WO1992003436A1 - Nouveau derive de methotrexate - Google Patents
Nouveau derive de methotrexate Download PDFInfo
- Publication number
- WO1992003436A1 WO1992003436A1 PCT/JP1991/001078 JP9101078W WO9203436A1 WO 1992003436 A1 WO1992003436 A1 WO 1992003436A1 JP 9101078 W JP9101078 W JP 9101078W WO 9203436 A1 WO9203436 A1 WO 9203436A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- compound
- added
- solvent
- stirred
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
- C07D475/06—Heterocyclic compounds containing pteridine ring systems with a nitrogen atom directly attached in position 4
- C07D475/08—Heterocyclic compounds containing pteridine ring systems with a nitrogen atom directly attached in position 4 with a nitrogen atom directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
Definitions
- the present invention relates to a novel methotrexate derivative, and more particularly, to a novel methotrexate derivative useful as an antirheumatic agent, a therapeutic agent for psoriasis, and an anticancer agent.
- Methotrexet has long been used as a treatment for leukemia
- the present inventors have conducted intensive studies in search of a better compound in this type of methotrexate derivative, and have accomplished the present invention.
- the present invention has the following general formula (I)
- R 1 represents a member selected from the group consisting of CH 2 , CH 2 CH 2 , CH 20 , CH 2 .S and CH 2 SO;
- R 2 represents a hydrogen atom or a lower alkyl group having 1 to 4 carbon atoms or a benzyl group
- n an integer from 1 to 4.
- R 3 represents a general formula C00R 4 (where R 4 represents a hydrogen atom or a lower alkyl group having 1 to 4 carbon atoms) or a general formula NHC0R 5 (where R 5 represents a phenyl group which may be substituted) Or a general formula CONR 6 R 7 (where R 6 represents a hydrogen atom or a lower alkyl group having 1 to 4 carbon atoms; R 7 represents a lower alkyl group having 1 to 4 carbon atoms or a substituted or unsubstituted fluorine atom).
- Rumotoma other is a group carboxyalkyl represents an alkyl group or a lower alkyl sulfonyl Le group) or represented by P0 3 H 2, S0 3 H ;
- the compound of the present invention represented by the above general formula (I) has an excellent antirheumatic effect, a psoriasis therapeutic effect and an anticancer effect, and has the feature of having lower toxicity than conventional methotrexate. [Brief description of the drawings]
- 1 to 3 show the uptake (ratio) of 3 H—UdR at each concentration of the test drug.
- FIG. 4 shows the “rat keratinocyte proliferation inhibitory action”
- FIG. 5 shows the “human keratinocyte proliferation inhibitory action”.
- the absorbance at each concentration of the test drug is shown as a percentage when the absorbance without drug addition is 100%.
- FIG. 6 shows the “proliferation inhibitory effect of P388 cells”
- FIG. 7 shows the “growth inhibitory effect of colon26”.
- the absorbance at each concentration of the test drug is shown as a percentage when the absorbance when no drug is added is 100%.
- FIG. 8 shows changes in rat body weight when methotrexate and the compound of the present invention were administered
- FIG. 9 shows changes in white blood cell count (WBC) and red blood cell count (RBC)
- FIG. Shows the change in TG content in the liver.
- the compounds of the present invention are all novel compounds described at the end of the literature, and are synthesized, for example, as follows.
- R 3 is a general formula
- R ' a hydrogen atom or a lower alkyl group having 1 to 4 carbon atoms
- A represents an atom or a lower alkyl group having 1 to 4 carbon atoms
- a 1 and A 2 represent a protecting group
- X represents a halogen atom.
- the compound of the general formula (1) is suspended in an acid halogenating agent such as thionyl chloride, oxalyl chloride and the like.
- the reaction is carried out by stirring at room temperature in the coexistence of dimethylformamide and the like.
- examples of the protecting group represented by A 1 include a carbobenzoxy group, a tosyl group, an acetyl group and the like.
- the reaction for obtaining the compound of the general formula (4) from the compound of the general formula (2) and the compound of the general formula (3) is performed by dissolving the compound of the general formula (2) in a solvent such as dichloromethane, and cooling the mixture with ice.
- the reaction is carried out by stirring at room temperature in the presence of an inorganic base such as potassium carbonate, sodium hydroxide or sodium hydrogen carbonate in addition to an aqueous solution of the compound of the general formula (3) under water or under water cooling.
- the reaction for obtaining the compound of the general formula (5) from the compound of the general formula (4) is carried out by adding the compound of the general formula (4) to a solution of anisol difunol or the like in a hydrogen bromide monoacetic acid solution, C. to 60.degree. C., preferably by stirring at room temperature.
- the reaction for obtaining the compound of the general formula (5) from the compound of the general formula (4) is as follows:-The compound of the general formula (4) is dissolved in a solvent such as methanol, ethanol or acetic acid, and palladium-carbon is added. Thereafter, stirring may be performed at room temperature under a hydrogen atmosphere.
- the reaction for obtaining the compound of the general formula (7) from the compound of the general formula (6) and the compound of the general formula (5) is performed by converting the compound of the general formula (6) and the compound of the general formula (5) to dimethylacetamide and dimethyl In a solvent such as formamide, the reaction is carried out with stirring at 0 ° C to 100 ° C, preferably at 50 to 60 ° C. Especially when R 2 is a hydrogen atom, Then, a 1N aqueous solution of sodium hydroxide is added to a solvent such as methanol-ethanol, and the mixture is stirred at O ° C to 60 ° C, preferably 35 ° C, to obtain the desired product.
- examples of the halogen atom represented by X include a bromine atom and a chlorine atom.
- the compound of the general formula (6) and the compound of the general formula (8) are dimethyl It is carried out by stirring at 0 to 100 ° C, preferably at 55 ° C in a solvent such as acetoamide-dimethylformamide.
- the compound of the general formula (9) is obtained by converting the compound of the general formula (9) to cyanide getyl phosphate or dicyclohexylcarbodiimide.
- a solvent such as dimethylformamide, dimethylacetamide, N-methylpyrrolidinone and the like in the presence of 1-hydroxybenzotriazole and the like
- the compound of the general formula (3) is added, and the mixture is added at 0 ° C to 200 ° C. C., preferably by stirring at 10 to 80.degree.
- R 2 is a hydrogen atom
- a 1 N aqueous sodium hydroxide solution is further added in a solvent such as methanol or ethanol, and the mixture is stirred at O ° C to 60 ° C, preferably at room temperature, to obtain the desired product.
- the reaction of obtaining the compound of the general formula (12) from the compound of the formula (10) and the compound of the formula (11) by converting the compound of the general formula (10) into chloroform, dichloromethane, tetrahydrofuran This is carried out by dissolving in an aprotic solvent such as dioxane, adding the compound of the formula ⁇ ), water and, for example, potassium carbonate, triethylamine, sodium hydrogen carbonate, pyridine and the like, and stirring at room temperature.
- examples of the protecting group represented by A 2 include a carbobenzoxy group, a tosyl group, and an acetyl group.
- the reaction for obtaining the compound of the general formula as) from the compound of the general formula (12) is carried out in a solvent such as methanol at 160 ° C. to 120 ° C., preferably at 130 ° C., and the thionyl chloride is removed. After addition, the reaction is carried out by refluxing.
- the reaction for obtaining the compound of the general formula (14) from the compound of the general formula (13) is carried out by dissolving the compound of the general formula (13) in ethanol, methanol, tetrahydrofuran, dioxane, etc., and coexisting with palladium-carbon, in a hydrogen atmosphere This is done by stirring at room temperature.
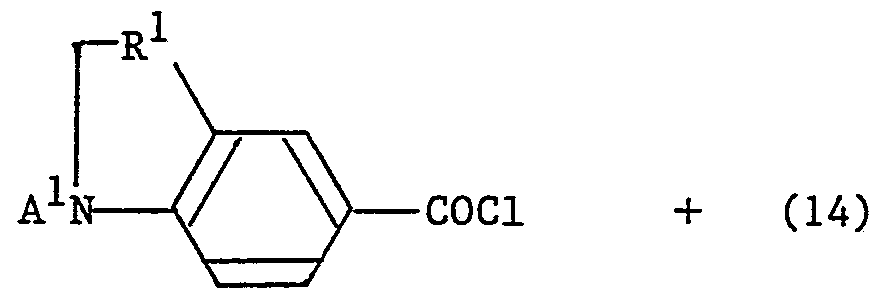
- the reaction for obtaining the compound of the general formula (15) from the compound of the general formula (14) and the compound of the general formula (2) is carried out by dissolving the compound of the general formula (2) in dichloromethane or the like.
- the compound (14) and potassium carbonate or triethylamine and water are added, and the mixture is stirred at room temperature.
- the amidation may be performed by a mixed acid anhydride method, an active ester or an active amide method.
- the reaction for obtaining the compound of the general formula (16) from the compound of the general formula (15) is carried out by adding a hydrogen bromide monoacid in which phenol or anisol is dissolved in the compound of the general formula (15) at room temperature. This is performed by stirring.
- the reaction for obtaining the compound of the general formula (17) from the compound of the general formula (6) and the compound of the general formula (16) is carried out in an aprotic polar solvent such as dimethylacetamide and dimethylformamide. after the 100 ° C preferably stirring at 50 e C ⁇ 65 ° C, carried out for example Toriechiruamin, by stirring in water with a carbonate force potassium or sodium hydrogen carbonate.
- an aprotic polar solvent such as dimethylacetamide and dimethylformamide.
- reaction for obtaining the compound of the general formula (I) from the compound of the general formula (17) is carried out by adding an aqueous solution of sodium hydroxide in a solvent such as ethanol and stirring at room temperature.
- the compound represented by the general formula (I) obtained by the present invention has an antirheumatic effect, a psoriasis therapeutic effect and an anticancer effect. It is also less toxic than methotrexate. This effect is shown in the following experimental example
- Lymphocytes were separated using a Ficoll-Paque R from human peripheral blood, moderately diluted drug and its lymphocyte 10 5 were cultured in PHA (0.3 zg / ml) together with 2 days in 96-well culture plate. Five hours before the end of the culture, 3 H—UdR ⁇ i / we 11) was added, and the uptake of 3 H—UdR into lymphocytes was measured with a scintillation counter. (Here, PHA stands for phytohaemagg lutinin and UdR stands for deoxyuridine.) The drugs used are as follows.
- Figures 1 to 3 show the ratios of 3H-UdE uptake of drug-free PHA-stimulated lymphocytes as 100%. As is clear from FIGS. 1 to 3, it was confirmed that the compound of the present invention had a superior lymphocyte proliferation inhibitory action (anti-rheumatic action) than the control compound.
- Rat keratinocytes which were primarily cultured using Swiss 3T3 cells as a feeder layer, were converted into TypelV using MCDB153 ZDMEZ 10% FCS medium containing growth factors (insulin, hydrocortisone, EGF, cholera toxin).
- FCS medium containing growth factors (insulin, hydrocortisone, EGF, cholera toxin).
- a 24-well plate treated with collagen was seeded at a concentration of 4 xlO 4 cells / mlZwell, and cultured at 5% CO 2 95% Air 37 ° C.
- Human keratinocytes purchased from Kurabo Industries, Ltd. were converted to thymidine-adenine-free MCDB 153 medium (0.15 ml Ca) containing growth factors (inulin, hydrocortisone, rhEGF, ethanolamine, and phosphoethanolamine). The cells were seeded at a concentration of 2 x 10 4 cells / mL well on a 24-well plate treated with Type eW collagen using 5% CO 2 95% Air 37. C.
- the medium was aspirated, and the dye (formazane) produced by the living cells was dissolved by adding 250 ⁇ 1 / well of DMSO (dimethyl sulfoxide).
- the absorbance (A550) of the dissolved dye was measured, and the average of 6 wells was calculated as a relative value of the number of cells.
- Figure 4 shows the results of the “rat keratinocyte proliferation inhibition experiment”
- Figure 5 shows the results of the “human keratinocyte proliferation inhibition experiment”. In each case, the absorbance when no drug was added was shown as a percentage with 100%.
- BPM11640 medium fetal calf serum 5%, 2-mercaptoethanol and 1 0 6 M and becomes as those added
- FIG. 6 Example using P388 cells, Figure 7 using co 10n26 An example in the case of the above is shown.
- the data show the percentage of cell growth, ie, the absorbance, in cells without drug added, and the extent to which growth was suppressed by the addition of drug in%.
- the compound of the present invention has a superior cancer cell growth inhibitory activity (anticancer activity) as compared with the control drug.
- MTX or a compound of the present invention was intraperitoneally administered to 8-week-old male SD rats once a day for 5 weeks at a rate of 5 times a week.
- the dose is 0.225, 0.5 mgZkg for MTX and the compound of the present invention in two doses.
- the control group received a solvent (phosphate buffer, pH 7.4) in the same manner.
- the number of animals per group is five.
- the inspection items are as follows.
- liver weight and triglyceride (TG) content in liver liver After final administration, dissection was performed to measure liver weight and liver TG content.
- the 0.25 mg Zkg group suffered short duration during the administration period, no deaths occurred, and no abnormal symptoms were observed.
- the 0.5 mg / kg group deterioration of nutritional status, anemia, decreased fecal volume, swelling around the mouth, etc. were observed from the 4th week, and 1 of 5 patients died.
- Figure 8 shows the weight change.
- the compound of the present invention was weaker in suppressing weight gain than MTX.
- Figure 9 shows the blood test results.
- the reduction in WBC and RBC was clearly weaker with the compounds of the present invention than with MTX.
- FIG. 10 shows the liver weight and the TG content in the liver.
- the compound of the present invention was weaker than MTX in increasing the weight of the liver and the TG content in the body.
- the foam layer was washed with a 1N hydrochloric acid solution and dried over sodium sulfate.
- Target C140nig The foam layer was washed with a 1N hydrochloric acid solution and dried over sodium sulfate.
- Target C140nig
- the compound of Reference Example 10 (330 mg) was added to a 30% solution of fuynol (300 mg) in 30% hydrogen bromide monoacetic acid (8 ml), and the mixture was stirred at room temperature for 4 hours. Next, a large amount of ether was added to the reaction solution, and a red-brown oily substance precipitated. After removing most of the ether layer, the oily substance was suspended in chloroform, and the suspension was washed with saturated aqueous sodium hydrogen carbonate solution and extracted with chloroform. The port-form layer was dried over sodium sulfate, and the solvent was distilled off under reduced pressure to obtain the desired product (147 mg). Was.
- reaction solution was poured into water and extracted with chloroform.
- the foam layer was washed with 1N-hydrochloric acid and dried over sodium sulfate.
- the solvent was distilled off under reduced pressure.
- the obtained residue was subjected to silica gel chromatography, and the desired product (310 mg) was obtained using chloroform: methanol 100: 3 as the elution solvent.
- the compound of Reference Example 20 (1.7 g) was dissolved in trifluoroacetic acid (8 ml) under ice cooling, and the mixture was stirred for 30 minutes. The solvent was distilled off under reduced pressure, and the residue was dissolved in chloroform. The black-mouthed form layer was washed with a saturated aqueous solution of sodium hydrogen carbonate and dried over sodium sulfate. The solvent was distilled off under reduced pressure to obtain the desired product (1.3 g).
- reaction solution was washed successively with 1 N hydrochloric acid, a saturated aqueous solution of sodium hydrogen carbonate and water, dried over sodium sulfate, and then the solvent was distilled off under reduced pressure.
- the resulting residue was subjected to silica gel chromatography, and the desired product (317 mg) was obtained using dichloromethane as the elution solvent and then using chloroform.
- the compound of Reference Example 23 (470 mg) was dissolved in dry methanol (20 ml), trimethylsilyldiazomethane (2 ml) was added, and the mixture was stirred for 10 hours. Trimethylsilyldiazomethane (3 ml) was further added, and the mixture was stirred for 20 hours. After evaporating the solvent under reduced pressure, the obtained residue was subjected to silica gel chromatography, and the desired product (252 mg) was purified by using chloroform as the elution solvent, and then using methanol with methanol: 199: 1. Obtained.
- Reference Example 29 10% paradigm carbon (l.lg) was added to a methanol solution (30 ml) of the compound of Example 9 (5.2 g), and the mixture was stirred at room temperature for 15 hours under a hydrogen atmosphere. Palladium-carbon was removed by filtration using celite, and the solvent was distilled off under reduced pressure. The obtained residue was dissolved in a saturated aqueous solution of sodium hydrogen carbonate, and washed with a black hole form. After the aqueous layer was separated, the pH was adjusted to 4 using 5% citric acid, and extracted with chloroform. After drying the form layer at the mouth, the solvent was distilled off under reduced pressure to obtain the desired product (3.9 g).
- reaction solution was washed sequentially with 2N hydrochloric acid, a saturated aqueous solution of sodium hydrogen carbonate and water, dried over sodium sulfate, and then the solvent was distilled off under reduced pressure.
- N-potassium L-benzoxyl L-glutamic acid To a solution of benzylester (508 mg) and triethylamine (0.19 ml) in tetrahydrofuran (2 ml) was added ethyl chloride chlorocarbonate (0.14 ml) at 20 ° C under a nitrogen atmosphere. 3 0 min ⁇ the c then, after addition of dimethyl hydrochloride and (112ing) Toriechiruamin the dichloro Rometan solution (0. 19ml) (3 ml) , and stirred for 1 hour. The temperature was gradually returned to room temperature, and the mixture was further stirred for 24 hours. The solvent was distilled off under reduced pressure, and the obtained residue was dissolved in chloroform.
- the compound of Example 19 (70 mg) was dissolved in methanol (5 ml), a 1N aqueous solution of sodium hydroxide (0.15 ml) was added, and the mixture was stirred at room temperature for 12 hours. Water (2 ml) was added, and the mixture was further stirred for 5 hours. The solvent was distilled off under reduced pressure while maintaining the temperature of the water bath at 30 ° C or lower. The obtained residue was dissolved in a saturated aqueous solution of sodium hydrogen carbonate, and the pH was adjusted to 3.7 with 1N hydrochloric acid. The deposited orange precipitate was collected by filtration to obtain the desired product (16 mg).
- Triethylamine (60 mg) and getyl phosphorocyanidate (98 mg) were suspended in anhydrous dimethylformamide (6 ml), and the compound of Reference Example 41 (60 mg) was further added and stirred. After dissolution, the reaction solution was stirred at 80 ° C. for 3 minutes, then returned to room temperature and stirred for 10 minutes. Next, triethylamine (20 mg) and glutamic acid getyl ester hydrochloride (40 mg) were added, and the mixture was stirred at 80 ° C for 2 hours. After the reaction, the solvent was distilled off under reduced pressure, and chloroform was added to the residue. The chloroform layer was washed with saturated aqueous sodium hydrogen carbonate.
- the compound of Reference Example 45 (6.0 g) was suspended in ethanol (300 ml), and the suspension was added with potassium titanate (5. Og) and refluxed for 90 minutes under a nitrogen stream. After cooling the reaction solution, the precipitate was collected by filtration and dried under vacuum to obtain the desired product (5.2 g).
- the tetrahydrofuran-borane complex (1 [ ⁇ ] solution, 10 ml) and the compound of Reference Example 46 (440 mg) were added to tetrahydrofuran (30 ml) at 0 ° C, and the mixture was stirred at room temperature for 15 minutes. Thereafter, the mixture was refluxed for 4 hours. The reaction solution was cooled to room temperature, and 6N hydrochloric acid (2.7 ml) was added. After the reaction solution was concentrated under reduced pressure, it was poured into water and made alkaline with a 2N aqueous solution of sodium hydroxide.
- the compound of Reference Example 48 (1.79 g) was suspended in ethanol (50 ml), and an IN-sodium hydroxide aqueous solution (8.2 ml) was added to the suspension, followed by stirring overnight. After evaporating the solvent under reduced pressure, the obtained residue was dissolved in water (20 ml). Next, 11 ⁇ 'monohydrochloric acid was slowly added to adjust the pH to 2, and the deposited precipitate was collected by filtration and dried under vacuum to obtain the desired product (1.39 g).
- Hydrogen chloride gas was passed into a methanol solution (20 ml) of 4-amino-n-butyric acid (l.Og) for 10 minutes, and the mixture was stirred at room temperature for 5 hours. The solvent was distilled off under reduced pressure to obtain the desired product (1.5 g).
- Triethylamine (8701) and chlorotrimethylsilane (6301) were added to a suspension of homocysteine hydrobromide (330 ml) in benzene (6 ml) under a nitrogen atmosphere, and the mixture was stirred at room temperature for 3 days. The precipitate was separated by filtration, and the filtrate was concentrated to obtain silylated homocystinic acid (450 mg). Under a nitrogen atmosphere, disoyl cynophosphonate (213 ⁇ 1) and triethylamine 721) were dissolved in dimethylformamide (18 ⁇ 1), and 1-[(2,4-diamino) -6-pteridinyl) methyl] indoline was dissolved at room temperature.
- the obtained mouth layer was washed with a saturated saline solution and dried over anhydrous sodium sulfate.
- the residue obtained by evaporating the solvent under reduced pressure was dissolved in methanol, hydrogen chloride gas was passed through the solution for 10 minutes, and the mixture was stirred at room temperature overnight.
- the obtained organic layer was washed with saturated saline and dried over anhydrous sodium sulfate.
- Example 32 The compound of Example 32 (10 mg) was suspended in water (1 ml) and dissolved by adding a 1 N aqueous sodium hydroxide solution. The mixture was cooled on ice, 0.5M sodium metaperiodate (50 ⁇ 1) was added, and the mixture was stirred at the same temperature for 5 hours. The pH was adjusted to 3.5 with 1N hydrochloric acid, and the precipitated precipitate was collected by filtration to obtain the desired product (4 mg).
- N, N'-carbonyldiimidazole (13.6 g) and [N- (t-butoxycarbonyl) glutamic acid] -monobenzyl ester (25 g) were dissolved in tetrahydrofuran (268 ml), and the mixture was stirred under ice cooling for 1 hour. Then, the solution was added dropwise to a solution of methanesulfonamide (20.5 g) and 1,8-diazabicyclo [5,4,0] -7-indene (32.9 g) in tetrahydrofuran (132 ml) under ice cooling. After the dropwise addition, the mixture was returned to room temperature and stirred for 4 days.
- Example 34 The compound of Example 34 (25 mg) was suspended in ethanol (5 ml), 1N aqueous sodium monohydroxide solution (2001) was added, and the mixture was stirred at room temperature overnight. Then, water (0.5 ml) was added to the reaction solution, and ethanol was distilled off under reduced pressure. The obtained residue was dissolved in water (6 ml), the pH was adjusted to 3.7 with 1N hydrochloric acid under ice-water cooling, and the mixture was allowed to stand overnight in a cool place. The deposited precipitate was collected by filtration to obtain the desired product (20 mg).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description




























Claims
















Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES91914615T ES2141710T3 (es) | 1990-08-14 | 1991-08-14 | Derivado de metotrexato con una actividad antirreumatica. |
| US07/971,773 US5354753A (en) | 1990-08-14 | 1991-08-14 | Methotrexate derivative |
| EP91914615A EP0543997B1 (en) | 1990-08-14 | 1991-08-14 | Derivatives of methotrexate with antirheumatic activity |
| DE69131835T DE69131835T2 (de) | 1990-08-14 | 1991-08-14 | Methotrexatderivate mit antirheumatischer aktivität |
| HU9300264A HU223948B1 (hu) | 1990-08-14 | 1991-08-14 | Metotrexát-származékok, ilyeneket tartalmazó gyógyszerkészítmények és eljárás előállításukra |
| DK91914615T DK0543997T3 (da) | 1990-08-14 | 1991-08-14 | Methotrexatderivat med antirheumatisk aktivitet |
| RU93005141A RU2109016C1 (ru) | 1990-08-14 | 1991-08-14 | Производные метотрексата, способы их получения и фармацевтическая композиция |
| CA002088665A CA2088665C (en) | 1990-08-14 | 1991-08-14 | Methotrexate derivative |
| GR20000400402T GR3032704T3 (en) | 1990-08-14 | 2000-02-18 | Novel methotrexate derivative. |
Applications Claiming Priority (22)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2/214691 | 1990-08-14 | ||
| JP21469190 | 1990-08-14 | ||
| JP2/215639 | 1990-08-15 | ||
| JP21563990 | 1990-08-15 | ||
| JP2/253466 | 1990-09-21 | ||
| JP25346690 | 1990-09-21 | ||
| JP29310790 | 1990-10-30 | ||
| JP2/293107 | 1990-10-30 | ||
| JP33184590 | 1990-11-29 | ||
| JP2/331845 | 1990-11-29 | ||
| JP18062691 | 1991-04-19 | ||
| JP3/180626 | 1991-04-19 | ||
| JP18594391 | 1991-04-23 | ||
| JP3/185943 | 1991-04-23 | ||
| JP3228158A JPH04352785A (ja) | 1991-05-30 | 1991-05-30 | 新規メトトレキセート誘導体 |
| JP3/228158 | 1991-05-30 | ||
| JP3/247141 | 1991-06-12 | ||
| JP3247141A JPH04368385A (ja) | 1991-06-12 | 1991-06-12 | 新規メトトレキセート誘導体 |
| JP3/258301 | 1991-07-03 | ||
| JP25830191 | 1991-07-03 | ||
| JP3/279047 | 1991-07-30 | ||
| JP27904791 | 1991-07-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1992003436A1 true WO1992003436A1 (fr) | 1992-03-05 |
Family
ID=27582227
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1991/001078 Ceased WO1992003436A1 (fr) | 1990-08-14 | 1991-08-14 | Nouveau derive de methotrexate |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US5354753A (ja) |
| EP (1) | EP0543997B1 (ja) |
| CN (1) | CN1032858C (ja) |
| AT (1) | ATE187455T1 (ja) |
| AU (1) | AU8333291A (ja) |
| CA (1) | CA2088665C (ja) |
| DE (1) | DE69131835T2 (ja) |
| DK (1) | DK0543997T3 (ja) |
| ES (1) | ES2141710T3 (ja) |
| GR (1) | GR3032704T3 (ja) |
| HU (2) | HU223948B1 (ja) |
| SG (1) | SG49917A1 (ja) |
| WO (1) | WO1992003436A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996030019A1 (en) * | 1995-03-27 | 1996-10-03 | Chugai Seiyaku Kabushiki Kaisha | Drug containing methotrexate derivative |
| US5728692A (en) * | 1992-12-25 | 1998-03-17 | Chugai Seiyaku Kabushiki Kaisha | Methotrexate derivative |
| WO2001087834A1 (en) * | 2000-05-16 | 2001-11-22 | Takeda Chemical Industries, Ltd. | Melanin-concentrating hormone antagonist |
| US6559149B1 (en) * | 1992-01-27 | 2003-05-06 | Chugai Seiyaku Kabushiki Kaisha | Methotrexate derivatives |
| US9000208B2 (en) | 2010-03-24 | 2015-04-07 | Ajinomoto Co., Inc. | Glutamate derivatives or salts thereof |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0897724A4 (en) * | 1996-03-19 | 2003-05-07 | Chugai Pharmaceutical Co Ltd | MEDICINE AGAINST UVEITY |
| AU2001239549A1 (en) * | 2000-03-24 | 2001-10-03 | Akzo Nobel N.V. | Keratinocyte growth inhibitors and hydroxamic acid derivatives |
| US7998967B2 (en) | 2008-03-03 | 2011-08-16 | Tosk, Incorporated | Methotrexate adjuvants to reduce toxicity and methods for using the same |
| WO2009137081A2 (en) * | 2008-05-07 | 2009-11-12 | Massachusetts Institute Of Technology | Small molecule inhibitors of plasmodium falciparum dihydroorotate dehydrogenase |
-
1991
- 1991-08-14 HU HU9300264A patent/HU223948B1/hu not_active IP Right Cessation
- 1991-08-14 CN CN91105803A patent/CN1032858C/zh not_active Expired - Fee Related
- 1991-08-14 SG SG1996009165A patent/SG49917A1/en unknown
- 1991-08-14 US US07/971,773 patent/US5354753A/en not_active Expired - Fee Related
- 1991-08-14 DE DE69131835T patent/DE69131835T2/de not_active Expired - Fee Related
- 1991-08-14 WO PCT/JP1991/001078 patent/WO1992003436A1/ja not_active Ceased
- 1991-08-14 DK DK91914615T patent/DK0543997T3/da active
- 1991-08-14 AT AT91914615T patent/ATE187455T1/de not_active IP Right Cessation
- 1991-08-14 AU AU83332/91A patent/AU8333291A/en not_active Abandoned
- 1991-08-14 EP EP91914615A patent/EP0543997B1/en not_active Expired - Lifetime
- 1991-08-14 CA CA002088665A patent/CA2088665C/en not_active Expired - Fee Related
- 1991-08-14 ES ES91914615T patent/ES2141710T3/es not_active Expired - Lifetime
-
1995
- 1995-06-29 HU HU95P/P00529P patent/HU211481A9/hu unknown
-
2000
- 2000-02-18 GR GR20000400402T patent/GR3032704T3/el not_active IP Right Cessation
Non-Patent Citations (1)
| Title |
|---|
| J. Med. Chem., Vol. 21, No. 4, April 1978, D.F. WORTH et al. "Folate Antagonists. 10. Synthesis and Antimalarial Effects of 6-(((Aryl and aralyl)amino)methyl)-2,4-pteridinediamines and -pteridin-ediamine 8-Oxides", pages 331-337. * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6559149B1 (en) * | 1992-01-27 | 2003-05-06 | Chugai Seiyaku Kabushiki Kaisha | Methotrexate derivatives |
| US5728692A (en) * | 1992-12-25 | 1998-03-17 | Chugai Seiyaku Kabushiki Kaisha | Methotrexate derivative |
| WO1996030019A1 (en) * | 1995-03-27 | 1996-10-03 | Chugai Seiyaku Kabushiki Kaisha | Drug containing methotrexate derivative |
| WO2001087834A1 (en) * | 2000-05-16 | 2001-11-22 | Takeda Chemical Industries, Ltd. | Melanin-concentrating hormone antagonist |
| US7229986B2 (en) | 2000-05-16 | 2007-06-12 | Takeda Pharmaceutical Company Ltd. | Melanin-concentrating hormone antagonist |
| US9000208B2 (en) | 2010-03-24 | 2015-04-07 | Ajinomoto Co., Inc. | Glutamate derivatives or salts thereof |
| JP5796570B2 (ja) * | 2010-03-24 | 2015-10-21 | 味の素株式会社 | グルタミン酸エステル誘導体又はその塩 |
Also Published As
| Publication number | Publication date |
|---|---|
| HU211481A9 (en) | 1995-11-28 |
| ATE187455T1 (de) | 1999-12-15 |
| HUT64974A (en) | 1994-03-28 |
| ES2141710T3 (es) | 2000-04-01 |
| EP0543997B1 (en) | 1999-12-08 |
| DE69131835D1 (de) | 2000-01-13 |
| HU9300264D0 (en) | 1993-04-28 |
| SG49917A1 (en) | 1998-06-15 |
| CN1059725A (zh) | 1992-03-25 |
| GR3032704T3 (en) | 2000-06-30 |
| DE69131835T2 (de) | 2000-06-15 |
| AU8333291A (en) | 1992-03-17 |
| HU223948B1 (hu) | 2005-03-29 |
| CA2088665C (en) | 2001-05-01 |
| US5354753A (en) | 1994-10-11 |
| CA2088665A1 (en) | 1992-02-15 |
| DK0543997T3 (da) | 2000-04-10 |
| EP0543997A4 (en) | 1993-12-29 |
| EP0543997A1 (en) | 1993-06-02 |
| CN1032858C (zh) | 1996-09-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5874441A (en) | Carbocyclic and hetertocyclic fused-ring quinolinecarboxylic acids useful as immunosuppressive agents | |
| JP2623800B2 (ja) | チエノジアゼピン化合物 | |
| WO2001014378A1 (en) | PYRROLOTRIAZINE DERIVATIVES HAVING sPLA2-INHIBITORY ACTIVITIES | |
| JP2002513013A (ja) | グアニジン誘導体 | |
| JPWO1999059999A1 (ja) | sPLA2阻害作用を有するピロロ[1,2−b]ピリダジン誘導体 | |
| CN109678815B (zh) | N-苄基苯甲酰胺类衍生物及其制备方法与制药用途 | |
| WO1992003436A1 (fr) | Nouveau derive de methotrexate | |
| AU755654B2 (en) | Novel amide compounds and drugs containing the same | |
| WO2019085441A1 (zh) | 一种免疫细胞迁徙抑制剂 | |
| KR0140886B1 (ko) | 세팔로스포린 유도체 및 이의 제법 | |
| CN101910128A (zh) | 新的二醇二氮烯*化合物、其制备方法以及含有它们的药用组合物 | |
| JPH03505205A (ja) | 縮合ピラゾル3―オキソ―プロパンニトリル誘導体及びその製造方法 | |
| JPH03505204A (ja) | 3環式3―オキソ―プロパンニトリル誘導体およびその調製方法 | |
| WO1999054303A1 (en) | Optically active tetrahydrobenzindole derivatives | |
| JP2959598B2 (ja) | 光学活性なチエノトリアゾロジアゼピン化合物 | |
| JPS62138494A (ja) | イミダゾピリダジンアルケン酸アミド類 | |
| WO1998023606A1 (en) | Cyclic dithio derivatives, remedies for diabetic kidney diseases, hypoglycemic agents, hypolipidemic agents, and lenitives for digestive disorders | |
| JP3281099B2 (ja) | 抗リウマチ剤 | |
| WO1994017075A1 (en) | Diazepin derivatives and antiviral compositions | |
| KR100192023B1 (ko) | 신규 메토트렉세이트 유도체 | |
| CN103086958A (zh) | NFκB通路激活抑制剂、其制备方法、药物组合物及用途 | |
| WO2022122044A1 (zh) | 作为gls1抑制剂的杂环化合物 | |
| JPH10503510A (ja) | ストレプトグラミン誘導体、その製造およびそれを含む製薬組成物 | |
| JP3124592B2 (ja) | 新規メトトレキセート誘導体 | |
| RU2109016C1 (ru) | Производные метотрексата, способы их получения и фармацевтическая композиция |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AT AU BB BG BR CA CH DE DK ES FI GB HU KR LK LU MC MG MW NL NO PL RO SD SE SU US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE BF BJ CF CG CH CI CM DE DK ES FR GA GB GN GR IT LU ML MR NL SE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2088665 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1991914615 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1991914615 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1991914615 Country of ref document: EP |