WO1994012165A2 - Enzyme inhibitors - Google Patents
Enzyme inhibitors Download PDFInfo
- Publication number
- WO1994012165A2 WO1994012165A2 PCT/GB1993/002437 GB9302437W WO9412165A2 WO 1994012165 A2 WO1994012165 A2 WO 1994012165A2 GB 9302437 W GB9302437 W GB 9302437W WO 9412165 A2 WO9412165 A2 WO 9412165A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- isothiourea
- ethyl
- group
- methyl
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/32—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D207/33—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms with substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D207/333—Radicals substituted by oxygen or sulfur atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/155—Amidines (), e.g. guanidine (H2N—C(=NH)—NH2), isourea (N=C(OH)—NH2), isothiourea (—N=C(SH)—NH2)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/02—Non-specific cardiovascular stimulants, e.g. drugs for syncope, antihypotensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C335/00—Thioureas, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C335/30—Isothioureas
- C07C335/32—Isothioureas having sulfur atoms of isothiourea groups bound to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/28—Radicals substituted by singly-bound oxygen or sulphur atoms
- C07D213/32—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/08—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D277/12—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/18—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/40—Unsubstituted amino or imino radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/14—Radicals substituted by singly bound hetero atoms other than halogen
- C07D333/18—Radicals substituted by singly bound hetero atoms other than halogen by sulfur atoms
Definitions
- the present invention relates to isothiourea derivatives, to methods for their manufacture, to pharmaceutical compositions containing them and to their use in therapy, in particular their use as nitric oxide synthase inhibitors.
- endothelium-derived relaxing factor a labile humoral factor termed endothelium-derived relaxing factor (EDRF).
- NO nitric oxide
- NO is the endogenous stimulator of the soluble guanylate cyclase and is involved in a number of biological actions in addition to endothelium-dependent relaxation including, cytotoxicity of phagocytic cells and cell-to-cell communication in the central nervous sytem (see Moncada elal, Biochemical Pharmacology, 2-S 1709-1715 (1989) and Moncada et al, Pharmacological Review, 42, 109-142 (1991)). It is now thought that excess NO production may be involved in a number of conditions, particularly conditions which involve systemic hypotension such as septic (toxic) shock and therapy with certain cytokines.
- L-NMMA L-arginine analogue L-N- monomemyl-arginine
- L-NMMA L-N- monomemyl-arginine
- the therapeutic use of certain other NO synthase inhibitors apart from L- NMMA for the same purpose has also been proposed in WO 91/04024 and in EP-A- 0446699.
- a constitutive, Ca "H” /calmodulin dependent enzyme located in the endothelium, that releases NO in response to receptor or physical stimulation.
- a constitutive, Ca ⁇ 'calmodulin dependent enzyme located in the brain, that releases NO in response to receptor or physical stimulation.
- the NO released by the constitutive enzymes acts as a transduction mechanism underlying several physiological responses.
- the function of the NO produced by the inducible enzyme is as a cytotoxic molecule for fighting tumour cells and invading microorganisms (Wright et al., Card. Res. 26, 48-57 (1992) and Moncada si al, Pharmacological Review, 41, 109-142 (1991)). It also appears that the adverse effects of excess NO production, in particular pathological vasodilation and tissue damage, may result largely from the effects of NO synthesised by the inducible NO synthase.
- the NO synthase inhibitors proposed for therapeutic use so far, and in particular L-NMMA, are non-selective in that they inhibit both the constitutive and the inducible NO synthase enzymes.
- Use of such a non-selective NO synthase inhibitor requires that great care be taken in order to avoid the potentially serious consequences of over-inhibition of the constitutive NO-synthase enzyme including hypertension, thrombosis, CNS toxicity and tissue damage.
- L-NMMA for the treatment of septic shock it has been recommended that the patient must be subject to continuous blood pressure monitoring throughout the treatment.
- NO synthase inhibitors which are selective in the sense that they inhibit the inducible NO synthase enzyme to a considerably greater extent than the constitutive NO synthase enzyme would be of even greater therapeutic benefit and much easier to use.
- isothioureas are inhibitors of NO synthase, and are useful in the treatment of systemic hypotension, and. in particular, the treatment of septic shock.
- many of these compounds possess selectivity for the inducible NO synthase enzyme as compared with the constitutive NO synthase enzymes.
- the present invention provides a method of treatment of conditions requiring inhibition of the nitric oxide synthase enzyme, which comprises administering to a mammal in need thereof an effective amount of an isothiourea derivative having an inhibitory effect against the NO synthase enzyme, or a pharmaceutically acceptable salt thereof.
- the present invention provides the use of an isothiourea having an inhibitory effect against the NO synthase enzyme for the manufacture of a medicament for the treatment of conditions where there is an advantage in inhibiting the NO synthase enzyme.
- a method of treatment of systemic hypotension and/or septic shock which comprises administering to a mammal in need thereof an effective amount of an isothiourea derivative having an inhibitory effect against the NO synthase enzyme, or a pharmaceutically acceptable salt thereof.
- an isothiourea derivative having an inhibitory effect against the NO synthase enzyme for the manufacture of a medicament for the treatment of systemic hypotension and/or septic shock.
- cytokines such as TNF, IL-1 and IL-2
- cytokine- inducing agents for example 5, 6-dimethylxanthenone acetic acid
- compounds which inhibit NO synthesis may be of use in reducing the NO concentration in patients suffering from inflammatory conditions in which an excess of NO contributes to the pathophysiology of the condition, for example adult respiratory distress syndrome (ARDS) and myocarditis.
- ARDS adult respiratory distress syndrome
- an NO synthase enzyme may be involved in the degeneration of cartilage which takes place in autoimmune and/or inflammatory conditions such as arthritis, rheumatoid arthritis, chronic bowel disease and systemic lupus erythematosis (SLE). It is also thought that an NO synthase enzyme may be involved in insulin- dependent diabetes mellitis.
- a yet further aspect of the present invention provides an isothiourea derivative or salt thereof in the manufacture of a medicament for use in cytokine or cytokine-inducing therapy, as an adjuvant to short term immunosuppression in transplant therapy, for the treatment of patients suffering from inflammatory conditions in which an excess of NO contributes to the pathophysiology of the condition, in autoimmune and/or inflammatory indications and in insulin-dependent diabetes mellitis.
- a still further aspect provides a method of treatment of adverse effects associated with cytokine therapy, of short term immunosuppression in transplant therapy, of patients suffering from inflammatory conditions in which an excess of NO contributes to the pathophysiology of the condition, of autoimmune and/or inflammatory indications and of insulin-dependent diabetes mellitis, which comprises administering to a mammal in need thereof an effective amount of an isothiourea derivative having an inhibitory effect against the NO synthase enzyme or a pharmaceutically acceptable salt thereof.
- treatment of a patient is intended to include prophylaxis; the term “mammal” is intended to include a human or an animal.
- Preferred isothioureas include those of formula (I)
- R is (1) a C ⁇ _i4 hydrocarbyl group
- each group R being optionally substituted by one or two groups independently selected from:
- X is oxygen, C(O) m wherein m is 1 or 2, S(O) n wherein n is 0, 1, or 2, or NR2 wherein R2 is hydrogen, C ⁇ . alkyl or C3.6 cycloalkyl or R2 is linked to R! to form a C2- alkylene group;
- R! is hydrogen; or C ⁇ . alkyl, C2-6 alkenyl, C3.6 cycloalkyl, C7.9 aralkyl, C6-10 aryl, or a 5- or 6- membered heterocyclic group, each group optionally substituted by one or two groups independently selected from C ⁇ _3 alkyl, hydroxy, C1-.3 alkoxy, amino, C1.3 alkylamino, halo, nitro, or a group C(O) m > R ⁇ b wherein m' is 1 or 2 and R ⁇ b is hydrogen or C 1-4 alkyl; or R-- is a group NR ⁇ R4 wherein R ⁇ and R ⁇ are the same or different and each is hydrogen or C 1-4 alkyl or R ⁇ and R 4 are linked to form a C2- alkylene group;
- Y is oxygen, S(O) n wherein n is as hereinbefore defined, or NR-5 wherein
- R 5 is hydrogen or C ⁇ _4 alkyl; w is O or l; Q is C2-4 hydrocarbyl
- R is
- R? and R ⁇ are each independently selected from hydrogen, C ⁇ _4 alkyl, C2-4 alkenyl, Cj_4 alkoxyalkyl; or R? and R ⁇ are linked to form a 5- or 6-membered heterocyclic ring;
- R is N
- R ⁇ b and R ⁇ b are independently selected from hydrogen or Cj_ alkyl, preferably hydrogen, methyl or ethyl;
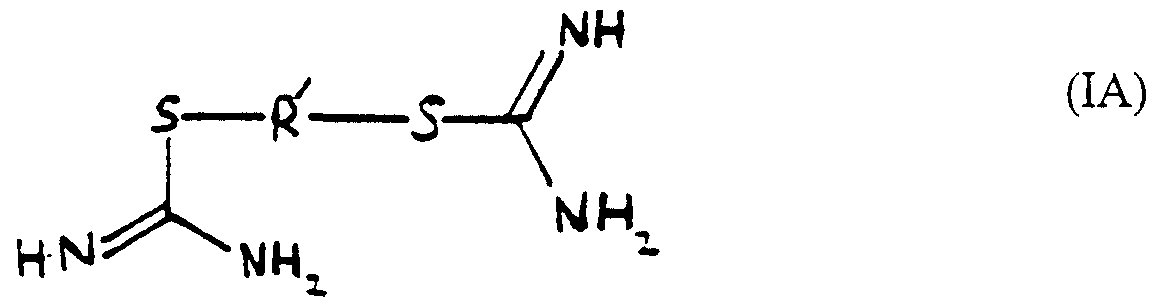
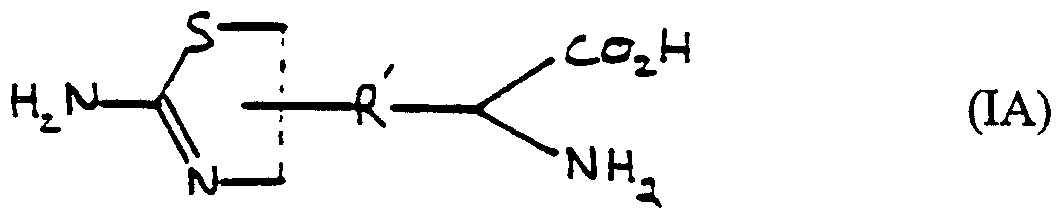
- Formula (I) includes isothiourea derivatives of formula (IA), (IB) and (IC)
- R' is a Cj.g alkylene group or a C2-8 alkenylene or alkynylene group each optionally containing a phenyl ring, a 5- or 6-membered heterocyclic ring or a group X as hereinbefore defined, and the dotted line represents a double or a single bond.
- Formula I also includes compounds of formula (II)
- R a is a C _$ hydrocarbyl or 5- or 6-membered heterocyclic ring or a 9-membered bicyclic heterocyclic ring system each optionally substituted by halo or by one or two groups -X a R la wherein R la is hydrogen, C ⁇ .
- t is 0 to 4 and w 3 - is 0 or 1
- Y a is oxygen, sulphur and NR? a wherein R? a is hydrogen or C ⁇ _4 alkyl:
- R a links the sulphur atom to one of the nitrogen atoms in the compound of the formula (I) to form a 5- or 6-membered heterocyclic ring, with the proviso that R a is not methyl.
- One preferred group of compounds are those wherein R is not methyl, ethyl, propyl or isopropyl.
- Preferred compounds of die formula (I) include:
- Especially preferred compounds include S,S'-(l,3-phenylenebis(l,2- ethanediyl))diisothiourea, S,S'-(l,4-phenylenebis(l,2-ethanediyl)) diisothiourea, S-(2-(5- antidinot_do)methyl)-2-thienyl)ethyl)isothiourea, S-(3-(5-(2-amidinothio)ethyl)-2- thienyl)propyl)isothiourea and S-(2'-(3-methoxyphenyl) ethyl)isothiourea.
- S-ethylisothiourea S-propylisothiourea and S-isopropylisothiourea, particularly S-ethylisothiourea and S- isopropylisothiourea, and especially S-ethylisothiourea.
- hydrocarbyl group is meant a group that contains only carbon and hydrogen atoms but may contain double and/or triple bonds and which may be cyclic or aromatic in nature.
- heterocyclic ring a cyclic compound containing one to three hetero atoms selected from oxygen, sulphur and nitrogen, and preferably nitrogen or sulphur.
- halo is meant fluoro, chloro, bromo or iodo, and preferably bromo.
- the compounds of formula (I) may include a number of asymmetric centres in the molecule depending on the precise meaning of the various groups and formula (I) is intended to include all possible isomers.
- the present invention provides an isothiourea of the formula (I) other than benzylisothiourea, S.S-(l,4-phenylenebis(methylene))diisothiourea and S-(2- (dimethylamino)ethyl)isothiourea, or a pharmaceutically acceptable salt thereof having an inhibitory effect against the NO synthase enzyme for use in medicine.
- Such compounds include:
- the present invention includes isothioureas in the form of salts, in particular acid addition salts.
- Suitable salts include those formed with both organic and inorganic acids.
- Such acid addition salts will normally be pharmaceutically acceptable although salts of non- pharmaceutically acceptable salts may be of utility in the preparation and purification of the compound in question.
- preferred salts include those formed from hydrochloric, hydrobromic, sulphuric, citric, tartaric, phosphoric, lactic, pyruvic, acetic, trifluoroacetic, succinic, oxalic, fumaric, maleic, oxaloacetic, methanesulphonic, ethanesulphonic, p- toluenesulphonic, benzenesulphonic and isethionic acids.
- Salts of isothioureas can be made by reacting the appropriate compound in the form of the free base with the appropriate acid.
- the isothioureas of the present invention Whilst it may be possible for the isothioureas of the present invention to be administered as the raw chemical, it is preferable to present them as a pharmaceutical formulation.
- the present invention provides a pharmaceutical formulation comprising an isothiourea of the present invention or a pharmaceutically acceptable salt or solvate thereof, together with one or more pharmaceutically acceptable carriers therefor and optionally one or more other therapeutic ingredients, for example an antibiotic, and/or a volume replacement liquid.
- the carriers must be "acceptable" in the sense of being compatible with the other ingredients of the formulation and not deleterious to die recipient thereof.
- the formulations include tiiose suitable for oral, parenteral (including subcutaneous, intradermal, intramuscular, intravenous and intraarticular), rectal and topical (including dermal, buccal, sublingual and intraocular) administration although the most suitable route may depend upon for example the condition and disorder of the recipient.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing into association a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof ("active ingredient”) with the carrier which constitutes one or more accessory ingredients.
- formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and then, if necessary shaping the product into the desired formulation.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid: or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion.
- the active ingredient may also be presented as a bolus, electuary or paste.
- a tablet may be made by compression or moulding, optionally witii one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, lubricating, surface active or dispersing agent.
- Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein.
- Formulations for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render die formulation isotonic with the blood of die intended recipient; and aqueous and. non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of die sterile liquid carrier, for example, saline, water- for-injection, immediately prior to use. Altematively, the formulations may be presented for continuous infusion.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Formulations for rectal administration may be presented as a suppository with the usual carriers such as cocoa butter or polyethylene glycol.
- Formulations for topical administraton in the mouth include lozenges comprising the active ingredient in a flavoured basis such as sucrose and acacia or tragacanth, and pastilles comprising the active ingredient in a basis such as gelatin and glycerin or sucrose and acacia.
- Preferred unit dosage formulations are those containing an effective dose, as herein below recited, or an appropriate fraction thereof, of the active ingredient.
- formulations of this invention may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
- the compounds of the invention may be administered orally or via injection at a dose of from 0.1 to 250mg/kg per day.
- the dose range for adult humans is generally from 5mg to 17.5g/day, preferably 5mg to 2g/day and most preferably lOmg to lg/day.
- Tablets or other forms of presentation provided in discrete units may conveniently contain an amount of compound of the invention which is effective at such dosage or as a multiple of the same, for instance, units containing 5mg to 500mg, usually around lOmg to 200mg.
- the compounds of formula (I) are preferably administered orally or by injection (intravenous or subsutaneous).
- the precise amount of compound administered to a patient will be the responsibility of the attendant physician. However the dose employed will depend on a number of factors, including the age and sex of die patient, the precise disorder being treated, and its severity. Also the route of administration may vary depending on the condition and its severity.
- the present invention also provides processes for the preparation of novel compounds as hereinbefore defined, analagous to those known in the art for preparing isothiourea derivatives.
- compounds of formula (I) or protected derivatives thereof may be prepared by the reaction of thiourea with a compound RL(L') r wherein R is as hereinbefore defined, L and L' are both leaving groups, for example a halo atom such as bromo, and r is 0 or 1, followed by deprotection if necessary.
- compounds of formula (IA) as hereinbefore defined may be prepared by the reaction of thiourea with a compound LR'L' wherein L.L' and R' are as hereinbefore defined.
- the reaction is carried out in a polar solvent, such as ethanol, at a temperature of from 20°C to the refluxing solvent temperature.
- R 1 and th defined, and P and P' are the same or different and are both protecting groups such as benzyl, benzyloxycarbonyl or tert- butoxycarbonyl.
- the reaction may be carried out in trifluoroacetic acid at a non-extreme temperature of from -20°C to 100°C such as 0°C in the presence of scavenger molecules such as thioanisole and 1, 2-ethanedithiol.
- compounds of formula (IB1) may be prepared from a compound of formula (IB2)
- R, P and P' are as hereinbefore defined, by displacement of tosylate with azide anion in a polar solvent such as dimethyl formamide at non-extreme temperature of from - 20°C to 200°C such as the refluxing solvent temperature.
- R 1 , P' and P are as hereinbefore defined, by die addition of ti iocyanate anion to yield a ring-opened alkoxide which may be trapped as the tosyl derivative (IB3) by the addition of para-toluenesulphonyl chloride.
- Compounds of formula (IB4) may be prepared by methods known to a person skilled in the art.
- Compounds of formula (IB5) may be prepared form the corresponding epoxide by ring opening with azide anion followed by trapping of the alkoxide by para-toluenesulphonyl chloride in a polar solvent such as dimethylformamide at non-extreme temperatures of from -20°C to 200°C such as 100°C (Tetrahedron Lett. 1990, 21 (2), 221).
- a polar solvent such as dimethylformamide
- compounds of formula (IBl) may be prepared by die cyclisation of chloroacetals of formula (IB6)
- R', P and P* are as hereinbefore defined and R ⁇ is a C1-4 alkyl group, with thiourea.
- the reaction may be carried out in a polar solvent such as acetone or ethanol at a non-extreme temperature of from -20°C to 200°C (Chem. Abs. 54:14230d).
- compounds of formula (IBl) may be prepared by methods known in the art, for example ⁇ -N-ben_ ⁇ loxycarbonyl- ⁇ -(2'-amino-4'-thiazolyl) alanine benzyl ester (Syndietic Communications 1990, 20. (30), 3097-3102).
- the cyclisaton reaction may be carried out in a polar solvent such as acetone at a non-extreme temperature of from -20°C to 200°C such as 20°C.
- Compounds of formula (IC1) may be prepared from compounds of formula HO2C-R-CO2H, wherein R' is as hereinbefore defined by methods " known in the art (J. Chem. Soc. 1940, 1304-7; Chem. Abs. 35: 113 3 ).
- ⁇ -(2'-amino-4'-thiazolyl)-L homoalanine was prepared from ⁇ -N-t-butoxycarbonyl- ⁇ -(2'- amino-4'-d_iazolyl)-L-homoalanine benzyl ester (Patt et al. Synth. Commun. 1990, 2-0 (20), 3097-3102) in 9.6% yield, according to the method of Example 6.
- Ci2H2 ⁇ Br2N S 2 C, 32.44; H, 4.54; Br, 35.97; N, 12.61; S, 14.43. Found: C, 32.52; H, 4.49; Br, 36.04; N, 12.61; S, 14.35.
- the concentrated filtrate (oil) was taken up into metiianol/ethanol and treated with ethanolic hydrogen chloride until no further precipitation was observed and the mixture was filtered.
- the oil resulting from concetration of the filtrate was purified by ion-exchange chromatography (Dowex 50X8. strongly acidic) eluting with 0.1N ammonium hydroxide.
- the ninhydrin positive fractions were pooled and freeze-dried to yield 0.453g of a light tan-coloured solid contaminated with dimethylformamide.
- t-Boc and t-butyl ester protecting groups were removed as follows: To a solution of 1.68g 4-((2-amino-4-thiazolyl)methyl)-N ⁇ -t-Boc-L-homoalanine t-butyl ester in 35 mL dioxane was added 1.1 mL triethylsilane and 8 mL 4N hydrochloric acid in dioxane solution. The mixture was filtered and the solids rinsed with dioxane after stirring for 16 hours at 22°C. The NMR of a crude sample indicated incomplete reaction. Redissolved the crude solid in 20 mL dioxane and treated with 4N hydrochloric acid (5 mL) for 4 hours.
- the dibromide product (1.10g, 3.87 mmol) in 50 mL ethanol was treated with 0.59 g (7.75 mmol) thiourea.
- the solution was stirred at reflux for 2 h.
- the concentrated solution was purified by prepative HPLC (Waters C18 BondaPak PrepPak cartridge) witii a memanol/water/trifluoroacetic acid gradient (5/95/0.1 to 90/10/0.1).
- 5-Formyl-2-thiopheneacetic acid etiiyl ester was prepared as described in example 13. To a solution of 2.0g (10.09 mmol) of this ester in 100 mL tetrahydrofuran was added 3.87 g (11.10 mmol) carbethoxymethylenetriphenylphosphorane. The solution was refluxed overnight and concentrated. The crude product was combined with a second 2.0g reaction utilizing 10.54g (30.27mmol) carboethoxymethylenetriphenyl- phosphorane refluxed in 100 mL tetrahydrofuran for 3h.
- Examples 15-17 were prepared by the method of example 2.
- the crude products were purified by preparative HPLC (Waters, C18 BondaPak PrepPak cartridge). Gradient elutions with methanol/water/trifluoroacetic acid (5/95/0.1 to 90/10/0.1) followed by freeze- drying provided the target amino acids as trifluoroacetic acid addition salts.
- the crude bromide was dissolved in 95% ethanol (10ml), and thiourea (251mg, 3.30 mmol) was added. The reaction mixture was warmed to reflux for 16 hr, cooled to room temperature, and the solvent was removed in vacuo . The crude yellow solid was suspended in acetone, and die mixture was warmed to reflux for 10 min. The hot solution was filtered to give a white solid.
- NO synthase inhibition was determined by me following procedure:
- Amion and chorion were removed from fresh placenta, which was then rinsed witii 0.9% NaCl.
- the tissue was homogenized in a Waring blender in 3 volumes of HEDS buffer (20 mM Hepes pH 7.8, 0.1 mM EDTA, 5mM DTT, 0.2M sucrose) plus 0.1 mM PMSF.
- the homogenate was filtered through cheesecloth and then centrifuged at lOOOg for 20 min. The supernatant was recentrifuged at 27,500g for 30 min. Solid ammonium sulfate was- added to die supernatant to give 32% saturation.
- Precipitated protein was pelleted at 25,000g and then redissolved in a minimal volume of HEDS buffer plus 0.1 mM PMSF, lO ⁇ g/ml leupeptin and soybean trypsin inhibitor, and l ⁇ g/ml pepstatin. The redissolved pellet was centrigued at 15,000g for 10 min. To the supernatant was added 1/20 volume at 2,5' ADP agarose resin (Sigma), and me slurry was mixed slowly overnight. In the morning, slurry was packed into a column. The resin was sequentially washed with HEDS, 0.5M NaCl in HEDS, HEDS, and then NOS was eluted with 10 mM NADPH in HEDS. The enzyme could be concentrated by ultrafiltration and quick frozen and stored at -70°C without loss in activity for at least 3 months.
- NOS was assayed for the formation of citrulline following the procedure of Schmidt et al (PNAS 88 365-369, 1991) with these modifications: 20 mM Hepes, pH 7.4, lO ⁇ g/ml calmodulin. 2.5 mM CaC , 2.5 mM DTT, 125 ⁇ M NADPH, lO ⁇ M tetrahydrobiopterin, 0.5mg/ml BSA. and l ⁇ M L-[ 14 C]arginine (New England Nuclear). Linearity of NOS- catalyzed rate was confirmed prior to kinetic studies mat used single time point determination of rate. Purification of NOS from cvtokine-induced human colorectal adenocarcinoma DLD-1 cells.
- DLD-1 (ATCC No. CCL 221) were grown at 37°C, 5% CO2 in RPMI 1640 medium supplemented witii L- glutamine, penicillin, streptomycin, and 10% heat-inactivated fetal bovine serum.
- Cells were grown to confluence and then die following cocktail of cytokines were added: 100 units/ml interferon-gamma, 200 units/ml interleukin-6, 10 ng/ml tumor necrosis factor, and 0.5 ng/ml interleukin- IB.
- cytokines 100 units/ml interferon-gamma, 200 units/ml interleukin-6, 10 ng/ml tumor necrosis factor, and 0.5 ng/ml interleukin- IB.
- cytokines 100 units/ml interferon-gamma
- 200 units/ml interleukin-6 10 ng/ml tumor necrosis factor
- 0.5 ng/ml interleukin- IB
- citrulline were assayed as described above except that 10 ⁇ M FAD was included and calmodulin and CaCb excluded from die assay mix.
- Human brain NOS Human brain NOS was prepared using variations of the procedures of Schmidt et al. (PNAS US 365-369, 1991), Mayer et al. (Fed. Eur. Biochem. Soc. 288 187-191, 1991), and Bredt and Snyder, (PNAS 87 682-685, 1990). Briefly, fresh whole brains (3 with myelinated tissue disected away, 1050g) were homogenized in cold buffer A (50 mM HEPES, pH 7.5 (pH at RT) and 0.5 mM EDTA, 10 mM DTT, 3.6 L total volume) with a polytron. The mixture was centrifuged at 13,000g for 1 hour and die supernatant fluid was removed (about 2050ml).
- cold buffer A 50 mM HEPES, pH 7.5 (pH at RT) and 0.5 mM EDTA, 10 mM DTT, 3.6 L total volume
- the column was washed with 100ml buffer A, 200ml buffer A with 500 mM NaCl, 100ml Buffer A, then 30ml buffer A with 5 mM NADPH.
- To the enzyme solution was added tetrahydrobiopterin to lO ⁇ M, FAD and FMN to l ⁇ M, and Tween to 0.1%. This solution was concentrated by Centriprep-30 to a volume of approximately 500 ⁇ l. Enzyme activity was determined as described by Schmidt et al. 1991, except that lO ⁇ M tetrahydrobiopterin was included in the assay.
- Values are inhibition constants (Ki) obtained from measuring percent inhibition at three or more concentrations of inhibitor and assuming competitive inhibition with respect to arginine.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Communicable Diseases (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Oncology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Pyrrole Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description














Claims





Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP94900266A EP0670720A1 (en) | 1992-11-27 | 1993-11-26 | Enzyme inhibitors |
| AU55330/94A AU5533094A (en) | 1992-11-27 | 1993-11-26 | Enzyme inhibitors |
| JP6512923A JPH08503940A (en) | 1992-11-27 | 1993-11-26 | Enzyme inhibitors |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB929224948A GB9224948D0 (en) | 1992-11-27 | 1992-11-27 | Nitric oxide synthase inhibitors |
| GB9224948.1 | 1992-11-27 | ||
| GB9315159.5 | 1993-07-22 | ||
| GB939315159A GB9315159D0 (en) | 1993-07-22 | 1993-07-22 | Nitric oxide synthase inhibitors |
| GB939319663A GB9319663D0 (en) | 1993-09-23 | 1993-09-23 | Enzyme inhibitors |
| GB9319663.2 | 1993-09-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1994012165A2 true WO1994012165A2 (en) | 1994-06-09 |
| WO1994012165A3 WO1994012165A3 (en) | 1994-12-08 |
Family
ID=27266488
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB1993/002437 Ceased WO1994012165A2 (en) | 1992-11-27 | 1993-11-26 | Enzyme inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP0670720A1 (en) |
| JP (1) | JPH08503940A (en) |
| CN (1) | CN1095710A (en) |
| AU (1) | AU5533094A (en) |
| IL (1) | IL107771A0 (en) |
| SI (1) | SI9300616A (en) |
| WO (1) | WO1994012165A2 (en) |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995005363A1 (en) * | 1993-08-12 | 1995-02-23 | Astra Aktiebolag | Amidine derivatives with nitric oxide synthetase activities |
| WO1995011231A1 (en) * | 1993-10-21 | 1995-04-27 | G. D. Searle & Co. | Amidino derivatives useful as nitric oxide synthase inhibitors |
| WO1996009286A1 (en) * | 1994-09-20 | 1996-03-28 | Astra Aktiebolag | Isothiourea derivatives as no synthase inhibitors |
| WO1996014842A1 (en) * | 1994-11-15 | 1996-05-23 | Merck & Co., Inc. | Substituted heterocycles as inhibitors of nitric oxide synthase |
| EP0717040A1 (en) | 1994-12-14 | 1996-06-19 | Japan Tobacco Inc. | Thiazine or thiazepine derivatives which inhibit NOS |
| WO1996018617A1 (en) * | 1994-12-12 | 1996-06-20 | Merck & Co., Inc. | Substituted 2-acylamino-pyridines as inhibitors of nitric oxide synthase |
| FR2728261A1 (en) * | 1994-12-14 | 1996-06-21 | Japan Tobacco Inc | THIAZINE OR THIAZEPINE DERIVATIVES USEFUL AS INHIBITORS OF NO-SYNTHETASE |
| EP0718294A1 (en) * | 1994-12-16 | 1996-06-26 | Hoechst Aktiengesellschaft | 2-Amino-1,3-thiazepines and their use as inhibitors of the nitrogene oxide synthase |
| FR2730733A1 (en) * | 1995-02-17 | 1996-08-23 | Hoechst Lab | Novel nitro ethylenic sulphur derivatives |
| WO1996030350A1 (en) * | 1995-03-27 | 1996-10-03 | Fujisawa Pharmaceutical Co., Ltd. | Amidine derivatives |
| WO1996033175A1 (en) * | 1995-04-20 | 1996-10-24 | G.D. Searle & Co. | Cyclic amidino agents useful as nitric oxide synthase inhibitors |
| US5674907A (en) * | 1995-03-24 | 1997-10-07 | Children's Hospital Medical Center | Mercapto derivatives as inhibitors of nitric oxide synthase |
| US5786364A (en) * | 1995-02-11 | 1998-07-28 | Astra Aktiebolag | Bicyclic isothiourea derivatives useful in therapy |
| US5807886A (en) * | 1994-05-07 | 1998-09-15 | Astra Aktiebolag | Bicyclic amidine dervatives as inhibitors of nitric oxide synthetase |
| US5849782A (en) * | 1995-01-13 | 1998-12-15 | The General Hospital Corporation | Methods of inhibiting neurodegenerative diseases |
| US5908842A (en) * | 1995-12-08 | 1999-06-01 | Merck & Co., Inc. | Substituted 2-acylamino-pyridines as inhibitors of nitric oxide synthase |
| US5929063A (en) * | 1995-03-24 | 1999-07-27 | Children's Hospital Medical Center | Mercapto and seleno derivatives as inhibitors of nitric oxide synthase |
| US5945408A (en) * | 1996-03-06 | 1999-08-31 | G.D. Searle & Co. | Hydroxyanidino derivatives useful as nitric oxide synthase inhibitors |
| US5958958A (en) * | 1997-07-22 | 1999-09-28 | G.D. Searle & Co. | 1,2,4-oxa diazolino and 1,24-oxa diazolidion heterocycles as useful nitric oxide synthase inhibitors |
| WO1999051215A3 (en) * | 1998-04-06 | 2000-03-09 | Fujisawa Pharmaceutical Co | Immunosuppressive imidazole derivatives and their combination preparations with tacrolimus or cyclosporins |
| US6090839A (en) * | 1996-12-23 | 2000-07-18 | Merck & Co., Inc. | Antidiabetic agents |
| WO2000048591A1 (en) * | 1999-02-16 | 2000-08-24 | Angiogene Pharmaceuticals Ltd. | Combinations for the treatment of diseases involving angiogenesis |
| US6160000A (en) * | 1996-12-23 | 2000-12-12 | Merck & Co., Inc. | Antidiabetic agents based on aryl and heteroarylacetic acids |
| WO2001074351A1 (en) * | 2000-03-31 | 2001-10-11 | Universitair Medisch Centrum | Composition for the prevention and/or treatment, in newborn babies, of the effects of complications during childbirth |
| WO2001093867A1 (en) * | 2000-06-09 | 2001-12-13 | Aventis Pharma S.A. | 4,5-dihydro-thiazo-2-ylamine derivatives and their use as no-synthase inhibitors |
| WO2001094325A1 (en) * | 2000-06-09 | 2001-12-13 | Aventis Pharma S.A. | 2-aminothiazoline derivatives and their use as no-synthase inhibitors |
| US6344473B1 (en) | 2000-08-07 | 2002-02-05 | G.D. Searle & Co. | Imidazoles useful as nitric oxide synthase inhibitors |
| US6403830B2 (en) | 2000-03-24 | 2002-06-11 | Pharmacia Corporation | Amidino compound and salts thereof useful as nitric oxide synthase inhibitors |
| WO2001053257A3 (en) * | 2000-01-19 | 2002-06-27 | Cadila Healthcare Ltd | Compounds having hypolipedemic and hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them |
| US6420566B2 (en) | 2000-06-09 | 2002-07-16 | Aventis Pharma S.A. | Pharmaceutical compositions containing a 4, 5-dihydro-1, 3-thiazol-2-ylamine derivative, novel derivatives and preparation thereof |
| US6451821B1 (en) | 2000-06-09 | 2002-09-17 | Aventis Pharma S.A. | Use of 2-aminothiazoline derivatives as inhibitors of inducible no-synthase |
| US6465686B2 (en) | 2000-04-13 | 2002-10-15 | Pharmacia Corporation | Halogenated 2-amino-5,6 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6465518B2 (en) | 2000-04-13 | 2002-10-15 | Pharmacia Corporation | Halogenated 2-amino-4, 5 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6489323B1 (en) | 1998-06-10 | 2002-12-03 | G.D. Searle & Co. | Heterobicyclic and tricyclic nitric oxide synthase inhibitors |
| US6545170B2 (en) | 2000-04-13 | 2003-04-08 | Pharmacia Corporation | 2-amino-5, 6 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6552052B2 (en) | 1998-06-10 | 2003-04-22 | Monsanto/G.D. Searle | Pyrrolo[2,1-c][1,2,4] thiadiazoles and Pyrollo[2,1-c][1,12,4]oxadiazoles useful as nitric oxide synthase inhibitors |
| WO2003040142A1 (en) * | 2001-11-09 | 2003-05-15 | Aventis Pharma S.A. | 2-amino-4-heteroarylethyl thiazoline derivatives and their use an inhibitors of inducible no-synthase |
| WO2003040115A1 (en) * | 2001-11-09 | 2003-05-15 | Aventis Pharma S.A. | 2-amino-thiazoline derivatives and their use as inhibitors of inducible no-synthase |
| FR2832150A1 (en) * | 2001-11-09 | 2003-05-16 | Aventis Pharma Sa | New 2-amino-4-(pyridylmethyl) thiazoline derivatives having inducible NO-synthase inhibiting activity, useful for treating of Parkinson's disease, cerebral disorders, migraines, depression, diabetes |
| FR2832152A1 (en) * | 2001-11-09 | 2003-05-16 | Aventis Pharma Sa | New 2-amino-thiazoline derivatives having inducible NO-synthase inhibiting activity, useful for treating Parkinson's, cerebral disorders, migraines, depression, diabetes |
| FR2832151A1 (en) * | 2001-11-09 | 2003-05-16 | Aventis Pharma Sa | New 2-amino-4-(heteroarylethyl) thiazoline derivatives having inducible NO-synthase inhibiting activity, useful for treating Parkinson's, cerebral disorders, migraines, depression, diabetes |
| US6586471B1 (en) | 2000-04-13 | 2003-07-01 | G. D. Searle | Halogenated 2-amino-3, 4 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| WO2003039446A3 (en) * | 2001-11-09 | 2003-11-27 | Aventis Pharma Sa | Use of 2-amino-4-pyridylmethyl-thiazoline derivatives as inhibitors of inducible no-synthase |
| US6787668B2 (en) | 2000-04-13 | 2004-09-07 | Pharmacia Corporation | 2-amino-4,5 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6956131B2 (en) | 2000-04-13 | 2005-10-18 | Pharmacia Corporation | 2-amino-3, 4 heptenoic compounds useful as nitric oxide synthase inhibitors |
| US7012098B2 (en) | 2001-03-23 | 2006-03-14 | Pharmacia Corporation | Inhibitors of inducible nitric oxide synthase for chemoprevention and treatment of cancers |
| US7087633B2 (en) | 2000-03-31 | 2006-08-08 | Universitair Medisch Centrum | Method for treating perinatal asphyxia in a human or animal neonate |
| WO2007108004A3 (en) * | 2006-03-23 | 2008-01-17 | Meditor Pharmaceuticals Ltd | S-alkylisothiouronium derivatives for the treatment of inflammatory diseases |
| WO2010113848A1 (en) | 2009-03-31 | 2010-10-07 | 塩野義製薬株式会社 | Isothiourea derivative or isourea derivative having bace1 inhibitory activity |
| US8168630B2 (en) | 2007-04-24 | 2012-05-01 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives substituted with a cyclic group |
| US8173642B2 (en) | 2005-10-25 | 2012-05-08 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| US8637504B2 (en) | 2008-06-13 | 2014-01-28 | Shionogi & Co., Ltd. | Sulfur-containing heterocyclic derivative having beta secretase inhibitory activity |
| US8653067B2 (en) | 2007-04-24 | 2014-02-18 | Shionogi & Co., Ltd. | Pharmaceutical composition for treating Alzheimer's disease |
| US8703785B2 (en) | 2008-10-22 | 2014-04-22 | Shionogi & Co., Ltd. | 2-aminopyrimidin-4-one and 2-aminopyridine derivatives both having BACE1-inhibiting activity |
| US8927721B2 (en) | 2010-10-29 | 2015-01-06 | Shionogi & Co., Ltd. | Naphthyridine derivative |
| US8999980B2 (en) | 2009-12-11 | 2015-04-07 | Shionogi & Co., Ltd. | Oxazine derivatives |
| US9758513B2 (en) | 2012-10-24 | 2017-09-12 | Shionogi & Co., Ltd. | Dihydrooxazine or oxazepine derivatives having BACE1 inhibitory activity |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101372471B (en) * | 2008-10-08 | 2010-12-22 | 中国科学院化学研究所 | Novel use of alkyl isourea compound and analogues thereof |
| EP2634188A4 (en) | 2010-10-29 | 2014-05-07 | Shionogi & Co | Fused aminodihydropyrimidine derivative |
| US8883779B2 (en) | 2011-04-26 | 2014-11-11 | Shinogi & Co., Ltd. | Oxazine derivatives and a pharmaceutical composition for inhibiting BACE1 containing them |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1178242A (en) * | 1966-02-05 | 1970-01-21 | Wellcome Found | Novel Biologically Active Bis-Isothioureas |
| US3790600A (en) * | 1972-04-06 | 1974-02-05 | Uniroyal Inc | 2-(3-benzo(b)thenyl)-2-thiopseudourea and its pharmaceutically acceptable salts |
| US3954982A (en) * | 1972-04-20 | 1976-05-04 | Smith Kline & French Laboratories Limited | Pharmaceutical compositions and method of inhibiting H-1 and H-2 histamine receptors |
| ZA771408B (en) * | 1976-03-29 | 1978-04-26 | Smith Kline French Lab | Pharmaceutical compositions |
| US4208430A (en) * | 1979-03-15 | 1980-06-17 | Smithkline Corporation | Pharmaceutical compositions and method of inhibiting phenylethanolamine N-methyltransferase |
| US4262125A (en) * | 1979-07-23 | 1981-04-14 | American Home Products Corporation | (1H-Imidazol-5-ylmethyl)isothioureas |
| EP0245669B1 (en) * | 1986-05-14 | 1993-12-01 | Medopharm Arzneimittelwerk Dr. Zillich GmbH & Co. | Pharmaceutical preparation for preventing damage to living cells by free radicals, or for increasing the efficacy of organic sulphur compounds, and process for increasing the life span of isolated organs |
| GB8916947D0 (en) * | 1989-07-25 | 1989-09-13 | Smith Kline French Lab | Medicaments |
| CA2066728C (en) * | 1989-09-19 | 2001-12-25 | Jan K. Hellstrand | Anti-tumor preparation comprising interleukin-2 and histamine, analogs thereof or h2-receptor agonists |
| ATE191847T1 (en) * | 1991-12-16 | 2000-05-15 | Univ Washington | USE OF AMINOGUAMIDINE FOR THE PRODUCTION OF A MEDICINAL PRODUCT FOR SUPPRESSING NITROGEN OXIDE FORMATION |
| KR100892685B1 (en) * | 2007-11-09 | 2009-04-15 | 주식회사 하이닉스반도체 | EAC system |
-
1993
- 1993-11-26 EP EP94900266A patent/EP0670720A1/en not_active Withdrawn
- 1993-11-26 JP JP6512923A patent/JPH08503940A/en active Pending
- 1993-11-26 CN CN93121643A patent/CN1095710A/en active Pending
- 1993-11-26 AU AU55330/94A patent/AU5533094A/en not_active Abandoned
- 1993-11-26 SI SI9300616A patent/SI9300616A/en unknown
- 1993-11-26 IL IL10777193A patent/IL107771A0/en unknown
- 1993-11-26 WO PCT/GB1993/002437 patent/WO1994012165A2/en not_active Ceased
Cited By (100)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6030985A (en) * | 1993-08-12 | 2000-02-29 | Astra Aktiebolag | Amidine derivatives with nitric oxide synthetase activities |
| US5807885A (en) * | 1993-08-12 | 1998-09-15 | Astra Aktiebolag | Amidine derivatives with nitric oxide synthetase activities |
| WO1995005363A1 (en) * | 1993-08-12 | 1995-02-23 | Astra Aktiebolag | Amidine derivatives with nitric oxide synthetase activities |
| EP0897912A1 (en) * | 1993-10-21 | 1999-02-24 | G.D. Searle & Co. | Amidino derivatives useful as nitric oxide synthase inhibitors |
| WO1995011231A1 (en) * | 1993-10-21 | 1995-04-27 | G. D. Searle & Co. | Amidino derivatives useful as nitric oxide synthase inhibitors |
| US5854234A (en) * | 1993-10-21 | 1998-12-29 | G. D. Searle & Co. | Amidino dervatives useful as nitric oxide synthase inhibitors |
| US6046211A (en) * | 1993-10-21 | 2000-04-04 | G.D. Searle & Co. | Amidino derivatives useful as nitric oxide synthase inhibitors |
| AU688811B2 (en) * | 1993-10-21 | 1998-03-19 | G.D. Searle & Co. | Amidino derivatives useful as nitric oxide synthase inhibitors |
| US6448286B1 (en) | 1993-10-21 | 2002-09-10 | G.D. Searle & Co. | Imino pyrrolidine derivatives useful as nitric oxide synthase inhibitors |
| US6071906A (en) * | 1993-10-21 | 2000-06-06 | G. D. Searle & Co. | Imidino piperidine derivatives useful as nitric oxide synthase inhibitors |
| US5807886A (en) * | 1994-05-07 | 1998-09-15 | Astra Aktiebolag | Bicyclic amidine dervatives as inhibitors of nitric oxide synthetase |
| US6117898A (en) * | 1994-05-07 | 2000-09-12 | Astra Aktiebolag | Bicyclic amidine derivatives as inhibitors of nitric oxide synthetase |
| US5721247A (en) * | 1994-09-20 | 1998-02-24 | Astra Aktiebolag | Isothiourea derivatives useful in therapy |
| WO1996009286A1 (en) * | 1994-09-20 | 1996-03-28 | Astra Aktiebolag | Isothiourea derivatives as no synthase inhibitors |
| WO1996014842A1 (en) * | 1994-11-15 | 1996-05-23 | Merck & Co., Inc. | Substituted heterocycles as inhibitors of nitric oxide synthase |
| WO1996018617A1 (en) * | 1994-12-12 | 1996-06-20 | Merck & Co., Inc. | Substituted 2-acylamino-pyridines as inhibitors of nitric oxide synthase |
| FR2728261A1 (en) * | 1994-12-14 | 1996-06-21 | Japan Tobacco Inc | THIAZINE OR THIAZEPINE DERIVATIVES USEFUL AS INHIBITORS OF NO-SYNTHETASE |
| EP0717040A1 (en) | 1994-12-14 | 1996-06-19 | Japan Tobacco Inc. | Thiazine or thiazepine derivatives which inhibit NOS |
| EP0718294A1 (en) * | 1994-12-16 | 1996-06-26 | Hoechst Aktiengesellschaft | 2-Amino-1,3-thiazepines and their use as inhibitors of the nitrogene oxide synthase |
| US6133306A (en) * | 1995-01-13 | 2000-10-17 | The General Hospital Corporation | Methods of inhibiting neurodegerative diseases |
| US5849782A (en) * | 1995-01-13 | 1998-12-15 | The General Hospital Corporation | Methods of inhibiting neurodegenerative diseases |
| US5786364A (en) * | 1995-02-11 | 1998-07-28 | Astra Aktiebolag | Bicyclic isothiourea derivatives useful in therapy |
| FR2730733A1 (en) * | 1995-02-17 | 1996-08-23 | Hoechst Lab | Novel nitro ethylenic sulphur derivatives |
| US5985917A (en) * | 1995-03-24 | 1999-11-16 | Children's Hospital Medical Center | Mercapto and seleno derivatives as inhibitors of nitric oxide synthase |
| US5674907A (en) * | 1995-03-24 | 1997-10-07 | Children's Hospital Medical Center | Mercapto derivatives as inhibitors of nitric oxide synthase |
| US5929063A (en) * | 1995-03-24 | 1999-07-27 | Children's Hospital Medical Center | Mercapto and seleno derivatives as inhibitors of nitric oxide synthase |
| US5952385A (en) * | 1995-03-24 | 1999-09-14 | Children's Hospital Medical Center | Mercapto derivatives as inhibitors of nitric oxide synthase |
| WO1996030350A1 (en) * | 1995-03-27 | 1996-10-03 | Fujisawa Pharmaceutical Co., Ltd. | Amidine derivatives |
| AU712315B2 (en) * | 1995-04-20 | 1999-11-04 | G.D. Searle & Co. | Cyclic amidino agents useful as nitric oxide synthase inhibitors |
| WO1996033175A1 (en) * | 1995-04-20 | 1996-10-24 | G.D. Searle & Co. | Cyclic amidino agents useful as nitric oxide synthase inhibitors |
| US5883251A (en) * | 1995-04-20 | 1999-03-16 | G. D. Searle & Co. | Azepine derivatives useful as nitric oxide synthase inhibitors |
| US5908842A (en) * | 1995-12-08 | 1999-06-01 | Merck & Co., Inc. | Substituted 2-acylamino-pyridines as inhibitors of nitric oxide synthase |
| US5945408A (en) * | 1996-03-06 | 1999-08-31 | G.D. Searle & Co. | Hydroxyanidino derivatives useful as nitric oxide synthase inhibitors |
| US6160000A (en) * | 1996-12-23 | 2000-12-12 | Merck & Co., Inc. | Antidiabetic agents based on aryl and heteroarylacetic acids |
| US6090839A (en) * | 1996-12-23 | 2000-07-18 | Merck & Co., Inc. | Antidiabetic agents |
| US5958958A (en) * | 1997-07-22 | 1999-09-28 | G.D. Searle & Co. | 1,2,4-oxa diazolino and 1,24-oxa diazolidion heterocycles as useful nitric oxide synthase inhibitors |
| US6136829A (en) * | 1997-07-22 | 2000-10-24 | G.D. Searle & Co. | Oxathiadiazole derivatives usful as iNOS inhibitors |
| US5981556A (en) * | 1997-07-22 | 1999-11-09 | G.D. Searle & Co. | 1,3-diazolino and 1,3-diazolidino heterocycles as useful nitric oxide synthase inhibitors |
| WO1999051215A3 (en) * | 1998-04-06 | 2000-03-09 | Fujisawa Pharmaceutical Co | Immunosuppressive imidazole derivatives and their combination preparations with tacrolimus or cyclosporins |
| US6489323B1 (en) | 1998-06-10 | 2002-12-03 | G.D. Searle & Co. | Heterobicyclic and tricyclic nitric oxide synthase inhibitors |
| US6552052B2 (en) | 1998-06-10 | 2003-04-22 | Monsanto/G.D. Searle | Pyrrolo[2,1-c][1,2,4] thiadiazoles and Pyrollo[2,1-c][1,12,4]oxadiazoles useful as nitric oxide synthase inhibitors |
| WO2000048591A1 (en) * | 1999-02-16 | 2000-08-24 | Angiogene Pharmaceuticals Ltd. | Combinations for the treatment of diseases involving angiogenesis |
| US7087627B1 (en) | 1999-02-16 | 2006-08-08 | Angiogene Pharmaceuticals Ltd. | Combinations for the treatment of diseases involving angiogenesis |
| EA007959B1 (en) * | 2000-01-19 | 2007-02-27 | Кадила Хелзкэр Лтд. | Compounds having hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them |
| WO2001053257A3 (en) * | 2000-01-19 | 2002-06-27 | Cadila Healthcare Ltd | Compounds having hypolipedemic and hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them |
| CZ304346B6 (en) * | 2000-01-19 | 2014-03-19 | Cadila Healthcare Ltd. | Substituted pyrrole derivative exhibiting hypolipidemic and hypocholesteremic activity , process for its preparation and pharmaceutical composition containing thereof |
| HRP20020643B1 (en) * | 2000-01-19 | 2015-04-24 | Cadila Healthcare Ltd Zydus Tower, Satellite Cross Roads, Rrd Center | NEW COMPOUNDS CONTAINING HYPOLYPEDEMIC, HYPOCOLESTEREMIC ACTIVITIES, THE PROCEDURE OF THEIR PREPARATION AND THE PHARMACEUTICAL PREPARATIONS CONTAINING THEM |
| US7102013B2 (en) | 2000-03-24 | 2006-09-05 | Pharmacia Corporation | Methods of making amidino compounds useful as nitric oxide synthase inhibitors |
| US6403830B2 (en) | 2000-03-24 | 2002-06-11 | Pharmacia Corporation | Amidino compound and salts thereof useful as nitric oxide synthase inhibitors |
| US6914158B2 (en) | 2000-03-24 | 2005-07-05 | Pharmacia Corporation | Amidino compounds useful as nitric oxide synthase inhibitors |
| US6586474B2 (en) | 2000-03-24 | 2003-07-01 | Pharmacia Corporation | Amidino compounds useful as nitric oxide synthase inhibitors |
| US7087633B2 (en) | 2000-03-31 | 2006-08-08 | Universitair Medisch Centrum | Method for treating perinatal asphyxia in a human or animal neonate |
| WO2001074351A1 (en) * | 2000-03-31 | 2001-10-11 | Universitair Medisch Centrum | Composition for the prevention and/or treatment, in newborn babies, of the effects of complications during childbirth |
| US6894069B2 (en) | 2000-03-31 | 2005-05-17 | Universitair Medisch Centrum | Method for treating perinatal asphyxia in a human or animal neonate |
| US6586471B1 (en) | 2000-04-13 | 2003-07-01 | G. D. Searle | Halogenated 2-amino-3, 4 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6545170B2 (en) | 2000-04-13 | 2003-04-08 | Pharmacia Corporation | 2-amino-5, 6 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6465686B2 (en) | 2000-04-13 | 2002-10-15 | Pharmacia Corporation | Halogenated 2-amino-5,6 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6465518B2 (en) | 2000-04-13 | 2002-10-15 | Pharmacia Corporation | Halogenated 2-amino-4, 5 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| US6956131B2 (en) | 2000-04-13 | 2005-10-18 | Pharmacia Corporation | 2-amino-3, 4 heptenoic compounds useful as nitric oxide synthase inhibitors |
| US6787668B2 (en) | 2000-04-13 | 2004-09-07 | Pharmacia Corporation | 2-amino-4,5 heptenoic acid derivatives useful as nitric oxide synthase inhibitors |
| AU2001266123B2 (en) * | 2000-06-09 | 2007-01-25 | Aventis Pharma S.A. | 4,5-dihydro-thiazo-2-ylamine derivatives and their use as no-synthase inhibitors |
| WO2001093867A1 (en) * | 2000-06-09 | 2001-12-13 | Aventis Pharma S.A. | 4,5-dihydro-thiazo-2-ylamine derivatives and their use as no-synthase inhibitors |
| US6451821B1 (en) | 2000-06-09 | 2002-09-17 | Aventis Pharma S.A. | Use of 2-aminothiazoline derivatives as inhibitors of inducible no-synthase |
| US6699895B2 (en) | 2000-06-09 | 2004-03-02 | Aventis Pharma S.A. | 2-aminothiazoline derivatives and process for preparing the same |
| WO2001094325A1 (en) * | 2000-06-09 | 2001-12-13 | Aventis Pharma S.A. | 2-aminothiazoline derivatives and their use as no-synthase inhibitors |
| FR2810037A1 (en) * | 2000-06-09 | 2001-12-14 | Aventis Pharma Sa | Inducible nitrogen monoxide synthase inhibitors comprising new or known 4- and/or 5-substituted 4,5-dihydro-thiazol-2-ylamine compounds, useful e.g. for treating neurodegenerative or inflammatory diseases |
| FR2810036A1 (en) * | 2000-06-09 | 2001-12-14 | Aventis Pharma Sa | Compositions containing new or known 4-substituted 4,5-dihydro-thiazol-2-ylamines, as inducible nitrogen monoxide synthase inhibitors useful e.g. for treating neurodegenerative or inflammatory diseases |
| US6420566B2 (en) | 2000-06-09 | 2002-07-16 | Aventis Pharma S.A. | Pharmaceutical compositions containing a 4, 5-dihydro-1, 3-thiazol-2-ylamine derivative, novel derivatives and preparation thereof |
| US6344473B1 (en) | 2000-08-07 | 2002-02-05 | G.D. Searle & Co. | Imidazoles useful as nitric oxide synthase inhibitors |
| US7012098B2 (en) | 2001-03-23 | 2006-03-14 | Pharmacia Corporation | Inhibitors of inducible nitric oxide synthase for chemoprevention and treatment of cancers |
| US7227022B2 (en) | 2001-11-09 | 2007-06-05 | Aventis Pharma Sa | Use of 2-amino-thiazoline derivatives as inhibitors of inducible no-synthase |
| FR2832150A1 (en) * | 2001-11-09 | 2003-05-16 | Aventis Pharma Sa | New 2-amino-4-(pyridylmethyl) thiazoline derivatives having inducible NO-synthase inhibiting activity, useful for treating of Parkinson's disease, cerebral disorders, migraines, depression, diabetes |
| US6953796B2 (en) | 2001-11-09 | 2005-10-11 | Aventis Pharma S.A. | Use of 2-amino-thiazoline derivatives as inhibitors of inducible No-synthase |
| FR2832152A1 (en) * | 2001-11-09 | 2003-05-16 | Aventis Pharma Sa | New 2-amino-thiazoline derivatives having inducible NO-synthase inhibiting activity, useful for treating Parkinson's, cerebral disorders, migraines, depression, diabetes |
| US6872740B2 (en) | 2001-11-09 | 2005-03-29 | Aventis Pharma S.A. | Use of 2-amino-4-heteroarylethyl-thiazoline derivatives as inhibitors of inducible no-synthase |
| FR2832151A1 (en) * | 2001-11-09 | 2003-05-16 | Aventis Pharma Sa | New 2-amino-4-(heteroarylethyl) thiazoline derivatives having inducible NO-synthase inhibiting activity, useful for treating Parkinson's, cerebral disorders, migraines, depression, diabetes |
| WO2003040115A1 (en) * | 2001-11-09 | 2003-05-15 | Aventis Pharma S.A. | 2-amino-thiazoline derivatives and their use as inhibitors of inducible no-synthase |
| US6762196B2 (en) | 2001-11-09 | 2004-07-13 | Aventis Pharma S. A. | Use of 2-amino-4-pyridylmethyl-thiazoline derivatives as inhibitors of inducible no-synthase |
| CN1314674C (en) * | 2001-11-09 | 2007-05-09 | 安万特医药股份有限公司 | 2-Amino-dihydrothiazole derivatives and their use as inducible NO-synthase inhibitors |
| US6699867B2 (en) | 2001-11-09 | 2004-03-02 | Aventis Pharma S.A. | Use of 2-amino-thiazoline derivatives as inhibitors of inducible NO-synthase |
| WO2003040142A1 (en) * | 2001-11-09 | 2003-05-15 | Aventis Pharma S.A. | 2-amino-4-heteroarylethyl thiazoline derivatives and their use an inhibitors of inducible no-synthase |
| WO2003039446A3 (en) * | 2001-11-09 | 2003-11-27 | Aventis Pharma Sa | Use of 2-amino-4-pyridylmethyl-thiazoline derivatives as inhibitors of inducible no-synthase |
| US8546380B2 (en) | 2005-10-25 | 2013-10-01 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| US8815851B2 (en) | 2005-10-25 | 2014-08-26 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| US9029358B2 (en) | 2005-10-25 | 2015-05-12 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| US8173642B2 (en) | 2005-10-25 | 2012-05-08 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| US8633188B2 (en) | 2005-10-25 | 2014-01-21 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives |
| WO2007108004A3 (en) * | 2006-03-23 | 2008-01-17 | Meditor Pharmaceuticals Ltd | S-alkylisothiouronium derivatives for the treatment of inflammatory diseases |
| US8653067B2 (en) | 2007-04-24 | 2014-02-18 | Shionogi & Co., Ltd. | Pharmaceutical composition for treating Alzheimer's disease |
| US8884062B2 (en) | 2007-04-24 | 2014-11-11 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives substituted with a cyclic group |
| US8168630B2 (en) | 2007-04-24 | 2012-05-01 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives substituted with a cyclic group |
| US8541408B2 (en) | 2007-04-24 | 2013-09-24 | Shionogi & Co., Ltd. | Aminodihydrothiazine derivatives substituted with a cyclic group |
| US8637504B2 (en) | 2008-06-13 | 2014-01-28 | Shionogi & Co., Ltd. | Sulfur-containing heterocyclic derivative having beta secretase inhibitory activity |
| US9650371B2 (en) | 2008-06-13 | 2017-05-16 | Shionogi & Co., Ltd. | Sulfur-containing heterocyclic derivative having beta secretase inhibitory activity |
| US8703785B2 (en) | 2008-10-22 | 2014-04-22 | Shionogi & Co., Ltd. | 2-aminopyrimidin-4-one and 2-aminopyridine derivatives both having BACE1-inhibiting activity |
| WO2010113848A1 (en) | 2009-03-31 | 2010-10-07 | 塩野義製薬株式会社 | Isothiourea derivative or isourea derivative having bace1 inhibitory activity |
| US8999980B2 (en) | 2009-12-11 | 2015-04-07 | Shionogi & Co., Ltd. | Oxazine derivatives |
| US9656974B2 (en) | 2009-12-11 | 2017-05-23 | Shionogi & Co., Ltd. | Oxazine derivatives |
| US8927721B2 (en) | 2010-10-29 | 2015-01-06 | Shionogi & Co., Ltd. | Naphthyridine derivative |
| US9758513B2 (en) | 2012-10-24 | 2017-09-12 | Shionogi & Co., Ltd. | Dihydrooxazine or oxazepine derivatives having BACE1 inhibitory activity |
Also Published As
| Publication number | Publication date |
|---|---|
| IL107771A0 (en) | 1994-02-27 |
| SI9300616A (en) | 1994-06-30 |
| AU5533094A (en) | 1994-06-22 |
| JPH08503940A (en) | 1996-04-30 |
| CN1095710A (en) | 1994-11-30 |
| WO1994012165A3 (en) | 1994-12-08 |
| EP0670720A1 (en) | 1995-09-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1994012165A2 (en) | Enzyme inhibitors | |
| US6803365B2 (en) | Imidazo[1,3,5]triazinones and the use thereof | |
| WO1995009619A2 (en) | Substituted urea and isothiourea derivatives as no synthase inhibitors | |
| EP0705257B1 (en) | Aminoacid derivatives as no synthase inhibitors | |
| MXPA05005406A (en) | Novel chemical compounds. | |
| CA3105506A1 (en) | Dimeric immuno-modulatory compounds against cereblon-based mechanisms | |
| EP0138464A2 (en) | 2-Amino-5-hydroxy-4-methylpyrimidine derivatives | |
| AU2017339104B2 (en) | 2-amino-N-(arylsulfinyl)-acetamide compounds as inhibitors of bacterial aminoacyl-tRNA synthetase | |
| JPH10508847A (en) | Aminotetrazole derivatives useful as nitric oxide synthase inhibitors | |
| KR20010034598A (en) | Halogenated amidino amino acid derivatives useful as nitric oxide synthase inhibitors | |
| US5063240A (en) | Novel compounds | |
| JPH10505862A (en) | Isothiourea derivatives as NO synthase inhibitors | |
| US6683081B2 (en) | Triazolotriazinones and the use thereof | |
| JP2025156455A (en) | Thiazolo[5,4-b]pyridine MALT-1 inhibitors | |
| HU181960B (en) | Process for producing substituted imidazolylmethylthio-compounds | |
| EP0251453A2 (en) | Substituted amino-dihydrooxazoles, -thiazoles and -imidazoles, process for their preparation and pharmaceutical compositions containing them | |
| EP1178963A1 (en) | Ion channel modulating agents | |
| AU2003238915A1 (en) | (halo-benzo carbonyl)heterobicyclic p38 kinase inhibiting agents | |
| KR20090024705A (en) | Ruthenium II Compound | |
| CN103435562B (en) | 6-replaces Benzodiazepine-2,4-cyclohexadione compounds and uses thereof | |
| JP7815213B2 (en) | Benzylamine derivatives, their preparation method and uses | |
| RU2026288C1 (en) | Derivatives of pyrrolo-(2,1-b)-thiazole and derivatives of 2-thioxopyrrolidine as intermediate products for synthesis of pyrrolo-(2,1-b)-thiazole derivatives | |
| US6090846A (en) | Substituted urea and isothiourea derivatives as no synthase inhibitors | |
| KR20020079963A (en) | Medicament for Viral Diseases | |
| US6225305B1 (en) | Substituted urea and isothiorea derivatives as no synthase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AT AU BB BG BR BY CA CH CZ DE DK ES FI GB HU JP KP KR KZ LK LU LV MG MN MW NL NO NZ PL PT RO RU SD SE SK UA US UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AT AU BB BG BR BY CA CH CZ DE DK ES FI GB HU JP KP KR KZ LK LU LV MG MN MW NL NO NZ PL PT RO RU SD SE SK UA US UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994900266 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: US Ref document number: 1995 436465 Date of ref document: 19950719 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994900266 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| NENP | Non-entry into the national phase |
Ref country code: CA |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994900266 Country of ref document: EP |