WO1994008983A1 - Amide derivatives - Google Patents
Amide derivatives Download PDFInfo
- Publication number
- WO1994008983A1 WO1994008983A1 PCT/GB1993/002090 GB9302090W WO9408983A1 WO 1994008983 A1 WO1994008983 A1 WO 1994008983A1 GB 9302090 W GB9302090 W GB 9302090W WO 9408983 A1 WO9408983 A1 WO 9408983A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- amide
- give
- defined above
- carbon atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *O[C@](C(O*)=O)c1ccccc1 Chemical compound *O[C@](C(O*)=O)c1ccccc1 0.000 description 2
- YGCKKQVHQRXACX-UHFFFAOYSA-N CC(CCO)C(N1CCCCCC1)=O Chemical compound CC(CCO)C(N1CCCCCC1)=O YGCKKQVHQRXACX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/02—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C233/11—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to carbon atoms of an unsaturated carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/145—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
Definitions
- This invention relates to a novel asymmetric synthesis for preparing amide derivatives and to intermediates useful in the synthesis.
- the invention particularly relates to a process for preparing optically active amides of the general formula
- I I X represents -N- or -CH- ,
- R represents a mono or bicyclic aryl or heteroaryl group
- R is an aryl or heteroaryl radical
- R is hydrogen or lower alkyl
- R is hydrogen, an alkyl group of 1 to 10 carbon atoms, cycloalkyl of 3 to 12 carbon atoms, cycloalkyl- (lower)alkyl , aryl or aryl( lower)alkyl or
- R and R together with the nitrogen atom to which they are both attached represent a saturated heterocyclic ring which may contain a further hetero atom [eg an azetidino, pyrro idino, piperidino, hexahydroazepino, heptamethyleneimino, orpholino or piperazino ring which may be optionally substituted by, for example, lower alkyl, aryl, aryl( lower)alkyl , lower alkoxy, halogen or haloClower)alkyl ] .
- a further hetero atom eg an azetidino, pyrro idino, piperidino, hexahydroazepino, heptamethyleneimino, orpholino or piperazino ring which may be optionally substituted by, for example, lower alkyl, aryl, aryl( lower)alkyl , lower alkoxy, halogen or
- lower as used herein means that the radical referred to contains 1 to 6 carbon atoms . Preferably such radicals contain 1 to 4 carbon atoms. Examples of “lower alkyl” radicals are methyl, ethyl, propyl , isopropyl, butyl, tert. -butyl, pentyl and isopentyl . When R is an alkyl group a particularly preferred radical is a tertiary alkyl radical such as tert.butyl
- a cycloalkyl group can contain 3 to 12 carbon atoms.
- a cycloalkyl group is cyclopentyl, cyclohexyl or cycloheptyl, most preferably cyclohexyl .
- Cycloalkyl groups also include bicyclic, tricyclic and tetracyclic groups, eg adamantyl.
- aryl means an aromatic radical having 6 to 12 carbon atoms (eg phenyl or naphthyl) which optionally may be substituted by one or more substituents commonly used in medical chemistry, eg substituents such as lower alkyl, lower alkoxy (eg methoxy, ethoxy, propoxy, butoxy), loweralkylthio, halogen, haloClower)alkyl (eg trifluoromethyl ) , nitro, cyano, carboxamido, (lower)alkoxycarbonyl, amino, (lower)alkylamino or di ( lower)alkylamino substituents.
- R may be a bicyclic oxygen-containing radical such as a optionally substituted radical of the formula
- heteroaryl refers to an aromatic radical containing one or more hetero atoms (eg oxygen, nitrogen, sulphur) and which may be optionally substituted by one or more substituents. Some examples of suitable substituents are given above in connection with “aryl” radicals.
- the heteroaryl radical may, for example, contain up to 10 ring atoms; for example the heteroaryl radical may be a monocyclic radical containing 5 to 7 ring atoms.
- the hetero ring contains a nitrogen hetero atom with or without one or more further hetero atoms.
- R is a heteroaryl radical it is preferably an optionally substituted pyrimidyl (particularly 2-pyrimidyl) , quinolinyl or indolyl [particularly indol-4-yl which may be optionally substituted eg by (lower)alkoxycarbonyl ] radical.
- heteroaryl When R is a heteroaryl or heteroaryl-lower alkyl the "heteroaryl” group is preferably a nitrogen containing heteroaryl radical (eg an optionally substituted pyridinyl, pyrimidinyl or pyrazinyl radical) or a heteroaryl radical containing an oxygen or sulphur atom as a hetero atom eg an optionally substituted thienyl or furyl group.
- Preferred compounds of formula A have the following characteristics either singly or in any possible combination: -
- X is -N- Cb)
- R is optionally substituted phenyl, eg o-alkoxy-phenyl (particularly o-methoxyphenyl )
- R 2 and R3 together wi•th the ni•trogen atom to which they are both attached represent a saturated heterocyclic ring, particularly hexahydroazepino.
- Compounds of formula (A) are useful because of their pharmacological activity, eg as 5-HT- -antagonists.
- the compounds and their uses are disclosed, for example, in GB 2230780 A, GB 2230781 A, GB 2248836 A, GB 2254324 A, GB 2262093 A and WO-GB 93/01542.
- the prior specifications refer to the preparation of enantiomers by, for example, resolution of the racemates.
- the process of the present invention avoids the inconvenient resolution step.
- the invention particularly relates to a process for preparing (-)-(R)-2 , 3 , 4 , 5 , 6, 7-hexahydro- -l-[4-[4- (2-methoxyphenyl)piperazin-l-yl ]-2-phenyl 3-butanoyl-lH- azepine and the pharmaceutically acceptable acid addition salts thereof.
- the compound, in its free base form, has the formula
- GB-A-2248836 The compound and its use as a 5-HT. -antagonist is disclosed in GB-A-2248836.
- Example 2(a) of GB-A-2248836 describes the preparation of the compound and its salts by resolution of a corresponding racemate .
- compound (I) can be prepared in good yield by an asymmetric synthesis from readily available starting materials thus avoiding an inconvenient resolution step.
- An essential step in the synthesis of the present invention which forms the first aspect of the present invention is a process which comprises condensation of an aldehyde of formula
- the condensation of the aldehyde of formula (B) with the piperazine of formula (C) may be carried out, for example, in presence of a reducing agent such as sodium triacetoxyborohydride or sodium cyanoborohydride .
- a reducing agent such as sodium triacetoxyborohydride or sodium cyanoborohydride .
- the aldehyde of formula (B) may be prepared by a process which comprises hydrolysing a diester of formula
- R are each lower alkyl groups of 3 to 6 carbon atoms) to give a diacid amide of formula
- R and R are both branched chain alkyl groups such as isopropyl or, more preferably, tertiary butyl .
- the hydrolysis of the diester can be effected with an acid, e.g. formic acid, trifluoroacetic acid.
- an acid e.g. formic acid, trifluoroacetic acid.
- the diacid amide need not be isolated before carrying out the decarboxylation process.
- the decarboxylation may be carried out by heating the diacid amide in an inert solvent, e.g. acetonitrile , optionally in presence of a catalytic amount of Cu_0.
- the monacid amide (F) may be reduced directly to the aldehyde (B) with, for example, an aminoalane, but it is prefered to reduce the monoacid amide to the alcohol of formula
- the reduction to the alcohol may be effected by using a reducing agent that does not reduce the amide group, e.g. reduction with Me S.BH.,, preferably in the presence of BF,.Et_0, or alternatively activation of the monoacid amide by reaction with bis-succinimido carbonate followed by reduction with NaBH .
- the oxidation of the alcohol (G) to the aldehyde (B) may be effected with, for example, tetra-n-propylammonium per-ruthenate, DMSO, oxalyl chloride triethvlamine.
- the diesters of formula CD) are novel compounds provided by the invention. Particularly preferred are the diesters of formula
- the diesters of formula (D) may be prepared by a novel process which comprises reacting an activated ⁇ -hydroxy amide of formula
- R R ' and R3 are as defi.ned above and R6 i.s an activating group which maintains chirality such as an arylsuphonyl group, e.g. p-toluenesulphonyl ) with a dialkylmalonate of formula
- the dialkylmalonate is preferably reacted in the form of its sodium or potassium salt. It has been found that the reaction is stereospecific with inversion of the centre at the benzylic position to give the desired stereochemistry in the diester (D) .
- the stereospecif icity of the process is surprising since the compound of formula (H) has an aryl or heteroaryl group and an amide group which would be expected to produce a much more acidic proton (in ⁇ e benzylic position) than the corresponding proton in the prior art compound in which equivalent groups are respectively alkyl and ester (M. Larcheveque et al. Synthesis, February 1991, 162-164). The more labile proton would have been expected to give rise to a racemic product.
- the activated ⁇ -hydroxy amides of formula (H) can be prepared from the S- ( + ) -mandelic acid derivatives of formula
- the prefered protecting groups, R , for S-(+ )-mandelic acid are trimethysilyl or tertbutyldimethylsilyl .
- the mandelic acid may, for example, be reacted with
- the protected derivative (VI) may be halogenated with, for example, oxalyl chloride to give an acyl halide (VII) where Z is chlorine or, more preferably with triphenyl phosphine/bromine to give an acyl halide ( VII ) where Z is bromine.
- the acyl halide (VII) need not be isolated before reaction with the amine (eg hexamethyleneimine) .
- the protecting group R may be removed from the protected hydroxy amide. (VIII) with an acid e.g. citric acid.
- the hydroxyamide (VIII) may be activated with a reagent that does not destroy the chirality of the compound.
- Suitable reagents include arylsulphonic anhydrides (e.g. p-toluenesulphonic anhydride) and methanesulphonic anhydride.
- the processes desribed above may be carried out to give a product in the form of a free base or as an acid addition salt. If the product is obtained as an acid addition salt, the free base can be obtained by basifying a solution of the acid addition salt.
- an acid addition salt particularly a pharmaceutically acceptable acid addition salt
- a suitable organic solvent may be used to dissolve the free base in a suitable organic solvent and treating the solution with an acid, in accordance with conventional procedures for preparing acid addition salts from base compounds.
- acid addition salts are those formed from inorganic and organic acids, such as sulphuric, hydrochloric, hydrobromic, phosphoric, tartaric, fumaric, maleic, citric, acetic, formic, methanesulphonic, p-toluenesulphonic, oxalic and succinic acids .
- reaction mixture was stirred under argon at room temperature until it became homogeneous.
- the cooling bath was returned and hexamethyleneimine (143ml, 1.27mol) added dropwise over forty five minutes. Following addition the cooling bath was removed and the reaction mixture allowed to attain room temperature. After a further hour the solvent was removed and the solid extracted with hexane (2 x 1.51). This was filtered by suction through Celite. The solvent was removed and the residue distilled under vacuum. At 0.5m bar: 125-155 ° C discarded
- the OTMS ether obtained in Example 2 (86.77g, 0.284mol) was dissolved in methanol (200ml) and catalytic citric acid added. The solvent was removed and the residue dissolved in dichloromethane. This was washed with sodium bicarbonate, saturated sodium chloride solution, dried (MgSO.)and reduced under vacuum. The colourless oil was distilled under vacuum.
- the di-tert-butyl ester from Example 5 (9.64g, 22.3mmol) was added to formic acid (75ml) cooled to 0 ° C with stirring. The cooling bath was removed and stirring continued for three hours. The solvent was removed under reduced pressure (bath temperature less than 40°C). Dichloromethane was used to co-evaporate the formic acid and a stable white foam was obtained. The foam was dissolved in dry acetonitrile and refluxed under argon for three hours. The solution was allowed to cool overnight.
- Example 7 The alcohol from Example 7 (226mg, 0.86mmol) was dissolved in dry dichloromethane (8ml) at room temperature under argon, N-methylmorpholine N-oxide (152mg, 1.29mmol) and 4 A-molecular sieves added with stirring. After ten minutes tetra-n-propylammonium per-ruthenate (15mg, 0.043mmol) was added.
- reaction mixture was stirred for three hours.
- the reaction mixture was washed with saturated sodium bicarbonate (10ml), saturated sodium chloride solution (10ml), dried (MgSO.), and reduced in vacuo.
- the resulting oil was purified by flash chromatrography eluting with dichloromethane/methanol (20/1) to give the title compound.
- the material was shown to be identical to an authenittiicc ssaammppllee bbyy IH, C NMR, IR and chiral stationary phase HPLC.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Hydrogenated Pyridines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Indole Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Pyridine Compounds (AREA)
Abstract
Description


















Claims








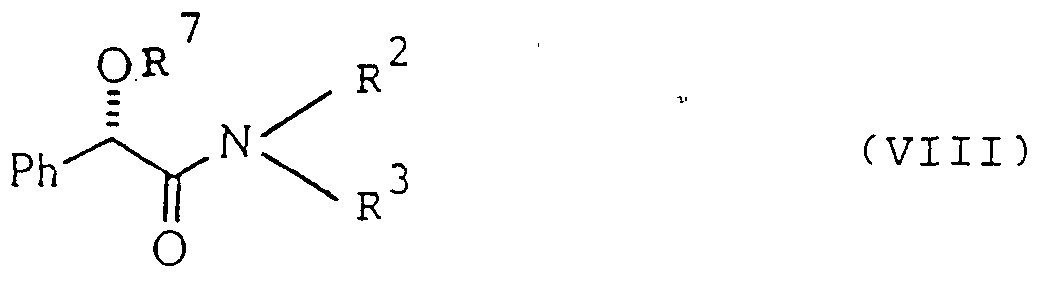



Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP93922576A EP0664799B1 (en) | 1992-10-17 | 1993-10-08 | Amide derivatives |
| CA002147155A CA2147155C (en) | 1992-10-17 | 1993-10-08 | Amide derivatives |
| US08/411,601 US5637701A (en) | 1992-10-17 | 1993-10-08 | Process for preparing optically active amide derivatives |
| DK93922576T DK0664799T3 (en) | 1992-10-17 | 1993-10-08 | amide derivatives |
| AU51524/93A AU5152493A (en) | 1992-10-17 | 1993-10-08 | Amide derivatives |
| DE69325660T DE69325660T2 (en) | 1992-10-17 | 1993-10-08 | AMID DERIVATIVES |
| KR1019950701459A KR950703544A (en) | 1992-10-17 | 1993-10-08 | Amide derivatives |
| JP50974494A JP3478827B2 (en) | 1992-10-17 | 1993-10-08 | Amide derivatives |
| GR990402205T GR3031123T3 (en) | 1992-10-17 | 1999-08-31 | Amide derivatives. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9221931.0 | 1992-10-17 | ||
| GB929221931A GB9221931D0 (en) | 1992-10-17 | 1992-10-17 | Piperazine derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994008983A1 true WO1994008983A1 (en) | 1994-04-28 |
Family
ID=10723688
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB1993/002090 Ceased WO1994008983A1 (en) | 1992-10-17 | 1993-10-08 | Amide derivatives |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US5637701A (en) |
| EP (1) | EP0664799B1 (en) |
| JP (1) | JP3478827B2 (en) |
| KR (1) | KR950703544A (en) |
| AT (1) | ATE182144T1 (en) |
| AU (1) | AU5152493A (en) |
| CA (1) | CA2147155C (en) |
| DE (1) | DE69325660T2 (en) |
| DK (1) | DK0664799T3 (en) |
| ES (1) | ES2133414T3 (en) |
| GB (2) | GB9221931D0 (en) |
| GR (1) | GR3031123T3 (en) |
| MX (1) | MX9306421A (en) |
| PH (1) | PH31583A (en) |
| TW (1) | TW340844B (en) |
| WO (1) | WO1994008983A1 (en) |
| ZA (1) | ZA937516B (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5525600A (en) * | 1994-11-28 | 1996-06-11 | American Home Products Corporation | (Thiophen-2-yl)-piperidin or tetrahydropyridin carboxamides |
| WO1997028140A1 (en) * | 1996-02-02 | 1997-08-07 | Pierre Fabre Medicament | NOVEL PIPERIDINES DERIVED FROM 1-/(PIPERAZIN-1-YL)ARYL(OXY/AMINO)CARBONYL/-4-ARYL-PIPERIDINE AS SELECTIVE 5-HT1Db RECEPTOR ANTAGONISTS |
| US5710155A (en) * | 1995-04-14 | 1998-01-20 | Boehringer Ingelheim Kg | Arylglycinamide derivatives, processes for the manufacture thereof and pharmaceutical compositions containing these compounds |
| EP0783498A4 (en) * | 1994-09-30 | 1998-04-15 | Merck & Co Inc | ARYL SUBSTITUTED PIPERAZINES NEUROKININ ANTAGONISTS |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2757158B1 (en) * | 1996-12-18 | 1999-04-02 | Lipha | NOVEL 4- (1-PIPERAZINYL) BENZOIC ACID DERIVATIVES, PROCESS FOR THEIR PREPARATION AND THERAPEUTIC APPLICATIONS |
| US6900228B1 (en) | 1998-03-10 | 2005-05-31 | Research Triangle Institute | Opiate compounds, methods of making and methods of use |
| US20040072839A1 (en) * | 2002-06-14 | 2004-04-15 | Amedeo Leonardi | 1-Phenylalkylpiperazines |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2776282A (en) * | 1954-05-04 | 1957-01-01 | Searle & Co | Cyclic amides of alpha-toluic acids and derivatives thereof |
| EP0395312A2 (en) * | 1989-04-22 | 1990-10-31 | JOHN WYETH & BROTHER LIMITED | Piperazine derivatives |
| GB2230780A (en) * | 1989-04-22 | 1990-10-31 | American Home Prod | Tertiary alkyl functionalised piperazine derivatives |
| EP0481744A1 (en) * | 1990-10-19 | 1992-04-22 | JOHN WYETH & BROTHER LIMITED | Piperazine derivatives |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8909209D0 (en) * | 1989-04-22 | 1989-06-07 | Wyeth John & Brother Ltd | Piperazine derivatives |
-
1992
- 1992-10-17 GB GB929221931A patent/GB9221931D0/en active Pending
-
1993
- 1993-10-08 DK DK93922576T patent/DK0664799T3/en active
- 1993-10-08 EP EP93922576A patent/EP0664799B1/en not_active Expired - Lifetime
- 1993-10-08 US US08/411,601 patent/US5637701A/en not_active Expired - Lifetime
- 1993-10-08 KR KR1019950701459A patent/KR950703544A/en not_active Abandoned
- 1993-10-08 ES ES93922576T patent/ES2133414T3/en not_active Expired - Lifetime
- 1993-10-08 AT AT93922576T patent/ATE182144T1/en not_active IP Right Cessation
- 1993-10-08 GB GB9320819A patent/GB2272436B/en not_active Expired - Fee Related
- 1993-10-08 DE DE69325660T patent/DE69325660T2/en not_active Expired - Fee Related
- 1993-10-08 AU AU51524/93A patent/AU5152493A/en not_active Abandoned
- 1993-10-08 CA CA002147155A patent/CA2147155C/en not_active Expired - Fee Related
- 1993-10-08 WO PCT/GB1993/002090 patent/WO1994008983A1/en not_active Ceased
- 1993-10-08 JP JP50974494A patent/JP3478827B2/en not_active Expired - Fee Related
- 1993-10-11 ZA ZA937516A patent/ZA937516B/en unknown
- 1993-10-15 MX MX9306421A patent/MX9306421A/en not_active IP Right Cessation
- 1993-10-15 PH PH47088A patent/PH31583A/en unknown
- 1993-10-15 TW TW082108553A patent/TW340844B/en active
-
1999
- 1999-08-31 GR GR990402205T patent/GR3031123T3/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2776282A (en) * | 1954-05-04 | 1957-01-01 | Searle & Co | Cyclic amides of alpha-toluic acids and derivatives thereof |
| EP0395312A2 (en) * | 1989-04-22 | 1990-10-31 | JOHN WYETH & BROTHER LIMITED | Piperazine derivatives |
| GB2230780A (en) * | 1989-04-22 | 1990-10-31 | American Home Prod | Tertiary alkyl functionalised piperazine derivatives |
| GB2230781A (en) * | 1989-04-22 | 1990-10-31 | Wyeth John & Brother Ltd | Piperazine derivatives as 5-ht(1a) antagonists |
| EP0481744A1 (en) * | 1990-10-19 | 1992-04-22 | JOHN WYETH & BROTHER LIMITED | Piperazine derivatives |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0783498A4 (en) * | 1994-09-30 | 1998-04-15 | Merck & Co Inc | ARYL SUBSTITUTED PIPERAZINES NEUROKININ ANTAGONISTS |
| US5525600A (en) * | 1994-11-28 | 1996-06-11 | American Home Products Corporation | (Thiophen-2-yl)-piperidin or tetrahydropyridin carboxamides |
| US5710155A (en) * | 1995-04-14 | 1998-01-20 | Boehringer Ingelheim Kg | Arylglycinamide derivatives, processes for the manufacture thereof and pharmaceutical compositions containing these compounds |
| WO1997028140A1 (en) * | 1996-02-02 | 1997-08-07 | Pierre Fabre Medicament | NOVEL PIPERIDINES DERIVED FROM 1-/(PIPERAZIN-1-YL)ARYL(OXY/AMINO)CARBONYL/-4-ARYL-PIPERIDINE AS SELECTIVE 5-HT1Db RECEPTOR ANTAGONISTS |
| FR2744448A1 (en) * | 1996-02-02 | 1997-08-08 | Pf Medicament | NOVEL PIPERIDINES DERIVED FROM ARYL PIPERAZINE, AND PROCESS FOR PREPARING THEM, PHARMACEUTICAL COMPOSITIONS AND THEIR USE AS MEDICAMENTS |
Also Published As
| Publication number | Publication date |
|---|---|
| GB2272436B (en) | 1997-06-18 |
| GB2272436A (en) | 1994-05-18 |
| KR950703544A (en) | 1995-09-20 |
| PH31583A (en) | 1998-11-03 |
| EP0664799A1 (en) | 1995-08-02 |
| GB9221931D0 (en) | 1992-12-02 |
| GB9320819D0 (en) | 1993-12-01 |
| JP3478827B2 (en) | 2003-12-15 |
| TW340844B (en) | 1998-09-21 |
| JPH08502738A (en) | 1996-03-26 |
| ES2133414T3 (en) | 1999-09-16 |
| US5637701A (en) | 1997-06-10 |
| GR3031123T3 (en) | 1999-12-31 |
| DE69325660D1 (en) | 1999-08-19 |
| ZA937516B (en) | 1995-04-21 |
| EP0664799B1 (en) | 1999-07-14 |
| AU5152493A (en) | 1994-05-09 |
| CA2147155A1 (en) | 1994-04-28 |
| ATE182144T1 (en) | 1999-07-15 |
| DE69325660T2 (en) | 2000-03-09 |
| CA2147155C (en) | 2005-01-04 |
| MX9306421A (en) | 1994-04-29 |
| DK0664799T3 (en) | 1999-11-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR970009728B1 (en) | Process for the preparation of -2-(2-(4-((4chlorophenyl)phenylmethyl-1-piperazinyl)ethoxy)-acetic acid and tis dihydrochloride | |
| KR0173310B1 (en) | Piperazine derivatives, their preparation method and pharmaceutical composition comprising thereof | |
| EP0434561B1 (en) | 1-Naphthyl-piperazine derivatives, process for their preparation and pharmaceutical compositions containing them | |
| EP0664799B1 (en) | Amide derivatives | |
| US5965734A (en) | Processes and intermediates for preparing 2-substituted piperidine stereoisomers | |
| JP4157766B2 (en) | Process for producing substituted imidazopyridine compounds | |
| CA2038962A1 (en) | Aminobenzene compounds, their production and use | |
| JP4763954B2 (en) | Process for the production of {2- [4- (α-phenyl-p-chlorobenzyl) piperazin-1-yl] ethoxy} acetic acid and novel intermediates therefor | |
| KR20010042750A (en) | Method for producing enantiomer-free n-methyl-n-[(1s)-1-phenyl-2-((3s)-3-hydroxypyrrolidine-1-yl)ethyl]-2,2-diphenyl acetamide | |
| WO2001044184A1 (en) | Synthesis of indole-containing spla2 inhibitors | |
| US4767767A (en) | 2-pyrrolidinylethyl-2-(7-trifluoromethyl-4-quinolyl-aminobenzoate having analgesic, antipyretic and anti-inflammatory activities | |
| GB2305171A (en) | Diester intermediates | |
| US4091095A (en) | Phosphinyl compounds | |
| JPH05239005A (en) | N- (2-aminoethyl) benzamides and novel intermediates thereof | |
| RU2225859C2 (en) | Synthesis of 3-amino-3-arylpropanoates | |
| US5561233A (en) | Process for the preparation of an intermediate of a benzo[a]quinolizinone derivative | |
| RU2176639C2 (en) | New heteroaryloxyethyl amines, method of preparing thereof, pharmaceutical composition comprising said amines having affinity with 5ht1a receptors and intermediate compounds | |
| JPH08333340A (en) | Method for producing aminoethylpiperidine derivative | |
| US6150417A (en) | Phenoxyethylamine derivatives, method of preparation application as medicine and pharmaceutical compositions containing same | |
| US6355804B1 (en) | Process for producing piperidinecarboxylic acid amide derivatives | |
| SU523634A3 (en) | Method for preparing benzylamine derivatives or their salts | |
| KR100468936B1 (en) | Substituted [2- (1-piperazinyl) ethoxy] methyl compound | |
| KR810000293B1 (en) | Method for preparing substituted amino quinazoline derivatives | |
| JP2024511422A (en) | 5-{5-chloro-2-[(3S)-3-[(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1H)-carbonyl]phenyl}-1,2-dimethyl- Novel production method for synthesizing 1H-pyrrole-3-carboxylic acid derivatives and its application for producing pharmaceutical compounds | |
| JPH03148276A (en) | Optically active pyridonecarboxylic acid compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CZ FI HU JP KP KR KZ LK MG MN MW NO NZ PL RO RU SD SK UA US VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1993922576 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08411601 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2147155 Country of ref document: CA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993922576 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1993922576 Country of ref document: EP |