WO1994019318A1 - Compose de 5-aminoacetylaminosulfonanilide - Google Patents
Compose de 5-aminoacetylaminosulfonanilide Download PDFInfo
- Publication number
- WO1994019318A1 WO1994019318A1 PCT/JP1994/000228 JP9400228W WO9419318A1 WO 1994019318 A1 WO1994019318 A1 WO 1994019318A1 JP 9400228 W JP9400228 W JP 9400228W WO 9419318 A1 WO9419318 A1 WO 9419318A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- acid
- compound
- methanesulfonamide
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/08—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/145—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
- C07D295/15—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings to an acyclic saturated chain
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B11/00—Electrodes; Manufacture thereof not otherwise provided for
- C25B11/04—Electrodes; Manufacture thereof not otherwise provided for characterised by the material
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- the present invention has an anti- action, antipyretic action, analgesic action and anti-allergic action
- the present inventors have conducted intensive studies with the aim of solving the above problems, and as a result, have found that the 5-aminoacetylaminosulfonanilide compound shown below can achieve the object, and have completed the present invention. did.
- the present invention provides a compound of the formula (I)
- R 1 represents a phenyl group, a halophenyl group or a cycloalkyl group having 3 to 8 carbon atoms
- R2 represents a hydrogen atom or an alkyl group having 1 to 5 carbon atoms
- R3 represents a hydrogen atom, an alkyl group having 1 to 7 carbon atoms, a cycloalkyl group having 3 to 8 carbon atoms, an alkenyl group or a benzyl group having 3 to 5 carbon atoms, or R2 And R3 together represent a group that forms a 5- to 5-membered heterocyclic ring.
- the halophenyl group represented by R 1 is a phenyl group substituted by one or two identical or different fluorine atoms, chlorine atoms or bromine atoms, and a cycloalkyl group having 3 to 8 carbon atoms is cyclobutyl. Mouth bil group, cyclopibutyl group, cyclopentino group, cyclohexyl group, cycloheptinol group or cyclooctyl group.
- the alkyl group having 1 to 7 carbon atoms of R 3 is a linear or branched alkyl group, for example, a methyl group, an ethyl group, an n-bromo group, an isobromo group, an n-butyl group.
- Group, isobutyl group, n-bentyl group, n-hexyl group, n-hexyl group, etc., and the cycloalkyl group having 3 to 8 carbon atoms is cyclobutyl C3 building group, cyclobutyl group, cyclobenzyl group.
- a cyclohexyl group, a cyclohexyl group or a cyclooctynole group, and an alkenyl group having 3 to 5 carbon atoms includes an aryl group, a 2-buteninole group, a 3-butenyl group, and a 3-methyl-2- group. Buteninole group and the like.
- the group in which R2 and R3 become " ⁇ to form a 5- to 7-membered heterocyclic ring includes, for example, pyrrolidino, biberidino, hexamethyleneimino, morpholino, thiomorpholino, biverazino, 4 -Methylbiverazino group.
- Salts are salts with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, or acetic acid, brobionic acid, succinic acid, fumaric acid, maleic acid, benzoic acid, phthalic acid, keichic acid It is a salt with organic acids such as glycolic acid, lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid and p-toluenesulfonic acid.
- the method for producing the compound of the present invention is as follows.
- the compound of the formula (I) of the present invention can be obtained by, for example, a production method shown in the following reaction route (i) using 2-fluoro-5-dinitroaline as a starting material. Reaction path (i)
- R1, R2 and R3 are as defined above, and X is a chlorine atom, a bromine atom or an iodine atom.
- N- (2-fluoro-5-nitrophenyl) methanesulfonamide and a compound represented by the formula (II) are subjected to an etherification reaction in the presence of a base to give a compound represented by the formula (III.).
- a compound that can be obtained can be obtained.
- Examples of the base in this reaction include lithium metal hydroxides such as lithium hydroxide, sodium hydroxide, and hydroxide hydroxide, sodium carbonate, lithium carbonate such as lithium carbonate, sodium hydrogen carbonate, and the like.
- Alkali metal bicarbonate such as hydrogen carbonate bicarbonate, sodium hydride, alkaline metal hydride such as hydrogen hydride realm, inorganic base or triethylamine such as metal sodium, sodium amide, tri-n-butylamine, 1,5-Diazabicyclo [4.3.0] 1-5-Nonene, 1,8-Diazabicyclo [5.4.0] — 7-Pendecene, pyridine, organic bases such as dimethylaminoviridine Can be
- This reaction can be carried out without solvent or with dioxane, tetrahydrofuran, ethyl ether, petroleum ether, n-hexane, cyclohexane, benzene, toluene, xylene, cyclobenzene, pyridine, N, N-dimethylformamide
- the solvent can be arbitrarily selected such as solvent, dimethyl sulfoxide, water, dichloromethane, and chloroform.
- This reaction is usually carried out in a solvent, but the solvents include dichloromethane, chloroform, dihydroxane, tetrahydrofuran, ethyl ether, benzene, toluene, xylene, acetone, acetonitrile, water, pyridine, ', N — Dimethylformamide, dimethylsulfoxide and the like.
- the compound of the formula (VI) is nitrated by using a nitrile or nitrite or the like, and the compound of the formula (VII) can be obtained.
- the agent include sodium nitrate, nitric acid rim, iron nitrate, and rare nitrate.
- the solvent used in this reaction is preferably selected arbitrarily according to the nitrating agent.Sulfuric acid, acetic anhydride, trifluoroacetic acid, sulfuric acid, dichloromethane, methanol, chloroform, benzene, Dioxane, ethanol and the like.
- the halogen atom of the compound represented by the formula (VI I) is replaced with the compound represented by the formula (VI II) or an inorganic acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid or formic acid Acetic acid, brobionic acid, oxalic acid, succinic acid, fumaric acid, maleic acid, benzoic acid, phthalic acid, keichic acid, glycolic acid, lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, p-toluenesulfonic acid, etc.
- the compound of the present invention [the compound represented by the formula (I)] can be obtained by performing a substitution reaction using a salt with an organic acid.
- This reaction is preferably performed in the presence of a base.
- the base include inorganic bases such as lithium carbonate, sodium carbonate, carbonated lime, sodium hydrogencarbonate, and hydrogencarbonate, triethylamine, and tri-n-butylamine. , 1, 5-diazabicyclo [4.3.0] -5-nonene, 1,8-diazabicyclo [5.4.0] one 7-ndecene, four-methylmorpholine, 1-methylbiberidine, pyridine, N, Organic bases such as N-dimethylaminoviridine, and a substituted compound represented by the formula (VI II) can be used as the base.
- This reaction is carried out without solvent or with acetone, acetonitrile, ethyl acetate, diisopropyl ether, tetrahydrofuran, dioxane, benzene, toluene, xylene, benzene, nitrobenzene, pyridine, N, N-dimethylformamide, It is preferable to perform the reaction in a solvent such as dimethyl sulfoxide.
- potassium iodide tris [2- (2-methoxyethoxy) ethyl] amine, tetra-n-butylammonium Muchloride, tetra-n-butyl Quaternary ammonium salts such as ammonium bromide, benzyltriethylammonium chloride, benzyltriethylammonium bromide, and tricabutylmethylammonium chloride; crown ethers such as 18-crown-16 ether; Can also accelerate the reaction.
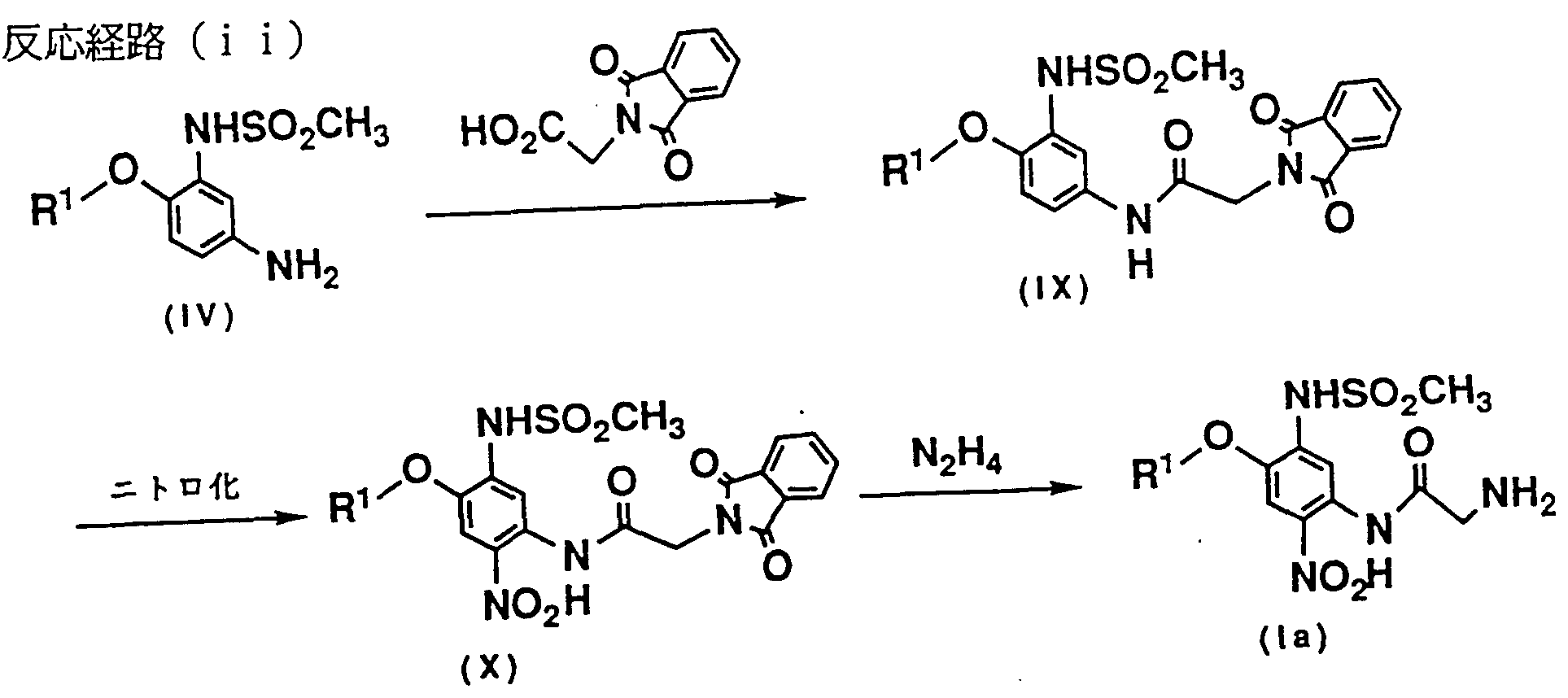
- the compound of the formula (IX) can be obtained by nitrating the compound of the formula (IX) in the same manner as in the step (e) of (1).
- Inorganic acids in this reaction include hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids include acetic acid, propionic acid, succinic acid, fumaric acid, maleic acid, benzoic acid, and the like. Examples include phthalic acid, keichic acid, glycolic acid, lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, and p-toluenesulfonic acid.
- the reaction is preferably carried out in a solvent.
- solvent examples include acetone, acetonitrile, ethyl sulphate, disobrovir ether, tetrahydrofuran, dioxane, benzene, toluene, xylene, benzene, nitrobenzene, and dichloromethane. , Such as black mouth Holm.
- the compounds of the present invention can be administered orally or parenterally in conventional dosage forms.
- Example 1 In Example 1 (6), N- (5-chloroacetylamino-4-nitro-12-phenoxyphenyl) methanesulfonamide obtained by the method of (1) to (5) was used as a raw material.
- the compounds of the present invention shown in Table 1 were obtained in the same manner as in Example 1 (6) except that the following amines were used in place of the n-butylamine used.
- n-Broviramine n-Bentilamine, Isobroviramine, Cyclobutyramine, Cyclobutylamine, Cyclobentylamine, Cyclohexylamine, Arylamine, Benzylamine, Jethylamine, Jibulin Vilamin, Viveridine, Morpholine ⁇ table 1 ⁇
- N- [5-Chloroacetylamino-2- (2-chlorophenoxy) -14-nitrophenyl] methanesulfonamide is used in the case of C6)
- n-: Butylamine The following compound of the present invention was obtained in the same manner as in Example 1 (6) except that n-proviramine, cyclobutyramine or arylamine was used instead.
- Example 1 (6) N- (5-chloroacetylamino-2-cyclohexyloxy-1-2-trophenyl) methanesulfonamide was replaced with n- bromoviramine or cyclopropyl in place of n-butylamine used in Example 1 (6).
- the following compound of the present invention was obtained in the same manner as in Example 1 (6) except that amine was used.
- Example 1 N— (5-chloroacetylamino-4-nitro-12-phenoxyphenyl) methanesulfonamide obtained by the method of (1) to (5)].
- N, '' Pam 1.2 g, monomethylamine hydrochloride 0.5 s and triei ';.
- a dimethylformamide 1 Om1 solution containing 6s was stirred at room temperature for 10 hours. reaction To the solution was added 5 ml of 3N hydrochloric acid, and then neutralized with a saturated aqueous solution of sodium hydrogen carbonate. The precipitate was collected by filtration, washed with ethyl acetate, and dried to give yellow crystals of N- (5-methylaminoacetyl). 4-amino-2-aminophenol) 0.40 g of methyl sulfonamide was obtained.
- CD i In a 140 ml solution of ethyl acetate containing 7.0 g of N- (5-amino-phenoxyphenyl) methanesulfonamide and 6.2 g of phthaloylglycine obtained by the method of (1) to (3). At room temperature, 1-ethyl 3- (3-dimethylaminobuguchi) carbopimid hydrochloride was added, and the mixture was stirred for 19 hours. Water was added to the reaction solution, and the precipitate obtained by filtration and the filtrate were extracted with ethyl acetate.
- Example 23 In place of the 1 ⁇ -[5- (n-butylaminoacetylamino) 1-42-trou 2-phenoxyphenyl] methanesulfonamide used in Example 23, obtain it by the method of The same procedure as in Example 23 was carried out except that N- [4-212trough 5- (n-butyl ⁇ -biraminoacetinolamino) -2—phenoxyphenyl] methanesulfonamide was used. [4-Nitro 5- ( ⁇ -propylaminoacetylamino) -12-phenoxyphenyl] methanesulfonamide hydrochloride was obtained.
- the compound of the present invention exhibits anti-inflammatory, antipyretic, analgesic, and antiallergic effects and has little side effects such as gastrointestinal tract disorders, so that it is useful as an antipyretic, antipyretic, analgesic, and antiallergic agent. is there.
- Test Example 1 Force lagenin foot edema test
- Adjuvant arthritis was induced by injecting 0.7% mycobacterium tuberculosis suspended in liquid paraffin subcutaneously into the left leg of Lewis rats (10 rats per group). 15-18 days after adjuvant administration, using arthritic rats showing a vocal reaction to flexion and extension stimulation pain of the right hind ankle joint, a sample suspended in a 5% aqueous solution of arabia gum [Compounds of the present invention a, b, k and m And a control drug were orally administered at a dose of 1 ml per 100 g body weight. The presence or absence of a vocal reaction was measured over time up to 5 hours after administration, and the analgesic effect was examined by determining the suppression rate (%). The administered dose of the sample was 1. Omg Zkg.
- Table 4 shows the results. [Table 4] Test example Inhibition rate (%) a 50, 6 b 4 7.5 k 4 1.9 m 5 2. 1. Control drug 19.4
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Description






Claims

Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP94907051A EP0685461A4 (en) | 1993-02-19 | 1994-02-16 | 5-AMINOACETYLAMINOSULPHONE ANILIDE COMPOUND. |
| US08/501,029 US5521311A (en) | 1993-02-19 | 1994-02-16 | 5-aminoacetylaminosulfonanilide compounds |
| KR1019950703038A KR960700361A (ko) | 1993-02-19 | 1994-02-16 | 5-아미노아세틸아미노술폰아닐리드 화합물 |
| AU60445/94A AU672285B2 (en) | 1993-02-19 | 1994-02-16 | 5-Aminoacetylaminosulfonanilide compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2955093 | 1993-02-19 | ||
| JP5/29550 | 1993-02-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994019318A1 true WO1994019318A1 (fr) | 1994-09-01 |
Family
ID=12279255
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1994/000228 Ceased WO1994019318A1 (fr) | 1993-02-19 | 1994-02-16 | Compose de 5-aminoacetylaminosulfonanilide |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US5521311A (ja) |
| EP (1) | EP0685461A4 (ja) |
| KR (1) | KR960700361A (ja) |
| AU (1) | AU672285B2 (ja) |
| CA (1) | CA2156303A1 (ja) |
| WO (1) | WO1994019318A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU661906B2 (en) * | 1992-06-12 | 1995-08-10 | Taisho Pharmaceutical Co., Ltd. | 5-amino-2-phenoxysulfonanilide compound |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1847528A1 (en) * | 2006-04-18 | 2007-10-24 | DyStar Textilfarben GmbH & Co. Deutschland KG | Coupling compounds and hair dyeing compositions containing them |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63313765A (ja) * | 1987-05-29 | 1988-12-21 | Fujisawa Pharmaceut Co Ltd | 新規アルカンスルホンアニリド誘導体、その製造法およびそれを含有する医薬組成物 |
| JPH04283555A (ja) * | 1991-03-12 | 1992-10-08 | Taisho Pharmaceut Co Ltd | 4置換スルホンアニリド化合物 |
| JPH05201956A (ja) * | 1992-01-28 | 1993-08-10 | Taisho Pharmaceut Co Ltd | 5−オキサリルアミノスルホンアニリド化合物 |
| JPH05201957A (ja) * | 1991-09-25 | 1993-08-10 | Taisho Pharmaceut Co Ltd | 5−アミノ−2−フェノキシスルホンアニリド化合物 |
| JPH06100525A (ja) * | 1992-09-21 | 1994-04-12 | Taisho Pharmaceut Co Ltd | オキサミドを有するスルホンアニリド化合物 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3856857A (en) * | 1970-02-13 | 1974-12-24 | En Nom Collectif Science Union | Amino acids and their derivatives |
| DE69210629T2 (de) * | 1991-08-08 | 1996-09-19 | Taisho Pharma Co Ltd | 5-aminosulfonanilid-verbindungen |
| KR0150826B1 (ko) * | 1992-06-12 | 1998-10-15 | 우에하라 아키라 | 5-아미노-2-페녹시술폰아닐리드 화합물 |
| HU210922B (en) * | 1993-05-24 | 1995-09-28 | Europharmaceuticals Sa | Nimesulide alkali salt cyclodextrin inclusion complexes their preparation and pharmaceutical compositions containing them |
-
1994
- 1994-02-16 WO PCT/JP1994/000228 patent/WO1994019318A1/ja not_active Ceased
- 1994-02-16 AU AU60445/94A patent/AU672285B2/en not_active Ceased
- 1994-02-16 CA CA002156303A patent/CA2156303A1/en not_active Abandoned
- 1994-02-16 EP EP94907051A patent/EP0685461A4/en not_active Ceased
- 1994-02-16 US US08/501,029 patent/US5521311A/en not_active Expired - Fee Related
- 1994-02-16 KR KR1019950703038A patent/KR960700361A/ko not_active Withdrawn
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63313765A (ja) * | 1987-05-29 | 1988-12-21 | Fujisawa Pharmaceut Co Ltd | 新規アルカンスルホンアニリド誘導体、その製造法およびそれを含有する医薬組成物 |
| JPH04283555A (ja) * | 1991-03-12 | 1992-10-08 | Taisho Pharmaceut Co Ltd | 4置換スルホンアニリド化合物 |
| JPH05201957A (ja) * | 1991-09-25 | 1993-08-10 | Taisho Pharmaceut Co Ltd | 5−アミノ−2−フェノキシスルホンアニリド化合物 |
| JPH05201956A (ja) * | 1992-01-28 | 1993-08-10 | Taisho Pharmaceut Co Ltd | 5−オキサリルアミノスルホンアニリド化合物 |
| JPH06100525A (ja) * | 1992-09-21 | 1994-04-12 | Taisho Pharmaceut Co Ltd | オキサミドを有するスルホンアニリド化合物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0685461A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU661906B2 (en) * | 1992-06-12 | 1995-08-10 | Taisho Pharmaceutical Co., Ltd. | 5-amino-2-phenoxysulfonanilide compound |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0685461A1 (en) | 1995-12-06 |
| KR960700361A (ko) | 1996-01-20 |
| US5521311A (en) | 1996-05-28 |
| CA2156303A1 (en) | 1994-09-01 |
| AU6044594A (en) | 1994-09-14 |
| AU672285B2 (en) | 1996-09-26 |
| EP0685461A4 (en) | 1996-08-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| FI64154C (fi) | Foerfarande foer framstaellning av en terapeutiskt verkande 5-etyl-isoxazol-4-karboxylsyra-(4-trifluormetyl)-anilid | |
| Jakubkien et al. | Synthesis and anti-inflammatory activity of 5-(6-methyl-2-substituted 4-pyrimidinyloxymethyl)-1, 3, 4-oxadiazole-2-thiones and their 3-morpholinomethyl derivatives | |
| JP2722250B2 (ja) | 新規なジアミン化合物及びこれを含有する脳機能障害改善剤 | |
| TW201111374A (en) | Compounds which selectively modulate the CB2 receptor | |
| JP3939246B2 (ja) | インドロキナゾリノン類 | |
| BRPI0713590A2 (pt) | derivados de pirrol com atividade de modulador de receptor de crth2 | |
| BRPI0610833A2 (pt) | derivados de acetileno | |
| Sañudo et al. | A diastereoselective synthesis of pseudopeptidic hydantoins by an Ugi/cyclization/Ugi sequence | |
| US4431663A (en) | Treatment of psychosis with meta-sulfonamido-benzamide derivatives | |
| EP1877367A1 (en) | Acetylene derivatives | |
| EP0623023A1 (en) | Retroreverse pyrrole-amidino oligopeptide anticancer agent analogues, preparation of same, and pharmaceutical compositions containing such analogues | |
| WO1994019318A1 (fr) | Compose de 5-aminoacetylaminosulfonanilide | |
| JPH03866B2 (ja) | ||
| Alsafi et al. | Synthesis, characterization and acute anti-inflammatory evaluation of new mefenamic acid derivatives having 4-thiazolidinone nucleus | |
| JPH06298722A (ja) | 5−アミノアセチルアミノスルホンアニリド化合物 | |
| WO1993003008A1 (fr) | Compose 5-aminosulfonanilide | |
| US5449826A (en) | 5-amino-2-phenoxysulfonanilide compound | |
| JPH05262718A (ja) | 5−置換アミノ−2−フェノキシスルホンアニリド化合物 | |
| JPH06100525A (ja) | オキサミドを有するスルホンアニリド化合物 | |
| JPS649989B2 (ja) | ||
| JP3721583B2 (ja) | 抗炎症剤 | |
| JPH06206859A (ja) | N−アシル−5−アミノスルホンアニリド化合物 | |
| IE58577B1 (en) | Novel benzamides and their preparation and therapeutic application | |
| JPH05246978A (ja) | 5−カルボキシアルカノイルアミノスルホンアニリド化合物 | |
| GB2080791A (en) | 1,4-Benzoxazines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 08501029 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2156303 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994907051 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994907051 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1994907051 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994907051 Country of ref document: EP |