WO1995017405A1 - Indoline derivatives, method of preparation and their use as pharmaceuticals - Google Patents
Indoline derivatives, method of preparation and their use as pharmaceuticals Download PDFInfo
- Publication number
- WO1995017405A1 WO1995017405A1 PCT/EP1994/004220 EP9404220W WO9517405A1 WO 1995017405 A1 WO1995017405 A1 WO 1995017405A1 EP 9404220 W EP9404220 W EP 9404220W WO 9517405 A1 WO9517405 A1 WO 9517405A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- compounds
- pharmaceutically acceptable
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Definitions
- This invention relates to tricyclic indoline derivatives, to processes for their preparation, to pharmaceutical compositions containing them and to their medical use.
- R1 is hydrogen, halogen or C-
- R 2 is a group of formula -CR 3 R 4 (CH 2 ) p NR 5 COR 6 ;
- R 3 , R 4 and R 5 which may be the same or different, are hydrogen or C-
- an alkyl group may be a straight chain or branched chain alkyl group.
- suitable alkyl groups include C-
- a preferred alkyl group is methyl.
- a halogen substituent may be, for example, fluorine, chlorine, bromine or iodine.
- R 2 preferably represents a group -CR 3 R (CH2) p NHCOR 6 wherein R 3 and R 4 each independently represent hydrogen or C-1.3 alkyl (e.g. methyl), p is an integer of 1 or 2, especially 1, and R 6 is C-
- R 1 examples include hydrogen, halogen (e.g. chlorine) and C -3 alkyl (e.g. methyl).
- R 1 examples include hydrogen, halogen (e.g. chlorine) and C -3 alkyl (e.g. methyl).
- a preferred group of compounds of the invention are compounds of formula
- Particular compounds according to the present invention include: N-[2-(2,3,8,9-Tetrahydro-7H-pyrano[2 I 3-g]indol-1-yl)-ethyl]-acetamide; N-[2-(2 I 3,7,8-Tetrahydro-1H-furo[2,3-g]indol-1-yl)-ethyl]-acetamide; N-[2-(5-Chloro-2,3 I 7,8-tetrahydro-1H-furo[2,3-g]indol-1-yl)-ethyl]-acetamide; Cyclopropanecarboxylic acid [2-(2,3.7.8-tetrahydro-1H-furo[2.3-g]indol-1-yl)- ethyl]-amide; and pharmaceutically acceptable salts and solvates thereof.
- a particularly suitable compound according to the present invention is N-[2-(2,3,7,8-Tetrahydro-1 H-furo[2,3-g]indol-1 -yl)-ethyl]-acetamide, and pharmaceutically acceptable salts and solvates thereof.
- Pharmaceutically acceptable salts of the compounds of formula (I) include those derived from pharmaceutically acceptable inorganic and organic acids.
- suitable acids include hydrochloric, hydrobromic, sulphuric, nitric, perchloric, fumaric, rnaleic, phosphoric, glycollic, lactic, salicylic, succinic, toluene-p-sulphonic, tartaric, acetic, citric, methanesulphonic, formic, benzoic, malonic, naphthalene-2-sulphonic and benzenesulphonic acids.
- a particularly suitable pharmaceutically acceptable salt of the compounds of formula (I) is the hydrochloride salt.
- Other acids such as oxalic, while not, in themselves pharmaceutically acceptable, may be useful as intermediates in obtaining the compounds of the invention and their pharmaceutically acceptable acid addition salts.
- references hereinafter to a compound of formula (I) includes the compound and its pharmaceutically acceptable salts.
- the compounds of formula (I) may contain at least one asymmetric carbon atom and may exist as stereoisomers.
- the compounds of formula (I) thus include the d- and l-isomers and mixtures, for example racemic mixtures, thereof.
- the compounds of formula (I) are of use in the treatment of disorders which arise from a disturbed functioning of the melatonin system.
- the compounds of formula (I) may be used in the treatment of chronobiological disorders, especially in the elderly population, glaucoma, cancer, psychiatric disorders, osteoporosis, neurodegenerative diseases or neuroendocrine disorders arising as a result of or influenced by the melatonin system.
- Chronobiological disorders include seasonal affective disorders (SAD), primary and secondary insomnia disorders, primary and secondary hypersomnia disorders, sleep-wake schedule disorders (including • advanced phase type, delayed phase type, disorganised type and frequently-changing type) and other dyssomnias, especially those caused by ageing, dementias, blindness shift work and by rapid time-zone travel, commonly known as jet lag.
- Cancers which may be treated with a compound of formula (I) include solid tumours, e.g. melanomas and breast carcinomas.
- Psychiatric disorders which may be related to altered melatonin function or influenced by melatonin and circadian rhythms include mood disorders (including bipolar disorders of all types, major depression, dysthymia and other depressive disorders), psychoactive substance dependence and abuse, anxiety disorders (including panic disorder, agoraphobia, social phobia, simple phobia, obsessive-compulsive disorder, post-traumatic stress disorder and generalised anxiety disorder), schizophrenia, epilepsy and epileptic seizures (including grand mal, petit mal, myoclonic epilepsy and partial seizures), disorders of involuntary movement (including those due to Parkinson's disease, and drug- induced involuntary movements) and dementias (including primary degenerative dementia of the Alzheimer type).
- mood disorders including bipolar disorders of all types, major depression, dysthymia and other depressive disorders
- psychoactive substance dependence and abuse anxiety disorders (including panic disorder, agoraphobia, social phobia, simple phobia, obsessive-compulsive disorder, post-traumatic stress disorder and generalised
- Neurodegenerative diseases which may be related to altered melatonin function or influenced by melatonin and biological rhythms include multiple sclerosis and stroke.
- Neuroendocrine disorders which may be related to altered melatonin function or influenced by melatonin and biological rhythms include peptic ulceration, emesis, psoriasis, benign prostatic hyperplasia, hair condition and body weight.
- Particular neuroendocrine disorders which may be treated include those relating to the regulation of reproductive maturation and function include idiopathic delayed puberty, sudden infant death, premature labour, infertility, antifertility, premenstrual syndrome (including late luteal phase dysphoric disorder) and sexual dysfunction (including sexual desire disorders, male erectile disorder, post-menopausal disorders and orgasm disorders).
- the compounds may also be used to manipulate breeding cycles, body weight, coat colour and oviposition of susceptible hosts, including birds, insects and mammals.
- the compounds of formula (I) may also have sedative, anti- inflammatory and analgesic effects, effects on the microcirculation and immunomodulant effects and may be useful for the treatment of hypertension, migraine, cluster headache, arthritis, regulation of appetite and in the treatment of eating disorders such as obesity, anorexia nervosa and bulimia nervosa.
- a compound of formula (I) for use in therapy, in particular in human medicine. It will be appreciated that use in therapy embraces but is not necessarily limited to use of a compound of formula (I) as an active therapeutic substance.
- a compound of formula (I) for use in the preparation of a medicament for use in the treatment of conditions associated with a disturbed functioning of the melatonin system.
- a method for the treatment of a mammal, including man comprising administration of an effective amount of a compound of formula (I), in particular for the treatment of conditions associated with a disturbed functioning of the melatonin system.
- a compound of formula (I) may be administered as the raw chemical it is preferable to present the active ingredient as a pharmaceutical formulation.
- the invention thus further provides a pharmaceutical formulation comprising a compound of formula (I) together with one or more pharmaceutically acceptable carriers therefor.
- the carrier(s) must be 'acceptable' in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- a process of preparing a pharmaceutical formulation which process comprises mixing a compound of formula (I) with one or more pharmaceutically acceptable carriers therefor.
- compositions include those suitable for oral, rectal, vaginal, nasal, topical or parenteral (including intramuscular, subcutaneous and intravenous) administration or in a form suitable for administration by inhalation or insufflation.
- the formulations may, where appropriate, be conveniently presented in discrete dosage units and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing into association the active compound with liquid carriers or finely divided solid carriers or both and then, if necessary, shaping the product into the desired formulation.
- the pharmaceutical compositions may take the form of, for example, tablets or capsules prepared by conventional means with pharmaceutically acceptable excipients such as binding agents (e.g. pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g. lactose, microcrystalline cellulose or calcium phosphate); lubricants (e.g. magnesium stearate, talc or silica); disintegrants (e.g. potato starch or sodium starch glycollate); or wetting agents (e.g. sodium lauryl sulphate).
- binding agents e.g. pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose
- fillers e.g. lactose, microcrystalline cellulose or calcium phosphate
- lubricants e.g. magnesium stearate, talc or silica
- disintegrants e.g. potato starch or sodium starch glycollate
- Liquid preparations for oral administration may take the form of, for example, solutions, syrups or suspensions, or they may be presented as a dry product for constitution with water or other suitable vehicle before use.
- Such liquid preparations may be prepared by conventional means with pharmaceutically acceptable additives such as suspending agents (e.g. sorbitol syrup, methyl cellulose or hydrogenated edible fats); emulsifying agents (e.g. lecithin or acacia); non-aqueous vehicles (e.g. almond oil, oily esters or ethyl alcohol); and preservatives (e.g. methyl or propyl-g-hydroxybenzoates or sorbic acid).
- suspending agents e.g. sorbitol syrup, methyl cellulose or hydrogenated edible fats
- emulsifying agents e.g. lecithin or acacia
- non-aqueous vehicles e.g. almond oil, oily esters or ethyl alcohol
- preservatives e.g
- compositions may take the form of buccal or sub-lingual tablets, drops or lozenges formulated in conventional manner.
- the compounds may be formulated as creams, gels, ointments or lotions or as a transdermal patch.
- Such compositions may for example be formulated with an aqueous or oily base with the addition of suitable thickening, gelling, emulsifying, stabilising, dispersing, suspending and/or colouring agents.
- the compounds of the invention may be formulated for parenteral administration by injection, conveniently intravenous, intramuscular or subcutaneous injection, for example by bolus injection or continuous intravenous infusion.
- Formulations for injection may be presented in unit dosage form e.g. in ampoules or in multi-dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilising and/or dispersing agents.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g. sterile pyrogen-free water, before use.
- the compounds of the invention may also be formulated in rectal compositions such as suppositories or retention enemas, e.g. containing conventional suppository bases such as cocoa butter or other glyceride.
- Pessaries for vaginal administration may be formulated in a similar manner.
- the compounds of the invention may be used, for example, as a liquid spray, as a powder or in the form of drops.
- the compounds according to the invention are conveniently delivered in the form of an aerosol spray presentation from pressurised packs or a nebuliser, with the use of a suitable propellant, e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- a suitable propellant e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- Capsules and cartridges of e.g. gelatin for use in an inhaler or insufflator may be formulated containing a powder mix of a compound of the invention and a suitable powder base such as lactose or starch.
- compositions described above may be presented in a conventional manner associated with controlled release forms.
- the active ingredient may conveniently be presented in unit dose form.
- a convenient unit dose formulation contains the active ingredient in an amount of from about 0.01 mg to about 200mg.
- the compound may be administered in single or divided doses and may be administered one or more times, for example 1 to 4 times per day.
- a proposed dose of the compounds of the invention for oral, rectal, vaginal, intranasal, topical or parenteral administration to man (of approximately 70kg bod weight) for the treatment of conditions associated with a disturbed functioning of the melatonin system is 0.01 to 200mg of the active ingredient per unit dose which could be administered, for example, 1 to 4 times per day.
- a unit dose will preferably contain from 0.1 to 200mg of the active ingredient.
- a unit dose for parenteral administration will preferably contain 0.1 to 5 mg of the active ingredient.
- Aerosol formulations are preferably arranged so that each metered dose or 'puff delivered from a pressurised aerosol contains 0.2 mg to 2 mg of a compound of the invention, and capsules and cartridges delivered from an insufflator or an inhaler, contain 0.2 mg to 20 mg of a compound of the invention.
- the overall daily dose by inhalation with an aerosol will be within the range 1 mg to 100 mg. Administration may be several times daily, for example from 2 to 8 times, giving for example 1 , 2 or 3 doses each time.
- Dosages of the compounds of the invention for rectal, vaginal, intranasal or topical administration are similar to those for oral administration.
- the compounds of the invention may, if desired, be administered in combination with one or more other therapeutic agents such as a hypnotic or antidepressant agent, or an anti-cancer agent such as tamoxiphen, or in combination with radiation therapy to treat cancer.
- a hypnotic or antidepressant agent such as a hypnotic or antidepressant agent, or an anti-cancer agent such as tamoxiphen
- radiation therapy to treat cancer.
- the combinations referred to above may conveniently be presented for use in the form of a pharmaceutical formulation and thus pharmaceutical formulations comprising a compound of formula (I) together with at least one other therapeutic agent and one or more pharmaceutically acceptable carriers therefor comprise a further aspect of the invention.
- the compounds of formula (I) may be administered either sequentially or simultaneously by any convenient route.
- each component of the combination will in general be that employed for each component when used alone.
- Compounds of formula (I) and pharmaceutically acceptable salts and solvates (e.g. hydrates) thereof may be prepared by methods known in the art for the preparation of analogous compounds.
- the compounds of formula (I) may be prepared by the methods outlined below and which form a further aspect of the invention.
- R1, R 3 , R 4 , R ⁇ , n and p are as defined for formula (I).
- a compound of formula (I) may be prepared by acylation of a compound of formula (II)
- Suitable acylating agents which may conveniently be used in the above process include acid anhydrides and acid halides.
- the reaction is conveniently effected in a suitable solvent such as an ether (e.g. diethyl ether, tetrahydrofuran or dioxan), a hydrocarbon such as toluene or a halogenated hydrocarbon (e.g. dichloromethane), preferably in the presence of a base such as pyridine or a tertiary amine (e.g, diisopropylethylamine), at a temperature in the range of 0 to 100°C, preferably 0 to 20°C.
- a suitable solvent such as an ether (e.g. diethyl ether, tetrahydrofuran or dioxan), a hydrocarbon such as toluene or a halogenated hydrocarbon (e.g. dichloromethane), preferably in the presence of a base such as pyridine or a ter
- the reduction may conveniently be effected using a boron hydride reducing agent such as borane- tetrahydrofuran complex in an ether solvent (e.g. tetrahydrofuran) optionally in the presence of a suitable acid (e.g. trifluoroacetic acid, hydrochloric acid or the like) at a suitable temperature, for example from 0° to 100°C.
- a suitable acid e.g. trifluoroacetic acid, hydrochloric acid or the like
- the reduction may employ catalytic hydrogenation in the presence of a noble metal catalyst, such as platinum, palladium or the like, in a suitable organic solvent, such as an alcoholic solvent, e.g. ethanol, conveniently at a temperature in the range of 0° to 100°C, aptly at room temperature.
- Compounds of formula (II) in which R 5 is C ⁇ _ ⁇ alkyl may be prepared by N- alkylation of compounds of formula (II) in which R 5 is hydrogen using standard procedures.
- HalCR 3 R 4 (CH2) p - CN in which Hal is a halogen atom (fluorine, bromine, chlorine or iodine), suitably in the presence of a base.
- the alkylation may be carried out under standard conditions.
- the reaction may be effected in a ketonic solvent in the presence of an alkali or alkaline earth metal carbonate (e.g. potassium carbonate) at an elevated temperature (e.g. under reflux).
- the reaction may be effected in dimethylformamide in the presence of an alkali metal hydride (e.g. sodium hydride) at about ambient temperature.
- an alkali metal hydride e.g. sodium hydride
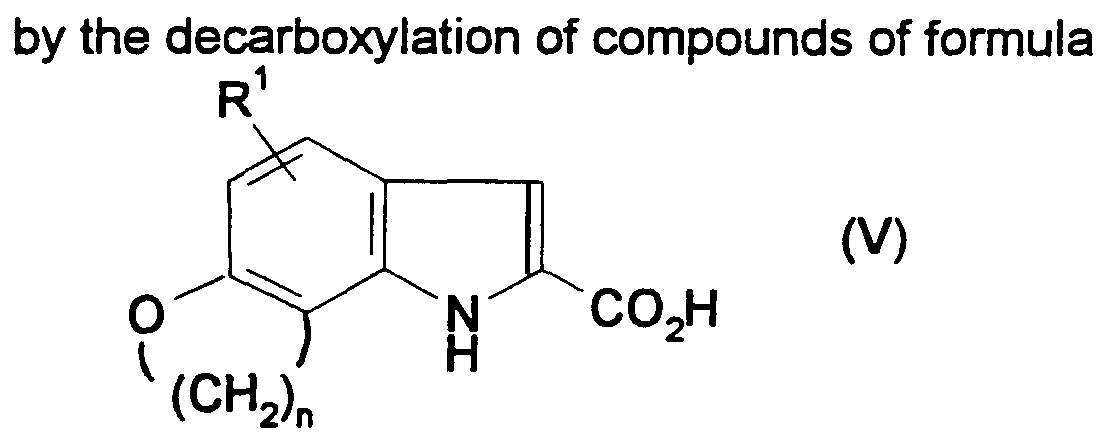
- compounds of formula (V) may be decarboxylated by heating the compounds at a very high temperature (e.g. at about 250°C), optionally in the presence of copper and a suitable copper salt, such as copper (I) oxide, cuprous oxide and the like.
- a very high temperature e.g. at about 250°C
- copper and a suitable copper salt such as copper (I) oxide, cuprous oxide and the like.
- the cyclisation reaction may conveniently be effected by heating (VI) to reflux in an aromatic hydrocarbon solvent (e.g. xylene).
- Conversion of the so-formed ester to the corresponding acid of formula (V) involves routine hydrolysis, for example using a base such as a hydroxide (e.g. sodium hydroxide) at an elevated temperature (e.g. under reflux).
- a base such as a hydroxide (e.g. sodium hydroxide) at an elevated temperature (e.g. under reflux).
- alkyl azidoacetate in the presence of a strong base (e.g. potassium tert- butoxide) at a temperature of from -20° to +10°C.
- a strong base e.g. potassium tert- butoxide
- X represents a halogen atom (e.g. fluorine) and R is a d- ⁇ alkyl group, such as methyl or ethyl.
- Preparation of compounds of formula (IV) typically involves addition of a solution of compounds of formula (VIII) (suitably in a chlorinated organic solvent such as dichloromethane, dichloroethane or the like) to an acidic medium, such as halogenated acetic acid and/or acetic anhydride optionally in a chlorinated organic solvent as described above.
- an acidic medium such as halogenated acetic acid and/or acetic anhydride optionally in a chlorinated organic solvent as described above.
- the addition is carried out at 0°C under an inert atmosphere such as nitrogen.
- the reaction may be progressed by allowing the reagents to reach room temperature, and stirring for about 18 to 20 hours.
- the resulting mixture is generally treated with a base, such as an alkali metal hydrox
- acylating agents such as acid anhydrides and acid halides.
- a halogenated acetic anhydride (aptly trif luoroacetic anhydride) in a chlorinated organic solvent as described above is added to a solution of a compound of formula (IX) in a basic solvent, such as triethylamine and the like.
- a basic solvent such as triethylamine and the like.
- the addition is carried out at 0°C under an inert atmosphere such as nitrogen.
- acetal derivatives are aptly reacted with a suitable acetal derivative, conveniently in the presence of a base (an alkali metal carbonate being an example of an appropriate base), with heating over a prolonged period of time (such as 40 to 65 hours) at an elevated temperature in the range of 90° to 110°C, in order to yield compounds of formula (IX).
- a base an alkali metal carbonate being an example of an appropriate base
- R 1 represents halogen
- compounds of formula (I) in which R 1 represents halogen may be prepared via compounds of formulae (II), (III) and (IV) wherein R 1 represents halogen employing process steps substantially as hereinbefore described.
- compounds of formula (IV) in which R 1 represents halogen are prepared from compounds of formula (XI) wherein R 1 represents halogen and the dotted line indicates an optional double bond.
- a compound of formula (XI) is dissolved in an organic solvent, such as an alcoholic solvent, acidified, and the mixture subjected to stirring and refluxing for a suitable length of time to yield a corresponding compound of formula (IV).
- an organic solvent such as an alcoholic solvent
- compounds of formula (XI) wherein R 1 represents hydrogen as described above may be prepared from compounds of formula (IV) wherein R 1 represents hydrogen by reaction of the latter with an appropriate anhydride in an acidic medium.
- a compound of formula (I) may be prepared from a compound of formula (XII) suitably by stirring for several hours (17 to 19 hours) in a basic medium, conveniently an alkali metal hydroxide or the like, under an inert atmosphere such as nitrogen, followed by refluxing for 1 to 2 hours.
- a compound of formula (XII) may be prepared by acylation of a compound of formula (II) employing acylation techniques substantially as hereinbefore described.
- a compound of formula (I) may be prepared by alkylating a saturated compound of formula (IV).
- alkylation is achieved by refluxing a compound of formula (IV) together with an alkylating agent over several days.
- Suitable alkylating agents include
- HalCR 3 R 4 (CH 2 ) p NR 5 COR 6 (wherein Hal, R 3 , R 4 , R 5 , R 6 and p are as hereinbefore defined), ANR 5 COR 6 wherein A represents a 2-membered alkyl chain or the like.
- a compound of formula (I) may be prepared by subjecting a protected derivative of a compound of formula (I) or a salt thereof to reaction to remove the protecting group or groups.
- the following reactions may according to process (D), if desirable and/or if necessary, be carried out in any appropriate sequence: (i) removal of any protecting groups; and (ii) conversion of a compound of formula (I) or a salt thereof into a pharmaceutically acceptable salt thereof.
- the protecting groups used in the preparation of compounds of formula (I) may be used in conventional manner. See for example 'Protective Groups in Organic Chemistry' Ed.J.F.W. McOmie (Plenum Press 1973) or 'Protective Groups in Organic Synthesis' by Theodora W Greene (John Wiley and Sons 1991).
- compounds of formula (I) may be prepared from other compounds of formula (I) by interconversion reactions.
- acid addition salts of compounds of formula (I) may be prepared from a corresponding compound of formula (I) by suitable acid treatment, for example addition of a suitable acid, such as hydrochloric acid, generaly in the presence of an organic solvent such as an alcohol or ester.
- a suitable acid such as hydrochloric acid
- an organic solvent such as an alcohol or ester.
- an acid may be added dropwise to a solution of a compound of formula (I) in an appropriate organic solvent as described above, optionally under an inert atmosphere such as nitrogen.
- a compound of the invention for example as an acid addition salt
- this may be achieved by treating the free base of general formula (I) with an appropriate acid, preferably with an equivalent amount, or with creatinine sulphate in a suitable solvent (e.g. ethanol).
- a suitable solvent e.g. ethanol
- Compounds of the invention may be isolated in association with solvent molecules by crystallisation from or evaporation of an appropriate solvent.
- Individual enantiomers of the compounds of the invention may be prepared from racemates by resolution using methods known in the art for the separation of racemic mixtures into their constituent enantiomers, for example using chiral HPLC.
- the general methods indicated above for the preparation of the compounds of the invention may also be used for the introduction of the desired groups at an intermediate stage in the preparation of the required compound. It should therefore be appreciated that in such multi-stage processes, the sequence of reactions should be chosen in order that the reaction conditions do not affect groups present in the molecule which are desired in the final product.
- THF means tetrahydrofuran.
- EtOH means ethanol.
- EtOAc means ethyl acetate.
- DMF means dimethylformamide.
- NH 3 means commercially available aqueous ammonium hydroxide.
- TFA means trifluoroacetic acid.
- TFAA means trifluoroacetic anhydride.
- Dried means dried over anhydrous sodium sulphate (unless otherwise stated). Chromatography was performed on silica (Merck 9385 unless otherwise stated). System A is dichloromethane/ethanol/aqueous ammonia.
- T.l.c. means thin layer chromatography on silica gel. The n.m.r. analysis was conducted at 250mHz.
- Bromoacetaldehyde diethyl acetal (11.8ml) was added to a mixture of chroman- 5-yl-amine (5.85g) (prepared according to J. Heterocyclic Chem.. (1973), Vol 10 (4) page 623), and potassium carbonate (10.84g) in dry DMF (70ml) at room temperature under N 2 .
- the mixture was heated at 100°C for 60h.
- the cooled mixture was partitioned between water (800ml) and ether (3x200ml).
- the combined organic extracts were washed with brine water 1:1 (2x200ml) and dried.
- the solvent was evaporated and the residue purified by flash column chromatography on silica.
- Trifluoroacetic anhydride (4.12ml) was added dropwise to a cooled (0 ⁇ C) stirring solution of the intermediate 20 (6.67g) and triethylamine (4.06ml) in dichloromethane (100ml) under nitrogen. The mixture was warmed to room temperature and stirred for V ⁇ h. The reaction mixture was partitioned between water and dichloromethane. The aqueous phase was re-extracted with dichloromethane.
- Example 3 The title compound of Example 3 (334mg) was dissolved in ethyl acetate (20ml) and was treated with ethereal HCl (1.35ml). This was stirred at room temperature for 2h and then the solvent was evaporated to give the title compound as a pale green powder (383mg), m.p. 152-154°C.
- T.l.c. System A 100:8:1, Rf 0.41.
- Example 9 Compounds of formula (I) have been included in pharmacy formulations, and details of such formulations are given below. TABLETS FOR ORAL ADMINISTRATION A. Direct Compression
- the active ingredient was sieved and blended with the excipients.
- the resultant mix was compressed into tablets using a tablet machine fitted with appropriately sized concave punches.
- the active ingredient was sieved through a suitable sieve and blended with lactose, starch and pregelatinised maize starch. Suitable volumes of purified water were added and the powders were granulated. After drying, the granules were screened and blended with the magnesium stearate. The granules were then compressed into tablets using suitable diameter punches.
- Tablets of other strengths may be prepared by for example altering the ratio of active ingredient to lactose or the compression weight and using punches to suit.
- the active ingredient and lactose were mixed together and granulated by the addition of purified water.
- the granules obtained after mixing were dried and passed through a screen, and the resulting granules were then mixed with the other tablet core excipients. The mix is compressed into tablets.
- the tablets may be film coated with suitable film-forming materials, such as hydroxypropyl methylcellulose, using standard techniques.
- suitable film-forming materials such as hydroxypropyl methylcellulose
- the tablets may be sugar coated, or enteric coated.
- Opaspray white is a proprietory film coating suspension, obtainable from Colorcon Ltd, UK, which contains hydroxypropyl methylcellulose and titanium dioxide.
- the tablets were film coated using the coating suspension in conventional film coating equipment.
- the active ingredient, anhydrous monosodium citrate, sodium bicarbonate and aspartame were mixed together and granulated by the addition of a solution of the polyvinylpyrrolidone in the alcohol.
- the granules obtained after mixing were dried and passed through a screen, and the resulting granules were then mixed with the sodium benzoate and flavourings.
- the granulated material was compressed into tablets using a machine fitted with 20mm punches.
- a rotary machine fitted with 20mm punches may also be used for tabletting.
- Liquid formulations were prepared by slow addition of active ingredient into the other ingredients at 35-50°C with constant mixing (amounts are given as percentage w/w).
- liquid formulations were filled into hard gelatin capsules, each capsule containing 25mg of active ingredient.
- a form of directly compressible starch is A form of directly compressible starch.
- the active ingredient was sieved and blended with the excipients.
- the mix was filled into size No. 2 hard gelatin capsules using suitable machinery.
- Other doses may be prepared by altering the fill weight and if necessary changing the capsule size to suit.
- the hydroxypropylmethylcellulose was dispersed in hot water, cooled and then mixed with an aqueous solution containing the active ingredient and the other components of the formulation. The resultant solution was adjusted to volume and mixed. The syrup was clarified by filtration.
- the aluminium monostearate was dispersed in about 90% of the fractionated coconut oil.
- the resulting suspension was heated to 115°C while stirring and then cooled.
- the sweetening agent, flavour and colour were added and the active ingredient was suitably dispersed.
- the suspension was made up to volume with the remaining fractionated coconut oil and mixed.
- Active ingredient/lactose granule* 49.0 Compressible sugar NF 50.5 Magnesium Stearate BP 0.5 Compression Weight 100.0
- the active ingredient was sieved through a suitable sieve, blended with the excipients and compressed using suitable punches. Tablets of other strengths may be prepared by altering either the ratio of active ingredient to excipients or the compression weight and using punches to suit.
- a suspension of the active ingredient in molten Witepsol was prepared and filled using suitable machinery, into 1g size suppository moulds.
- the active ingredient was dissolved in a portion of the Sodium Chloride Intravenous Infusion, the solution made to volume with the Sodium Chloride Intravenous Infusion, and the solution thoroughly mixed.
- the solution was filled into clear, Type 1, glass 1ml ampoules and sealed by fusion of the glass under a nitrogen or air headspace.
- the ampoules were sterilised by autoclaving at 121 °C for not less than 15 minutes. Alternatively the solution may be sterilised by filtration prior to filling aseptically into ampoules.
- the active ingredient was micronised in a fluid energy mill to a fine particle size range prior to blending with normal tabletting grade lactose in a high energy mixer.
- the powder blend was filled into No 3 hard gelatin capsules on a suitable encapsulating machine.
- the contents of the cartridges were administered using a powder inhaler such as the Glaxo Rotahaler.
- the active ingredient was micronised in a fluid energy mill to a fine particle size range.
- the oleic acid was mixed with the trichloromethane at a temeprature of 10-15°C and the micronised drug was mixed into the solution with a high shear mixer.
- the suspension was metered into aluminium aerosol cans and suitable metering valves, delivering 85mg of suspension, were crimped onto the cans and the dichlorodifluoromethane was pressure filled into the cans through the valves.
- Active ingredient 7.0 Sodium Chloride BP 0.9 Purified Water BP to 100 Shot Weight 100mg (equivalent to 7mg active ingredient)
- the active ingredient and sodium chloride were dissolved in a portion of the water, the solution made to volume with the water and the solution thoroughly mixed.
- the pH may be adjusted to facilitate solution of the active ingredient, using acid or alkali and/or subsequently adjusted ideally to near neutrality taking into account the pH for optimum stability.
- suitable buffer salts may be used.
- the solution may be preserved with, for example, benzalkanium chloride and phenylethyl alcohol, for a multi-dose nasal spray.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Anesthesiology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Psychiatry (AREA)
- Obesity (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Indole Compounds (AREA)
- Furan Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description












Claims





Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU12743/95A AU684877B2 (en) | 1993-12-22 | 1994-12-20 | Indoline derivatives, method of preparation and their use as pharmaceuticals |
| JP7517174A JPH09507057A (en) | 1993-12-22 | 1994-12-20 | Indoline derivatives, their production and their use as a medicine |
| US08/652,460 US5633276A (en) | 1993-12-22 | 1994-12-20 | Indoline derivatives, method of preparation and use |
| EP95903817A EP0736028A1 (en) | 1993-12-22 | 1994-12-20 | Indoline derivatives, method of preparation and their use as pharmaceuticals |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB939326192A GB9326192D0 (en) | 1993-12-22 | 1993-12-22 | Chemical compounds |
| GB9326192.3 | 1993-12-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995017405A1 true WO1995017405A1 (en) | 1995-06-29 |
Family
ID=10747030
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1994/004220 Ceased WO1995017405A1 (en) | 1993-12-22 | 1994-12-20 | Indoline derivatives, method of preparation and their use as pharmaceuticals |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US5633276A (en) |
| EP (1) | EP0736028A1 (en) |
| JP (1) | JPH09507057A (en) |
| AP (1) | AP535A (en) |
| AU (1) | AU684877B2 (en) |
| CA (1) | CA2179402A1 (en) |
| CO (1) | CO4340623A1 (en) |
| GB (1) | GB9326192D0 (en) |
| IL (1) | IL112097A (en) |
| IS (1) | IS4244A (en) |
| MY (1) | MY131641A (en) |
| SV (1) | SV1994000083A (en) |
| TW (1) | TW291479B (en) |
| WO (1) | WO1995017405A1 (en) |
| ZA (1) | ZA9410056B (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2741799A1 (en) * | 1995-12-04 | 1997-06-06 | Oreal | USE OF MELATONIN IN A COMPOSITION FOR TREATING SKIN SIGNS OF FATIGUE CONDITIONS |
| WO1997032871A1 (en) * | 1996-03-08 | 1997-09-12 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| WO1998052935A1 (en) * | 1997-05-16 | 1998-11-26 | Adir Et Compagnie | Substituted heterocyclic compounds, method for preparing and compositions containing same |
| US5856529A (en) * | 1996-12-10 | 1999-01-05 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US5948817A (en) * | 1997-03-05 | 1999-09-07 | Bristol-Myers Squibb Company | Polycyclic ethyl alkylamide melatonergic agents |
| US6034239A (en) * | 1996-03-08 | 2000-03-07 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| US6211225B1 (en) | 1999-06-30 | 2001-04-03 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US6214869B1 (en) | 1998-06-05 | 2001-04-10 | Bristol-Myers Squibb | Heterocyclic cis cyclopropane derivatives as melatonergic agents |
| EP0990650A4 (en) * | 1997-06-13 | 2002-02-13 | Yamanouchi Pharma Co Ltd | Tricyclic pyrrole or pyrazole derivatives |
| EP1199304A1 (en) * | 1997-03-05 | 2002-04-24 | Takeda Chemical Industries, Ltd. | Bicyclic compounds and pharmaceutical composition containing tricyclic compound for treating or preventing sleep disorders |
| AU774337B2 (en) * | 1999-08-11 | 2004-06-24 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2C, receptor ligands |
| US7008940B1 (en) | 1999-08-20 | 2006-03-07 | Takeda Pharmaceutical Company Limited | Dihydrobenzofuran derivatives, process for the preparing thereof and agents |
| WO2009053440A1 (en) * | 2007-10-25 | 2009-04-30 | Ferrer Internacional S.A. | Indoline compounds |
| WO2010012789A1 (en) * | 2008-07-30 | 2010-02-04 | Ferrer Internacional S. A. | 1,6-dihydro-2h-3-oxa-6-aza-as-indacene compounds |
| WO2018076090A1 (en) | 2016-10-24 | 2018-05-03 | Aché Laboratórios Farmacêuticos S.A. | Compounds, process for obtaining the compounds, pharmaceutical composition, use of the compounds and method for treating psychiatric disorders and/or sleep disorders |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7012090B1 (en) | 2000-03-17 | 2006-03-14 | Alcon, Inc. | Pyranoindoles for treating glaucoma |
| WO2001070745A1 (en) * | 2000-03-17 | 2001-09-27 | Alcon, Inc. | Pyranoindoles for treating glaucoma |
| ES2172415B2 (en) | 2000-07-28 | 2003-11-16 | Univ Madrid Complutense | TREATMENT OF GLAUCOMA AND OCULAR HYPERTENSION THROUGH A MELATONINE ANALOG. |
| US6743448B2 (en) * | 2000-12-11 | 2004-06-01 | Abraham H. Kryger | Topical testosterone formulations and associated methods |
| WO2002079153A1 (en) * | 2001-03-28 | 2002-10-10 | Biocatalytics, Inc. | Method for producing tryptamine derivatives |
| AR059935A1 (en) * | 2006-03-20 | 2008-05-07 | Takeda Pharmaceutical | PHARMACEUTICAL COMPOSITION FOR THE PREVENTION OR TREATMENT OF IRRITABLE COLON SYNDROME |
| KR20090086641A (en) * | 2006-12-08 | 2009-08-13 | 다케다 야쿠힌 고교 가부시키가이샤 | Tricyclic Compounds and Medical Uses thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0043752A1 (en) * | 1980-06-26 | 1982-01-13 | SOCIETE DE RECHERCHES INDUSTRIELLES S.O.R.I. Société anonyme dite: | Pyranoindole derivative and process for its preparation |
| EP0207605A1 (en) * | 1985-05-21 | 1987-01-07 | Pfizer Inc. | Hypoglycemic thiazolidinediones |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4738972A (en) * | 1985-05-21 | 1988-04-19 | Pfizer Inc. | Hypoglycemic thiazolidinediones |
-
1993
- 1993-12-22 GB GB939326192A patent/GB9326192D0/en active Pending
-
1994
- 1994-12-08 TW TW083111439A patent/TW291479B/zh active
- 1994-12-16 IS IS4244A patent/IS4244A/en unknown
- 1994-12-19 MY MYPI94003410A patent/MY131641A/en unknown
- 1994-12-19 ZA ZA9410056A patent/ZA9410056B/en unknown
- 1994-12-20 WO PCT/EP1994/004220 patent/WO1995017405A1/en not_active Ceased
- 1994-12-20 EP EP95903817A patent/EP0736028A1/en not_active Ceased
- 1994-12-20 CA CA002179402A patent/CA2179402A1/en not_active Abandoned
- 1994-12-20 AU AU12743/95A patent/AU684877B2/en not_active Ceased
- 1994-12-20 JP JP7517174A patent/JPH09507057A/en active Pending
- 1994-12-20 AP APAP/P/1994/000706A patent/AP535A/en active
- 1994-12-20 US US08/652,460 patent/US5633276A/en not_active Expired - Fee Related
- 1994-12-21 IL IL112097A patent/IL112097A/en not_active IP Right Cessation
- 1994-12-21 SV SV1994000083A patent/SV1994000083A/en unknown
- 1994-12-21 CO CO94057739A patent/CO4340623A1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0043752A1 (en) * | 1980-06-26 | 1982-01-13 | SOCIETE DE RECHERCHES INDUSTRIELLES S.O.R.I. Société anonyme dite: | Pyranoindole derivative and process for its preparation |
| EP0207605A1 (en) * | 1985-05-21 | 1987-01-07 | Pfizer Inc. | Hypoglycemic thiazolidinediones |
Non-Patent Citations (1)
| Title |
|---|
| P. T. KIM ET AL: "Synthèse de pyranno(f et g)indoles", CANADIAN JOURNAL OF CHEMISTRY., vol. 60, no. 16, 1982, OTTAWA CA, pages 2093 - 2098 * |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2741799A1 (en) * | 1995-12-04 | 1997-06-06 | Oreal | USE OF MELATONIN IN A COMPOSITION FOR TREATING SKIN SIGNS OF FATIGUE CONDITIONS |
| CN100441574C (en) * | 1996-03-08 | 2008-12-10 | 武田药品工业株式会社 | Tricyclic compounds and their preparation and use |
| CN100443480C (en) * | 1996-03-08 | 2008-12-17 | 武田药品工业株式会社 | Tricyclic compounds, their preparation and use |
| KR100494214B1 (en) * | 1996-03-08 | 2005-11-25 | 다케다 야쿠힌 고교 가부시키가이샤 | Tricyclic compounds, their production and use |
| US6034239A (en) * | 1996-03-08 | 2000-03-07 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| WO1997032871A1 (en) * | 1996-03-08 | 1997-09-12 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| EP1550655A1 (en) * | 1996-03-08 | 2005-07-06 | Takeda Pharmaceutical Company Limited | Bicyclic intermediates |
| US5856529A (en) * | 1996-12-10 | 1999-01-05 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US5981571A (en) * | 1996-12-10 | 1999-11-09 | Bristol-Myers Squibb Company | Benzodioxa alkylene ethers as melatonergic agents |
| US6060506A (en) * | 1996-12-10 | 2000-05-09 | Bristol-Myers Squibb Company | Benzopyran derivatives as melatonergic agents |
| EP1199304A1 (en) * | 1997-03-05 | 2002-04-24 | Takeda Chemical Industries, Ltd. | Bicyclic compounds and pharmaceutical composition containing tricyclic compound for treating or preventing sleep disorders |
| US5948817A (en) * | 1997-03-05 | 1999-09-07 | Bristol-Myers Squibb Company | Polycyclic ethyl alkylamide melatonergic agents |
| WO1998052935A1 (en) * | 1997-05-16 | 1998-11-26 | Adir Et Compagnie | Substituted heterocyclic compounds, method for preparing and compositions containing same |
| EP0990650A4 (en) * | 1997-06-13 | 2002-02-13 | Yamanouchi Pharma Co Ltd | Tricyclic pyrrole or pyrazole derivatives |
| EP1082115A4 (en) * | 1998-06-05 | 2002-10-24 | Bristol Myers Squibb Co | Heterocyclic cis cyclopropane derivatives as melatonergic agents |
| US6214869B1 (en) | 1998-06-05 | 2001-04-10 | Bristol-Myers Squibb | Heterocyclic cis cyclopropane derivatives as melatonergic agents |
| US6211225B1 (en) | 1999-06-30 | 2001-04-03 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7166613B2 (en) | 1999-08-11 | 2007-01-23 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2c, receptor ligands |
| US7166632B2 (en) | 1999-08-11 | 2007-01-23 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2C, receptor ligands |
| US7173056B2 (en) | 1999-08-11 | 2007-02-06 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2C, receptor ligands |
| US7323486B2 (en) | 1999-08-11 | 2008-01-29 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2C, receptor ligands |
| US7323487B2 (en) | 1999-08-11 | 2008-01-29 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2C, receptor ligands |
| US7323473B2 (en) | 1999-08-11 | 2008-01-29 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5-HT, in particular 5-HT2C, receptor ligands |
| US6962939B1 (en) | 1999-08-11 | 2005-11-08 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2C, receptor ligands |
| AU774337B2 (en) * | 1999-08-11 | 2004-06-24 | Vernalis Research Limited | Condensed indoline derivatives and their use as 5HT, in particular 5HT2C, receptor ligands |
| US7008940B1 (en) | 1999-08-20 | 2006-03-07 | Takeda Pharmaceutical Company Limited | Dihydrobenzofuran derivatives, process for the preparing thereof and agents |
| WO2009053440A1 (en) * | 2007-10-25 | 2009-04-30 | Ferrer Internacional S.A. | Indoline compounds |
| ES2331274A1 (en) * | 2007-10-25 | 2009-12-28 | Ferrer Internacional, S.A. | Indoline compounds |
| ES2331274B1 (en) * | 2007-10-25 | 2010-10-21 | Ferrer Internacional, S.A. | INDOLINE COMPOUND |
| WO2010012789A1 (en) * | 2008-07-30 | 2010-02-04 | Ferrer Internacional S. A. | 1,6-dihydro-2h-3-oxa-6-aza-as-indacene compounds |
| CN102112480A (en) * | 2008-07-30 | 2011-06-29 | 菲尔若国际公司 | 1, 6-dihydro-2H-3-oxa-6-aza-as-indacene compounds |
| US8227501B2 (en) | 2008-07-30 | 2012-07-24 | Ferrer Internacional, S.A. | 1,6-dihydro-2H-3-oxa-6-aza-as-indacene compounds |
| WO2018076090A1 (en) | 2016-10-24 | 2018-05-03 | Aché Laboratórios Farmacêuticos S.A. | Compounds, process for obtaining the compounds, pharmaceutical composition, use of the compounds and method for treating psychiatric disorders and/or sleep disorders |
| EP3741760A1 (en) | 2016-10-24 | 2020-11-25 | Aché Laboratórios Farmacêuticos S.A. | Compounds, process for obtaining the compounds, pharmaceutical composition, use of the compounds and method for treating psychiatric disorders and/or sleep disorders |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0736028A1 (en) | 1996-10-09 |
| AU684877B2 (en) | 1998-01-08 |
| TW291479B (en) | 1996-11-21 |
| IS4244A (en) | 1995-06-23 |
| US5633276A (en) | 1997-05-27 |
| CA2179402A1 (en) | 1995-06-29 |
| CO4340623A1 (en) | 1996-07-30 |
| JPH09507057A (en) | 1997-07-15 |
| SV1994000083A (en) | 1995-07-24 |
| ZA9410056B (en) | 1995-10-18 |
| AP535A (en) | 1996-09-16 |
| AP9400706A0 (en) | 1995-01-31 |
| GB9326192D0 (en) | 1994-02-23 |
| MY131641A (en) | 2007-08-30 |
| AU1274395A (en) | 1995-07-10 |
| IL112097A (en) | 1998-06-15 |
| IL112097A0 (en) | 1995-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AP535A (en) | Indoline derivatives, method of preparation and use. | |
| JP2941309B2 (en) | Compound | |
| KR930003759B1 (en) | Process for the preparation of an indole derivatives | |
| EP0690843B1 (en) | Formyl- or cyano- substituted indole derivatives having dopaminergic activity | |
| JP2000511518A (en) | Benzofurans and benzopyrans as chronobiological agents | |
| JPH10508297A (en) | Substituted arylpiperazines as neurokinin antagonists | |
| JP2000510153A (en) | Spiro-piperidine derivatives and their use as therapeutics | |
| JPH08506823A (en) | Aza cyclic compounds, compositions containing the compounds and use of the compounds as tachykinin antagonists | |
| LU85743A1 (en) | HETEROCYCLIC COMPOUNDS | |
| JPS6277383A (en) | Heterocyclic compound | |
| US7060710B2 (en) | Isoxazole pyrazoloindane derivatives as cognition enhancing GABAA α5 subtype ligands | |
| JPH0419221B2 (en) | ||
| JPH10502336A (en) | Naphthalene derivatives, production method and use | |
| CH667454A5 (en) | DERIVATIVES OF INDOLES. | |
| RU2169147C2 (en) | DERIVATIVES OF CARBOXYLIC ACID AMIDES WITH HETEROCYCLIC SUBSTITUENTS AND COMPOSITION SHOWING ABILITY TO INHIBIT 5-HT1A- AND/OR alpfa- AND/OR alpfa- AND/OR alpfa- AND/OR α1-RECEPTORS | |
| US20030073832A1 (en) | Novel aminophenyl ketone derivatives | |
| JPS62195363A (en) | Chemical compound | |
| JPH05262766A (en) | Piperidylmethyl-substituted chroman derivatives | |
| JPH0358987A (en) | New compound, preparation thereof, and drug composition containing same | |
| WO2001014384A1 (en) | Tricyclic dihydrobenzofuran derivatives, process for the preparation thereof and agents | |
| JPH09512034A (en) | Indole derivatives as 5HT1-like agonists | |
| JP2791069B2 (en) | Cyclooctane neuroprotective agent | |
| JPS6333362A (en) | Indole derivative | |
| CN101318943A (en) | Substituted piperazine compounds, preparation method and application thereof, and pharmaceutical composition containing compounds | |
| JPH10101644A (en) | Pineal hormone agonist |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AM AT AU BB BG BR BY CA CH CN CZ DE DK ES FI GB GE HU JP KE KG KP KR KZ LK LT LU LV MD MG MN MW NL NO NZ PL PT RO RU SD SE SI SK TJ TT UA US UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE MW SD SZ AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1995903817 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08652460 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2179402 Country of ref document: CA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1995903817 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1995903817 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1995903817 Country of ref document: EP |