PROCESS FOR THE PREPARATION OF 7α-ALKOXYCEPHEM DERIVATIVES
The present invention relates to a new process for the preparation of 7α-alkoxycephem derivatives.
it is known that some 7α-alkoxycephem sulphones are potent protease inhibitors, in particular human leucocyte elastase (HLE) inhibitors (see, for example, J.B. Doherty et al., Nature 1986, 322:192; W.K. Hagmann et al., Eur. J. Med. Chem. 1989, 24:599; R.J. Bonney et al., Journal of Cellular Biochemistry, 1989, 39:47).
The 7β-aminocephems, namely 70-aminocephalosporanic acid (7-ACA) and 70-amino-3-desacetoxy-cephalosporanic acid (7-ADCA), are the most convenient starting materials for the preparation of 7α-alkoxy cephems. The 7α-alkoxy cephems are useful intermediates in the synthesis of the above mentioned elastase inhibitors and β-lactamase inhibitors, as described in, for example, EP-A-337,704.
The known synthetic sequences for preparing 7α- alkoxy cephems include:
- protection of the C4-carboxylic function of the starting material;
- diazotization to an unstable 7-diazo compound;
- reaction of the 7-diazo compound with the desired alcohol under suitable conditions, especially in the presence of a rhodium catalyst; and
- optionally deprotection of the C4-carboxylic function.
The above mentioned procedure is complex, dangerous and low yielding.
We have found a new, straightforward, simple, safe and high yielding process for transforming 7-ACA, 7-ADCA and their
oxidized derivatives (cheap and easily available compounds), into their 7α-alkoxy analogues by a direct conversion in one single step, without using such unstable intermediates as the 7-diazo compounds and without accumulating a hazardous by-product.
According to the present invention there is provided a process for preparing a compound of the formula (I):
R1 represents:
(a) a straight or branched alkyl group having from 1 to 20
carbon atoms,
(b) a straight or branched alkenyl group having from 2 to 6
carbon atoms,
(c) a straight or branched alkynyl group having from 2 to 6
carbon atoms, or
(d) an aryl C1-C4 alkyl group such as a phenyl (C1-C4 alkyl) group, each of the groups defined in (a) to (d) above being unsubstituted or substituted by one or more of:
(i ) a halogen atom ,
( ii ) a C1-C4 alkoxy group ,
( iii ) a cyanc group, and
( iv) a C1 -C4 alkyl thio group,
R2 represents :
1) a hydrogen atom,
2) a chlorine atom,
3) a methoxy or ethoxy group, or
4) an acetoxy group and
n represents zero, one or two;
the process comprises reacting a compound of the formula (II):
wherein R2 and n are as defined above, with an inorganic or organic nitrite in an alcohol R1OH wherein R1 is as defined above, or in a mixture of the alcohol R1OH with an organic solvent, in the presence of an inorganic or organic acid, and optionally converting a resulting compound of the formula (I) wherein n is zero, into a compound of the formula (I) wherein n is one or two by oxidation.
The above mentioned reaction is usually performed at a temperature of from 0°C to about 60°C; preferably the reaction is carried out at a temperature of from 10ºC to about 40ºC.
Moreover, the reaction may be carried out also in the presence of a transition metal catalyst such as, for example, rhodium or copper salts.
R1 preferably, represents:
(a') a straight or branched alkyl group having from 1 to 6 carbon atoms,
(b') a straight or branched alkenyl group having from 2 to 5 carbon atoms,
(c') a straight or branched alkynyl group having from 2 to 4 carbon atoms, or
(d') benzyl,
each of the groups defined in (a') to (d') above, is preferably unsubstituted or substituted by one or more:
(i') chlorine or fluorine atom
(ii') methoxy or ethoxy group,
(iii') cyano group, or
(iv') methylthio group;
R2 preferably represents:
1') a hydrogen atom,
2') a chlorine atom,
3') a methoxy group, or
4') an acetoxy group and
n preferably represents one or two.
More preferably, R1 represents:
methyl, ethyl, n-prcpyl, n-butyl, n-pentyl, n-hexyl, isopropyl, sec
butyl, tert-butyl, allyl, 2-methyl-2-propenyl, 2-butenyl, 3-methyl-2-butenyl, propargyl, 2-butynyl, 2-chloroethyl, 2-fluoroethyl, 2-methoxyethyl, 2-ethoxyethyl, cyanomethyl, 2-methylthiomethyl or benzyl, and R2 represents a hydrogen atom or an acetoxy group.
The inorganic or organic nitrites are preferably of the formula R3ONO wherein R3 is an alkali metal, an
alkaline-earth metal, a C1-C6 alkyl group, an ammonium group or a tetra C1-C4 alkylammonium group.
More preferably R3 is sodium, potassium, butyl, terbutyl, amyl or tetrabutylammonium.
Preferred inorganic acids are: perchloric acid, sulphuric acid, nitric acid, fluoboric acid, chlorosulfonic acid, boron trifluoride (BF3). Preferred organic acids are sulfonic acids such as, e.g., p-toluenesulfonic acid, methaneεulfonic acid, trifluoromethanesulfonic acid.
When the alcohol R1OH is mixed with an organic solvent, the organic solvent is typically tetrahydrofuran (THF), acetonitrile, dimethoxyethane, hexamethyl- phosphoramide (HMPA), dimethylformamide (DMF) or N-methyl- pyrrolidone, preferably THF, DMF or HMPA.
The optional oxidation of a compound of the formula (I) wherein n is zero to give a compound of the formula (I) wherein n is one or two, may be carried out by means of an inorganic or organic peracid or a salt thereof, such as, e.g., peracetic acid, m-chloroperoxybenzoic acid (MCPBA), monoperphthalic acid
or monoperoxysulphate acid, usually in a mixture of an inorganic and an organic solvent.
The oxidation preferably is carried out by means of potassium monoperoxysulphate (OxoneR), usually in acetonitrile/water, methanol/water, ethanol/water, dimethylforroamide/water or acetone/water; more preferably in acetonitrile/water or methanol/water.
The oxidation is typically carried out at a temperature of from 10° to about 100°C preferably at a temperature of from 30° to about 70°C.
The starting compounds of the formula II are known compounds or can be prepared from known compounds by known methods.
In the formulae of this specification the dotted line (""") indicates a substituent in the a configuration, i.e. below the plane of the ring, and the wedged line
indicates a substituent in the β configuration, i.e. above the plane of the ring.
The following examples illustrate but do not limit the invention.
Example 1
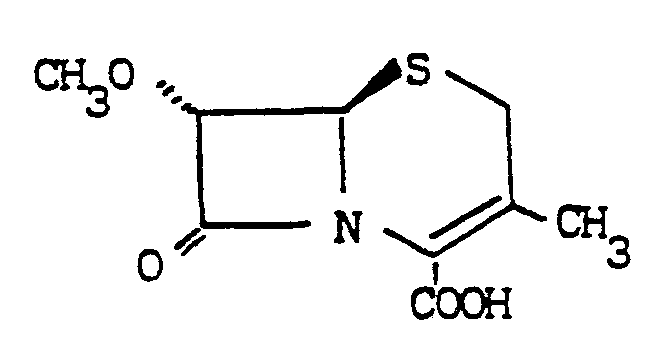
7α-Methoxy-3-methyl-3-cephem-4-carboxylic acid
To a solution of 7β-amino-3-desacetoxycephalosporanic acid (7-ADCA) (43 g), 70% perchloric acid (85 ml) and methanol (1400 ml) at room temperature, sodium nitrite (60 g) was added and the resulting mixture was stirred at 25-27ºC for 6 hours.
After pouring into H2O (1500 ml) and CH2Cl2 (800 ml), the organic phase was washed with brine then dried over Na2SO4 and eventually concentrated to dryness.
The reddish residue was purified by chromathography on LiChroprepR RP18 eluting with water-acetonitrile mixtures.
The title compound was obtained as a waxy solid (18.5 g).
NMR (CDCl3, 90 MHz)
δ 2.22 (3H, S)
3.37 (2H, ABq, J=18.1 Hz)
3.57 (3H, s)
4,52 (1H,d, J<2Hz)
4.72 (1H,d J<2Hz)
8.85 (1H, bs, exch. with D2O)
IR (CHCl3)δmax 1775, 1725 cm-1
Example 2
By following a procedure similar to that described in example 1 and substituting the proper alcohol for the methanol as reaction solvent, the below reported compounds were prepared:
7α-Ethoxy-3-methyl-3-cephem-4-carboxylic acid
NMR (CDCl
3+D
2O, 200 MHz),
δ1.27 (6H, , J=7.0 Hz),
2.21 (3H, s),
3.21 (1H,d, J=18.3 Hz),
3.54 (1H, dd, J=0.9 and 18.3 Hz),
3.6-3.9 (4H, m),
4.54 (1H,d, J= 1.5 Hz),
4.68 (1H, d, J=1.5 Hz).
IR (CHCl3)δ max 1770, 1725 cm-1;
7α-Isopropoxy-3-methyl-3-cephem-4-carboxylic acid
NMR (CDCl3+D2O, 200 NHz)
δ 1.26 (3H, d, J= 6.2 Hz)
2.21 (3H, S)
3.20 (1H, d, J=18.1 Hz)
3.54 (1H, dd, J=0.9 and 1801 Hz)
3.82 (1H, m)
4.52 (1H, d, J=1.6 Hz)
4.61 (1H, d, J=1.6 Hz)
IR (CHCl3)δ max 1780, 1725 cm-1;
7α-Butoxy-3-methyl-3-cephem-4-carboxylic acid
NMR (CDCl3+D2O, 200 MHz)
δ 0.92 (3H, t, J=7.2 Hz)
1.3-1.5 (2H, m)
1.5-1.8 (2H, m)
2.22 (3H, s)
3.23 (1H, d, J=18.1 Hz)
3.53 (1H, dd, J= 1Hz and 18.1 Hz)
3.5-3.8 (2H, m)
4.56 (1H,d, J=1.6 Hz)
4.69 (1H,d, J=1.6 Hz)
IR (CHCl3)δ max 1780, 1725, cm-1;
7α-(2-Methoxyethoxy)-3-methyl-3-cephem-4-carboxylic acid
NMR (CDCl3+D2O, 200 NHz)
δ 2.20 (3H, s)
3.19 (1H, d, J=18.1 Hz)
3.39 (3H, s)
3.52 (1H, d, J=18.1 Hz)
3.5-4.0 (4H, m)
4.61 (1H,d, J=1.5 Hz)
4.72 (1H,d, J=1.5 Hz)
IR (CHCl3)δ max 1785, 1720 cm-1;
7α-Allyloxy-3-methyl-3-cephem-4-carboxylie acid
NMR (CDCl3+D2O, 200 NMz)
δ 2.20 (3H,s)
3.20 (1H,d,J=18.2 Hz)
3.53 (1H,d, J=18.2 Hz)
4.0-4.4 (2H,m)
4.57 (1H,d, J=1.6 Hz)
4.68 (1H,d, J=1.6 Hz)
5.29 (1H, dd, J=1.1 and 10.2 Hz)
5.36 (1H, dd, J=1.4 and 7.1 Hz)
5.8-6.0 (1H,m)
IR (CHCl3)δ max 1775 and 1725 cm-1.
Example 3
7α-Methoxy-3-acetoxymethyl-3-cephem-4-carboxylic acid
By following a procedure similar to that described in Example 1 and using 7β-aminocephalosporanic acid (7-ACA) instead of
7β-amino-3-desacetpxycephalosporanic acid (7-ADCA) the title compound was obtained as a white solid.
IR (CHCl3) 1785, 1740-1720 cm-1
Example 4
7α-Methoxy-3-methyl-3-cephem-4-carboxylic acid 1,1-dioxide
A mixture of 7β-amino-3-desacetoxycephalosporanicacid (43g), 70% perchloric acid (80 ml), methanol (1300 ml) and sodium nitrite (45 g) was stirred at 25º for 6 hours, then poured into H2O/CH2Cl2. The organic phase was washed with brine, then concentrated in vacuo.
The residue was taken up with acetonitrile (300 ml) and water (300 ml). OxoneR (potassium peroxymonosulfate) (90 g) was added and the mixture was heated at 55° for 2 hours under stirring. The mixture was filtered and the filtrate was poured into H2O/ethyl acetate. The organic phase was washed with brine then concentrated under vacuum.
Treatment of the residue with diethyl ether allowed the isolation of the title compound as white crystals (12 g).
IR (KBr) 1788, 1731 cm-1.
Example 5
7α-methoxy-3-methyl-3-cephem-4-carboxylic acid-1,1-dioxide
Step a
A solution of tert-butyl 7β-amino-3-desacetoxycephalosporate (25 g) in dichloromethane/dioxane 1:1 (600 ml) was treated with tert-butyl percarbonate (32.7 g) and triethylamine (14 ml).
After standing overnight at room temperature, the solution was concentrated and the residue was purified by flash chromatography, affording tert-butyl 7β-tert - . butoxycarbonylamino-3-desacetoxycephalosporanate (27 g) as 3:1 mixture of Δ3 and Δ2 isomers.
This product was dissolved in dichloromethane (400 ml) and treated at -10°C with 55% MCPBA (65 g), then stirred 6 hours at room temperature. The mixture was filtered and the filtrate washed sequentially with 4% ag. NaHSO3, 4% aq. NaHCO3 and sat. ag. NaCl solutions. The organic phase was dried over Na2SO4 and concentrated in vacuo.
The residue was treated with diethyl ether affording tert-butyl 7β-tert-butoxycarbonyl-3-desacetoxycephalosporate 1,1-dioxide
(Δ3 isomer) as white crystals (30 g). This compound (11 g) was dissolved in dichloromethane (50 ml) and treated with anisole (10 ml) and trifluoroacetic acid (100 ml) for 3 hours at room temperature. The mixture was concentrated under vacuum to a small volume, then treated with diethyl ether.
A yellowish solid was formed, which was filtered and washed with diethyl ether; it was then poured into water (300 ml).
The pH value was adjusted to 4 by adding NaHCO3 portionwise.
After stirring at pH 4 for 1 hour at 10ºC, the mixture was filtered and the white solid washed with water then acetone.
After drying under vacuum, there were obtained as a white powder 5.5 g of 7β-amino-3-desacetoxycephalosporanic acid sulphone.
IR (KBr) 1810, 1640, 1550 cm-1
NMR (200 MHz, CF3COOD) δ 22.33 (3H,S)
4.12 (1H,d,J=.18.7 Hz)
4.28 (1H,d,J=18.7 Hz)
5.37 (1H,d,J=4.6 Hz)
5.63 (1H,d,J=4.6 HZ)
Step b
To a stirred solution of 7B-amino-3-desacetoxycephalosporanic acid sulphone (2.5 g) in methyl alcohol (70 ml) and 70% HC10„
(2 ml) at 20ºC, sodium nitrite (3 g) was added.
The resulting mixture was stirred at room temperature for 4 hours, then poured into water/ethyl acetate.
The organic phase was dried over Na2SOH4 and concentrated in vacuo.
Purification of the residue by reversed-phase chromatography
(LichroprepR RP 18) eluting with water/acetonitrile mixtures allowed the isolation of the title product as a white powder
(450 mg).
NMR (DMSO-d6)δ 1.91 (3H,s)
3.52 (3H,s)
4.20 (2H,s)
5.13 (1H,d,J=1.2 Hz)
5.35 (1H,S)
Example 6
7α-methoxy-3-methyl-3-cephem-4-carboxylic acid
To a solution of 7β-amino-3-de6acetoxycephalosporanic acid (4.3 g), methanesulphonic acid (3.88 ml) and methanol (140 ml) at room temperature, potassium nitrite (3.5 g) was added and the resulting mixture was stirred for 12 hours at room temperature. The reaction mixture was worked up as described in Example 1 affording the title product as a colourless oil (1.6 g) which solidified in the fridge.
IR (CHCl3)δ max 1775, 1725 cm-1.
Example 7
7α-methoxy-3-methyl-3-cephem-4-carboxylic acid
7β-Amino-3-desacetoxycephalosporanic acid (7-ADCA) (5.25 g) was added to a solution of boron trifluoride etherate (d20
1.13; 22ml) in methanol (170 ml) at 10° C. After 1 min. sodium nitrite (NaNO2) (5.0 g) was added and the resulting mixture was stirred for 12 h at 15° C, then poured into
CH2Cl2/water. The organic phase was dried (Na2SO4) and concentrated "in vacuo".
The crude product was purified by ion-exchange chromato- graphy (Amberlite IRA-458, elution with pH 7 phosphate buffer).
The product containing fractions (HPLC monitoring) were salted with NaCl, acidified with 20% aqueous HCl and extracted with ETOAc.
After evaporation of the dried ETOAc solution, the title product was obtained as a waxy solid (3.2 g) with the same physico-chemical characteristics of the compound prepared in
Example 1.
Example 8
7α-methoxy-3-methyl-3-cephem-4-carboxylic acid 1,1-dioxide
96% H2SO4 (15 ml) was slowly added to a mixture of 7-ADCA
(10.5 g) in methanol (350 ml) while keeping the temperature under 0° C. Sodium nitrite (NaNO2) (15 g) was then added and the mixture was stirred for 15 h at 18° C. After pouring into ETOAc/water, the organic phase was dried (Na2SO4) and rotoevaporated.
The residue (10g ) was dissolved in acetonitrile (200 ml) and water (200 ml). Potassium monoperoxy sulphate (Oxone ®)
(50 g) was then carefully added, and the mixture was heated at 55° C, under vigorous stirring, for 1.5 h; the reaction mixture was poured into ETOAc/water. The organic phase was washed with aqueous NaHSO3 then brine and eventually it was dried and concentrated. The residue was taken up with diethyl ether - diisopropyl ether and let stand at 4° C overnight.
The title product obtained as a white solid (4.1 g) showed the same physico-chemical characteristics of the compound prepared in Example 5.