steps 1-3 are repeated with 4.77 kg of 4-[4-fluorobenzenesulfonyl)- 1-[4-nitrobenzoyl]piperazine combining the f iltrates and washes from both runs in the 50-gal wash tank.
5. The combined filtrates are treated with 2. 10 I of concentrated hydrochloric acid , the mixture is stirred 10 min and the solution drawn into nitrogen-inerted reaction vessel . The wash tank is rinsed with 2.0 gal of glacial acetic acid.
6. The reaction mixture is heated to 65° C for 3 hours and then the solution is concentrated to 15.0 gal by vacuum distillation (jacket temp. 85° C , jet vac) .
7. 190 Liters of ethyl acetate are slowly blown into the hot concentrate in the reaction vessel while permitting any cloudiness to dissipate as crystals.
8. The mixture is chilled to 20° C with cold water.
9. The distillate is diluted with 30 gal of absolute methanol and discarded.
10. The crystals from step 8 are collected on a grounded stainless steel filter (the filtrate is saved) , the crystals are rinsed with 3.0 gal of ethyl acetate and dried on the filter.
11. The crystals are assayed by TLC (Analtech GF, 6% methanol/methylene chloride-uv). The crystals contained two major components (Rf-0.43, 85%; Rf-0.31, 15%).
12. The filtrate from step 10 is discarded after TLC establishes the absence of significant product.
Recrystallization 13. A nitrogen-inerted reaction vessel is charged with the crystals from step 10.
14. 140 I of absolute methanol are drawn into the reaction vessel from a grounded drum.
15. 140 l of methylene chloride are added.
16. The mixture is heated at reflux (44° C) with stirring. All solids are dissolved.
17. 120 I of ethyl acetate are blown from a grounded drum into the refluxing solution. (Note: Very little crystalline solid is present at the end of the ethyl acetate addition.)
18. The mixture is distilled and concentrated to approximately 25 gal (62° C pot temperature) at atmospheric pressure. The mixture is cooled to 20° C while maintaining the stirring.
19. The distillate from step 18 is discarded.
20. The crystalline product is collected by filtration through a stainless steel filter. The filter cake is rinsed with 3.0 gal of ethyl acetate.
21. The solids are dried in a vacuum oven at 50° C, wt. 6.72 kg (73.5%).
22. The filtrate is concentrated to about 3 gal and 3 gal of ethyl acetate are blown into the hot solution.
23. The mixture is cooled and the crystals are collected by filtration, and the crystals are rinsed with ethyl acetate. The filtrate is discarded. The crystals are dried in a vacuum oven at 50° C, wt. 450 g (5%).
24. TLC, (Analtech GF, 6% methanol/methylene chloride, uv) analysis shows the first crop >99.9% one spot; second crop >98% one spot.
Rf-0.43.
Example 1 1 -C ( 4-fluorophenyl)sulfonyl]-4-[4-[[7(trifluoromethyl )- 4-quinolinyl]amino]phenyl]benzoyl piperazine monohydrochloride hemi hydrate Refer to Chart C (conversion of C-5 to C-7) .
A solution of 7-trifluoromethyl-4-chloroquinoline (0.002 mole) Preparation 3 ( 1-( 4-aminobenzoyl)-4-(4-fluorophenyl )sulfonyl piperazine) , ( 0. 002 mole ) and ethanolic hydrochloric acid (0.7 ml , 4 molar ) in 15 ml of ethanol is heated to reflux for 18 hours under nitrogen. The above solution is then concentrated in vacuo and the residue is crystallized from methanol/methylene chloride to gi ve 0. 8 g ( 66%) of yellow crystalline titled product , mp 298-300° .
Example 2 Preparation of 1 -[ ( 4-Fluorophenyl)sulfonyl]-4[ 4-[[7- (trifluoromethyl )-4-quinolinyl]amino]benzoyl]pi perazine monohydrochlor ide Refer to Chart C (conversion of C-5 to C-7) .
Materials 100 gal glass-lined reaction vessels 200 gal glass-lined wash tank 24" square stainless steel filter Sethco in-line filter
Procedure 1. From a grounded drum, 256 liters of chloroform are drawn into the 100 gallon vessel , which is maintained under nitrogen.
2. With good stirring, 6400 g of 1-[4-fluorobenzenesulfonyl]-4-[4- aminobenzoyl] piperazine hydrochloride are added . The reaction vessel is again flushed with nitrogen .
3. 672.5 g of sodium hydroxide pellets are dissolved in 3.5 gal of water.
4. The sodium hydroxide solution is added to the chloroform suspension through the port all at once.
5. Stirring is continued until essentially all of the solids are dissolved.
6. The pH of the aqueous portion is monitored , and is basified via the addition of more sodium hydroxide if necessary .
7. The layers are then separated. The chloroform (lower ) layer is separated into the 200 gal wash tank. The aqueous layer is discarded after pH is adj usted to 7.
8. The organic layer is washed with 187 l of water .
9. The organic (lower) layer is drawn into the second 100 gallon vessel which is clean and dry.
10. The aqueous layer is monitored for the presence of residual starting material by TLC . When no significant starting material is present , the pH is adj usted to 6.5-8.5 and the material is discarded.
The TLC assay is performed with Analtech GF plates , using 6% methanol/-methylene chloride , developed with ultraviolet light (uv) .
11 . 64.0 l of absolute ethanol are drawn into the reaction vessel through a Sethco filter .
12. 40-60 I of the solvent are distilled off at atmospheric pressure to dry the mixture. The reaction mixture is then cooled to 30° c.
13. From a grounded container and through a Sethco in-line filter , a solution of 4-chloro-7-trifluoromethylquinoline ( 3708 g ) in chloroform ( 15.0 I) is drawn into the 100 gal reactor .
14. In a laboratory hood, a solution of anhydrous hydrogen chloride is prepared by bubbling gaseous hydrogen chloride into 900 ml of pre-weighed absolute ethanol until 58.4 g have been absorbed.
15. The ethanolic hydrogen chloride solution is then drawn into the reaction mixture. Beware of corrosive hydrogen chloride fumes .
16. The mixture is heated at reflux for 24 hr . ( The solution should be clear . It may be necessary to add more chloroform. )
17. The hot reaction mixture is assayed by treating a sample with a few drops of 10% sodium hydroxide solution. The organic portion is analyzed by TLC comparing with the starting material and the product , using Analtech GF plates , and 6% methanol/methyl ene chloride and developing with ultraviolet light .
18. When the reaction i s approximately 95% complete , the mixture is concentrated under reduced pressure to about 22 2, ( 6.0 gal) . The chloroform distillate is discarded.
19. While the solution i3 still hot , 150 I of ethyl acetate is slowly drawn into GD-1006 from a grounded drum and through a Sethco in-line filter . The rate of addition should be slow enough to avoid a gummy precipitate.
20. The mixture is chilled to 18° C with cold water cooling.
21 . The resulting yellow solids are collected on a grounded stainless steel filter. The chloroform containing filtrate by TLC is assayed as described above . If no significant product remains , the filtrate is discarded. If si gnificant product does remain , the filtrate is concentrated and a second crop is collected .
22. The crystalline filter cake is dried in a vacuum oven at 50° C.
23. TLC (analysis Analtech GF, 6% methanol/methylene chloride, u.v.) shows one spot with an Rf value of 0.51.
24. A clean, dry nitrogen inerted reaction vessel is charged with the dry product of Step 22.
25. The reaction vessel is re-inerted with nitrogen 75 % of methanol is drawn into the vessel from a grounded drum through a Sethco filter.
26. Through the Sethco filter, 230 I of methylene chloride are added.
27. With good stirring, heat the solution to reflux. (All solids should be dissolved.)
28. While refluxing, blow in 150 I of toluene from a grounded drum through the Sethco filter.
29. The mixture is concentrated to about 20 gal. Jacket temperature is 128° C and head temperature is 105° C when the distillation is stopped.
30. The solution is cooled to 20° C with cold water and the solids are collected on a grounded 24" stainless steel filter and rinsed with Skellysolve B (a commercial mixture of essentially n-hexane).
31. The product is dried overnight in vacuum oven at 50° c, WT. 8.92 G (94%).
32. TLC analysis (Analtech GF, 6% methanol/methylene chloride, u.v.) indicates one spot. Analysis C27H23F4CIN4SO3
Calcd. C, 54.50; H, 3-90; N, 9.40; Cl, 5.96; S, 5.39; F, 12.77.
Found: C, 54.38; H, 3.97; N, 9.44; Cl, 6.04; S, 5.50, F, 12.35.
HPLC analysis indicates total impurities are 0.10%. Additional Notes
1. A Sethco (in-line) filter is used to remove any find articulate matter.
2. It has not been necessary to add additional base when the stated molar ratio of sodium hydroxide is used. The pH check and adjustment is a precautionary measure to insure completion of reaction.
3. This distillation is required to azeotrope off any water and is critical for the preparation of anhydrous product.
4. The filtrate should have no appreciable quantity of desired product remaining therein.



J

Claims
CLAIMS 1 . A process for preparing a compound of the Formula C-7 :

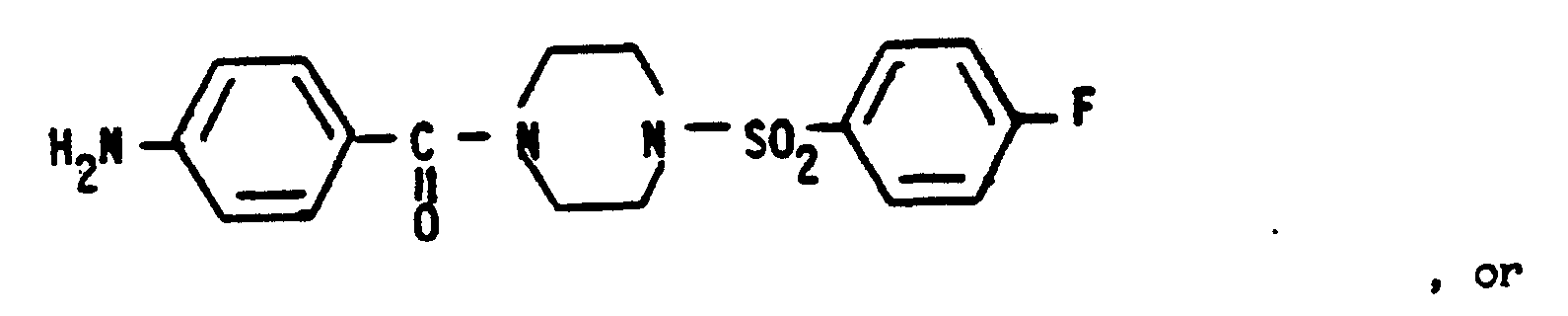

to yield the Formula C-7 compound.
2. A process of claim 1 , wherein the reaction is conducted in cosolvent , the temperature of the reaction is maintained at reflux , and the Formula C-6 compound is a hydrochloride salt.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71134285A | 1985-03-13 | 1985-03-13 | |
| US711,342 | 1985-03-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1986005489A1 true WO1986005489A1 (en) | 1986-09-25 |
Family
ID=24857712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1986/000484 Ceased WO1986005489A1 (en) | 1985-03-13 | 1986-03-11 | Process for 4-amino quinolines |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP0215902A1 (en) |
| JP (1) | JPS62502339A (en) |
| ES (1) | ES8707187A1 (en) |
| WO (1) | WO1986005489A1 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998054164A1 (en) | 1997-05-30 | 1998-12-03 | Takeda Chemical Industries, Ltd. | Sulfonamide derivatives, their production and use |
| WO1999040075A1 (en) | 1998-02-05 | 1999-08-12 | Takeda Chemical Industries, Ltd. | Sulfonamide derivatives, process for producing the same and utilization thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4025629A (en) * | 1974-01-07 | 1977-05-24 | The Upjohn Company | P-(trifluoromethylquinolylamino)benzamides, pharmaceutical dosage forms and method of treatment |
| US4159331A (en) * | 1978-05-05 | 1979-06-26 | The Upjohn Company | Antihypertensive 4-aminoquinolines |
| US4167567A (en) * | 1978-05-05 | 1979-09-11 | The Upjohn Company | Antihypertensive 4-aminoquinolines |
-
1986
- 1986-03-11 JP JP61502163A patent/JPS62502339A/en active Pending
- 1986-03-11 WO PCT/US1986/000484 patent/WO1986005489A1/en not_active Ceased
- 1986-03-11 EP EP19860902141 patent/EP0215902A1/en not_active Withdrawn
- 1986-03-12 ES ES552937A patent/ES8707187A1/en not_active Expired
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4025629A (en) * | 1974-01-07 | 1977-05-24 | The Upjohn Company | P-(trifluoromethylquinolylamino)benzamides, pharmaceutical dosage forms and method of treatment |
| US4159331A (en) * | 1978-05-05 | 1979-06-26 | The Upjohn Company | Antihypertensive 4-aminoquinolines |
| US4167567A (en) * | 1978-05-05 | 1979-09-11 | The Upjohn Company | Antihypertensive 4-aminoquinolines |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0215902A1 (en) | 1987-04-01 |
| ES552937A0 (en) | 1987-08-01 |
| JPS62502339A (en) | 1987-09-10 |
| ES8707187A1 (en) | 1987-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0260817B1 (en) | Quinazolinediones and pyridopyrimidinediones | |
| JP2638752B2 (en) | 1-aminoethylindole derivative | |
| FI88504C (en) | Process for Preparing Therapeutically Useful 4-Benzyl-1- (2H) -phthalazinone Derivatives | |
| NO822643L (en) | PROCEDURE FOR THE MANUFACTURE OF NEW PIPERAZINONES AND THEIR USE. | |
| ZA200005881B (en) | Amide derivatives and nociceptin antagonists. | |
| EP1651214A2 (en) | Benzimidazole derivatives as mek inhibitors | |
| DE60304695T2 (en) | 7-ARYLSULFONAMIDO-2,3,4,5-TETRAHYDRO-1H-BENZO [D] AZEPINE DERIVATIVES WITH 5-HAT-6-RECEPTOR AFFINITY FOR THE TREATMENT OF DISEASES OF THE CENTRAL NERVOUS SYSTEM | |
| EP0144986A2 (en) | Indole-3-carboxamide derivatives | |
| JP2002533445A (en) | Synthesis of histamine dihydrochloride | |
| DK151017B (en) | METHOD OF ANALOGUE FOR PREPARING SUBSTITUTED N- (4-INDOLYL-PIPERIDINO-ALKYL) -BENZIMIDAZOLONES OR PHYSIOLOGICALLY ACCEPTABLE ACID ADDITION SALTS THEREOF | |
| JP2001518906A (en) | Indazole amide compounds as serotonin-like agents | |
| EP0937715B1 (en) | Tetrahydrobenzindole compounds | |
| NZ284851A (en) | 1-[3-(piperazin-1-yl)-prop-1-yl] benzimidazole derivatives and their use in the treatment of cns disorders | |
| WO1986005489A1 (en) | Process for 4-amino quinolines | |
| US4148796A (en) | γ-Piperidinobutyrophenones | |
| NZ243337A (en) | 1-piperidyl substituted quinoline derivatives and pharmaceutical compositions | |
| NO136713B (en) | ||
| NO833181L (en) | PROCEDURE FOR PREPARING 6 - ((CYCLIC AMINO) ALKYL-AMINO) -TETRAHYDROTRIAZOLE (3,4-A) PHTHALAZINES | |
| US5753658A (en) | Quinolonecarboxylic acid derivatives | |
| CN111253315A (en) | Imidapril hydrochloride organic impurities and preparation method thereof | |
| EP0735030A1 (en) | Piperazine derivative having antiallergic activity | |
| US6229015B1 (en) | Acridone derivatives and method of preparation of 8-hydroxy imidazoacridinone derivatives | |
| EP0221541A2 (en) | Quinolonecarboxylic acid derivatives and their preparation | |
| JPS624263A (en) | Novel derivative of hydroxyalkoxy-4-phenylpropyl indole, itssalt, its production, its intermediate, its use as drug and pharmaceutical composition containing the same | |
| US4840950A (en) | 4-arylcarbonyl-1-[(4-morpholinyl)-lower-alkyl]-1H-indoles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 1986902141 Country of ref document: EP |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE FR GB IT LU NL SE |
|
| WWP | Wipo information: published in national office |
Ref document number: 1986902141 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1986902141 Country of ref document: EP |