m * 発明の名称
4 H - 3 , 1 一べンゾォキサジン一 4 一オン化合物およ びそのエラスターゼ阻害用医薬組成物
技術分野
本発明は 4 H一 3', 1 一べンゾォキサジン— 4 —ォン化 合物およびそれを有効成分とする医薬組成物に閔するも のである。
更に詳しく述べるならば、 本発明は動物のエラスチン などのようなタンパク質に対するプロテアーゼに起因す る疾患の予防 · 治療、 特にエラスタ一ゼの分解作闬によ つて惹起される組織の損傷および種々な炎症または変性 状態を抑制するためのプロテアーゼ阻害効果、 特にェラ スクーゼに対し選択的な阻害効果を有する 4 H - 3 , 1 一 ベンゾォキサジン一 4 一オン化合物、 およびそれを有効 :;分とするセリンプロテアーゼに起因する疾患の予防 - 治療、 特にヒ ト白血球エラスターゼに起因する疾患の予 防 ■ 治療に用いることのできる医薬組成物に関するもの である
背景技術
エラスチンは結合組織中にある弾性繊維の主成分を形 成し、 かつゴム状の弾性を有する繊維状タンパクであつ て、 肺, 気管支, 大動脈などに多く含まれている , エラ
スターゼはエラスチンを加水分解し得るタンパク分解酵 素の一群であり、 哺乳動物の腾臓, 多形核白血球, ある いは微生物が産生する 白血球のエラスターゼは、 貧食 した細菌などを分解する消化酵素と して重要な働きを持 つが、 いったん細胞外へ漏出すると組織中のエラスチン を分解して組織の損傷および種々の炎症または変性状態 を惹起する。 このようなエラスターゼによるエラスチン の過度の分解は肺気腫, 成人呼吸窮迫症候群, 肺線維症 気管支炎, 肺炎, リウマチ関節炎, 動脈硬化, 敗血症, ショ 、 '/ク, 滕炎, 腎炎, ある種の皮膚病などの原因と考 えられている。 徒って、 エラスターゼ阻害剤はこれらの 疾患の治療剤、 あるいは予防剤と して有用であると考え られる。
エラスターゼ酵素阻害作用を有する化合物と して、 下 5匚 ;
o
4
0
L、
N
の化学構造を基本構造とする 4 H— 3, i 一べンゾォキサ ジン— 4一才ン化合物が知られている:
例えば、 テシマ他、 「 J. Bioi. Cheirし 」 , 25了 卷, 5085〜5091頁, δ2年には、 種々の 2—アルキル一 4 Η
3, 1 一べンゾ才キサジン一 4一オンが報告されており へドストロム他、 「 Biochemistry , 23卷、 1753〜Π59 頁、 19δ4年には 2—エトキシー 4 Η— 3, 1 —ベンゾォキ サジン一 4—オンが報告されている また、 スペンサー 他, 「Biochern. Bi ophys. Res, Commun.」 , 140 卷、 928 〜933 頁、 年には、 2—アルキル一 4 H— 3, 1 -ベンゾォキサジン一 4一オンの 5—メチル置換体が、 優れたエラスタ一ゼ阻害作用を示寸ことが報告されてい る -, また、 スティン他、 「 Bioclieiriistry」 , 26卷、 412fe 〜4130頁、 1987年、 及びラダク リシュナン他、 「 J. Μοί ο 1. 」 , 19δ 卷、 417-424 頁、 1987年には、 ベンゾォ キサジノン誘導体のエラスターゼ阻害機構が報告されて いる 更に、 特開昭 60-1694 号には、 Η -3, 1 -ベ ンゾォキサジン一 4—オンの 2—アミ ノ誘導体が開示さ れており、 特開昭 62- 30Π0号には、 4 Η— 3, 1 —べンゾ ォキサジン一 4一オンの 2—ォキシ誘導体が開示されて いる。
これらの化合物は、 多くの場合、 エラスターゼに対寸 る阻害よりも、 キモト リブシンに対してより強い阻害活 性を示すことが知られている。
また、 キモト リァシンよりェラスタ一ゼに対してより 強い阻害活性を示す 4 Η - 3, 1 —ベンゾォキサジン一 4 一オン誘導体と して 2,? —ジアミノ誘導体が本発明者ら によって提示されている (W088/ 790号) ,,
本発明者らによりすでに開示されている 4 H— 3 , 1 — ベンゾォキサジン一 4 一オン誘導体 ( W088/ 0 9 7 9 0号〉 は エラスターゼに対する強い阻害活性と高い選択性を有寸 る、·. しかしながらこの誘導体は水に対する溶解度が低い こと、 動物の気道内に投与した場合に短時間で肺組織か ら消失しやすいこと等の改善の余地があることがわかつ てきた
発明の開示
本発明の目的は、 セリンプロテアーゼに対し、 強い阻 害作用を示寸新規 4 H— 3 , 1 —べンゾォキサジン一 4 - オン化合物、 およびそれを有効成分とする医薬組成物を 提供することにある。
本発明の他の目的は、 セリンプロテアーゼのなかでも 特に、 ヒト白血球エラスターゼに対し選択的かつ強い阻 害作用を示す新規 4 H— 3 , i 一べンゾォキサジン一 4 一 オン化合物、 およびそれを有効成分とする医薬組成物を 提供することにある
更に本発明の他の目的は、 ヒト白血球エラスタ一ゼに 対し強い阻害活性と高い選択性を持ち、 かつ、 水に対す る溶解性, 肺組織での滞留性に優れた新規 4 H— 3 , 1 - ベンゾォキサジン— 4 一オン化合物、 およびそれを有効 成分とする医薬組成物を提供することにある。
上記目的は、 本発明の化合物により達成される すな わち本発明は、 下記式 [ I ]
ノ酸残基またはこれらアミノ酸から構成されるジべ \ ァチドを表わし、 A 3 はリジン, グルタミン酸およ ': びァスパラギン酸から選ばれるアミノ酸残基を表わ ; し 〈これらのアミノ酸残基の側鎖は保護基により保 護されていてもよい) 、 Y i はァミノ基の保護基を ! 表わし、 Y 2 は水素原子またはスルホ二ル基を表わ ; し (但し、 Α 3 のアミノ酸残基の側鎖が保護基によ , り保護されている場合は水素原子を表わす〉 、 mは : 0または 1 を表わし ;
(Π) ¾は水素原子または低級アルキル基を表わし ; ( および は
(a) Xが式 [A] を表わす場合には、 ¾は 1ないし 2個のカルボキシル基を有する低級アルキル基、 ¾は水素原子, 低級アルキル基または 1ないし 2 個のカルボキシル基を有する低級アルキル基を表 ; わすか、 または と ¾は互いに結合して 1ないし ; 2個のカルボキシル基あるいはカルボキシル基を ; 有する低級アルキル基で置換された 5 , 6もしく ; は 7員環を表わし、
! (b) Xが式 [ B ] を表わす場合には、 R2は低級アル ! キル基を表わし、 は水素原子を表わす。
で表わされる 4 H— 3, 1 —ベンゾォキサジン一 4 _オン 化合物、 その塩類、 及びその医薬組成物である。
木発明の医薬組成物は、 前記式 [ I ] の 4 H— 3, ί - ベンゾォキサジン一 4一オン化合物または、 その医薬的 有効量と、 医薬的に許容されるキヤリヤーとの混合物を 含有するものである。
発明を実施するための最良の形態
式 [ I ] において は、 式 [ Α]
または式 [ B ]
Y 2 - { A 2 ) m - A 3 - [ B ] で表わされる置換基を示す。
ここで、 はアミノ酸残基または 2ないし 3ァミノ 酸残基で構成されるペアチドを表わす 〈これらのァミノ 酸残基の側鎖は保護基によ り保護されていてもよい〉 。
A i により表わされるアミノ酸残基、 又はペプチドを構 成するアミノ酸残基は、 a —, /3—およびァーァミノ力 ルボン酸の!)および L光学異性体、 およびラセミ体残基 を包舍するもので、 このよ うなアミノ酸は、 例えばァラ ニン, ァスパラギン, ァスパラギン酸, システイ ン シ スチン, グルタミン酸、 グルタミン, グリシン, ヒスチ ジン, イ ソロイ シン, ロイシン, リジン, メチォニン, フエ二ルァラニン, ァロリン, セリン, スレオニン, ト リプトフアン, チロシン, ノくリン, ホモセリン, ホモシ スティ ン, ヒ ドロキシルプロリン, オル二チン, チロキ シン, ノルバリン, ノルロイシン, フエニルグリシン, /3 —ァラニン, およびァーァミノ酪酸の Dおよび L光学 異性体又はラセミ体から選ばれることが好ましい。
更に好ましいアミノ酸は、 Lーァラニン, グリシン, L一イソロイシン, L一口イシン, L—フエ二ルァラ二 ン, L一プロリン, L—バリン, L一ノルバリン, L— ノルロイ シン, L—フエニルグリシン, カルボべンゾキ シ基で保護された ε —アミノ基を有する L一リジン, ベ ンジルエステルと して保護されている ^ 一カルボキシル 基を有する Lーァスパラギン酸、 およびァ一ベンジルェ ステルと して保護されているカルボキシル基を有する L
一グルタミン酸などである :,
式 [ A ] において、 によ り表わされるアミノ酸残 基の側鎖は、 保護基によ り保護されていてもよいく 式 [ Α ] において、 このような A i によ り表わされるアミ ノ酸残基の側鎖 (ァミノ基, カルボキシル基, グァニジ ノ基, イ ミダゾリル基, メルカアト基, または水酸基〉 を保護する保護基と しては、 例えば、 ァミ ノ基の保護基 と しては、 カルボベンゾキシ基, スクシ二ル基, メ キ シスクシニル基, ァセチル基, ト リフル才ロアセチ/レ基 t e r t—ブトキシカルボニル基, イソニコチニルォキシ力 ルポ二ル基, およびトシル基などが知られている また カルボキシル基の保護基と してはべンジルエステルおよ び 4—ピコ リルエステル等が知られており 、 グァニジノ 基およびィ ミダゾリル基の保護基と して力ルボベンゾキ シ基およびトシル基などが知られており、 メル力ァ ■··基 の保護基と しては S—べンジル基などが知られており、 水酸基の保護基と しては 0—ベンジル基等が知られてい る;. しかし、 本発明に有用なこれらの保護基は、 いずれ も上述のものに限定されるものではない。
式 [ A ] において、 はアミノ基の保護基を表わす が、 係る保護基と しては例えば、 上述のァミノ基の保護 基をあげることができる これらのなかでも保護基は力 ルボベンゾキシ基, t e r t—ブトキシカルボニル基, ト キシスクシニル基, およびァセチル基から選ばれること
が好ましい。
また式 [ I ] において Xが式 [ B ] で表わされる場合 A 2 はグリシン, アラこン, バリン, およびロイシンか ら選ばれるアミノ酸残基またはこれらアミノ酸から構成 されるジペプチドを表わし、 A 3 はリジン , グルタミン 酸およびァスパラギン酸から選ばれるアミノ酸残基を表 わす (これらのアミ ノ酸残基の側鎖は保護基によ り保護 されていてもよい) . mは 0または 1 を表わす
これらのアミノ酸残基は、 前述の A のアミ ノ酸残基 の場合と同様に、 その D , L光学異性体、 ラセ ミ体残基 を含むものである。
A 2 と しては、 これらのなかでもグリシン, ァラニン バリンから選ばれるアミノ酸残基が好ま しい:、
また、 A 3 と しては、 これらのなかでも リジン, ダル ク ミン酸残基が好ましい .
A s のアミノ酸残基の側鎖が保護基によ り保護されて いる場合の保護基と して,は、 前述の のアミノ酸残基 の側鎖の保護基と同様のものをあげることができる。
式 [ B ] において、 Y 2 は水素原子またはスルホニル 基を表わす 〈但し、 Α 3 のアミノ酸残基の側鎖が保護基 によ り保護されている場合は水素原子を表わす)
スルホニル基と しては、 置換または非置換の芳香族ス ルホニル基をあげることができる。 かかる置換基と して は、 1 または 2以上の同一または異なってハロゲン原子
(フッ素原子, 塩素原子, 臭素原子等〉 、 低級アルキル 基、 d 〜C 6 の低級アルコキシ基 (メ トキシ, ェトキ シ、 イソプロポキシ等〉 をあげることができる
これらのなかでも置換基としては塩素原子, メチル基 メ トキシ基等が好ましい。 芳香族スルホニル基と しては ベンゼンスルホニル基, α —ナフチルスルホニル基, β 一ナフチルスルホニル基をあげることができるが、 なか でもベンゼンスルホニル基が好ましくあげられる。
式 [ I ] において、 は水素原子または低級アルキル 基を表わすが、 かかる低級アルキル基は、 1〜6個の炭 素原子を有するものであることが好ましく、 このような アルキル基は、 メチル, ェチル, プロピル, ブチル, ぺ ンチル, およびへキシル並びに、 それらの異性体形から 選ばれることが好ましい。
一般に、 式 [ I ] において、 Riが水素原子, メチル基 乂はェチル基を表わすことがより好ましい。
式 [ I ] において、 R2および は、 Xが式 [ A ] を表 わす場合には、 は 1ないし 2個のカルボキシル基を有 する低級アルキル基、 ¾は水素原子, 低級アルキル基ま たは 1ないし 2個のカルボキシル基を有する低級アルキ ル基を表わすか、 または と は互いに結合して 1ない し 2個のカルボキシル基あるいはカルボキシル基を有す る低級アルキル基で置換された 5, 6もしくは 7員環を 表わす。
式 [ ェ ] において R2'によ り表わされる 1ないし 2個の カルボキシル基を有する低級アルキル基は、 1〜 6個の 炭素原子を有するものであることが好ましく 、 例えば、 カルボキシメチル基, 1 一カルボキシェチル基, 3—力 ルボキシァロピル基, 1 , 2 —ジカルボキシェチル基ある いは 1 , 3 ージカルボキシプロピル基などである
式 [ I ] において ¾によ り表わされる低級アルキル基 は、 1〜 6個の炭素原子を有するものであることが好ま しく 、 このようなアルキル基は、 メチル, ェチル, フ'口 ピル, プチル, ペンチルおよびへキシル並びに、 それら の異性体形から選ばれることが好ましい,
式 [ ェ ] において ¾によ り表わされる 1ないし 2個の カルボキシル基を有する低級アルキル基は、 1〜 6個の 炭素原子を有するものであることが好ま しく 、 例えば力 ノンボキシメチル基, 1 一力ルボキシェチル基, 3 -力ル ボキシプロピル基, 1 , 2 —ジカルボキシェチル基あるい は 1, 3 —ジカルボキシプロピル基などである。
一般に、 式 [ I ] において ¾が水素原子, メチル基, ェチル基, カルボキシメチル基を表わすことが好ま しい
^ [ I ] において と が互いに結合して、 R2と が 結合している窒素原子と と もに 1ないし 2個のカルボキ シル基あるいは 1ないし 2個のカルボキシルを有する低 級アルキル基で置換された 5, 6又は 7員環を形成する ものと して、 2 —カルボキシピロリジンもしくは 3 —力
ルポキシピロリジン又は 4 一カルボキシピペリジンなど あるいはカルボキシ低級アルキルピロリジン, カルボキ シ低級ピぺリジンなどをあげることができる。
また式 [ェ ] において、 Xが式 [ B ] を表わす場合に は、 R2は低級アルキル基を表わし、 ¾は水素原子を表わ す R2が低級アルキル基を表わす場合、 かかるアルキル 基の定義は上述の場合と同じである
式 [ I ] の化合物の塩類としては、 または ¾に含ま れているカルボキシル基あるいは、 アミノ酸残基または ベプチド中のアミノ酸残基中の側鎖に付随するカルボキ シル基から形成され得る無機および有機塩基の塩類、 お よび、 ァミ ノ, グァニジノ, またはイ ミダゾリル基から 形成され得る無機および有機酸の酸付加塩類がある .. 無 機塩基から誘導される塩と しては、 アンモニゥム, カリ ゥム, ナト リウム, カルシウム, マグネシウムなどの塩 がある。 また有機塩基から誘導される塩と しては、 ジニ
、 チルァミン, ィ、 /プロピルァミン, エタノールァ ン またはピペリジン等の塩がある。 酸付加塩と しては塩酸 臭化水素酸, 硫酸, 硝酸およびリン酸などの無機酸を用 いて形成されたもの、 および酢酸, プロピ才ン酸, グリ コール酸, ピルビン酸, しゅう酸, りんご酸, マロン'酸 コハク酸, マレイン酸, フマール酸, 酒石酸, クェン酸 安息香酸, 桂皮酸, メタンスルホン酸, エタンスルホン 酸, p —トルエンスルホン酸, およびサリチル酸などの
ような有機酸を用いて形成されたものがある。
本発明の [ I ] 式において Xが式 [ A ] を表わす場合 の 4 H— 3, 1 —べンゾォキサジン一 4一オン化合物は、 例えば下記合成経路により合成することができる。
02 N
O2 N
¾ Pd— C
C O O H
D C C
反応 )
02 N N - C N
!:
H O
( D )
Ri O
O Y i 一 A 〇 H 入
N R2 反応 ie:'
N
( E )
Ri O
0 肌休 5¾ 反応 ί ί )
V A N , R2
N
、.、¾
( F )
Ri Ο
Ο
A
Υ 1 A Ν Ν R2
Η ( I Α )
但し、 ' , ' はそれぞれ R2, におけるカルボキ シル基が保護された基を表わす。
反応(a> において、 式 ( A ) の置換あるいは非置換 4 一二トロアントラニル酸ベンジルをビスト リクロロメチ ルカーボネートと反応させ、 式 ( B ) のフ X二ルイ ソシ ァネート誘導体に変換する。 これらの方法は、 ェ、' /力一 ト他 [ Anew. C enu Int. Ed. Engl. ] 26巻、 894-895 頁, 1987年に記載されている。
応(b) において、 式 ( B ) のフエ二ルイ ソシァネー ト誘導体を下記式 [ Π ]
Η - Ν [ Π ]
ヽ '
但し、 ¾' , ¾' はそれぞれ R2, におけるカルボキ : シル基が保護された基を表わす
で表わされるァミンと反応させ式 ( C ) の 2—ゥレイ — 4一二トロアントラニル酸ベン ジル誘導体を合成する この反応は、 不活性有機溶媒の存在下、 両化合物を接触 させることによって行うことができる 好ま しい有機溶 媒と してテトラヒ ドロフラン, エーテル, ベンゼンなど あげることができる。 反応は室温で 1 〜48時間、 好ま しくは 3〜 20時間反応させることによ り行うことができ る
上記式 [ ] で表わされるァミンにおいて は
れぞれ R2 , ¾におけるカルボキシル基が保護された基 を表わす これらカルボキシル基の保護基と しては t 一 ブチル基, トリクロ口ェチル基などが用いられる。
反応(c ;' において、 式 ( C ) の 2 —ゥレイ ドー 4一二 トロアントラニル酸ベンジル誘導体を、 公知の方法によ り、 式 ( D ) の 2—ゥレイ ド一 4 一アミノアントラニル 酸誘導体に変換する 反応(c ) は好ましくはパラジウム 炭素触媒存在下水素ガス接触還元により達成することが できる。
反応(cU において、 式 ( D ) の 2 —ゥレイ ドー 4 —ァ ミノアントラニル酸誘導体を、 不活性有機溶媒中脱水縮 合剤を用いて環化反応させ、 式 ( E ) の 7—アミ ノー 4 I I - 3 , 1 一べンゾォキサジン一 4 一オン誘導体を合成ォ る . 好ましい有機溶媒と して酢酸ェチル, 塩化メチレ ン ベンゼンなどがあげられる: また、 好ましい脱水縮合剤 と して N, N ' ージシクロへキシルカルボジイ ミ ド ( D C C ) , 1 - ( 3 -ジメチルァミ ノア口ピル) 一 3 —ェ チルカルボジィ ミ ド ( E D C ) などをあげることができ る。 反応は室温で 1〜48時間、 好ましくは 3〜20時間反 応させることにより行うことができる。
反応 ) において、 式 ( E〉 の 7—アミノー 4 H— 3 , ] —ベンソ"ォキサジン— 4—オン誘導体を Y 1 一 A 1 -- 0 Hで表わされる保護されているアミノ基を有するアミ ノ酸あるいはぺプチドのカルボキシル基と縮合反応させ
式 ( F〉 の 7— [ Y i - A i ] —アミノー 4 H— 3, 1 ― ベンゾォキサジン一 4—オン誘導体を合成する この縮 合反応はぺプチド合成におけるぺプチド結合の生成と し て様々な公知方法を用いることができる (泉屋信夫他著 「ペプチド合成の基礎と実験」 1985年、 丸善) 。 この縮 合反応(e> において、 保護されたアミノ基を有するアミ ノ酸、 あるいはべプチドのカルボキシル基を活性化させ 反応 ) で得られた式 ( E〉 の 7—アミノー 4 H— 3, 1 一べンゾォキサジン一 4一オン誘導体と縮合させること が好ましい。 よ り好ましい縮合方法においては、 保護さ れたアミノ基を有する 7:ミノ酸あるいはペアチドと、 炭 酸モノアルキルエステル を反応させて混合酸無水物を 調製し、 この混合酸無水物と、 式 〈 E〉 の化合物とを反 応させる。 この方法に用いられる好ま しい炭酸モ ノアル キルエステル塩化物はクロ口蟻酸ィ ソブチル ( I F である ..
応(Π において式 ( F ) の 7— 「 Y — A,- ] 一ァ ミノー 4 H— 3, 1 一べンゾォキサジン一 4一才ン誘導体 の 2位置換基 R2' , ¾' に含まれるカルボキシル基の保 護基を除去し、 本発明の式 [ I A ] の 4 H— 3, 1 一ベン ゾォキサジン一 4一才ン化合物を得る。 この脱保護反応 は !¾', ' に含まれるカルボキシル基の保護基によ り異 なるが、 例えば保護基と して t一ブチル基を用いた場合 には、 酸で処理することによ り容易に除去することがで
きる。
Λ ( Α ) において、 が水素原子を表わす場合の出発 化合物は市販品と して容易に入手することができる: ま た、 カルボべ Cンゾキシ基ぁるいは tert—ブトキシカルボ ニル基で保護されたアミノ基を有するアミノ酸、 あるい は保護された側鎖を有するアミノ酸は、 その多くが市販 品と して容易に入手でき< oるノ .:, 更に、 メ トキシスクシニル 基で保護されたアミノ基を有. 0するァミノ酸は、 コバク酸 モノメチルエステル (市販品と して容易に入手可能) と アミ ノ酸とから公知のペプチド結合生成反応により製造 することができる。
本発明の式 [ I A ] の化合物の出発化合物と して用い られる式 〈 A ) の化合物において、 ¾がアルキル基を表 わす化合物、 すなわち、 4一 二トロー ό一アルキルアン トラニル酸ベンジルは下記の方法で製造できる
Ri
〇 H Ρ 0 C U
ジェチルァ ノ- 、
反応
02 N
G )
Rt
NaO e, HMPA
反応(h
02N NO2
H )
¾ S O 4
反応 (j
( J
Na2 C03,
ベンジルアルコール
( A )
すなわち、 反応(g) において、 式 〈 G ) のフエノール 誘導体は、 ブースロイ ド他 [J. Chem. Soc. ] 1504- 1508 頁、 1953年記載の方法により対応する式 ( H〉 のクロ口 化合物に変換される。 次に、 反応 ) において、 ガンビ 一他、 [J. Indian Chem. Soc. ] 41卷、 43-46 頁, 1¾4 年記載の方法により、 式 ( H ) の化合物をペンタン一 2, ージオンおよびナトリウムメ トキシドと反応させて式 ( J ) の ( 2—アルキル— 4, 6 —ジニトロフエニル〉 ジ
ァセチルメタンに変換し、 次に、 反応(j) において式 ( J ) の化合物を、 溏硫酸により環化させて式 ( K〉 の 化合物に変換し、 最後に反応( においてこの式 ( K 〉 の化合物を炭酸ナトリゥムおよびべンジルアルコール存 在下、 テトラヒドロフラン中で加熱することにより、 式 ( A ) の 4一二トロ— 6—アルキルアントラニル酸ベン ジルが製造される。
本発明の [ I ] 式において Xが式 【 B ] を表わす場合 の 4 H _3, 1 —べンゾォキサジン— 4一オン化合物は、 例えば下記の 3つの合成経路により合成することができ る。
(U 式 [ B ] において Y 2 が水素原子を表わす場合
O Y- (A2 ) m - A ? -OH 人 反応 ( 】 )
H
(し )
Ri O
I
H z -、、、
, Q 脱保 δ§
A 入一メ m )
Y-(A2 ) m — A' 、N 反応 (
N ― R2
H
( M )
NHi
但し、 Yはアミノ基の保護基を表わし、 A ' 3 は A 3 ' において側鎖が保護されたアミノ酸残基を表わ寸
(i i)式 [ B ] において Y 2 がスルホ二ル基を表わォ場合
H2N
(し )
Ri 〇 广ヽ 〇 脱 ノ、 , 反応 (n ϊ 2 (A: A' N N N R2
H H
N
Ri O
^く丫 '人0
く z
Y' (A2 ) m -A: N -R2
H X.
H H
[ I B 2 ] 又-
P 〇
O スルホ二 レ化 反応 ( o )
H- (A2 ) ni -A3 一 N '
2
H
B l ]
但し、 Y' 2 はスルホ二ル基を表わし A' 3 は A3 ^ おいて側鎖が保護されたアミノ酸残基を表わす。
反応 ( 1 〉 において式 ( L ) の 7—ァミノ— 4 H— 3, 1 一べンゾォキサジン一 4一オン誘導体を下記式 [ H ]
Y — ( A 2 ) m — A ' -OH [I] 但し Yはァミノ基の保護基を表わし、 A' 3 は Α3 に I j おいて側鎖が保護されたアミノ酸残基を表わす。 : で表わされるアミノ酸又はペプチドと縮合反応させ、 式
〈 M〉 の 7— [Y— ( A2 〉 m — A' 3 ] —アミノー 4 H— 3, 1 —ベンゾォキサジン一 4—オン誘導体を合成す る。 この縮合反応は反応 ( e ) と同様の方法で実施する ことができる。
反応 ( m ) において式 ( M〉 の 7— [Y— ( A2 ) m — A' 3 ] —アミノー 4 H— 3, 1 —ベンゾォキサジン一 4一オン誘導体の保護基 Yを除去し、 本発明の式 [ I B1] の 4 H— 3, 1 一べンゾォキサジン— 4一オン化合物を得
る。 この脱保護反応は反応 ( f ) と同様の方法で実施す ることができる。 またこの反応において、 A' 3 のアミ ノ酸残基の側鎖保護基を同時に除去してもよい。
反応 〈ί ' )において式 〈し〉 の 7—アミノー 4 Η— 3, 1 —ベンゾ才キサジン一 4一オン誘導体を下記式 [IV]
Y' 2 - ( A2 ) m - A' 3 — ΟΗ … [IV]
但し、 Y' 2 はスルホ二ル基を表わし、 A' 3 は A3 I I において側鎖が保護されたアミノ酸残基を表わす。 _j で表わされるアミノ酸又はペプチドと縮合反応させ、 式
( N ) の 7— [Υ' 2 - { A2 ) m - A' 3 ] —ァミノ 一 4 H— 3, 1 —ベンゾォキサジン— 4—オン誘導体を合 成する。 この縮合反応は反応 ( e〉 と同様の方法で実施 することができる。
反応 ( n〉 において式 ( N〉 の 7— [ Y' 2 - ( A2 ) ra — A' 3 ] —ァミノ一 4 H— 3, 1 —べンゾォ キサジン一 4—オン誘導体の A' 3 におけるアミノ酸残 基の側鎖保護基を除去し、 本発明の式 [ I B2] の 4 H—
3, 1 —ベンゾォキサジン一 4 _オン化合物を得る . この 脱保護反応は、 反応 ( f ) と同様の方法で実施すること ができる。
また、 反応 ( o〉 において式 [ Ι ΒΪ] の 4 H— 3, 1 一 ベンゾォキサジン一 4一オン化合物をスルホ二ル化し、
本発明の式 [ I B 2 ] の 4 H— 3 , 1 —べンゾォキサジン一 4 一オン化合物を得る。 このスルホニル化反応は例えば P —クロ口ベンゼンスルホニルクロリ ド、 p—トルエン スルホ二ルクロリ ド等のスルホニルクロリ ドを有機溶媒 中で反応させることにより行なうことができる。 好まし い有機溶媒としては塩化メチレン、 テトラヒドロフラン ベンゼンなどがあげられる。 また、 トリェチルァミン、 N—メチルモルホリン等の塩基を加えることが好ましい 反応は室温で 1〜48時間、 好ましくは 3〜20時間反応さ せることにより行なうことができる。
本発明の式 [ I B 2 ] の化合物の出発化合物として用い られる式 〈し) で表わされる化合物は、 本発明者らが提 示した方法により合成することができる。 88/09790 口 )
このようにして、 本発明の化合物及び得られた化合物 を、 例えば無機もしくは有機塩基または無機酸もしくは 有機酸との塩生成反応に付すことにより、 その塩類を得 ることができる。
本発明の式 [ I ] で示される化合物の好適な具体例と しては次の表に示される置換基を有するものが挙げられ る。
なお、 化合物構造式中に不斉炭素を有するときは、 そ のすベての光学異性体を含み、 またこれら化合物の塩類 を含む。
X-j
Xが式 [A] (Yi -Ai -)で ¾bされる
LZ
£8l00/l6df/JOd
、?
X— 、N 2
Xが式 [B] (Y2一(Α2 ) » — Α3—)で^される^
Uii ^において、略号の ϋ¾ίάΰΙ下のとおりである,
本発明の式 [ I ] の 4 H— 3, 1 ベンゾォキサジン一 4一オン化合物は、 セリンプロテア一ゼに対し阻害作用 を示すものであるが、 特にエラスターゼ、 更に特に tト 血球エラスターゼに対し他のセリンァロテア一ゼ類 〈例えばキモトリプシンなど〉 に比べて、 より強い阻害 作用を示すものである。
本発明の化合物の酵素反応阻害作用に鬨するィンビト uにおける試験は次のように行われる。
ヒト膿性痰エラスタ一ゼは、 ヒト白血球エラスタ一ゼ と同一物質と考えられており ( ト ウマシ他、 [ J. Biol.
Cliein. ] 、 252 卷、 1917-1924 頁, 1977年) 、 市販品と して容易に入手することができる。
また、 ヒト白血球エラスターゼに対し高度に選択的な 合成基質の一"" と して、 メ トキシスクシ二ルー Lーァラ ニル一 L—ァラニル一 L—プロリルー L一バリル一パラ ニトロァニリ ド ( AAPVpNA)が知られており (ナカジマ他
[ J. Biol. Chem. ] 254 卷, 4027-4032 頁, 1979年〉 、 市販品と して容易に入手することができる。 ヒト膿性痰 エラスターゼによる AAPVpNA の加水分解の程度は、 それ により放出されるパラ二トロア二リンを、 分光光度計で 検出定量することにより容易に測定することができる。 そこで、 ヒト膿性痰エラスターゼによる AAPVpNA の加水 分解の程度を、 供試化合物の非存在下あるいは様々な濃 度での存在下で追跡し、 この酵素反応を 50%阻害する供
試化合物の濃度 ( I c 5 0〉 を求めることができる。
上記と同様のインビトロ試験を、 キモ卜リアシンに対 しても適用することができる。
キモトリアシンとしては、 市販品と して容易に入手で きる牛腌臓の α —キモトリブシンを用い、 キモトリアシ ンの高度に選択的な合成基質の一つであるスクシ二ルー
L—ァラニル一 Lーァラニル一 Lーフ。口リル一 L—フエ 二ルァラ二ルーパラニトロァニリ ド ( AAPFp NA)の加水分 解を、 前記と同様に供試化合物の非存在下あるいは様々 な濃度での存在下で追跡し、 この酵素反応を 50 %阻害す る供試化合物の濃度 ( I C 5 0 ) を求めることができる。 木発明の式 [ I ] の 4 H— 3, 1 一べンゾォキサジン一 4 一オン化合物の、 経気道投与時の肺内滞留性は、 次の 方法で測定できる。 一定量の本発明化合物溶液を、 動物 に経気道的に投与し、 各種時間後に脱血屠殺する 肺内 に滞留する本発明化合物量は、 気管支肺胞洗浄液あるい は肺組織ホモジネートから有機溶媒抽出した試料を高速 液体クロマトグラフィーにより測定することができる。
本発明化合物の i n v i v oにおける薬効は、 次の方法で 評価できる。 一定量の本発明化合物溶液または分散液を 経気道投与し、 一定時間後にヒト膿性痰エラスターゼを 経気道的に投与する。 エラスターゼ投与の一定時間後に 気管支肺胞洗浄を行い、 気管支肺胞洗浄液中のへモグロ ビン量を測定する。 本発明化合物の代わりに、 化合物を
舍まない溶解液を投与した場合の肺出血に対する、 本発 明化合物の肺出血の抑制効果を測定する ( Bonney他、 [J. Cellular Biochemistry], 39: 47~53 (1989) ) c 本発明の 4 H-3, 1 一べンゾォキサジン— 4—オン化 合物、 または、 その塩類は、 セリンプロテアーゼ阻害用 医薬組成物の有効成分と して使用される。
前記一般式 [ I ] の 4 H— 3, 1 —べンゾォキサジン一 4一才ン化合物をセリンァロテアーゼ阻害用医薬と して 用いる場合、 これらの化合物は遊離型と して、 または薬 学的に許容される酸あるいは塩基の付加塩と して使用さ れる。 塩として使用する際 付加される酸あるいは塩基 は無機化合物、 有機化合物のいずれでもよく、 塩にした 時にも十分な効果を発揮し、 生体に無毒性あるいは低毒 件のものであればなんら限定されるものではない。
前記一般式 [ I ] の化合物またはその塩は、 医薬組成 物の有効成分として薬学的に許容される担体、 賦形剤、 溶剤、 稀釈剤、 着色剤、 保存剤、 中和剤、 安定化剤と混 合して所望の剤型として経口的、 非経口的または経気道 的に投与することができる。
経口投与剤は、 錠剤: 顆粒剤、 散剤、 カプセル剤など の固型製剤あるいはシ ップ剤、 エリキシル剤、 乳化剤 懸濁剤などの液状製剤とすることができる。 また非経口 投与剤は注射剤、 座剤、. 皮廣外用剤などとすることがで きる。 これらの製剤は、 前記一般式 [ I ] の化合物また
はその塩に薬学的に許容される製造補助剤を加え常法に よってつくられる。 さらに公知の方法により持続性製剤 にすることもできる。
'経口投与のための固型製剤は、 前記一般式 [ I ] の化 合物またはその塩と乳糖、 デンアン、 結晶セルロース、 メチルセルロース、 グリセリン、 アルギン酸ナトリウム アラビアゴム、 リン酸水素カルシウム、 メタケイ酸アル ミン酸マグネシウム、 乳酸カルシウム、 炭酸カルシウム, 塩化ナトリウム、 カオリンなどの賦形剤とを混合して散 剤にするか、 必要に応じてヒドロキシプロピルセル口一 ス、 ポリビニルピロリ ドン、 白糖、 アルギン酸ナトリウ ム、 炭酸水素ナトリウムなどの崩壌剤を加えて造粒し顆 粒剤とする。 錠剤は、 これらの散剤、 顆粒剤をそのまま、 あるいは滑沢剤と してタルク、 ステアリン酸マグネシゥ ムなどを加えて打錠してつくる。 さらに上述の顆粒また は錠剤をメタアクリル酸メチルコポリマー、 ヒドロキシ ァロピルメチルセル口一スフタレ一トなどの基剤で被覆 して腸溶製剤、 あるいはェチルセルロース、 硬化油など で被覆して持続性製剤にすることも可能である。 カァセ ル剤は、 散剤または顆粒剤を硬カプセルに充填するか、 前記一般式 [ I ] の化合物またはその塩をグリセリン、 ポリエチレングリコ一ル、 ォリーブ油などに懸濁あるい は溶解した後ゼラチン膜で覆い、 軟カァセル剤とするこ ともできる。
経口投与のための液状製剤は、 前記一般式 [ I ] の化 合物またはその塩と白糖、 グリセリン、 ソルビトールな どの甘味剤とを水に溶かしてシロップ剤にする力、 さら にエタノール、 精油などを加えてエリキシル剤とするか またはポリソルべ一卜 80、 カルボキシメチルセルロース ナトリウム、 アラビアゴムなどを添加し、 乳化剤あるい は懸濁剤とする。
注射剤は、 前記一般式 [ I ] の化合物またはその塩に リン酸一水素ナトリウム、 リン酸二水素ナトリウム、 水 酸化ナトリウム、 塩酸、 乳酸、 乳酸ナトリウムなどの PH 調整剤、 ブドウ糖、 塩化ナトリウムなどの等張化剤、 ァ スコルビン酸などの酸化防止剤とともに注射用蒸留水に 溶解し、 無菌沪過してアンプルまたはポリエチレン製、 ガラス製の容器などに充填し、 皮下、 筋肉内、 静脈内、 動脈内への単回用または長 · 短時間の持続用注入剤とす る。 また用時調製型の注射剤はさらにデキストリン、 シ クロデキストリン、 マンニトール、 ゼラチンなどを加え た後真空凍結乾燥してつくることができる。 また公知の 方法に従って前記一般式 [ I ] の化合物またはその塩を リボソームやマイクロスフェア一に封入した注射剤にす ることもできる。
座剤は、 前記一般式 [ I ] の化合物またはその塩をポ リエチレングリコール、 ラノリン、 脂肪酸のモノ、 ジぁ るいはトリグリセリ ド、 カカオ脂と共に加温, 溶融し、
冷却加塑するか、 大豆油、 ボリエチレ ン グリコールなど に懸濁あるいは溶解した後ゼラチンなどで膜被覆してつ くることができる
皮廣外用剤は、 前記一般式 [ I ] の化合物またはその 塩をポリエチレングリコ一ル、 白色ワセリン、 流動パラ フィンなどに加え、 軟膏、 クリーム、 ゲルなどのいずれ の形状のものであつてもよい。
経気道投与剤は、 前記一般式 [ I ] の化合物またはそ の塩を通常吸入法で微粒子と して投与する . 薬剤を有効 成分と して含有する微粒子はエアロゾル、 粉体などの形 体で粒径が 0. 5 〜5 0 mのものが好ましい, エアロゾル を発生させる装置と しては、 例えば超音波式ゃジ Xッ ト 式のネブライザ一、 低級アルカンあるいはフッ素化アル 力ンなどを噴射剤に使ったスアレーなどが使える。 また 粉体は、 自発的あるいは強制的呼吸に連動させた簡易型 の吸入器などを使って投与される。
かかる医薬製剤中の一般式 [ェ ] で表わされる化合物 またはその塩の瀵度には特に限定はないが、 一般には製 剤中に 0. 01〜50重量 、 好ましくは 0. 1 〜10重量_%程度 が適当である:
またその用量にも限定はないが、 0 . 1 〜1 0 0 0 m g 日 患者、 好ましくは 1〜2 0 0 m g/ 日 Z患者程度が適当であ り、 投与回数は通常 1 日当り 1〜4回である。
以下本発明を参考例及び実施例により更に詳細に説明
する。 参考例
2—ィ ソプロピルァ ノー 5—メチルー 7—アミノー 4
H-3, 1 —ベンゾ才キサジン一 4一オンの合成
(i) _ ニトロ一 6—メチルアントラニル酸 —メチルの 合成
40· Ogの 4—メチルー 6—二トロアントラニルを 700 mlのメタノールに溶解後、 4.00 gの炭酸力リウムを加え 3時間還流した。 メダノールを減圧下濃縮した残渣に水 酢酸ェチルを加え抽出した。 有機層を飽和食塩水で洗浄 後乾燥した。 沪過瀵縮して得られた粗生成物をメタノー ルより結晶化し 3 0 gの題記化合物を得た。
本化合物の N M Rを m下に示す。
H - NMR ( C D C 1 3 , ppm )
2.94(3H, s), 3.94(3H, s), 5.1_5.4 (2H, m) ,
7.34{2H, s).
(i i) 2 - 〈 3—イ ソプロピルウレイ ド)_一 4一二トロー
6—メチル安息香酸 メチルの合成
30.0 gの 4一二トロー 6 —メチルアントラニル酸 メ チルを 300 mlのテトラヒ ドロフランに溶解した, miの 酢酸及び 38gのィソプロピルイソシァネートを加え 19時 間攪拌した。 析出した結晶を沪取、 乾燥し 31.6 gの題記 化合物を得た。
mp. 208 - 209 。C
本化合物の N M Rを以下に示す。
- N M R ( C D C 1 3 , pm 〉
i.23 (6H, d, J = 6.4Hz) , 2.53 (3H, s) ,
3.98(3H, s), 3.75-4.15 (lH, m),
4.3-4.55 (lH, ffi) , 7.66 {1H, d, J = 2.0Hz),
δ*. ζζ-8.7 (lH, m) , 9.03 (1H, d, J = 2.0Hz).
(Hi) 2— ( 3—ィソァロピルゥレイ ド (一 4 一アミ ノ - 6—メチル安息香酸 メチルの合成
7. G3 gの 2— ( 3—イソァロピルウレイ ド〉 一 4 —二 トロ— 6—メチル安息香酸 メチルを lOOOffilの酢酸ェチ ルに溶解した。 1.00 gの 10%パラジウム一炭素を加え水 素雰囲気下、 室温で 4時間撹拌した。 沪過濃縮後得られ た粗生成物をへキサン一酢酸ェチルより結晶化し 5. 58g の題記化合物を得た。
ntp. 173- 175 °C
本化合物の N M Rを以下に示す。
i— N M R ( C D C 1 3 , pm 〉
1.18{6H, d, J = 6. Hz}, 2.38(3H, s),
3.83(3H, s), 3.7-4.2(3H,m),
4.4-4.65(lH,m), 6.10(1H, d, J = 2.4Hz),
7.65(1H, d, J = 2.4Hz), 9.8-10.1 (1H, a) .
Uv) 2—ィソプロピルアミノー 5—メチルー 7ーァミノ - 4 H - 3, 1 —ベンゾォキサジン一 4—オンの合成
9.57gの 2— ( 3—イ ソァロピルウレイ ド) —4—ァ ミノ— 6—メチル安息香酸 メチルを 55mlの瀵硫酸に溶 解し室温で 2時間攪拌した。 反応液を 130 gの炭酸水素 ナトリウム、 水及び酢酸ェチルの混合物中に氷冷下撹拌 しながら徐々に滴下し.た。 滴下後さらに炭酸水素ナトリ ゥムを加え中和し酢酸ェチルで抽出した。 有機層を飽和 食塩水で洗浄後乾燥した。 沪過濃縮して得られた粗生成 物をへキサン一酢酸ェチルより結晶化し 6.84gの題記化 合物を得た。
rap. 203 - 205 。C
本化合物の N MRを以下に示す。
一 NMR (ピリジン一 d 5 , -ό- pm )
1.24(6H, d, J = 6.4Hz), 2.75{3H, s},
4.0-4.4(lH,m), 6.2-6.6(3H, br s),
6.65-6.8(1H, br s), 8.0-8.3(lH,m).
実施例 1
7— ( N—ベンジフレオキシカルボ二ルー L—フヱニルァ ラニル) アミノー 5—メチルー 2— ( 1一カルボキシェ
チル) アミノー 4 H— 3, 1 —べンゾ才キサジン一 4ーォ ンの合成 (化合物 No.101)
( i ) 2一べンジル才キシカルボニニル— 3—メチル— 5— ニトロフエ二ルイソシァネート 〈前記合成経路中式 ( B ) の化合物〉 の合成
4一二トロー 6—メチルアントラニル酸べンジル 3.55 gを 120 mlの四塩化炭素に溶解した。 触媒量のトリェチ ルァミンおよび 1.23 gのビストリクロロメチルカーボネ ートを加え 2時間還流した。 更に 1.23 gのビストリクロ ロメチルカ一ボネートを加え 1時間還流した。 室温まで 冷却し、 生成晶を浐別後、 母液を濃縮し、 3.51 gの題記 [J的物を得た。
1 H - N M R ( C D Cl3 , δ· pm )
2.38(3H, s), 5.43(2H, s}( 7.41 (5H, s),
I R ( neat, cm一1〉
2280, 1730, 1535, 1350, 1270
(ii) 1 — N— ( 2—ベンジルォキシカルボニル— 3一メ チル— 5—二トロフエニル〉 力ルバモイルー L一ァラニ ンー t一ブチルエステル 〈前記合成経路中式 ( C ) の化 合物〉 の合成
Lーァラニン— t _ブチルエステル塩酸塩 606 を 10
mlの乾燥 TH F中に懸濁させた。 窒素雰囲気下水冷しな がら 371 のトリエチルァミンを加え◦ °Cで 15分間、 更 に室温で 15分間攪拌した。 この中に前記(i) で得られた 2—ベンジルォキシカルボ二 レー 3—メチル一 5—二ト 口フエ二ルイソシァネート 521 ragを 10mlの T H Fに溶解 した溶液を加え 14時簡室温で撹拌した。 水を加え酢酸ェ チルより抽出した。 飽和食塩水で洗浄後、 乾燥
、 濃縮し得'られた粗生成物をシリカゲルカラ ムで精製し 476 の題記目的物を得た。 N MRは以下の とおり。
XH -NMR ( C D Cl3 , ppffi )
1.40(3H, d, J = 7.3Hz), 1.52{9H, s),
2.34(3H, s), 4.25 -4.65 (lH,m),
5.38(2H, s), 6. lOilH, d, J = 7.7Hz},
7.4 (5H, s), 7.4-7. 5 (1H, DI) , 8. 77 (1H, brs}
8.91 (1H, d, J = 2.4Hz)
(iii) N - 〈 5—メチル一 7 -アミノー 4 H— 3, 1 一べ ンゾォキサジン一 4一オン一 2—ィル) — Lーァラニン 一 t一ブチルエステル (前記合成経路中式 ( E ) の化合 物) の合成
1 - N - ( 2—ベンジル才キシカルボ二ルー 3—メチ ルー 5—ニトロフエニル〉 力ルバモイル一 Lーァラニン - t—プチルエステル 3.66 eを 300 mlのエタノールに溶
解後 810 の 10%Pd-Cを加え水素雰囲気下、 室温で 6時 間激しく攪拌した。 セライ トカラムで沪過後濃縮するこ とにより 2.80gの 1 — N— ( 2—カルボキシー 3—メチ ル一 5—アミノフエニル〉 力ルバモイノレ一 Lーァラニン - t 一ブチルエステルを得た。 このものを 200 mlの酢酸 ェチルに溶解した。 この溶液に、 2.57 の^ヽ N—ジシ クロへキシルカルボジィ ミ ドを加え室温で 15時間撹拌し た。 生成晶を沪別し、 母液を濃縮後得られた粗生成物を シリカゲルカラムで精製し 2.20 gの題記目的物を得た。 N MRを示す。
^-NMR ( C D C , ' δ-ρριη )
1.46(3H, d, J = 7.3Hz), 1.48{9H, s),
2.58(3H, s), 4.0-4.5{4H,m), 6.25(2H, s)
(iv) 7 - ( N—ベンジルォキシカルボ二ルー L一フエ二 ルァラニル) アミノー 5—メチルー 2— ( 1 一 t —ブト キシカルボニルェチルァミノ〉 一 4 H— 3, 1 —ベンゾォ キサジン一 4一オン (前記合成経路中式 ( F ) の化合物) の合成
94mffの N—べンジルォキシカルボニル一 L一フエニル ァラニンおよび 32 の N—メチルモルホリンを窒素雰囲 気下、 5 mlの無水 TH Fに溶解した。 一 18でに冷却後ィ ソブチルクロルホルメート in?を加え 2分間撹拌した。 N - ( 5ーメチルー 7—アミノー 4 H— 3, 1 —ベンゾォ
キサジン— 4一オン— 2—ィル〉 一 Lーァラニン一 t— ブチルエステル 100 および N—メチルモルホリン 32 ( 1 eq) の TH F溶液 ( 6 ml〉 を滴下した。 一 18°Cから 室温で一晩攪拌後水を加え酢酸ェチルより抽出した。 1 N—塩酸、 飽和炭酸水素ナトリウム水溶液、 飽和食塩水 で洗浄後乾燥した。 得られた粗生成物をシリ力ゲルカラ ムで精製し 101 の題記目的物を得た。 NMRを次に示 した。
w . 108 〜11Q 。C 〈へキサン一酢酸ェチル〉
1 H - N M R ( C D C , d ppm )
i.45(3H, d, J = 7.2Hz},
1.56(9H, s), 2.52{-3H, s}, 2.95-3.2 (2H, m) ,
4.2-5.15(3H,m), 5.11 (2H, s),
5.80 (1H, br, d, J = 8.4Hz),
7.10-7.35(2H,ir
1)
I 7.17(5H, s), 7.30{5H, s),
(v) 7— ( N—ベンジル才キシカルボ二ルー L一フエ二 ルァラニル〉 アミノー 5—メチル一 2— ( 1一カルボキ シェチルァミノ) — 4 H— 3, 1 —べンゾォキサジン一 4 一オン (前記合成経路中:式 〈 I 〉 の化合物) の合成
7— ( N—べンジルォキシカルボ二ルー L—フ ニル ァラニル) アミノー 5 メチルー 2— ( 1— tーブトキ シカルボニルェチルアミ.ノ) 一 4 H— 3, 1 —ベンゾォキ
サジン— 4一オン 188 ra?を の 4 M HCi/'ジ才キサ ン溶液に溶解し、 室温で 2時間攪拌した。 溶媒を濃縮後 得られた粗生成物をシリカゲルカラムで精製し、 153 の題記目的物を得た。 N MRは以下のとおり。
XH - N M R ( d & - acetone , ppm )
1.58{3H, d, J = 7.3Hz), 2.59(3H, s),
2.9-3.4(2H, m), 4.4-4. 3H, m) '
4.9-5.4(lH, m), 5.03(2H, s),
7.1-7.5(12H)m}) 9.5-9.9(iH,m) 実施例 2
7— ( N—ベンジルォキシカルボ二ルー L—フエニルァ ラニル) アミノー 5—メチル一 2— 〈 4一カルボキシピ ペリジノ) —4 H— 3, 1 —べンゾ才キサジン一 4—オン の合成 (化合物 No.105)
( i } 1 一 N— ( 2—ベンジルォキシカルボニ レ一 3—メ チル— 5—ニトロフエニル〉 力ルバモイルー 4一ピぺリ ジンカルボン酸一 t一ブチルエステル (式 ( C〉 の化合 物〉 の合成
実施例 1 , (ii)において、 Lーァラニン一 t一ブチル エステル塩酸塩に代わりに 4ーピぺリジンカルボン酸一 t一ブチルエステルを用いる以外は実施例 1 , と同 様にして、 題記目的物を得た。 本化合物の N MRは以下
4フ のとおりであった。
1.46(9H, s), 1.5-2.05(4H,m},
2.2-2.5(lH,m}, 2.48(3H, s)
2.8-3.2(2H,m)) 3· 75-4.1 (2H, m) ,
5.40(2H, s), 7.40(5H, s), ?.63(1H, brs),
8.97(1H, brs), 9.29(1H, brs)
(ii)N——( 5—メチルー 7—アミノー 4 H— 3 , 丄 ペン ゾォキサジン一 4—オン一 2—ィル〉 一 4ーピペリジニン カルボン酸— t一ブチルエステル (式 ( E〉 の化合物) の合成
実施例 1 , (i i U において、 1— N— ( 2—ベンジル ォキシカルボニル— 3—メチルー 5—二トロフエニル〉 カルバモィルー L—ァラニン一 t一ブチルエステルの代 わりに上記 (ί)で得られた目的物を使用する以外は同様の 方法により、 題記目的物を合成した。 本目的物の N MR を以下に示した。
1 H - N M R ( C D C 13 , ^ ιη )
1.45(9H, s), 1.5-2.05(4H,m},
2.2-2.5(lH,m), 2.60(3H, s),
2.9-3.25{2H,m), 4.05-4.45 (4H, m) ,
6.23(2H, s)
\i i i) 7 - ( N—べンジル才キシカルボ二ルー L一フエ
二ルァラ二ル) アミノー 5—メチル一 2— ( 4— tーブ キシカルボ二ルビペリジノ) 一 4 H— 3, 1 —ベンゾォキ サジン一 4一オン (式 ( F ) の化合物〉 の合成
実施例 1、 (iv)において、 N— ( 5—メチルー 7—ァ ミノー 4 H— 3, 1 —ベンゾォキサジン— 4一オン一 2— ィル) 一 Lーァラニン一 t—ブチルエステルの代わりに (ii)で得られた 2—置換一 5—メチルー 7—アミノー 4 H— 3, 1 —べンゾォキサジン一 4—オン誘導体を使用し それ以外は同様の方法によって上記目的物を合成した。 N MRを以下に示す。
1 H - N M R ( C DC , ppm )
1. 5{9H, s), 1.5-2.0(4H,m})
2.2-2.6 (1H, in), 2.58(3H, s),
2.9-3.25(4H,m), 4.05-4. ό (3H, m) ,
5.09 (2H, s), 5.4-5. 6 (1H, ID} ,
6.76(1H, brs}, 7.23 (5H, s), 7.30(5H, s),
7.2-7.35(lH,ffi), 8.14(lH,brs).
(iv) 7 - ( N—ベンジルォキシカルボ二ルー L—フエ二 ルァラニル) ァミノ一 5—メチルー 2— ( 4一カルボキ シピペリジノ〉 一 4 H— 3, 1 —ベンゾォキサジン一 4— オン (式 ( I 〉 の化合物) の合成
実施例 1, (V) において、 7— ( N—べンジルォキシ
カルボ二ルー L—フヱ二ルァラニル) アミノー 5—メチ - 2— ( 1 — t 一ブトキシカルボニルェチルァミノ〉 - 4 H - 3, 1 一べンゾォキサジン一 4 一オンの代わりに (i i i) で合成した 7 — ( N—べンジルォキシカルボニル 一 L—フエ二ルァラニル〉 アミノー 5—メチルー 2—置 換— 4 H— 3, 1 一べンゾォキサジン一 4 一オン誘導体を 使用する以外は同様の方法により、 題記目的物を合成し た。 N M Rは以下のとおりであった。
1 H— N M R ( d 6 - acetone , ό piri )
1. 5-2.3 (4H,m}, 2, 58 (3H, s),
2.4-2.9 (lH, ffi) , 2.9-3.4 (4H, nil ,
4.1-4.7 (3H, m} , 5.04 (2H, s},
6. 5-6.8(1H, mi , 7.00 (1H, brs),
7, 26 (5H, s} , 7.30 (5H, s), 7.54 (1H, brs)、
9, 39 (1H, brs) 実施例 3
7 — ( N—ベンジルォキシカルボ二ルー L一フエニルァ ラニル) アミノー 5—メチルー 2— ( N—メチルー 3 — カルボキシプロヒ。ルァミノ) 一 4 H— 3, 1 —ベンゾォキ サジン— 4—オンの合咸 (化合物 . 110 )
( i } 1 — N—メチスレ一 1 — N— ( 2—ベンジルォキシカ ルボニル一 3—メチル一 5—二トロフヱニル) カルバモ
ィルー 4—アミノ酪酸一 t 一ブチルエステル (式 ( C.〉 の化合物) の合成
実施例 1 , (ii)において、 Lーァラニン一 t —ブチル エステル塩酸塩の代わりに 4ーメチルァ ^_ミノ酪酸一 t 一 ブチルエステルを用いる以外は実施例]^と同様にして、 題記目的物を得た。 本化合物の N M Rを以下に示した。
XH - N M R { C D Cls , ^ pm )
].45 (9H, s), 1. -1.95 (2H, m),
2.1-2.4 (2H, m) , 2.48(3H, s), 2.96 {3H, s) ,
3.25-3.45 (2H, m}, 5, 40 (2H, s),
7.40 (5H, s), 7.62 (1H, brs),
9.02 (1H, brs), 9.20 (1H, brs)
( i i } 2 - ( N—メチル— 3— t 一ブトキシカルボニルプ πピルァミノ〉 一 5—メチルー 7—アミノー 4 H— 3, 1 一べンゾォキサジン— 4 一オン (式 ( E ) の化合物) の 合成
実施例 1 , (ii i) において、 1 一 N— 〈 2—べンジル ォキシカルボ二ルー 3—メチル一 5—二トロフエニル〉 力ルバモィル— L—ァラニン— t —ブチルエステルの代 hりに上記( i ) で得られた目的物を使用する以外は同様 の方法により、 題記目的物を合成した。 本目的物の Rを以下に示した。
1 H - N M R ( C D C , d ppm )
1.44(9H, s), 1.65-2.05{2H,m}I
2.05-2.35 ( 2H,m), 2.60(3H, s},
3.09(3H, s), 3.4-3.6(2H, in},
4.15 (2H, brs), 6.24 (2H, brs)
(i i i) 7— < N—べンジ /レオキジカノレボニルーし一フエ 二ルァラ二ル〉 ァミノ— 5—メチル— 2— ( N—メチル • 3— t—ブトキシカルボニルプロピル) アミ ノー 4 H - 3, 1 一べンゾォキサジン一 4一オン (式 ( F〉 の化合 物) の合成
実施例 1 , (iv)において、 N— ( 5—メチルー 7—ァ ミノ一 4 H— 3, 1 —べンゾォキサジン一 4—オン一 2— ィル〉 一 Lーァラニン一 t一ブチルエステルの代わりに (ii)で得られた 2—置換— 5—メチルー 7—アミノー 4 H 一 3, 1 一べンゾォキサジン一 4一オン誘導体を使用し、 それ以外は同様の方法によって上記目的物を合成した . N MRを以下に示す。
XH - N M R ( C D C , δ· ppm )
ί.44(9H, s}, 1.65-2.05(2H1m),
2.1-2.4(2H,m), 2.59(3H, s},
3.09(3H, s), 3.0-3.2(2H,ffi),
3.35 - 3.65
4.4-4.7(lH,m),
h.09(2H, s), 5.35 - 5.55 (lH(in),
6.76(1H, brs), 7.20 - 7.35 (lH,m},
(ϊ v) 7— ( N—べンジルォキシカルボニル一 L一フエ二 ルァラニル) アミノー 5—メチル一 2— ( N—メチ —
3—カルボキシプロピルアミノ) — 4 H— 3.1 —べン」ノ" ォキサジン一 4一オン (式 ( I 〉 の化合物〉 の合成
実施例 1 , (V) において、 S 7— ( N—べンジルォキシ カルボニル一 Lーフヱニヌレアラニル > アミノー 5—メチ ルー 2— ( 1一 t—ブトキシカルボニルェチルァミノ、 - 4 H— 3, 1 —ベンゾォキサジン一 4一オンの代わりに ⑩で合成した 7— ( N一ベンジルォキジカルボ二ルー L —フエ二ルァラニル) ァミノ— 5—メチルー 2—置換一
4 H - 3, 1 一べンゾォキサジン一 4一オン誘導体を使用 する以外は同様の方法により、 題記目的物を合成した
N MRを次に示したぐ
1 H— N M R ( d 6 - acetone , d ra )
1.7-2. l(2H,ffi), 2.25-2.5(2H,ffi),
2, 59(3H, s}, 2.95-3.3 (2H, a), 3.14(3H, s),
3.5- 3· δ(2Η, m), 4.4-4.7 (1H, m) ,
5.04 (2H, s), 6.5-6.7(lH,in),
7.04(1H, brs), 7.26 (5H, s), 7.30 {5H, s},
7.52(1H, brs}, 9, 38(1H, brs)
実施例 4
7 - ( ベンジルォキシカルボ二 レー: L—フエ二ルァ ラニル〉 ァミノー 5—メチルー 2 - - 〈 3—カルボキシフ" 口ピルァ ノ) 一 4 H— 3, 1 一ベン 'ゾ才キサジン一 4一
^ンの合成 (化合物 No.109)
(i) 1— N— ( 2—ベン.ジルォキシカルボ二ルー 3—メ チル一 5—二トロフエ二ル、 力ルバモイルー 4一アミ ノ 酪酸一 t—プチルエステル (式 ( C ) の化合物) の合成 実施例 1 , (ii)において、 L一アラこンー t一ブチル エステル塩酸塩の代わりに 4—アミノ酪酸一 t 一ブチル エステルを用いる以外は実施例" τ¾同様にして、 題記目 的物を得た。 本化合物の N M.Rは以下のとおり:
1 Η - N M R ( C D C 13 , ^ppm )
1.45(9H, s), 1.6-1, 95 (2H, m),
2.1-2.5{2H,m}, 2,.44(3H, s),
3. l-3.4(2H,m), 4.8-5.05(lH,m),
5.38(2H, s), 7.41 (5H, s), 7.61 {1H, brs) ,
8.50(1H, brs), 8.'92 (1H, brs)
(ii) 2— ( 3— t—ブトキシカルボニルァ口ピルァミ ノ: Ϊ — όーメチルー 7—ァミノ一 4 Η - 3, i —ベンゾォキサ ジン一 4—オン 〈式 ( E ) の化合物〉 の合成
施例 1 , (Πί) において、 ユ ー N— ( 2—べンジル
ォキシカルボ二ルー 3—メチルー 5—二トロフエニル、 カルバモィルー; L—ァラニン— t一ブチルエステルの代 わりに上記( i ) で得ちれた目的物を使用する以外は同様 の方法により、 題記目的物を合成した : 本目的物の NM Rを以下に示した。
ί.45 (9H, s) , 1.7-2.0 (2Η, πι) ,
2. 1-2.4 (2H, iri) , 2.6! (3H, s),
3.25-3.55 (2H, m;', 3. 9-4.2 (2H, m),
4.8-5. ί (IB, nij, 6.26 (2K, b sl
(iii) 7— ( N—ベンジルォキシカルボ二ルー L—フエ 二ルァラニル) アミノー 5—メチル— 2— 〈 3— t一ブ トキシカルボ二ルァロピル) アミノー 4 H— 3, 1 -ベン- ゾォキサジン一 4一才ン (式 ( F ) の化合物) ク:) '
実施例 1 , ( )において、 N— 〈 5—メチルー 7—ァ ミノ一 4 H— 3, 1 —べンゾォキサジン一 4一オン一 2— イスレ〉 一 Lーァラニン一 i: 一ブチルエステルの代わりに { i i }で得られた 2―置換— 5—メチルー 7—アミ ノー 4 H— 3, 1 一べンゾォキサジン一 4一オン誘導体を使 ¾し それ以外は同様の方法によつて上記目的物を合成した: MRを以下に示寸:
1, 44 (9H, si, 1.65-2.0(2H,mi,
2.15-2.45(2H,m}, 2.62(3H' s), 3.0-3.2 (2H, m) ,
3.3-3.55(2H,m)) 4.05-4.25 (lH,m),
4.5-4.75(lH,m)> 5. lKlH.m}, 5.4-5.7(1^ 111) , 6.86(lH, brs), 7.10-7.30 (1H, m) ,
7.18(5H, s), 7.31 (5H, s},
8.2-8.6(lH,m}
\iv) 7— ( N—べンジル才キシカルボ二ルー Lーフェ二 ルァラニル〉 アミノー 5—メチル一 2— ( 3—カルボキ シプロピルァミノ) 一 4 H— 3, 1 —ベンゾォキサジン一 4一オン 〈式 〈 I ) の化合物) の合成
実施例 1 , (V) において、 7— ( N—べンジルォキシ カルボニル— L一フエ二ルァラニル〉 アミノー 5—メチ ゾレー 2— ( 1— t一ブトキシカルボニルェチルアミノ) 4 H - 3, 1 一べンゾォキサジン一 4一オンの代わりに で合成した 7— ( N—べンジル才キシカルボ二ルー L 一フエ二ルァラニル〉 アミノー 5—メチルー 2—置換一 4 H -3, 1 —ベンゾォキサジン— 4一オン誘導休を使用 する以外は同様の方法により、 題記目的物を合成した .. NMRを以下に示す。
1 H - N M R ( d 6 - acetone , d ppm )
1.7-2.1 (2H, ml, 2.2-2.6(2H,ffi),
2.9-3.2 (2H, m), 3.25-3.ョ (2H, mj,
3.6-3.8(^, 111) , 4.5- 4. S5(lH,m),
5.07(2H, s)., 5.7-6. ΟίΙΗ, ιη),
6.90(lH, brsi, 7.10-7. BOilH.m),
7.19(5H, s),7.27(5H, s
8.8-9.1 (1H, in!
実施例 5
7— 〈 N—べンジル才キシカルボニル—し—フエニルァ ラニル) ァミ ノー 5—メチ/レー 2— ( 2—カルボキシェ チル Ύミん) - 4 H - 3^1 一ベンゾォキサジン 一 4 -ォ ンの合成 (化合物 No.107)
( i ) 1 一 N— ( 2—ベンジルォキシカルボ 23
sニル: 3 -メ— w 2
チルー 5—二トロフエニル) iiルバモィル一 /3 K i— ァラニ s ンー tニズチルェステ (式 ( C ') ク、化合物 ) 0 )合成 実施例ュ , ( i i )において、 L一 ァラニン — t -- ブ ルニステ /レ塩酸塩の代わりに; 3—ァラニン一 tーブ手 エステル塩酸塩を用いる以外は実施例 1 (ii)と同様に して、 題記目的物を得た。
本化合物の N M Rを以下に示す ..
1 H - M R ( C D C 1 3 , d ppiri )
i.47 i ^H, s;, 2.44 (3H, si, 2.4-2
3.3-3.6{2H, in), 4.2-4.35(ϊΗ, ηΐί,
/.40 (5H, si , 7.62(1H, d, J = 0.7Hz)
8.53 (1H, s:',
D.7Hz)
(ii) 2— ( 2 - t -ブトキシカルボニルェチル) ァミノ — 5—メチルー 7—アミノー 4 H— 3 , 1 —ベンソォキサ ジン— 4一オン (式 ( E ) の化合物〉 の合成
実施例 1 , U ) において、 1 — N— ( 2—べンジル ォキシカルボ二ルー 3—メチル一 5—二トロフ ニル) 力ルバモイルー Lーァラニン一 t一ブチルエステルの代 わりに上記( i ) で得られた目的物を使用する以外は同様 の方法により、 題記目的物を得た.,
本化合物の N M Rを以下に示す。
' K - N MR ( C D C 1 3 , ό、 ppm )
1.4u( ^H, s}, 2.51 (5H, a), 3.62{4H, m),
4.75(lH,ffi), 6.28(2H, s).
( i i i ) 7— ( N—べンジルォキシカルボ二ルー L一フユ 二ルァラニル) アミ ノー 5—メチルー 2— ( 2 - 1 ーブ トキシカルボニルェチル) アミノー 4 H— 3, 1 —べンゾ ォキサジン一 4一オン (式 ( F〉 の化合物) の合成
実施例 1 , (iv)において、 N— ( 5 -メチルー 7—ァ ミノ一 4 H— 3, 1 一べンゾォキサジン一 4一オン一 2— ィル〉 一 Lーァラニン一 t一ブチルエステルの代わりに 上記 i)で得られた 2—置換一 5—メチルー 7 --アミ ノ 一 4 H— 3, 1 —べンゾォキサジン一 4一オン誘導体を使 用し、 それ以外は同様の方法により、 題記目的物を得た 本化合物の N M Rを以下に示す .
^-NMR ( C D C 1 3 , ό· ρηι )
1.46(9H, s), 2.45-2.65(2H, m), 2.67(3H, s}, 3.15(2H, d, J = 6.7Hz), 3.63(2H,m),
L 55(2H, t, J = 6.7Hz), 5.11 (2H, s),
6.88(1H, br s), 7.2-7.35(2H,m),
?.27(5H,m), 7, 31 (5H, s), 8.02(1H, br s).
(iv) 7 - ( N—べンジルォキシカルボ二ルー L—フエ二 ルァラニル) アミノー 5—メチルー 2 - ( 2—カルボキ シェチル) アミノー 4 H— 3, 1 —べンゾォキサジン一 4 一オン (式 ( 1 〉 の化合物) の合成
実施例 1, (V) において、 7— ( N—べンジルォキシ カルボ二ルー L一フエ二ルァラニル〉 アミノー 5—メチ ヌレー 2— ( 1 - t一ブトキシカルボニルェチルアミノ)
4 H— 3, 1 —ベンゾォキサジン— 4—オンの代わりに 上記(iii) で合成した 7— ( N—べンジルォキシカルボ 二ルー L—フヱニルァラニル〉 アミノー 5—メチルー 2 一置換— 4 H-3, 1 —ベンゾォキサジン— 4一オン誘導 体を使用する以外は同様の方法により、 題記目的物を得 た。
本化合物の N M Rを以下に示す。
3 H - N M R ( d 6 - acetone , d ppm )
2.4-2. π , 2.65(3H, s),
3.13(2H, d, J = 6.7Hz), 3.69(2H,m),
4.54(2H, t, J = 6.7Hz), 5.12(2H, s},
6.94 (1H, br s), 7.2-7.35(2H,m),
7.27(5H,m 7.32(5H, s), 9.03(1H, br s). 実施例 6
7— J N—ベンジルォキシカルボニル— L—フヱニルァ
、
ラニル) ァ、ノー 5—メチルー 2— 〈 1, 2 —ジカルボキ シェチル〉 ァミノー 4 H— 3, 1 一べンゾォキサジン一 4 一オンの合成 (化合物 No.113)
(i) 1 - N - 〈 2—べンジル才キシカルボ二ルー 3—メ チルー 5—ニトロフヱニル) 力ルバモイルー Lーァスパ ラギン酸— , /3—ジ— t—ブチルエステル (式 ( C ) の化合物) の合成
実施例 1, Ui)において、 Lーァラニン一 ΐ一ブチル エステル塩酸塩の代わりに Lーァスパラギン酸一 a , /3 ージー t一ブチルエステル塩酸塩を用いる以外は実施例 1 , Ui)と同様にして、. 題記目的物を得た。
本化合物の N MRを以下に示す。
— N M R ( C D C 1 3 , ppm 〉
1.46(9H, s), 1.49(9H, sj, 2.44(3H. s),
2.75-2.9(2Η,ιη), 4.5-4.75 (1H, m) , 5.40(2H, s), 5· 7-5. lH,m), 7.41 (5H, s), 7.64(lH,m),
8.72{1H, br s), 8.96 (1H, m).
(ill 2 - ( 1, 2 —ジー t一ブトキシカルボニルェチル) アミノー 5—メチルー 7—アミノー 4 H— 3, 1 —ベンゾ' ォキサジン一 4一オン 〈式 ( E ) の化合物〉 の合成
実施例 1 , (ΠΠ において、 1 — N— ( 2—べン ジル ォキシカルボニル一 3—メチル一 5—二トロフエニル) カルバモィル— Lーァラニン一 t 一ブチルエステ/レの代 わりに上記( i ) で得ちれた目的物を使用する以外は同様 の方法によ り、 題記目的物を得た:
本化合物の N M Rを以下に示す:
1 H - N M R ( C I) C 1 3 , p m )
1.44{9H, s}, 1.47(9H, si, 2.6K3H, s),
2.90(2H. d, J = 4, 4Hz). 4.12(2H, br si,
6-4.8(1H, mi, 5.6-5. SdH.ml, D.25(2H, S ; . に i i ) 7 - ( N—ベンジ レ才キシカルボ二 /レ --- 1 -- ―' 二 二ルァラニル〉 ァミノ一 5—メチルー 2— ( 1, 2 —ジ— t—ブトキシカルボニルェチル〉 アミノー 4 H— 3, 1 - ベンゾォキサジン一 4一才ン (式 ( F ) の化合物) の合 実施例 1 , (iv)において、 N— ( 5—メチルー 7—ァ ミノー 4 H— 3, 1 —ベンゾォキサジン一 4 _オン一 2— ィ /レ) 一: L—ァラニン一 t—ブチルエステルの代わりに 上記(ii)で得られた 2—置換一 5—メチル— 7—ァミノ - 4 H— 3, 1 —ベンゾォキサジン— 4一オン誘導体を使
し、 それ以外は同様の方法により、 題記目的物を得た 本化合物の N MRを以下に示す。
- N M R ( C D C 1 3 , ^p m )
1.44 (9H, s 1. 9(9H, s), 2.59(3H, s
Ί.91 (2Η, d, J = 4.6Hz), 3.13 (2H, d, J = 7.0Hz),
4.4-4.8{2H,m), 1K2H, s),
〕.35 - 5.55 ( 1 H, in) , 5.85-6.05 (1H, m) ,
D.91 Ϊ1Η, br s), 1.20-?.35 (1H, s、に
7.23 (5H, s), 7.31 (ョ H, si, 8.16 (1H, br s) . iivi 7— ( N—べンジルォキシカルボニル L一フ_ェ二 ルァラニル) アミノー 5—メチルー 2— ( L 2 ージカル ポ'キシェチル) アミノー 4 H— 3, 1 _一ベ _ン ゾォ_キサジン 一 4一オン (式 ( I ) の化合物) の合成
実施例 1 , において、 7— —べン ジ / オキシ えルボニルー L一フエニルァラニル) ァミ ノー 5—メチ ルー 2— ( I - t -ブトキシカルボ二 /レエチルァミ ノ ) - 4 H - 3, 1 —ベンゾォキサジン一 4ーォンの代わりに 上記 ii) で合成した 7— ( N—ベンジルォキシカルボ 二ルー L—フエ二ルァラニル〉 アミノー 5—メチルー 2 一置換一 4 H— 3, 1 —べンゾォキサジン一 4一オン誘導 体を使用する以外は同様の方法により、 題記目的物を得 た - 本化合物の N M Rを以下に示す t
:H - NMR ( d 6 一 acetone , d ppm )
2.61 (3H, s), 3.05(2H, d, J=4. 6Hz),
3.21 (2H, d, J = 7.0Hz), 4.45-4 • 85(2H, m}!
} ,
5.85-6.05(lH,in)( 6.9K1H, br s),
7.2-7.35(lH,m), 7.23(5H, s)
ヽ 実施例 7
7— ( Ν—べンジルォキシカルボ二ルー L—フエニルァ ラニル〉 アミノー 5—メチル一 2 - ( 1, 3 ージカルボキ シァロピル〉 アミノー 4 Η— 3, 1 一べンゾォキサジン一
4一オンの合成 (化合物 Νο· 115〉
{i) 1 — N— ( 2一べンジルォキシカルボ二ルー 3 -メ チル— 5 _ニトロフヱニル) 力ルバモイルー L一 ルタ— ミン酸一 " , ァ—ジ一 tーブチルェステ ( ¾ ( C ) の 化合物) の合成
実施例 1 , (ii)において、 Lーァラニン一 t—ブチル エステル塩酸塩の代わりに L—グルタミン酸— α , r - ジ— t一ブチルエステル塩酸塩を用いる以外は実施例 1 (ii)と同様にして、 題記目的物を得た。
本化合物の N MRを以下に示す。
XH-NMR ( C D C 1 3 , d ppm )
1. 5 (9H, s) , 1. 51 (9H, s}, 1. -2.45 (4H, m) ,
2.42 (3H, s) , 4.3-4. 6 UH, m}, 5.41 (2H, s) ,
.5-5.7 (lH, nii, 1.42 (5H, s 7. ?7 (1H, m) ,
8.77 (1H, br si , 8.97 (lH, in} .
(i i) 2 - ( 1, 3 —ジー tープトキシカルボ二ルブロピル アミノー 5—メチル— 7—アミノー 4 H - 3, I 一ベンゾ ォキサジン一 4 一オン (式 ( E〉 の化合物〉 ク 合成
実施例 1 , (ΐ ϋ) において、 1 — N— 〈 2—ベンジル ォキシカルボ二ルー 3—メチルー 5—二トロフヱニル〉 力ルバモイルー Lーァラニン一 t 一ブチルエステルの代 わりに上記( Π で得られた目的物を使用する以外は同様 の方法によ り、 題記目的物を得た:
本化合物の N M Rを以下に示す
1 H ~ M R ( C D C i 3 , ppm )
1.44 (9H, s) , 1.48(9H, si , i.8-2.4 (4H, mi ,
2. 59 (3H, s 4, 23 (2K, br s), 4.3-4.6 (lH, mi , 5. 3 - 5. 6 ( 1 H, DI) , 6. 25 ( 2 H . s). ii i i) 7 — ( N—べンジルォキシカルボ二ルー- L—フエ 二/'レアラニル) アミ ノー 5—メチルー 2 — ( 1, 3 -ジ— t -ブトキシカルボニルプロピル) ァミ ノ一 4 H - 3 , 1 一べンゾォキサジン一 4一オン 〈式 〈 F ) の化合物 .) の 合成
実施例 1, (iv)において、 N— ( 5—メチルー 7—ァ ミノー 4 H— 3, 1 —ベンゾ才キサジン— 4—オン一 2— ィル) — Lーァラニン— t 一ブチルエステルの代わりに 上記(i i)で得られた 2—置換一 5—メチルー 7 —ァミノ - 4 H - 3, 1 一べンゾォキサジン— 4一オン誘導体を使 用し、 それ以外は同様の方法により、 題記目的物を得た 本化合物の N M Rを以下に示す。
^ - N M R ( C D C 1 3 , d ppm ) .
1.44 (9H, s), 1. 56 (9H, s), 1.8-2.4 (4H, m},
2, 53 (3H, s), 3.0- 3.2 (2H,m),
4.25-4.8 (2H, m), 5.11 (2H, s},
5.6-5.8(lH, m} , 15-6.4 (1H, m),
6.72 (1H, br s), 7.1-7.3 (1H, s) ,
7.19 (5H, s), 7.30 (5H, s), 8.65 (1H, br s}.
(iv) 7 - ( N—ベンジルォキシカルボ二ルー; L一フエ二 ルァラニル〉 アミノー 5 —メチルー 2— ( 1, 3 —ジカル ボキシァロピル) アミノー 4 H— 3, 1 —ベンゾォキサジ ンー 4 一オン 〈式 ( I ) の化合物〉 の合成
実施例 1 , (V) において、 7— ( N—べンジルォキシ カルボニル— L—フエ二ルァラニル〉 アミノー 5—メチ ルー 2— ( 1 - t -ブトキシカルボニルェチルァミノ〉 - 4 H - 3, 1 —ベンゾォキサジン一 4一オンの代わりに 上記 Uii) で合成した 7— ( N—ベンジルォキシカルボ
二ルー L一フエ二ルァラ二ル) アミノー 5 —メチルー 2 置換— 4 H— 3, 1 一べンゾォキサジン— 4 一オン誘導 体を使用する以外は同様の方法により、 題記目的物を得 た。
本化合物の N M Rを以下に示す。
1 H - N M R ( d 6 - acetone , δ" pm )
1. 9-2.5 (4H, m) , 2. 59 (3H, s) ,
3. 0-3. 25 (2H, m) , 4. 3-4. 8 (2H, m},
5. 17 (2H, s) , 5. 7- 5. 9 ( 1 H, III) )
6.3-6. 55 (lH, ffi) , 6.82 {1H, br s) , 7. 1-?· 3 (1H, s) ,
7. 24 (5H, s) , 7. 3K5H, s) , 9. 32 (1H, br s) . 実施例 8
7 — ( N—ベンジルォキシカルボ二ルー Lーァラニル) アミ ノー 5 —メチルー 2 — ( 1 —カルボキシェチルァミ ノ) 一 4 H— 3, 1 —べンゾォキサジン— 4 一オンの合成 (化合物 NG. 102 )
(i) 7 — ( N—べンジルォキシカルボ:!ルー L一ァラニ ル〉 アミノー 5 —メチル— 2 — ( 1 — t—ブトキシカル ボニルェチル〉 アミノー 4 H— 3, 1 —べンゾォキサジン 4 一オン (式 ( F〉 の化合物) の合成
実施例 1 , (iv)にお て、 N—ベンジルォキジカルボ 二ルー L—フェニルァラニンの代わりに N—ベンジルォ
キシカルボ二ルー Lーァラニンを使用し、 それ以外は同 様の方法により、 題記目的物を得た
本化合物の N M Rを以下に示す
1 H - N M R ( C D C 1 3 , ό- ιη )
i.42 (3H, d, J = 7. li ) , 1.50 (3H. d, J = 6.5Hz).
J. 53 (9H, s), 2. 5δ(3Η, s), 4.2_ョ.1 (3H, m),
5. 12 (2H, s 5. 7 '1H, br s
;. 1-7.4 (2H, iri), 7.32 i5H, s),
8.8-8. 95 ilH, m). ii i ) 7 — 〈 N—ベンジルォキシカルボニル— L一ァラニ ル) アミ ノー 5—メチルー 2— ( 1 —カルボキシェチル ) アミ ノー 4 H— 3, 1 —べンゾォキサジン一 4 一オン ': ¾ ( I ) の化合物) の合成
実施例 1 . (V) において、 Ί — ( X—ベン ジル'ォキン 力ルボニルー L一フヱ二ルァラニル) アミノー ーメチ ル一 2— ( 1 — tーブトキシカルボニルェチルァミ ノ :' • · 4 H - 3, 1 —ベンゾォキサジン一 4一オンの代わりに 上記( で合成した 7 — ( N—べンジルォキシカルボ二 ル一 Lーァラニル〉 ァミノ一 5—メチルー 2— ( 1 - t 一ブトキシカルボニルェチルァミノ〉 一 4 H— 3 , i —べ ンゾォキサジン一 4—オンを使用する以外は同様の方法 によ り、 題記目的物を得た。
本化合物の N M Rを以下に示す-
H - N M R ( d 6 - acetone , piri )
1. 8(3H, d, J = 7. IHz) , 1. 51 (3 H, d, J = 6. 5Hz)
2.60 (3H( s) , 4. 5-5. Ο ΒΗ, πι) ,
5.16 (2H, s), 5. 5 (1H, br s) ,
7, 1-7.35 (2H, m) , 7.30 (5H, s),
9.35 (1H, br s) . 実施例 9
7— 〈 N—べンジル才キシカルボ ルー L一バリル一 L ーァロリル〉 アミノー 5—メチル— 2— 〈 3—カルボキ シァロピル〉 ァミノ一 4 H— 3, 1 —ベンゾォキサジン一 一オンの合成 (化合物 No, 112)
(i) 7 - ( N_—べンシル才キシカルホニルー L一ノくリル 一 Lーァロリル〉 アミノー 5—メチル— 2— ( 3 - - ブトキシカルボ:!ルァロピル〉 アミノー 4 H— 3, 1 —べ ンゾォキサジン一 4 一オン (式 ( F ) の化合物) の合成 実施例 1 , (iv)において、 N— ( 5—メチルー 7 —ァ ミノー 4 H— 3, 1 —ベンゾォキサジン一 4一オン一 2— ィル) 一 L—ァラニン一 t一ブチルエステルの代わりに 実施例 4 , (i i)で得られた 2—置換一 5—メチルー 7 — アミノー 4 H— 3, 1 —ベンゾォキサジン— 4 —オン誘導 体を使用し、 N—べンジルォキシカルボ二ルー Lーフヱ 二ルァラニンの代わりに N—べンジル才キシカルボニル
- L—バリルー L一プロリンを使用し、 それ以外は同様 の方法によって、 題記目的物を得た。
本化合物の N MRを以下に示す:
1 H - N M R ( C D C 1 3 , όゝ ppm )
0.8-1. ll (6H,m), 1.44(9H, si,
i.7-2.45(9K,ffl}, 2.62ί3Η, s),
3.35-3.9(4K, m), 4. G5_4.2 (1H, m) ,
4.25-4.6(2Η, ιη1, 5.09I2H, s).
5.3-5.5 iiH, m) , 6.86 (1H, r s
(i i) 7— ( N—べンジルォキシカルボ二ルー L—バリル — L—プロリル) アミノー 5—メチルー 2— ( 3—カル ボキシプロピル) アミノー 4 H— 3, 1 —べンゾォキサジ ン ー 4一オン (式 〈 ェ ) の化合物) の合成
実施例 1 , (v) において、 7— ( N—ベンジルォキジ カルボ二ルー L—フエニルァラニル〉 アミノー 5—メチ ルー 2— ( 1 一 t—ブトキシカルボニルェチルァミ ノ) 一 4 H— 3 , i —ベン'ゾォキサジン一 4一オンの代わりに 上記 UUで合成した 7— ( 一べンジルォキシカルボ二 ル一 Lーノくリル一 L一プロリル〉 ァミノ一 5—メチル'一- 2— ( 3— t—ブトキシカルボニルプロピル〉 ァミ ノー 4 H - 3, 1 一べンゾォキサジン一 4一オン誘導体 使^ する以外は同様の方法によ り、 題記目的物を得た
本化合物の N M Rを以下に示す。
1 H - N M R ( d 6 -acetone , d ppm )
0.8-1.11 (6H, in), 1.7-2.4 (9H, m) ,
2.60 (3H, s), 3.4-3.9(4H,ffi),
4.05-4.2(lH,m}, 4.3~4.6 (2H, m) ,
5.11 (2H, s), 5.3-5.5(lH,m),
6.9(1H, br s), 7.1-7.3(lH,br s),
7.30(6H, s), 8.8(1H, br s).
実施例 1 0
7— ( £— N—べンジルォキシカルボ二ルー L—リジル ァミノ一 5—メチルー 2—ィソプロピルァミノー 4 H - 3, 1 —ベンゾ才キサジン一 4 一オン (化合物 NQ.202 ) 塩 酸塩の合成
{ ΐ } 7 -_( α - Ν一 _t—ブ卜キシカルボ二ルー ε— Ν— ベンジルォキシカルボ二ルー L—リジル) アミノー 5_ - メチルー 2—ィソプロピルアミノー 4 Η― 3, 1 —ベンゾ ォキサジン一 4一オンの合成
732mg の "— N— t—ブトキシカルボ二ルー ε - - N— ベンジルォキシカルボ二ルー L—リジンを 15mlの乾燥 Τ H Fに溶かし、 210 1の N—メチルモルホリンを加え た。 反応混合物を約一 15Cに冷却し、 981 1 のイソブ チル クロ口ホルメートを加えた。 5分間 で冷却を 続けながら攪拌し、 300 mgの 2—ィソプロピルァミノー
5—メチルー 7—アミノー 4 H— 3, 1 —ベンゾ才キサジ ンー 4一オンを溶解した乾燥 TH F溶液を滴下した: 一 15て:で約 3時間攪拌を続けた後ゆつく りと室温にもどし 約 12時間撹拌した。 反応混合物に 10G miの水を加え約 100 mlの酢酸ェチルで 2回抽出し、 集めた有機層を無水 硫酸マグネシウムで乾燥し溶媒を留去した 残渣をシリ 力ゲルクロマトグラフィ一で精製し、 813 mgの題記目的 物を得た Λ
本化合枸の N M Rを以下に示す ..
11 - M R ( C D C 1 3 , pffi )
1.21 (6H, d, J = 6.4Ηζ), ί.42(9H, s),
i.4-1, 8(6H, m), 2.67 (3H, s;', 3.24 (2K, m),
4.00-4.42 (2H, m;', 4.75 (IK, mi. 5.20 (1H, mi.
m; ,
( i i } 7— ( £一 N—ベンジルォキシカルボニル一 L -リ ジル) アミノー 5—メチルー 2—ィ ゾブロピルァミ ノー 4 H - 3.1 一べンゾォキサジン一 4一オン 塩酸塩の合
7 — ( — N— t—ブトキシカルボ二ル一 ε'— X—べ ンジルォキシカルボニル一 L一 リジル) アミ ノー 5—メ チル一 2—ィソプロピルァミノー 4 H— 3, 1 —ベンゾォ キサジン一 4一才ン 65 G iri gを 5 Offi iの 4 N H ; 一ジォ
キサンに溶解し室温で 1時間攪拌した後、 溶媒及び塩酸 を留去した。 残渣にベンゼンを加え減圧濃縮、 乾固させ る操作を 3回繰り返し、 640 mgの題記化合物を得た。
本化合物の N M Rを以下に示す。
^-NMR ( ¾0 , ppm 〉
1.32(6H, d, J = 6.4Hz), 1.5-2.2(6H,m))
2.77(3H, s), 3.0-3.15(lH,m),
4.0-4.5 (3H, m), 5.24(2H, s), 7.0-7.2 (1H, m) , 7.40(5H, br s), 7.3-7.5(lH,m), 突施例 1 1
7— L一グルタミルアミノー 5—メチルー 2—イソァロ ピルアミノー 4 H— 3, 1 —ベンゾォキサジン一 4—オン (化合物 No.204 ) 塩酸塩の合成
(i) Ί_- ( Ν— t—ブトキシカルボニル一ァ一 t—プチ ルー L―グルタミル) ァミノ一 5—メチルー 2—イ ソプ 口ピルアミノー 4 H— 3, 1 —べンゾォキサジン一 4—ォ ンの合成
実施例 1 〇 (U において、 a— N— t—ブトキシカル ボニルー ε—べンジルォキシカルボ二ルー L—リジンの 代わりに Ν— t一ブトキシカルボ二ルー L一グルタミン 酸ーァー t一ブチルエステルを用いる以外は実施例 1 0 (i) と同様にして、 題記化合物を得た。
本化合物の N M Rを以下に示す。
1 H - N M R ( C D C 1 3 , ό ρριτι )
J.27 (6H, d, J = 6.4Hz) , ί.45 (9K, s),
1. 7 (9H, s), 1.85-2.15 (2H, iri}t
2.25-2. 54 ( 2H, iri), 2.68 (3H, s),
3.9-4.7 (2H, in}, 5.0-5.3 (1H, m) ,
. 5-5.6 (lH, ni! , 7.08 (1H. br s
7.43 (1H, br s 9.03 (1H, s i i ) 7 — Lーグ/レタミブレアミノー 5—メチル一 2 —ィ ソ ァロピルアミノー 4 H— 3, 1 —べンゾォ ¾サ_ジン_一 _4二 オン 塩酸塩の合成
実施例 1 0 (i Uにおいて、 7— ( a - N - t -ブトキ シカゾレボニノレ一 ε — Ν—べンジル才キシカルボ二ル - し 一 リジル) ァ ノ一 5—メチル '一 2—イ ソマ口レ -> ノー 4 Η— 3, 1 一べンゾォキサジン一 4一才ンの代わり に上記(Π で得られた 7— ( Ν— t 一ブトキシ力/レポ二 ノレーア一 t 一プチ/レ一 L一グヌレタミゾレ) ァミ ノ一 5—メ チル— 2 —イソプロピルアミノー 4 H— 3, ί —べンゾォ キサジン一 4一オンを用いる以外は実施例: I ϋ (Η)と同 様の方法により、 題記化合物を得た:
本化合物の N M Rを以下に示す:
1 Η - N M R ( D 2 0, d ppiri )
1.28 (6H, d, j = 6.4Hzj, 1. -2.2 (2H, m) ,
2.3-2.6(2H,m}) 2.73(3H, s),
5.3-5. S(lH,m), 7.0-7.2 ( 1H, m),
7.3-7.5(lH,m).
突施例 1 2
7 - ( N— p—クロ口べンゼンスルホニルー L—グル夕 ミル) アミノー 5—メチルー 2—ィソァロピルアミノー 4 H— 3, 1 —べンゾォキサジン一 4一オン (化合物 205 ) の合成 '
200 ffigの 7— L—グルタミルアミノー 5—メチルー 2 一ィソプロピルァミノ一 4 H— 3, 1 —ベンゾォキサジン 一 4一オン 塩酸塩を 30mlの塩化メチレンに懸濁させ、 490 u 1 のへキサメチルジシラザンを加え室温で 2時間 攪拌した。 110 の P—クロ口ベンゼンスルホユルク口 ライ ドと、 70 1 のトリェチルァミンを加えた。 室温で 2時間撹拌後、 140 ; u 1 のトリエチルァミンを加え室温 で更に一晩攪拌した。 反応混合物を 50ffliの 10%クェン酸 溶液にあけ、 50mlの酢酸ェチルで 2回抽出した。 有機層 を集め、 無水硫酸マグネシウムで乾燥し、 過後溶媒を 留去した。 残澄をシリカゲルクロマトグラフィ一で精製 し、 175 mgの題記目的物を得た。
本化合物の N MRを以下に示す。
XH - N M R ( d 6 - acetone , & ppm )
1.29 (6H, d, J = 6.6Hz), 2.02(2H,m),
2.4 (2H, t, J = 8.1Hz), 2.59 (3H, s),
4.09(lH,m), 6.56(1H, brs), 6.94(1H, s),
7.36(2H, d, J = 6.8Hz), 7.4K1H, s),
?.67(2H, d, J = 6.8Hz), 9.38(1H, s). 実施例 1 3
7— (グリシルー L一グルタミル〉 アミノー 5—メチル 一 2—イソプロピルアミノー 4 H— 3, 1 —ベンゾ才キサ ジン一 4一オン (化合物 No, 213) 塩酸塩の合成
(i) N— t—ブトキシカルボニルダリシルー L一グルタ ミン酸ーァ— t一ブチル エステルの合成
12.4 の1^—七一ブトキシカルボ二ルグリシンを 150 mlの塩化メチレンに溶解し 11· 4gの N, N' —カルボ二 ルジイ ミダゾールを加えた 室温で 15分間攪拌後、 13.9 gのグ /レタミン酸一ァ ー t―ブチル エステル一ひ 一メ チル エステルを加え一晩攪拌した。 反応混合物に 100 mlの水及び 10%クェン酸水溶液を加え、 水層の pHを約 2.0 とし分液した。 さらに、 水層から 100 mlの酢酸ェチ ルで抽出を行い有機層を集め、 無水硫酸マグネシウムで 乾燥 過後溶媒を留去した。 残渣をシリカゲルクロマト グラフィ一によって精製し 21.1 gの N— t—ブトキシカ ルボニルダリシル— L一グルタミン酸ーァ一 t—ブチル エステル一ひ 一メチル エステルを得た。
本化合物の N MRを以下に示す。
^ - N M R ( C D C ] 3 , ^ p m )
1.43 (9H, s) , 1.47 (9H, s),
1.83-2. 1 (2H, HI) , 2.2-2. 5 {2H, m) ,
3.75 (3H, s) , 3.85 (2H, s) , 4.0-4.3 (1H, m),
4.6-4.8(1H, m), 4.95- 5.20 (lH, ni) . このようにして得られた N— t 一ブトキシカルボニル グリシルー L一グルタミン酸 γ 一 1 一ブチル ェステ ルー《—メチル エス子ル 21. 1 gを O miのメタノール に溶解し、 26.9ffiiの 2 N 一水酸化ナトリゥム水溶液と 20.9inlの水を加え室温で 3· 5 時間攪拌した,. メタノール を留去し 50miの水を加え 250 miのェチルエーテルで洗浄 した、· 水層を分離し 10%コハク酸水溶液を加え溶液の pH を 3程度に調整した。 さらに、 50iniの酢酸ェチルで 3 [m 抽出を行い、 有機層を集め、 無水硫酸マグネシウムで乾 燥し、 沪過後溶媒を留去することによ り 20. 1 gの題記目 的物を得た <
本化合物の N M Rを以下に示す。
3 H— N M R ( C D C 1 3 , d pm )
ί.43 (9H, s) , 1. 7 (9Η, s) , 1.8-2. l (2H, iri) ,
2.2-2. 5 (2H, in) , 3.88 (2H, s
4.0-4.35 (lH, m) , 4.6-4.78 (lH, iri) .
5. 15-5.23 (1H, m) , 8. 2-8.33 (1H, m) .
(ϊ i) 7 - ( N— t —ブトキシカルボニルグリシル—ァ— し一ブチルー L一グルタミル) ァミ_ノ一 5—メチルー 2
-ィゾプロピルアミノー 4 H— 3, 1 —べンゾォキサジン 一 4 一オンの合成
実施例 1 0 (U において、 ひ — N— t ブトキシ力ル ボニル一 ε — N—ベンジルォキシカルボ ルー L一リジ ンの代わりに上記( で得られた Ν— t ブトキシカル ,ニ―ルダリシル— L一グル夕ミン i Ύ ί. 一ブチル エステルを用いる以外は実施例 1 0 (Π と同様にして、 題記化合物を得た。
本化合物の N M Rを以下に示す。
^ - M R ( C D s O D, d ppiri )
I.27 (6H, d, J=6.4Hz) , I.45 C9H, si .
1.47 (9H. sL 1.80-2.1? (2H, m
2.2-2. 58 (2H, in), 2. 50 ί3Κ, s),
3.88-4. 10 (3H, mi , 4.15-4.60 (1H, nu,
7.0δ(1Η, br s), 7.43 (lH, br s).
(iii) 7— (グリシル一 Lーグルタミル) アミ ノー 5 — メチルー 2—イ ソプロピルアミノー 4 H— 3, 1 —べンゾ ォキサジン一 4 一オン 塩酸塩の合成
実施例 1 0 (i i )において、 7— ( α — — t ーブトキ シカルボニゾレ一 ε 一 Ν—ベンジルォキシカ /レボニルー L ―リジル〉 ァミノ一 5—メチルー 2—ィ 、ノブロピ /レアミ
ノー 4 H— 3, 1 —べンゾォキサジン一 4 —オンの代わり に上記( i i )で得られた 7 — 〈 N— t —ブトキシカルボ二 ルグリシル—ァ— t—ブチルー L―グルタミル) ァミノ — 5—メチルー 2—イソプロピルアミノー 4 H— 3, 1 - ベンゾ才キサジン一 4一才ンを用いる以外は実施例 1 〇 ( i i )と同様にして、 題記化合物を得た。
本化合物の N M Rを以下に示す。
— N M R ( D 2 O , δ· ppm )
1.30 (6H, d, J = 6.4Hz) , 1.80~2.2 (2H, m) ,
2.25-2.6 (2H, ffi), 2.65 (3H, s
3.9-4.1 (2H, in), 4· 2-4. 5 (2H, m),
7.0-7.2<lH, m), 7.3-7.5 (lH, m) . 実施例 1 4
7 一 ( N— p—クロ口ベンゼンスルホニルグリシル― L - -グルタミル) ァミノ— 5—メチル— 2—ィソァロピル ァミノ一 4 H— 3, 1 —べンゾォキサジン一 4 一オン (化 合物 No.214 ) の合成
実施例 1 2において、 7 — L—グルタミルアミノー 5 —メチル一 2—イソプロピルアミノー 4 H— 3, 1 —ベン ゾォキサジン— 4 一オン 塩酸塩の代わりに 7— (ダリ シルー L一グルタミル) アミノー 5—メチルー 2—イソ プロピルアミノー 4 H— 3, 1 一べンゾォキサジン一 4 一 オン 塩酸塩を用いる以外は実施例 1 2と同様にして、
題記化合物を得た。
本化合物の N MRを以下に示す。
^ - N M R ( d 6 一 acetone , ό- ppm )
1.22(6H, d, J = 6.3Hz), 2.02-2.20 {2H, m) ,
2.3-2.53(2H)m), 2.60(3H, s),
3.77-3.82 (2H,m), 3.90-4.45 (2H, m) ,
4.5-4.76{lH,m), 6.90-7. O^dH.m),
6.95(1H, brs), ?.33(2H, d, J = 6.8Hz),
7.48(1H, brs), ?.70 (2H, d, J = 6.8Hz),
9.38(1H, s). 実施例 1 5
7— ( 3—ベンジルー L—ァスパルチルァミノ )一— 5 - メチルー 2—ィソプロピルアミノ— 4 H— 3, 1 —べンゾ ォキサジン一 4一オン (化合物 No.208 ) 塩酸塩の合成
(i) 7— ( N— t一ブトキシカルボ二ルー /3—べンジル — L一ァスパルチル〉 ァミノ一 5—メチルー 2—イソァ uピルァミノ一 4 H— 3, 1 —べンゾォキサジン一 4ーォ ンの合成
実施例 1 0 (i) において、 α— N— t—ブトキシカル ボニル— ε - Ν—べンジルォキシカルボ二ルー L -リジ ンの代わりに Ν— t—ブトキシカルボ二ルー L -ァスパ ラギン酸—^一べンジル エステルを用いる以外は実施
例 l O U) と同様にして、 題記化合物を得た。
木化合物の N MRを以下に示す。
1H- N MR ( C D C 1 3 ) d w^ )
1.26(6H, d, J = 6.6Hz), 1.48(9H, s),
2.67(3H, s}, 2.85-3. l (2H,m})
3.9-4.3(lH,m), 4.5- 4.9 (2H, m),
5.16(2H, s), 5.65-5.9(lH,m},
7.05-7. HdH. m}, 7.33(5H, s),
7.25-7.35(lH,m), 8.69 (1H, brs) .
(ii) 7 - ( ^—ベンジルー Lーァスパルチルァミノ〉 一 5—メチルー 2—イソァロピルアミノー 4 H— 3, 1 —べ ンゾォキサジン一 4一オン 塩酸塩の合成
実施例 1 0 (ii)において、 7— ( α— N— t—ブトキ シカルボニル— ε— N—べンジルォキシカルボ二ルー L リジル) ァミノ一 5—メチル— 2—イソプロピルアミ ノー 4 Η— 3, 1 —ベンゾォキサジン一 4一オンの代わり に上記(i) で得られた 7— 〈 N— t一ブトキシカルボ二 ルー;3—ベンジル一 L一ァスパルチル〉 ァミノ一 5—メ チルー 2—ィソプロピルァミノ— 4 H— 3, 1 —べンゾォ キサジン一 4一オンを用いる以外は実施例 1 0 (ii;'と同 様にして、 題記化合物を得た。
本化合物の N M Rを以下に示す。
XH - N M R ( D 2 0, d ppm )
1.30 (6H, d, J = 6.6Hz) , 2.75 {3H, s),
2.9-3.2 (2H, m), 4.3-4.9 (2H, m),
5. 2 (2H, s), 7. 0-7. 15 (1H, ID) ,
7.25-7.35 (lH, in), 7.40 (5H, br s). 実施例 1 6
7― _( 一 N— p一 ロ_口ベンゼンスルホニル一 L一リ ジル) ァミノ一 5 _メチル— 2—イソァロピルァミノ— 4 H - 3, 1 一べンゾォキサジン一 4一オン (化合物 No. 203 ) 塩酸塩の合成
(i ) a - N - p 一クロ口ベンゼンス レホニ/レー ε — Ν— ί —ブトキシカルボ二ルー L一リジンの合成
2.00 gの ε — Ν— t —ブトキシカルボニル— Lーリジ ンーメチルエステル 塩酸塩を、 40mlの塩化メチレンに 懸濁させ 2· 05gのトリエチルアミンを加えた。 更に 1, Π gの P —クロロベンゼンスルホニルク口リ ドを加え室温 で一晩攪拌した。 溶媒を減圧下濃縮し 1 N—塩酸を加え 酢酸ェチルより抽出した。 有機層を飽和炭酸水素ナトリ ゥム水溶液、 飽和食塩水で洗浄後乾燥した。 浐過濃縮後 得られた粗生成物をシリカゲルカラムクロマトで精製し 2 83 gの"一 N— p—クロ口ベンゼンスノレホニル— ε — Ν - t一ブトキシカルボニル— L—リジン一メチルエス テルを得た。
本化合物の N M Rを以下に示す。
XH - N M R ( C D C 1 3 , δ ppra )
1.3- 1.8(6H,m), 1.44(9Η,ιη),
2.9-3.2(2H, m), 3.52(3H, s), 3.7-4.2(lH,m},
4.3-4.65(lH,m), 5.1-5.35(lH,m),
7.46 (2H, d, J=8.8Hz}, 7.77 (2H, d, J=8.8Hz). このようにして得られた α— Ν— p—クロ口ベンゼン スルホニル— ε— N— t一ブトキシカルボ二ルー L一リ ジン一メチルエステル 2, 83gを、 δθηιΐのメタノールに溶 解し 7.4 mlの 1 N—水酸化ナトリウム水溶液を加え室温 で一晩攪拌した。 メタノールを留去し 20mlの水を加え 50 Hi 1のェチルエーテルで洗浄した。 水層を分離し 10 %コノ、 ク酸水溶液を加え溶液の PHを 3程度に調整した。 さらに 50miの酢酸ェチルで 3回抽出を行い、 有機層を集め、 無 水硫酸マグネシウムで乾燥し、 沪過後溶媒を留去するこ とにより 2.5 gの題記目的物を得た。
本化合物の N MRを以下に示す。
2H - N M R ( C D C 1 3 , ppiii )
1.4- 2.0(6H, m}, 1,43 (9H, ID) ,
2.9-3.2(2Η,ιιι)) 3.8-4.1 (1H, m),
5.0-5.5 (1H, m), 5.7-6. OdH.m),
7.46 (2H, d, J=8.8Hz), 7.77 (2H, d, J=8.8Hz).
{ i i } 7— ( ひ 一 N— p—クロロベンゼンスルホ二ルー ε 一 Ν— t —ブトキシカルボ二ルー Lーリジル) ァミノ一 5—メチル一 2—ィソァロピルアミ ノー 4 H— 3, 1 — ンゾォキサジン一 4一オンの合成
実施例 1 0 (i) において、 — t—ブトキシカル ボニルー ε 一 Ν—べンジルォキシカルボ二ルー L—リジ ンの代わりに上記(U で得られた α— N— p —クロロべ ンゼンスルホニル— ε — Ν— t —ブトキシカルボ二ル- L一リジンを用いる以外は実施例 1 ◦ (i) と同様にして 題記化合物を得た。
本化合物の N M Rを以下に示す。
— N MR ( C D C 1 3 , d ppm )
1.27 (6H, d, J = 6.4Hz), 1. -2.0 (6H, m) ,
1.43 (9H, in), 2.67 (3H, s) , 2· 9 .2 (2H, m) ,
3, 8-4.4 (3H,m), 5. 1-5. 5 (1H, m) ,
5.75-6. O dH. in}, 7.0-7.4 (2H, m),
7.48 (2H, d, J = 8.8Hz), 7.79 (2H, d, J = 8.8Hz) .
(i i i } 7— ( α _— N— p —クロ口ベンゼンスゾレホニスレ一 L—リジル〉 ァミノ— 5—メチルー 2—イソプロピルァ ミノー 4 H— 3, 1 —ベンゾォキサジン— 4—オン 塩酸 塩の合成
実施例 1 〇 (ii)において、 7— ( ひ 一 N— t —ブトキ シカルボニル— ε — N—べンジルォキシカルボ二ル - L
—リジル〉 ァミノ _ 5—メチル一 2—イ ソプロピルアミ ノ— 4 H — 3 , 1 —べンゾォキサジン一 4 一オンの代わり に上記(ii)で得られた 7— ( α— N— p—クロ口べンゼ ンスルホニルー ε — Ν— t一ブトキシカルボ二ルー L一 リジル〉 アミノー 5—メチルー 2—イソプロピルアミノ •4 H-3, 1 —ベンゾォキサジン一 4一オンを用いる以 外は実施例 1 0 (ii)と同様にして、 題記化合物を得た。
本化合物の N MRを以下に示す。
XH - N M R ( D a 0 , ^ p m )
1.30(6H, d, J = 6.4 H 2 ) , 1, 2.1 (6H, m) ,
2.70(3H, s), 3.0-3:.3(2H,m)(
3.9-4.5(2H,m), 7.0-7.4(2H,m},
7.50 (2H, br s 7.80 (2H, r si. 実施例 1 Ί
7— ( L—ァラニルー L—グルタミル) アミノー 5一—メ チル一 2—ィ ソァロピルアミノー 4 H— 3, 1 —ベンゾォ キサジン一 4 一オン (化合物 N Q . 217 ) 塩酸塩の合成
U) N— t—ブトキシカルボ二ルー L一ァラニルー L一 グルタミン酸—ァ一 t一ブチル エステルの合成
実施例 1 3 (i) において、 N— t—ブトキシカルボ二 ルグリシンの代わりに N— t一ブトキシカルボ二ルァラ ニンを用いる以外は実施例 1 3 (i) と同様にして、 題記
化合物を得た。
本化合物の N M Rを以下に示す,
1 H— N M R ( C D C 1 3 , 0s m )
1.40 (3H, d, J = 6. 5Hz}, ί.43 (9H, s) ,
i.4? (9H, s)、 ί, 8-2. l (2H,m},
2.2-2.5 (2H, mi , 4.0-4.4 (2H, in) ,
\ i l ; I —— 〈 —— t. —— ノ 卜キシカヌレ rj、一 レ—— L—— 厂一)二ヌレ
- r - -ブチル L—グルタ ミル〉 アミ ノー 5 —メチ ー 2—ィ ソプロピルァミ ノー 4 H— 3, 1 —ベンゾォキ サジン一 4一オンの合成
-: 施例 1 0 ( U において、 《— N— † 一一ブト、キシ力 /レ ボ二ルー £' 一 N—ベンジルォキシ力ルボ二ル - ί . -. り' ジ ンの代わりに上記 ίϋ で得られた \一 t -- ブ' ;-、キ i ι , ボニスレー L—ァラニ /レ一 Lーグゾレタ ミ ン酸一 ?'一 1 一ブ チル エステルを用いる以外は実施例〗 0 (i ; と同様に して、 題記化合物を得た,-.
本化合狗の N M Rを以下に示す
1 H - M R ( C D 3 O D , d ppni )
1.27 (6K, d, J = 6.4Hz) , ].40 (3H, d, J = 6. 5Hz.i .
1.45 {9H. s) , 1.47 (9H, s;, 1.8-2. 17 (2H, m; .
3.8 - 4. 5 ( 3 H , Hi / , 7. I ( 1 H , b r si ,
7.43dH. br s).
(ίϋ) 7 ( Lーァラニル一 L—ダルタミル〉 アミノー 5—メチルー 2—ィソァロピルアミノー 4 H— 3,】 一べ ンゾォキサジン一 4一オン 塩酸塩の合成
実施例 1 〇 (ii)において、 7— 〈 α— N— t—ブトキ シカルボ二ルー ε — N—ベンジルォキシカルボニル— L —リジル〉 アミノー 5—メチル一 2—ィソプロピルァミ ノ - 4 Η— 3, 1 一べンゾォキサジン— 4一オンの代わり に上記(ii)で得られた 7— ( N— t—ブトキシカルボ二 ルー L—ァラニル一ァー t—ブチル一 L一グルタミノレ ) ァミノ一 5—メチルー 2—ィソァロピルァミノ一 4 H— 3, 1 一べンゾ才キサジン一 4一オンを用いる以外は実施 例 1 0 (Π)と同様にして、 題記化合物を得た、.
木化合物の N MRを以下に示す。
XH - NMR ( D 2 0 , ^ m )
1.30(6H, d, J = 6.4Hz), 1.53(3H, d, J = 6.5Hz),
1.8-2.17(2H, m}, 2.2-2.6 (2H, m) ,
2.59(3H, s), 3.9-4.6(3H,m),
7.0-7.2(lH,m) , 7, 3_7.5(lH,m). 実施例 1 8
7 - ( N一 p—クロロベンゼンスルホ二ル— L一ァラニ ル一 Lーグルタミル) アミノー 5—メチルー 2——ィ Vプ
口ピルアミノー 4 H— 3, 1 —べンゾォキサジン一 4一才 ン (化合物 NQ.218) の合成
実施例 1 2において、 7— L—ダルタミルアミノー 5 —メチルー 2—ィソァロピルアミノー 4 H— 3, 1 —ベン ゾォキサジン— 4一オン 塩酸塩の代わりに 7— ( L - ァラニルー Lーグルタミル) ァミノ— 5—メチル— 2— イソプロピルアミノー 4 H— 3, 1 —べンゾォキサジン一 4 一オン 塩酸塩を用いる以外は実施例 1 2と同様にし て、 題記化合物を得た。
本化合物の N MRを以下に示す。
3 II - N M R ( d 6 一 acetone , δ" pm )
1.2K6H, d, J = 6.3Hz}, 1.44(3H, d, J = 6.2Hz),
2.0-2.25(2H,m), 2.3-2.53(2H,m),
2.58(3H, s), 3, 90-4.45 ( 3H,m),
4.5-4.70(lH,m), 6.90-7.05(lH,m),
6.9(1H, brs), 7.36 (2H, d, J = 6.8Hz},
7.45(1H, brs), 7.73(2H, d, J = 6.8Hz),
5. K1H, s). 実施例 1 9
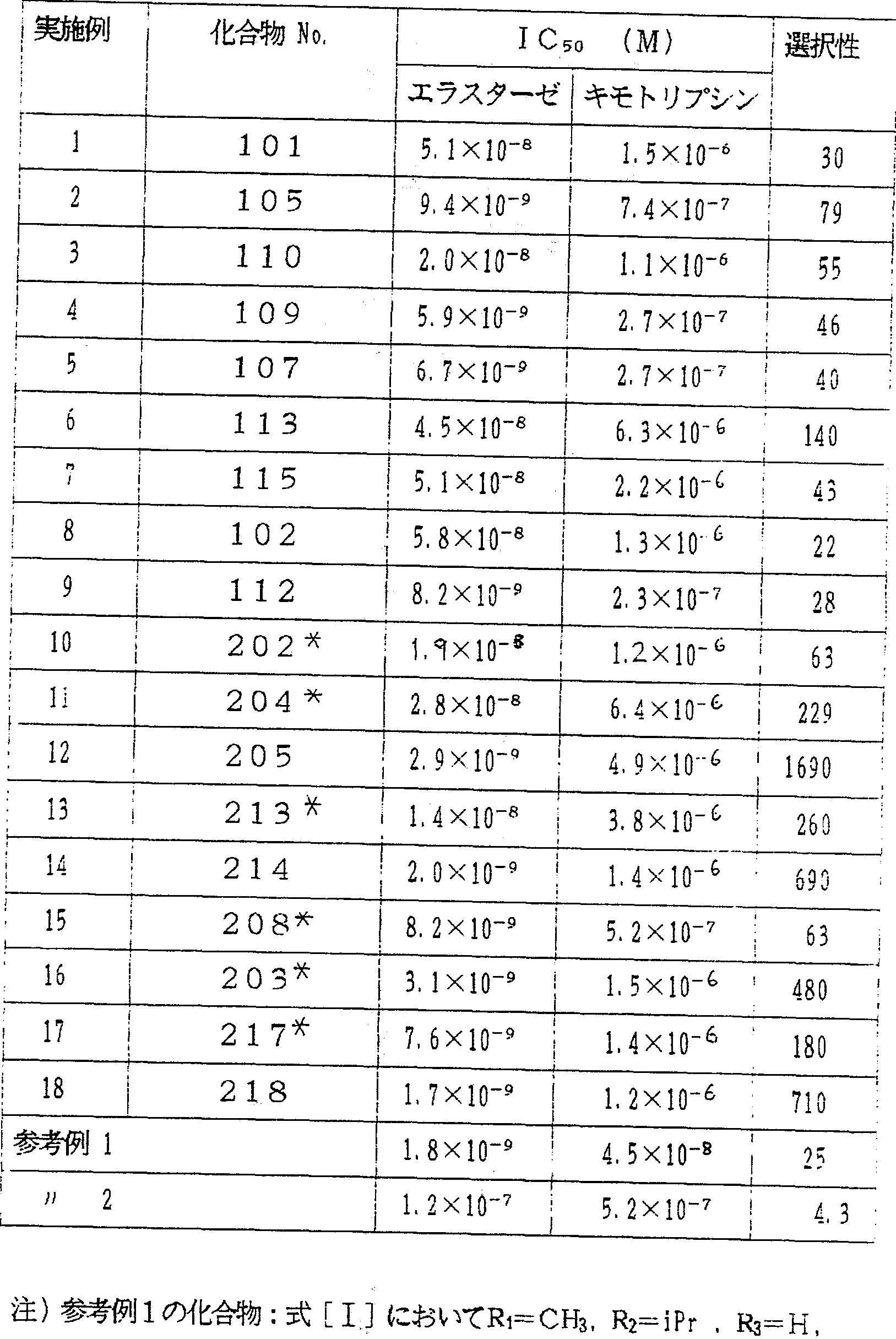
本発明の 4 H - 3, 1 —ベンゾォキサジン— 4一オン化合 物 (実施例 1〜 1 8 , 参考例 1および 2 ) のプロテア一 ゼ阻害活性
4 H - 3, 1 —ベンゾォキサジン一 4一オン化合物の、
ヒ ト膿性痰エラスターゼ、 《—キモト リプシン阻害活性 を下記の方法によ り測定した: その結果を第 1表に示す A ) ヒ ト膿性痰エラスターゼ阻害活性の測定
測定用緩衝液
C 1 M N— 2—ヒド口キシェチルピぺラジン一 N— 2—エタンスルホン酸, 1 M塩化ナトリウム, 0.1 %ボ リエチレンダリコール 6000を成分とする測定用緩衝液, P H . -)
酵 素
ヒ ト膿性痰エラスターゼはエラスチン · ァロダク ト - カンパニーよ り入手し、 測定用緩衝液で、 2 Χ 1Γ8Μに 調製した。
基 質
メ トキシスクシニルー Lーァラ二ルー L一ァラニルー j ,一プロリル一 L—バリ /レーパラ トロァニリ ドは ) \ »./ ケム社よ り入手し、 ジメチルスルホキシドで、 ΙΰηιΜに調 製した。
方 法
恒温セルホルダー付の島津 ϋ V- 2100 型分光光度計にセ ッ トしたセル中に入れた 2.4 miの測定用緩衝液、 37でに, 25 ϋ の基質溶液と 、 阻害剤を含むかあるいは含まない ジメチルスルホォキシドを加え、 攆拌した。 50M ^ の酵 素溶液を加えることにより反応を開始し、 基質の加水分 解を 410nm における吸光度変化によ り測定した ヒ ト膿
性痰ェラスターゼの活性を 50%阻害する阻害剤濃度
( I C 5。) を、 定常状態の基質加水分解速度から求めた ) aーキモ卜リプシン阻害话性の測定
酵素溶液と して牛脾臓なーキモトリアシン (シグマよ り入手) の測定用緩衝液溶液 ( 2 >:: 1Γ9Μ ) 、 および基 質スクシニルー L一ァラニルー Lーァラ二ルー L -プロ リル一 L—フエ二ルァラ二ルーパラニトロア二リ ド ぃバ 、νケム社よ り入手) を使用したこと以外は、 上記ヒ 藤 性痰エラスターゼに鬨する測定方法 A ) と同様の方法で ( -キモトリプシンに対する I C 5。を決定した ,,
X -Z-Phe を»す。 例 2c ^匕^):式 [ェ]において RFC¾. R2=iPr . Ra-H.
X = Acを表わす,.
:
表 Φ' 「選択性」 は 牛キモトリアシンに対する I C il tト膿性痰エラスターゼに対する I (:^ 対寸る比 を示すもので、 この比の値の大きいもの程、 ェラスタ一 ゼに対する阻害作用の選択性が高いことを示す,. 本発明 化合物は、 本発明者らによる WO88/09790号に開示された 化合物の 1つである参考例 1 と、 同程度のエラスターゼ 阻害活性、 及び同程度あるいはそれ以上のエラス夕ーゼ に対する選択性を示した また、 本発明化合物は、 Krantzちによる特開昭 69 号に開示された化合物 の 1つである参考例 2に比べ、 より高いエラスターゼ阻 害话性及び選択性を示した ..' 実施例 2 0
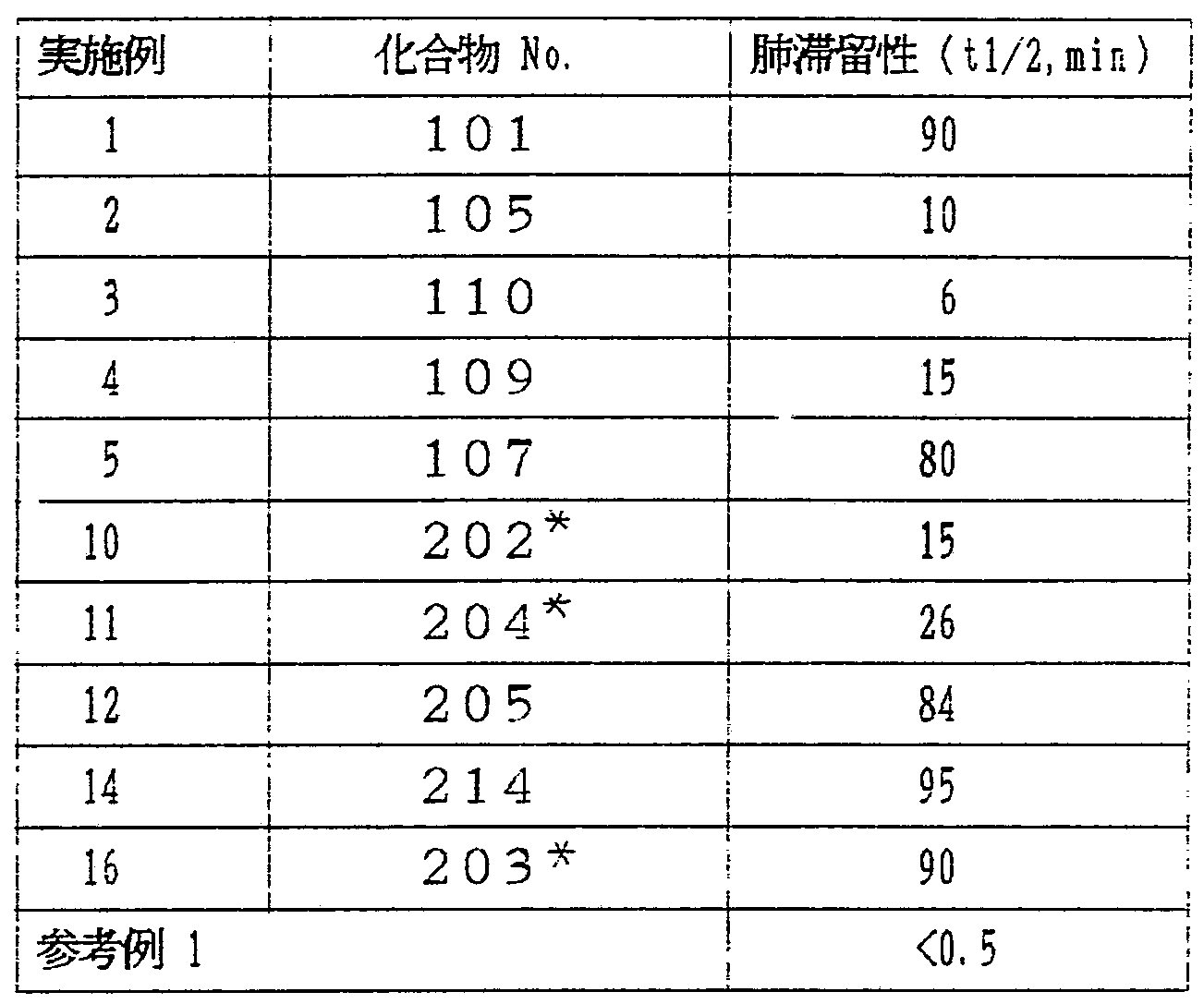
ハムスターにおける本発明の 4 H— 3, 1 —ベン γ'ォキサ ジン一 4一オン化合物の経気道投与時の肺内滞留性
第 2表に示す本発明化合物をシリアン · ゴールデン ハ ムスタ一 (体重約 100 ε ) に 200 kg経気道投与し 経時的に肺組織濃度を定量した . 化合物を l(img.' miの^ 度にジメチルスルホキシドに溶解し、 投与前に生理食塩 水で 50倍に希釈した、, この 200 g . mlの化合物溶液を ウレタン麻酔下気管力ニューレを施したハムスタ一に 1 ml -'kg ( 200 μ. g g ) 投与した .
一定時間後に脱血後、 化合物が酸性条件下、 クロロホ ルム ''メタノール二 2 ,' 1に抽出される場合、 肺 〈湿重
量約 0. 5 ε ) を摘出し、 クロ口ホルム Ζメタノール = 2 1 ( v/v) 10ml , 生理食塩水 4.5 ml , 酢酸 0, 1 mlおよび 内部標準を含んだ抽出用混合液中でホモジナイズした。 クロ口ホルム層を分離し、 減圧乾固させた後 0.5 mlのァ セトニトリルで再抽出した。 不溶物を 0.45 u mフィルタ 一で除去後、 逆相高速液体クロマトグラフィーにより定 量した。 有機溶媒抽出ができない場合には、 生理食塩水 4 mlで気管支肺胞洗浄を行い、 不溶物を遠心除去後、 直 接逆相高速液体クロマトグラフィーにより定量した、 検 出には化合物自身のもつ蛍光 (励気波長 26( m , 測定波 長 430 nni)を用いた。 半減期は肺組織中化合物量の対数を 時間に対してプロッ トし、 直線の傾きから求めた。 結果 を肺組織中での半減期と して第 2表に示した。 参考 WO88/ 09790 号に開示された化合物の一つであり、 本発 明の化合物が半減期が長く、 肺内滞留性が大幅に改善さ れていることを示している。
第 2 表
注) !11 匕^]:式 [I]において Ri=C , R
2=iPr , ί¾=Η,
X =Z-Phe を表わす < 第 3 表
' m 化^) No. 肺出 Μ$率(%)
\ 1 101 66.4
! 4 109 81.6 !
! 10 202 ^ 76.2 !
! 11 204 ォ 60.6 !
! 12 205 71.3 !
! 14 214 79.5 1
I 16 203 ^ 68.5 · i 例 1 23.8 注) ^Jl^ f匕^]:第 2表のものと同じ
: ί^ *
実施例 2 1
本発明 4 Η - 3, 1 一べンゾォキサジン一 4一才ン化合物 のエラスターゼ投与ハムスターにおける肺出血抑制効果 第 3表記載の本発明化合物を 0.01Mリン酸緩衝生理食 塩水 ( ΡΗ7· )に溶解または分散させ、 シリアン · ゴール デンハムスター (体重約 100 g ) に 1 ' kg経気道投与 し、 30分後にヒ ト膿性痰エラスターゼを 1 mg kg経気道 投与した エラスターゼ投与 3時間後に脱血屠殺し、 m 胸後生理食塩水で気管支肺胞洗浄を行った: 気管支肺胞 洗浄液中のヘモグロビン量を、 シアンメ トヘモグロビン 法により測定した
結果を本発明化合物の代わりに、 0.01Mリン酸緩衝生 理食塩水 (PH7. 5)を投与した動物の肺出血に対寸る、 本 ¾明化合物の肺出血抑制率と して第 3表に示した . 参考 は、 'ϋδ8/ 09790号に開示された化合枸の ] つであり、 肺出血抑制効果が弱かった— 一方、 本発明の化合物はェ ラスターゼ投与ハムスターに対して、 優れた肺出血仰制 効果を示した。 '