WO1994007873A1 - Prodedimiento para la obtencion de benzoxazinas utiles para la sintesis de ofloxacina, levofloxacina y derivados - Google Patents
Prodedimiento para la obtencion de benzoxazinas utiles para la sintesis de ofloxacina, levofloxacina y derivados Download PDFInfo
- Publication number
- WO1994007873A1 WO1994007873A1 PCT/ES1993/000080 ES9300080W WO9407873A1 WO 1994007873 A1 WO1994007873 A1 WO 1994007873A1 ES 9300080 W ES9300080 W ES 9300080W WO 9407873 A1 WO9407873 A1 WO 9407873A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ofloxacin
- obtaining
- methyl
- formula
- difluoro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*C(*)=C(C)NC1=CC*C(C)=C1OCC(CC*I)O Chemical compound C*C(*)=C(C)NC1=CC*C(C)=C1OCC(CC*I)O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/34—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings
- C07D265/36—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings condensed with one six-membered ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Definitions
- This invention describes a new method for the synthesis of benzoxazine derivatives of general formula (I)
- R 1 is H, an alkyl radical of up to 6 carbon atoms, preferably methyl, an alkenyl radical of up to 6 carbon atoms or an aryl group;
- Xb is a halogen atom.
- Ofloxacin [9-fluoro-3-methyl-10- (4-methyl-1-piperazinyl) -7-oxo-2,3 dihydro-7H-pyrido (l, 2,3-de) (1,4) -benzoxazine-6-carboxylic acid] responds to the formula: described in the patents: Japanese n ⁇ 1,444,043; US 4,382,892 and European ⁇ n n ⁇ EP-B-0,047, 005.
- the Ofloxacin is an excellent agent synthetic antimicrobial, as mentioned in Japanese Patent Application 46986/92 ns (OPI), which has been marketed in large number of countries
- Ofloxacin has an asymmetric carbon atom at position 3 and is usually obtained as a racemate by known procedures. However, by means of the process of the present invention, any of its optically active forms can be obtained directly without the need for any resolution step.
- the 3- (S) - of the Ofloxacin isomer known as Levofloxacin
- Levofloxacin possesses (i) an antimicrobial activity approximately two times greater than that of the racemic, confirming the aforementioned in European Patent ns EP-A-0,206,283 and (ii) an acute toxicity (LD 50 ) weaker than that of the racemic compound, determined in mice by intravenous administration.
- the isomer (R) has (i) an antimicrobial activity between ten and one hundred times less than that of the racemic compound and (ii) has an acute toxicity substantially equal to that of said racemic.
- the form (S) of Ofloxacin (Levofloxacin) has very interesting properties, such as antimicrobial activity increased and reduced toxicity, so presumably it will be a pharmaceutically more useful compound than the racemic. Furthermore, it has been determined that both the (R) - Ofloxacin form and the (S) -Ofloxacin form in free form have a more pronounced water solubility than that of the compound (R, S), so they can be used In injectable preparations.
- Spanish Patent No. 553520 describes a process for obtaining Ofloxacin by hydrolysis of the precursor nitrile 9-fluorine-3-methyl-10- (4-methyl-l-piperazinyl) -7-oxo-2,3-dihydro-7H- pyrido (l, 2,3-de) -l, 4-benzoxazine-6-carbonitrile.
- the present invention provides a process for the preparation of benzoxazine derivatives of formula (I):
- R 1 is H, an alkyl radical of up to 6 carbon atoms, preferably methyl, an alkenyl radical of up to 6 carbon atoms or an aryl group;
- Xb is a halogen atom, preferably fluorine.
- alkyl radical of up to 6 carbon atoms means, in the sense used in this description, any radical derived from a straight or branched chain alkane of up to 6 carbon atoms inclusive, such as methyl, ethyl, propyl, isopropyl.
- alkenyl radical of up to 6 carbon atoms means any radical derived from a linear or branched chain alkene of up to 6 carbon atoms, even having one or more degrees of unsaturation at any position in the chain.
- aryl should be understood as a phenyl group, optionally substituted with one or more alkyl groups or with one or more halogen atoms.
- R 1 and Xb have the meanings indicated above.
- An object of the present invention is to provide these intermediates of formula (I) by a new synthetic route, which allows obtaining racemic Ofloxacin and its enantiomers.
- a further object of the present invention is the preparation of these intermediates by a new process, for use in the synthesis of the aforementioned antimicrobial compounds.
- Another object of the present invention is the use of racemic or enantiomerically pure epoxides which, in a particular and preferred case of the present invention, may be (R, S) -propylene oxide or (R) -propylene oxide in one of the stages of the synthesis, which would lead, in a particular preferred case, to the preparation of intermediate (3,4-difluoro-2- (2- Enantiomerically pure diethyl hydroxypropoxy) anilino) methylemalmalonate, which incorporated into the synthetic route of Ofloxacin, would lead to the formation of Levofloxacin, no step of enantiomer resolution being necessary.
- the compounds of formula (I) of the present invention can be prepared by the process which, applied to the particular case mentioned above, is described below.
- the 3,4-difluoroaniline compound can be converted into benzoxazine derivatives (identified as compound [6] in The reaction scheme corresponds to a compound of formula (I) in which Xb is F and R 1 is methyl), and subsequently in Ofloxacin and other quinolone structurally related by a series of reactions, which appear in the attached scheme.
- the products obtained can be isolated or purified, by known methods, such as: extraction, recrystallization, flash chromatography or combinations thereof.
- Step a The protection of the 3,4-difluoroaniline amino group is carried out with a suitable protecting group, such as (BOC) 2 O (Diterbutyldicarbonate) in low polar solvents, such as ethers, chlorinated solvents, and the like, at temperatures comprised between 30 ⁇ and ioosc.
- a suitable protecting group such as (BOC) 2 O (Diterbutyldicarbonate) in low polar solvents, such as ethers, chlorinated solvents, and the like, at temperatures comprised between 30 ⁇ and ioosc.
- BOC is a suitable protecting group, such as (BOC) 2 O (Diterbutyldicarbonate) in low polar solvents, such as ethers, chlorinated solvents, and the like, at temperatures comprised between 30 ⁇ and ioosc.
- the BOC group acts not only as a blocking agent, but also to achieve an "ortho director" effect, which the
- this step can also be performed advantageously and preferably using n-heptane as a solvent and heating at the reflux temperature of the solvent.
- Step b To carry out this step, a strong base, usually an alkyl lithium, is required to cause orthometalation, which is completely regioselective in position 2 at low temperatures, continuing the sequence with. the addition of trimethyl borate, also at low temperatures. Generally, this step can be performed at a temperature between -50 ⁇ and -90 ⁇ C, preferably at -78 ⁇ C. Subsequently, oxidation in an acid medium of arylborate with hydrogen peroxide leads to the corresponding phenol [2]. In the reaction, inert and low polar solvents are used.
- Step c Deprotection of the amino group is carried out in a conventional manner, known per se, for example, by treatment with dilute acids.
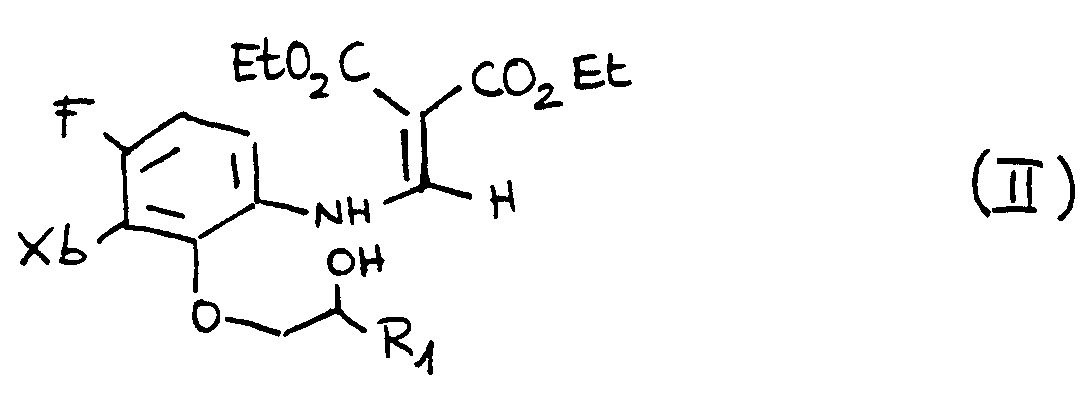
- Step d It is carried out by reaction of 6-amino-2,3-difluorophenol [3] with a compound of the type:
- Y-CH C- (COOEt) 2 where Y represents an alkoxy group.
- the reaction can be carried out in the absence or in the presence of solvents that are inert, such as n-hexane or the like.
- the reaction can be carried out in a temperature range between 90 ⁇ and 180 ⁇ C, if it is carried out in the absence of solvent, or at temperatures close to the boiling point of the solvent when it is carried out in the presence of solvents.
- Step e Formation of the derived oxybenzene (II) by opening epoxides of formula where R 1 is the one defined above (in the particular case shown in the reaction scheme R 1 is methyl). It is a reaction in which a catalyzed opening of the epoxide occurs, under mild temperature conditions and adequate catalyst proportions.
- this step can be carried out at a temperature between 0 ⁇ and 60 ⁇ C, preferably between 20 ⁇ and 50 ⁇ C, and more preferably at 40 ⁇ C, in the presence of a catalyst that can be a Lewis acid, such as boron trifluoride, zinc bromide, magnesium bromide, lithium perchlorate and in the presence of a base such as, preferably, sodium hydride.
- a catalyst that can be a Lewis acid, such as boron trifluoride, zinc bromide, magnesium bromide, lithium perchlorate
- a base such as, preferably, sodium hydride.
- Molar ratios of the reagents (starting product / catalyst / epoxide) involved in this step can be 1-1.5 / 2-6 / 1.5-3, respectively.
- this reaction step if the optically pure suitable chiral epoxide is used, the formation of the enantiomerically pure compound [5] is achieved, which by continuing the synthetic route provides the two Ofloxacin enantiomers, no resolution step being necessary by this method .
- this reaction step can be performed in an aprotic solvent.
- Step f Cyclization of the derived oxybenzene [5] is carried out stereoselectively using as triphenylphosphine (TPP) and ethyl azodicarboxylate reagents in a low polar inert organic solvent, such as tetrahydrofuran, at a temperature between 0 ⁇ and 60 ⁇ c, preferably between 20 ⁇ and 30 ⁇ c, and more preferably at room temperature, for a period of time between 12 and 30 hours, preferably 18 hours.
- TPP triphenylphosphine
- ethyl azodicarboxylate reagents in a low polar inert organic solvent, such as tetrahydrofuran
- Steps g, h They are described in European Patent Application EP 0206283 A2 ⁇ n, whereby the core of quinolones and Ofloxacin is obtained. Additionally, obtaining quinolone ⁇ derivatives (optionally substituted in position 7 with piperazine) from methylenemalonate derivatives can be carried out following the Gould-Jacobs sequence (thermal or catalyzed cyclization followed by hydrolysis of the ester and replacement of fluorine by piperazine) [ Hayakawa et al. , Chem. Pharm. Bull., 1984, 32, 4907; Koga et al., J. Med. Chem., 1980, 23, 1358; Albrecht R., Prog. Drug. Res., 1977, 21, 9-104].
- Step i The 9,10-difluoro-3-methyl-7-oxo-2,3-dihydro-7H-pyrido [l, 2,3-de] benzoxazine-6-carboxylic acid [8] obtained is treated with N-methylpiperazine in an inert polar suitable as DMSO (dimethyl sulfoxide) or acetonitrile, preferably DMSO, at a temperature between 50 ⁇ and 150 ⁇ c, preferably at a temperature between 100 ⁇ and 120 ⁇ C solvent, and optionally, in the presence of a base such as triethylamine, whereby Ofloxacin [9] is obtained which may be in its racemic form if (R, S) -propylene oxide or in its pure enantiomeric forms such as as, for example, in the form 3- (S) - or Levofloxacin if (R) -propylene oxide has been used.
- DMSO dimethyl sulfoxide
- this step (i) can be advantageously carried out, in the absence of base, using DMSO as solvent and heating at a temperature of about 100 ° C for about two hours.
- the product obtained is dissolved in 100 ml of DMSO, adding 14.2 ml of triethylamine and
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description








Claims








Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP6508738A JPH07501835A (ja) | 1992-10-07 | 1993-10-06 | オフロキサジン,レボフロキサジン及び誘導体の合成に使用されるベンゾキサジンの製造法 |
| CA002125287A CA2125287A1 (en) | 1992-10-07 | 1993-10-06 | Process to obtain benzoxazines to be used for the synthesis of ofloxazine, levofloxazine and derivatives |
| KR1019940701925A KR0131914B1 (ko) | 1992-10-07 | 1993-10-06 | 오플로옥사진, 레보플로옥사진과 이 유도체의 합성에 이용될 수 있는 벤조옥사진의 수득과정 |
| AU51118/93A AU674542B2 (en) | 1992-10-07 | 1993-10-06 | Process for obtaining benzoxazines useful for the synthesis of ofloxacin, levofloxacin and derivatives thereof |
| US08/244,455 US5521310A (en) | 1992-10-07 | 1993-10-06 | Process to obtain benzoxazines to be used for the synthesis of ofloxazine, levofloxazine and derivatives |
| EP93921930A EP0619311A1 (en) | 1992-10-07 | 1993-10-06 | Process for obtaining benzoxazines useful for the synthesis of ofloxacin, levofloxacin and derivatives thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES9201983A ES2055656B1 (es) | 1992-10-07 | 1992-10-07 | Procedimiento para la obtencion de benzoxazinas utiles para sintesis de ofloxacina, levofloxacina y derivados. |
| ESP9201983 | 1992-10-07 | ||
| ES9302080A ES2069500B1 (es) | 1992-10-07 | 1993-10-04 | Perfeccionamientos introducidos en el objeto de la patente espa¦ola n 9201983 por "procedimiento para la obtencion de benzoxazinas utiles para la sintesis de oflexacina, levofloxacina y derivados. |
| ESP9302080 | 1993-10-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994007873A1 true WO1994007873A1 (es) | 1994-04-14 |
Family
ID=26154661
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/ES1993/000080 Ceased WO1994007873A1 (es) | 1992-10-07 | 1993-10-06 | Prodedimiento para la obtencion de benzoxazinas utiles para la sintesis de ofloxacina, levofloxacina y derivados |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US5521310A (es) |
| EP (1) | EP0619311A1 (es) |
| JP (1) | JPH07501835A (es) |
| KR (1) | KR0131914B1 (es) |
| AU (2) | AU674542B2 (es) |
| CA (1) | CA2125287A1 (es) |
| WO (1) | WO1994007873A1 (es) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5811550A (en) * | 1992-11-20 | 1998-09-22 | Rhone-Poulenc Rorer, S.A. | Process for the preparation of a 1,3-oxazolidine-5-carboxylic acid |
| US7902227B2 (en) | 2007-07-27 | 2011-03-08 | Janssen Pharmaceutica Nv. | C-7 isoxazolinyl quinolone / naphthyridine derivatives useful as antibacterial agents |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1055927C (zh) * | 1997-11-18 | 2000-08-30 | 中国科学院上海药物研究所 | 左旋氧氟沙星类似物的合成及其用途 |
| JP3201998B2 (ja) * | 1998-12-16 | 2001-08-27 | サムソン ジェネラル ケミカルズ カンパニー リミテッド | (s)−ベンゾオキサジン誘導体の製造方法及び(r)−ベンゾオキサジン誘導体のラセミ化方法 |
| CA2380359C (en) | 1999-09-08 | 2011-11-29 | Daiichi Pharmaceutical Co., Ltd. | Process for producing benzoxazine derivative and production intermediate thereof |
| US7425628B2 (en) * | 2001-10-03 | 2008-09-16 | Teva Pharmaceutical Industries Ltd. | Methods for the purification of levofloxacin |
| WO2003028664A2 (en) * | 2001-10-03 | 2003-04-10 | Teva Pharmaceutical Industries Ltd. | Preparation of levofloxacin and forms thereof |
| KR100440192B1 (ko) * | 2001-11-22 | 2004-07-12 | 이수화학 주식회사 | 광학 활성 퀴놀론카르복실산 유도체의 제조방법 |
| AU2002365416A1 (en) * | 2001-11-29 | 2003-06-10 | Teva Pharmaceutical Industries Ltd. | Methods for the purification of levofloxacin |
| CN102101865A (zh) * | 2009-12-22 | 2011-06-22 | 江苏九寿堂生物制品有限公司 | 一种喹诺酮类化合物盐酸盐的结晶形式 |
| CN101880288B (zh) * | 2010-07-12 | 2012-02-22 | 浙江东亚药业有限公司 | 一种氧氟沙星的合成方法 |
| CN105037388A (zh) * | 2015-08-28 | 2015-11-11 | 安徽环球药业股份有限公司 | 一种安妥沙星的制备方法 |
| CN109627407B (zh) * | 2018-12-26 | 2021-03-23 | 四川理工学院 | 一种原位还原氧化石墨烯/苯并噁嗪复合材料的制备方法及其产品 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0322815A2 (en) * | 1987-12-25 | 1989-07-05 | Daiichi Pharmaceutical Co., Ltd. | Propoxybenzene derivatives and process for preparing the same |
| JPH02218648A (ja) * | 1989-02-17 | 1990-08-31 | Dai Ichi Seiyaku Co Ltd | プロポキシニトロベンゼン類の製法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5746986A (en) * | 1980-09-02 | 1982-03-17 | Dai Ichi Seiyaku Co Ltd | Pyrido(1,2,3-de)(1,4)benzoxazine derivative |
| NO166131C (no) * | 1985-06-20 | 1991-06-05 | Daiichi Seiyaku Co | Analogifremgangsmaate for fremstilling av s(-)-pyridobenzoksazinforbindelser. |
| DE3522406A1 (de) * | 1985-06-22 | 1987-01-02 | Bayer Ag | Verfahren zur herstellung von 1,8-verbrueckten 4-chinolon-3-carbonsaeuren |
| DE3639465A1 (de) * | 1986-11-18 | 1988-05-19 | Hoechst Ag | Optisch aktive gyrasehemmer, ihre herstellung und verwendung als antibiotika |
-
1993
- 1993-10-06 AU AU51118/93A patent/AU674542B2/en not_active Ceased
- 1993-10-06 US US08/244,455 patent/US5521310A/en not_active Expired - Fee Related
- 1993-10-06 EP EP93921930A patent/EP0619311A1/en not_active Withdrawn
- 1993-10-06 KR KR1019940701925A patent/KR0131914B1/ko not_active Expired - Fee Related
- 1993-10-06 JP JP6508738A patent/JPH07501835A/ja active Pending
- 1993-10-06 CA CA002125287A patent/CA2125287A1/en not_active Abandoned
- 1993-10-06 WO PCT/ES1993/000080 patent/WO1994007873A1/es not_active Ceased
-
1996
- 1996-09-27 AU AU65878/96A patent/AU686955B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0322815A2 (en) * | 1987-12-25 | 1989-07-05 | Daiichi Pharmaceutical Co., Ltd. | Propoxybenzene derivatives and process for preparing the same |
| JPH02218648A (ja) * | 1989-02-17 | 1990-08-31 | Dai Ichi Seiyaku Co Ltd | プロポキシニトロベンゼン類の製法 |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 114, no. 9, 1991, Columbus, Ohio, US; abstract no. 81231y, T. FUJIWARA ET AL.: "Preparation of 2-propoxynitrobenzene derivatives as intermediates for DR-3355, (S)-ofloxacin" page 674; * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5811550A (en) * | 1992-11-20 | 1998-09-22 | Rhone-Poulenc Rorer, S.A. | Process for the preparation of a 1,3-oxazolidine-5-carboxylic acid |
| US7902227B2 (en) | 2007-07-27 | 2011-03-08 | Janssen Pharmaceutica Nv. | C-7 isoxazolinyl quinolone / naphthyridine derivatives useful as antibacterial agents |
Also Published As
| Publication number | Publication date |
|---|---|
| AU6587896A (en) | 1996-12-12 |
| AU5111893A (en) | 1994-04-26 |
| AU686955B2 (en) | 1998-02-12 |
| AU674542B2 (en) | 1997-01-02 |
| EP0619311A1 (en) | 1994-10-12 |
| JPH07501835A (ja) | 1995-02-23 |
| KR0131914B1 (ko) | 1998-04-17 |
| CA2125287A1 (en) | 1994-04-14 |
| US5521310A (en) | 1996-05-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1994007873A1 (es) | Prodedimiento para la obtencion de benzoxazinas utiles para la sintesis de ofloxacina, levofloxacina y derivados | |
| US4777253A (en) | Process for preparation of racemate and optically active ofloxacin and related derivatives | |
| KR0125115B1 (ko) | (-)피페라진 벤즈옥사진 유도체의 제조방법 | |
| CA2338498C (en) | Process for the synthesis of (1h)-benzo[c]quinolizin-3-ones derivatives | |
| KR100309871B1 (ko) | (-)피리도벤즈옥사진 카르복실산 유도체의 제조방법 | |
| JPH0144717B2 (es) | ||
| ES2409740T3 (es) | Procedimiento de preparación de monoclorhidrato de 8-hidroxi-5-[(1R)-1-hidroxi-2[[(1R)-2-(4-metoxifenil)-1- metiletil]amino]-etil]-2(1H)-quinolinona | |
| KR900004145B1 (ko) | 트리사이클릭 화합물의 제조방법 | |
| US4895944A (en) | Benzoxazine intermediates | |
| US4826985A (en) | Intermediates for preparation of racemate and optically active ofloxacin and related derivatives | |
| US4183943A (en) | Organic compounds | |
| KR100880654B1 (ko) | 광호변성 옥사진 화합물의 제조 방법 | |
| ES2292776T3 (es) | Sintesis total de la galantamina, de sus analogos y de sus derivados. | |
| Okada et al. | Synthesis and antibacterial activities of novel dihydrooxazine and dihydrothiazine ring‐fused tricyclic quinolonecarboxylic acids: 9‐Fluoro‐3‐methylene‐10‐(4‐methylpiperazin‐1‐yl)‐7‐oxo‐2, 3‐dihydro‐7h‐pyrido [1, 2, 3‐de][1, 4] benzoxazine‐6‐carboxylic acid and its 1‐thia congener | |
| IE45729B1 (en) | Rifamycin sv derivatives | |
| KR100355756B1 (ko) | S-오플로삭신의 제조방법 | |
| KR100494881B1 (ko) | 에틸 2-[2,3,5-트리플루오로-4-(4-메틸-1-피페라지닐)]벤조일-3(s)-(1-히드록시프로프-2-일아미노)아크릴레이트 및 그의 제조방법 | |
| JP2724383B2 (ja) | (s)−ベンゾオキサジン誘導体の製法 | |
| KR960004825B1 (ko) | 신규한 퀴놀론계 화합물 및 그의 제조방법(ⅲ) | |
| SAIGA et al. | Synthesis of 1, 2, 3, 4-Tetrahydro-β-carboline Derivatives as Hepatoprotective Agents. II. Alkyl 1, 2, 3, 4-Tetrahydro-β-carboline-2-carbodithioates | |
| EP0675880B1 (en) | Quinoline disulfides as intermediates | |
| KR940008422B1 (ko) | 신규한 3, 6-디아자비시클로[3, 1, 0]헥산 화합물과 그의 제조방법 | |
| JP2004035502A (ja) | オクタヒドロシクロペンタ〔c〕ピリジン誘導体およびその製造法 | |
| EP0768303A1 (en) | Optically active quinolinecarboxylic acid derivative and process for producing the same | |
| EP1939206A1 (en) | Process for the preparation of an antibacterial quinolone compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AT AU BB BG BR CA CH CZ DE DK FI GB HU JP KP KR LK LU MG MN MW NL NO NZ PL PT RO RU SD SE SK UA US VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2125287 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993921930 Country of ref document: EP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 08244455 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993921930 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993921930 Country of ref document: EP |