WO1994005672A1 - PROCESS FOR PREPARING CERTAIN PYRROLO[3,4- b]QUINOLINES, CERTAIN 1H-PYRANO[3',4':6,7]INDOLIZINO[1,2-b]QUINOLIN-3,14(4H,12H)-DIONES, AND CERTAIN 8-METHYL-7-(OXOPROPYL)-INDOLIZINO[1,2-b]QUINOLIN-9(11H)-ONES - Google Patents
PROCESS FOR PREPARING CERTAIN PYRROLO[3,4- b]QUINOLINES, CERTAIN 1H-PYRANO[3',4':6,7]INDOLIZINO[1,2-b]QUINOLIN-3,14(4H,12H)-DIONES, AND CERTAIN 8-METHYL-7-(OXOPROPYL)-INDOLIZINO[1,2-b]QUINOLIN-9(11H)-ONES Download PDFInfo
- Publication number
- WO1994005672A1 WO1994005672A1 PCT/US1993/008434 US9308434W WO9405672A1 WO 1994005672 A1 WO1994005672 A1 WO 1994005672A1 US 9308434 W US9308434 W US 9308434W WO 9405672 A1 WO9405672 A1 WO 9405672A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- quinolin
- process according
- indolizino
- compound
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/12—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains three hetero rings
- C07D493/14—Ortho-condensed systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to a process for preparing certain pyrrolo-[3,4-b]quinolines, certain 1H-pyrano[3',4':6,7] indolizino[3,4-b]quinolin-3,14(4H,12H)-diones, specifically camptothecin and its analogs, and certain 8-methyl-7-(oxopropyl)-indolizino[3,2-b]quinolin-9(11H)-ones, specifically mappicine ketones and mappicines.
- camptothecin itself is known to be cytotoxic at levels which inhibit tumor growth
- certain water-soluble camptothecin analogs which exhibit litde or no cytotoxicity are efficacious against solid tumor types normally refractory to known treatments.
- (S)-10-[(dimethylamino)methyl]-4-ethyl-4,9-dihydroxy-1H-pyrano[3',4':6,7] indolizino[1,2-b]quinolin-3,14(4H,12H)-dione commonly known as topotecan (Formula lb), which is disclosed in U.S. Pat No.5,004,758, issued to Boehm et al.
- tumors include, but are not limited to, ovarian, esophageal, non-small cell lung and colorectal carcinomas.
- topotecan is also useful for the preparation of a combination chemotherapeutic pharmaceutical composition which also comprises a platinum coordination compound, most preferably cis-platin.
- a platinum coordination compound most preferably cis-platin.
- the 4-(piperidino)-piperidinyl carbamate analogue of 7-ethyl-10-hydroxycamptothecin, commonly known as irinotecan (or CPT- 11) (Formula Ic) has also been shown to be efficacious against certain tumors. Both compounds are presently undergoing human clinical testing in refractory tumor types. Additionally, the 9-amino and 10,11-methylenedioxy analogs: of camptothecin (Formula Id) have shown promising antitumor activity in preclinical testing.
- camptothecin molecule results in compounds possessing antiviral activity while exhibiting little or no cytotoxicity.
- examples of such compounds include derivatives in which the E ring lactone has been replaced by some other functionality, i.e., two broad classes of compounds commonly called mappicine ketones and mappicines, of Formulas IIa and IIb respectively.
- mappicine ketones and mappicines of Formulas IIa and IIb respectively.
- These compounds which are useful in treating infections in humans and animals caused by a variety of viruses, (for example, Herpes simplex virus types 1 and 2, cytomegalovirus, and Varicella zoster virus) are disclosed in U.S. Ser. No. 07/606,216, filed by S. Petteway et al. on October 31,1990; U.S. Ser. No.
- Camptothecins have also been recently shown to possess antiretroviral activity, specifically the ability to inhibit the replication of Human
- HTV-1 Immunodeficiency Virus
- HTV-1 at dosages non-cytotoxic to mammalian cells, and thus may be useful in the treatment of patients suffering from Acquired Immune Deficiency Syndrome (AIDS, see AIDS Res. Hum. Retr., 1991, 7, 65).
- topoisomerase I is a monomeric enzyme with a molecular weight of approximately 100,000.
- Camptothecin is known to exert its antitumor activity by stabilization of the covalently bound topoisomerase I - DNA complex. As a result, progression of the DNA replication sequence proceeds only as far as induction of a single strand break. The ultimate result of this inhibition of the DNA transcription/replication process is cell death. Camptothecin and a few of its close congeners are the only agents in clinical drug development which inhibit topoisomerase I.
- Topoisomerase II consists of two identical subunits of molecular weight 170,000. Topoisomerase II induces transient breaks of both strands of the DNA helix and passes another double-stranded segment through the break.
- Several commercially important oncolytic agents e.g., etoposide, doxorubicin and mitoxantrone
- camptothecin does not inhibit topoisomerase ⁇
- mappicines and mappicine ketones are known to inhibit topoisomerase II As such these compounds are of interest both as potential antineoplastic and antiviral agents.
- camptothecin discloses a semisynthetic method of preparing certain water-soluble camptothecin analogs from camptothecin.
- the only practical method for obtaining preparative- and commercial-scale quantities of camptothecin is by extraction of the tissues of the Camptotheca acuminata tree indigenous to the People's Republic of China or the Nothapodytes foetida shrub indigenous to India.
- reliance on such natural sources has certain inherent disadvantages, including very high starting material cost (about $28,000/kg) and lack of a dependable source. Camptothecin has also been prepared from readily available materials by various totally synthetic approaches.
- strategy A a tricyclic CDE ring system is prepared by known methods and is coupled with ortho-aminobenzaldehyde via a Friedlander quinoline synthesis.
- the critical bond disconnection strategy is shown by the dashed line labelled A.
- strategy B tricyclic ABC and bicyclic DE ring components are prepared separately by known methods and are coupled using a two-step, standard procedure to form the D ring pyridone.
- the critical bond disconnection strategy is shown by the dashed line labelled B.
- the tricyclic ABC ring fragment for strategy B is ultimately prepared via a Friedlander quinoline synthesis requiring several steps. The Friedlander quinoline synthesis is therefore a basic component of many of the known approaches.
- the present invention provides an efficient general route for the facile synthesis of pyrrolo[3,4-b]quinolines, 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-ones and 1H-pyrano[3',4':6,7]indolizmo[1,2-b]qumolin-3,14(4H,12H)-diones, preferably camptothecin analogs.
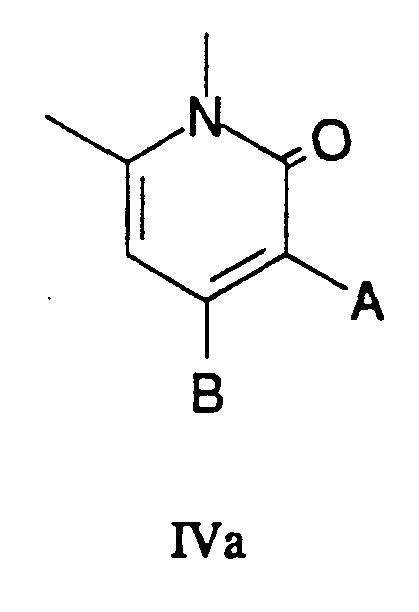
- the present invention provides a process for preparing pyrrolo[3,4-b]quinolines, 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-ones; and 1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-3,14(4H,12H)-diones, preferably camptothecin analogs , more preferably water-soluble camptothecin analogs, yet more preferably topotecan and irinotecan, most preferably topotecan, said process comprising the step of intramolecular [4+2] cycloaddition of the N-arylimidate portion of a compound of Formula IV with the unactivated acetylene portion of said compound, the compound of Formula IV being generated from compounds of Formula III (shown in Scheme 2 below and having the same substitutions as compounds of Formula IV), which are derived from generally available compounds.
- R 1 H, OH, or OR, where R is an ester protecting group
- R 2 H, NO 2 , or a protected amine function
- R 3 H, C 2 H 5 , or a trialkylsilyl
- R 4 H or CH 2 COOEt
- R 5 COOMe or tosyl
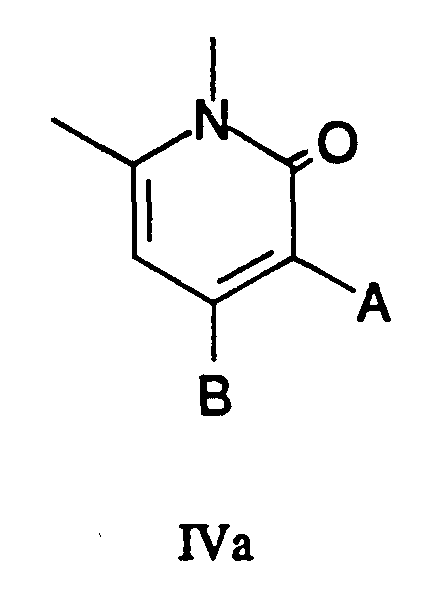
- R 4 and R 5 are joined together to form a substituted pyridone IIa:
- A H, COOR, or a functionality for preparation of the hydroxymethyl (C-17) portion of an E ring lactone
- B H, OH, an appropriate leaving group such as halide or
- the present invention also provides a process for the preparation of 7-(1,1-bis-alkoxycarbonyl)propyl-8-methoxycarbonyl-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-ones, 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-ones; and 1H- pyrano[3 , ,4 , :6,7]indolizino[1,2-b]quinolin-3,14(4H,12H)-diones, preferably camptothecin analogs, more preferably water-soluble camptothecin analogs, yet more preferably topotecan and irinotecan, most preferably topotecan, said process comprising the step of coupling a vinyl triflate derived from a 7-hydroxy-5,6-dihydroindolizino[1,2-b]quinolin-9(11
- the resulting 7-(1,1-bis-alkoxycarbonyl)propyl-8-methoxycarbonyl-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-one may be conveniently elaborated into a 1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-3,14(4H,12H)-dione or a 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-one as disclosed elsewhere in the present application.
- the present invention also provides a process for the preparation of pyrrolo[3,4-b]quinolines, 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-ones; and 1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-3,14(4H,12H)-diones, preferably camptothecin analogs, more preferably water-soluble camptothecin analogs, yet more preferably topotecan and irinotecan, most preferably topotecan, said process comprising the steps of:
- the present invention also provides a process for the preparation of camptothecin analogs which lack the lactone E ring, preferably 8-methyl-7-(oxopropyl)-indolizino[1,2-b]quinolin-9(11H)-ones, most preferably mappicines and mappicine ketones, said process comprising the steps of:
- the present invention provides a process for the total synthesis of, depending upon choice of termination step, 4-ethyl-4-hydroxy-9-methoxy-1H-pyrano[3',4':6,7] indolizino[1,2-b]quinolin-3,14(4H, 12H)-dione, more commonly known as 10-methoxycamptothecin, 4-ethyl-4,9-dihydroxy-1H-pyrano[3',4':6,7] indolizino[1,2-b]quinolin-3,14(4H,12H)-dione, more commonly known as 10-hydroxycamptothecin, as well as of 10-[(dimethylamino)methyl]-4-ethyl-4,9-dihydroxy-1H-pyrano[3',4 , :6,7]indolizino[1,2-b]quinolin-3,14(4H,12H)-dione, more commonly known as topotecan, said process comprising the
- tetrahydrofuran N,N-dimethylformamide, acetonitrile, acetone or N-methylpyrrolidinone, most preferably acetonitrile;at a temperature of about 20 -85°C, preferably about 60 - 85°C, most preferably at about the reflux temperature of acetonitrile; in the presence of a strong alkylatmg agent or such agent as is capable of transforming an amide into its corresponding O-alkylimidate or imidate ester, imidoyl halide, or a strong acid (such as aluminum chloride) capable of initiating a cyclodehydration, preferably trifluoromethanesulfonic anhydride, dimethylsulfate, alkyloxonium tetrafluoroborates, aluminum chloride, O-benzyltrichloroacetimidate, triphenylphosine/carbon tetrachloride, or triphenylphosphine/carbon tetrabromid
- a camptothecin includes camptothecin and and any derivative thereof the structure of which is based on the 1H-pyrano[3',4':6,7] indolizino-[1,2-b]quinolin-3,14(4H,12H)-dione ring system.
- pyrrolo[3,4-b]quinoline refers generally to compounds based on these ring systems.
- camptothecin analog includes camptothecins as defined above and also includes derivatives of camptothecin wherein the E ring has been replaced with another functionality.
- esteer protecting group is defined to include C 1 -C 6 alkyl groups as well as allyl, benzyl, phenyl and ⁇ , ⁇ , ⁇ -trichloroethyl.
- amine protecting group is defined to include alkyl and arylsulfonate esters, carbamates of common alkyl groups such as methyl, ethyl, ⁇ , ⁇ , ⁇ -trichloroethyl, allyl, tert-butyl and phenyl, amides such as acetamido and propionamido, as well as common alkyl groups such as methyl and benzyl.
- trimerkylsilyl is defined to include trimethyl, triethyl, triisopropyl and tripropylsilyl, as well as phenyldimethylsilyl and tert-butyldimethylsilyl.
- the present invention provides a process for preparing pyrrolo[3,4-b]quinolines, 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-ones; and 1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-3,14(4H,12H)-diones, preferably camptothecin analogs, more preferably water-soluble camptothecin analogs, yet more preferably topotecan and irinotecan, most preferably topotecan, the process comprising the step of intramolecular [4+2] cycloaddition of the N-arylimidate portion of a compound of Formula IV, generated from a starting material of Formula III, with the unactivated acetylene portion of said compound.
- Scheme 2 illustrates one embodiment of the present process wherein R 4 and R 5 of the compound of Formula IV are not joined together to form a pyridone
- X, R 1 -R 5 and Y are as defined for Formula IV;
- the starting material of Formula III is heated in a polar solvent, preferably methylene chloride, 1,2-dichloroethane, 1,2-dimethoxyethane tetrahydrofuran, N,N-dimethylformamide, acetonitrile, acetone or N-methylpy ⁇ olidinone, most preferably acetonitrile; at a temperature of about 20 - 85 °C, preferably 60 - 85 °C, most preferably at the reflux temperature of acetonitrile; in the presence of a strong alkylating agent or such agent as is capable of producing either an O-alkylimidate or imidate ester, imidoyl halide, or such imide derivative as is capable of
- cyclodehydration preferably trifluoromethanesulfonic anhydride, dimethylsulfate, alkyloxonium tetrafluoroborates, aluminum chloride, O-benzyltrichloroacetimidate, triphenylphosine/carbon tetrachloride, or triphenylphosphine/carbon tetrabromide, most preferably trimethyloxonium tetrafluoroborate, thereby generating the compound of Formula IV which then undergoes [4 + 2] cycloaddition to yield a pyrrolo[3,4-b]quinoline.
- Compounds of Formula IV have not been isolated, but have been detected.
- the products of the present process are either pharmaceutically useful themselves, e.g. topotecan, or are intermediates, e.g. 10-methoxycamptothecin, useful for elaboration into pharmaceutically useful compounds, particularly camptothecin analogs.
- the present invention also provides a process for the preparation of 7-(1,1-bis-alkoxycarbonyl)propyl-8-methoxycarbonyl-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-ones, 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-ones; and1H-pyrano [3',4': 6,7] indolizino [1,2-b] quinolin - 3,14 (4H, 12H - diones, preferably camptothecin analogs, more preferably water-soluble camptothecin analogs, yet more preferably topotecan and irinotecan, yet more preferably topotecan and irinotecan, most preferably topotecan, said process, referring to Scheme 3, comprising the step of coupling a vinyl triflate dervived from a 7-hydroxy-5,6-dihydroindoli
- the resulting 7-(1,1-bis-alkoxycarbonyl)propyl-8-methoxycarbonyl-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-one may be conveniently elaborated into a 1H-pyrano[3',4':6,7]indolizino-[1,2-b]quinolin-3,14(4H,12H)-dione or a 8-methyl-7-(oxopropyl)indolizino[1,2-b]quinolin-9(11H)-one as disclosed elsewhere in the present application.
- R 4 H or CH 2 COOEt
- R 5 COOMe, toluenesulfonyl, benzyl, COCH 3 ; or COCH 2 CH 3
- Y M.
- this embodiment of the present process may be conveniently used to provide a variety of useful
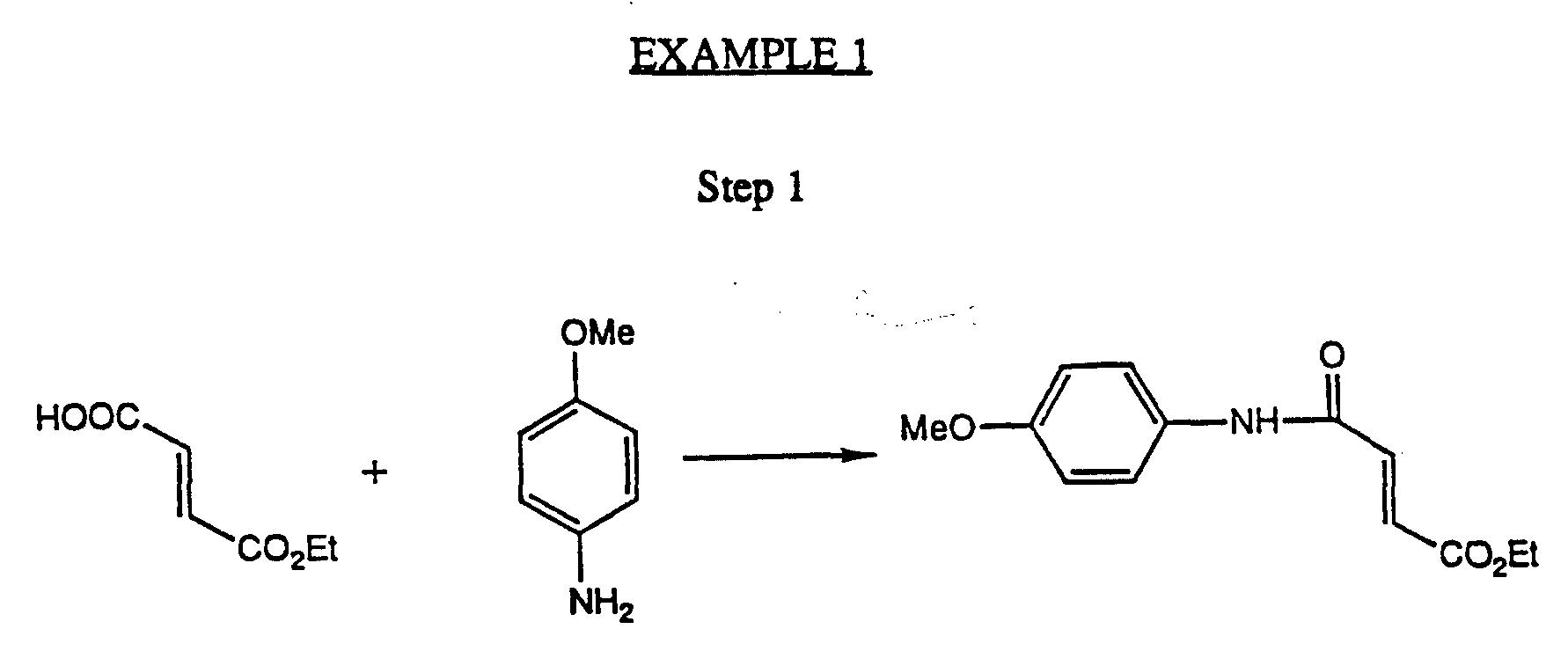
- the compounds of Formula IV are readily prepared by known methods, for example as shown in Scheme 5 and Steps 1-3 of Scheme 7, below, and as is exemplified in me Example (Steps 1-3 ).
- a polar solvent such as methylene chloride, 1,2-dichl ⁇ roethane, 1,2-dimethoxyethane tetrahydrofuran, N,N-dimethylformamide, acetonitrile, acetone or N-methylpyrrolidinone, but preferably acetonitrile; at a temperature of about 20 - 85 °C, preferably at 60 - 85 °C, most preferably at the reflux temperature of acetonitrile; in the presence of a strong alkylating agent or an agent capable of transforming an amide into its corresponding O-alkylimidate or imidate ester, or an imidoyl halide, e.g.
- trifluoromethanesulfonic anhydride dimethylsulfate, alkyloxonium tetrafluoroborates, aluminum chloride, O-benzyltrichloroacetimidate, triphenylphosine/carbon tetrachloride, or
- triphenylphosphine/carbon tetrabromide preferably trimethyloxonium
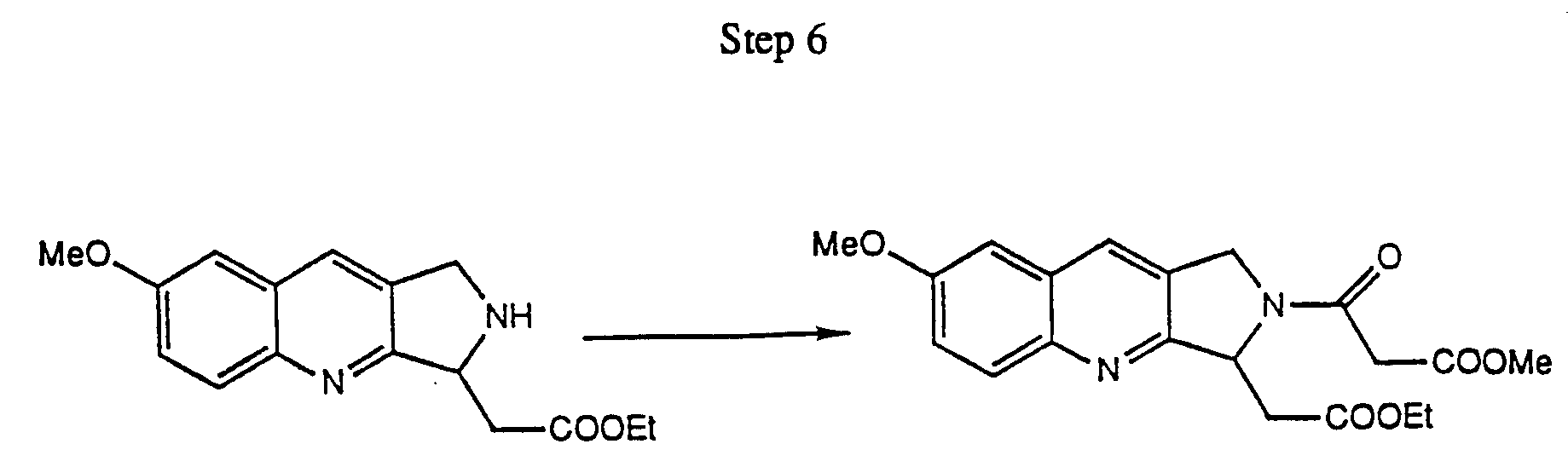
- step (b) the substituted pyrrolo[3,4-b]quinoline resulting from step (a) is cyclized to a 7-hydroxy-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-one as follows.
- the carbamate function protecting the C-ring nitrogen is hydrolytically cleaved, preferably with acetic acid saturated with HBr, to give the resulting tricyclic amine.
- the amine is then coupled at the C-ring nitrogen with a monoalkyl malonyl chloride, preferably monomethyl malonyl chloride, to give the resulting malonate half-amide.
- the D ring is formed when the malonate half-amide undergoes a Dieckmann condensation in the presence of base, preferably methoxide, to give a 7-hydroxy-8-methoxycarbonyl-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-one.
- step (c) the vinyl triflate of the 7-hydroxy-8-methoxycarbonyl-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-one is coupled with a tertiary malonate anion to form a 7-(1,1-di-tert-butoxycarbonyl)propyl-8-methoxycarbonyl-5,6-dihydroindolizino[1,2-b]quinolin-9(11H)-one.
- step (d) the diester is cyclized to form the lactone E ring of an indolizino[1,2-b]quinolinone, preferably a camptothecin analog.
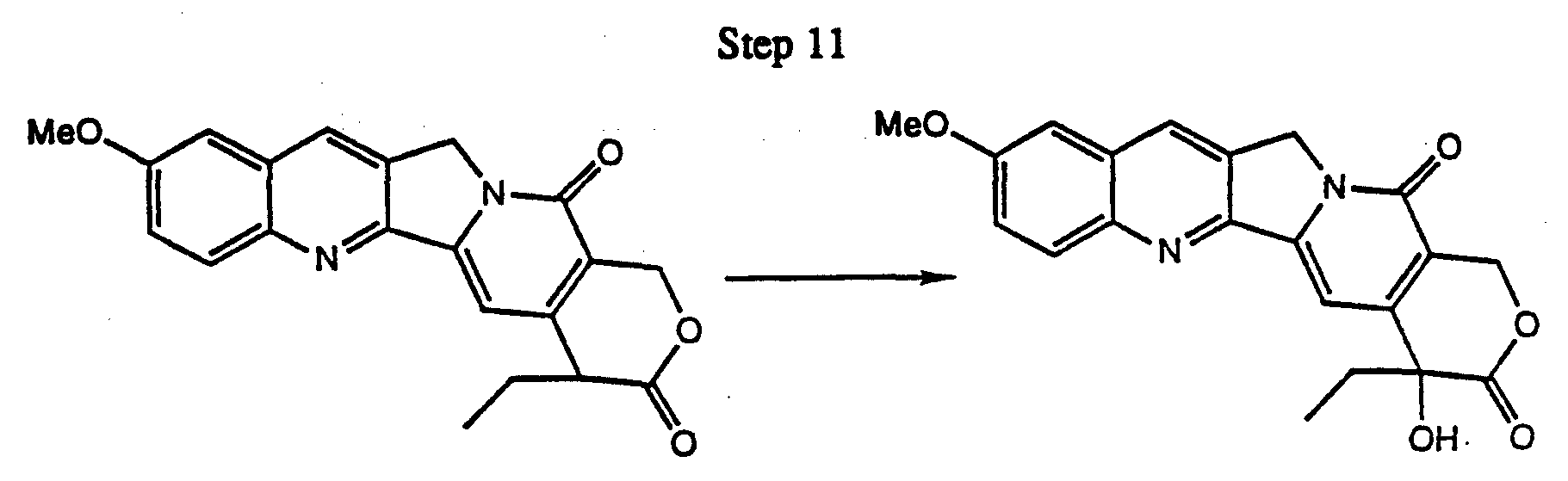
- 1,2 elimination of the tertiary hydrogen ⁇ to the quinolino nitrogen by oxidation in the presence of sodium nitrite or other oxidizing agents such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gives the analogous indolizino[1,2-b]quinolin-9(11H)-one, followed by reduction of the carbomethoxy group at C-8 to a hydroxymemyl function and hydrolysis/decarboxylation of the malonate diester to give the resulting 4-ethyl- 1H- pyrano[3',4':6,7] indolizino[1,2-b]quinolin-3,14(4H,12H)-dione.
- DDQ 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- the 4-ethyl-1H-pyrano[3',4':6,7] indolizino[1,2-b]quinolin-3,14(4H,12H)-dione may be further reacted to form a tertiary alcohol ⁇ to the E ring carbonyl by air-oxidation, thus giving a camptothecin.
- camptothecin may be further elaborated to give a variety of
- camptothecin analogs For instance, cleavage of the methoxy group of 4-ethyl-4-hydroxy-9-methoxy-1H-pyrano[3',4 , :6,7] indolizino[1,2-b]quinolin-3,14(4H,12H)-dione (10-methoxycamptothecin) with HBr yields 10-hydroxycamptothecin, which is itself pharmaceutically useful. 10-Hydroxycamptothecin may be conveniently converted to topotecan by reaction with N.N.N'.N'-tetramethyldiaminomemane (BDAM).
- BDAM N.N.N'.N'-tetramethyldiaminomemane
- camptothecin products of the present process may further be converted to camptothecin analogs in which the E ring has been replaced by some other functionality, e.g., mappicines and mappicine ketones, for instance by the process described in our recently allowed U.S. Ser. No. 07/589,848, which discloses a semisynthetic method of preparing certain 8-methyl-7-(-oxopropyl)indolizino[1,2-b]quinolin-9(11H)-ones from camptothecin or derivitized camptothecins.
- some other functionality e.g., mappicines and mappicine ketones
- the starting material is a compound of Formula in wherein R 4 and R 5 are joined together to form a substituted pyridone.
- the present invention provides a process for the preparation of camptothecin analogs which lack the lactone E ring, preferably 8-methyl-7-(oxopropyl)-indolizino [1,2-b]quinolin-9(11H)-ones, most preferably mappicines (1ib) and mappicine ketone (11a), said process comprising the steps of:
- camptothecin analog which lacks the lactone E ring, preferably a 8-methyl-7-(oxopropyl)-indolizino[1,2-b]quinolin-9(11H)-one, most preferably a mappicine or mappicine ketone.
- the present invention provides a process, as shown in Scheme 7, for the total synthesis of, depending upon choice of termination step, 4-ethyl-4-hydroxy-9-memoxy-1H-pyrano[3',4':6,7] indolizino[1,2-b]quinolin-3,14(4H, 12H)-dione, more commonly known as 10-methoxycamptothecin, 4-ethyl-4,9-dihydroxy-1H-pyrano[3',4 , :6,7] indolizino[1,2-b]quinolin-3,14(4H,12H)-dione, more commonly known as 10-hydroxycamptothecin, as well as of 10-[(dimethylamino)methyl]-4-ethyl-4,9-dihydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-3,14(4H,12H)-dione, more commonly known as topotecan, said
- tetrahydrofuran N,N-dimeti ⁇ ylformamide, acetonitrile, acetone or N-methylpyrrolidinone, most preferably acetonitrile;at a temperature of about 20 -85°C, preferably about 60 - 85°C, most preferably at about the reflux temperature of acetonitrile; in the presence of a strong alkylating agent or such agent as is capable of transforming an amide into its corresponding O-alkylimidate or imidate ester, imidoyl halide, or a strong acid (such as aluminum chloride) capable of initiating a cyclodehydration, preferably trifluoromethanesulfonic anhydride, dimethylsulfate, alkyloxonium tetrafluoroborates, aluminum chloride, O-benzyltrichloroacetimidate, triphenylphosine/carbon tetrachloride, or triphenylphosphine/carbon tetra
- step (b) hydrolyzing the carbamate function of the C-2 substituted pyrrolo[3,4-b]quinoline from step (a) with acetic acid saturated with HBr to give a tricyclic amine (X);
- Soduim borohydride (200 mg, 3 eq.) was added in small portions to this stirred solution and the resulting mixture was stirred for 15 minutes after addition was complete.
- the solvent was removed on a rotavapor and dilute hydrochloric acid (50 ml of 0.4% solution) was added to the residue.
- the mixture was extracted with 3 ⁇ 100 ml of methylene chloride.
- the combined organic layers were dried over magnesium sulfate and concentrated to give a dark solid to which trifluoroacetic acid (20 ml) was added.
- the mixture was stirred at ambient temperature for 1 h. Trifluoroacetic acid was removed on a rotavapor and water (50 ml) was added to the residue.
- the overall yield of topotecan for the total synthesis in 13 steps was about 5- 6%.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description























Claims





Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU51023/93A AU5102393A (en) | 1992-09-08 | 1993-09-08 | Process for preparing certain pyrrolo(3,4- b)quinolines, certain 1H-pyrano(3',4':6,7)indolizino(1,2-b)quinolin-3,14(4H,12H) -diones, and certain |
| EP93920496A EP0660835A4 (en) | 1992-09-08 | 1993-09-08 | Process for preparing certain pyrrolo 3,4- -i(b))quinolines, certain 1h-pyrano 3',4':6,7)indolizino 1,2--i(b))quinolin-3,14(4h,12h)-diones, and certain 8-methyl-7-(oxopropyl)-indolizino 1,2--i(b))quinolin-9(11h)-ones. |
| KR1019950700883A KR950702987A (en) | 1992-09-08 | 1993-09-08 | Specific pyrrolo [3,4-b] quinoline, specific 1H-pyrano [3 ', 4': 6,7] indolizino [1,2-b] quinoline-3,14 (4H, 12H) -dione And PROCESS FOR PREPARING CERTAIN PYRROLO [3,4-b], for the preparation of certain 8-methyl-7- (oxopropyl) -indolinino [1,2-b] quinolin-9- (11H) -ones. QUINOLINES, CERTAIN 1H-PYRANO [3 ′, 4 ′: 6,7] INDOLIZINO [1,2-b] QUINOLIN-3,14 (4H, 12H) -DIONES, AND CERTAIN 8-METHYL-7- (OXOPROPYL)- INDOLIZINO [1,2-b] QUINOLIN-9 (11H) -ONES |
| JP6507518A JPH08501104A (en) | 1992-09-08 | 1993-09-08 | Certain pyrrolo [3,4-b quinolines, certain 1H-pyrano [3 ', 4': 6,7 indolizino [1,2-b quinoline-3,14 (4H, 12H) -diones and Process for the production of certain 8-methyl-7- (oxopropyl) -indolidino [1,2-bquinolin-9 (11H) -ones |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US94149692A | 1992-09-08 | 1992-09-08 | |
| US07/941,496 | 1992-09-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994005672A1 true WO1994005672A1 (en) | 1994-03-17 |
Family
ID=25476581
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1993/008434 Ceased WO1994005672A1 (en) | 1992-09-08 | 1993-09-08 | PROCESS FOR PREPARING CERTAIN PYRROLO[3,4- b]QUINOLINES, CERTAIN 1H-PYRANO[3',4':6,7]INDOLIZINO[1,2-b]QUINOLIN-3,14(4H,12H)-DIONES, AND CERTAIN 8-METHYL-7-(OXOPROPYL)-INDOLIZINO[1,2-b]QUINOLIN-9(11H)-ONES |
Country Status (11)
| Country | Link |
|---|---|
| EP (1) | EP0660835A4 (en) |
| JP (1) | JPH08501104A (en) |
| KR (1) | KR950702987A (en) |
| CN (1) | CN1096297A (en) |
| AU (2) | AU5102393A (en) |
| CA (1) | CA2144048A1 (en) |
| MX (1) | MX9305517A (en) |
| NZ (1) | NZ255942A (en) |
| TW (1) | TW246674B (en) |
| WO (1) | WO1994005672A1 (en) |
| ZA (1) | ZA936580B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6660861B1 (en) * | 2003-03-27 | 2003-12-09 | Council Of Scientific And Industrial Research | Process for preparing Topotecan from 10-hydroxy-4-(S) camptothecin |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN2757510Y (en) | 2004-12-06 | 2006-02-08 | 鸿富锦精密工业(深圳)有限公司 | Radiator buckle |
| US7520313B2 (en) | 2006-03-16 | 2009-04-21 | Fu Zhun Precision Industry (Shen Zhen) Co., Ltd. | Locking device for heat sink |
| CN105859716B (en) * | 2016-05-06 | 2018-03-09 | 华东师范大学 | In a kind of synthesis camptothecine compounds 56 and ring structure method |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS49117491A (en) * | 1973-03-26 | 1974-11-09 | ||
| JPS5217499A (en) * | 1975-07-31 | 1977-02-09 | Nippon Chemiphar Co Ltd | Preparation of mappicine |
| DE2534601A1 (en) * | 1975-08-02 | 1977-02-17 | Basf Ag | Camptothecin analogues prepn. - involving alkylation prior to reduction and lactonization of chloro-camptothecin precursors |
| US4914205A (en) * | 1987-06-25 | 1990-04-03 | Kabushiki Kaisha Yakult Honsha | Camptothecin derivatives |
| JPH0383986A (en) * | 1989-08-29 | 1991-04-09 | Yakult Honsha Co Ltd | New quinoline derivative and its production |
| US5155225A (en) * | 1990-09-28 | 1992-10-13 | Smithkline Beecham Corporation | Method for making certain pyrano[3',4':6,7]indolizino-[1,2-B]quinolinones |
-
1993
- 1993-09-07 ZA ZA936580A patent/ZA936580B/en unknown
- 1993-09-08 WO PCT/US1993/008434 patent/WO1994005672A1/en not_active Ceased
- 1993-09-08 NZ NZ255942A patent/NZ255942A/en unknown
- 1993-09-08 AU AU51023/93A patent/AU5102393A/en not_active Abandoned
- 1993-09-08 MX MX9305517A patent/MX9305517A/en unknown
- 1993-09-08 CN CN93119287A patent/CN1096297A/en active Pending
- 1993-09-08 CA CA002144048A patent/CA2144048A1/en not_active Abandoned
- 1993-09-08 EP EP93920496A patent/EP0660835A4/en not_active Withdrawn
- 1993-09-08 KR KR1019950700883A patent/KR950702987A/en not_active Withdrawn
- 1993-09-08 JP JP6507518A patent/JPH08501104A/en active Pending
- 1993-11-02 TW TW082109133A patent/TW246674B/zh active
-
1997
- 1997-07-11 AU AU28591/97A patent/AU2859197A/en not_active Abandoned
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS49117491A (en) * | 1973-03-26 | 1974-11-09 | ||
| JPS5217499A (en) * | 1975-07-31 | 1977-02-09 | Nippon Chemiphar Co Ltd | Preparation of mappicine |
| DE2534601A1 (en) * | 1975-08-02 | 1977-02-17 | Basf Ag | Camptothecin analogues prepn. - involving alkylation prior to reduction and lactonization of chloro-camptothecin precursors |
| US4914205A (en) * | 1987-06-25 | 1990-04-03 | Kabushiki Kaisha Yakult Honsha | Camptothecin derivatives |
| JPH0383986A (en) * | 1989-08-29 | 1991-04-09 | Yakult Honsha Co Ltd | New quinoline derivative and its production |
| US5155225A (en) * | 1990-09-28 | 1992-10-13 | Smithkline Beecham Corporation | Method for making certain pyrano[3',4':6,7]indolizino-[1,2-B]quinolinones |
Non-Patent Citations (6)
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6660861B1 (en) * | 2003-03-27 | 2003-12-09 | Council Of Scientific And Industrial Research | Process for preparing Topotecan from 10-hydroxy-4-(S) camptothecin |
Also Published As
| Publication number | Publication date |
|---|---|
| AU5102393A (en) | 1994-03-29 |
| KR950702987A (en) | 1995-08-23 |
| EP0660835A4 (en) | 1995-08-16 |
| MX9305517A (en) | 1994-05-31 |
| TW246674B (en) | 1995-05-01 |
| NZ255942A (en) | 1996-11-26 |
| AU2859197A (en) | 1997-10-23 |
| JPH08501104A (en) | 1996-02-06 |
| EP0660835A1 (en) | 1995-07-05 |
| CA2144048A1 (en) | 1994-03-17 |
| CN1096297A (en) | 1994-12-14 |
| ZA936580B (en) | 1994-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5244903A (en) | Camptothecin analogs as potent inhibitors of topoisomerase I | |
| FI91872C (en) | Process for the preparation of therapeutically active pyranoindolizinoquinoline derivatives | |
| US5106742A (en) | Camptothecin analogs as potent inhibitors of topoisomerase I | |
| US4981968A (en) | Synthesis of camptothecin and analogs thereof | |
| ES2254600T3 (en) | NEW ANALOGS OF CAMPTOTECHINE, PREPARATION PROCEDURES, ITS APPLICATION AS MEDICATIONS AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. | |
| WO1998035940A1 (en) | Highly lipophilic camptothecin derivatives | |
| US5364858A (en) | Camptothecin analogs as potent inhibitors of topoisomerase I | |
| US5405963A (en) | Process for asymmetric total synthesis of camptothecin analogues | |
| WO1996021666A1 (en) | Camptothecin derivatives and its manufacturing method | |
| NZ520835A (en) | Alkylated imidazopyridine derivatives | |
| EP0555347A1 (en) | SUBSTITUTED INDOLIZINO 1,2-b]QUINOLINONES | |
| JP2003528879A (en) | Tricyclic imidazopyridine | |
| WO1994005672A1 (en) | PROCESS FOR PREPARING CERTAIN PYRROLO[3,4- b]QUINOLINES, CERTAIN 1H-PYRANO[3',4':6,7]INDOLIZINO[1,2-b]QUINOLIN-3,14(4H,12H)-DIONES, AND CERTAIN 8-METHYL-7-(OXOPROPYL)-INDOLIZINO[1,2-b]QUINOLIN-9(11H)-ONES | |
| NZ248841A (en) | Pyrido[4,3-b]carbazole derivatives (or ellipticines) and medicaments | |
| MXPA04011682A (en) | Camptothecins with a modified lactone ring. | |
| CA2161318A1 (en) | New anthraquinonic derivatives having an antitumor activity, and applications thereof | |
| Albright et al. | Synthesis of 1, 4, 5, 6‐tetrahydropyrazolo [3, 4‐d] pyrido [3, 2‐b] azepine | |
| IL144987A (en) | Homocamptothecin derivatives and medicaments containing them | |
| US6169080B1 (en) | Highly lipophilic camptothecin derivatives | |
| US6500953B1 (en) | Preparation of camptothecin and nothapodytine derivatives | |
| Hepburn et al. | A concise synthesis of further thiophene analogues of kuanoniamine a | |
| Groundwater et al. | Synthesis of pyrido [2, 3-c] acridines | |
| Hermecz et al. | A ring transformation of 6, 7-dihydro-4H-pyrido [1, 2-a] pyrimidin-4-ones | |
| MXPA01000767A (en) | Preparation of camptothecin and nothapodytine derivatives | |
| JPH0517479A (en) | Fluoroethylcamptothecin derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CZ FI HU JP KP KR KZ LK LV MG MN MW NO NZ PL RO RU SD SK UA US VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 255942 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2144048 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993920496 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 1995 397214 Country of ref document: US Date of ref document: 19950407 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993920496 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993920496 Country of ref document: EP |