WO1994000480A1 - Steroidal glycosides for treating hypercholesterolemia - Google Patents
Steroidal glycosides for treating hypercholesterolemia Download PDFInfo
- Publication number
- WO1994000480A1 WO1994000480A1 PCT/US1993/004092 US9304092W WO9400480A1 WO 1994000480 A1 WO1994000480 A1 WO 1994000480A1 US 9304092 W US9304092 W US 9304092W WO 9400480 A1 WO9400480 A1 WO 9400480A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cellobiosyl
- carbonyl
- methylene
- compound
- oxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J71/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton is condensed with a heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J71/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton is condensed with a heterocyclic ring
- C07J71/0005—Oxygen-containing hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/58—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids containing heterocyclic rings, e.g. danazol, stanozolol, pancuronium or digitogenin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- This invention relates to steroidal glycosides and methods of using the same, particularly as hypocholesterolemic agents and antiatherosclerosis agents, in mammals.
- cross-linked synthetic polymer derivatives for example of polystyrene.
- cross-linked, water-insoluble, bile-acid-binding polystyrene-based resins e.g., Cholestyramine ® agents
- these resin beads typically have a low in vivo efficiency.
- the effective hypocholesterolemic dose of these materials is excessive, typically 18-24 grams of formulated product per day.
- hypocholesterolemic activity include the natural product chitosan and chitosan derivatives as described in European Application pub. no. 0212145. However, the effective hypocholesterolemic dose of these materials is also high.
- hypercholesterolemia controlling agents include plant extracts such as "alfalfa saponins".
- plant extracts are of variable composition and contain significant amounts of nonuseful chemical substances. Due to the variations in composition, it is difficult to set a standard dosage or predict the impurities present. Thus, such extracts are not well suited for use by humans.
- pure "sapogenin-derived” compounds e.g., substances compounded from spirostane, spirostene or sterol-derived compounds depress cholesterol absorption more effectively than alfalfa extracts on a weight basis and thus can be administered in reasonable sized doses. Because the chemical compositions of these substances are known and because they can be synthesized at a high degree of purity, they are suitable for use by any warmblooded animal, including humans. However, unless administered in massive amounts, pure sapogenins do not significantly inhibit cholesterol's absorption. It is only when compounded with another moiety that sapogenins have the desired effect.
- sapogenin compounds examples are compounds of tigogenin and diosgenin, particularly glycosides thereof.
- P.K. Kintia, lu. K. Whynko, G.M. Gorianu, V.A. Bobeiko, I.V. Suetina, ⁇ .E. Mashchenko, Kim. Pharm. Zh., 1981 , 15(9), 55 discloses 3-O-(ß-D-galactopyranosyl)hecogenin and its use as a hypocholesterolemic agent.
- 4,602,003 and 4,602,005 disclose certain steroidal glycosides, in particular 3-O-(ß-D-glucopyranosy ⁇ )tigogenin and 3-O-(ß-D-cellobiosyl)tigogenin and their use for the control of hypercholesterolemia.
- 3-O-(ß-D-cellobiosyl)tigogenin has superior hypocholesterolemic activity when compared to, for example, cholestyramine.
- hypocholesterolemic compounds described above make a significant contribution to the art there is a continuing search in this field of art for improved hypocholesterolemic pharmaceuticals.
- This invention is directed to steroidal glycosides, particularly spirostanyl glycosides, that are useful as hypocholesterolemic agents and antiatherosclerosis agents.
- the compounds of this invention have the formula
- Q 1 is carbonyl, or ;
- Q 2 is carbonyl, methylene, or ;
- Q 1 , Q 4 and Q 5 are all methylene
- Q 2 is or ;
- Q 3 is , ,
- Q 2 is carbonyl
- Q 3 is , ,
- Q 1 , Q 2 , Q 4 and Q 5 are each methylene
- Q 1 , Q 2 , and Q 5 are each methylene
- Q 4 is carbonyl or ;
- C 5 is alpha
- Q 1 , Q 2 , and Q 4 are each methylene; Q 5 is carbonyl or ;
- C 5 is alpha
- a first group of preferred compounds of Formula IA consists of these compounds wherein Q 1 is carbonyl, or , Q 2 is methylene, Q 3 is , Q 4 is methylene, Q 5 is methylene, the C 5 hydrogen is alpha and C 25 has
- R 1 is ß-D-cellobiosyl, ⁇ -D-cellobiosyl, ß-D-glucopyranosyl, ß-D- galactopyranosyl, ß-D-lactosyl, ß-D-maltosyl or ß-D-maltotriosyl.
- Q 1 is carbonyl and R 1 is ß-D-cellobiosyl, ⁇ -D-cellobiosyl, ß-D-glucopyranosyl, ß-D- galactopyranosyl, ß-D-lactosyl, ß-D-maltosyl or ß-D-maltotriosyl.
- Q 1 is and R 1 is ß-D- cellobiosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is and R 1 is ß-D-cellobiosyl.
- a second group of preferred compounds of Formula IA are compounds wherein Q 1 is methylene, Q 2 is or , Q 3 is , Q 4 is methylene, Q 5 is methylene, the C 5 hydrogen is alpha and C 25 is (R).
- Q 1 is methylene
- Q 2 is or
- Q 3 is a compound wherein Q 4 is methylene
- Q 5 is methylene
- the C 5 hydrogen is alpha
- C 25 is (R).
- Especially preferred within this second group is a compound wherein Q 2 is and R 1 is ß-D-cellobiosyl.
- a third group of preferred compounds of Formula IA are compounds wherein
- Q 1 is carbonyl, or , Q 2 is carbonyl, or , Q 3 is
- Q 4 is methylene
- Q 5 is methylene
- the C 5 hydrogen is alpha
- C 25 is (R).
- Especially preferred within this group is a compound wherein Q 1 is carbonyl, Q 2 is carbonyl and R 1 is ß-D-cellobiosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is carbonyl, Q 2 is and R 1 is ß-D- cellobiosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is carbonyl, Q 2 is and R 1 is ß-D-lactosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is , Q 2 is carbonyl and R 1 is ß-D-cellobiosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is , Q 2 is carbonyl and R 1 is ß-D-cellobiosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is , Q 2 is carbonyl and R 1 is ß-D-cellobiosyl.
- R 1 is ß-D-cellobiosyl.
- a fourth group of preferred compounds of Formula IA are compounds wherein Q 1 is methylene, Q 2 is carbonyl, Q 3 is , Q 4 is methylene, Q 5 is methylene, the C 5 hydrogen is alpha and C 25 is (R).
- R 1 is ß-D-lactosyl or ß-D-cellobiosyl.
- a fifth group of preferred compounds of Formula IA are compounds wherein
- Q 1 and Q 2 are each methylene
- Q 3 is
- Q 4 and Q 5 are each methylene
- C 25 is (R).
- Especially preferred within this fifth group is a compound wherein the C 5 hydrogen is alpha and R 1 is ß-D-gentiobiosyl.
- Another especially preferred compound within this group is a compound wherein the C 5 hydrogen is beta and R 1 is ß-D-cellobiosyl.
- a sixth group of preferred compounds of Formula IA are compounds wherein Q 1 , Q 2 and Q 5 are each methylene, Q 3 is , Q 4 is carbonyl, the C 5 hydrogen is alpha and C 25 is (R). Especially preferred within this group is a compound wherein R 1 is ß-D-cellobiosyl.
- a seventh group of preferred compounds of Formula IA are compounds wherein Q 1 , Q 2 and Q 4 are each methylene, Q 3 is , Q 5 is carbonyl, the C 5 hydrogen is alpha and C 25 is (R). Especially preferred within this group is a compound wherein R 1 is ß-D-cellobiosyl.
- Yet another aspect of this invention is directed to a method for controlling hypercholesterolemia or atherosclerosis in a mammal by administering to a mammal suffering from hypercholesterolemia or atherosclerosis a hypercholesterolemia or atherosclerosis controlling amount of a Formula I spirostanyl glycoside
- Q 1 is methylene, carbonyl, or ;
- Q 2 is methylene, carbonyl, or ;
- Q 3 is , or ;
- ß-D-gentiobiosyl 3-O-ß-D-galactopyranosyl- ⁇ -D-arabanopyranosyl, or ß-maltotriosyl;
- Q 1 , Q 2 , and Q 5 are each methylene
- Q 4 is carbonyl or ;
- C 5 is alpha
- Q 1 , Q 2 and Q 4 are each methylene; Q 3 is or ;
- Q 5 is carbonyl or ;
- C 5 is alpha
- a first group of preferred compounds of Formula I are compounds wherein Q 1 , Q 2 , Q 4 and Q ⁇ are methylene, C 25 is (R) and Q 3 is .
- C 5 hydrogen is alpha and R 1 is ß-D-glucopyranuronosyl, ß-D-maltosyl, ß-D-lactosyl, ß-D-gentiobiosyl or ß-D-galactopyranosyl.
- R 1 is ß-D-glucopyranuronosyl
- ß-D-maltosyl ß-D-lactosyl
- ß-D-gentiobiosyl ß-D-galactopyranosyl
- Another especially preferred compound within this group is a compound wherein the C 5 hydrogen is beta and R 1 is ß-D-cellobiosyl.
- a second group of preferred compounds of Formula I are compounds wherein Q 1 is carbonyl, or , Q 2 is methylene, Q 3 is , Q 4 and Q 5
- Especially preferred within this second group are compounds wherein Q 1 is carbonyl and R 1 is ß-D-cellobiosyl, ⁇ -D-cellobiosyl, ß-D-glucopyranosyl, ß-D-galactopyranosyl, ß-D-lactosyl, ß-D-maltosyl or ß-D-maltotriosyl.
- Another especially preferred compound within this second group is a compound wherein Q 1 is and R 1 is ß-D-cellobiosyl.
- Another especially preferred compound within this second group is a compound wherein Q 1 is and R 1 is ß-D-cellobiosyl.
- a third group of preferred compounds of Formula I are compounds wherein Q 1 is methylene, Q 2 is carbonyl or , Q 3 is , Q 4 and Q 5 are
- each methylene, C 25 is (R) and the C 5 hydrogen is alpha.
- Especially preferred within this third group are compounds wherein Q 2 is carbonyl and R 1 is ß-D-cellobiosyl or ß-D-lactosyl.
- Other especially preferred compounds within this third group are compounds wherein Q 2 is and R 1 is ß-D-cellobiosyl or ß-D-galactopyranosyl.
- a fourth group of preferred compounds of Formula I are compounds wherein Q 1 is carbonyl, or , Q 2 is carbonyl, or , Q 3 is
- Q 4 and Q 5 are each methylene, the C 5 hydrogen is alpha and C 25 is (R).
- Especially preferred within this fourth group is a compound wherein Q 1 is carbonyl, Q 2 is carbonyl and R 1 is ß-D-cellobiosyl.
- Especially preferred within this fourth group are compounds wherein Q 1 is carbonyl, Q 2 is and R 1 is ß-D-cellobiosyl or ß-D- lactosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is , Q 2 is carbonyl, and R 1 is ß-D-cellobiosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is , Q 2 is carbonyl, and R 1 is ß-D-cellobiosyl.
- Another especially preferred compound within this group is a compound wherein Q 1 is , Q 2 is carbonyl, and R 1 is ß-D-cellobiosyl.
- preferred compound within this group is a compound wherein Q 1 is , Q 2 is
- R 1 is ß-D-cellobiosyl.
- a fifth group of preferred compounds of Formula I are compounds wherein Q 1 , Q 2 and Q 5 are each methylene, Q 3 is , Q 4 is carbonyl, the C 5 hydrogen is alpha and C 25 is (R). Especially preferred within this group is a compound wherein R 1 is ß-D-cellobiosyl.
- a sixth group of preferred compounds of Formula I are compounds wherein Q 1 , Q 2 and Q 4 are each methylene, Q 3 is , Q 5 is carbonyl, the C 5 hydrogen is
- alpha and C 25 is (R).
- R 1 is ß-D-cellobiosyl.
- compositions for the control of hypercholesterolemia or atherosclerosis in mammals which comprise a
- Yet another aspect of this invention is directed to a composition comprising a hydrate of a compound of the Formula 1 A.
- the Formula IA compounds are a subset of the Formula I compounds.
- reaction Schemes I, II & III describe how to make the Formula I compounds wherein Q 4 and Q 5 are both methylene.
- the desired Formula I compounds wherein Q 1 , Q 2 and Q 3 are as defined above may be prepared by deacetylating the appropriate alpha peracetylated Formula III compound or beta peracetylated
- the deacetylation is accomplished by combining the Formula III or IV compound with a nucleophilic base such as sodium methoxide or potassium cyanide in a solvent such as methanol, tetrahydrofuran, n-propanol or mixtures thereof at elevated temperatures of about 40° C to about 100°C (typically at reflux) and pressures of 0.5 psi to about 50 psi (typically ambient) for about 0.25 hour to about 2 hours.
- a nucleophilic base such as sodium methoxide or potassium cyanide
- a solvent such as methanol, tetrahydrofuran, n-propanol or mixtures thereof
- pressures 0.5 psi to about 50 psi (typically ambient) for about 0.25 hour to about 2 hours.
- Formula I compounds when the sugar is
- the resultant deacetylated compound is further hydrolyzed by, for example, exposure to sodium hydroxide.
- those compounds wherein either Q 1 or Q 2 are carbonyl may be reduced to yield the corresponding alcohols in an alternative process to performing the reduction prior to coupling (described in Reaction Scheme IV and the accompanying text).
- those compounds wherein either Q 1 or Q 2 are hydroxy may be oxidized to yield the corresponding carbonyl in an alternative process to performing the oxidation prior to coupling.
- the desired Formula III compound wherein Q 1 and Q 2 are as defined above may be prepared by anomerizing the appropriate Formula IV compound wherein Q 1 and Q 2 are as defined above.
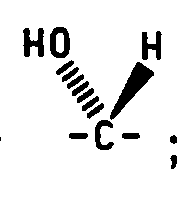
- the stereochemical terms alpha and beta refer to the configuration of the attachment carbon of the sugar.
- the anomerization is performed by treatment with a mineral acid such as hydrobromic acid in an anhydrous aprotic solvent such as methylene chloride at temperatures of 20° C to about 40° C (typically ambient) for at least 24 hours, typically to several days.
- a mineral acid such as hydrobromic acid
- an anhydrous aprotic solvent such as methylene chloride
- the alpha anomer is obtained directly from the saccharide-steroid coupling described below and the beta anomer from the above process (i.e., the nomenclature reverses).
- the desired Formula IV compounds wherein Q 1 and Q 2 are as defined above may be prepared by coupling the appropriate acetylated sugar halide (e.g., bromide) and steroid. More specifically, for those Formula IV compounds where the sugar is other than ß-D-maltosyl, ß-D-gentiobiosyl or ß-D-2-acetamido-2-deoxy-glucopyranosyl, a zinc fluoride promoted coupling of the appropriate Formula V compound (wherein Q 1 and Q 2 are as defined above and X is either a bond or alkylene-O-) and peracetylated sugar halide is used and for those Formula IV compounds where the sugar is ß-D-maltosyl, ß-D-gentiobiosyl or ß-D-2-acetamido-2-deoxy-glucopyranosyl, a mercuric bromide and mercuric cyanide promoted coupling of the appropriate Formula V compound (wherein
- the zinc fluoride promoted coupling of the Formula V compound and the peracetylated sugar bromide occurs in a non-protic, anhydrous reaction-inert solvent (e.g., acetonitrile) at a temperature of about 20° C to about 100°C for about 0.5 to about 12 hours.
- a non-protic, anhydrous reaction-inert solvent e.g., acetonitrile
- zinc fluoride is used and about 0.5 to about 3 equivalents acetylated sugar bromide is used.
- the coupling is acid catalyzed and it is especially preferred that hydrohalic acid generated during the reaction is used as the acid catalyst.
- the desired compounds may be prepared at pressures of 0.5 to 50 psi, although typically ambient pressures are used.
- the glycosides may be precipitated from the crude filtered reaction mixture (e.g., acetonitrile product solution) by the addition of about 25% to 75% water and the remainder alcohol (e.g., methanol).
- the crude filtered reaction mixture e.g., acetonitrile product solution
- the remainder alcohol e.g., methanol
- the mercuric bromide and mercuric cyanide promoted coupling of the Formula VI compound and the acetylated sugar bromide is performed in an aprotic, anhydrous solvent such as methylene chloride at a temperature of about 20°C to about 100°C for about 0.5 to about 6 hours.
- aprotic, anhydrous solvent such as methylene chloride
- about 0.5 to about 4 equivalents (based on Formula IV compound) mercuric bromide and mercuric cyanide is used and about 0.5 to about 3 equivalents peracetylated sugar bromide (e.g., ß-D-maltosyl, ß-D-gentiobiosyl or ß-D-2-acetamido-2-deoxy-glucopyranosyl) is used.
- the desired compounds may be prepared at pressures of 0.5 to 50 psi, although typically ambient pressures are used. Preferably they are isolated as described for the zinc fluoride promoted coupling of
- the desired Formula VI compounds wherein Q 1 and Q 2 are as defined above and X is either a bond or alkylene-O- may be prepared by silylating the appropriate Formula V compound wherein Q 1 and Q 2 are as defined above and X is either a bond or alkylene-O-.
- a base such as triethylamine and an activated trialkylsilyl compound (e.g., trimethylsilyl trifluoromethane sulfonate or trimethylsilyl chloride) are reacted in an aprotic, anhydrous solvent such as methylene chloride at a temperature less than about 10°C for about 0.5 hour to about 2 hours.
- an activated trialkylsilyl compound e.g., trimethylsilyl trifluoromethane sulfonate or trimethylsilyl chloride
- the reduction is performed by reaction of the Formula VII compound with lithium aluminum hydride in an anhydrous solvent such as tetrahydrofuran at temperatures of less than about 10°C for about 0.5 hour to about 3 hours.
- anhydrous solvent such as tetrahydrofuran
- the desired Formula VII compounds wherein Q 1 and Q 2 are as defined above may be prepared by coupling the appropriate Formula VIII compound where Q 1 and Q 2 are as defined above with ethyl diazoacetate in the presence of rhodium acetate dimer.
- the Formula VIII compound and ethyl diazoacetate are reacted in an aprotic solvent such as methylene chloride in the presence of rhodium acetate dimer at ambient temperature for about 0.5 hour to about 3 hours.
- Q 1 is methylene and Q 2 and the stereochemistry of the C 5 hydrogen and C 25 carbon are as defined below) are described in Table I.
- Formula VIII compounds wherein Q 1 is methylene, Q 2 is either methylene or carbonyl and the C 3 hydroxy is beta may be converted to the corresponding Formula VIII compounds where the C 3 hydroxy is alpha by the following two procedures. These preparative methods may be used independent of the C 25 stereochemistry.
- the carbonyl is protected as a ketal (e.g., ethylene ketal), by reacting the steroid with ethylene glycol and an acid catalyst according to the procedure of Engel and Rakhit, Can. J. Chem, 40, 2153, 1962.
- the C 5 hydrogen is alpha
- the C 3 hydroxy group is oxidized to the ketone using pyridinium chloro chromate (PCC) in methylene chloride at ambient conditions.
- PCC pyridinium chloro chromate
- the C 3 ketone is reduced with a sterically hindered reducing agent such as K-Selectride ® reducing agent, at low temperature in tetrahydrofuran to give the C 3 alpha alcohol according to Gondos and Orr, J. Chem. Soc. Chem. Commun. 21, 1239, 1982.
- the Q 2 protecting group is removed with acid, such as hydrochloric acid, in an appropriate solvent such as acetone.
- Reaction Scheme IV illustrates the reaction pathways to achieve the Formula VIII compounds wherein Q 1 and Q 2 are defined above starting from the Formula VIII compound wherein Q 1 is methylene and Q 2 is carbonyl.
- Reaction Scheme IV Method 1
- the starting material is acetylated and bromininated according to the procedure described in J. Chem. Soc. 1956, 4344.
- This intermediate is then reduced with lithium aluminum hydride and treated with silver oxide by a procedure similar to that described in Helv. Act. Chim.. 1953, 36, 1241.
- the resulting ß-11 ,12-epoxide is opened with trichloroacetic acid, saponified and reduced with zinc and acetic acid using the procedure described in J. Chem. Soc. 1956, 4330 to give the product shown for method 1.
- the starting material is selectively acetylated using the procedure described in J. Chem. Soc., 1956, 430.
- the resulting product is oxidized with chromium trioxide and pyridine.
- the resulting product is saponified with potassium cyanide in water, methanol and THF to give the product shown for method 2.
- the starting material is converted to the corresponding toluenesulfonylhydrazone which is in turn treated with sodium methoxide using a procedure similar to that described in J. Am. Chem. Soc. 1954, 76, 4013.
- the resulting 11-ene product is oxidized with osmium tetroxide and N-methylmorpholine-N-oxide according to the procedure describe in Tetrahedron Letters, 1976, 1973 to give the product shown for method 3.
- the starting material is reduced with lithium and ammonia according to the procedure described in J. Am. Chem. Soc. 1953, 75, 1282 to give the product shown.
- the starting material is acetylated according to the procedure described in J. Am. Chem. Soc. 1955, 77, 1632 to give a mixture of acetates from which the 3,11 -diacetate can be isolated.
- the unprotected 12-alcohol is then oxidized with chromium trioxide and pyridine according to the procedure described in Org. Syn., 1976, 55, 84. Saponification of the acetates gives the product shown for method 9.
- the starting material is diacetylated using the procedure described in J. Chem. Soc. 1956, 4330.
- the diacetate is reduced with calcium and ammonia using the procedure described in J. Chem. Soc. 1956, 4334 to give the product shown for method 10.
- the starting material is reduced with lithium aluminum hydride according to the procedure described in J. Am. Chem. Soc. 1951 , 73, 1777 to give the product shown.
- the starting material is selectively protected at the 3-alcohol with t-butyldimethylchlorosilane and imidazole using the procedure described in J. Am. Chem. Soc. 1972, 94, 6190.
- the product is oxidized with chromium trioxide and pyridine.
- the 3-alcohol is then desilylated with hydrofluoric acid in acetonitrile using the procedure described in J. Am. Chem. Soc., 1972, 94, 6190 to give the product shown for method 13.
- the starting material is selectively protected at the 3-alcohol with t-butyldimethylchlorosilane and imidazole using the procedure described in J, Am. Chem. Soc., 1972, 94, 6190.
- the resulting intermediate is reduced with lithium aluminum hydride using the procedure described in J. Am. Chem. Soc., 1951 , 73, 1777.
- the resulting intermediate is selectively acetylated on the 12-alcohol, silylated on the 11-alcohol with trimethylsilyltriflate and 2,6-lutidine using the procedure described in Tetrahedron Letters.
- the desired Formula X compound can be prepared by the oxidation of tigogenin IX. Generally the oxidation is performed by reaction of tigogenin with pyridinium chlorochromate in a reaction inert solvent such as methylene chloride at 0°C to ambient temperature for about 2 hours to about 10 hours.
- a reaction inert solvent such as methylene chloride
- the desired Formula XI compound can be prepared by bromination of the Formula X compound followed by an elimination reaction. Typically the bromination is performed by reaction of the Formula X compound with bromine in
- tetrahydrofuran at a temperature of about -78° C, followed by warming to ambient temperature for about 1 hour to about 3 hours.
- the elimination reaction is performed by reaction of the brominated product prepared above with lithium bromide and lithium carbonate in a polar, aprotic solvent such as dimethyl formamide at a temperature of about 100°C to about 140°C for about 1 hour to about 4 hours.
- the desired Formula XII compound can be prepared by epoxidation of the appropriate Formula XI compound followed by lithium aluminum hydride reduction.
- the epoxidation is performed by reaction of the Formula XI compound with hydrogen peroxide and sodium hydroxide in a polar, protic solvent such as methanol at ambient temperature for about 2 hours to about 6 hours.
- the reduction is performed by reaction of the epoxide prepared above with lithium aluminum hydride in a reaction inert solvent such as tetrahydrofuran at ambient temperature for 2 hours to about 6 hours.
- the desired Formula IA compound as described in (E) above wherein Q 4 contains a hydroxy group may then be prepared from the appropriate Formula XII compound by a zinc fluoride catalyzed coupling followed by deacetylation with sodium methoxide as described previously.
- Those compounds wherein Q 4 is carbonyl may be prepared in an analogous manner, with the addition of oxidation with pyridinium chlorochromate (as described for Formula X compounds ) prior to the deacetylation.
- the desired Formula IA compounds wherein Q 1 ,Q 2 ,Q 3 ,Q 4 Q 5 , C 5 and C 25 are as described in (F) above may be prepared by the following procedures.
- the desired Formula (XIV) compound can be obtained by protection (as denoted by P) of the alcohol function in diosgenin (XIII) followed by hydroboration of the oiefin.
- the alcohol is protected as an ethoxymethyl ether by reaction of diosgenin with ethoxymethyl chloride and diisopropylethyl amine in an anhydrous solvent such as methylene chloride at ambient temperature for about 2 hours to 6 hours.
- the hydroboration is performed by reaction of the compound prepared above with borane-tetrahydrofuran complex in a reaction-inert solvent such as tetrahydrofuran at ambient temperature for about 1 hour to about 6 hours.
- the desired Formula XV compound can be prepared by oxidation of the appropriate Formula XIV compound followed by removal of the alcohol protecting group.
- the oxidation is performed by reaction of the Formula XIV compound with pyridinium chlorochromate in an anhydrous solvent such as methylene chloride at ambient temperature for about 2 hours to about 8 hours.
- the removal of the alcohol protecting group can be accomplished by reaction of the oxidized product prepared above with concentrated hydrochloric acid in a mixed solvent containing methanol and tetrahydrofuran at a temperature of about 40 °C to about 65° C for about 5 minutes to about 1 hour.
- the desired Formula IA compound as described in (F) above wherein Q 5 is carbonyl may then be prepared from the appropriate Formula XV compound by a zinc fluoride catalyzed coupling reaction followed by deacetylation using sodium methoxide as described previously.
- Those compounds where in Q 5 contain a hydroxy group may be prepared in an analogous manner with the addition of reduction prior to deacetylation. Typically the reduction is performed by reaction with sodium borohydride in a mixed solvent of ethanol and dichloromethane at ambient temperature for about 1 hour to about 6 hours.
- the compounds of Formula I which have been obtained and have
- asymmetric carbon atoms can be separated into their diastereomers on the basis of their physical chemical differences by methods known ⁇ er se., for example, by chromatography and/or fractional crystallization.
- the compounds of this invention where the sugar is ß-D-glucopyranuronosyl are acidic and they form base salts. All such base salts are within the scope of this invention and they can be prepared by conventional methods. For example, they can be prepared simply by contacting the acidic and basic entities, usually in a stoichiometric ratio, in either an aqueous, non-aqueous or partially aqueous medium, as appropriate. The salts are recovered either by filtration, by precipitation with a non-solvent followed by filtration, by evaporation of the solvent, or, in the case of aqueous solutions, by lyophilization, as appropriate.
- the compounds of this invention are potent inhibitors of cholesterol absorption and thus are all adapted to therapeutic use as hypercholesterolemia controlling agents in mammals, particularly humans. Since hypercholesterolemia is closely related to the development of generalized cardiovascular, cerebral vascular or peripheral vascular disorders, secondarily these compounds prevent the development of atherosclerosis particularly arteriosclerosis.
- the hypercholesterolemia controlling activity of these compounds may be demonstrated by methods based on standard procedures.
- the in vivo activity of these compounds in inhibiting intestinal absorption of cholesterol may be determined by the procedure of Melchoir and Harwell (J. Lipid Res., 1985, 26, 306-315).
- Activity can be determined by the amount of hypocholesterolemic agent that reduces the cholesterol absorption, relative to the control, in male golden Syrian hamsters.
- Male golden Syrian hamsters are administered either a cholesterol-free diet (control animals) or a diet supplemented with 1% cholesterol and 0.5% cholic acid for 4 days. The following day the animals are fasted for 18 hours, then administered a 1.5 ml oral bolus of water containing 0.25% methylcellulose, 0.6% Tween 80 and 10% ethanol (control animals) or an oral bolus that contains, in addition, the desired concentration of the compound to be tested.
- the animals receive a second 1.5 ml oral bolus of liquid hamster diet containing 1% [ 3 H] cholesterol (2.0 ⁇ Ci/animal; 210 dpm/nmol) and 0.5% cholic acid, and are fasted for an additional 24 hours.
- animals are sacrificed livers are excised, saponified and aliquots are decolorized by addition of hydrogen peroxide, and assessed for radioactivity.
- Total hepatic radioactivity is calculated based on measured liver weights. The degree of cholesterol absorption is expressed as a percentage of the total radioactivity administered as an oral bolus that is present in the liver 24 hours following bolus administration.
- Anti-atherosclerosis effects of the compounds can be determined by the amount of agent that reduces the lipid deposition in the rabbit aorta.
- Male New Zealand white rabbits are fed a diet containing 0.4% cholesterol and 5% peanut oil for 1 week (meal-fed once a day). After 1 week, the rabbits are dosed daily with the desired concentration of the compound to be tested. After 8.5 weeks, drug treatment is discontinued and the animals are maintained on the cholesterol containing diet for an additional 2 weeks and then switched to a cholesterol free diet for 5 weeks. The animals are sacrificed, and the aortas removed from the thoracic arch to the branch of the iliacs.
- the aortas are cleaned of adventitia, opened longitudinally and then stained with Sudan IV as described by Holman et al. (Lab. Invet. 1958, 7, 42-47). The percent of the surface area stained is quantitated by densitometry using an Optimas Image Analyzing System (Image Processing
- Reduced lipid deposition is indicated by a reduction in the percent surface area stained in the drug treated group in comparison with the control rabbits.
- Administration of the compounds of this invention can be via any method which delivers the compounds to the intestinal lumen. These methods include oral routes, intraduodenal routes etc.
- an effective dosage is in the range of 0.71 to 200 mg/kg/day, preferably 2 to 50 mg/kg/day, most preferably 2 to 7 mg/kg/day. For an average 70 kg human, this would amount to 0.05 to 14 g/day, preferably 0.14 to 3.5 g/day, most preferably 0.14 to 0.5 g/day.
- a pharmaceutical composition can take the form of solutions, suspensions, tablets, pills, capsules, powders, sustained release formulations and the like.
- the pharmaceutical compositions may be in the form of solid, semi-solid or liquid dosage forms, such as, for example, tablets, pills, capsules, powders, liquids, suspensions, or the like, preferably in unit dosage forms suitable for single administration of precise dosages.
- the pharmaceutical compositions will include a conventional pharmaceutical carrier or excipient and a compound according to the invention as an active ingredient, in addition, it may include other medicinal or pharmaceutical agents, carriers, adjuvants, etc.
- compositions according to the invention may contain 0.1%-95% of the compound, preferably 1%-70%.
- the composition or formulation to be administered will contain a quantity of a compound according to the invention in an amount effective to alleviate the signs of the subject being treated, i.e., hypercholesterolemia or atherosclerosis.
- conventional non-toxic solid carriers include, for example, pharmaceutical grades of mannitol, lactose, starch,
- magnesium stearate sodium saccharin, talcum, cellulose, glucose, sucrose, magnesium carbonate, and the like.
- Liquid pharmaceutically administrable compositions can be prepared by dissolving or dispersing, or otherwise preparing a compound according to this invention and mixing it optionally with a pharmaceutical adjuvant in a carrier, such as, for example, water, saline, aqueous dextrose, glycerol, ethanol, and the like, to thereby form a solution or suspension.
- a carrier such as, for example, water, saline, aqueous dextrose, glycerol, ethanol, and the like, to thereby form a solution or suspension.
- Powdered 4A molecular sieves (1 g) were added to a solution of trimethylsilyl tigogenin (1.17 g, 2.4 mmol) and acetobromo lactose (3.36 g, 4.8 mmol) in CH 2 CI 2 (15 mL) and CH 3 CN (5 mL) at room temperature. After stirring for 15 minutes Hg(CN) 2 (2.4 g, 9.6 mmol) and HgBr 2 (3.4 g, 9.6 mmol) were added and the mixture stirred at room temperature for three hours. The mixture was diluted with ethyl acetate (50 mL) and filtered.
- Trimethylsilyl trifluoromethanesulfonate (4 mL, 22.1 mmol) was added dropwise to a solution of tigogenin (6 g, 14.4 mmol) and triethyl amine (6 mL, 45 mmol) in CH 2 CI 2 (50 mL) at 0°C. After 1 hour, the mixture was diluted with ether (100 mL) and washed with saturated NaHCO 3 solution (2 x 50 mL) and brine (1 ⁇ 50 mL), dried (Na 2 SO 4 ) filtered and concentrated in vacuo. Upon addition of methanol, a precipitate formed which was filtered and washed with methanol and dried to afford 6.2 g product as a colorless solid.
- Lithium aluminum hydride (0.285 g, 7.5 mmol) was added to a solution of tigogenin-O-acetic acid ethyl ester (2.5 g, 4.98 mmol) in THF (50 mL) at 0°C. After 1 hour, the reaction was quenched by the sequential addition of H 2 O (0.285 mL), 15% NaOH (0.285 mL) and H 2 O (0.85 mL). The mixture was diluted with ether (25 mL) and dried with MgSO 4 , filtered and concentrated in vacuo to afford 2.1 g product as a colorless solid. MP 207-208°C. MS 461 (M+H) + .
- (3ß,5 ⁇ ,12ß,25R)spirostan-3,12-diol-11-one (3ß,5 ⁇ ,12B,25R)-3,12-di(acetoxy)spirostan-11-one (purchased from Steraloids, Inc., or see preparation G13) was saponified with potassium carbonate in water, methanol and THF to provide the title compound.
- (3ß,5 ⁇ ,11ß,25R)-spirostan-3,11-diol-12-one The title compound was synthesized from (3ß,5 ⁇ ,11ß,25R)-3-(t-butyldimethylsilyloxy)-11-(trimethylsilyloxy)spirostan-12-one was desiiylated with hydrofluoric acid in acetonitrile according to the procedure described in J, Am, Chem, Soc. 1972, 94, 6190. The title compound must be carefully handled because it will rearrange to (3ß,5 ⁇ ,12ß,25R)-spirostan-3,12-diol-11-one if exposed to base.
- (3ß,5 ⁇ ,25R)spirostan-3-ol-12-tosylhydrazone (3ß,5 ⁇ ,25R)-spirostan-3-ol-12-one(8.00g) was dissolved in glacial acetic acid (200 mL) and warmed to 50° C.
- Paratoluenesulfonylhydrazide (6.928 g) was added and the solution was stirred at 50° C for 30 min. After an additional 2 hours of stirring at room temperature, water (200 mL) was added. The resulting solid was collected, washed with water (100 mL), dried, triturated with refluxing acetone (300 mL), filtered hot and dried to give 3.903 g of the title compound.
- (3B,5 ⁇ ,11 ⁇ ,12 ⁇ ,25R)spirostan-3,11 ,12-triol (3ß,5 ⁇ ,25R)spirost-11-en-3-ol was oxidized to the title compound with osmium tetroxide and N-methylmorpholine-N-oxide in water, t-butanol and acetone according to the procedure describe in Tetrahedron Letters. 1976, 1973.
- the reaction was diluted with 50 gallons of water and stirred for 10 minutes. After separation of layers, the aqueous layer was extracted twice with 30 gallons of methylene chloride. The three combined organic extracts were washed twice with 30 gallons of water, once with 30 gallons of saturated brine, then dried using 7.0 Kg of magnesium sulfate. The drying agent was removed by filtration on a 30 inch Lapp followed by two 3 gallon methylene chloride washes. The filtrate and washes combined were atmospherically distilled to a 7 gallon total volume. Two 10 gallon methanol charges were made followed by continued distillation. When a final volume of ⁇ 10 gallons had been reached the mixture was cooled to room temperature.
- the resulting suspension was granulated for 2 hours, filtered on a 30 inch Lapp, and the filter cake was washed twice with 3 gallons of methanol. Vacuum drying the filter cake at 45-50° C yielded 12.6 Kg (58.6% yield) of the title compound.
- the reaction was distilled to remove the t-butanol. This was accomplished both by vacuum and atmospheric distillation. During the concentration two 32.5 gallon charges of water were added. Once the t-butanol had been removed, the aqueous suspension was cooled to room temperature and granulated for 2 hours. The suspension was filtered on a 30 inch Lapp, washed twice with 3 gallons of water, and the filter cake was air dried at 60 °C. This afforded 11.1 Kg of the title compound.
- the mixture was diluted with 87 gallons of water and cooled to room temperature. After 5 hours of granulation, the titled compound was isolated by filtration on a 30 inch Lapp followed by two 3 gallon water washes. The filter cake was dried at 60° C under vacuum to yield 12.2 Kg (93.3%).
- Lithium aluminum hydride (0.43 g, 15.38 mmol) was added to a solution of (1 ⁇ ,2 ⁇ ,5 ⁇ ,25R)-1 ,2-epoxy-spirostan-3-one in THF (20 mL) at 0°C.
- the reaction was gradually warmed to room temperature and after 3 hours, additional lithium aluminum hydride (0.10 g, 3.58 mmol) was added.
- the reaction was cooled to 0°C and quenched by the sequential addition of H 2 O (0.75 mL), 15% NaOH (0.75 mL), and H 2 O (1.50 mL).
- the mixture was dried with MgSO 4 , filtered, and concentrated in vacuo.
- the product was purified by flash chromatography (50% EtOAc/50% hexane to 95% EtOAc/5%MeOH) to afford 0.460 g, (34%) of the title compound.
- the reaction was quenched at 0°C by the addition of saturated sodium bisulfite solution.
- the mixture was diluted with ethyl acetate and washed with ammonium chloride solution (1x), brine (1x), dried (sodium sulfate), filtered and concentrated in vacuo to give 0.11 g of a mixture of the 6 ⁇ -alcohol and the 6ß-alcohol.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cardiology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Steroid Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Description


























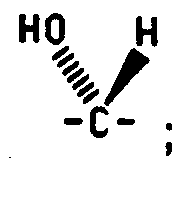

































































Claims







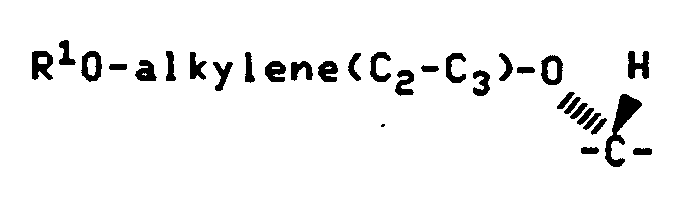




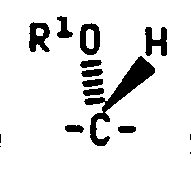
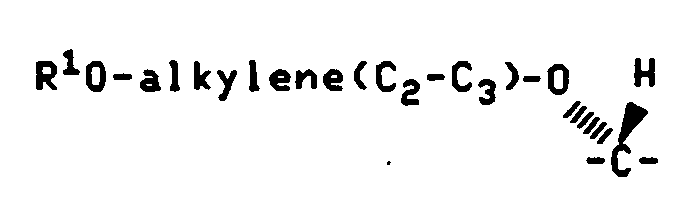





















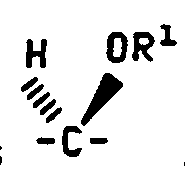













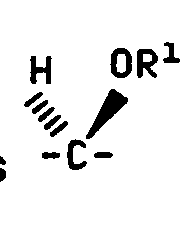























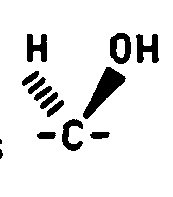











Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR9306619A BR9306619A (en) | 1992-06-26 | 1993-05-06 | Steroidal glycosides for the treatment of hypercholesterolemia |
| JP6502331A JPH07504921A (en) | 1992-06-26 | 1993-05-06 | Steroid glycosides to treat hypercholesterolemia |
| SK1583-94A SK158394A3 (en) | 1992-06-26 | 1993-05-06 | Steroidal glycosides |
| US08/351,470 US5629295A (en) | 1992-06-26 | 1993-05-06 | Steroidal glycosides for treating hypercholesterolemia |
| AU42265/93A AU4226593A (en) | 1992-06-26 | 1993-05-06 | Steroidal glycosides for treating hypercholesterolemia |
| EP93910951A EP0647234A1 (en) | 1992-06-26 | 1993-05-06 | Steroidal glycosides for treating hypercholesterolemia |
| BG99261A BG99261A (en) | 1992-06-26 | 1994-12-13 | Steroid glycosides for the treatment of hyperchlesterolemia |
| NO945001A NO945001L (en) | 1992-06-26 | 1994-12-23 | Steroid glycosides for the treatment of hypercholestrolemia |
| KR1019940704728A KR950702203A (en) | 1992-06-26 | 1994-12-24 | STEROLDAL GLYCOSIDES FOR TREATING HYPERCHOLESTEROLEMIA |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90491492A | 1992-06-26 | 1992-06-26 | |
| US07/904,914 | 1992-06-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994000480A1 true WO1994000480A1 (en) | 1994-01-06 |
Family
ID=25419965
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1993/004092 Ceased WO1994000480A1 (en) | 1992-06-26 | 1993-05-06 | Steroidal glycosides for treating hypercholesterolemia |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US5629295A (en) |
| EP (4) | EP0647234A1 (en) |
| JP (2) | JPH07504921A (en) |
| KR (1) | KR950702203A (en) |
| CN (1) | CN1085561A (en) |
| AP (1) | AP489A (en) |
| AU (1) | AU4226593A (en) |
| BG (1) | BG99261A (en) |
| BR (1) | BR9306619A (en) |
| CA (1) | CA2139104A1 (en) |
| CZ (1) | CZ331094A3 (en) |
| HR (1) | HRP930994A2 (en) |
| IL (1) | IL106055A0 (en) |
| MX (1) | MX9303826A (en) |
| OA (1) | OA10121A (en) |
| RU (1) | RU94046294A (en) |
| SK (1) | SK158394A3 (en) |
| WO (1) | WO1994000480A1 (en) |
| YU (1) | YU44793A (en) |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994025479A1 (en) * | 1993-04-28 | 1994-11-10 | Pfizer Inc. | Spirostanyl glycosidal crystalline monohydrate |
| WO1995000018A1 (en) * | 1993-06-21 | 1995-01-05 | Medical Research Foundation Of Oregon | Cholesterol sequestrant glycosides that inhibit intestinal cholesterol absorption |
| WO1995018144A1 (en) * | 1993-12-28 | 1995-07-06 | Pfizer Inc. | Steroidal glycosides |
| WO1995018143A1 (en) * | 1993-12-28 | 1995-07-06 | Pfizer Inc. | Hypocholesterolemic agents |
| ES2074006A1 (en) * | 1993-07-05 | 1995-08-16 | Pfizer | Steroid glycosides for treating hypercholesterolaemia |
| FR2722690A1 (en) * | 1994-07-21 | 1996-01-26 | Universite Montpellier Ii | COMPOSITIONS OF SAPONINS AND / OR THEIR AGLYCONIC FORMS AND THEIR APPLICATIONS AS MEDICINAL PRODUCTS |
| WO1996006854A1 (en) * | 1994-08-30 | 1996-03-07 | Pfizer Inc. | Spirostanyl glycosidal crystals |
| US5563259A (en) * | 1992-06-26 | 1996-10-08 | Pfizer Inc. | Process for making β-O-cellobiosyl steroid derivatives and trimethyl silyl steroid intermediates used therein |
| WO1996038466A1 (en) * | 1995-05-29 | 1996-12-05 | Pfizer Inc. | Steroidal glycosides |
| US5698527A (en) * | 1995-08-08 | 1997-12-16 | Merck & Co., Inc. | Steroidal glycosides as antihyperlipidemic agents |
| US5698789A (en) * | 1993-10-26 | 1997-12-16 | Ilmari Paakkinen | Method and device for measuring the properties of granular earth materials |
| US5756470A (en) * | 1996-10-29 | 1998-05-26 | Schering Corporation | Sugar-substituted 2-azetidinones useful as hypocholesterolemic agents |
| US5807834A (en) * | 1994-09-20 | 1998-09-15 | Pfizer Inc. | Combination of a cholesterol absorption inhibitor and a cholesterol synthesis inhibitor |
| WO2001032679A3 (en) * | 1999-11-01 | 2001-10-25 | Forbes Medi Tech Inc | Novel glycosides comprising pentose mono-, di-, tri-, or oligosaccharides and phytosterols and/or phytostanols |
| US6982251B2 (en) | 2000-12-20 | 2006-01-03 | Schering Corporation | Substituted 2-azetidinones useful as hypocholesterolemic agents |
| US7030106B2 (en) | 2001-01-26 | 2006-04-18 | Schering Corporation | Sterol absorption inhibitor compositions |
| US7053080B2 (en) | 2001-09-21 | 2006-05-30 | Schering Corporation | Methods and therapeutic combinations for the treatment of obesity using sterol absorption inhibitors |
| US7056906B2 (en) | 2001-09-21 | 2006-06-06 | Schering Corporation | Combinations of hormone replacement therapy composition(s) and sterol absorption inhibitor(s) and treatments for vascular conditions in post-menopausal women |
| US7071181B2 (en) | 2001-01-26 | 2006-07-04 | Schering Corporation | Methods and therapeutic combinations for the treatment of diabetes using sterol absorption inhibitors |
| US7132415B2 (en) | 2001-09-21 | 2006-11-07 | Schering Corporation | Methods and therapeutic combinations for the treatment of xanthoma using sterol absorption inhibitors |
| US7192944B2 (en) | 2003-03-07 | 2007-03-20 | Schering Corp. | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7208486B2 (en) | 2003-03-07 | 2007-04-24 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| WO2007062314A2 (en) | 2005-11-23 | 2007-05-31 | Bristol-Myers Squibb Company | Heterocyclic cetp inhibitors |
| US7235543B2 (en) | 2003-03-07 | 2007-06-26 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| WO2008070496A2 (en) | 2006-12-01 | 2008-06-12 | Bristol-Myers Squibb Company | N- ( (3-benzyl) -2, 2- (bis-phenyl) -propan-1-amine derivatives as cetp inhibitors for the treatment of atherosclerosis and cardiovascular diseases |
| US7417039B2 (en) | 2001-01-26 | 2008-08-26 | Schering Corporation | Use of substituted azetidinone compounds for the treatment of sitosterolemia |
| US7446121B2 (en) | 2004-11-23 | 2008-11-04 | Pfizer Inc. | Pyrazole-based HMG CoA reductase inhibitors |
| US7459442B2 (en) | 2003-03-07 | 2008-12-02 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7560449B2 (en) | 2002-11-06 | 2009-07-14 | Schering Corporation | Methods and therapeutic combinations for the treatment of demyelination |
| WO2011145022A1 (en) | 2010-05-21 | 2011-11-24 | Pfizer Inc. | 2-phenyl benzoylamides |
| EP2392567A1 (en) | 2005-10-21 | 2011-12-07 | Bristol-Myers Squibb Company | Benzothiazine derivatives and their use as lxr modulators |
| WO2012120414A2 (en) | 2011-03-04 | 2012-09-13 | Pfizer Inc. | Edn3-like peptides and uses thereof |
| WO2014170786A1 (en) | 2013-04-17 | 2014-10-23 | Pfizer Inc. | N-piperidin-3-ylbenzamide derivatives for treating cardiovascular diseases |
| JP2015506929A (en) * | 2011-12-27 | 2015-03-05 | セントロ デ インベスティガシオン イ デサロリョ デ ロス メディカメントス(サイデム)Centro De Investigacion Y Desarrollo De Los Medicamentos (Cidem) | Spirosteroids acting as neuroprotective and anti-inflammatory agents |
| WO2016055901A1 (en) | 2014-10-08 | 2016-04-14 | Pfizer Inc. | Substituted amide compounds |
| WO2020150473A2 (en) | 2019-01-18 | 2020-07-23 | Dogma Therapeutics, Inc. | Pcsk9 inhibitors and methods of use thereof |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9923076D0 (en) * | 1999-09-29 | 1999-12-01 | Phytopharm Plc | Sapogenin derivatives and their use |
| BR9909110A (en) * | 1998-03-26 | 2000-12-12 | Phytopharm Plc | Uses of smilagenin and anzurogenin d, pharmaceutical composition having cognitive function enhancing properties, process to intensify cognitive function, and composition for use thereof |
| JP3370610B2 (en) * | 1998-08-21 | 2003-01-27 | 武田薬品工業株式会社 | Pest control agent |
| US20030060425A1 (en) * | 1998-11-24 | 2003-03-27 | Ahlem Clarence N. | Immune modulation method using steroid compounds |
| GB9923078D0 (en) * | 1999-09-29 | 1999-12-01 | Phytopharm Plc | Sapogenin derivatives and their use |
| US6343258B1 (en) | 1999-08-13 | 2002-01-29 | ALEXIS Brian | Method for testing for readiness for harvesting of tribulus terrestris l. having high steroidal saponin content |
| GB9923077D0 (en) * | 1999-09-29 | 1999-12-01 | Phytopharm Plc | Sapogenin derivatives and their use |
| GB0000228D0 (en) * | 2000-01-06 | 2000-03-01 | Phytopharm Plc | Fluoro substituted sapogenins and their use |
| US6207638B1 (en) | 2000-02-23 | 2001-03-27 | Pacifichealth Laboratories, Inc. | Nutritional intervention composition for enhancing and extending satiety |
| US6429190B1 (en) | 2000-12-15 | 2002-08-06 | Pacifichealth Laboratories, Inc. | Method for extending the satiety of food by adding a nutritional composition designed to stimulate cholecystokinin(CCK) |
| AU2002232575A1 (en) | 2000-12-15 | 2002-06-24 | Pacifichealth Laboratories, Inc. | Nutritional composition for improving the efficacy of a lipase inhibitor |
| GB0107822D0 (en) * | 2001-03-28 | 2001-05-23 | Phytopharm Plc | Sapogenin derivatives their synthesis and use methods based upon their use |
| US6703530B2 (en) * | 2002-02-28 | 2004-03-09 | General Electric Company | Chemical reactor system and process |
| US20050130948A1 (en) * | 2002-03-27 | 2005-06-16 | Daryl Rees | Therapeutic methods and uses of sapogenins and their derivatives |
| KR101130212B1 (en) * | 2002-03-27 | 2012-04-13 | 파이토팜 피엘씨 | Theraputic methods and uses of SAPOGENIN and their derivatives |
| CA2503899C (en) * | 2002-10-28 | 2011-12-20 | Phytopharm Plc | Stereospecific reduction of sapogen-3-ones |
| CA2721665C (en) * | 2008-04-18 | 2017-01-24 | Reata Pharmaceuticals, Inc. | Compounds including an anti-inflammatory pharmacore and methods of use |
| ES2449396T3 (en) * | 2008-07-22 | 2014-03-19 | Trustees Of Dartmouth College | Monocyclic cyanoenones and methods of use thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0159431A2 (en) * | 1984-04-20 | 1985-10-30 | Medical Research Foundation Of Oregon | Tigogenin-cellobioside and its heptaacetate, methods for their preparation, and pharmaceutical compositions containing tigogenin-cellobioside |
| EP0403150A2 (en) * | 1989-06-13 | 1990-12-19 | Pfizer Inc. | Process for the preparation of tigogenin beta-cellobioside |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3178418A (en) | 1963-02-26 | 1965-04-13 | Glaxo Lab Ltd | Monobromination at the 11-position of hecogenin and 3-lower alkanoyl esters thereof |
| DE2926463A1 (en) * | 1978-07-05 | 1980-01-24 | Roecar Holdings Nv | SPIROKETALINE AND THEIR USE |
| US4260603A (en) | 1979-01-02 | 1981-04-07 | Pegel Karl H | Sterol glycoside with activity as prostaglandin synthetase inhibitor |
| NZ193564A (en) * | 1979-05-02 | 1985-01-31 | Aruba Pty Ltd | Steroid alkaloids from solanum sodomeum and pharmaceutical compositions |
| US4602003A (en) | 1982-05-17 | 1986-07-22 | Medical Research Foundation Of Oregon | Synthetic compounds to inhibit intestinal absorption of cholesterol in the treatment of hypercholesterolemia |
| ES524614A0 (en) * | 1982-08-12 | 1984-05-01 | Nativelle Sa Ets | PROCEDURE FOR THE PREPARATION OF NEW AMINO-14 STEROIDS. |
| ES524796A0 (en) * | 1982-08-20 | 1984-05-01 | Nativelle Sa Ets | PROCEDURE FOR THE PREPARATION OF NEW AMINO-14 STEROID DERIVATIVES |
| IT1188184B (en) | 1985-08-14 | 1988-01-07 | Texcontor Ets | QUATERNARY AMMONIC SALTS OF POLYESACCHARIDES WITH HYPO-COLESTEROLEMIZING ACTIVITY |
| US5017562A (en) * | 1987-02-11 | 1991-05-21 | Regents Of The University Of Minnesota | Crystalline saponin-containing complex |
| FR2621316B1 (en) * | 1987-10-02 | 1991-06-21 | Nativelle Sa Ets | NOVEL ANDROSTANE 17-CARBOXYLIC ACID ESTERS, PROCESS FOR THEIR PREPARATION, AND MEDICAMENTS CONTAINING THEM |
| ES2107548T3 (en) * | 1991-07-23 | 1997-12-01 | Schering Corp | SUBSTITUTED BETA-LACTAMA COMPOUNDS USEFUL AS HYPOCHOLESTEROLEMIC AGENTS AND PROCEDURES FOR THEIR PREPARATION. |
| WO1993005790A1 (en) * | 1991-09-27 | 1993-04-01 | Procter & Gamble Pharmaceuticals, Inc. | Use of 14-aminosteroids for the manufacture of a medicament to slow the rate of progression of myocardial structural damages characteristic of congestive heart failure |
| ES2114948T3 (en) * | 1991-10-04 | 1998-06-16 | Procter & Gamble | CHOLESTEROL DECREASING COMPOUNDS AND PROCEDURE FOR PREPARING THEM. |
| JPH0826064B2 (en) * | 1991-11-25 | 1996-03-13 | ファイザー インク. | Method for producing steroid peracyl glycosides |
| US5530107A (en) * | 1992-10-15 | 1996-06-25 | Pfizer Inc. | Method for making steroidal peracyl glycosides |
| LT3595B (en) * | 1993-01-21 | 1995-12-27 | Schering Corp | Spirocycloalkyl-substituted azetidinones useful as hypocholesterolemic agents |
-
1993
- 1993-05-06 EP EP93910951A patent/EP0647234A1/en not_active Withdrawn
- 1993-05-06 RU RU94046294/04A patent/RU94046294A/en unknown
- 1993-05-06 US US08/351,470 patent/US5629295A/en not_active Expired - Fee Related
- 1993-05-06 WO PCT/US1993/004092 patent/WO1994000480A1/en not_active Ceased
- 1993-05-06 BR BR9306619A patent/BR9306619A/en not_active Application Discontinuation
- 1993-05-06 AU AU42265/93A patent/AU4226593A/en not_active Abandoned
- 1993-05-06 SK SK1583-94A patent/SK158394A3/en unknown
- 1993-05-06 EP EP97200454A patent/EP0796862A2/en not_active Withdrawn
- 1993-05-06 EP EP97200455A patent/EP0796863A2/en not_active Withdrawn
- 1993-05-06 CZ CZ943310A patent/CZ331094A3/en unknown
- 1993-05-06 CA CA002139104A patent/CA2139104A1/en not_active Abandoned
- 1993-05-06 JP JP6502331A patent/JPH07504921A/en active Pending
- 1993-05-06 EP EP97200456A patent/EP0796864A2/en not_active Withdrawn
- 1993-06-17 AP APAP/P/1993/000539A patent/AP489A/en active
- 1993-06-17 IL IL106055A patent/IL106055A0/en unknown
- 1993-06-24 HR HR07/904,914A patent/HRP930994A2/en not_active Application Discontinuation
- 1993-06-25 YU YU44793A patent/YU44793A/en unknown
- 1993-06-25 MX MX9303826A patent/MX9303826A/en unknown
- 1993-06-25 CN CN93107620A patent/CN1085561A/en active Pending
-
1994
- 1994-12-13 BG BG99261A patent/BG99261A/en unknown
- 1994-12-23 OA OA60598A patent/OA10121A/en unknown
- 1994-12-24 KR KR1019940704728A patent/KR950702203A/en not_active Ceased
-
1995
- 1995-06-06 US US08/470,554 patent/US5703052A/en not_active Expired - Fee Related
-
1997
- 1997-02-14 JP JP9030588A patent/JPH09309897A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0159431A2 (en) * | 1984-04-20 | 1985-10-30 | Medical Research Foundation Of Oregon | Tigogenin-cellobioside and its heptaacetate, methods for their preparation, and pharmaceutical compositions containing tigogenin-cellobioside |
| EP0403150A2 (en) * | 1989-06-13 | 1990-12-19 | Pfizer Inc. | Process for the preparation of tigogenin beta-cellobioside |
Non-Patent Citations (5)
| Title |
|---|
| ANNALS OF THE NEW YORK ACADEMY OF SCIENCES (ATHEROSCLEROSIS) vol. 454, 1985, NEW YORK, US pages 23 - 27 M. R. MALINOW 'Effects of Synthetic Glycosides on Cholesterol Absorption' * |
| CHEMICAL ABSTRACTS, vol. 107, no. 3, 20 July 1987, Columbus, Ohio, US; abstract no. 22075, G. S. SIDHU ET AL 'Effects of Soy Saponins and Tigogenin-cellobioside on Intestinal Uptake of Cholesterol, Cholate and Glucose' page 499 ;column 1 ; * |
| JOURNAL OF PHARMACEUTICAL SCIENCES vol. 67, no. 11, 1978, WASHINGTON D.C., US pages 1589 - 1592 R. SEGAL ET AL 'Hemolytic Properties of Synthetic Glycosides' * |
| JUSTUS LIEBIGS ANNALEN DER CHEMIE vol. 699, 1966, WEINHEIM DE pages 212 - 222 R. TSCHESCHE ET AL ']ber Parillin, ein Saponin mit stark verzweigter Zuckerkette' * |
| STEROIDS: STRUCTURE, FUNCTION, AND REGULATION vol. 48, no. 3-4, 1986, STONEHAM, MA US pages 197 - 211 M. R. MALINOW ET AL 'Effects of alpha- and beta-Tigogenin Cellobiosides on Cholesterol Absorption' * |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5563259A (en) * | 1992-06-26 | 1996-10-08 | Pfizer Inc. | Process for making β-O-cellobiosyl steroid derivatives and trimethyl silyl steroid intermediates used therein |
| US5760009A (en) * | 1993-04-28 | 1998-06-02 | Pfizer Inc. | Spirostanyl glycosidal crystalline monohydrate |
| WO1994025479A1 (en) * | 1993-04-28 | 1994-11-10 | Pfizer Inc. | Spirostanyl glycosidal crystalline monohydrate |
| WO1995000018A1 (en) * | 1993-06-21 | 1995-01-05 | Medical Research Foundation Of Oregon | Cholesterol sequestrant glycosides that inhibit intestinal cholesterol absorption |
| US5502038A (en) * | 1993-06-21 | 1996-03-26 | Medical Research Foundation Of Oregon | Cholesterol sequestrant glycosides that inhibit intestinal cholesterol absorption |
| ES2074006A1 (en) * | 1993-07-05 | 1995-08-16 | Pfizer | Steroid glycosides for treating hypercholesterolaemia |
| US5698789A (en) * | 1993-10-26 | 1997-12-16 | Ilmari Paakkinen | Method and device for measuring the properties of granular earth materials |
| WO1995018144A1 (en) * | 1993-12-28 | 1995-07-06 | Pfizer Inc. | Steroidal glycosides |
| WO1995018143A1 (en) * | 1993-12-28 | 1995-07-06 | Pfizer Inc. | Hypocholesterolemic agents |
| FR2722690A1 (en) * | 1994-07-21 | 1996-01-26 | Universite Montpellier Ii | COMPOSITIONS OF SAPONINS AND / OR THEIR AGLYCONIC FORMS AND THEIR APPLICATIONS AS MEDICINAL PRODUCTS |
| WO1996006854A1 (en) * | 1994-08-30 | 1996-03-07 | Pfizer Inc. | Spirostanyl glycosidal crystals |
| US5807834A (en) * | 1994-09-20 | 1998-09-15 | Pfizer Inc. | Combination of a cholesterol absorption inhibitor and a cholesterol synthesis inhibitor |
| US6150336A (en) * | 1995-05-29 | 2000-11-21 | Pfizer Inc. | Steroidal glycosides |
| WO1996038466A1 (en) * | 1995-05-29 | 1996-12-05 | Pfizer Inc. | Steroidal glycosides |
| US5698527A (en) * | 1995-08-08 | 1997-12-16 | Merck & Co., Inc. | Steroidal glycosides as antihyperlipidemic agents |
| US5756470A (en) * | 1996-10-29 | 1998-05-26 | Schering Corporation | Sugar-substituted 2-azetidinones useful as hypocholesterolemic agents |
| WO2001032679A3 (en) * | 1999-11-01 | 2001-10-25 | Forbes Medi Tech Inc | Novel glycosides comprising pentose mono-, di-, tri-, or oligosaccharides and phytosterols and/or phytostanols |
| US6982251B2 (en) | 2000-12-20 | 2006-01-03 | Schering Corporation | Substituted 2-azetidinones useful as hypocholesterolemic agents |
| US7030106B2 (en) | 2001-01-26 | 2006-04-18 | Schering Corporation | Sterol absorption inhibitor compositions |
| US7612058B2 (en) | 2001-01-26 | 2009-11-03 | Schering Corporation | Methods for inhibiting sterol absorption |
| US7417039B2 (en) | 2001-01-26 | 2008-08-26 | Schering Corporation | Use of substituted azetidinone compounds for the treatment of sitosterolemia |
| US7071181B2 (en) | 2001-01-26 | 2006-07-04 | Schering Corporation | Methods and therapeutic combinations for the treatment of diabetes using sterol absorption inhibitors |
| US7132415B2 (en) | 2001-09-21 | 2006-11-07 | Schering Corporation | Methods and therapeutic combinations for the treatment of xanthoma using sterol absorption inhibitors |
| US7056906B2 (en) | 2001-09-21 | 2006-06-06 | Schering Corporation | Combinations of hormone replacement therapy composition(s) and sterol absorption inhibitor(s) and treatments for vascular conditions in post-menopausal women |
| US7053080B2 (en) | 2001-09-21 | 2006-05-30 | Schering Corporation | Methods and therapeutic combinations for the treatment of obesity using sterol absorption inhibitors |
| US7560449B2 (en) | 2002-11-06 | 2009-07-14 | Schering Corporation | Methods and therapeutic combinations for the treatment of demyelination |
| US7459442B2 (en) | 2003-03-07 | 2008-12-02 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7208486B2 (en) | 2003-03-07 | 2007-04-24 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7368562B2 (en) | 2003-03-07 | 2008-05-06 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7378518B2 (en) | 2003-03-07 | 2008-05-27 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7368563B2 (en) | 2003-03-07 | 2008-05-06 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7235543B2 (en) | 2003-03-07 | 2007-06-26 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7192944B2 (en) | 2003-03-07 | 2007-03-20 | Schering Corp. | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| US7446121B2 (en) | 2004-11-23 | 2008-11-04 | Pfizer Inc. | Pyrazole-based HMG CoA reductase inhibitors |
| EP2392567A1 (en) | 2005-10-21 | 2011-12-07 | Bristol-Myers Squibb Company | Benzothiazine derivatives and their use as lxr modulators |
| WO2007062314A2 (en) | 2005-11-23 | 2007-05-31 | Bristol-Myers Squibb Company | Heterocyclic cetp inhibitors |
| WO2008070496A2 (en) | 2006-12-01 | 2008-06-12 | Bristol-Myers Squibb Company | N- ( (3-benzyl) -2, 2- (bis-phenyl) -propan-1-amine derivatives as cetp inhibitors for the treatment of atherosclerosis and cardiovascular diseases |
| WO2011145022A1 (en) | 2010-05-21 | 2011-11-24 | Pfizer Inc. | 2-phenyl benzoylamides |
| WO2012120414A2 (en) | 2011-03-04 | 2012-09-13 | Pfizer Inc. | Edn3-like peptides and uses thereof |
| JP2015506929A (en) * | 2011-12-27 | 2015-03-05 | セントロ デ インベスティガシオン イ デサロリョ デ ロス メディカメントス(サイデム)Centro De Investigacion Y Desarrollo De Los Medicamentos (Cidem) | Spirosteroids acting as neuroprotective and anti-inflammatory agents |
| WO2014170786A1 (en) | 2013-04-17 | 2014-10-23 | Pfizer Inc. | N-piperidin-3-ylbenzamide derivatives for treating cardiovascular diseases |
| WO2016055901A1 (en) | 2014-10-08 | 2016-04-14 | Pfizer Inc. | Substituted amide compounds |
| WO2020150473A2 (en) | 2019-01-18 | 2020-07-23 | Dogma Therapeutics, Inc. | Pcsk9 inhibitors and methods of use thereof |
| EP4470609A2 (en) | 2019-01-18 | 2024-12-04 | Astrazeneca AB | Pcsk9 inhibitors and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0796864A2 (en) | 1997-09-24 |
| HRP930994A2 (en) | 1996-08-31 |
| AP9300539A0 (en) | 1993-07-31 |
| BG99261A (en) | 1995-06-30 |
| MX9303826A (en) | 1994-02-28 |
| EP0796862A2 (en) | 1997-09-24 |
| JPH09309897A (en) | 1997-12-02 |
| CN1085561A (en) | 1994-04-20 |
| EP0647234A1 (en) | 1995-04-12 |
| US5629295A (en) | 1997-05-13 |
| CA2139104A1 (en) | 1994-01-06 |
| US5703052A (en) | 1997-12-30 |
| AU4226593A (en) | 1994-01-24 |
| BR9306619A (en) | 1998-12-08 |
| YU44793A (en) | 1996-10-09 |
| CZ331094A3 (en) | 1995-09-13 |
| AP489A (en) | 1996-05-02 |
| EP0796863A2 (en) | 1997-09-24 |
| OA10121A (en) | 1996-12-18 |
| SK158394A3 (en) | 1995-05-10 |
| RU94046294A (en) | 1996-10-10 |
| IL106055A0 (en) | 1993-10-20 |
| KR950702203A (en) | 1995-06-19 |
| JPH07504921A (en) | 1995-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AP489A (en) | Steroidal glycosides for treating hypercholesterolemia. | |
| US5939398A (en) | Hypocholesterolemic agents | |
| JPH04257599A (en) | Novel preguna-1,4-diene-3,20-dione-16,17- acetal-21-ester and preparation and use thereof | |
| JP3279574B2 (en) | Triterpene derivative and therapeutic agent for liver disease | |
| US5760009A (en) | Spirostanyl glycosidal crystalline monohydrate | |
| GB1599863A (en) | Pharmaceutical compositions for use in the inhibition of the biosynthesis of mevalonic acid | |
| US5698526A (en) | Steroidal glycosides | |
| US6150336A (en) | Steroidal glycosides | |
| EP0072546B1 (en) | Hydrocortisone derivatives, their preparation and their utilization | |
| WO1996006854A1 (en) | Spirostanyl glycosidal crystals | |
| US4260464A (en) | Process for the preparation of 17α-(3-iodobenzoyloxy)-9α-chloro-4-pregnene-3,20-diones and their D-homo homologs | |
| EP1422235B1 (en) | Stereoselective method of producing 6alpha-fluoropregnanes and intermediaries | |
| EP0149222A2 (en) | 6-Alpha, 16 alpha-dimethyl corticoids | |
| NO945001L (en) | Steroid glycosides for the treatment of hypercholestrolemia | |
| US3484436A (en) | Bicyclo(2.2.2)octane - 1 - -carboxylate esters of delta**4 - pregnene corticoid steroids | |
| JPS6246557B2 (en) | ||
| US4127589A (en) | 4,5-Seco-steroids |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BG BR CA CZ DE JP KR NO NZ RO RU SK UA US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1993910951 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 252495 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08351470 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 158394 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2139104 Country of ref document: CA Ref document number: 94-02092 Country of ref document: RO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1994-3310 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993910951 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1994-3310 Country of ref document: CZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993910951 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: PV1994-3310 Country of ref document: CZ |