DESCRIPTION
-PYRR0LIDINYLTHI0-CARBAPENEM DERIVATIVES AND THEIR ANTIMICROBAL ACTIVITY
TECHNICAL FIELD
The present invention relates to novel azabicyclo compounds and pharmaceutically acceptable salts thereof.
More particularly, it relates to novel
3-pyrrolidinylthio-l-azabicyclo[3.2 .0]hept-2-ene-2- carboxylic acid compounds and pharmaceutically acceptable salts thereof, which have antimicrobial activity to processes for the preparation thereof, to a pharmaceutical composition comprising the same, and to a use of the same as a medicament and in the treatment of infectious diseases in human being or animal.
INDUSTRIAL APPLICABILITY
Accordingly, one object of the present invention is to provide novel 3-pyrrolidinylthio-l-azabicyclo- [3.2.0]hept-2-ene-2-carboxylic acid compounds and pharmaceutically acceptable salts thereof, which are highly active against a number of pathogenic microorganisms and are usef l as antimicrobial agents.
Another object of the present invention is to provide processes for the preparation of novel 3-pyrrolidinylthio- l-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid compounds and salts thereof.
A further object of the present invention is to provide a pharmaceutical composition comprising, as an active ingredient, said 3-pyrrolidinylthio-l-azabicyclo- [3.2.0]hept-2-ene-2-carboxylic acid compounds and pharmaceutically acceptable salts thereof.
Still further object of the present invention is to provide a use of said 3-pyrrolidinylthio-l-azabicyclo- [3.2.0]hept-2-ene-2-carboxylic acid compounds and pharmaceutically acceptable salts thereof as a medicament and in the treatment of infectious diseases by pathogenic microorganisms in human being or animal.
DISCLOSURE OF INVENTION
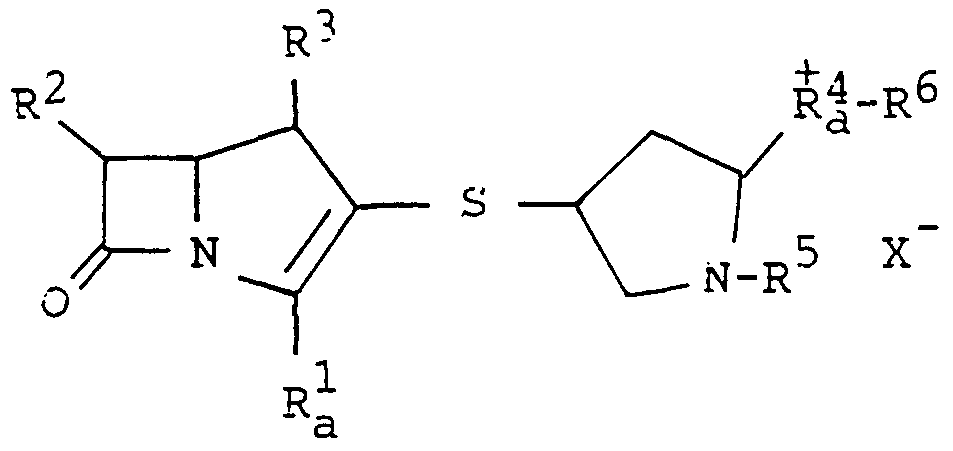
The object 3-pyrrolidinylthio-l-azabicyclo[3.2.0]- hept-2-ene-2-carboxylic acid compounds are novel and can be represented by the following general formula:
in which R1 is carboxy, COO- or protected carboxy,
R-2 is hydroxy(lower)alkyl or protected hydroxy(lower)alkyl, R-***" is hydrogen or lower alkyl, R^ is 2(or 3)-methylpyridin-4-ylmethyl, 2- cyanopyridin-4-ylmethyl, 2-
carbamoylpyridin-4-ylmethyl, [1,2(or 1,3)- di ethyl-4-pyridinio]methyl, (l-methyl-2- cyano-4-pyridinio) ethyl, (l-methyl-2- carbamoyl-4-pyridinio)methyl, [l-(2- 5 procected hydroxyethy1)-4-pyridinio]methyl,
[l-(2-hydroxyethyl)-4-pyridinio] ethyl, [1- (carbamoylmethyl)-4-pyridinio]methyl, [2- (hydroxymethyl)pyridin-4-yl] ethyl, [2- (hydroxymethyl)-l-methyl-4-
10 pyridinio]methyl, 2-(imidazol-l-yl)propyl,
2-(l-methylimidazol-5-yl)ethyl, 2-(1,3- dimethyl-4-imidazolio)ethyl, 2-(3-methyl-l- imidazolio)propyl, [l-methyl-2- (hydroxymethyl)imidazol-5-yl]methyl, (1-
15 methyl-2-carbamoylimidazol-5-yl)methyl,
[1,3-dimethyl-2-(hydroxymethyl)-4- imidazolio]methyl, (l,3-dimethyl-2- carbamoyl-4-imidazolio)methyl, 2-(1- carbamoylmethyl-3-methyl-4-
20 imidazolio)ethyl, [3-methyl-l-(2- hydroxyethyl)-4-imidazolio]methyl, [3- methyl-1-(carbamoylmethyl)-4- imidazolio]methyl, [l-methyl-4- {hydroxymethyl)pyrazol-5-yl]methyl, [1-
25 methyl-5-( ydroxymethyl)pyrazol-3- yl] ethyl, [2-methyl-l-(carbamoylmethyl)-3- pyrazolio]methyl, [l-(2- hydroxyethyDpyrazol-4-yl]methyl, [2- methyl-l-(2-hydroxyethyl)-3-
30 pyrazolio] ethyl, [1,2-dimethyl-4-
(hydroxymethyl)-3-pyrazolio]methyl, [1,2- dimethyl-5-(hydroxymethyl)-3- pyrazolio]methyl, [l-methyl-5- (hydroxymethyl)pyrazol-4-yl]methyl, [1,2-
35 dimethyl-3-(hydroxymethyl)-4-
pyrazolio]methyl, [l-methyl-2- (carbamoylmethyl)-4-pyrazolio]methyl, [1- methyl-2-(2-hydroxyethyl)-4- pyrazolio]methyl, 2-[2-(protected imino)-3- methylimidazolin-1-yl]ethyl, 2-(2-imino-3- methylimidazolin-1-yl)ethyl, (1-methγl- 1,2,3-triazol-5-yl)methyl, [1,3-dimethyl-4- (1,2,3-triazolio) ]methyl, or 6- (pyrazolidino[1,2-a]pyrazolio)methyl, and R^ is hydrogen or imino-protective group, or pharmaceutically acceptable salt thereof.
Suitable pharmaceutically acceptable salts of the object compound (I) are conventional non-toxic salts and may include a salt with a base such as an inorganic base salt, for example, an alkali metal salt (e.g. sodium salt, potassium salt, etc.), an alkaline earth metal salt (e.g. calcium salt, magnesium salt, etc.), an ammonium salt, an organic base salt, for example, an organic amine salt (e.g. triethylamine salt, pyridine salt, picoline salt, ethanolamine salt, triethanolamine salt, dicyclohexylamine salt, N,N'-dibenzylethylenediamine salt, etc.); a salt with an acid such as inorganic acid addition salt (e.g. hydrochloride, hydrobromide , sulfate, fluorosulfate, phosphate, etc.), an organic acid addition salt (e.g. formate, acetate, trifluoroacetate, maleate, tartrate, methanesulfonate, benzenesulfonate, trifluoromethanesulfonate etc.); a salt with a basic or acidic amino acid (e.g. arginine, aspartic acid , glutamic acid, etc.); an intermolecular or intramolecular quaternary salt; and the like.
The said intermolecular quaternary salt can be formed, for example, between the quaternary nitrogen atom in R4 and counter anion such as halide (e.g. iodide,
chloride, etc.), trihalo(lower)alkyl (e.g. trifluoro ethyl, etc.), and the like.
The said intramolecular quaternary salt can be formed, for example, between the quaternary nitrogen atom in R4 and COO" group of R1.
In the object compound (I) and the intermediary compounds mentioned below, it is to be understood that there may be one or more stereo-isomeric pair(s) such as optical isomers due to asymmetric carbon atom(s), and such isomers are also included within the scope of the present invention.
According to the present invention, the object compound (I) or pharmaceutically acceptable salts thereof can be prepared by the processes as illustrated by the following reaction schemes.
Process 1 :
(ID or a reactive derivative at the oxo group thereof or salts thereof
(I) or salts thereof
Process 2
Removal reaction of the carboxy-protective group on R
(I-a) or salts thereof
COOH
(I-b) or salts thereof
Process 3
Removal reaction of the
(I-C) or salts thereof
(I-d) or salts thereof
Process 4
(I-e) or salts thereof
- o -
(I-f) or salts thereof
Process 5
Removal reaction of the imino or hydroxy-protective group
(i-g) or salts thereof
(I-h) or salts thereof
Process 6
(I-i) or salts thereof
(I-j) or salts thereof
Process 7
Removal reaction of the hydroxy-protective group on τR.2
R '
[ I -i ) or salts thereof
R R 3, R4 and R5 are each as defined above,
R is protected carboxy, is protected hydroxy(lower)alkyl,
cyanopyridin-4-ylmethyl, 2- carbamoylpyridin-4-ylmethyl, [2- (hydroxymethyl)pyridin-4- 1]methyl, 2- (imidazol-l-yl)propyl, 2-(1-methylimidazol- 5-yl)ethyl, [1-methyl-2- (hydroxymethyl)imidazol-5-yl]methyl, (1- methyl-2-carbamoylimidazol-5-yl)methyl, [1- methyl-4-(hydroxymethyl)pyrazol-5- yl]methyl, [1-methyl-5-
(hydroxymethyl)pyrazol-3-yl]methyl, [l-(2- hydroxyethyl)pyrazol-4-yl]methyl, [1- methyl-5-(hydroxymethyl)pyrazol-4- yl]methyl, 2-[2-(protected imino)-3-methyl- 4-imidazolin-l-yl]ethyl, 2-(2-imino-3- methyl-4-imidazolin-l-yl)ethyl, or (1- methyl-lH-1,2,3-triazol-5-yl)methyl,
R + is [1,2(or 1,3)-dimethyl-4- pyridinio]methyl, (l-methyl-2-cyano-4- pyridinio)methyl, (l-methyl-2-carbamoyl-4- pyridinio)methyl, [1-(2-protected
hydroxyethyl)-4-pyridinio]methyl, [1-(2- hydroxyethyl)-4-pyridinio]methyl, [1- (carbamoylmethyl)-4-pyridinio]methyl, [2- (hydroxymethyl)-l-methyl-4- pyridinio]methyl, 2- ( 1,3-dimethyl-4- imidazolio)ethyl, 2- (3-methyl-l- imidazolio)propyl, [1,3-dimethyl-2- (hydroxymethyl)-4-imidazolio]methyl, (1,3- dimethyl-2-carbamoyl-4-imidazolio)methyl, 2-(1-carbamoylmethyl-3-methyl-4- imidazolio)ethyl, [3-methyl-l-( 2- hydroxyethyl)-4-imidazolio]methyl, [3- methyl-1-(carbamoylmethyl)-4- imidazolio]methyl, [2-methyl-l- (carbamoylmethyl)-3-pyrazolio]methyl, [2- methyl-1-(2-hydroxyethyl)-3- pyrazolio]methyl, [1,2-dimethyl-4- (hydroxymethyl)-3-pyrazolio]methyl, [1,2- dimethyl-5-(hydroxymethyl)-3- pyrazolio]methyl, [1, 2-dimethyl-3-
(hydroxymethyl)-4-pyrazolio]methyl, [1- methyl-2-(carbamoylmethyl)-4- pyrazolio]methyl, [l-methyl-2-(2- hydroxyethyl)-4-pyrazolio]methyl or [1,3- dimethyl-4-(lH-l,2,3-triazolio) ]methyl,
R4 is [l-(2-protected hydroxyethyl)-4- pyridinio]methyl, or 2-(2-protected i ino- 3-methylimidazolin-l-yl)ethyl, Rc is [1-(2-hydroxyethyl)-4-pyridinio]methyl, or 2-(2-imino-3-methylimidazolin-l-yl)ethyl,
Rg is imino-protective group,
R° is methyl, carbamoylmethyl, 2-hydroxyethyl, 2- protected hydroxyethyl, and
X is an acid residue.
The compound (III) used in the Process 1 is new and can be prepared, for example, by the following methods or a conventional manner.
Method A :
(IV) (Ill-a) or a reactive derivative or salts thereof at the hydroxy group thereof or salts thereof
Method B :
Elimination reaction
( III-a) (III) or a salt thereof oorr salts thereof
in which R 4 and R5 are each as defined above, and R 7 .s mercapto-protective group.
In the above and subsequent descriptions of the present specification, suitable examples and illustrations of the various definitions which the present invention includes within the scope thereof are explained in detail
as follows .
The term "lower" is intended to mean 1 to 6, preferably 1 to 4 carbon atom(s), unless otherwise indicated.
Suitable "hydroxy(lower)alkyl" may include straight or branched lower alkyl having hydroxy group such as hydroxymethyl, hydroxyethyl, hydroxypropyl, 1- (hydroxymethyl)ethyl, 1-hydroxy-1-methylethyl, hydroxybutyl, hydroxypentyl, hydroxyhexyl, and the like, in which more preferable example may be hydroxyfC^-C^alkyl and the most preferable one may be 2- hydroxyethyl.
Suitable "lower alkyl" may include straight or branched one such as methyl, ethyl, propyl, isopropyl, butyl, t-butyl, pentyl, hexyl, and the like, in which more preferable example may be C..-C. alkyl and the most preferable one may be methyl.
Suitable " ercapto-protective group" may include acyl such as aliphatic acyl, aromatic acyl, heterocyclic acyl and aliphatic acyl substituted with aromatic or heterocyclic group(s) derived from carboxylic, carbonic, sulfonic and carbamic acids.
The aliphatic acyl may include saturated or unsaturated, acyclic or cyclic ones, for example, alkanoyl such as lower alkanoyl (e.g. formyl, acetyl, propionyl, butyryl, isobutyryl, valeryl, isovaleryl, pivaloyl, hexanoyl, etc.), alkylsulfonyl such as lower alkylsulfonyl (e.g. mesyl, ethylsulfonyl, propylsulfonyl, isopropylsulfonyl, butylsulfonyl, isobutylsulfonyl, pentylsulfonyl, hexylsulfonyl, etc.), carbamoyl, N-alkylcarbamoyl (e.g. methylcarba oyl, ethylcarbamoyl, etc.), alkoxycarbonyl such as lower alkoxycarbonyl (e.g. methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, butoxycarbonyl, t-butoxycarbonyl, etc.),
alkenyloxycarbonyl such as lower alkenyloxycarbonyl (e.g. vinyloxycarbonyl, allyloxycarbonyl, etc.), alkenoyl such as lower alkenoyl (e.g. acryloyl, ethacryloyl, crotonoyl, etc.), cycloalkanecarbonyl such as cyclo(lower)alkanecarbonyl (e.g. cyclopropanecarbonyl, cyclopentanecarbonyl, cyclohexanecarbonyl, etc.), and the like.
The aromatic acyl may include Cb,-C_1,A1) aroyl (e.g. benzoyl, toluoyl, xyloyl, etc.), N-(Cg-C10)arylcarbamoyl (e.g. N-phenylcarbamoyl, N-tolylcarbamoyl,
N-naphthylcarbamoyl, etc.), Cg-C-0 arenesulfonyl (e.g. benzenesulfonyl, tosyl, etc.), and the like.
The aliphatic acyl substituted with aromatic group(s) may include aralkoxycarbonyl such as phenyl(lower)alkoxycarbonyl (e.g. benzyloxycarbonyl, phenethyloxycarbonyl, etc. ) , and the like.
These acyl groups may be further substituted with one or more suitable substituent(s) such as nitro, and the like, and preferable acyl having such substituent(s) may be nitroaralkoxycarbonyl (e.g. nitrobenzyloxycarbonyl, etc. ) , and the like.
More preferable example of "mercapto-protective group" thus defined may be C..-C, alkanoyl and C8-C. Q aroyl and the most preferable one may be acetyl and benzoyl. Suitable "acid residue" may include an inorganic acid residue such as azido, halogen (e.g. chlorine, bromine, fluorine or iodine), and the like, an organic acid residue such as acyloxy (e.g. benzenesulfonyloxy, tosyloxy, methanesulfonyloxy, trihalomethanesulfonyloxy, etc.), and the like, in which more preferable example may be halogen and trihalomethanesulfonyloxy, and the most preferable one may be iodine and trifluoromethanesulfonyloxy.
Suitable "protected carboxy" may include esterified carboxy wherein "esterified carboxy" can be referred to
the ones as mentioned below.
Suitable examples of the ester moiety of an esterified carboxy may be the ones such as lower alkyl ester (e.g. methyl ester, ethyl ester, propyl ester, isopropyl ester, butyl ester, isobutyl ester, t-butyl ester, pentyl ester, hexyl ester, etc.) which may have at least one suitable substituent(s) , for example, lower alkanoyloxy(lower)alkyl ester [e.g. acetoxymethyl ester, propionyloxymethyl ester, butyryloxymethyl ester, valeryloxymethyl ester, pivaloyloxymethyl ester, hexanoyloxymethyl ester, l-(or 2-)acetoxyethyl ester, l-(or 2- or 3-)acetoxypropyl ester, l-(or 2- or 3- or 4-)acetoxybutyl ester, l-(or 2-)propionyloxyethyl ester, l-(or 2- or 3-)propionyloxypropyl ester, l-(or 2-)- butyryloxyethyl ester, l-(or 2-)isobutyryloxyethγl ester, l-(or 2-)pyvaloyloxyethyl ester, l-(or 2-)hexanoyloxyethyl ester, isobutyryloxymethyl ester, 2-ethylbutyryloxymethyl ester, 3,3-dimethylbutyryloxymethy1 ester, l-(or 2-)- pentanoyloxyethyl ester, etc.], lower alkanesulfonyKlower)alkyl ester (e.g. 2-mesylethyl ester, etc.), mono(or di or tri)halo(lower)alkyl ester (e.g. 2-iodoethyl ester, 2,2,2-trichloroethyl ester, etc.); lower alkoxycarbonyloxy(lower)alkyl ester [e.g. methoxycarbonyloxymethyl ester, ethoxycarbonyloxymethyl ester, propoxycarbonyloxymethyl ester, t-butoxycarbonyl- oxymethyl ester, l-(or 2-)methoxycarbonyloxyethyl ester, l-(or 2-)ethoxycarbonyloxyethyl ester, l-(or 2-) isopropoxycarbonyloxyethyl ester, etc.], phthalidylidene(lower)alkyl ester, or (5-lower alkyl-2- oxo-l,3-dioxol-4-yl) (lower)alkyl ester [e.g. (5-methyl-2- oxo-l,3-dioxol-4-yl)methyl ester, (5-ethyl-2-oxo-l,3- dioxol-4-yl)methyl ester, (5-propyl-2-oxo-l,3-dioxol-4- yl)ethyl ester, etc.]; lower alkenyl ester (e.g. vinyl ester, allyl ester, etc.); lower alkynyl ester (e.g. ethynyl ester, propynyl ester,
etc.); ar(lower)alkyl ester which may have at least one suitable substituent(s) (e.g. benzyl ester, 4-methoxybenzyl ester, 4-nitrobenzyl ester, phenethyl ester, trityl ester, benzhydryl ester, bis(methoxyphenyl)methyl ester, 3,4-dimethoxybenzyl ester, 4-hydroxγ-3,5-di-t-butylbenzyl ester, etc.); aryl ester which may have at least one suitable substituent(s) (e.g. phenyl ester, 4-chlorophenyl ester, tolyl ester, t-butylphenyl ester, xylyl ester, mesityl ester, cumenyl ester, etc.); phthalidyl ester; and the like.
More preferable example of the protected carboxy thus defined may be C--C. alkenyloxycarbonyl and phenyl(or nitrophenyl) (C--C.)alkoxycarbonyl, and the most preferable one may be allyloxycarbonyl.
Suitable "imino-protective group" may include acyl as mentioned in the explanation of mercapto-protective group, in which more preferable example may be C~-C. alkenyloxycarbonyl and the most preferable one may be allyloxycarbonyl.
Suitable hydroxy-protective group may include aforementioned acyl, tri(lower)alkylsilyl, and the like, in which more preferable example may be lower alkenyloxycarbonyl and tri(lower)alkylsilyl, and the most preferable one may be allyloxycarbonyl and t-butyldimethylsilyl.
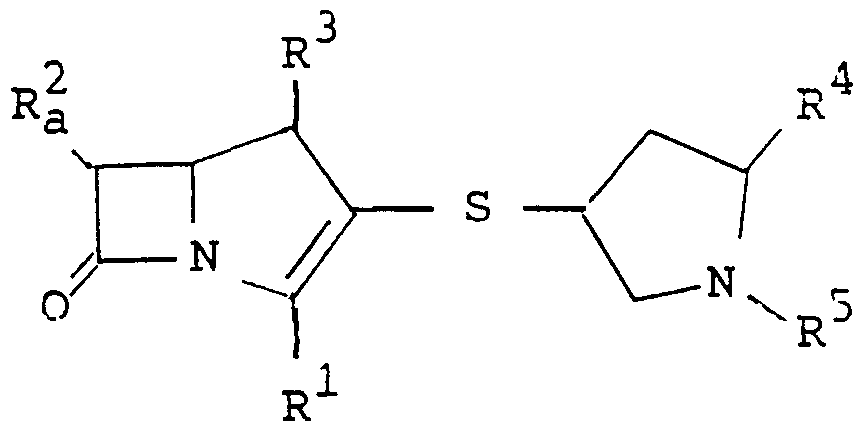
Preferable examples of R 1, R2, R3, R4 and R5 are as follows.
R is carboxy or esterified carboxy,
2
R is 1-hydroxyethyl,
3 R is methyl, R4 is 2(or 3)-methylpyridin-4-ylmethyl, 2-cyanopyridin-4-
ylmethyl, 2-carbamoylpyridin-4-ylmethyl, [1,2(or l,3)-dimethyl-4-pyridinio]methyl, (1-methyl-2-cyano- 4-pyridinio)methyl, (l-methyl-2-carbamoyl-4- pyridinio) ethyl, [l-(2-protected hydroxyethyl)-4- pyridinio]methyl, said protected hydroxy being acyl or tri(lower)alkylsilyloxy, [l-(2-hydroxyethyl)-4- pyridinio] ethyl, [1-(carbamoylmethyl)-4- pyridinio] ethyl, [2-(hydroxymethyl)pyridin-4- yl]methyl, [2-(hydroxymethyl)-l-methyl-4- pyridinio]methyl, 2-(imidazol-l-yl)propyl, 2-(l- methylimidazol-5-yl)ethyl, 2-(1,3-dimethyl-4- imidazolio)ethyl, 2-(3-methyl-l-imidazolio)propyl, [1-methyl-2-(hydroxymethyl)imidazol-5-yl]methyl, (1- methyl-2-carbamoylimidazol-5-yl)methyl, [1,3- dimethyl-2-(hydroxymethyl)-4-imidazolio]methyl,
(1,3-dimethyl-2-carbamoyl-4-imidazolio)methyl, 2-(1- carbamoylmethyl-3-methyl-4-imidazolio)ethyl, [3- methyl-l-(2-hydroxyethyl)-4-imidazolio]methyl, [3- methyl-1-(carbamoylmethyl)-4-imidazolio]methyl, [1- methyl-4-(hydroxymethyl)pyrazol-5-yl]methyl, [1- methyl-5-( ydroxymethyl)pyrazol-3-yl]methyl, [2- methyl-l-(carbamoylmethyl)-3-pyrazolio]methyl, [1- (2-hydroxyethyl)pyrazol-4-yl]methyl, [2-methyl-l-(2- hydroxyethyl)-3-pyrazolio]methyl, [1,2-dimethyl-4- (hydroxymethyl)-3-pyrazolio]methyl, [1,2-dimethyl-5-
(hydroxymethyl)-3-pyrazolio]methyl, [l-methyl-5- (hydroxymethyl)pyrazol-4-yl]methyl, [1,2-dimethyl-3- (hydroxymethyl)-4-pyrazolio] ethyl, [1-methyl-2- (carbamoylmethyl)-4-pyrazolio]methyl, [l-methyl-2- (2-hydroxyethyl)-4-pyrazolio]methyl, 2-[2-(protected imino)-3-methylimidazolin-l-yl]ethyl, said protected imino being acylimino, 2-(2-imino-3- methylimidazolin-1-yl)ethyl, (l-methyl-1,2,3- triazol-5-yl)methyl, [1,3-dimethyl-4-(1,2,3- triazolio) ]methyl, or 6-(pyrazolidino[l,2-a]-
pyrazolio)methyl,
5 R is hydrogen or esterified carboxy.
More preferable examples of R 1, R2, R3, R4 and R5 are as follows.
R is carboxy,
2
R is 1-hydroxyethyl,
3
R is methyl,
4 R is 2(or 3)-methylpyridin-4-ylmethyl, 2-cyanopyridin-4- ylmethyl, 2-carbamoylpyridin-4-ylmethyl [1,2(or
1,3)-dimethyl-4-pyridinio]methyl, (l-methyl-2-cyano-
4-pyridinio)methyl, (l-methyl-2-carbamoyl-4- pyridinio) ethyl, [l-(2-t- butyldi ethylsilyloxyethyl)-4-pyridinio] ethyl, [1-
(2-hydroxyethyl)-4-pyridinio] ethyl, [1-
(carbamoylmethyl)-4-pyridinio] ethyl, [ 2-
(hydroxymethyl)pyridin-4-yl] ethyl, [2-
(hydroxymethyl)-l-methyl-4-pyridinio]methyl, 2- (imidazol-l-yl)propyl, 2-(l-methylimidazol-5- yl)ethyl, 2-(l,3-dimethyl-4-imidazolio)ethyl, 2-(3- methyl-l-imidazolio)propyl, [l-methyl-2-
(hydroxymethyl)imidazol-5-yl]methyl, (l-methyl-2- carbamoylimidazol-5-yl)methyl, [1,3-dimethyl-2- (hydroxymethyl)-4-imidazolio]methyl, (1,3-dimethyl-
2-carbamoyl-4-imidazolio)methyl, 2-(1- carbamoylmethyl-3-methyl-4-imidazolio)ethyl, [3- methyl-1-(2-hydroxyethyl)-4-imidazolio]methyl, [3- methyl-1-(carbamoylmethyl)-4-imidazolio]methyl, [1- methyl-4-(hydroxymethyl)pyrazol-5-yl]methyl, [1- methyl-5-(hydroxymethyl)pyrazol-3-yl]methyl, [2- methyl-l-(carbamoylmethyl)-3-pyrazolio]methyl, [1-
(2-hydroxyethyl)pyrazol-4-yl]methyl, [2-methyl-l-(2- hydroxyethyl)-3-pyrazolio]methyl, [1,2-dimethyl-4- (hydroxymethyl)-3-pyrazolio]methyl, [1,2-dimethyl-5-
(hydroxymethyl)-3-pyrazolio]methyl, [1-methyl-5-
(hydroxymethyl)pyrazol-4-yI]methyl, [1,2-dimethyl-3-
(hydroxymethyl)-4-pyrazolio]methyl, [l-methyl-2-
(carbamoylmethyl)-4-pyrazolio]methyl, [l-methyl-2- (2-hydroxyethyl)-4-pyrazolio]methyl, 2-[2-
(allyloxycarbonylimino)-3-methylimidazolin-l- yl]ethyl, 2-{2-imino-3-methylimidazolin-l-yl)ethyl,
(l-methyl-l,2,3-triazol-5-yl)methyl, [1,3-dimethyl-
4-(1,2,3-triazolio) ]methyl, or 6-(pyrazolidi"no[l,2- a]pyrazolio)methyl,
5 R is hydrogen.
The processes for the preparation of the object compound (I) of the present invention are explained in detail in the following.
(1) Process 1 :
The compound (I) or salts thereof can be prepared by reacting the compound (II) or a reactive derivative at the oxo group thereof or salts thereof with the compound (III) or salts thereof.
Suitable salts of the compound (II) may be salts with bases such as those given for the compound (I) .
The reactive derivative at the oxo group of the compound (II) can be represented by the following formula (II1), which is preferably used in this reaction and can be prepared by reacting the compound (II) or salts thereof with an acylating agent.
iO -
(ID (II') or salts thereof or salts thereof
1 in which R , R 2 and R3 are each as defined above, and
R^ is acyl as exemplified for the imino-protective group and further 0,0-substituted phosphono derived from, for example, organic phosphoric acid mentioned hereinbelow.
Suitable acylating agents may include conventional ones which can introduce the acyl group as mentioned above into the compound (II) , and preferable acylating agents may be organic sulfonic or phosphoric acid or its reactive derivative such as acid halide, acid anhydride, and the like, for example, arenesulfonyl halide (e.g. benzenesulfonyl chloride, p-toluenesulfonyl chloride, p-nitrobenzenesulfonyl chloride, p-bromobenzenesulfonyl chloride, etc.), arenesulfonic anhydride (e.g. benzenesulfonic anhydride, p-toluenesulfonic anhydride, p-nitrobenzenesulfonic anhydride, etc.), lower alkanesulfonyl halide which may have additional halogen (e.g. methanesulfonyl chloride, ethanesulfonyl chloride, trifluoromethanesulfonyl chloride, etc.), lower alkanesulfonic anhydride which may have halogen (e.g. methanesulfonic anhydride, ethanesulfonic anhydride,
trifluoromethanesulfonic anhydride, etc.), di(lower)alkyl phosphbrohaloridate (e.g. diethyl phosphorochloridate, etc.), diaryl phosphorohaloridate (e.g. diphenyl phosphorochloridate, etc.), and the like. This acylation reaction is usually carried out in a conventional solvent which does not adversely influence the reaction such as acetone, dioxane, acetonitrile, chloroform, dichloromethane, hexamethylphosphoramide, dichloroethane, tetrahydrofuran, ethyl acetate, dimethyl sulfoxide, N,N-dimethylformamide, pyridine, etc., or a mixture thereof.
When the acylating agent is used in a free acid form or its salt form in this reaction, the reaction is preferably carried out in the presence of a conventional condensing agent such as carbodiimide compound [e.g. N,N'- diethylcarbodiimide, N,N'-diisopropylcarbodiimide, N,N'- dicyclohexylcarbodiimide, N-cyclohexyl-N'- morpholinoethylcarbodiimide, N-cyclohexyl-N'-(4-diethyl- aminocyclohexyl)carbodiimide, N-ethyl-N'-(3-dimethyl- aminopropyDcarbodiimide, etc.]; N,N'-carbonyldiimidazole, N,N'-carbonylbis(2-methylimidazole) ; keteneimine compound (e.g. pentamethyleneketene-N-cyclohexγlimine, diphenyl- ketene-N-cyclohexylimine, etc.); ethoxyacetylene; 1-alkoxy-1-chloroethylene; ethyl polyphosphate; isopropylpolyphosphate; phosphorus oxychloride; phosphorus trichloride; thionyl chloride; oxalyl chloride; a combination of triphenylphosphine with carbon tetrachloride or diazenedicarboxylate; 2-ethyl-7-hydroxy- benzisoxazolium salt; 2-ethyl-5-(m-sulfophenyl)- isoxazolium hydroxide intramolecular salt; l-(p-chloro- benzenesulfonyloxy)-6-chloro-lH-benzotriazole; so-called Vilsmeier reagent prepared by the reaction of N,N- dimethylformamide with thionyl chloride, phosgene, phosphorus oxychloride, etc. ; and the like. This acylation reaction may be carried out in the
presence of an inorganic or organic base such as an alkali metal bicarbonate (e.g. sodium bicarbonate, potassium bicarbonate, etc.), alkali metal carbonate (e.g. sodium carbonate, potassium carbonate, etc.), alkaline earth metal carbonate (e.g. magnesium carbonate, calcium carbonate, etc.), tri(lower)alkylamine (e.g. trimethylamine, triethylamine, N,N-diisopropyl-N- ethylamine, etc.), pyridine compound [e.g. pyridine, picoline, lutidine, N,N-di(lower)alkylaminσpyridine such as N,N-dimethylaminopyridine, etc.], quinoline, N-lower alkylmorpholine (e.g. N-methylmorpholine, etc.), N,N- di(lower)alkylbenzylamine (e.g. N,N-dimethylbenzylamine, etc.), alkali metal alkoxide (e.g. sodium methoxide, sodium ethoxide, potassium butoxide, etc.), and the like.
The reaction temperature of this acylation reaction is not critical and the reaction is usually carried out under from cooling to warming.
With regard to the compound (II), it is to be noted that the 3,7-dioxo-l-azabicyclo[3.2.0]heptane ring system of the following formula (IIA) is well known to lie to tautomeric relation with the 3-hydroxy-7-oxo-l- azabicyclo[3.2.0]hept-2-ene ring system of the following formula (IIB), and accordingly, it is to be understood that both of these ring systems are substantially the same.
Tautomerism
(HA) (IIB)
The compound (II') or salts thereof can be used with
or without isolation for the subsequent reaction with the compound (III) or salts thereof.
Suitable salts of the compound (III) may be the same as those for the compound (I) and silver salt. The reaction of the compound (II) or its reactive derivative or salts thereof with the compound (III) or salts thereof can be carried out in the presence of an organic or inorganic base such as those given in the explanation of the acylation reaction as stated above. This reaction can be carried out in a conventional solvent which does not adversely influence the reaction such as those given in the explanation of the acylation reaction.
The reaction temperature is not critical and the reaction is usually carried out under from cooling to warming.
{2) Process 2 :
The compound (I-b) or salts thereof can be prepared by subjecting the compound (I-a) or salts thereof to a removal reaction of the carboxy-protective group on Ra-
Suitable salts of the compounds (I-a) and (I-b) may be the same as those for the compound (I) .
The present reaction is usually carried out by a conventional method such as hydrolysis, reduction, and the like.
(i) Hydrolysis :
Hydrolysis is preferably carried out in the presence of a base or an acid. Suitable base may include an alkalimetal hydroxide (e.g. sodium hydroxide, potassium hydroxide, etc.), an alkaline earth metal hydroxide (e.g. magnesium hydroxide, calcium hydroxide, etc.), alkali metal hydride (e.g. sodium hydride, potassium hydride, etc.), alkaline earth metal hydride (e.g. calcium hydride.
etc.), alkali metal alkoxide (e.g. sodium methoxide, sodium ethoxide, potassium t-butoxide, etc.), an alkali metal carbonate (e.g. sodium carbonate, potassium carbonate, etc.), and alkaline earth metal carbonate (e.g. magnesium carbonate, calcium carbonate, etc.), an alkali metal bicarbonate (e.g. sodium bicarbonate, potassium bicarbonate, etc.), and the like.
Suitable acid may include an organic acid (e.g. formic acid, acetic acid, propionic acid, trifluoroacetic acid, benzenesulfonic acid, p-toluenesulfonic acid, etc.) and an inorganic acid (e.g. hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, etc.). The acidic hydrolysis using trifluoroacetic acid is usually accelerated by addition of cation trapping agent (e.g. phenol, anisole, etc.).
In case that the hydroxy-protective group is tri(lower)alkylsilyl, the hydrolysis can be carried out in the presence of tri(lower)alkylammonium halide (e.g. tributylammonium fluoride, etc.). This reaction is usually carried out in a conventional solvent which does not adversely influence the reaction such as water, dichloromethane, alcohol (e.g. methanol, ethanol, etc.), tetrahydrofuran, dioxane, acetone, etc., or a mixture thereof. A liquid base or acid can be also used as the solvent.
The reaction temperature is not critical and the reaction is usually carried out under from cooling to heating.
(ii) Reduction :
The reduction method applicable for this removal reaction may include, for example, reduction by using a combination of a metal (e.g. zinc, zinc amalgam, etc.) or a salt of chrome compound (e.g. chromous chloride, chromous acetate, etc.) and an organic or inorganic acid
(e.g. acetic acid, propionic acid, hydrochloric acid, sulfuric acid, etc.); and conventional catalytic reduction in the presence of a conventional metallic catalyst such as palladium catalysts (e.g. spongy palladium, palladium black, palladium oxide, palladium on carbon, colloidal palladium, palladium on barium sulfate, palladium on barium carbonate, palladium hydroxide on carbon, etc.), nickel catalysts (e.g. reduced nickel, nickel oxide, Raney nickel, etc.), platinum catalysts (e.g. platinum plate, spongy platinum, platinum black, colloidal platinum, platinum oxide, platinum wire, etc.), and the like.
In case that the catalytic reduction is applied, the reaction is preferably carried out around neutral condition.
This reaction is usually carried out in a conventional solvent which does not adversely influence the reaction such as water, alcohol (e.g. methanol, ethanol, propanol, etc.), dioxane, tetrahydrofuran, acetic acid, buffer solution (e.g. phosphate buffer, acetate buffer, etc.), and the like, or a mixture thereof.
The reaction temperature is not critical and the reaction is usually carried out under from cooling to warming. In case that the carboxy-protective group is allyl group, it can be deprotected by hydrogenolysis using a palladium compound.
Suitable palladium compound used in this reaction may be palladium on carbon, palladium hydroxide on carbon, palladium chloride, a palladium-ligand complex such as tetrakis(triphenylphosphine)palladiu (0) , bis(dibenzylideneacetone)palladium(0) , di[l,2-bis(diphenyl phosphino)ethane]palladium(0) , tetrakis(triphenyl phosphite)palladium(0) , tetrakis(triethyl phosphite)- palladium(0) , and the like.
The reaction can preferably be carried out in the presence of a scavenger of allyl group generated in situ, such as amine (e.g. morpholine, N-methylaniline, etc.), an activated methylene compound (e.g. dimedone, benzoylacetate, 2-methyl-3-oxovaleric acid, etc.), a cyanohydrin compound (e.g. α-tetrahydropyranyloxybenzyl cyanide, etc.), lower alkanoic acid or a salt thereof (e.g. formic acid, acetic acid, ammonium formate, sodium acetate, etc.), N-hydroxysuccinimide, and the like. This reaction can be carried out in the presence of a base such as lower alkyla ine (e.g. butylamine, triethyamine, etc.), pyridine, and the like.
This reaction can also be carried out in the presence of a conventional reducing agent such as sodium borohydride, tributyltin hydride, and the like.
When palladium-ligand complex is used in this reaction, the reaction can preferably be carried out in the presence of the corresponding ligand (e.g. triphenylphosphine, triphenyl phosphite, triethyl phosphite, etc.).
This reaction is usually carried out in a conventional solvent which does not adversely influence the reaction such as water, methanol, ethanol, propanol, dioxane, tetrahydrofuran, acetonitrile, chloroform, dichloromethane, dichloroethane, ethyl acetate, acetic acid, etc., or a mixture thereof.
The removal reaction can be selected according to the kind of carboxy-protective group to be removed.
The present process includes within the scope thereof a case that the i ino-protective group of R is removed at the same time during the reaction.
(3) Process 3 :
The compound (I-d) or salts thereof can be prepared by subjecting the compound (I-c) or salts thereof to a
removal reaction of the imino-protective group on R .
Suitable salts of the compounds (I-c) and (I-d) may be the same as those for the compound (I) .
This reaction is usually carried out by a conventional method such as hydrolysis, reduction and the like.
The method of hydrolysis and reduction, and the reaction conditions (e.g. reaction temperature, solvent, etc.) are substantially the same as those illustrated for removal reaction of the carboxy-protective group of the compound (I-a) in Process 2, and therefore are to be referred to said explanation.
The present process includes within the scope thereo)ff aa ccaassee tthhaatt tthhee ccaarrbbooxxyy--pprrootteeccttiivvee ggnroup on R is removed at the same time during the reaction.
(4) Process 4 :
The compound (I-f) or salts thereof can be prepared by reacting the compound (I-e) or salts thereof with the compound (IV) .
Suitable salts of the compounds (I-e) and (I-f) may be the same as those for the compound (I) .
This reaction is usually carried out in a conventional solvent which does not adversely influence the reaction such as water, dioxane, tetrahydrofuran, acetone, acetonitrile, etc., or a mixture thereof.
The reaction temperature is not critical, and the reaction is usually carried out under from cooling to warming.
(5) Process 5 :
The compound (I-h) or salts thereof can be prepared by subjecting the compound (I-g) or salts thereof to a
4 removal reaction of the hydroxy-protective group on Rb.
Suitable salts of the compounds (I-g) and (I-h) may be the same as those for the compound (I) .
This reaction is usually carried out by a conventional method such as hydrolysis, reduction and the like.
The method of hydrolysis and reduction, and the reaction conditions (e.g. reaction temperature, solvent, etc.) are substantially the same as those illustrated for removal reaction of the carboxy-protective group of the compound (I-a) in Process 2, and therefore are to be referred to said explanation.
The present process includes within the scope thereof a case that the carboxy-protective group on R and/or imino-protective group of R are removed at the same time during the reaction.
(6 ) Process 6 :
The compound (I-j) or salts thereof can be prepared by reacting the compound (I-i) or salts thereof with acid anhydride.
Suitable salts of the compounds (I-i) and (I-j) may be the same as those for the compound (I) .
Said acid anhydride is used only for preparing reactive derivative at the hydroxy group of the compound (I-i).
Therefore, suitable reagent, which can prepare following reactive derivatives at the hydroxy group of the compound (I-i), can be used in this reaction. Suitable reactive derivative at the hydroxy group of the compound (I-i) may include a conventional one such as halide (e.g. iodide, etc.), trifluoromethanesulfonate prepared by, for example, reactive with trifluoromethanesulfonic anhydride, and the like. This reaction can be carried out in the presence of
a base such as those mentioned in the explanation of Process 2.
This reaction is usually carried out in a conventional solvent which does not adversely influence the reaction such as water, dichloromethane, alcohol (e.g. methanol, ethanol, etc.), tetrahydrofuran, dioxane, acetone, etc., or a mixture thereof. A liquid base can be also used as the solvent.
The reaction temperature is not critical and the reaction is usually carried out under from cooling to warming.
(7) Process 7 :
The compound (I- ) or salts thereof can be prepared by subjecting the compound (I-k) or salts thereof to a
2 removal reaction of the hydroxy-protective group on Rc„l.
Suitable salts of the compounds (I-k) and (I-i) may be the same as those for the compound (I) .
This reaction is usually carried out by a conventional method such as hydrolysis, reduction and the like.
The method of hydrolysis and reduction, and the reaction conditions (e.g. reaction temperature, solvent, etc.) are substantially the same as those illustrated for removal reaction of the carboxy-protective group of the compound (I-a) in Process 2, and therefore are to be referred to said explanation.
The present process includes within the scope thereof a case that the protective groups on R , R4 and/or R***-1 is(are) removed at the same time during the reaction.
Method A and B for preparing the new starting compound (III) or salts thereof are explained in detail in the following.
Method A
The compound (Ill-a) or salts thereof can be prepared by reacting the compound (IV) or a reactive derivative at the hydroxy group thereof or salts thereof with the compound (V) or salts thereof.
Suitable salts of the compounds (III-a) and (IV) may be the same as those for the compound (I).
Suitable salts of the compound (V) may be salts with bases such as those given for the compound (I) . Suitable reactive derivative at the hydroxy group of the compound (IV) may include a conventional one such as halide (e.g. chloride, bromide, iodide, etc.), sulfonate (e.g. ethanesulfonate, benzenesulfonate, toluenesulfonate, etc.), and the like, in which more preferable example may be sulfonate.
The starting compound (IV) of this method is new and can be prepared by the methods described in the Preparations mentioned below.
Preferable example of the compound (V) may be ar(lower)alkanethiol such as mono- or di- or triphenyKlower)alkanethiol (e.g. phenylmethanethiol, diphenylmethanethiol, triphenylmethanethiol, etc.), thio(lower)alkanoic S-acid (e.g. thioacetic S-acid, etc.) or salts thereof, thioarenoic S-acid or salts thereof (e.g. thiobenzoic S-acid, etc.), and the like, in which more preferable example may be triphenyl(C.,- C.)alkanethiol, thio(C,-C.)alkanoic S-acid or alkali metal salts thereof and thio(C8-C.0)arenoic S-acid or alkali metal salts thereof, and the most preferable one may be triphenylmethanethiol, thioacetic S-acid and potassium thioacetate.
In case that the compound (V) may be ar(lower)- alkanethiol, the starting compound (IV) of the present reaction is preferably used in the form of its reactive derivative at the hydroxy group, and in, such a case, this
reaction is usually carried out in the presence of an organic or inorganic base such as those exemplified in the explanation of Process 2.
In case that suitable example of compound (V) may be thio(lower)alkanoic S-acid or thioarenoic S-acid, this reaction is preferably carried out in the presence of a conventional condensing agent such as combination of triarylphosphine (e.g. triphenylphosphine, etc.) and di(lower)alkyl azodicarboxylate (e.g. diethyl azodicarboxylate, etc.).
This reaction is usually carried out in a conventional solvent which does not adversely influence the reaction such as dichloromethane, methanol, ethanol, propanol, pyridine, N,N-dimethylformamide, 4-methyl-2- pentanone, tetrahydrofuran, etc., or a mixture thereof.
The reaction temperature is not critical and the reaction is usually carried out under from cooling to warming.
In this method, the configuration on the carbon atom substituted with the hydroxy group of the compound (IV) is inverted in the compound (Ill-a).
(B) Method B
The compound (III) or salts thereof can be prepared by subjecting the compound (III-a) or salts thereof to elimination reaction of the mercapto-protective group.
This elimination reaction can be carried out by a conventional method as described below, which can be selected according to the kind of mercapto-protective group to be eliminated.
In case that the protective groups may be ar(lower)alkyl group, it can generally be eliminated by treating, for example, with a silver compound (e.g. silver nitrate, silver carbonate, etc.). The reaction with the silver compound as stated
above is preferably carried out in the presence of an organic base (e.g. pyridine, etc.).
The resultant silver salt of compound (III) can be transformed into its alkali metal salt, if necessary, by reacting with alkali metal halide (e.g. sodium iodide, potassium iodide, etc.).
Further, in case that the protective groups may be acyl group, it can generally be eliminated by solvolysis such as hydrolysis using an acid or base, alcoholysis using a base, and the like.
Suitable acid or base used in these reactions may be the same such as those given in the explanation of hydrolysis of the Process 2.
The hydrolysis is usually carried out in a conventional solvent which does not adversely influence the reaction such as water, alcohol (e.g. methanol, ethanol, etc.), pyridine, N,N-dimethylformamide, etc., or a mixture thereof, and further in case that the base or acid to be used is in liquid, it can also be used as a solvent.
The alcoholysis is usually carried out in a conventional alcohol such as methanol, ethanol, and the like.
The reaction temperature is not critical and the reaction is usually carried out under from cooling to warming.
The object compounds obtained according to the above Processes can be isolated and purified in a conventional manner, for example, extraction, precipitation, fractional crystallization, recrystallization, chromatography, and the like.
The object compound (I) and pharmaceutically acceptable salts thereof of the present invention are novel and exhibit high antimicrobial activity, inhibiting the growth of a wide variety of pathogenic microorganisms
including Gram-positive and Gram-negative microorganisms and are useful as antimicrobial agents.
In the present invention, the object compound (I) possessing more potent antimicrobial activity can be represented by the following formula :
COOH
in which R 2, R3 and R4 are each as defined above, and pharmaceutically acceptable salts thereof.
Particularly, the compound (I) possessing the most potent antimicrobial activity can be represented by the following formula:
COOH
in which R 3 and R4 are each as defined above, and pharmaceutically acceptable salts thereof.
Now in order to show the utility of the object compound (I) , the test data on antimicrobial activity of the representative compound of the compound (I) of this invention is shown in the following.
in vitro Antimicrobial Activity
Test Method :
in vitro Antimicrobial Activity was determined by the two-fold agar-plate dilution method as described below.
One loopful of an overnight culture of a test strain in Trypticase-soy broth (10 viable cells per ml) was streaked on heart infusion agar (Hl-agar) containing graded concentrations of the test compound, and the minimal inhibitory concentration (MIC) was expressed in terms of μg/ml after incubation at 37°C for 20 hours.
Test Compound
The compound of Example 4-4)
Test Result :
Test Strain MIC (μg/ml)
K. pneumoniae 1690 0.1
For therapeutic administration, the object compound (I) and the pharmaceutically acceptable salts thereof of the present invention are used in the form of conventional pharmaceutical preparation which contains said compound, as an active ingredient, in admixture with pharmaceutically acceptable carriers such as an organic or inorganic solid or liquid excipient which is suitable for oral, parenteral and external administration. The pharmaceutical preparations may be in solid form such
as tablet, granule, powder, capsule, or liquid form such as solution, suspension, syrup, emulsion, lemonade, and the like.
If needed, there may be included in the above preparations auxiliary substances, stabilizing agents, wetting agents and other commonly used additives such as lactose, stearic acid, magnesium stearate, terra alba, sucrose, corn starch, talc, gelatin, agar, pectin, peanut oil, olive oil, cacao butter, ethylene glycol, tartaric acid, citric acid, fumaric acid, and the like.
While the dosage of the compound (I) may vary from and also depend upon the age, conditions of the patient, a kind of diseases, a kind of the compound (I) to be applied, etc. In general, amount between 1 mg and about 4,000 mg or even more per day may be administered to a patient. An average single dose of about 1 mg, 10 mg, 50 mg, 100 mg, 250 g, 500 mg, 1000 mg, 2000 mg, of the object compound (I) of the present invention may be used in treating diseases infected by pathogenic microorganisms.
EXAMPLES
The following Preparations and Examples are given for the purpose of illustrating this invention in more detail.
(continued on the next page)
Preparation 1-1
To a solution of pyridine (2.305 g) in dichloromethane (100 ml), under nitrogen atmosphere, at -20 ~ -25°C was added dropwise trifluoromethanesulfonic anhydride (7.83 g) . After 5 minutes, methyl glycolate (2.50 g) was added dropwise. After 15 minutes at -20°C the mixture was warmed slowly to room temperature over 1.5 hours. The mixture was then washed with water, 0.1 M-hydrochloric acid, water, dried over magnesium sulfate and evaporated under reduced pressure to give methoxycarbonyl ethyl trifluoromethanesulfonate (4.82 g) as a liquid.
NMR (CDC13, δ) : 3.88 (3H, s), 4.92 (2H, s)
Preparation 2-1)
To a solution of (2R,4S)-l-allyloxycarbonyl-2-(1- methyl-5-pyrazolyl)methyl-4-benzoylthiopyrrolidine (1 g) in tetrahydrohydrofuran-methanol (1:1) (10 ml) at 0°C was added 28% sodium methoxide in methanol solution (549 μl) dropwise. After 60 minutes, to the reaction mixture was added trityl chloride (760.3 mg) as a solid in a single portion and the mixture was stirred for 1 hour. The mixture was then evaporated under reduced pressure and then diluted with ethyl acetate, washed with brine, water, dried over magnesium sulfate and evaporated to give an oil. Purification by silica gel (30 g) column (eluent : ethyl acetate - isopropyl ether = 2:3) gave (2R,4S)-1- allyloxycarbonyl-2-(1-methyl-5-pyrazolyl)methyl-4- tritylthiopyrrolidine as a white solid (1.08 g) . NMR (CDCI3, 6) : 1.45-1.70 (IH, m) , 1.85-2.15 (IH, ), 2.60-3.00 (3H, ) , 3.05-3.40 (2H, m), 3.70- 3.90 (4H, ), 4.40-4.70 (2H, m) , 5.20-5.35 (2H, ), 5.79-5.99 (IH, m) , 5.99 (IH, br s), 7.08- 7.70 (16H, ) APCI-MS (m/z) : 524 (MH+)
Preparation 2-2)
To a solution of (2R,4S)-l-allyloxycarbonyl-2-(1- methyl-5-pyrazolyl)methyl-4-tritylthiopyrrolidine (204 mg) in 1,2-dichloroethane (2 ml) was added methoxycarbonyl ethyl trifluoromethanesulfonate (104 mg) . After 4 hours at room temperature the solution was evaporated under reduced pressure to give an amorphous solid. This solid was dissolved in methanol (2 ml) and treated with 28% aqueous ammonia solution (0.5 ml). After 1 hour the reaction mixture was evaporated under reduced pressure then azeotroped with toluene (3 ml x 5) and finally dried in vacuo over phosphorus pentoxide to give (2R,4S)-l-allyloxycarbonyl-2-[ (2-methyl-1-carbamoylmethyl- 3-pyrazolio)methyl]-4-tritylthiopyrrolidine trifluoro- methanesulfonate as an amorphous brown solid (284 mg). NMR (DMSO-d6, δ) : 1.40-1.60 (IH, ) , 2.10-2.35 (IH, m), 2.60-2.90 (2H, m) , 2.95-3.13 (IH, m) , 3.20-3.40 (IH, m), 3.80-4.00 (5H, ) , 4.38-4.48 (2H, m), 5.15-5.37 (4H, m) , 5.80-5.98 (IH, m) , 6.75 (IH, d, J=3Hz), 7.10-7.40 (15H, m) , 7.69
(IH, s), 7.95 (IH, s), 8.42 (IH, d, J=3Hz) FAB-MS : 581 (free cation, M+)
Preparation 3-1) {2R,4S)-1-Allyloxyearbon 1-2-(l-methyl-4- pyrazolyl)methyl-4-tritylthiopyrrolidine (1.15 g) was obtained by the same procedure as Preparation 2-1).
NMR (CDCI3, δ) : 1.25-1.70 (IH, m) , 1.90-2.15 (IH, m), 2.50-3.30 (5H, m) , 3.60-3.90 (4H, ) , 4.40- 4.70 (2H, m), 5.10-5.40 (2H, m) , 5.80-6.00 (IH, m) , 7.00-7.50 (17H, m) APCI-MS (m/z) : 524 (MH+)
Preparation 3-2) (2R,4S)-l-Allyloxycarbonyl-2-(l-methyl-4-
pyrazolyl)methyl-4-tritylthiopyrrolidine (870 mg) was dissolved in 1,2-dichloroethane (8.7 ml) and treated with methoxycarbonylmethyl trifluoromethanesulfonate (406.5 mg) . After standing overnight, concentration under reduced pressure gave (2R,4S)-l-allyloxycarbonyl-2-(1- methy1-2-methoxycarbonylmeth l-4-pyrazolio)methyl-4- tritylthiopyrrolidine trifluoromethanesulfonate as an amorphous solid (1.24 g) .
NMR (CDC13, δ) : 1.33-1.54 (IH, m) , 2.00-2.30 (IH, m), 2.40-3.50 (5H, complex m) , 3.68-3.85 (IH, m) ,
3.78 (3H, s), 4.05 (3H, s) , 4.30-4.60 (2H, ) , 5.10-5.35 (2H, m) , 5.62 (2H, m) , 5.80-6.00 (IH, m), 7.05-7.40 (IH, m) , 8.27 (IH, s), 8.42 (IH, s) APCI-MS (m/z) : 596 (M+-CF3S03 ~)
Preparation 3-3)
(2R,4S)-l-Allyloxycarbonyl-2-(l-methyl-2- carbamoylmethyl-4-pyrazolio)methyl-4-tritylthiopyrrolidine trifluoromethanesulfonate (1.22 g) was obtained by a similar procedure to that of Preparation 2-2).
NMR (DMS0-d6, δ) : 1.35-1.56 (IH, ) , 2.09-2.25 (IH, m) , 2.40-3.20 (5H, complex m) , 3.60-3.85 (IH, ) , 4.00 (3H, s), 4.30-4.60 (2H, m) , 5.10-5.35 (4H, m) , 5.80-6.00 (IH, m) , 7.00-7.40 (15H, m) , 7.70 (IH, s), 7.98 (IH, s), 8.29 (IH, s), 8.35 (IH, s)
APCI-MS (m/z) : 581 (M+-CF3S03 ~)
Preparation 4-1)
To a solution of l-[ (2S,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyloxy)pyrrolidin-2-yl]-l-(l-methylpyrazol-4- yDmethanol (121 g) , imidazole (240 mg) and carbon disulfide (55.5 ml) in tetrahydrofuran (1.2 Q ) was added sodium hydride (about 60% oil suspension, 13 g) at 0 - 5°C and the mixture was stirred for 30 minutes. To the mixture was added methyl iodide (38 ml) and the mixture was stirred for
1.5 hours at the same temperature then stirred for 4.5 hours at room temperature. To the reaction mixture was added water (300 ml) and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (800 ml), and the combined organic layer was washed with water (200 ml) and brine, dried over magnesium sulfate and evaporated under reduced pressure. The residue was column chromatographed on silica gel (3.5 kg, eluting with n-hexane - ethyl acetate = 1:1) to give (2S,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyloxy)-2-[1-(l-methylpyrazol-4-yl)-1- (methylthiothiocarbonyloxy) ethyl]pyrrolidine. IR (Neat) : 1690, 1640, 1400, 1100 cirT1 NMR (CDC13, δ) : 0.01, 0.05 (total 6H, each s),
0.84, 0.85 (total 9H, each s), 1.75-2.10 (2H, m) , 2.41, 2.43 (total 3H, each s), 2.90-3.10, 3.30-
3.73 (total 2H, m) , 3.86, 3.87 (total 3H, each s), 4.30-4.80 (4H, ) , 5.15-5.70 (3H, m) , 5.82-6.10 (IH, m), 7.18-7.45 (2H, m) APCI-MS (m/z) : 486 (MH+)
Preparation 4-2)
To a solution of (2S,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyloxy)-2-[1-(l-methylpyrazol-4-yl)-1- (methylthiothiocarbonyloxy)methyl]pyrrolidine (92.3 g) in toluene (950 ml) was added tri-n-butyltin hydride (77 ml) and 2,2'-azobisisobutyronitrile (6.3 g) . Under nitrogen atmosphere the mixture was refluxed for 4 hours then cooled. To the mixture were added tri-n-butyltin hydride (26 ml) and 2,2'-azobisisobutyronitrile (3.1 g), then refluxed again for 6 hours. After cooling the solvent was removed under reduced pressure and the residue was column chromatographed on silica gel (3.5 kg, eluting with n-hexane - ethyl acetate = 1:1) to give (2R,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyloxy)-2-[ (l-methylpyrazol-4- yl)methyl]pyrrolidine (52.1 g).
IR (Neat) : 1680, 1400, 1095 cm"1
NMR (CDC13, δ) : 0.02 (6H, s), 0.84 (9H, s), 1.63-
2.00 (2H, m), 2.70-2.95 (2H, ) , 3.26 (IH, dd, =11.2, 4.8Hz), 3.28-3.45 (IH, m) , 3.84 (3H, s), 4.03-4.21 (2H, ) , 4.58-4.70 (2H, m) , 5.17-5.40
(2H, m), 5.86-6.06 (IH, m) , 7.12 (IH, s), 7.25
(IH, s) APCI-MS (m/z) : 380 (MH+)
Preparation 5-1)
(2S,4R)-l-Allyloxycarbonyl-4-(t-butyldimethyl- silyloxy)-2-[l-(l-methylpyrazol-5-yl)-l-(methylthiothio- carbonyloxy)methyl]pyrrolidine was obtained in substantially the same manner as that of Preparation 4-1). IR (Neat) : 1705, 1647, 1402, 1207 cm-1
NMR (CDCI3, δ) : 0.02-0.08 (total 6H, each s), 0.84, 0.87 (total 9H, each s), 1.83-2.55 (2H, m) , 2.56, 2.57 (total 3H, each s), 2.95-3.60 (2H, m), 3.85, 3.92, 4.02 (total 3H, each s), 4.05-4.33 (IH, m) , 4.50-4.73 (3H, m) , 5.17-5.42
(2H, m) , 5.80-6.08 (IH, ) , 6.14-6.25 (IH, m) , 6.85-7.22 (IH, m) , 7.40-7.43 (IH, m) APCI-MS (m/z) : 486 (MH+)
Preparation 5-2)
(2R,4R)-l-Allyloxycarbonyl-4-(t- butyldimethylsilyloxy)-2-[ (l-methylpyrazol-5- yDmethylpyrrolidine was obtained in substantially the same manner as that of Preparation 4-2). IR (Neat) : 1701, 1406, 1109 cm-1
NMR (CDCI3, δ) : 0.03 (6H, s), 0.84 (9H, s), 1.73-
2.00 (2H, m) , 2.65-2.83 (IH, m) , 3.03-3.50 (3H, m), 3.75-3.90 (3H, m) , 4.08-4.28 (2H, ) , 4.55-
4.68 (2H, m), 5.18-5.38 (2H, m) , 5.86-6.06 (IH, m), 6.01 (IH, d, J=1.8Hz), 7.39 (IH, d, J=1.8Hz)
APCI -MS ( m/ z ) : 380 ( MH+ )
Preparation 6-1)
To a solution of (2R,4R)-l-allyloxycarbonyl-2- (pyridin-4-yl)methyl-4-t-butyldimethylsilyloxypyrrolidine (359.7 g) in methanol (3.6 ϋ ) was added a solution of sodium bicarbonate (170.89 g in water (2 J> ) at room temperature, followed by addition of a solution of OXONE (Trademark, made by Aldrich Chemical Company, Inc.) (440.34 g) in water (2 ϋ). The resulting suspension was stirred at room temperature for 20 hours. To the mixture was added chloroform (9 Jl ) and the mixture was separated. The organic layer was washed with water and brine, dried over magnesium sulfate to give crude 4-[ (2R,4R)-1- allyloxycarbonyl-4-t-butyldimethylsilyloxypyrrolidin-2- yl]methylpyridine N-oxide (359.9 g) as an oil.
IR (Film) : 2955, 2930, 2855, 1700, 1655 crn"1 NMR (CDC13, δ) : 0.02 (6H, s) , 0.82 (9H, s),
1.8-2.0 (2H, m) , 2.8-3.6 (4H, m) , 4.1-4.3 (2H, m), 4.6-4.7 (2H, m) , 5.2-5.4 (2H, m) , 5.8-6.05
(IH, m) , 7.08 (2H, d, J=6.7Hz), 8.13 (2H, d, J=6.9Hz)
Preparation 6-2) To a solution of crude 4-[ (2R,4R)-1- allyloxycarbonyl-4-t-butyldimethyl(silyloxypyrrolidin-2- yl]methylpyridine N-oxide (359.9 g) in dichloromethane (1 J? ) was added trimethylsilyl cyanide (102.7 g) at room temperature, followed by dropwise addition of N,N- dimethylcarbamoyl chloride (111.3 g) over 1 hour. The mixture was stirred at room temperature for 16 hours and poured into a mixture of ice water (1 H ) and 5N aqueous sodium hydroxide (100 ml) and adjusted to pH 7 by addition of 5N aqueous sodium hydroxide. The organic layer was washed with brine, dried over magnesium sulfate and
evaporated in vacuo. The residue was purified by column chromatography on silica gel to give (2R,4R)-1- allyloxycarbonyl-2-(2-cyanopyridin-4-yl)methyl-4-t- butyldimethylsilyloxypyrrolidine (263.26 g) as a yellow oil.
IR (Film) : 2955, 2885, 2860, 1735, 1700, 1595 cm-1 NMR (CDC13, δ) : 0.04 (6H, s), 0.85 (9H, s),
1.6-2.0 (2H, m), 2.8-3.0 and 3.2-3.6 (4H, m) , 4.2-4.3 (2H, ), 4.6-4.7 (2H, m) , 5.25-5.4 (2H, ), 5.85-6.0 (IH, m), 7.3-7.4 (IH, ) , 7.54 (IH, br s), 8.63 (IH, d, J=4.5Hz)
Preparation 6-3
To a solution of (2R,4R)-l-allyloxycarbonyl-2-(2- cyanopyridin-4-yl)methyl-4-t-butyldimethylsilyloxy- pyrrolidine (140 g) in dimethyl sulfoxide (560 ml) was added powdered potassium carbonate (24.1 g) at 10°C, followed by dropwise addition of 30% aqueous hydrogen peroxide (47.5 ml) over 25 minutes. The mixture was stirred at room temperature for 3.5 hours and cooled to 5°C. Ethyl acetate (500 ml) and 10% aqueous sodium thiosulfate (200 ml) were added and the mixture was stirred at 5°C for 1 hour. Additional ethyl acetate was added and the separated organic layer was washed with water and brine, dried over magnesium sulfate and evaporated in vacuo to give (2R,4R)-l-allyloxycarbonyl-2- (2-carbamoylpyridin-4-yl)methyl-4-t-butyldimethylsilyloxy- pyrrolidine (132.78 g) as a yellow oil.
IR (Film) : 3450, 3315, 2950, 2930, 2850, 1695, 1605, 1560 era"1
NMR (CDCI3, δ) : 0.00 (6H, s), 0.82 (9H, s),
1.7-2.0 (2H, m), 2.8-3.0 and 3.3-3.6 (4H, m) , 4.15-4.35 (2H, ) , 4.6-4.7 (2H, m) , 5.2-5.45 (2H, m), 5.67 (IH, br s), 5.9-6.1 (IH, m) , 7.33 (IH, br s), 7.86 (IH, m) , 8.03 (IH, s), 8.47
( IH , d , J=4 . 9Hz )
Preparation 6-4
To a solution of (2R,4R)-l-allyloxycarbonyl-2-(2- carbamoylpyridin-4-yl)methyl-4-t-butyldimethylsilyloxy- pyrrolidine (132.7 g) was added dropwise cone, hydrochloric acid (52.7 ml) at 5°C and the mixture was stirred at room temperature for 2 hours. To the mixture was added carefully sodium bicarbonate (53.09 g) at 5°C and the mixture was stirred at room temperature for 1 hour and evaporated in vacuo. To the residue was added toluene (600 ml) and the solution was evaporated in vacuo to remove water under azeotropic condition. To the residue was added dichloromethane (600 ml) and the mixture was stirred for 20 minutes. The insoluble materials were filtered off and the filtrate was evaporated in vacuo. The residue was purified by column chromatography on silica gel to give (2R,4R)-l-allyloxycarbonyl-2-(2- carbamoylpyridin-4-yl)methyl-4-hydroxypyrrolidine (108.32 g) as an oil.
IR (Film) : 3450, 2940, 1685, 1600, 1560 cm"1 NMR (CDC13, δ) : 1.6-2.0 (2H, m) , 2.8-3.7 (4H, m) , 4.2-4.4 (2H, m), 4.6-4.8 (2H, ) , 5.2-5.4 (2H, m), 5.9-6.2 (2H, m) , 7.2-7.4 (IH, m) , 7.89 (IH, m), 8.02 (IH, s), 8.46 (IH, d, J=4.8Hz)
Preparation 6-5
To a solution of (2R,4R)-l-allyloxycarbonyl-2-(2- carbamoylpyridin-4-yl)methyl-4-hydroxypyrrolidine (108.3 g) in dichloromethane (500 ml) was added triethylamine (43.1 g) at 5°C, followed by dropwise addition of methanesulfonyl chloride (44.73 g) over 30 minutes at 5°C. The mixture was stirred at 5°C for 2 hours and poured into ice water (300 ml). The separated organic layer was washed with water, dried over magnesium sulfate, and
evaporated in vacuo to give crude (2R,4R)-1- allyloxycarbonyl-2-(2-carbamoylpyridin-4-yl)methyl-4- methylsulfonyloxypyrrolidine (129.58 g) as an oil.
IR (Film) : 3460, 3340, 3110, 2940, 1690, 1605, 1540 c "1
NMR (CDC13, δ) : 1.9-2.1 and 2.3-2.45 (2H, m) , 2.8- 3.9 (3H, ), 3.01 (3H, s), 4.0-4.15 and 4.3-4.45 (2H, m), 4.6-4.7 (2H, m), 5.25-5.4 (2H, m) , 5.9- 6.1 (2H, m), 7.2-7.4 (lH,.br s), 7.8-7.95 (IH, m), 8.03 (IH, s), 8.49 (IH, d, J=4.9Hz)
Preparation 6-6)
To a solution of potassium t-butoxide (45.48 g) in dimethylformamide (500 ml) was added dropwise thiobenzoic acid (56.01 g) at room temperature and the mixture was stirred for 30 minutes. To the mixture was added dropwise a solution of (2R,4R)-l-allyloxycarbonyl-2-(2- carbamoylpyridin-4-y1)methyl-4- methylsulfonyloxypyrrolidine (129.5 g) in dimethylformamide (400 ml) over 25 minutes at room temperature and the mixture was stirred at 80°C for 2.3 hours. The mixture was cooled to 5°C and poured into a mixture of ethyl acetate (1.2 β ) and ice water (2.7 0 ) . The separated organic layer was washed with water and brine, dried over magnesium sulfate (MgSθ4) and evaporated in vacuo. The residue was purified by column chromatography on silica gel to give (2R,4S)-1- allyloxycarbonyl-2-(2-carbamoylpyridin-4-yl)methyl-4- benzoylthiopyrrolidine (52.97 g) as a red amorphous solid. IR (Film) : 3450, 3060, 2940, 2895, 1675, 1600 cm"1
NMR (CDCI3, δ) : 1.7-1.9 and 2.4-2.6 (2H, m) , 2.8- 3.6 (4H, m), 4.1-4.35 (2H, ) , 4.6-4.7 (2H, m) , 5.2-5.45 (2H, m) , 5.9-6.1 (2H, m) , 7.3-7.7 (4H, m), 7.9-8.0 (3H, m) , 8.18 (IH, s), 8.48 (IH, d, J=4.9Hz)
Preparation 6-7)
To a solution of (2R,4S)-l-allyloxycarbonyl-2-(2- carbamoylpyridin-4-yl)methyl-4-benzoylthiopyrrolidine (52.96 g) in a mixture of tetrahydrofuran(THF) (265 ml) and methanol(MeOH) (265 ml) was added dropwise 28% sodium methoxide in methanol (23.9 ml) at 5°C and the mixture was stirred at 5°C for 50 minutes. To the mixture was added dropwise 6N hydrochloric acid (HCI) (20.7 ml) and the mixture was poured into ethyl acetate and ice water and adjusted to pH 2 by addition of 6N HCI. The separated organic layer was washed with water and brine, dried over MgS04 and evaporated in vacuo. The residue was purified by column chromatography on silica gel (300 g) (eluent : dichloromethane:methanol = 10:1) to give (2R,4S)-1- allyloxycarbonyl-2-(2-carbamoylpyridin-4-yl)methyl-4- mercaptopyrrolidine (33.95 g) .
IR (Film) : 3450, 2945, 2875, 1695, 1600, 1555 cm-1 NMR (CDC13, δ) : 1.6-1.9 and 2.3-2.5 (3H, m) ,
2.85-3.6 (4H, m) , 4.1-4.3 (2H, m) , 4.6-4.7 (2H, m), 5.2-5.4 (2H, m) , 5.8-5.9 (IH, br s), 5.9-6.1
(IH, m), 7.3-7.4 (IH, br s) , 7.85-7.95 (IH, m) , 8.07 (IH, s), 8.48 (IH, d, J=4.9Hz)
Preparation 7-1) The following compound was obtained by the similar manner to that of Preparation 6-4).
(2R,4R)-l-Allyloxycarbonyl-2-(2-cyanopyridin-4- y1)methyl-4-hydroxypyrrolidine. IR (Film) : 3420, 3055, 2945, 2885, 2235, 1685,
1650, 1600, 1555 crn-1 NMR (CDCI3, δ) : 1.7-2.1 (2H, m) , 2.8-3.0 and 3.3- 3.7 (4H, m), 4.2-4.4 (2H, m) , 4.6-4.7 (2H, m) , 5.25-5.45 (2H, m) , 5.9-6.1 (IH, m) , 7.3-7.4 (IH, m), 7.63 (IH, s), 8.61 (IH, d, J=4.8Hz)
Preparation 7-2)
The following compound was obtained by the similar manner to that of Preparation 6-5).
(2R,4R)-l-Allyloxycarbonyl-2-(2-cyanopyridin-4- yl)methyl-4-methylsulfonyloxypyrrolidine
IR (Film) : 3055, 2940, 2235, 1700, 1650, 1595,
1555 cm"1 NMR (CDC13, δ) : 1.8-2.0 and 2.3-2.5 (2H, m) , 3.06 (3H, s), 3.4-3.55 (2H, m) , 3.9-4.1 (IH, m) , 4.6-
4.7 (2H, m), 5.1-5.2 (IH, m) , 5.3-5.45 (2H, m) , 5.8-6.05 (IH, m), 7.35-7.45 (IH, m) , 7.64 (IH, s), 8.63 (IH, d, J=5.1Hz)
Preparation 7-3)
The following compound was obtained by the similar manner to that of Preparation 6-6).
(2R,4S)-l-Allyloxycarbonyl-2-(2-cyanopyridin-4- y1)methyl-4-benzoylthiopyrrolidine
IR (Film) : 3055, 2980, 2945, 2875, 2235, 1700,
1665, 1595 crn"1 NMR (CDCI3, δ) : 1.7-1.85 and 2.45-2.65 (2H, ) , 2.8-3.0 and 3.3-3.6 (4H, m) , 4.1-4.3 (2H, m) , 4.55-4.7 (2H, m) , 5.2-5.45 (2H, m) , 6.85-7.1
(IH, m), 7.4-7.7 (4H, m) , 7.9-8.05 (3H, m) , 8.62 (IH, d, J=5.0Hz)
Preparation 7-4) The following compound was obtained by the similar manner to that of Preparation 6-7).
(2R,4S)-l-Allyloxycarbonyl-2-(2-cyanopyridin-4- yl)methyl-4-mercaptopyrrolidine IR (Film) : 3055, 2940, 2875, 2235, 1695, 1600 cm-1
NMR (CDCI3, δ) : 1.5-1.8 and 2.1-2.3 (3H, m) , 2.9- 3.6 (4H, m), 4.05-4.2 (2H, m) , 4.6-4.7 (2H, ) , 5.25-5.4 (2H, m) , 5.85-6.1 (IH, m) , 7.35-7.45 (IH, m), 7.64 (IH, s), 8.63 (IH, d, J=4.9Hz)
Preparation 8-1)
To a solution of 1M methylmagnesiu bromide- tetrahydrofuran solution (91.6 ml) in tetrahydrofuran (100 ml) was added dropwise a solution of (2S,4R)-1- allyloxycarbonyl-4-(t-butyldimethylsilyl)oxy-2-
(formylmethyl)pγrrolidine (20 g) in tetrahydrofuran (60 ml) with stirring under atmospheric pressure of nitrogen at 0-5°C. A mixture was stirred at ambient temperature for 2 hours. To the reaction mixture was added saturated aqueous ammonium chloride (20 ml) and the solution was stirred at the same condition for 10 minutes. The resulting precipitates were filtered off. To the filtrate was added ethyl acetate (200 ml) and the solution was washed successively with IN hydrochloric acid, water, saturated aqueous sodium hydrogen carbonate and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (300 g) eluting with a mixture of n-hexane and ethyl acetate (2:1, V/V) . The first fractions were collected and evaporated in vacuo to give (2R,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyl)oxy-2-[ (2S)-2-hydroxypropyl]pyrrolidine (6.76 g).
NMR (CDCI3, δ) : 0.05 (6H, s), 0.85 (9H, s), 1.11 (3H, d, J=6.26Hz), 1.30-2.10 (4H, m) , 3.30-3.80
(3H, m) , 4.10-4.45 (2H, m) , 4.45-4.55 (2H, ) , 5.05-5.35 (2H, m) , 5.70-6.00 (IH, m)
The second fractions were collected and evaporated in vacuo to give (2R,4R)-l-allyloxycarbonyl-4-(t-
butyldimethylsilyl)oxy-2-[ (2R)-2-hydroxypropyl]pyrrolidine (5.30 g) .
NMR (CDCI3, δ) : 0.05 (6H, s), 0.84 (9H, s), 1.14 (3H, d, J=6.22Hz), 1.35-2.15 (4H, ) , 3.30-3.45 (2H, broad d, J=4.39Hz), 3.65-3.90 (IH, ) ,
4.00-4.15 (IH, ) , 4.20-4.40 (IH, ) , 4.45-4.60 (2H, ), 5.05-5.35 (2H, m) , 5.75-6.00 (IH, m)
Preparation 8-2) To a solution of (2R,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyl)oxy-2-[ ( 2R)-2-hydroxypropyl]pyrrolidine (19.34 g) and triethylamine (11.0 ml) was added dropwise methanesulfonyl chloride (5.23 ml) under ice-cooling. After stirring at the same temperature for 1 hour, the reaction mixture was washed with water and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo to give a residue. The residue was chromatographed on silica gel (300 g) eluting with a mixture of n-hexane and ethyl acetate (2:1, V/V) . The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- allyloxycarbonyl-4-(t-butyldimethylsilyl)oxy-2-[ (2R)-2- methylsulfonyloxypropyl]pyrrolidine (6.03 g). This compound was immediately used as the stirring compound for the next step.
Preparation 8-3)
A solution of (2R,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyl)oxy-2-[ (2R)-2- methylsulfonyloxypropyl]pyrrolidine (6.02 g), imidazole (1.26 g) and potassium t-butoxide (2.08 g) in N,N- dimethylformamide (60 ml) was stirred at 70-75°C for 3 hours. To the reaction mixture were added ethyl acetate (150 ml) and water (50 ml) with stirring and the organic layer was separated. The organic layer was washed with
saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (150 g) eluting with a mixture of chloroform and methanol (9:1, V/V) . The fractions containing the desired compound were collected and evaporated in vacuo to give a residue (2.90 g). The residue was dissolved in methanol (30 ml). To the solution was added cone, hydrochloric acid (1.23 ml) with stirring at ambient temperature and allowed to stand at the same temperature overnight. To the reaction mixture was added 28% sodium methoxide-methanol solution (2.83 ml) under ice-cooling. The resulting precipitates were filtered off. The filtrate was evaporated in vacuo to give a residue. The residue was chromatographed on silica gel (100 g) eluting with a mixture of chloroform and methanol (9:1, V/V) . The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-l-allyloxycarbonyl-4-hydroxy-2-[ (2S)-2-(imidazol- l-yl)propyl]pyrrolidine (1.06 g). NMR (CDC13, δ) : 1.48 (3H, d, J=6.81Hz), 1.55-2.60
(4H, m), 3.25-4.75 (8H, m) , 5.10-5.40 (2H, ) , ' 5.80-6.10 (IH, m), 6.96 (IH, s), 7.02 (IH, s), 7.55 (IH, s)
Preparation 8-4)
To a solution of (2R,4R)-l-allyloxycarbonyl-4- hydroxy-2-[ (2S)-2-(imidazol-1-yl)propyl]pyrrolidine (2.48 g) and triethylamine (1.73 ml) in ethyl acetate (30 ml) was added dropwise methanesulfonyl chloride (0.82 ml) with stirring under ice-cooling and the mixture was stirred at the same temperature for 1 hour. The reaction mixture was washed with water and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (100 g) eluting with a mixture of chloroform
and methanol (19:1, V/V). The fractions containing the lesired compound were collected and evaporated in vacuo to give (2R,4R)-l-allyloxycarbonyl-4-methylsulfonyloxy-2- [ (2S)-2-(imidazol-l-yl)propyl]pyrrolidine (2.60 g) . NMR (CDC13, δ) : 1.50 (3H, d, J=6.81Hz), 1.55-2.65
(4H, m), 2.99 (3H, s), 3.47 (IH, dd, J=3.88, 9.24Hz), 3.80-4.50 (3H, ) , 4.50-4.70 (2H, m) , 5.09 (IH, broad s), 5.15-5.41 (2H, ) , 5.80-6.10 (IH, m), 6.98 (IH, s), 7.08 (IH, s), 7.59 (IH, s)
Preparation 8-5)
To a solution of potassium t-butoxide (1.06 g) in N,N-dimethylformamide (5 ml) was added dropwise thioacetic S-acid (0.67 ml) with stirring at -10 -20°C. The mixture was stirred at the same temperature for 30 minutes. The solution was added to a solution of (2R,4R)- l-allyloxycarbonyl-4-methylsulfonyloxy-2-[ (2S)-2- (imidazol-l-yl)propyl]pyrrolidine (2.59 g) in N,N- dimethylformamide (15 ml) and the mixture was stirred at
85-90°C for 3 hours. The reaction mixture was poured into water (100 ml) and extracted twice with ethyl acetate (60 ml) . The extract was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (100 g) eluting with a mixture of chloroform and methanol (19:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4S)-4-acetylthio-l- allyloxycarbonyl-2-[ ( 2S) -2- (imidazol-1- yDpropyl] pyrrolidine (2.20 g) .
NMR (CDCI3, δ) : 1.48 (3H, d, J=6.81Hz), 1.75-2.25 (3H, m), 2.32 (3H, s), 2.33-2.55 (IH, m) , 3.15 (IH, dd, J=6.88, 11.4Hz), 3.75-4.50 (4H, m) , 4.55-4.70 (2H, ) , 5.15-5.40 (2H, m) , 5.80-6.10
( IH , m) , 6 . 97 ( IH , s ) , 7 . 07 ( IH , s ) , 7 . 57 ( IH , s )
Preparation 9-1) To a solution of bromine (0.14 ml) and sodium carbonate (0.55 g) in dichloromethane (10 ml) was added dropwise a solution of (2R,4R)-l-benzyl-4-(t- butyldimethylsilyl)oxy-2-[ (l-methylpyrazol-5- yl)methyl]pyrrolidine (1.0 g) in dichloromethane (2 ml) with stirring under ice-cooling. The mixture was stirred at the same temperature for 30 minutes. To the mixture were added saturated aqueous sodium thiosulfate (5 ml) and dichloromethane (20 ml) with stirring. The organic layer was separated and washed successively with saturated aqueous sodium thiosulfate and aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (50 g) eluting with a mixture of n-hexane and ethyl acetate (2:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-l-benzyl-2-(4-bromo-l-methylpyrazol-5- yl)methyl-4-[ (t-butyldimethylsilyl)oxy]pyrrolidine (791 mg) .
NMR (CDC13, 5) : 0.01 (6H, s), 0.85 (9H, s), 1.65- 1.88 (2H, m), 2.27-2.40 (IH, m) , 2.60-3.22 (4H, m), 3.50 (IH, d, J=13Hz), 3.79 (3H, s), 3.96 (IH, d, J=13Hz), 4.20-4.35 (IH, m) , 7.20-7.33 (5H, m), 7.39 (IH, s) APCI-MS : 466, 464 (M+)
Preparation 9-2)
To a solution of (2R,4R)-l-benzyl-2-(4-bromo-l- methylpyrazol-5-yl)methyl-4-[ (1- butyldimethylsilyDoxy]pyrrolidine (1.0 g) in diethyl ether (15 ml) was added dropwise n-butyl lithium (1.62 M
- o i - in hexane solution) (1.86 ml) with stirring at -10°C. The mixture was stirred at -10°C for 30 minutes and then at ambient temperature for 1 hour. The solution was cooled at -30°C. To the solution was added dropwise N,N- dimethylformamide (0.5 ml) at the same temperature and the mixture was stirred at -20 ~ 0°C for 1 hour. To the reaction mixture were added saturated aqueous ammonium chloride (10 ml) and ethyl acetate (50 ml). The organic layer was separated and then washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (50 g) eluting with a mixture of n-hexane and ethyl-acetate (2:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-l-benzyl-4-(t- butyldimethylsilyl)oxy-2-[ (4-formyl-l-methylpyrazol-5- yl)methyl]pyrrolidine (591 mg) .
NMR (CDCI3, δ) : 0.01 (6H, s), 0.85 (9H, s), 1.70- 1.81 (3H, m), 2.32 (IH, dd, J=4.8, 10.3Hz), 3.00-3.35 (4H, m) , 3.53 (IH, d, J=13.1Hz), 3.83
(3H, s), 3.93 (IH, d, J=13.1Hz), 4.20-4.35 (IH, m), 7.20-7.40 (5H, m) , 7.88 (IH, s), 9.86 (IH, s) APCI-MS : 414 (M+)
Preparation 9-3)
To a solution of (2R,4R)-l-benzyl-4-(t- butyldimethylsilyl)oxy-2-(4-formyl-l-methylpyrazol-5- yUmethylpyrrolidine (14.0 g) in a mixture of tetrahydrofuran (140 ml) and methanol (140 ml) was added portionwise sodium borohydride (1.28 g) with stirring under ice-cooling. After stirring at the same temperature for 1 hour, IN hydrochloric acid (33 ml) was added to the solution and evaporated in vacuo to give a residue. The residue was dissolved in ethyl acetate (300 ml). The
ylchsolution was washed successively with water, saturated aqueous sodium hydrogen carbonate and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated under reduced pressure. The resulting residue was chromatographed on silica gel (400 g) eluting with a mixture of n-hexane and ethyl acetate (1:2, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- benzyl-4-(t-butyldimethylsilyl)oxy-2-[ (4-hydroxymethyl-1- methylpyrazol-5-yl)methyl]pyrrolidine (7.91 g) .
NMR (CDCI3, δ) : 0.01 (6H, s), 0.86 (9H, s),
1.45-1.60 (IH, m), 1.80-1.95 (IH, m) , 2.45-2.85 (3H, m), 2.97 (IH, dd, J=4.7, 11.2Hz), 3.56 (IH, d, J=12.2Hz), 3.69 (3H, s), 3.73 (IH, d, J=12.2Hz), 4.15-4.30 (IH, m) , 4.41 (2H, s),
5.05-5.50 (IH, broad ), 7.19-7.33 (5H, m) , 7.38 (IH, s) APCI-MS : 416 (M+)
Preparation 9-4)
A mixture of (2R,4R)-l-benzyl-4-(t- butyldimethylsilyl)oxy-2-[ (4-hydroxymethyl-1- methylpyrazol-5-yl)methyl]pyrrolidine (7.91 g), methanol (80 ml), ammonium formate (3.60 g) and 10% palladium on carbon (50% wet) (3.0 g) was refluxed for 1 hour. The catalyst was filtered off and the filtrate was evaporated in vacuo to give a residue. The residue was dissolved in a mixture of tetrahydrofuran (80 ml) and water (40 ml). To a solution was added dropwise allyl chloroformate (2.83 ml) with stirring under ice-cooling while keeping the pH at 8-10 with 4N sodium hydroxide. After 1 hour, to the solution was added ethyl acetate (200 ml). The organic layer was separated, washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was
chromatographed on silica gel (200 g) eluting with a mixture of dichloromethane and methanol (9:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- allyloxycarbonyl-4-(t-butyldimethylsilyl)oxy-2-[ (4- hydroxymethyl-l-methylpyrazol-5-yl) ethyl]pyrrolidine (6.89 g) .
NMR (CDC13, δ) : 0.01 (6H, s), 0.81 (9H, s), 1.75- 1.90 (2H, m), 2.50-2.80 (IH, ) , 3.10-3.55 (3H, m), 3.85 (3H, s), 4.05-4.65 (6H, ) , 5.15-5.35
(2H, m), 5.80-6.05 (IH, m) , 7.41 (IH, s) APCI-MS : 410 (M+)
Preparation 9-5) To a solution of (2R,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyl)oxy-2-[ (4-hydroxymethyl-1- methylpyrazol-5-yl) ethyl]pyrrolidine (6.89 g) and triethylamine (3.28 ml) in ethyl acetate (3.28 ml) was added dropwise acetyl chloride (1.44 ml) under ice-cooling with stirring and the mixture was stirred at the same temperature for 1 hour. To the reaction mixture was added water (40 ml) with stirring and the organic layer was separated. This layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was dissolved in acetonitrile (70 ml). To the solution was added cone. hydrochloric acid (2.8 ml) and the mixture was stirred at ambient temperature for 1.5 hours . Ethyl acetate (140 ml) and saturated aqueous sodium hydrogen carbonate (50 ml) were added to the reaction mixture with stirring. The organic layer was separated, washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo successively. The resulting residue was dissolved in ethyl acetate (70 ml). To the solution were added triethylamine (3.28 ml)
and methanesulfonyl chloride (1.56 ml) with stirring under ice-cooling. After stirring for 1 hour, the reaction mixture was washed successively with water, saturated aqueous sodium hydrogen carbonate and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo to give (2R,4R)-2-(4- acetoxymethyl-l-methylpyrazol-5-yl)methyl-l- allyloxycarbonyl-4-methanesulfonyloxypyrrolidine (7.33 g). NMR (CDC13, δ) : 0.01 (6H, s), 0.81 (9H, s), 1.60- 2.00 (2H, ), 2.65-2.80 (IH, ) , 3.20-3.50 (2H, m), 3.80-3.90 (3H, ) , 4.00-5.40 (8H, ) , 5.80- 6.05 (IH, m), 7.45 (IH, s)
Preparation 9-6) To a solution of (2R,4R)-2-(4-acetoxymethyl-l- methylpyrazol-5-yl)methyl-l-allyloxycarbonyl-4- methylsulfonyloxypyrrolidine (7.6 g) in methanol (73 ml) was added dropwise 28% sodium methoxide-methanol solution (3.51 ml) under ice-cooling with stirring. The mixture was stirred at the same temperature for 15 minutes. To the reaction mixture was added cone. hydrochloric acid (1.46 ml) and the mixture was evaporated in vacuo. The resulting residue was dissolved in ethyl acetate (200 ml). The solution was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (250 g) eluting with a mixture of dichloromethane and methanol (20:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- allyloxycarbonyl-2-(4-hydroxymethyl-l-methylpyrazol-5- yl)methyl-4-methylsulfonyloxypyrrolidine (4.32 g) .
NMR (CDCI3, δ) : 2.00-2.50 (3H, m) , 2.70-2.90 (IH, ), 3.01 (3H, s), 3.30-4.75 (11H, ) , 5.10-5.45 (3H, ), 5.80-6.10 (IH, ) , 7.42 (IH, s)
APCI-MS : 374 ( M+)
Preparation 9-7)
To a solution of potassium t-butoxide (1.69 g) in N,N-dimethylformamide (20 ml) was added dropwise thiobenzoic S-acid (1.91 ml) at -10 ~ -20°C with stirring and the mixture was stirred at the same temperature for 30 minutes. The solution was added to a solution of (2R,4R)- l-allyloxycarbonyl-2-(4-hydroxymethyl-l-methylpyrazol-5- yl)methyl-4-methylsulfonyloxypyrrolidine (4.32 g) in N,N- dimethylformamide (45 ml) and the mixture was stirred at 85-90°C for 3 hours. To a reaction mixture were added ethyl acetate (100 ml) and water (50 ml). The organic layer was separated and then washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (150 g) eluting with a mixture of dichloromethane and methanol (19:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4S)-1- allyloxycarbonyl-4-benzoylthio-2-[ (4-hydroxymethyl-1- methylpγrazol-5-yl)methyl]pyrrolidine (5.38 g) .
NMR (CDC13, δ) : 1.60-2.70 (3H, m) , 2.90-3.55 (3H, ), 3.75-4.70 (10H, ) , 5.20-5.45 (2H, ) , 5.85-6.05 (IH, ) , 7.35-8.10 (6H, m)
APCI-MS : 416 (M+)
Preparation 10-1)
To a solution of 4-bromo-5-(t- butyldimethylsilyloxy)methyl-l-methylpyrazole (1.15 g) in diethyl ether (20 ml) was added dropwise n-butyl lithium (1.62 M solution in hexane) (2.71 ml) with stirring at -40 - -50°C. A mixture was stirred at -10°C for 30 minutes and then at 0°C for 30 minutes. To a reaction mixture was added dropwise a solution of (2S,4R)-1-benzyl-
4-t-butyldimethylsilyloxy-2-formylpyrrolidine (1.0 g) in diethyl ether (3 ml) at -30 ~ -40°C. The mixture was stirred at 0°C for 1 hour. To the. reaction mixture were added water (20 ml) and ethyl acetate (50 ml) and the organic layer was separated. The organic layer was washed with water and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (50 g) eluting with a mixture of n-hexane and ethyl acetate (2:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2S,4R)-l-benzyl-4-tert-butyldimethylsilyloxy-2-[l-{5-(t- butyldimethylsilyloxy)methyl-l-methylpyrazol-4-yl}-l- hydroxymethyl]pyrrolidine (754 mg) . APCI-MS : 546 (M+)
Preparation 10-2)
A mixture of (2S,4R)-l-benzyl-4-t-butyldimethyl- silyloxy-2-[l-{5-(t-butyldimethylsilyloxy)methyl-l- methylpyrazol-4-yl}-l-hydroxymethyl]pyrrolidine (8.05 g), ammonium formate (2.8 g), 10% palladium on carbon (4.0 g) and methanol was refluxed for 3 hours. The catalyst was filtered off and the filtrate was evaporated in vacuo to give a residue. To a solution of the residue in a mixture of tetrahydrofuran (80 ml) and water (40 ml) was added dropwise allyl chloroformate (2.2 ml) under ice-cooling, keeping the pH at 8-10 with 4N aqueous sodium hydroxide and the mixture was stirred for 1 hour. To the reaction mixture was added ethyl acetate (150 ml) and the organic layer was separated. The organic layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (200 g) eluting with a mixture of n-hexane and ethyl acetate (2:1, V/V). The fractions containing the desired compound were
collected and evaporated in vacuo to give (2S,4R)-1- allyloxycarbonyl-4-tert-butyldimethylsilyloxy-2-[l-{5-(t- butyldimethylsilyloxy)methyl-l-methylpyrazol-4-yl}-l- hydroxymethyl]pyrrolidine (6.39 g) . APCI-MS : 540 (M+)
Preparation 10-3)
(2S,4R)-l-Allyloxycarbonyl-4-t-butyldimethyl- silyloxy-2-[l-{5-(t-butyldimethylsilyloxy)methyl-l- methylpyrazol-4-yl}-l-(methylthiothiocarbamoyloxy)methyl]- pyrrolidine (5.13 g) was obtained in substantially the same manner as that of Preparation 4-1).
This compound was immediately used as the starting compound for the next step.
Preparation 10-4)
(2R,4R)-l-Allyloxycarbonyl-4-t-butyldimethyl- silyloxy-2-[5-(t-butyldimethylsilyloxy)methyl-l- methylpyrazol-4-yl]methylpyrrolidine (3.47 g) was obtained in substantially the same manner as that of Preparation 4-1).
NMR (CDC13, δ) : 0.00 (3H, s), 0.07 (3H, s), 0.83 (9H, s), 0.88 (9H, s) , 1.65-1.90 (2H, m) , 2.50-2.95 (2H, ) , 3.20-3.50 (2H, ) , 3.87 (3H, s), 4.00-4.20 (2H, m) , 4.50-4.70 (4H, m), 5.10-
5.40 (2H, m), 5.80-6.10 (IH, m) , 7.20 (IH, s) APCI-MS : 524 (M+)
Preparation 10-5) A solution of (2R,4R)-l-allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-[5-(t-butyldimethylsilyloxy)- methyl-l-methylpyrazol-4-yl]methylpyrrolidine (3.47 g) and cone, hydrochloric acid (1.7 ml) in acetonitrile (30 ml) was stirred at ambient temperature for 2 hours. To the reaction mixture was added dropwise 28% sodium methoxide-
methanol solution (2.07 ml) under ice-cooling. The resulting precipitates were filtered off. The filtrate was evaporated in vacuo to give a residue. The residue was chromatographed on silica gel (150 g) eluting with a mixture of dichloromethane and methanol (10:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- allyloxycarbonyl-4-hydroxy-2-[5-hydroxymethyl-l- methylpyrazol-4-yl]methylpyrrolidine (1.63 g) . NMR (CDC13, δ) : 1.85-2.05 (IH, m), 2.45-3.10 (3H, ), 3.40-3.60 (3H, m) , 3.86 (3H, s), 3.90-4.75 (6H, m), 5.10-5.40 (2H, m) , 5.80-6.05 (IH, m) , 7.19 (IH, m) APCI-MS : 296 (M+)
Preparation 10-6)
To a solution of (2R,4R)-l-allyloxycarbonyl-4- hydroxy-2-[5-hydroxymethyl-l-methylpyrazol-4- yl]methylpyrrolidine (1.62 g) in dichloromethane (20 ml) were added successively triethylamine (0.92 ml), N,N- dimethylaminopyridine (34 mg) and t-butyldimethyl- silylchloride (0.91 g) with stirring under ice-cooling. The mixture was stirred at the same temperature overnight. To the reaction mixture were added dichloromethane (50 ml) and water (20 ml) and the organic layer was separated. The organic layer was washed in turn with water and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (100 g) eluting with a mixture of dichloromethane and methanol (19:1,
V/V) . The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- allyloxycarbonyl-2-[5-(t-butyldimethylsilyloxy)methyl-l- methylpyrazol-4-yl]methyl-4-hydroxypyrrolidine (1.29 g) . NMR (CDCI3, δ) : 0.08 (6H, s), 0.84 (9H, s), 1.65-
2.00 (2H, ), 2.45-2.90 (2H, m) , 3.22 (IH, dd, J=4.41, 11.8Hz) , 3.25-3.55 (IH, ) , 3.79 (3H, s), 3.90-4.20 (2H, ) , 4.40-4.60 (4H, m) , 5.05- 5.30 (2H, ), 5.75-6.00 (IH, ), 7.12 (IH, s)
Prepara ion 10-7
To a solution of (2R,4R)-l-allyloxycarbonyl-2-[5-(t- butyldimethylsilyloxy)methyl-1-methylpyrazol-4-yl]methyl- 4-hydroxypyrrolidine (1.29 g) and triethylamine (0.62 ml) in ethyl acetate (20 ml) was added dropwise methanesulfonyl chloride (0.29 ml) under ice-cooling with stirring. After stirring for 1 hour, to the reaction mixture were added water (20 ml) and ethyl acetate (50 ml). The organic layer was successively separated, washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo to give a residue. The residue was dissolved in a mixture of acetonitrile (20 ml) and cone, hydrochloric acid (0.53 ml) and the solution was stirred at ambient temperature for 2 hours. To the reaction mixture was added 28% sodium methoxide-methanol solution (1.22 ml) under ice-cooling with stirring. The resulting precipitates were filtered off. The filtrate was evaporated in vacuo to give a residue. The residue was chromatographed on silica gel (100 g) eluting with a mixture of dichloromethane and methanol (9:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-l-allyloxycarbonyl-2-[5-hydroxymethyl-l- methylpyrazol-4-yl]methyl-4-methylsulfonyloxypyrrolidine (1.10 g) .
NMR (CDC13, δ) : 2.00-2.40 (2H, m) , 2.60-3.00 (2H, m), 3.01 (3H, s), 3.30-3.90 (2H, m) , 3.91 (3H, s), 4.00-4.30 (IH, m), 4.55-4.70 (4H, ) , 4.90- 5.45 (3H, ), 5.80-6.10 (IH, ) , 7.22 (IH, m) APCI-MS : 374 (M+)
Preparation 10-8)
(2R,4S)-l-Allyloxycarbonyl-4-benzoylthio-2-[ (5- hydroxymethyl-1-methylpy azol-4-y1)methyl]pyrrolidine (1.08 g) was obtained in substantially the same manner as that of Preparation 9-7).
NMR (CDC13, δ) : 1.80-1.95 (IH, m) , 2.45-2.80 (2H, m), 2.85-3.60 (4H, m) , 3.90 (3H, s), 3.90-4.20 (3H, m), 4.50-4.70 (4H, m) , 5.15-5.40 (2H, m) , 5.80-6.10 (IH, m), 7.26 (IH, s), 7.30-8.00 (5H, m)
APCI-MS : 416 (M+)
Preparation 11-1)
To a solution of oxalyl chloride (1.43 ml) and dichloromethane (50 ml) was added dropwise dimethyl sulfoxide (2.33 ml) at -40 - -50°C with stirring and the mixture was stirred at the same temperature for 5 minutes. To the solution was added dropwise a solution of (2R,4R)- l-benzyl-4-(t-butyldimethylsilyloxy)-2-(2- hydroxyethyl)pyrrolidine (5 g) in dichloromethane (25 ml) at -40 - -50°C. After 10 minutes with stirring, triethylamine (6,22 ml) was added dropwise to the solution and the mixture was stirred at 0-10°C for 30 minutes. The insoluble material was filtered off and the filtrate was washed successively with IN hydrochloric acid, water, saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo to give a residue. On the other hand, to a solution of 3-(t-butyldimethylsilyloxy)-1- propyne (3.02 ml) in tetrahydrofuran (25 ml) was dropwise added n-butyl lithium (1.62 M in hexane solution) (3.02 ml) at -70°C with stirring and the mixture was stirred at -60 ~ 20°C for 1 hour. The reaction mixture was dropwise added to a solution of the residue obtained above in tetrahydrofuran (15 ml) at -60 - -40°C and the mixture was
stirred at -40 ~ -20°C for 2 hours. To the reaction mixture were added ethyl acetate (100 ml) and water (30 ml) with stirring and the organic layer was separated. The organic layer was washed successively with water and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (200 g) eluting with a mixture of n-hexane and ethyl acetate (1:2, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2S,4R)-1- benzyl-4-t-butyldimethylsilyloxy-2-(5-t-butyldimethyl- silyloxy-2-hydroxy-3-pentynyl)pyrrolidine (5.06 g) .
NMR (CDC13, δ) : 0.01-0.10 (12H, m) , 0.82-0.98
(18H, ), 1.65-2.50 (5H, m) , 2.85-3.45 (3H, m) , 3.58 (IH, d, J=12.6Hz), 4.03 (IH, d, J=12.6Hz),
4.20-4.40 (4H, m) , 4.50-4.65 (IH, m) , 4.70-4.85 (IH, m) , 7.25-7.30 (5H, m) APCI-MS : 504 (M+)
Preparation 11-2)
To a solution of oxalyl chloride (0.96 ml) and dichloromethane (50 ml) was added dropwise dimethyl sulfoxide (1.57 ml) at -40 - -50°C with stirring. After stirring at the same temperature for 5 minutes, to the solution was added dropwise a solution of (2S,4R)-1- benzyl-4-t-butyldimethylsilyloxy-2-(5-t- butyldimethylsilyloxy-2-hydroxy-3-pentynyl)pyrrolidine (5.06 g) in dichloromethane (25 ml) at -40 ~ -50°C. After 10 minutes with stirring, triethylamine (4.19 ml) was added dropwise to the solution and the mixture was stirred at 0-10°C for 30 minutes. The reaction mixture was washed with water, dried over anhydrous magnesium sulfate and evaporated in vacuo to give a residue. To a solution of the residue in ethanol (25 ml) was dropwise added methyl hydrazine (0.54 ml) under ice-cooling with stirring and
the mixture was refluxed for 1.5 hours. The reaction mixture was evaporated in vacuo to give a residue. The residue was dissolved in ethyl acetate (150 ml). The solution was washed successively with water and saturated aqueous sodium chloride, dried over an hydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (150 g) eluting with a mixture of n-hexane and ethyl acetate (2:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-l-benzyl-4-t- butyldimethylsilyloxy-2-[5-(t-butyldimethylsilyloxy)- methyl-l-methylpyrazol-3-yl]methylpyrrolidine (3.74 g) .
NMR (CDC13, δ) : -0.81 (6H, s), 0.01 (6H, s), 0.78 (9H, s), 0.84 (9H, s), 1.55-1.90 (3H, m) , 2.11 (IH, dd, J=6.06, 9.5Hz), 2.40-2.60 (IH, m) ,
2.80-3.15 (3H, m) , 3.26 (IH, d, J=12.9Hz), 3.71 (IH, d, J=12.9Hz), 4.10-4.25 (IH, ) , 4.59 (2H, s), 5.92 (IH, s), 7.20-7.30 (5H, m) APCI-MS : 530 (M+)
Preparation 11-3)
(2R,4R)-l-Allyloxycarbonyl-4-t-butyldimethyl- silyloxy-2-[5-(t-butyldimethylsilyloxy)methy1-1-methyl- pyrazol-3-yl]methylpyrrolidine (3.20 g) was obtained in substantially the same manner as that of Preparation 9-4). NMR (CDCI3, δ) : 0.00 (6H, s), 0.04 (6H, s), 0.81 (9H, s), 0.87 (9H, s), 1.80-2.00 (2H, m) , 2.70- 3.45 (4H, m), 3.80 (IH, s), 4.05-4.30 (2H, ) , 4.50-4.70 (4H, m) , 5.10-5.40 (2H, m) , 5.80-6.10 (2H, m)
APCI-MS : 524 (M+)
Preparation 11-4 )
( 2R, 4R) -l-Allyloxycarbonyl-4-hydroxy-2- ( 5- hydroxymethyl-l-methylpyrazol-3-yl )methylpyrrolidine ( 1.96
- 54 - g) was obtained in substantially the same manner as that of Preparation 10-5).
NMR (CDC13, δ) : 1.80-2.05 (2H, m) , 2.55-3.50 (6H, m), 3.80 (3H, s), 4.10-4.35 (2H, m) , 4.50-4.70 (4H, m), 5.15-5.45 (2H, ) , 5.80-6.05 (2H, m)
APCI-MS : 296 (M+)
Preparation 11-5)
(2R,4R)-l-Allyloxycarbonyl-2-[5-(t-butyldimethyl- silyloxy)methyl-l-methylpyrazol-3-yl]methyl-4- hydroxypyrrolidine (2.65 g) was obtained in substantially the same manner as that of Preparation 10-6).
NMR (CDCI3, δ) : 0.04 (6H, s), 0.85 (9H, s),
1.80-2.05 (2H, m), 2.65-3.60 (4H, m) , 3.76 (3H, s), 4.00-4.30 (2H, m) , 4.50-4.70 (4H, m) ,
5.10-5.40 (2H, m), 5.80-6.10 (2H, m) APCI-MS : 410 (M+)
Preparation 11-6) (2R,4R)-l-Allyloxycarbonyl-2-[5-hydroxymethyl-l- methylpyrazol-3-yl]methyl-4-methylsulfonyloxypyrrolidine (2.05 g) was obtained in substantially the same manner as that of Preparation 10-7).
NMR (CDCI3, δ) : 1.80-2.35 (3H, m) , 2.70-3.00 (2H, m), 2.90 (3H, s), 3.15-3.45 (IH, m) , 3.60-3.70
(4H, m), 4.10-4.30 (IH, ) , 4.45-4.65 (4H, m) , 4.80-4.90 (IH, m), 5.10-5.30 (2H, m) , 5.75-6.00 (2H, m) APCI-MS : 374 (M+)
Preparation 11-7)
(2R,4S)-l-Allyloxycarbonyl-4-benzoylthio-2-(5- hydroxymethyl-l-methylpyrazol-3-yl)methylpyrrolidine (1.49 g) was obtained in substantially the same manner as that of Preparation 9-7).
NMR (CDCI3, δ) : 1.50-2.50 (5H, ) , 2.90-3.15 (2H, m), 3.74 (3H, s), 3.80-4.20 (2H, m) , 4.40-4.60 (4H, m), 5.05-5.35 (2H, m) , 5.70-6.00 (2H, m) , 7.20-7.90 (5H, m) APCI-MS : 416 (M+)
Preparation 12-1)
(2R,4R)-l-Allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-(2-ethoxycarbonyl-l- methylimidazol-5-yl)methylpyrrolidine (15.11 g) was obtained in substantially the same manner as that of Preparation 10-2).
NMR (CDCI3, δ) : 0.00 (6H, s), 0.82 (9H, s), 1.39 (3H, t, d=7.1Hz), 1.75-1.88 (2H, m) , 2.59-2.71 (IH, ), 3.23-3.46 (2H, m) , 3.89 (IH, br) , 3.96
(3H, s), 4.00-4.15 (IH, m) ,.4.18-4.30 (IH, m) , 4.36 (2H, q, J=7.1Hz), 4.56 (2H, br), 5.18-5.33 (2H, m), 5.82-6.02 (IH, ) , 6.90 (IH, s)
Preparation 12-2)
(2R,4R)-1-Allyloxycarbonyl-2-(2-ethoxycarbonyl-l- methylimidazol-5-yl)methyl-4-hydroxypyrrolidine (13.33 g) was obtained in substantially the same manner as that of
Preparation 10-5). This compound was immediately used as the starting compound for the next step.
Preparation 12-3)
(2R,4R)-l-Allyloxycarbonyl-2-(2-ethoxycarbonyl-l- methylimidazol-5-yl)methyl-4-methylsulfonyloxypyrrolidine (13.70 g) was obtained in substantially the same manner as that of Preparation 10-7).
NMR (CDCI3, δ) : 1.43 (3H, t, J=7.1Hz), 1.97-2.07 (IH, m), 2.35-2.45 (IH, m) , 2.70-2.82 (IH, ) , 3.02 (3H, s), 3.36-3.57 (2H, m) , 3.93 (IH, br) ,
3.99 (3H, s), 4.10-4.25 (IH, ) , 4.41 (2H, q, d=7.1Hz), 4.65 (2H, d, J=5.5Hz ) , 5.14-5.37 (3H, m), 5.86-6.05 (IH, m) , 6.95 (IH, s)
Preparation 12-4)
To a solution of (2R,4R)-l-allyloxycarbonyl-2-(2- ethoxycarbonyl-l-methylimidazol-5-yl)methyl-4- methanesulfonyloxypyrrolidine (13.70 g) in ethanol (137 ml) and tetrahydrofuran (67 ml) were added lithium chloride (2.79 g) and sodium borohydride (2.49 g) at an ambient temperature, and the mixture was stirred at 50-55°C for 2 hours. The reaction mixture was poured into ice water and ethyl acetate. The mixture was adjusted to pH 2.6 with 6N hydrochloric acid. After stirring for several minutes, the mixture was again adjusted to pH 9.5-10 with 6N aqueous sodium hydroxide. After separation, the organic layer was dried over magnesium sulfate and evaporated in vacuo. The residue was purified by silica gel chromatography eluting with a mixture of dichlorimethane and methanol (9:1, V/V) to give (2R,4R)-1- allyloxycarbonyl-2-(2-hydroxymethyl-l-methylimidazol-5- yl)methyl-4-methylsulfonylpyrrolidine (11.4 g).
NMR (CDC13, δ) : 1.98-2.04 (IH, m) , 2.30-2.45 (IH, m), 2.59-2.72 (IH, ) , 3.01 (3H, s), 3.24-3.67 (5H, ), 3.77-4.17 (2H, m) , 4.55-4.65 (4H, ) ,
5.10-5.37 (3H, m) , 5.86-6.05 (IH, ) , 6.63 (IH, s)
Preparation 12-5) (2R,4S)-l-Allyloxycarbonyl-4-benzoylthio-2-[ (2- hydroxymethyl-l-methylimidazol-5-yl)methyl]pyrrolidine
(8.94 g) was obtained in substantially the same manner as that of Preparation 9-7).
NMR (CDCI3, δ) : 1.85-1.95 (IH, m) , 2.40-2.60 (IH, ), 2.69-2.90 (IH, ) , 3.30-3.50 (2H, m),
3.60-3.70 (4H, m) , 4.05-4.25 (3H, m) , 4.61-4.64 (4H, m), 5.22-5.37 (2H, ) , 5.86-6.05 (IH, m) , 6.68 (IH, s), 7.15-7.30 (2H, m) , 7.43-7.64 (2H, m), 7.91-7.95 (2H, m)
Preparation 13-1)
To a solution of (2R,4R)-l-benzyl-4-t- butyldimethylsilyloxy-2-[ (l-methylimidazol-5- yl) ethyl]pyrrolidine (46.5 g) in tetrahydrofuran (468 ml) was added 1.64 M n-butyl lithium in n-hexane solution
(88.2 ml) dropwise at -75 65°C. After stirring for 50 minutes, to the mixture was added dropwise ethyl chloroformate (13.8 ml) in tetrahydrofuran (234 ml) at the same temperature. After stirring for 30 minutes, the reaction mixture was poured into saturated sodium hydrogen carbonate. After being extracted with ethyl acetate, the organic layer was washed with brine, dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by silica gel chromatography eluting with a mixture of dichloromethane and methanol (20:1, V/V) to give (2R,4R)-l-benzyl-4-t-butyldimethylsilyloxy-2-[ (2- ethoxycarbonyl-l-methylimidazol-5-yl)methyl]pyrrolidine (39.23 g).
NMR (CDC13, δ) : 0.00 (6H, s), 0.85 (9H, s), 1.43 (3H, t, d=7.lHz), 1.67-1.92 (2H, m) , 2.26-2.34
(IH, m), 2.51-2.63 (IH, m) , 2.81-2.91 (IH, m) , 3.07-3.22 (2H, m) , 3.47 (IH, d, J=13.0Hz), 3.86 (3H, s), 4.00 (IH, d, J=13.0Hz), 4.22-4.30 (IH, m), 4.40 (2H, q, d=7.1Hz), 7.00 (IH, s), 7.16-7.33 (5H, m)
Preparation 13-2)
To a solution of (2R,4R)-l-benzyl-4-t- butyldimethylsilyloxy-2-[ (2-ethoxycarbonyl-l- methylimidazol-5-yl)methyl]pyrrolidine (19.77 g) in
methanol (480 ml) was added 28% aqueous ammonium hydroxide (120 ml) at an ambient temperature, and the mixture was stayed at the same temperature for 4 days . Methanol was removed under reduced pressure, and to the residue was added ethyl acetate. After separation, the organic layer was dried over magnesium sulfate and evaporated under reduced pressure. The residue was purified by silica gel chromatography eluting with a mixture of dichloromethane and methanol (20:1, V/V) to give (2R,4R)-l-benzyl-4-t- butyldimethylsilyloxy-2-[ (2-carbamoyl-l-methylimidazol-5- yl)methyl]pyrrolidine (21.98 g).
NMR (CDC13, δ) : 0.00 (6H, s), 0.86 (9H, s), 1.68- 1.93 (2H, m), 2.26-2.33 (IH, ) , 2.49-2.61 (IH, m), 2.80-2.89 (IH, ) , 3.03-3.21 (2H, m) , 3.46 (IH, d, J=13.0Hz), 3.91 (3H, s), 3.99 (IH, d,
J=13.0Hz), 4.25-4.30 (IH, ) , 6.87 (IH, s), 7.15-7.33 (5H, m)
Preparation 13-3) (2R,4R)-l-Allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-[ (2-carbamoyl-l-methylimidazol-5- yl)methyl]pyrrolidine (14.02 g) was obtained in substantially the same manner as that of Preparation 10-2). NMR (CDCI3, δ) : 0.04 (6H, s), 0.85 (9H, s), 1.70-
1.95 (2H, ), 2.63-2.75 (IH, m) , 3.24-3.30 (IH, m), 3.41 (2H, d, J=4.0Hz) , 3.93-3.97 (3H, ) , 4.12-4.25 (2H, m) , 4.62 (2H, ) , 5.21-5.36 (3H, m), 5.88-5.97 (IH, ) , 6.81 (IH, s), 7.16 (IH, br)
Preparation 13-4)
(2R,4R)-l-Allyloxycarbonyl-2-[ (2-carbamoyl-l- methylimidazol-5-yl)methyl]-4-hydroxypyrrolidine (10.23 g) was obtained in substantially the same manner as that of
Preparation 10-5).
This compound was immediately used as the starting compound for the next step.
Preparation 13-5)
(2R,4R)-l-Allyloxycarbonyl-2-[ (2-carbamoyl-l- methylimidazol-5-yl)methyl]-4-methylsulfonyloxypyrrolidine (10.49 g) was obtained in substantially the same manner as that of Preparation 10-7). NMR (CDC13, δ) : 1.90-2.10 (IH, ) , 2.32-2.50 (IH, m), 2.68-2.80 (IH, m) , 3.02 (3H, s), 3.35-3.58 (2H, m), 3.90-4.25 (5H, m) , 4.65 (2H, d, J=5.6Hz), 5.16-5.38 (4H, m) , 5.88-6.02 (IH, m) , 6.82 (IH, s), 7.16 (IH, br)
Preparation 13-6)
(2R,4S)-l-Allyloxycarbonyl-4-benzoylthio-2-[ (2- carbamoyl-l-methylimidazol-5-yl)methyl]pyrrolidine (14.08 g) was obtained in substantially the same manner as that of Preparation 9-7).
NMR (CDCI3, δ) : 1.80-1.95 (IH, ) , 2.45-2.65 (IH, m), 2.78-2.95 (IH, m) , 3.35-3.50 (2H, m) , 3.93- 4.20 (6H, m), 4.64 (2H, d, J=5.7Hz), 5.23-5.39 (3H, ), 5.86-6.03 (IH, m) , 6.84 (IH, s), 7.15-7.30 (3H, m) , 7.41-7.65 (2H, m) , 7.91-8.02
(2H, m)
Preparation 14-1)
To a solution of ethylene glycol (10 g) in tetrahydrofuran (250 ml) was added sodium hydride (about 60% oil suspension, 64 g) at 0-5°C and stirred for 1.5 hours at room temperature. The mixture was cooled to 0- 5°C and a solution of t-butyldimethylsilyl chloride (24.3 g) in tetrahydrofuran (50 ml) was added dropwise. After stirring for 2 hours at room temperature, ice-water (150
ml) was added. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried over magnesium sulfate and evaporated under reduced pressure. The resulting residue was column chromatographed on silica gel (800 g, eluting with n-hexane - ethyl acetate = 5:1) to give 2-t- butyldimethylsilyloxyethanol (24 g) .
IR (Neat) : 3320, 1455, 1250, 1108 cm-1 NMR (CDC13, δ) : 0.09 (6H, s) , 0.91 (9H, s),
3.60-3.78 (4H, m) APCI-MS (m/z) : 177 (MH+)
Preparation 14-2) To a solution of 2-t-butyldimethylsilyloxyethanol
(1.0 g) and 2,6-lutidine (0.794 ml) in dichloromethane (20 ml) was added trifluoromethanesulfonic anhydride (1.05 ml) at 0-5°C. After stirring for 15 minutes, the reaction mixture was washed with water, 0.5N-hydrochloric acid (x 3), water (x 2) and brine, dried over magnesium sulfate and evaporated under reduced pressure to give 2-t- butyldimethylsilyloxyethyl trifluoromethanesulfonate (1.54 g).
IR (Neat) : 2920, 1400, 1240, 1200 cm-1 NMR (CDCI3, δ) : 0.09 (6H, s), 0.90 (9H, s), 3.92
(2H, t, J=4.4Hz), 4.55 (2H, t, J=4.4Hz)
Preparation 15-1)
To a solution of diethyl l-ethoxycarbonyl-2-methyl- 1,4-dihydropyridine-4-phosphonate (7.1 g) in tetrahydrofuran (50 ml) was added dropwise n-butyllithium (1.64 N in n-hexane) (15 ml) under -45°C. After stirring for 30 minutes at -50°C, a solution of (2S,4R)-1- allyloxycarbonyl-4-t-butyldimethylsilyloxy-2- (iodomethyl)pyrrolidine (5 g) in tetrahydrofuran (10 ml)
under -50°C. After stirring for 1 hour at -50°C, the reaction mixture was allowed to warm to room temperature and then quenched with water and ethyl acetate. The aqueous layer was separated and extracted twice with ethyl acetate. The combined organic layer was washed with water and brine, dried over magnesium sulfate and evaporated under reduced pressure to give (2S,4R)-l-allyloxycarbonyl- 4-t-butyldimethylsilyloxy-2-[ (l-ethoxycarbonyl-2-methyl-4- diethγlphosphoryl-1,4-dihydropyridin-4- yl)methyl]pyrrolidine (8.76 g) . IR (Neat) : 1690 cm"1
NMR (CDC13, δ) : 0.03 (6H, s), 0.78 (9H, s), 1.20- 1.40 (9H, m) , 1.50-1.80 (4H, ) , 4.33 (3H, d, J=5Hz), 3.35 (2H, ) , 3.99-4.54 (8H, ) , 4.54 (2H, m), 5.00-5.40 (2H, m) , 5.80-6.10 (IH, m) ,
6.90-7.00 (IH, m) MS : (M+l) = 601
Preparation 15-2) To a solution of (2S,4R)-l-allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-[ (l-ethoxycarbonyl-2-methyl-4- diethylphosphoryl-1,4-dihydropyridin-4- yl)methyl]pyrrolidine (8.76 g) in tetrahydrofuran (88 ml) was added dropwise n-butyllithium (1.64 N in n-hexane) (26.7 ml) under -65°C for 30 minutes. After stirring for 1 hour, the reaction mixture was allowed to warm to -30°C and quenched with water and ethyl acetate. The aqueous layer was separated and extracted twice with ethyl acetate. The combined organic layer was washed with water - and brine, dried over magnesium sulfate and evaporated under reduced pressure. The residue was column chromatographed on silica gel (eluent : n-hexane - ethyl acetate = 4:1-2:1) to give (2R,4R)-l-allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-(2-metnylpyridin-4- ylmethyl)pyrrolidine (355 g) .
IR (Neat) : 1690, 1600, 1400 cm-1
NMR (CDC13, δ) : 0.02 (6H, s), 0.88 (9H, s), 1.20- 1.45 (2H, ), 1.60-2.00 (2H, m) , 2.55 (3H, s), 3.35 (2H, ), 4.00-4.35 (2H, ) , 4.66 (2H, d, J=5.3Hz), 5.20-5.45 (2H, ) , 5.87-6.04 (IH, m),
6.90-7.05 (2H, ) , 8.39 (IH, d, J=5.0Hz) MS : (M+l) = 391
Preparation 15-3) (2R,4R)-1-Allyloxycarbonyl-4-hydroxy-2-( 2- methylpyridin-4-ylmethyl)pyrrolidine (2.7 g) was obtained in the same procedure as that of Preparation 10-5). IR (Neat) : 3350, 1690, 1670, 1605 cm-1 NMR (CDCI3, δ) : 1.70-2.10 (2H, ) , 2.52 (3H, s), 2.65-2.80 (IH, m) , 3.10-3.80 (3H, ) , 4.20-4.40
(2H, m), 4.65 (2H, d, J=5.5Hz), 5.20-5.45 (2H, m), 5.85-6.05 (IH, m) , 6.80-7.00 (2H, m) , 8.38 (IH, d, J=5.0Hz) MS : (M+l) = 277
Preparation 15-4)
(2R,4R)-l-Allyloxycarbonyl-4-methylsulfonyloxy-2-( 2- methylpyridin-4-ylmethyl)pyrrolidine (6.93 g) was obtained in the same procedure as that of Preparation 10-7). The product was allowed to transform to the next reaction.
Preparation 15-5)
(2R,4S)-l-Allyloxycarbonyl-4-benzoylthio-2-(2- methylpyridin-4-ylmethyl)pyrrolidine (5.61 g) was obtained in the same procedure as that of Preparation 9-7). IR (Neat) : 1690, 1670, 1610 cm"1
NMR (CDCI3, δ) : 1.70-2.00 (2H, m) , 2.52 (3H, s), 2.72 (IH, dd, J=9.7, 12.9Hz), 3.20-3.60 (2H, ) ,
4.00-4.40 (2H, m), 4.60-4.70 (2H, m) , 5.20-5.45 (2H, m), 5.85-6.10 (IH, m) , 6.80-7.10 (2H, m) , 7.40-7.70 (3H, ) , 7.90-8.00 (2H, m) , 8.40 (IH, d, J=5.2Hz) MS : (M+l) = 397
Preparation 16-1) lH-l,2,3-Triazole (100 g) and iodomethane (90 ml) were heated at 40°C for two hours. After cooling, the mixture was evaporated. The residue was column chromatographed on silica gel (1.2 kg, eluting with chloroform-methanol = 20:1), then the eluate was distilled in vacuo to afford l-methyl-l,2,3-triazole (23.46 g) . bp 71-72°C/6 mitiHg IR (Neat) : 3450, 1651, 1489, 1311, 1265, 1215 cnr1
NMR (CDC13, δ) : 4.14 (3H, s), 7.55 (IH, s),
7.72 (IH, s) APCI-MS (m/z) : 84 (MH+)
Preparation 16-2)
To a solution of 1-methyl-l,2,3-triazole (23.4 g) in tetrahydrofuran (350 ml) was added dropwise n-butyllithium (1.6 N) in n-hexane (194 ml) keeping the temperature below -50°C. The mixture was stirred for 30 minutes then warmed to -20°C over 30 minutes. The mixture was cooled to
-50°C. To the mixture was added a solution of (2S,4R)-1- allyloxycarbonyl-2-formyl-4-t- butyldimethylsilyloxypyrrolidine (88 g) in tetrahydrofuran (90 ml) keeping the temperature below -50°C. The reaction mixture was warmed to -20°C over one hour, and stirred for 30 minutes at the same temperature, then stirred for one hour at 0-5°C. The mixture was quenched with ice water (200 ml) and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (x 2), and the combined organic layer was washed with water and brine,
dried over magnesium sulfate and evaporated under reduced pressure. The residue was column chromatographed on silica gel (2.5 kg, eluting with ethyl acetate - n-hexane = 3:1) to give l-[ (2S,4R)-l-allyloxycarbonyl-4-(t- butyldimethylsilyloxy)pyrrolidin-2-yl]-1-(1-methyl-l,2,3- triazol-5-yl)methanol (68.6 g) .
IR (Nujol) : 3197, 1699, 1406, 1255, 1192 cm"1 NMR (CDC13, δ) : 0.02, 0.03, 0.05 (total 6H, each s), 0.84, 0.85 (total 9H, each s), 1.43-1.95 (2H, m), 3.27-3.73 (2H, m) , 4.03-4.90, 5.19-5.40
(total 10H, each m), 5.85-6.05 (IH, m) , 7.50, 7.53 (total IH, each s) APCI-MS (m/z) : 397 (MH+)
Preparation 16-3)
(2S,4R)-l-Allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-[1-(1-methyl-l,2,3-triazol-5-yl)- l-[ (methylthio)thiocarbonyloxy]methyl]pyrrolidine (85.0 g) was obtained in the same procedure as Preparation 4-1). IR (Neat) : 1703, 1651, 1404, 1201 cm"1
NMR (CDCI3, δ) : 0.02-0.08 (6H, m) , 0.84, 0.87 (total 9H, each s), 1.82-2.50 (2H, m) , 2.56, 2.58 (total 3H, each s), 2.85-3.00, 3.30-3.60 (total 2H, each m), 3.95-4.30 (4H, m) , 4.48-4.70 (3H, m), 5.15-5.38 (2H, m) , 5.80-6.05 (IH, m) ,
6.90-7.30 (IH, m), 7.59, 7.60 (total IH, each s) APCI-MS (m/z) : 487 (MH+)
Preparation 16-4) (2R,4R)-1-Allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-[(1-methyl-l,2,3-triazol-5- yl)methyl]pyrrolidine (65.7 g) was obtained in the same procedure as Preparation 4-2).
IR(Neat) : 1701, 1651, 1404, 1111 cm"1 NMR (CDCI3, δ) : 0.03 (6H, s), 0.84 (9H, s), 1.62-
2.06 (2H, ), 2.80-2.95 (IH, ) , 3.20-3.56 (3H, m), 3.94-4.30 (5H, m) , 4.57-4.68 (2H, m) , 5.18-
5.38 (2H, ), 5.85-6.05 (IH, ) , 7.45 (IH, s) APCI-MS (m/z) : 381 (MH+)
Preparation 16-5)
(2R,4R)-l-Allyloxycarbonyl-4-hydroxy-2-[ (1-methyl- l,2,3-triazol-5-yl)methyl]pyrrolidine (43.6 g) was obtained in the same procedure as Preparation 6-4). IR (Neat) : 3400, 1691, 1410 cπT1
NMR (CDC13, δ) : 1.73-1.87 (IH, ) , 2.00-2.20 (IH, m), 2.20-2.32 (IH, m) , 2.80-2.95 (IH, m) , 3.10-
3.76 (3H, ), 3.92-4.10 (3H, m) , 4.10-4.43 (2H, m), 4.56-4.70 (2H, ), 5.18-5.38 (2H, ) , 5.85- 6.05 (IH, m), 7.45 (IH, s)
APCI-MS (m/z) : 267 (MH+)
Preparation 16-6)
To a solution of (2R,4R)-l-allyloxycarbonyl-4- hydroxy-2-{1-methy1-1,2,3-triazol-5-yl)methyl}pyrrolidine (43.6 g), triphenylphosphine (55.9 g) in tetrahydrofuran (520 ml) was added diethyl azodicarboxylate (33.6 ml) at -30°C. After 30 minutes thiobenzoic acid (27.2 ml) was added. Then the mixture was warmed up to room temperature over 30 minutes and stirred for 3 hours. After evaporation of tetrahydrofuran, the resulting residue was diluted with ethyl acetate (1 β), washed with saturated sodium bicarbonate solution (100 ml x 3), water, and brine, dried over magnesium sulfate and evaporated under reduced pressure. The residue was column chromatographed on silica gel (3 kg, eluting with ethyl acetate - n-hexane = 3:1) to give (2R,4S)-l-allyloxycarbonyl-4-benzoylthio-2- {(1-methyl-l,2,3-triazol-5-yl)methyl}pyrrolidine (52.3 g) . IR (Neat) : 1701, 1664, 1404, 1207 cm-1 NMR (CDCI3, δ) : 1.73-1.93 (IH, m) , 2.48-2.70 (IH,
m), 2.99 (IH, dd, J=14.7, 9.7Hz), 3.20-3.55 (2H, ), 3.85-4.30 (6H, m) , 4.55-4.70 (2H, ) , 5.18- 5.40 (2H, m) , 5.85-6.05 (IH, m) , 7.35-8.18 (6H, m) APCI-MS (m/z) : 387 (MH+)
Preparation 17-1)
To a solution of (2R,4R)-l-allyloxycarbonyl-2-[2-(2- phthalimidoimidazol-l-yl)ethyl]-4- methanesulfonyloxypyrrolidine (13.7 g) in dichloromethane (140 ml) was added methyl trifluoromethanesulfonate (3.81 ml) with stirring under ice-cooling. The mixture was stirred at the same temperature for 1 hour. To the reaction mixture was added Amberlist A-26 (Cl~ type, Trademark, made by Rohm and Haas Co., Ltd.) (70 ml) and the mixture was stirred for 5 minutes. The resin was filtered off and washed with a mixture of ethanol (50 ml) and dichloromethane (50 ml). The filtrate and washing were collected and evaporated in vacuo to give a residue. To a solution of the residue in ethanol (140 ml) was added hydrazine hydrate (4.1 ml) at ambient temperature and the mixture was stirred at the same temperature for 1 hour. The resulting precipitates were filtered off and washed with ethanol (20 ml). The filtrate and washing were combined and evaporated in vacuo to give a residue. The residue was dissolved in ethyl acetate (100 ml). The solution was washed three times with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. To a solution of the residue in a mixture of tetrahydrofuran (100 ml) and water (100 ml) was added dropwise allyl chloroformate (4.2 ml) keeping the pH to 10-13 with aqueous 4N sodium hydroxide with stirring under ice-cooling. The mixture was stirred at the same temperature for 1 hour. The reaction mixture was extracted eight times with a mixture of ethyl acetate (60
ml) and tetrahydrofuran (60 ml) and the extract was evaporated in vacuo to give a residue. The residue was chromatographed on silica gel (300 g) eluting with a mixture of dichloromethane and methanol (9:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- allyloxycarbonyl-2-[2-(2-allyloxycarbonylimino-3- methylimidazolin-1-yl)ethyl]-4- ethylsulfonyloxypyrrolidine (6.79 g) . NMR (CDC13, δ) : 1.80-2.60 (5H, m) , 3.05 (3H, s),
3.52 (3H, s), 3.55-3.65 (IH, ) , 3.70-4.15 (4H, m), 4.50-4.65 (4H, ) , 5.10-5.40 (5H, m) , 5.75- 6.15 (2H, ), 6.57 (d), 6.81 (broad s) (total 2H) APCI-MS : 457 (M+)
Preparation 17-2)
(2R,4S)-1-Allyloxycarbonyl-2-[2-(2- allyloxycarbonylimino-3-methylimidazolin-l-yl)ethyl]-4- benzoylthiopyrrolidine (5.02 g) was obtained in substantially the same manner as that of Preparation 9-7). NMR (CDCI3, δ) : 2.30-2.80 (2H, m) , 3.20-3.40 (IH, m), 3.51, 3.46 (3H, each s), 3.80-4.30 (5H, m) , 4.50-4.65 (4H, m) , 5.05-5.40 (4H, m) , 5.80-6.10 (2H, m), 6.50-7.00 (2H, m) , 7.30-8.10 (5H, m) APCI-MS : 499 (M+)
Preparation 18-1)
To a solution of oxalyl chloride (12.2 ml) in dichloromethane (400 ml) was added dropwise dimethyl sulfoxide (19.8 ml) at -40 ~ -50°C with stirring and the mixture was stirred at the same temperature for 5 minutes.
To the solution was added dropwise a solution of (2S,4R)- l-allyloxycarbonyl-4-t-butyldimethylsilyloxy-2-
(hydroxymethyl)pyrrolidine (40 g) in dichloromethane (160 ml) at -40 - -50°C. After 10 minutes with stirring,
triethylamine (53 ml) was added dropwise to the solution and the mixture was stirred at 0-10°C for 30 minutes. The insoluble material was filtered off and the filtrate was washed successively with IN hydrochloric acid, water, saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo to give a residue. On the other hand, to a solution of 4-bromo-l-{3-(t- butyldimethylsilyloxy)propyl}pyrazole (52.6 g) in tetrahydrofuran (500 ml) was added dropwise n-butyl lithium (1.62 M hexane solution) (109.6 ml) at -40°C with stirring and the mixture was stirred at -10°C ~ 15°C for 1 hour. The reaction mixture was dropwise added to a solution of the residue obtained above in tetrahydrofuran (200 ml) at -40°C and the mixture was stirred at -40°C - -20°C for 1 hour. To the reaction mixture were added ethyl acetate (2 I) and water (500 ml) with stirring and the organic layer was separated. The layer was washed in turn with water and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (1 kg) eluting with a mixture of n-hexane and ethyl acetate (2:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2S,4R)-l-allyloxycarbonyl-4-t-butyldimethylsilyloxy- 2-[l-{l-(3-t-butyldimethylsilyloxypropyl)pyrazol-4-yl}-l- hydroxy ethyl]pyrrolidine (11.93 g) .
NMR (CDC13, δ) : 0.00-0.03 (12H, m) , 0.74-0.86
(18H, m), 1.55-1.65 (2H, m) , 1.90-2.10 (2H, m) , 3.20-4.30 (10H, m) , 4.50-4.70 (3H, m) , 5.10-5.40
(3H, ), 5.80-6.05 (IH, m) , 7.25-7.60 (2H, m)
Preparation 18-2)
(2S,4R)-1-Allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-[l-{l-(3-t-
butyldimethylsilyloxypropyl)pyrazol-4-yl}-1- { (methylthio)thiocarbonyloxy}methyl]pyrrolidine (5.47 g) was obtained in substantially the same manner as that of Preparation 4-1). This compound was immediately used as the starting compound for the next step.
Preparation 18-3)
(2R,4R)-1-Allyloxycarbonyl-4-t- butyldimethylsilyloxy-2-[l-(3-t- butyldimethylsilyloxypropyl)pyrazol-4-yl]methylpyrrolidine (3.35 g) was obtained in substantially the same manner as that of Preparation 4-2).
NMR (CDC13, δ) : 0.01 (6H, s), 0.03 (6H, s), 0.80 (9H, s), 0.88 (9H, s), 1.60-2.10 (4H, m) , 2.60-
2.85 (2H, m), 3.15-3.60 (4H, m) , 4.00-4.20 (4H, m), 4.50-4.65 (2H, m) , 5.10-5.35 (2H, m) , 5.80- 6.00 (IH, m), 7.12 (IH, s), 7.25 (IH, ε)
Preparation 18-4)
(2R,4R)-1-Allyloxycarbonyl-4-hydroxy-2-[1-(3- hydroxypropyl)pyrazol-4-yl]methylpyrrolidine (1.74 g) was obtained in substantially the same manner as that of Preparation 10-5). NMR (CDCI3, δ) : 1.70-2.10 (3H, ) , 2.45-3.65 (9H, m), 4.05-4.30 (4H, ) , 4.50-4.70 (2H, m) , 5.15- 5.45 (2H, m), 5.80-6.10 (IH, m) , 7.20 (IH, s), 7.28 (IH, s) APCI-MS : 310 (M+)
Preparation 18-5)
(2R,4R)-1-Allyloxycarbonyl-2-[1-(3-t- butyldimethylsilyloxypropyl)pyrazol-4-yl]methyl-4- hydroxypyrrolidine (2.66 g) was obtained in substantially the same manner as that of Preparation 10-6).
NMR (CDCI3, δ) : 0.02 (6H, s), 0.88 (9H, s), 1.70- 2.10 (5H, m), 2.65-2.90 (2H, ) , 3.23 (IH, dd, J=4.36, 11.9Hz), 3.40-3.60 (3H, m) , 4.05-4.30 (4H, m), 4.50-4.65 (2H, m) , 5.10-5.40 (2H, m) , 5.80-6.05 (IH, m) , 7.12 (IH, s), 7.25 (IH, s)
APCI-MS : 424 (M+)
Preparation 18-6)
(2R,4R)-l-Allyloxycarbonyl-2-[1-(3-hydroxypropyl)- pyrazol-4-yl]methyl-4-methylsulfonyloxypyrrolidine (1.44 g) was obtained in substantially the same manner as that of Preparation 10-7).
NMR (CDCI3, δ) : 1.90-2.15 (3H, m) , 2.20-2.45 (IH, m), 2.75-2.95 (3H, m) , 3.01 (3H, s), 3.20-4.00 (4H, m), 4.10-4.30 (3H, m) , 4.55-5.50 (5H, ) ,
5.80-6.10 (IH, m), 7.21 (IH, s), 7.29 (IH, s) APCI-MS : 388 (M+)
Preparation 18-7) {2R,4S)-l-Allyloxycarbonyl-4-benzoylthio-2-[1-(3- hydroxypropyDpyrazol-4-yl]methylpyrrolidine (1.75 g) was obtained in substantially the same manner as that of Preparation 9-7).
NMR (CDCI3, δ) : 1.70-2.10 (4H, m) , 2.35-2.90 (3H, m), 3.05-3.25 (IH, m) , 3.50-3.65 (2H, ) , 3.90-
4.30 (5H, m), 4.50-4.70 (2H, m) , 5.20-5.45 (2H, ), 6.80-6.15 (IH, m) , 7.20-7.95 (7H, )
Preparation 19-1) To a solution of 4-formylimidazole (20 g) and bis(trimethylsilyDacetamide (102.9 ml) in tetrahydrofuran (200 ml) was added methyl iodide (50.9 ml) with stirring at ambient temperature and the mixture was stirred at the same temperature for 6 hours. To the reaction mixture was added water (50 ml) and the mixture was concentrated in
vacuo to give a residue. The residue was chromatographed on silica gel (500 g) eluting with a mixture of chloroform and methanol (9:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give l-methyl-5-formylimidazole (5.67 g) .
NMR (CDC13, δ) : 3.96 (3H, s), 7.66 (IH, s), 7.80
(IH, s), 9.78 (IH, s)
Preparation 19-2) To a solution of l-methyl-5-formylimidazole (6.38 g) in a mixture of methanol (60 ml) and tetrahydrofuran (60 ml) was added portionwise sodium borohydride (2.19 g) under ice-cooling with stirring. The mixture was stirred at the same temperature for 1 hour. The reaction mixture was adjusted to pH 8 with 6N hydrochloric acid and evaporated in vacuo. The resulting residue was chromatographed on silica gel (200 g) eluting with a mixture of chloroform, methanol and cone, ammonia (9:1:0.1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give 1- methyl-5-hydroxymethylimidazole (5.04 g) .
NMR (CDCI3, δ) : 3.65 (3H, s) , 4.55 (2H, s), 4.87 (IH, broad s), 6.76 (IH, s), 7.30 (IH, s)
Preparation 19-3)
To a solution of l-methyl-5-hydroxymethylimidazole (5.04 g) in benzene (50 ml) were added thionyl chloride (4.27 ml) and N,N-dimethylformamide (0.2 ml) at 0-5°C with stirring and the mixture was stirred at ambient temperature for 2 hours. To the reaction mixture was added diisopropyl ether (100 ml) with stirring. The supernatant solution was removed by decantation and the resulting residue was washed twice with diisopropyl ether (50 ml) and dried in vacuo for 2 hours to give l-methyl-5- chloromethylimidazole hydrochloride (8.12 g) .
This compound was immediately used as the starting compound for the next step.
Preparation 19-4) solution of l-methyl-5-chloromethγlimiazole hydrochloride (8.12 g) , triphenylphosphine (12.8 g) and sodium iodide (50 mg) in N,N-dimethylformamide (50 ml) was stirred at 80-90°C for 4 hours and cooled under ice-water. The resulting precipitates were collected by filtration and washed successively with N,N-dimethylformamide (20 ml), ethyl acetate (20 ml) and diisopropyl ether (100 ml) and then dried in vacuo to give (l-methylimidazol-5- yDmethyltriphenylphosphonium chloride hydrochloride (8.52 g). NMR (DMSO-d6, δ) : 3.28 (3H, s), 5.75 (2H, d,
J=14.9Hz), 7.21 (IH, s), 7.70-8.10 (15H, m) , 9.10 (IH, s)
Preparation 19-5) To a solution of oxalyl chloride (1.80 ml) in dichloromethane (70 ml) was dropwise added dimethyl sulfoxide (3.08 ml) at -40 ~ -50°C with stirring and the mixture was stirred at the same temperature for 5 minutes. To the solution was added dropwise a solution of (2S,4R)- l-benzyloxycarbonyl-4-(t-butyldimethylsilyl)oxy-2- hydroxymethylpyrrolidine (7.2 g) in dichloromethane (35 ml) at -40 - -50°C. After 10 minutes with stirring, triethylamine (8.23 ml) was added dropwise to the solution and then the mixture was stirred at ambient temperature for 30 minutes. The insoluble material was filtered off and the filtrate was washed successively with IN hydrochloric acid, water, saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo to give a residue. On the other hand, to a solution of
(l-methylimidazol-5-yl)methyltriphenylphosphonium chloride hydrochloride (8.51 g) in a mixture of tetrahydrofuran (40 ml) and dimethyl sulfoxide (40 ml) was added portionwise potassium t-butoxide (4.42 g) at 0-5°C with stirring and then stirred at the same temperature for 30 minutes. The reaction mixture was dropwise added to a solution of the residue obtained above in tetrahydrofuran (70 ml) at 0°C and the mixture was stirred at the same temperature for 2 hours. To the reaction mixture was added ethyl acetate. The organic layer was separated and then washed with successively with IN hydrochloric acid, saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was dissolved in methanol (100 ml). To the solution was added cone, hydrochloric acid (4.92 ml) with stirring at ambient temperature and the mixture was allowed to stand at the same temperature overnight. To the reaction mixture was added 28% sodium methoxide-methanol solution (11.4 ml) under ice-cooling with stirring and the insoluble material was filtered off. The filtrate was evaporated in vacuo to give a residue. The residue was chromatographed on silica gel (200 g) eluting with a mixture of chloroform and methanol (9:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2S,4R)-l-benzyloxycarbonyl-4-hydroxy-2-[2-(1- methylimidazol-5-yl)vinyl]pyrrolidine (6.53 g) .
NMR (CDC13, δ) : 1.80-2.10 (IH, m) , 2.15-2.45 (IH, m), 3.10-3.80 (7H, m) , 4.40-6.40 (5H, m) , 7.07 (IH, s), 7.10-7.45 (6H, m)
Preparation 19-6)
To a solution of (2S,4R)-l-benzyloxycarbonyl-4- hydroxy-2-[2-(1-methylimidazol-5-yl)vinyl]pyrrolidine (6.52 g) in a mixture of ethyl acetate (70 ml) and triethylamine (3.61 ml) was added dropwise methanesulfonyl
/10520
- 84 - chloride (1.70 ml) under ice-cooling with stirring and the mixture was stirred at the same temperature for 1 hour. To the reaction mixture were added ethyl acetate (100 ml) and water (50 ml). The organic layer was washed successively with saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (150 g) eluting with a mixture of chloroform and methanol (19:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2S,4R)-1- benzyloxycarbonyl-4-methylsulfonyloxy-2-[2-(l- methylimidazol-5-yl)vinyl]pyrrolidine (6.92 g) .
NMR (CDC13, δ) : 1.90-2.20 (IH, ) , 2.45-2.70 (IH, ), 3.04 (3H, s), 3.10-4.15 (5H, m) , 4.50-6.50
(5H, ), 7.00-7.50 (7H, )
Preparation 19-7)
A solution of (2S,4R)-l-benzyloxycarbonyl-4- methylsulfonyloxy-2-[2-(l-methylimidazol-5- yl)vinyl]pyrrolidine (6.91 g), cone, hydrochloric acid (2.84 ml) and 10% palladium on carbon (50% wet) (4.0 g) in methanol (140 ml) was stirred under atmospheric pressure of hydrogen at ambient temperature for 4 hours. The catalyst was filtered off and filtrate was concentrated in vacuo. The resulting residue was dissolved in a mixture of tetrahydrofuran (70 ml) and water (70 ml). To the solution was added dropwise allyloxycarbonyl chloride (2.35 ml) keeping the pH to 8-10 with 4N sodium hydroxide under ice-cooling and then stirred at the same temperature for 1 hour. The reaction mixture was concentrated in vacuo to give a residue. The residue was dissolved in ethyl acetate (150 ml) and then the solution was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The
resulting residue was chromatographed on silica gel (200 g) eluting with chloroform and methanol (9:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4R)-1- allyloxycarbonyl-4-methylsulfonyloxy-2-[2-(l- methylimidazol-5-yl)ethyl]pyrrolidine (6.10 g) .
NMR (CDC13, δ) : 1.65-2.15 (2H, ) , 2.20-2.70 (4H, m), 3.04 (3H, s), 3.57 (4H, broad s), 3.85-4.20 (2H, ), 4.61 (2H, broad d, J=5.22Hz), 5.20-5.40 (3H, ), 5.80-6.10 (IH, ) , 6.81 (IH, s), 7.44
(IH, s)
Preparation 19-8)
To a solution of potassium t-butoxide (2.49 g) in N,N-dimethylfθ2_τnamide (30 ml) was added dropwise thioacetic acid (1.58 ml) with stirring at -10 ~ -5°C and the mixture was stirred at -5 ~ 0°C for 10 minutes. To a solution of (2R,4R)-l-allyloxycarbonyl-4- methylsulfonyloxy-2-[2-(l-methylimidazol-5- yl)ethyl]pyrrolidine (6.09 g) in N,N-dimethyIformamide (60 ml) was added the mixture obtained above with stirring at ambient temperature. The mixture was stirred at 80-90°C for 3 hours . The reaction mixture was poured into ice- water (200 ml) and extracted three times with ethyl acetate (200 ml). The extract was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (200 g) eluting with a mixture of chloroform and methanol (19:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give (2R,4S)-4-acetylthio-l- allyloxycarbonyl-2-[ 2-(l-methylimidazol-5- yl)ethyl]pyrrolidine (4.23 g).
NMR (CDCI3, δ) : 1.55-1.90 (2H, m) , 2.34 (3H, s), 2.34-2.70 (4H, m) , 3.20 (IH, dd, J=7.49,
- 36 -
11.3Hz), 3.57 (3H, s), 3.70-4.20 (3H, m) , 4.58 v'2H, broad d, J=5.32Hz), 5.15-5.40 (2H, m) , 5.80-6.10 (IH, m), 6.81 (IH, s), 7.44 (IH, s)
Preparation 20-1)
To a solution of 3-picoline (77.9 ml) in acetonitrile (700 ml) were added ethyl chloroformate (76.5 ml) at -20°C. After stirring at 0°C for 40 minutes, to the mixture was added triethyl phosphite (137.2 ml) at -20°C. After stirring at room temperature for 15 hours, the mixture was evaporated to give diethyl 1- ethoxycarbonyl-3-methyl-l,4-dihydropyridine-4-phosphonate (251 g).
IR (Neat) : 1718 c "1 NMR (CDC13, δ) : 1.2-1.4 (9H, ) , 1.8-1.9 (3H, m) ,
3.2-3.4 (IH, ), 4.0-4.3 (6H, m) , 4.8-5.1 (IH, m) , 6.8-7.0 (2H, m)
Preparation 20-2) (2R,4R)-l-Allyloxycarbonyl-4-t-butyldimethylsiloxy-
2-(3-methyl-4-pyridylmethyl)pyrrolidine was obtained in 42% yield in substantially the same manner as that of Preparation 5-1).
IR (Neat) : 1716 cm-1 NMR (CDCI3, δ) : 0.02 (6H, s), 0.84 (9H, s), 1.7-
1.9 (2H, ), 2.3-2.7 (2H, m) , 3.2-3.5 (3H, m) , 4.1-4.4 (2H, m), 4.6-4.8 (2H, m) , 5.2-5.4 (2H, m), 5.8-6.1 (IH, m), 7.0-7.1 (IH, m) , 8.3-8.4 (2H, m) FAB-MS : 391 (M+l)
Preparation 20-3)
(2R,4R)-l-Allyloxycarbonyl-4-hydroxy-2-(3-methyl-4- pyridylmethyl)pyrrolidine was obtained in 89% yield in substantially the same manner as that of
Preparation 10-5).
NMR (CDC13, δ) : 1.7-2.9 (7H, m) , 3.2-3.8 (2H, m) , 4.0-4.8 (4H, m), 5.1-5.5 (2H, m) , 5.8-6.1 (IH, ), 7.01 (IH, d, J=4.7Hz) , 8.2-8.4 (2H, m)
Preparation 20-4)
(2R,4R)-l-Allyloxycarbonyl-4-methylsulfonyloxy-2-(3- methyl-4-pyridylmethyl)pyrrolidine was obtained in 77.3% yield in substantially the same manner as that of Preparation 10-7).
NMR (CDCI3, δ) : 1.8-2.6 (7H, m), 3.01 (3H, s),
3.5-3.7 (IH, m), 3.8-4.4 (2H, m) , 4.5-4.7 (2H, m), 5.1-5.4 (3H, m) , 5.8-6.1 (IH, m) , 6.9-7.1 (IH, ), 8.3-8.5 (2H, m)
Preparation 20-5)
(2R,4R)-l-Allyloxycarbonyl-2-(3-methyl-4- pyridylmethyl)-4-benzoylthiopyrrolidine was obtained in 82.6% yield in substantially the same manner as that of Preparation 10-8).
NMR (CDCI3, δ) : 1.8-2.1 (2H, m) , 2.3-3.0 (5H, m) , 3.3-3.8 (2H, m), 4.1-4.4 (2H, ) , 4.03 (2H, d, J=5.7Hz), 5.2-5.4 (2H, m) , 5.8-6.2 (IH, m) , 7.0- 8.5 (8H, m)
Preparation 21-1)
To a cooled solution of (2R,4R)-l-Allyloxycarbonyl- 2-(2-methyl-4-pyridylmethyl)-4-t-butyldimethylsilyl- pyrrolidine (43.96 g) in methanol (440 ml) was added a solution of sodium bicarbonate (20.14 g) in water (250 ml), followed by addition of a solution of OXONE (Trademark, made by Aldrich Chemical Company, Inc.) (52.10 g) in water (250 ml). The resulting suspension was stirred at Room temperature for 3.5 hours . To the mixture was added chloroform (1.2 1) and the insoluble materials
were filtered off. The filtrate was separated and the organic layer was washed with water and brine, dried over magnesium sulfate, and evaporated in vacuo. The Residue was purified by column chromatography on silica gel (eluent; ethyl acetate : methanol = 10 : 1) to give 4- [ (2R,4R)-l-Allyloxycarbonyl-4-t-butyldimethylsilyloxy- pyrrolidin-2-yl]methyl-2-methylpyridine N-oxide (9.95 g) as an orange oil.
IR (film) : 2955, 2930, 2855, 1730 cm-1 NMR (CDC13, δ) : 0.02 (6H, s), 0.84 (9H, S), 1.6-
1.95 (2H, m), 2.50 (3H, S), 2.7-2.85 (IH, m) , 3.1-3.5 (3H, m), 4.1-4.25 (2H, m) , 4.6-4.7 (2H, m), 5.2-5.45 (2H, m), 6.85-6.95 (IH, ) , 7.07 (IH, s), 8.17 (IH, d, J=6.6Hz)
Preparation 21-2)
4-[ (2R,4R)-l-Allyloxycarbonyl-4-t-butyldimethyl- silyloxypyrrolidin-2-yl]methyl-2-methylpyridine N-oxide (8.56 g) was dissolved in acetic anhydride (6.43 g) and the solution was stirred at 150°C for 30 minutes. The mixture was cooled to 5°C and poured into a mixture of ethyl acetate and ice water and adjusted to pH 7 by addition of sat. aqueous sodium bicarbonate. The separated organic layer was washed with water and brine, dried over magnesium sulfate and evaporated in vacuo. The residue was purified by column chromraphy on silica gel (eluent; ethyl acetate : isopropyl ether = 2 : 1) to give (2R,4R)-l-Allyloxycarbonyl-2-(2-acetoxymethyl-4- pyridylmethyl)-4-t-butyldimethylsilyloxypyrrolidine (7.23 g) as on orange oil.
IR (film) : 2950, 2890, 1750, 1700, 1610 cm-1 NMR (CDCI3, δ) : 0.00 (6H, s), 0.83 (9H, S), 1.6- 2.0 (2H, m), 2.10 (3H, S), 2.75-2.9 (IH, m) , 3.1-3.5 (3H, m), 4.1-4.3 (2H, ) , 5.20 (2H, s), 5.25-5.4 (2H, ) , 5.85-6.1 (IH, m) , 7.05-7.25
( 2H , m) , 8 . 50 ( IH , d , J=5 . 0Hz )
Preparation 21-3)
To a solution of (2R,4R)-l-Allyloxycarbonyl-2-(2- acetoxymethyl-4-pyridylmethyl-4-t- butyldimethylsilyloxypyrrolidine (5.0 g) in methanol (40 ml) was added dropwise cone, hydrochloric acid (1.92 ml) at room temperature. The reaction mixture was stirred for 30 minutes. To the reaction mixture was added sodium hydrogen carbonate (2.05 g), and the mixture was stirred for 30 minutes and evaporated under reduced pressure. To the residue was added tetrahydrofuran (20 ml) and ethyl acetate (40 ml). The insoluble material was filtered off. The filtrate was evaporated under reduced pressure. The residue was column chromatographed on silica gel (eluent; ethyl acetate to dichlorometane : methanol = 20 : 1) to give (2R,4R)-l-Allyloxycarbonyl-4-hydroxy-2-(2- acetoxymethyl-4-pyridyl-methyl)pyrrolidine (2.47 g) as on oil. NMR (CDC13, δ) : 1.65-2.10 (3H, m) , 2.16 (3H, ε), 2.60-2.85 (IH, m) , 3.10-3.70 (3H, ) , 4.20- 4.40 (2H, ), 4.55-4.70 (2H, ) , 5.10-5.40 (2H, m), 5.18 (2H, s), 5.85-6.15 (IH, m) , 7.00-7.25 (2H, ), 8.46 (IH, d, J=5.0Hz) APCI-MS (m/z) : 335(MH+)
Preparation 21-4)
(2R,4S)-l-Allyloxycarbonyl-4-benzoylthio-2-(2- acetoxymethyl-4-pyridylmethyl)pyrrolidine (4.42 g) was obtained by the similar procedure as Preparation 16-7). NMR (CDCI3, δ) : 1.70-2.00 (2H, m) , 2.15 (3H, s), 2.70-2.90 (IH, m) , 3.20-3.60 (2H, m) , 4.00- 4.40 (3H, m), 4.55-4.75 (2H, m) , 5.15-5.20 (2H, ), 5.18 (2H, s), 5.85-6.15 (IH, ) , 7.00-7.30 (2H, ), 7.40-8.00 (5H, m) , 8.50 (IH, d,
J=5.0Hz) film) : 1747, 1700, 1662 cπT1
(continued on the next page)
Example 1-1)
To a solution of allyl (4R)-2-diazo-4-[ (2R,3S)-3- [ (IR)-1-hydroxyethyl]-4-oxoazetidin-2-yl]-3-oxopentanoate (36.62 g) in ethyl acetate (366 ml) was added rhodium(II) octanoate (483 mg) at room temperature and the mixture was refluxed for 40 minutes under nitrogen. The mixture was cooled and evaporated in vacuo. The residue was dissolved in acetonitrile (366 ml) and cooled to 5°C. To this solution were added diphenyl chlorophosphate (36.59 g) and 4-dimethylaminopyridine (757 mg), followed by dropwise addition of N,N-diisopropyl-N-ethylamine (19.23 g) at 5°C. The mixture was stirred at 5°C for 2 hours and N,N- dimethylacetamide (183 ml) was added. To the mixture was added a solution of (2R,4S)-l-allyloxycarbonyl-2-(2- carbamoylpyridin-4-yl)methyl-4-mercaptopyrrolidine (33.95 g) in acetonitrile (340 ml) at 5°C, followed by dropwise addition of N,N-diisopropyl-N-ethylamine (13.70 g) and the mixture was stirred at 5°C for 14 hours. The mixture was poured into a mixture of ethyl acetate and ice water, and the separated organic layer was washed with water and brine, dried over magnesium sulfate and evaporated in vacuo. The residue was purified by a column chromatography on silica gel to give allyl (4R,5S,6S)-3- [ (2R,4S)-1-allyloxycarbonyl-2-(2-carbamoylpyridin-4- ylmethyl)pyrrolidin-4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (32.59 g) as a foam.
IR ( Br) : 3450, 2970, 1780, 1720, 1660, 1600, 1555 cm-1 NMR (CDC13, δ) : 1.23 (3H, d, J=7.2Hz), 1.35 (3H, d, J=6.2Hz), 1.55-1.8 and 2.3-2.5 (2H, m) , 2.8- 3.0 and 3.2-3.7 (6H, m) , 4.2-4.35 (3H, m) , 4.6- 4.7 (2H, m), 4.6-4.9 (2H, m) , 5.2-5.5 (4H, m) , 5.8-5.9 (IH, br s), 5.9-6.1 (2H, m) , 7.25-7.4 (IH, br s), 7.87 (IH, d, J=4.9Hz), 8.25 (IH, s),
8.49 (IH, d, J=4.9Hz)
Example 1-2)
The following compound was obtained by the similar manner to that of Example 1-1).
Allyl (4R,5S,6S)-3-[ [ (2R,4S)-l-allyloxycarbonyl-2- (2-cyanopyridin-4-yl)methyl]pyrrolidin-4-yl]thio-6-[ ( IR)- l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate
IR (KBr) : 3450, 2970, 2875, 2235, 1770, 1700,
1650, 1600, 1550 cm"1 NMR (CDC13, δ) : 1.25 (3H, d, J=7.2Hz), 1.36 (3H, d, J=6.3Hz), 1.6-1.8 and 2.3-2.5 (2H, m) , 2.85- 3.05 and 3.3-3.8 (6H, m) , 4.1-4.35 (4H, m) , 4.5-
4.7 (2H, m), 4.6-4.9 (2H, m) , 5.25-5.5 (4H, ) , 5.9-6.1 (2H, ), 7.3-7.4 (IH, ) , 7.5-7.6 (IH, ), 8.63 (IH, d, J=5.0Hz)
Example 1-3)
To a solution of allyl (4R)-2-diazo-4-[ (2R,3S)-3- { (lR)-l-hydroxyethyl}-4-oxoazetidin-2-yl]-3-oxopentanoate (1.30 g) in ethyl acetate (13 ml) was added rhodium(II) octanoate (17 mg) under refluxing in a stream of nitrogen. After refluxing for 30 minutes, the reaction mixture was evaporated in vacuo to give a residue. The residue was dissolved in acetonitrile (13 ml) and cooled at 0-5°C under atmosphere of nitrogen. To the solution were successively added diphenyl phosphorochloridate (1.01 ml), N,N-diisopropyl-N-ethylamine (0.92 ml) and N,N- dimethyla inopyridine (27 mg) and the mixture was stirred at the same temperature for 2 hours. On the other hand, to a solution of (2R,4S)-4-acetylthio-l-allyloxycarbonyl- 4-[ (2S)-2-(imidazol-l-yl)propyl]pyrrolidine (2.20 g) in acetonitrile (20 ml) was added 28% sodium
methoxide-methanol solution (1.27 ml) at -20 ~ 0°C with stirring. After stirring for 10 minutes, the reaction mixture and N,N-dimethylacetamide (10 ml) were added to the obtained solution described above at -10 ~ 20°C. The mixture was stirred at 0°C for 2 hours. To the reaction mixture were added ethyl acetate (50 ml) and water (30 ml). The organic layer was separated and then washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (150 g) eluting with a mixture of chloroform and methanol (19:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{(2S)-2-(imidazol-l-yl)propyl}pyrrolidin- 4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate (1.31 g).
NMR (CDC13, δ) : 1.21 (3H, d, J=7.22Hz), 1.35 (3H, d, J=6.23Hz), 1.48 (3H, d, J=6.80Hz), 1.90-2.80 (4H, m), 3.15-3.60 (4H, ) , 3.70-4.50 (5H, m) , 4.50- 4.90 (4H, m), 5.20-5.55 (4H, m), 5.80-6.10 (2H, m), 6.99 (IH, s), 7.08 (IH, s), 7.56 (IH, s)
Example 1-4)
To a solution of allyl (4R)-2-diazo-4-[ (2R,3S)-3- {(lR)-l-hydroxyethyl}-4-oxoazetidin-2-yl]-3-oxopentanoate (2.8 g) in ethyl acetate (30 ml) was added rhodium(II) octanoate (37 mg) under refluxing in a stream of nitrogen. After refluxing for 30 minutes, the reaction mixture was evaporated in vacuo to give a residue. The residue was dissolved in acetonitrile (30 ml) and cooled at 0-5°C under atmosphere of nitrogen. To the solution were successively added diphenyl phosphorochloridate (2.07 ml), N,N- diisopropyl-N-ethylamine (1.82 ml) and 4-(N,N- dimethylamino)pyridine (12 mg) and the mixture was stirred at the same temperature for 2 hours. On the other hand, to
a solution of (2R,4S)-l-allyloxycarbonyl-4-benzoylthio-2-(4- hydroxymethyl-l-methylpyrazol-5-yl)methylpyrrolidine (5.38 g) in acetonitrile (30 ml) was added 28% sodium methoxide-methanol solution (2.50 ml) at -20 ~ 0°C with stirring. After stirring at the same temperature for 10 minutes, the reaction mixture and N,N-dimethylacetamide (15 ml) were added to the solution obtained above at -10°C ~ -20°C The mixture was stirred at 0°C for 2 hours. To the reaction mixture were added ethyl acetate (100 ml) and water (30 ml). The organic layer was separated and then washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (140 g) eluting with a mixture of dichloromethane and acetone (1:1, V/V) . The fractions containing the desired compound were collected and evaporated in vacuo to give allyl (4R,5S,6S)- 3-[ (2R,4S)-l-allyloxycarbonyl-2-(4-hydroxymethyl-l- methylpyrazol-5-yl)methylpyrrolidin-4-yl]thio-6-[ ( 1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene- 2-carboxylate (3.06 g).
IR (Neat) : 1770, 1697, 1649, 1406, 1329, 1279 cπr1 NMR (CDC13, δ) : 1.25 (3H, d, J=7.21Hz), 1.36 (3H, d, J=6.24Hz), 1.80-2.00 (IH, m) , 2.00-2.55 (3H, m), 2.85-4.35 (13H, m) , 4.50-4.95 (6H, m) , 5.20- 5.55 (4H, m) , 5.80-6.15 (2H, m) , 7.44 (IH, s)
The following compounds were obtained in substantially the same manner as that of Example 1-4).
Example 1-5)
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-(5- hydroxymethyl-l-methylpyrazol-4-yl)methylpyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclof3.2.0]hept-2-ene-2-carboxylate IR (Neat) : 1768, 1691, 1408, 1327, 1276 cm-1
NMR (CDCI3, δ) : 1.24 (3H, d, J=7.15Hz), 1.36 (3H, d, J=6.24Hz), 1.60-2.90 (5H, m) , 2.95-3.75 (5H, m), 3.91 (3H, s), 3.91-4.30 (4H, m) , 4.40-4.90 (6H, m), 5.15-5.60 (4H, m) , 5.80-6.10 (2H, m) , 7.27 (IH, s)
APCI-MS : 561 (M+)
Example 1-6)
Allyl (4R,5S,6S )-3-[ (2R,4S)-l-allyloxycarbonyl-2-(5- hydroxymethyl-1-methylpyrazol-3-yl)methylpyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate
IR (Neat) : 1772, 1675, 1652, 1448, 1417 cirr1 NMR (CDCI3, δ) : 1.23 (3H, d, J=8.69Hz), 1.34 (3H, d, J=6.23Hz), 1.70-2.55 (4H, m) , 2.80-3.60 (6H, m), 3.85 (3H, s), 3.85-6.10 (17H, m) APCI-MS : 561 (M+)
Example 1-7) Allyl (4R,5S,6S)-3-[ (2R,4S)-1-allyloxycarbonyl-2-(2- hydroxymethyl-l-methylimidazol-5-ylmethyl)pyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate
NMR (CDCI3, δ) : 1.21 (3H, d, J=7.0Hz), 1.35 (3H, d, J=6.2Hz), 1.75-1.90 (IH, ) , 2.35-2.55 (IH, m), 2.75-4.30 (15H, m), 4.62-4.87 (4H, m) , 5.23- 5.49 (4H, m), 5.90-6.03 (2H, m) , 6.70 (IH, s)
Example 1-8) Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-(2- carbamoyl-l-methylimidazol-5-ylmethyl)pyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate
NMR (CDCI3, δ) : 1.25 (3H, d, J=7.2Hz), 1.36 (3H, d, J=6.2Hz), 1.73-1.86 (2H, m) , 2.35-2.55 (IH,
m), 2.80-2.95 (IH, m) , 3.24-3.40 (3H, m) , 3.62- 3.69 (IH, m), 3.97-4.03 (6H, m) , 4.61-4.89 (4H, m), 5.24-5.50 (6H, ) , 5.88-6.07 (2H, m) , 6.85 (IH, s), 7.23 (IH, br)
Example 1-9)
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-(2- methylpyridin-4-ylmethyl)pyrrolidin-4-yl]thio-6-[ (IR)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate
IR (CHC13) : 1774, 1697, 1606 cm-1
NMR (CDCI3, δ) : 1.24 (3H, d, J=7.5Hz), 1.36 (3H, d, J=6.2Hz), 1.60-1.80 (IH, m) , 1.80-2.45 (2H, ), 2.54 (3H, s), 2.60-2.80 (IH, ) , 3.20-3.65 (4H, m), 3.90-4.35 (4H, ) , 4.60-4.90 (4H, m) ,
5.20-5.55 (4H, ) , 5.80-6.10 (2H, m) , 6.90-7.10 (2H, ), 8.40 (IH, d, J=5.0Hz) MS : (M+l) = 542
Example 2-1)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-(4-pyridylmethyl)pyrrolidin-4-yl]thio- 6-[ (IR)-1-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate (3.31 g) in dichloromethane (100 ml) was added 2-t- butyldimethylsilyloxyethyl trifluoromethanesulfonate (18.7 g) at room temperature. After stirring for 12 hours, the solution was concentrated. Allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-[l-{(2-t-butyldimethylsilyloxyethyl)-4- pyridinio}methyl]pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate trifluoromethanesulfonate (32 g) was obtained as an oil.
IR (CHCI3) : 1760, 1690 cm-1 NMR (CDCI3, δ) : 0.0 (6H, s), 0.84 (9H, s), 1.3-1.5
(6H, m), 1.6-1.7 (IH, m) , 2.5-2.7 (IH, m) , 3.1- 4.4 (12H, m), 4.6-4.9 (6H, ) , 5.3-5.1 (4H, m) , 5.9-6.1 (2H, m), 7.90 (2H, d, J=6.0Hz), 8.81 (2H, d, J=6.0Hz)
Example 2-2)
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2- {(l-carbamoylmethyl-4-pyridinio)methyl}pyrrolidin-4- yl]thio-6-[ (IR)-1-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate iodide was obtained in substantially the same manner as the former part of Example 4-4).
NMR (CDC13, δ) : 1.1-1.5 (6H, m) , 2.5-5.0 (16H, m) , 5.2-5.5 (4H, m), 5.7-6.0 (2H, ) , 7.9-8.0 (2H, m), 9.0-9.1 (2H, m)
Example 2-3)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-(2-carbamoylpyridin-4- ylmethyl)pyrrolidin-4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (1.51 g) in dichloromethane (10 ml) was added methyl trifluoromethanesulfonate (methyl triflate) (2.60 g) at 5°C and the mixture was stirred at 5°C for 4.5 hours. The mixture was diluted with dichloromethane (60 ml) and ion exchange resin amberlyst A-26 (Cl* type) (which is pretreated with methanol and dichloromethane, and dried) (30 ml) was added. The mixture was stirred at room temperature for 40 minutes and filtered. The resin was washed with a mixture of dichloromethane and methanol
(4:1) (100 ml) several times. The filtrate was evaporated in vacuo to give crude allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{ (l-methyl-2-carbamoyl-4- pyridinio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-
ene-2-carboxylate chloride (1.63 g).
This compound was immediately used as the starting compound of the next step.
Example 2-4)
The following compound was obtained by the similar manner to that of Example 2-3).
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2- (l-methyl-2-cyano-4-pyridinio)methyl}pyrrolidin-4- yl]thio-6-[ (IR)-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate chloride.
This compound was immediately used as the starting compound of the next step.
Example 2-5)
A solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{(2S)-2-(imidazol-1- yl)propyl}pyrrolidin-4-yl]thio-6-[ (IR)-1-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
(1.31 g) and methyl iodide (1.50 ml) in acetone (6 ml) was stirred at ambient temperature for 10 minutes and then allowed to stand at the same temperature overnight. The ■ reaction mixture was evaporated in vacuo to give allyl (4R,5S,6S)-3-[(2R,4S)-l-allyloxycarbonyl-2-{(2S)-2-(3- methyl-l-imidazolio)propyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate iodide (1.65 g).
This compound was immediately used as the starting compound of the next step.
Example 2-6)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{(4-hydroxymethyl-l-methylpyrazol-5- yl)methyl}pyrrolidin-4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4-
methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (1.0 g) in dichloromethane (10 ml) was added dropwise methyl trifluoromethanesulfonate (0.24 ml) under ice- cooling and the mixture was stirred at the same temperature for 1 hour. The reaction mixture was evaporated in vacuo to give allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{(4-hydroxymethyl-1,2-dimethyl-3- pyrazolio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate trifluoromethanesulfonate (1.39 g) .
This compound was immediately used as the starting compound for the next step.
Example 2-7) Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-
{(2-carbamoyl-l,3-dimethyl-4-imidazolio)methyl}pyrrolidin- 4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate trifluoromethanesulfonate was obtained in substantially the same manner as that of Example 2-6).
This compound was immediately used as the starting compound for the next step.
Example 2-8) (4R,5S,6S)-3-[ (2R,4S)-2-{(1,2-Dimethyl-4- pyridinio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride was obtained in the same procedure as that of Example 2-5). IR (KBr) : 3429, 1770, 1697, 1645 cm"1
NMR (CDC13, 6) : 1.25 (3H, d, J=4.7Hz) , 1.35 (3H, d, J=6.2Hz), 1.60-1.90 (3H, m) , 2.60-2.80 (IH, m), 2.90 (3H, s), 3.00-4.40 (8H, m), 4.43 (3H, s), 4.60-4.90 (4H, m) , 5.20-5.60 (4H, m), 5.85-6.05 (2H, m) , 7.70-8.00 (2H, m) ,
9 . 00-9 . 20 ( IH, m) MS : (M-l ) = 556
Example 3-1) To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-[{1-(2-t-butyldimethylsilyloxyethyl)-4- pyridinio}methyl]pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene- 2-carboxylate trifluoromethanesulfonate in tetrahydrofuran (80 ml) were added acetic acid (2.28 ml) and a solution of tetrabutylammonium fluoride in tetrahydrofuran (1.0 M, 38 ml) at room temperature. After stirring at room temperature for 2 hours, to the mixture were added ethanol (160 ml), triphenylphosphine (3.97 g) , morpholine (11.6 ml) and tetrakis(triphenylphosphine)-palladium at room temperature. After stirring at room temperature for 45 minutes, an addition of ethyl acetate (500 ml) gave precipitates and the precipitates were collected by filtration and dissolved in water (500 ml). The solution was washed with ethyl acetate (200 ml) and dichloromethane (200 ml x 2). The aqueous layer was concentrated and chromatographed on Diaion HP-20 (Trademark, made by Mitsubishi Kasei Co.) ( I t ) eluting with a mixture of water and acetonitrile (95:5). The fractions containing the desired compound were collected and passed through ion exchange resin (Amberlyst A-26 (Cl- type)) (30 ml). Obtained solution was concentrated and lyophilized. (4R,5S,6S)-3-[ (4S)-2-[l-(2-hydroxyethyl)-4- pyridinio]methylpyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (4.38 g) was obtained in 24% yield as a white powder. IR (Nujol) : 1740 cm_1 NMR (D20, δ) : 1.21 (3H, d, J=7.1Hz), 1.28 (3H, d, J=6.3Hz), 1.8-2.9 (2H, ) , 3.3-4.8 (14H, m) ,
8 . 07 ( 2H , d , J=6 . 5Hz ) , 8 . 83 ( 2H , d , J=6 . 6Hz ) MS ( FAB+ ) : 448 . 3 ( M-HCl+1 )
Example 3-2) (4R,5S,6S)-3-[ (4S)-2-[l-(Carbamoylmethyl)-4- pyridinio]methylpyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride was obtained in 11.3% yield in substantially similar manner as that of Example 3-8). IR (Nujol) : 1750, 1690 cm-1
NMR (D20, 6) : 1.21 (3H, d, J=7.1Hz), 1.28 (3H, d, J=6.8Hz), 1.8-2.9 (2H, m) , 3.3-4.5 (10H, m) , 5.50 (2H, s), 8.09 (2H, d, J=6.7Hz) , 8.78 (2H, d, J=6.8Hz) MS (FAB+) : 461.2 (M-HCl+1)
Example 3-3 )
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-[ (2-carbamoylpyridin-4- yl)methyl]pyrrolidin-4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (1.03 g) in a mixture of tetrahydrofuran (THF) (6 ml) and ethanol (6 ml) were added triphenylphosphine (189 mg) and morpholine (392 mg) at room temperature. To the mixture was added tetrakis(triphenylphosphine)palladium (166 mg) and the mixture was stirred at room temperature for 40 minutes. The precipitates were collected by filtration and washed with ethyl acetate. The powder was dissolved in cold water (80 ml) and the solution was washed with ethyl acetate twice. The solution was adjusted to pH 3.5 by addition of IN hydrochloric acid and chromatographed on Diaion HP-20 (80 ml) and eluted with 8% aqueous acetonitrile. The eluate was lyophilized and the crude product was purified by preparative High Performance Liquid Chromatography (HPLC) [C*-_g μ-Bondapak, resin
(Waters Associates Inc.) eluting with pH 3 phosphate buffer : CH3CN = 87:13 (V/V)] to give (4R,5S,6S)-3- [ (2R,4S)-2-[ (2-carbamoylpyridin-4-yl)methyl]pyrrolidin-4- yl]thio-6-[ (IR)-1-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid (169 mg) . IR (KBr) : 3430, 2940, 1755, 1680, 1600 cm-1 NMR (D20, δ) : 1.20 (3H, d, J=7.2Hz), 1.29 (3H, d, J=6.3Hz), 1.7-1.95 (2H, m) , 2.7-2.9 (IH, m) , 3.3-3.5 (5H, m), 3.65-3.8 (IH, ), 4.0-4.2 (2H, m), 4.2-4.35 (2H, m), 7.60 (IH, d, J=5.0Hz) ,
7.99 (IH, s), 8.60 (IH, d, J=5.0Hz)
Example 3-4)
To a solution of crude allyl (4R,5S,6R)-3-[ (2R,4S)- l-allyloxycarbonyl-2-[ (l-methyl-2-carbamoyl-4- pyridinio)methyl]pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate chloride (1.62 g) in a mixture of THF (16 ml) and ethanol (8 ml) were added triphenylphosphine (274 mg) and acetic acid (407 mg) at room temperature. To the mixture was added tetrakis(triphenylphosphine)- palladium (241 mg) and the mixture was stirred at room temperature for 20 minutes. To the resulting solution was added tributyltin hydride (1.82 g) and the mixture was stirred at room temperature for 30 minutes. The mixture was diluted with ethyl acetate (20 ml) and the precipitates were collected by filtration and washed with ethyl acetate. The powder was dissolved in cold water (50 ml) and the solution was washed with ethyl acetate. The solution was adjusted to pH 3.5 by addition of IN hydrochloric acid and chromatographed on SP-205 (Trademark, made by Mitsubishi Chemical Industries) (60 ml) and eluted with 5-10% aqueous acetonitrile. The eluate was lyophilized and the crude product was purified by preparative High Performance Liquid Chromatography [C*j_g
μ-Bondapak resin (Waters Associates Inc.) eluting with pH 3 phosphate buffer:CH3CN = 83:17 (V/V)] to give (4R,5S,6S)-3-[ (4S)-2-[ (l-methyl-2-carbamoyl-4- pyridinio)methyl]pyrrolidin-4-yl]thio-6-[ (IR)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (30 mg) .
IR (KBr) : 2970, 2750, 1755, 1700, 1635, 1580 crn"1 NMR (D20, δ) : 1.21 (3H, d, J=7.2Hz) , 1.28 (3H, d, J=6.4Hz), 1.8-1.95 and 2.3-2.45 and 2.7-2.9 (2H, m), 3.3-3.6 (5H, m), 3.6-3.95 (IH, ) , 4.0-4.3
(4H, ), 4.39 (3H, s), 8.12 (IH, d, J=6.4Hz), 8.24 (IH, d, J=1.9Hz), 8.88 (IH, d, J=6.4Hz)
Example 3-5) The following compound was obtained by the similar manner to that of Example 3-3).
(4R,5S,6S)-3-[ (2R,4S)-2-[ (2-Cyanopyridin-4- yl)methyl]pyrrolidin-4-yl]thio-6-[ (IR)-1-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
IR (KBr) : 3410, 2970, 2240, 1755, 1600 cm"1 NMR (D20, δ) : 1.21 (3H, d, J=7.2Hz) , 1.29 (3H, d, J=6.4Hz), 1.7-1.95 and 2.7-2.9 (2H, m) , 3.3-3.5 (5H, m), 3.65-3.8 (IH, m) , 4.0-4.4 (4H, m) , 7.71
(IH, d, J=5.1Hz), 7.93 (IH, s), 8.66 (IH, d, J=5.1Hz)
Example 3-6) The following compound was obtained by the similar manner to that of Example 3-4).
(4R,5S,6S)-3-[ (4S)-2-[ (1-Methyl-2-cyano-4- pyridinio)methyl]pyrrolidin-4-yl]thio-6-[ (IR)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-
ene-2-carboxylic acid chloride
IR (KBr) : 2975, 1755, 1630, 1585 cπr1 NMR (D20, δ) : 1.21 (3H, d, J=7.1Hz) , 1.28 (3H, d, J=6.4Hz), 1.8-2.0 (IH, ) , 2.3-2.5 (2H, m) , 2.7- 2.9 (IH, ), 3.3-3.7 (5H, m) , 3.7-3.9 (IH, ) ,
4.1-4.35 (4H, m), 4.59 (3H, s), 8.34 (IH, d, J=4.9Hz), 8.66 (IH, s), 9.13 (IH, d, J=4.9Hz)
Example 3-7) To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{ (2S)-2-(3-methyl-l- imidazolio)propyl}pyrrolidin-4-yl}thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate iodide (1.65 g), triphenylphosphine (126 mg), acetic acid (0.55 ml) and tetrakis(triphenylphosphine)palladium(0) (111 mg) was added tributyltin hydride (2.59 ml) with stirring at ambient temperature. After stirring for 1 hour, the resulting precipitates were filtrated, washed with tetrahydrofuran and dried in vacuo. The solid was dissolved in water (50 ml). The solution was washed twice with ethyl acetate (30 ml) and evaporated in vacuo. The resulting residue was chromatographed on nonionic adsorption resin, Diaion HP-20 (Trademark, made by Mitsubishi Kasei Co.) (40 ml) eluting in turn with water (200 ml) and 5% aqueous acetone (200 ml). The fractions containing the desired compound were collected and evaporated in vacuo. The resulting residue (20 ml) was passed through ion exchange resin, Amberlist A-26 (Cl" type, Trademark, made by Rohm and Haas Co., Ltd.) (10 ml) eluting with water (60 ml). The eluate was lyophilized to give (4R,5S,6S)-6-[ (lR)-l-hydroxyethyl]-3-[ (2R,4S)-2- { (2S)-2-(3-methyl-l-imidazolio)propyl}pyrrolidin-4- yl]thio-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2- carboxylic acid chloride (444 mg) .
IR (Nujol) : 1730-1720, 1560-1530 cm-1
NMR (D20, δ) : 1.20 (3H, d, J=7.16Hz), 1.28 (3H, d,
J=6.33Hz), 1.60 (3H, d, J=6.73Hz), 1.62-1.75
(IH, ), 2.30-2.75 (3H, m) , 3.25-3.80 (5H, m) , 3.92 (3H, s), 3.92-4.10 (IH, ) , 4.15-4.35 (2H, ), 4.55-4.80 (IH, m) , 7.51 (IH, s), 7.65 (IH, s), 8.91 (IH, s) FAB-MS : 435 (M+)
Example 3-8)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{ (4-hydroxymethyl-1,2-dimethyl-3- pyrazolio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate trifluoromethanesulfonate (1.39 g), triphenylphosphine (94 mg), acetic acid (0.61 ml) and tetrakis(triphenylphosphine)palladium(0) (82 mg) was added tributyltin hydride (1.92 ml) with stirring at ambient temperature. The mixture was stirred at the same temperature for 1 hour. The resulting precipitates were filtrated, washed with tetrahydrofuran and dried in vacuo to give a solid. The solid was dissolved in water (60 ml). The solution was washed twice with ethyl acetate (30 ml) and evaporated in vacuo. The resulting residue was chromatographed on nonionic adsorption resin, Diaion HP-20 (100 ml) eluting in turn with water (300 ml), 3% aqueous acetone and 5% aqueous acetone (600 ml). The fractions containing the desired compound were collected and evaporated in vacuo. The resulting residue (50 ml) was passed through ion exchange resin, Amberlist A-26 (Cl-
^YPe ) (10 ml) eluting with water (150 ml). The eluate was lyophilized to give (4R,5S,6S)-6-[ (IR)-1-hydroxyethyl]-3- [(2R,4S)-2-{(4-hydroxymethyl-1,2-dimethyl-3- pyrazolio)methyl}pyrrolidin-4-yl]thio-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid chloride
(140 mg) .
IR (Nujol) : 1755, 1460, 1377, 1286, 1259 cm-1 NMR (D20, δ) : 1.22 (3H, d, J=7.18Hz), 1.28 (3H, d, J=6.35Hz), 1.70-2.00 (IH, m) , 2.65-2.85 (IH, ) , 3.30-3.90 (7H, m) , 3.95-4.05 (IH, m) , 4.07 (3H, s), 4.11 (3H, s), 4.15-4.35 (2H, ) , 4.67 (2H, s), 8.24 (IH, s) FAB-MS : 451 (M+)
Example 3-9)
(4R,5S,6S)-3-[ (2R,4S)-2-{ (2-Carbamoyl-l,3-dimethyl- 4-imidazolio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (1.01 g) was obtained in substantially the same manner as that of Example 3-8). IR (Nujol) : 1700, 1756 cm-1
NMR (D20, δ) : 1.23 (3H, d, J=7.2Hz), 1.30 (3H, d, J=6.3Hz), 1.75-1.95 (IH, m) , 2.80-2.95 (IH, ) , 3.30-3.49 (5H, m) , 3.67-3.77 (IH, m), 3.91 (3H, s), 3.96 (3H, s), 4.09-4.26 (4H, ) , 7.59 (IH, s) FAB-MS : 464 (M+)
Example 3-10) (4R,5S,6S)-3-[ (2R,4S)-2-{ (1,2-Dimethyl-4- pyridinio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride was obtained in 24% yield in substantially the same manner as that of Example 3-8). IR (Nujol) : 1758 cm-1
NMR (CDC13, δ) : 1.21 (3H, d, J=7.2Hz), 1.27 (3H, d, J=6.3Hz), 1.6-2.9 (2H, m) , 2.78 (3H, s), 3.3-4.3 (10H, m), 4.20 (3H, s), 7.79 (IH, d, J=6.5Hz), 7.87 (IH, s), 8.65 (IH, d, J=6.5Hz) MS (FAB+) : 432.5 (M+1-HC1)
Example 4-1)
(4R,5S,6S)-6-[ (lR)-l-Hydroxyethyl]-3-[ (2R,4S)-2-{3- hydroxymethyl-l,2-dimethyl-4-pyrazolio)methyl}pyrrolidin- 4-yl]thio-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2- carboxylic acid chloride (103 mg) was obtained by reacting allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-{(5- hydroxymethyl-l-methylpyrazol-4-yl)methyl}pyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate (395 mg) with methyl trifluoromethanesulfonate (0.10 ml) in dichloromethane (4 ml) and then tetrakis(triphenylphosphine)palladium(0) (33 mg) and tributyltin hydride (0.76 ml) in substantially the same manner as those of Example 2-6) and Example 3-8). IR (Nujol) : 1749, 1652, 1578, 1458 cm"1
NMR (D20, 6) : 1.21 (3H, d, J=7.21Hz), 1.28 (3H, d, J=6.35Hz), 1.65-1.90 (IH, m) , 2.60-2.85 (IH, m), 3.10-3.55 (5H, m) , 3.60-4.05 (3H, m) , 4.11 (3H, ε), 4.20 (3H, s), 4.25-4.30 (2H, m) , 4.85 (2H, s), 8.22 (IH, s)
FAB-MS : 451 (M+)
Example 4-2)
(4R,5S,6S)-6-[ (lR)-l-Hydroxyethyl]-3-[ (2R,4S)-2-{(5- hydroxymethyl-l,2-dimethyl-3-pyrazolio)methyl}pyrrolidin- 4-yl]thio-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2- carboxylic acid chloride (58 mg) was obtained in substantially the same manner as that of Example 4-1). IR (Nujol) : 1740, 1625, 1445 cm-1 NMR (D20, δ) : 1.21 (3H, d, J=7.12Hz), 1.28 (3H, d,
J=6.38Hz), 1.80-1.95 (IH, m) , 2.70-3.90 (8H, ) , 4.00 (6H, s), 4.05-4.35 (5H, m) , 6.78 (IH, s)
Example 4-3) (4R,5S,6S)-3-[(2R,4S)-2-{(2-Hydroxymethyl-l,3-
dimethyl-4-imidazolio)methyl}pyrrolidin-4-yl]thio-6-[ (IR)- 1-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (485 mg) was obtained in substantially the same manner as that of Example 4-1). IR (Nujol) : 1730 crn-1
NMR (D20, δ) : 1.22 (3H, d, J=7.2Hz) , 1.29 (3H, d, J=6.4Hz), 1.75-1.90 (IH, m) , 2.75-2.95 (IH, ) , 3.25-3.50 (5H, m) , 3.66-3.76 (IH, ) , 3.84 (3H, s), 3.89 (3H, s), 4.05-4.30 (4H, m) , 4.93 (2H, s), 7.45 (IH, s)
FAB-MS : 451 (M+)
Example 4-4)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{ (l-methylimidazol-5- yl)methyl}pyrrolidin-4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (5.0 g) in acetone (10 ml) was added iodoacetamide (2.09 g) and the mixture was stirred for 20 hours. The solvent was removed under reduced pressure and the residue was dissolved in tetrahydrofuran (48 ml) and ethanol (48 ml). To the solution was added triphenylphosphine (469 mg), morpholine (1.72 ml), then tetrakis(triphenylphosphine)- palladium(O) (414 mg) at room temperature. The mixture was stirred for 60 minutes. To the reaction mixture was added tetrahydrofuran (48 ml) and resulting precipitate was collected by filtration. The precipitate was dissolved in water (100 ml) and washed with ethyl acetate (50 ml x 2). The aqueous layer was applied to Amberlist A-26 (Cl-) (30 ml) and the eluate was concentrated (250 ml). Obtained solution was chromatographed on nonionic adsorption resin, Diaion HP-20 (330 ml) eluting with water. The fractions containing the desired compound were collected and adjusted to pH 8.0 with 5% aqueous sodium carbonate. The solution was purified by High Performance
Liquid Chromatography (C^8 μ-Bondapak resin, Water Associates, Inc.) eluting with 10% aqueous acetonitrile. The solution was concentrated in vacuo. The resulting solution was adjusted to pH 3.5 with IN-hydrochloric acid, and lyophilized to give (4R,5S,6S)-3-[ (2R,4S)-2-{ (1- carbamoylmethyl-3-methyl-4-imidazolio)methyl}pyrrolidin-4- yl]thio-6-[ (IR)-1-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid chloride (590 mg) . IR (Nujol) : 3400-3100, 1750, 1680, 1570 cnT1
NMR (D20, 6) : 1.22 (3H, d, J=7.2Hz) , 1.28 (3H, d, J=6.4Hz), 1.72-1.95 (IH, m) , 2.79-2.98 (IH, m), 3.23-3.55 (5H, m) , 3.71 (IH, dd, J=6.6, 12.5Hz), 3.88 (3H, s), 4.02-4.35 (4H, m) , 5.08 (2H, s), 7.50 (IH, s), 8.84 (IH, s)
Example 4-5)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{ (l-methylimidazol-5- yl)methyl}pyrrolidin-4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (10 g) in dichloromethane (50 ml) was added 2-t- butyldi ethylsilyloxyethyl trifluoromethanesulfonate (6.39 g) and the mixture was stirred for 1.5 hours. The solvent was removed under reduced pressure and the residue was dissolved in tetrahydrofuran (100 ml). To the solution was added acetic acid (1.10 ml) and tetrabutylammonium fluoride (1.0 M solution in tetrahydrofuran) (18.4 ml). After 2 hours, to the stirred solution were added ethanol (100 ml), triphenylphosphine (962 mg) morpholine (4.02 ml), then tetrakis(triphenylphosphine)palladium(0) (848 mg) at room temperature. The mixture was stirred for 1.5 hours. To the reaction mixture was added tetrahydrofuran (100 ml) and resulting precipitates were collected by filtration. The precipitate was dissolved in water (200
ml) and washed with ethyl acetate (100 ml x 2). The aqueous layer was applied to a column of Amberlist A-26 (Cl-) (60 ml). The eluate and washing were concentrated in vacuo to ca. 500 ml. The obtained solution was adjusted to pH 6 with 5% aqueous sodium carbonate and the solution was chromatographed on nonionic adsorption resin, Diaion HP-20 (600 ml) eluting in turn with water and 10% aqueous acetonitrile. The fractions containing the desired compound were collected and adjusted to pH 8.0 with 5% aqueous sodium carbonate. The solution was purified by High Performance Liquid Chromatography (C*]_g μ- Bondapak resin) eluting with 8% aqueous acetonitrile. The solution was concentrated in vacuo. The resulting solution was adjusted to pH 3.5 with IN-hydrochloric acid, and lyophilized to give (4R,5S,6S)-3-[ (2R,4S)-2-[ {l-(2- hydroxyethyl)-3-methyl-4-imidazolio}methyl]pyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclof3.2.0]hept-2-ene-2-carboxylic acid chloride (2.48 g) . IR (Nujol) : 3400-3100, 1750, 1570 cm-1
NMR (D20, δ) : 1.22 (3H, d,. J=7.2Hz), 1.28 (3H, d, J=6.4Hz), 1.75-1.97 (IH, ) , 2.77-2.98 (IH, ) , 3.24-3.60 (5H, m) , 3.74 (IH, dd, J=6.6 and 12.5Hz), 3.85 (3H, s), 3.88-4.02 (2H, m) , 4.02- 4.40 (4H, ), 7.54 (IH, s), 8.81 (IH, m)
Example 4-6)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-l- allyloxycarbonyl-2-(l-methylpyrazol-4-ylmethyl)pyrrolidin- 4-yl]thio-6-[ (IR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate (6.2 g) in 1,2- dichloroethane (120 ml) was added 2-t- butyldimethylsilyloxyethyl trifluoromethanesulfonate (4.32 g) and the mixture was stirred for 4 days at room temperature. The solvent was removed under reduced
- Ill - pressure and the residue was dissolved in tetrahydrofuran (60 ml). To the solution were added acetic acid (0.702 ml) and tetrabutyla monium fluoride (1.0 M solution in tetrahydrofuran (1.7 ml). After one hour, to the stirred solution were added ethanol (60 ml), triphenylphosphine (1.23 g), morpholine (2.56 ml), then tetrakis(triphenylphosphine)palladium(0) (676 mg) at room temperature. After one hour tetrahydrofuran (90 ml) was added, and resulting precipitates were collected by filtration. The precipitate was dissolved in water (250 ml) and washed with dichloromethane (x 2). The aqueous solution was adjusted to pH 6 with IN-hydrochloric acid and chromatographed on nonionic adsorption resin, Diaion SP-205 (Trademark, made by Mitsubishi Kasei Co.) (620 ml) eluting in turn with water and 10% aqueous acetone. The fractions containing the desired compound were collected and concentrated in vacuo. The resulting residue was passed through ion exchange resin, Amberlist A-26 (Cl- tyPe) (60 ml) eluting with water. The eluate was lyophilized to give (4R,5S,6S)-3-[ (2R,4S)-2-[{l-(2- hydroxyethyl)-2-methyl-4-pyrazolio}methyl]pyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid chloride (2.2 g) . IR (Nujol) : 3300, 1750, 1590 cm"1
NMR (D20, δ) : 1.21 (3H, d, J=7.2Hz), 1.29 (3H, d, J=6.4Hz), 1.72-1.90 (IH, m) , 2.69-2.87 (IH, ) , 3.10-3.25 (2H, m) , 3.26-3.55 (3H, m) , 3.69 (IH, dd, J=12.5, 6.7Hz), 3.90-4.14 (4H, m) , 4.15 (3H, s), 4.16-4.33 (2H, m) , 4.61 (2H, t, J=4.9Hz),
8.25 (IH, s), 8.31 (IH, s) FAB-MS (m/z) : 451 (M-Cl")
Example 4-7) (4R,5S,6S)-3-[ (2R,4S)-2-[{1-(2-Hydroxyethyl)-2-
methyl-3-pyrazolio}methyl]pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (2.6 g) was obtained in the same procedure as that of Example 4-6). IR (Nujol) : 3250, 1750, 1590 cm-1
NMR (D20, δ) : 1.21 (3H, d, J=7.2Hz), 1.28 (3H, d, J=6.4Hz), 1.79-1.94 (IH, ) , 2.80-2.96 (IH, ) , 3.25-3.55 (5H, m) , 3.73 (IH, dd, J=12.5, 6.5Hz), 3.90-4.32 (6H, m) , 4.07 (3H, s), 4.64 (2H, t, J=4.9Hz), 6.83 (IH, d, J=3.0Hz), 8.27 (IH, d,
J=3.0Hz) FAB-MS (m/z) : 451 (M-Cl-)
Example 5-1) A solution of (2R,4S)-l-Allyloxycarbonyl~2-[ (2- methyl-l-carbamoylmethyl-3-pyrazoliomethyl]-4- (tritylthio)pyrrolidine trifluoromethanesulfonate (284 mg) in dichloromethane (3 ml) was cooled to 0°C and treated successively with trifluoroacetic acid (444.7 mg) and triethylsilane (54.4 mg) . After 10 minutes at 0°C and 30 minutes at ambient temperature the mixture was evaporated under reduced pressure. The residue was washed three times with n-hexane (5 ml) and concentrated in vacuo. The resulting residue was dissolved in acetonitrile (1 ml) and added to a solution of allyl (4R,5S,6S)-2- diphenylphosphoro-6-[ (R)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate, prepared in the usual manner from allyl (4R)-2-diazo-4-[ (2R,3S)-3-{ (1R)-1- hydroxyethyl}-4-oxoazetidin-2-yl]-3-oxopentanoate (76.8 mg) in acetonitrile (0.77 ml) at 0°C, followed by addition of N,N-diisopropyl-N-ethylamine (100.8 mg). After standing overnight in the refrigerator the solution was evaporated under reduced pressure then dissolved in tetrahydrofuran-ethanol (1:1) and treated successively with triphenylphosphine (27.3 mg), morpholine (56.6 mg),
and tetrakis(triphenylphosphine)palladium (12 mg) . After 3.5 hours, tetrahydrofuran (1 ml) was added and the precipitate was removed by filtration, washed thoroughly with THF and dried in vacuo to give white powder (80 mg). Purification by Sepabeads SP 205 (Mitsubishi Kasei Co.) column (20 ml) eluting with 5% acetone in the used manner followed by lyophilization gave (4R,5S,6S)-3-[ (2R,4S)-2- { (2-methyl-l-carbamoylmethyl-3- pyrazolio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (30 mg) as a white powder.
NMR (D20, δ) : 1.23 (3H, d, J=7.2Hz), 1.30 (3H, d,
J=6.4Hz), 1.80-1.98 (IH, m) , 2.80-2.98 (IH, m) ,
3.30-3.90 (7H, ) , 4.01 (3H, s), 3.90-4.30 (3H, m), 5.48 (2H, s), 6.90 (IH, d, J=3Hz) , 8.29 (IH, d, J=3Hz) FAB-MS (m/z) : 464 (M+-Cl)
Example 5-2) (4R,5S,6S)-3-[ (2R,4S)-2-{ (l-Methyl-2- carbamoylmethyl-4-pyrazolio)methyl}pyrrolidin-4-yl]thio-6- [ (IR)-1-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid chloride (100 mg) was obtained by the same procedure as Example 5-1) .
NMR (D20, δ) : 1.22 (3H, d, J=7.1Hz) , 1.29 (3H, d, J=6.3Hz), 1.70-1.90 (IH, m) , 2.65-2.90 (IH, m) , 3.10-3.80 (7H, complex m), 3.90-4.15 (IH, m) , 4.08 (3H, s), 4.17-4.30 (2H, m) , 5.44 (2H, s), 8.31 (IH, s), 8.35 (IH, s)
FAB-MS (m/z) : 421-(M+-C1)
Example 6-1)
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2- { (1-methyl-l,2,3-triazol-5-yl)methyl}pyrrolidin-4-yl]thio-
6-[ (IR)-1-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate (7.4 g) was obtained in substantially the same manner as that of Example 1-4) . IR (CHCI3) : 3350, 1765, 1690, 1400, 1320 cm"1
NMR (CDCI3, δ) : 1.26 (3H, d, J=7.3Hz), 1.36 (3H, d, J=6.2Hz), 1.50-1.90 (IH, m) , 2.30-2.60 (IH, m), 2.85-3.10 (IH, m) , 3.15-3.55 (4H, m) , 3.56- 3.75 (IH, m), 3.85-4.33 (7H, m) , 4.50-4.90 (4H, m), 5.20-5.52 (4H, m) , 5.80-6.10 (2H, m) , 7.50
(IH, s) FAB-MS (m/z) : 532 (MH+)
Example 6-2) To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{ (1-methyl-l,2,3-triazol-5- yl)methyl}pyrrolidin-4-yl]thio-6-[ (IR)-1-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (1.03 g) in dichloromethane (20 ml) was added methyl trifluoromethanesulfonate (0.264 ml) at room temperature. The mixture was stirred for 30 minutes. Dichloromethane was evaporated under reduced pressure. The residue was dissolved in tetrahydrofuran-ethanol (1:1, 20 ml). To the solution was added triphenylphosphine (204 mg), morpholine (0.42 ml), then tetrakis(triphenylphosphine)palladium(0) (112 mg) at room temperature. After one hour to the reaction mixture was added tetrahydrofuran (20 ml), and the organic layer was removed by decantation. The gummy residue was dissolved in water (60 ml) and washed with dichloromethane (x 2). The aqueous solution was adjusted to pH 6 with IN-hydrochloric acid and chromatographed on nonionic adsorption resin Diaion SP-205 (Trademark, made by Mitsubishi Kasei Co.) (50 ml) eluting in turn with water and 8% aqueous acetone. The fractions containing the desired compound were collected and concentrated in
vacuo. The resulting residue was passed through ion exchange resin, Amberlist A-26 (Cl- type, Trademark, made by Rohm and Haas Co. Ltd.) (5 ml) eluting with water. The eluate was lyophilized to give (4R,5S, 6S)-3-[ (2R,4S)-2- [{1,3-dimethyl-4-(1,2,3-triazolio)}methyl]pyrrolidin-4- yl]thio-6-[ (1R)-1-hydroxyethyl]-4-methyl-7-oxo-1- azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid chloride (429.4 mg) .
IR (Nujol) : 3300, 1757, 1585 cm-1 NMR (D20, 6) : 1.22 (3H, d, J=7.2Hz), 1.28 (3H, d,
J=6.3Hz), 1.75-1.95 (IH, ) , 2.75-2.95 (IH, m) , 3.25-3.60 (5H, ) , 3.74 (IH, dd, J=12.5, 6.6Hz) , 4.00-4.33 (4H, m) , 4.27 (3H, s), 4.31 (3H, s), 8.56 (IH, s) FAB-MS (m/z) : 422 (M-Cl-)
Example 7-1)
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-{2- (2-allyloxycarbonylimino-3-methylimidazolin-l- yl)ethyl}pyrrolidin-4-yl]thio-6-[ (lR)-l-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (2.95 g) was obtained in substantially the same manner as that of Example 1-4 ) .
IR (Neat) : 1772, 1699, 1635, 1550 cm-1 NMR (CDCI3, δ) : 1.24 (3H, d, J=7.14Hz), 1.34 (3H, d, J=6.22Hz), 1.50-2.70 (4H, m) , 3.10-4.30 (13H, m), 4.50-4.90 (7H, m) , 5.10-5.55 (6H, m) , 5.80- 6.15 (3H, m), 6.50-6.90 (2H, m) FAB-MS : 644 (M+)
Example 7-2)
(4R,5S,6S)-3-[ (2R,4S)-2-{2-(3-Methyl-2- iminoimidazolin-1-yl)ethyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid hydrochloride (514 mg) was obtained
in substantially the same manner as that of Example 3-3). IR (Nujol) : 1751, 1670, 1652 cm-1
NMR (D20, δ) : 1.21 (3H, d, J=7.2Hz), 1.28 (3H, d, J=6.34Hz), 1.60-1.80 (IH, m) , 2.15-2.45 (2H, m) , 2.65-2.85 (IH, m) , 3.25-3.48 (3H, ) , 3.49 (3H, s), 3.50-3.85 (2H, m) , 3.90-4.35 (4H, ) , 6.84 (IH, d, J=2.5Hz), 6.88 (IH, d, J=2.5Hz) FAB-MS : 436 (M+)
Example 8-1)
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-{l- (3-hydroxypropyl)pyrazol-4-yl}methylpyrrolidin-4-yl]thio- 6-[ (IR)-1-allyloxycarbonyloxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate (1.77 g) was obtained by reacting allyl (4R)-2-diazo-4-[ (2R,3S)-3-
{ (lR)-l-allyloxycarbonyloxyethyl}-4-oxoazetidin-2-yl]-3- oxopentanoate (1.03 g) successively with rhodium(II) octanoate (11 mg) in ethyl acetate (10 ml), diphenyl phosphorochloridate (0.59 ml) and (2R,4S)-1- allyloxycarbonyl-4-benzoylthio-2-[1-(3- hydroxypropyl)pyrazol-4-yl]methylpyrrolidine (1.74 g) in a mixture of acetonitrile (20 ml), 28% sodium methoxide-methanol solution (0.78 ml) and N,N- dimethylacetamide (5 ml) in substantially the same manner as that of Example 1-4). APCI-MS : 659 (M+)
Example 8-2)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbσnyl-2-{l-(3-hydroxypropyl)pyrazol-4- yl]methylpyrrolidin-4-yl]thio-6-[ (IR)-1- allyloxycarbonylethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate (245 mg) in a mixture of dichloromethane (1 ml) and pyridine (0.032 ml) was added dropwise trifluoromethanesulfonic anhydride
(0.066 ml) at -20 ~ -40°C with stirring. The mixture was stirred at the same temperature for 10 minutes and then stirred under ice-cooling for 30 minutes. The reaction mixture was evaporated in vacuo to give a residue. To a solution of the residue in a mixture of tetrahydrofuran (3 ml) and ethanol (3 ml) were successively added triphenylphosphine (39 mg), acetic acid (0.19 ml), tetrakis(triphenylphosphine)palladium(0) (26 mg) and tributyltin hydride (0.6 ml) with stirring at ambient temperature. After stirring for 1 hour, the resulting precipitates were filtrated, washed with tetrahydrofuran and dried in vacuo. The solid was dissolved in water (5 ml) and the solution was chromatographed on nonionic adsorption resin, Diaion HP-20 (Trademark, made by Mitsubishi Chemical Industries) (10 ml) eluting in turn with water (30 ml) and 5% aqueous acetone (60 ml). The fractions containing the desired compound were collected and evaporated in vacuo. The resulting residue (20 ml) was passed through ion exchange resin, Amberlist A-26 (Cl- type, Trademark, made by Rohm and Haas Co., Ltd.) (5 ml) eluting with water (40 ml). The eluate was lyophilized to give (4R,5S,6S)-6-[ (1R)-1-hydroxyethyl]-3-[ (2R,4S)-2-{6- (pyrazolidino[1,2-a]pyrazolio) ethyl}pyrrolidin-4-yl]thio- 4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid chloride (51 mg) .
IR (Nujol) : 1740, 1580, 1290 cm-1
NMR (D20, δ) : 1.22 (3H, d, J=7.15Hz), 1.29 (3H, d, J=6.28Hz), 1.70-1.90 (IH, ) , 2.60-2.85 (IH, m) , 2.90-3.55 (7H, m) , 3.55-4.35 (6H, m) , 4.45-4.65 (5H, m), 8.18 (2H, s)
FAB-MS : 433 (M+)
Example 9-1)
To a solution of allyl-(4R)-2-diazo-4-[ (2R,3S)-3- { (lR)-l-hydroxyethyl}-4-oxoazetidin-2-yl]-3-oxopentanoate
(3.08 g) in ethyl acetate (30 ml) was added rhodium(II) octanoate (41 mg) under refluxing in a stream of nitrogen. The mixture was refluxed for 30 minutes and evaporated in vacuo to give a residue. The residue was dissolved in acetonitrile (40 ml) and cooled at 0-5°C under atmosphere of nitrogen. To the solution was added diphenyl phosphorochloridate (2.27 ml) and N,N-diisopropyl-N- ethylamine (2.00 ml) successively and the mixture was stirred at the same condition for 3 hours . On the other hand, to a solution of (2R,4S)-4-acetylthio-l- allyloxycarbonyl-2-[2-(l-methylimidazol-5- yl)ethyl]pyrrolidine (4.22 g) in acetonitrile (40 ml) was added dropwise '28% sodium methoxide-methanol solution (2.41 ml) at -10 ~ 0°C and the mixture was stirred at the same temperature for 10 minutes. The reaction mixture and N,N-dimethylacetamide (30 ml) were added to the solution described above with stirring under ice-cooling. The mixture was stirred at the same temperature for 2 hours . To the reaction mixture were added ethyl acetate (100 ml) and water (30 ml) with stirring. The organic layer was separated, washed twice with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and evaporated in vacuo. The resulting residue was chromatographed on silica gel (200 g) eluting with a mixture of chloroform and methanol (19:1, V/V). The fractions containing the desired compound were collected and evaporated in vacuo to give allyl (4R,5S,6S)-3- [ (2R,4S)-l-allyloxycarbonyl-2-{2-(l-methylimidazol-5- yl)ethyl}pyrrolidin-4-yl]thio-6-[ (IR)-1-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (5.72 g) .
IR (Neat) : 1760, 1680, 1620, 1400 cm-1 NMR (CDC13, δ) : 1.26 (3H, d, J=7.23Hz), 1.36 (3H, d, J=6.22Hz), 1.50-1.90 (2H, m) , 2.10-2.70 (4H, m), 3.10-3.45 (3H, ) , 3.57 (4H, broad s), 3.85-
4.35 (4H, m), 4.50-4.90 (4H, m) , 5.15-5.50 (4H, m), 5.80-6.10 (2H, ), 6.81 (IH, s), 7.42 (IH, s)
Example 9-2)
To a solution of allyl (4R,5S,6S)-3-[(2R,4S)-1- allyloxycarbonyl-2-{2-(l-methylimidazol-5- yl)ethyl}pyrrolidin-4-yl]thio-6-[ (IR)-1-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (1.55 g) in acetone (7 ml) was added iodomethane (1.77 ml) with stirring at ambient temperature and then allowed to stand overnight. The reaction mixture was evaporated in vacuo and dried in vacuo for 1 hour to give allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-{2-(l,3- dimethyl-4-imidazolio)ethyl}pyrrolidin-4-yl]thio-6-[ (1R)- l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate iodide (1.95 g).
This compound was immediately used as the starting compound for the next step.
Example 9-3)
To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{2-(1,3-dimethyl-4- imidazolio)ethyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- • hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate iodide (1.95 g), triphenylphosphine (75 mg), acetic acid (0.65 ml) and tetrakis(triphenylphosphine)palladium(0) (98 mg) in a mixture of tetrahydrofuran (20 ml) and ethanol (20 ml) was added dropwise tributylthin hydride (3.06 ml) at ambient temperature with stirring. The mixture was stirred at the same temperature for 30 minutes. The resulting precipitates were collected by filtration, washed with tetrahydrofuran (40 ml), dried in vacuo and dissolved in water (30 ml). The solution was chromatographed on
nonionic adsorption resin, Diaion HP-20 (80 ml) eluting in turn with water (250 ml) and 5% aqueous acetone (600 ml). The fractions containing the desired compound were collected and evaporated in vacuo. The resulting residue was passed through ion exchange resin, Amberlist A-26 (Cl- typβ) (20 ml) eluting with water (80 ml). The eluate was lyophilized to give (4R,5S,6S)-3-[ (2R,4S)-2-{2-( 1,3- dimethyl-4-imidazolio)ethyl}pyrrolidin-4-yl]thio-6-[ (IR)- 1-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (520 mg) . IR (Nujol) : 1740, 1580-1570 cm-1
NMR (D20, δ) : 1.23 (3H, d, J=7.21Hz), 1.30 (3H, d, J=6.35Hz), 1.65-1.85 (IH, m) , 2.10-2.40 (2H, m) , 2.70-2.90 (3H, m) , 3.30-3.55 (3H, ) , 3.72 (IH, dd, J=6.80, 12.5Hz), 3.79 (3H, s), 3.85 (3H, s),
3.95-4.35 (3H, ) , 7.30 (IH, s), 8.61 (IH, s) FAB-MS : 435 (M+)
Example 9-4) To a solution of allyl (4R,5S,6S)-3-[ (2R,4S)-1- allyloxycarbonyl-2-{2-(l-methylimidazol-5- yl)ethyl}pyrrolidin-4-yl]thio-6-[ (IR)-1-hydroxyethyl]-4- methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (4.33 g) in acetone (20 ml) was added iodoacetamide (4.41 g) with stirring at ambient temperature and then allowed to stand overnight. The reaction mixture was evaporated in vacuo and dried in vacuo for 1 hour to give allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-{2-(1- carbamoylmethyl-3-methyl-4-imidazolio)ethyl}pyrrolidin-4- yl]thio-6-[ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate iodide (5.80 g) .
This compound was immediately used as the starting compound for the next step.
Example 9-5)
(4R,5S,6S)-3-[(2R,4S)-2-{2-(l-Carbamoylmethyl-3- methyl-4-imidazolio)ethyl}pyrrolidin-4-yl]thio-6-[ (IR)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride (201 mg) was obtained in substantially the same manner as that of Example 3-7). IR (Nujol) : 1730, 1670, 1550 cm-1
NMR (D20, δ) : 1.23 (3H, d, J=7.19Hz), 1.30 (3H, d, J=6.35Hz), 1.70-1.90 (IH, ) , 2.10-2.40 (2H, m) , 2.70-2.95 (3H, m) , 3.30-3.55 (3H, m) , 3.60-3.80 (2H, m), 8.35 (3H, s), 3.90-4.35 (3H, ) , 5.06
(2H, s), 7.37 (IH, s), 8.77 (IH, s) FAB-MS : 478 (M+)
Example 10-1) Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-(3- methyl-4-pyridylmethyl)pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylate was obtained in 42.7% yield in substantially the same manner as that of Example 1-9). IR (CHC13) : 1774, 1697 cm-1
NMR (CDCI3, δ) : 1.24 (3H, d, J=6.7Hz), 1.36 (3H, d, J=6.3Hz), 1.5-1.8 (IH, ) , 2.2-2.5 (4H, m) , 3.2-4.4 (10H, m), 4.4-4.9 (4H, ) , 5.2-5.6 (4H, m), 5.8-6.2 (2H, m) , 7.0-7.1 (IH, m) , 8.3-8.4 (2H, m)
Example 10-2)
Allyl (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2- { (l,3-dimethyl-4-pyridinio)methyl}pyrrolidin-4-yl]thio-6- [ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate iodide was obtained in 95% yield in substantially the same manner as that of Example 2-8).
IR (CH2C12) : 1772, 1699 cm-1 NMR (CDCI3, δ) : 1.27 (3H, d, J=6.7Hz), 1.36 (3H,
d, J=6.3Hz), 2.18 (3H, s), 1.8-4.4 (12H, m) , 4.4-5.0 (4H, m), 5.2-5.6 (4H, m) , 5.8-6.2 (2H, m), 7.8-7.9 (IH, ) , 8.7-8.8 (2H, )
Example 10-3)
(4R,5S,6S)-3-[ (2R,4S)-2-{(l,3-Dimethyl-4- pyridinio)methyl}pyrrolidin-4-yl]thio-6-[ (1R)-1- hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2- ene-2-carboxylic acid chloride was obtained in 28.5% yield in substantially the same manner as that of Example 3-10). IR (KBr) : 1755 cm-1 NMR (CDC13, δ) : 1.20 (3H, d, J=7.2Hz), 1.28 (3H, d, J=6.4Hz), 1.7-1.9 (IH, m) , 2.51 (3H, s), 2.7- 2.9 (IH, m), 3.3-4.2 (10H, m) , 4.31 (3H, s), 7.89 (IH, d, J=6.1Hz), 8.5-8.6 (2H, m)
Example 11-1)
Ally (4R,5S,6S)-3-[ (2R,4S)-l-allyloxycarbonyl-2-(2- hydroxymethyl-4-pyridylmethyl)pyrrolidin-4-yl]thio-6- [ (lR)-l-hydroxyethyl]-4-methyl-7-oxo-l- azabicyclo[3.2.0]hept-2-ene-2-carboxylate was obtained in 20.9% yield in substantially same manner as that of Example 1-4) from (4R)-2-diazo-4-[ (2R,3S)-3-[ (1R)-1- hydroxyethyl]-4-oxoazetidin-2-yl]-3-oxopentanoate and (2R,4S)-l-allyloxycarbonyl-4-benzoylthio-2-(2- acetoxymethyl-4-pyridylmethylpyrrolidine. IR (CH2C12) : 1772, 1700 cm-1
NMR (CDCI3, δ) : 1.24 (3H, d, J=7.4Hz), 1.36 (3H, d, J=6.2Hz), 1.6-1.8 (IH, m) , 1.8-2.1 (IH, ) , 2.2-2.5 (IH, ) , 2.7-2.9 (IH, m) , 3.2-3.4 (4H, m), 4.05-4.35 (4H, m), 4.6-4.95 (4H, m) , 4.79 (2H, m), 5.2-5.6 (4H), 5.9-6.1 (2H, m) , 7.00-7.2 (2H, m), 8.47 (IH, d, J=5.0Hz) FAB-MS (m/z) : 558.1 (M+)
Example 11-2)
(4R,5S,6S)-3-[ (2R,4S)-2-{(1-methyl-2-hydroxymethyl- 4-pyridinio)methyl}pyrrolidin-4-yl]thio-6-[ (IR)-1- hydroxyethyl-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene- 2-carboxylic acid chloride (180 mg) was obtained by the similar procedure as Example 2-5) followed by Example 3- 8).
IR (KBr) : 3500-3300, 2970, 1760, 1645, 1580 cm-1
NMR (CDC13, δ) : 1.21 (3H, d, J=7.2Hz), 1.29 (3H, d, J=6.4Hz), 1.75-1.95 (IH, m) , 2.65-2.90 (IH, m), 3.30-3.65 (5H, m) , 3.72 (IH, dd, J=6.9 and 12.5Hz), 4.00-4.15 (IH, m) , 4.15-4.35 (6H, m) , 4.22 (3H, s), 5.02 (2H, s), 7.85-7.95 (IH, m), 8.15-8.20 (IH, m), 8.72 (IH, d, J=6.5Hz) FAB-MS (m/z) : 448.2 (M-Cl")