WO1995020962A1 - Protease inhibitors - Google Patents
Protease inhibitors Download PDFInfo
- Publication number
- WO1995020962A1 WO1995020962A1 PCT/US1994/011350 US9411350W WO9520962A1 WO 1995020962 A1 WO1995020962 A1 WO 1995020962A1 US 9411350 W US9411350 W US 9411350W WO 9520962 A1 WO9520962 A1 WO 9520962A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- butyl
- pharmaceutically acceptable
- amino
- acceptable salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 ****C(C1)=C(**)CN(*)C1C(N(*)O*)=O Chemical compound ****C(C1)=C(**)CN(*)C1C(N(*)O*)=O 0.000 description 13
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- a retrovirus designated human immuno-deficiency virus is the causative agent of the complex disease termed Acquired Immune Deficiency Syndrome (AIDS) , -and is a member of the lentivirus family of retroviruses .
- the complex disease AIDS includes progressive destruction of the immune system and degeneration of the central and peripheral nervous systems.
- the HIV virus was previously known or referred to as LAV, HTLV-III or ARV.
- a common feature of retrovirus replication is the post-translational processing of precursor polyproteins by a virally encoded protease to generate mature viral proteins required for viral assembly and function. Interruption of this processing appears to prevent the production of normally infectious virus. Unprocessed structural proteins also have been observed in clones of non-infectious HIV strains isolated from human patients. The results suggest that the inhibition of HIV protease represents a viable method for the treatment or prevention of AIDS and/or treatment or prevention of infection by HIV.
- the HIV genome encodes structural protein precursors known as gag and pol, which are processed to afford the protease, reverse transcriptase and endonuclease/integrase.
- the protease further cleaves gag and gag-pol polyproteins to yield mature structural proteins of the virus core.
- HIV a currently used therapeutic
- AZT is an inhibitor of the viral reverse transcriptase. H. Mitsuya, NS. Broder, "Inhibition of the In Vitro Infectivity in Cytopathic Effects of HTLV III", Proc. Natl. Acad. Sci. USA, 83, 1911 (1986) .
- Research efforts have also been directed toward HIV protease inhibitors.
- EPA European Patent Application
- EPA 361 341 EPA 346 847; EPA 402 646; and EPA 337 714 all disclose compounds which are said to be useful as HIV protease inhibitors.
- many of the known HIV protease inhibitors suffer from toxicity problems, lack of bioavailability or short in vivo half-lives.
- oral bioavailability is a necessary characteristic of an HIV protease inhibitor due to the chronic nature of the disease.
- peptides and peptide mimetics are notorious for their inability to be orally absorbed.
- a viable therapeutic agent has not yet emerged.
- a primary object of the present invention is to provide novel HIV protease inhibitors that possess desirable biological properties relative to previous HIV protease inhibitors while retaining potent HIV protease inhibitory activity.
- these HIV protease inhibitors promise to be useful for inhibiting HIV replication in an HIV infected cell, a cell susceptible to HIV infection or a primate in need thereof, thus treating and/or preventing HIV infection.
- a further object of the present invention is to provide therapeutic compositions that are of value in the treatment and/or prevention of HIV infection.
- Still another object is to provide methods for the treatment and/or prevention of HIV infection.
- the present invention relates to compounds of formula (I) , below, and pharmaceutically acceptable salts thereof that inhibit the protease encoded by human immunodeficiency virus (HIV) type 1 (HIV-1) or type 2 (HIV-2) . These compounds are useful in the treatment and/or prevention of infection by HIV.
- the compounds, their pharmaceutically acceptable salts, and the pharmaceutical compositions of the present invention can be used alone or in combination with other antivirals, immunomodulators, antibiotics or vaccines. Methods of treating or preventing AIDS, methods of treating or preventing HIV infection and methods of inhibiting HIV replication are disclosed.
- the present invention relates to a method for inhibiting HIV replication in an HIV infected cell, a cell susceptible to HIV infection or a primate in need thereof, thus treating and/or preventing HIV infection, comprising administering an effective amount of a compound of formula (I) :
- R is a group having the formula:
- Z is hydrogen, carbamoyl, formyl, C 2 -C 6 alkanoyl, C ⁇ -C 4 alkoxycarbonyl, -C(0)CF 3 or -S(0) 2 -Zi;
- Zi is C ⁇ -C 6 alkyl, amino, C ⁇ -C 4 alkylamino, trifluoromethyl or di (C 1 -C 4 )alkylamino;
- Z 2 is quinolinyl-C(O) -, naphthyloxymethyl-C(0) -, substituted quinolinyl-C(0) -, or substituted naphthyloxymethyl-C(0) -; ⁇ , an asymmetric center, is in a non-naturally occurring configuration; ⁇ , an asymmetric center, is in a naturally occurring configuration;
- R2 is an amino acid side chain or - (CH 2 ) y -W 1 -R2a ; y is 0, 1 or 2; 1 is a bond, divalent(C 2 -C 4 )alkenyl, divalent(C 2 -C 4 )alkynyl, -C(0)-0-, -O-C(O)-, -C(0)-NR2b-, -NR2b-C(0)-, -NR2b-, -C(O)-, -O-, -S-, -S (O) - or -S(0) 2 -;
- R2a is aryl, unsaturated heterocycle, heterocycle, aryl (C -C 4 )alkyl, unsaturated heterocycle(C -C )alkyl, heterocycle(C -C 4 )alkyl, tetrazolyl, N- (C ⁇ -C 4 )alkyltetrazolyl or N- (aryl) tetrazolyl;
- R2b is hydrogen or C ⁇ -C 4 alkyl
- R2c is an amino acid side chain
- a 1 and A 2 are independently -C-, -0-, -S-, -S(O)-, -S(0) 2 -, -NH- or -N(CH 3 )-
- Bi is -0-, -S-, -CH 2 -, -CH 2 -CH 2 -, -NH-, or -N(CH 3 )-;
- R 4 is C ⁇ -C 6 alkyl; i is 0, 1, 2, 3, or 4; R 3a is aryl, -O-aryl, or -S-aryl; R° and R 1 are independently hydrogen, C -C 6 alkyl, or hydroxy(C !
- X and Y are independently -S- , -S (O) - , -S (0) 2 - , -0-, -NH-, or -N(R9)-;
- R 9 is C ⁇ -C 6 alkyl, aryl (C ⁇ -C 6 )alkyl, aryl, arylcarbonyl, formyl, or C 2 -C 6 alkanoyl; with the provisos that: b and d cannot both be 0; the sum of a, b, c, d and e must be 2, 3, 4 or 5; if R 5 is -CR 5 ⁇ R 5 *-, then R6 must be -CH 2 - or -CHR6 ⁇ -; and R 7 must be -CH 2 - or -CHR 7 ⁇ -; if R 6 is -CR 6 ⁇ R6 ⁇ -, then R5 must be -CH 2 - or -CHR5 ⁇ - ; and R 7 must be -CH 2 - or -CHR ⁇ -; if R 7 is -CR 7 ⁇ R 7 ⁇ -, then R5 must be -CH 2 - or -CHR5 ⁇ - ; and R 7
- the present invention also relates to a compound of formula (I) , or a pharmaceutically acceptable salt thereof, wherein R 0 , R, R 1 , R 3 , R 5 , R 6 , R 7 , a, b, c, d, and e are as defined above in formula (I) .
- the present invention further provides pharmaceutical formulations comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof, in combination with a pharmaceutically acceptable carrier, diluent or excipient therefor.
- the present invention provides new compounds of formula (I) , as described above, that are useful for treating and/or preventing HIV infection and/or AIDS. All temperatures stated herein are in degrees Celsius ( * C) . All units of measurement employed herein are in weight units except for liquids which are in volume units.
- C -C 6 alkyl represents a straight or branched alkyl chain having from one to six carbon atoms.
- Typical C ⁇ -C 6 alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, t-butyl, pentyl, neo-pentyl, hexyl, and the like.
- C ⁇ -C 6 alkyl includes within its definition the term "C ⁇ -C 4 alkyl”.
- Divalent (C 2 -C 4 )alkenyl represents a straight or branched divalent alkenyl chain having from two to four carbon atoms.
- Typical divalent (C 2 -C )alkenyl groups include ethenyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl and the like.
- Divalent(C 2 -C 4 )alkynyl represents a straight or branched divalent alkynyl chain having from two to four carbon atoms.
- Typical divalent(C 2 -C 4 )alkynyl groups include ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl and the like.
- halo represents chloro, fluoro, bromo or iodo.
- halo(C -C 6 )alkyl represents a straight or branched alkyl chain having from one to six carbon atoms with 1-3 halogen atoms attached to it.
- Typical halo(C -C 6 ) -alkyl groups include chloromethyl, 2-bromoethyl, 1-chloroisopropyl, 3-fluoropropyl, 2, 3-dibromobutyl, 3-chloroisobutyl, iodo- -butyl, trifluoromethyl, 6-bromo-hexyl, and the like.
- hydroxy(C ⁇ -C 6 )alkyl represents a straight or branched alkyl chain having from one to six carbon atoms with a hydroxy group attached to it.
- Typical hydroxy(C ⁇ -C 6 )alkyl groups include hydroxymethyl, 2-hydroxyethyl, 3-hydroxypropyl, 2-hydroxyisopropyl, 4-hydroxybutyl, 2-hydroxyhexyl, and the like.
- C ⁇ -C 6 alkylthio represents a straight or branched alkyl chain having from one to six carbon atoms attached to a sulfur atom.
- Typical C ⁇ -C 6 alkylthio groups include methylthio, ethylthio, propylthio, isopropylthio, butylthio, sec-butylthio, t-butylthio, pentylthio, hexylthio, and the like.
- C ⁇ -C 6 alkylthio(C ⁇ -C 6 )alkyl represents a straight or branched C ⁇ -C 6 alkyl chain having from one to six carbon atoms with a C ⁇ -C 6 alkylthio moiety attached to it.
- Typical C ⁇ -C 6 alkylthio(C ⁇ -C 6 ) alkyl groups include methylthiomethyl, ethylthiomethyl, propylthioe hyl, isopropylthiomethyl, butylthiopentyl, sec-butylthiomethyl, hexylthiopropyl, and the like.
- C ⁇ -C alkylamino represents a straight or branched alkyl chain having from one to four carbon atoms attached to an amino group.
- Typical C ⁇ -C 4 alkylamino groups include ethylamino, ethylamino, propylamino, isopropylamino, butyla ino, sec-butylamino, and the like.
- di(C ⁇ -C 4 )alkylamino represents two straight or branched alkyl chains, each having from one to four carbon atoms attached to a common amino group.
- Typical di (C -C 4 )alkylamino groups include dimethylamino, ethylmethylamino, methylpropylamino, ethylisopropylamino, butylmethylamino, sec-butylethylamino, and the like.
- C -C 6 alkoxy represents a straight or branched alkyl chain having from one to six carbon atoms attached to an oxygen atom.
- Typical C -C 6 alkoxy groups include methoxy, ethoxy, propoxy, isopropoxy, butoxy, sec-butoxy, t-butoxy, pentoxy, hexoxy, and the like.
- C -C alkoxycarbonyl represents a straight or branched alkoxy chain having from one to four carbon atoms attached to a carbonyl moiety.
- Typical C -C alkoxycarbonyl groups include methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, butoxycarbonyl, and the like.
- C 2 -C 6 alkanoyl represents a straight or branched alkyl chain having from one to five carbon atoms attached to a carbonyl moiety.
- Typical C 2 -C 6 alkanoyl groups include ethanoyl, propanoyl, isopropanoyl, butanoyl, t-butanoyl, pentanoyl, hexanoyl, 3-methylpentanoyl -and the like.
- aryl represents a phenyl or naphthyl ring which is optionally substituted with halo, hydroxy, or C ⁇ -C 4 alkoxy.
- aryl (C -C 6 )alkyl represents a straight or branched alkyl chain having from one to six carbon atoms with an aryl group attached to it.
- Typical aryl(C ⁇ -C ) -alkyl groups include phenylmethyl, 2-naphth-l-ylethyl, 3-naphth-2-ylpropyl, 2-phenylisopropyl, 4-naphth-l-ylbutyl, 3-phenylpentyl, and the like.
- aryl(C -C 6 )alkyl includes within its definition the term “aryl (Ci-C 4 ) -alkyl. "
- heterocycle represents an unsubstituted or substituted stable 5- to 7-membered monocyclic or 7- to 10-membered bicyclic heterocyclic ring which is saturated and which consists of carbon atoms and from one to three heteroatoms selected from the group consisting of nitrogen, oxygen or sulfur, and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quaternized and including a bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which affords a stable structure.
- the heterocycle is unsubstituted or substituted with 1, 2 or 3 substituents independently selected from halo, hydroxy, halo(C ⁇ -C 4 )alkyl, C ⁇ -C 4 alkyl.
- the term "unsaturated heterocycle” represents an unsubstituted or substituted stable 5- to 7-membered monocyclic or 7- to 10-membered bicyclic heterocyclic ring which has one or more double bonds and which consists of carbon atoms and from one to three heteroatoms selected from the group consisting of nitrogen, oxygen or sulfur, and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quarternized and including a bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the unsaturated heterocyclic ring may be attached at any heteroatom or carbon atom which affords a stable structure.
- the unsaturated heterocycle is unsubstituted or substituted with 1, 2 or 3 substituents independently selected from halo, halo(C -C )alkyl, hydroxy, C -C alkyl.
- heterocycles and unsaturated heterocycles include piperidinyl, piperazinyl, azepinyl, pyrrolyl, 4-piperidonyl, pyrrolidinyl, pyrazolyl, pyrazolidinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxazolidinyl, isoxazolyl, isoxazolidinyl, morpholinyl, thiazolyl, thiazolidinyl, isothiazolyl, quinuclidinyl, isothiazolidinyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl, thiadiazolyl, benzopyranyl, benzothiazolyl, benzoazolyl, furyl, t
- Unsaturated heterocycle (C -C 4 )alkyl represents a straight or branched alkyl chain having from one to four carbon atoms with an unsaturated heterocycle group attached to it.
- Typical unsaturated heterocycle(C ⁇ -C )alkyl groups include pyrrolylmethyl, quinolinylmethyl, 1-indolylethyl,
- Heterocycle(C -C 4 )alkyl represents a straight or branched alkyl chain having from one to four carbon atoms with a heterocycle group attached to it.
- Typical heterocycle(C ⁇ -C )alkyl groups include tetrahydrofuryl- methyl, tetrahydropyranylmethyl, 1-indolylethyl,
- amino acid side chain represents the distinctive atom or group bonded to an ⁇ -carbon atom also having bonded thereto a carboxyl group and an amino group.
- amino-protecting group refers to substituents of the amino group commonly employed to block or protect the amino functionality while reacting other functional groups on the compound.
- amino-protecting groups include formyl, trityl, phthalimido, trichloroacetyl, chloroacetyl, bromoacetyl, iodoacetyl; or urethane-type blocking groups such as benzyloxycarbonyl, 4-phenylbenzyloxycarbonyl, 2-methylbenzyloxycarbonyl, 4-methoxybenzyloxycarbonyl, 4-fluorobenzyloxycarbonyl, 4-chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, 2-chlorobenzyloxycarbonyl, 2, 4-dichlorobenzyloxycarbonyl, 4-bromobenzyloxycarbonyl, 3-bromobenzyloxycarbonyl, 4-nitrobenzyloxycarbonyl,
- amino-protecting group employed is not critical so long as the derivatized amino group is stable to the condition of subsequent reaction(s) on other positions of the intermediate molecule and can be selectively removed at the appropriate point without disrupting the remainder of the molecule including any other amino-protecting group(s) .
- Preferred amino-protecting groups are t-butoxycarbonyl (t-Boc) and benzyloxycarbonyl (CbZ) . Further examples of groups referred to by the above terms are described by J. .
- carboxy-protecting group refers to substituents of the carboxy group commonly employed to block or protect the carboxy functionality while reacting other functional groups on the compound.
- carboxy-protecting groups include methyl, p-nitrobenzyl, p-methylbenzyl, p-methoxybenzyl, 3, 4-dimethoxybenzyl, 2,4-dimethoxybenzyl, 2,4, 6-trimethoxybenzyl, 2,4, 6-trimethylbenzyl, pentamethylbenzyl, 3, 4- ⁇ .ethylenedioxybenzyl, benzhydryl, 4, 4'-dimethoxybenzhydryl, 2,2', 4, 4'-tetramethoxybenzhydryl, t-butyl, t-amyl, trityl, 4-methoxytrityl, 4, 4'-dimethoxy-trityl, 4,4',4"-trimethoxytrityl, 2-phenylprop-2-yl, trimethyl
- a preferred method of protecting the carboxy group involves converting the carboxy moiety to an amide moiety and then hydrolyzing the amide back to provide the desired carboxy substituent. Further examples of these groups are found in E. Haslam, "Protective Groups in Organic Chemistry", J.G.W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, Chapter 5, and T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N.Y. , 1981, Chapter 5.
- the compounds of the present invention have at least five asymmetric centers denoted by an asterisk in the formula below:
- the compounds of the present invention can occur as mixtures of diastereomers, racemic mixtures and as individual enantiomers. All asymmetric forms, individual isomers and combinations thereof, are within the scope of the present invention.
- a preferred stereochemistry for compounds of formula (I) includes:
- the invention includes the pharmaceutically acceptable salts of the compounds defined by formula (I) .
- a compound of this invention may possess a sufficiently acidic, a sufficiently basic, or both functional groups, and accordingly react with any of a number of inorganic bases, and inorganic and organic acids, to form a pharmaceutically acceptable salt.
- pharmaceutically acceptable salt refers to salts of the compounds of the above formula which are substantially non-toxic to living organisms .
- Typical pharmaceutically acceptable salts include those salts prepared by reaction of the compounds of the present invention with a mineral or organic acid or an inorganic base. Such salts are known as acid addition and base addition salts.
- Acids commonly employed to form acid addition salts are inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, and the like, and organic acids such as p-toluenesulfonic, methanesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, and the like
- organic acids such as p-toluenesulfonic, methanesulfonic acid, oxalic acid, p-bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- salts examples include the sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate, isobutyrate, caproate, heptanoate, propiolate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, butyne-1,4-dioate, hexyne-1, 6-dioate, benzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, phthalate, sulfonate, xylenesulfonate, phenylacetate, phenylpropionate, phenylbut
- Preferred pharmaceutically acceptable acid addition salts are those formed with mineral acids such as hydrochloric acid and hydrobromic acid, and those formed with organic acids such as maleic acid and methanesulfonic acid.
- Base addition salts include those derived from inorganic bases, such as ammonium or alkali or alkaline earth metal hydroxides, carbonates, bicarbonates, and the like.
- bases useful in preparing the salts of this invention thus include sodium hydroxide, potassium hydroxide, ammonium hydroxide, potassium carbonate, sodium carbonate, sodium bicarbonate, potassium bicarbonate, calcium hydroxide, calcium carbonate and the like.
- the potassium and sodium salt forms are particularly preferred.
- any salt of this invention is not of a critical nature, so long as the salt as a whole is pharmacologically acceptable and as long as the counterion does not contribute undesired qualities to the salt as a whole.
- Preferred compounds of formula (I) are those compounds of the formula:
- R is a group of the formula:
- Ai is -C- , -0- , -S- , -S (0) 2 - ;
- A is -C- , -0- , -S- , or -S (0) 2 - ;
- Bi is -0- , -CH 2 - , or -NH- ;
- R3 is aryl , or -S-aryl ;
- Ro and Ri are independently hydrogen, or C ⁇ -C 6 alkyl ;
- each of R 5 ⁇ , R 6 ⁇ , and R ⁇ is independently selected from the group consisting of halo, C -C 6 alkyl, hydroxy(C ⁇ -C )alkyl or C ⁇ -C 4 alkylthio; and
- X and Y are independently -S-, -0-, or -NH-; or a pharmaceutically acceptable salt thereof.
- A is -C-, -0-, -S-, or -S(0) 2 -; and Bi is -0-, or -CH 2 -; or a pharmaceutically acceptable salt thereof.
- the most preferred compounds are N-t-butyl-octahydro-5 t2R-hydroxy-3R-N(l', l'-dioxo-2'R- isopropyl-tetrahydrothiophen-3'R-yloxycarbonyl) amino-4- phenylthio-) -butyl] - (3aR, 7aS) -thieno[3 ,2-c] pyridine- (6S) -carboxamide;
- N-t-butyl-octahydro-5 [2R-hydroxy-3R-N(2'R- isopropyl-tetrahydrothiophen-3'R-yloxycarbonyl)amino-4- phenylthio-) -butyl] - (3aR, 7aS) -thieno[3 , 2-c] pyridine- (6S) -carboxamide; N-t-butyl-octahydro-5 [2R-hydroxy-3R-N(l', l'-dioxo- tetrahydrothiophen-3'R-yloxycarbonyl)amino-4-phenylthio) - butyl] - (3aR, 7aS) -thieno [3 ,2-c] pyridine- (6S) -carboxamide; N-t-butyl-octahydro-5 [2R-hydroxy-3R-N(l', l'-diox
- the reaction is carried out in the presence or absence of a promoting agent, preferably in the presence of a promoting agent, and in the presence of a coupling reagent.
- a promoting agent preferably in the presence of a promoting agent
- Typical aprotic solvents for this reaction are tetrahydrofuran and dimethylformamide, or a mixture of such solvents.
- the reaction is carried out at a temperature from about -30"C to about 25 * C.
- the amine reactant is generally employed in equimolar proportions relative to the carboxylic acid reactant, in the presence of an equimolar quantity to a slight excess of the coupling reagent.
- Typical coupling reagents include the carbodiimides such as dicyclohexylcarbodiimide (DCC) and N, -diethylcarbodiimide; the imidazoles such as carbonyldiimidazole; as well as reagents such as bis (2-oxo-3-oxazolidinyl)phosphinic chloride (BOP-Cl) or N-ethoxycarbonyl-2-ethoxy-l,2- dihydroquinoline (EEDQ) .
- a preferred coupling reagent for this reaction is DCC.
- a promoting agent is preferably included for this reaction; a preferred promoting agent is hydroxybenzotriazole hydrate (HOBT-H 2 0) .
- the compound may be isolated, if desired, by procedures known in the art, for example, the compound may be crystallized and then collected by filtration, or the reaction solvent may be removed by extraction, evaporation or decantation.
- the compound may be further purified, if desired, by common techniques such as crystallization or chromatography over solid supports such as silica gel or. alumina.
- the compounds of formula (I) where Z is carbamoyl, formyl, C 2 -C 6 alkanoyl, C -C alkoxycarbonyl or -S(0) 2 -Zi, where Z is as defined above for formula (I) may be prepared by deprotectmg a compound of formula (IA) , and then reacting the resultant amine with an amino-protected compound having the formula,
- R A is an amino-protecting group.
- the amino-protecting group is then removed from the resulting compound according to procedures and methods known in the art to provide the compound of formula (I) , where Z or Z 2 is hydrogen.
- the resulting compound is then acylated or sulfonylated using procedures known in the art.
- the amine compounds may be acylated by reaction with a suitable acyl halide, isocyanate or chloroformate, preferably in the presence of an acid scavenger such as a tertiary amine, preferably triethylamine.
- the reaction is carried out at a temperature of from about -20"C to about 25'C.
- Typical solvents for this reaction include ethers and chlorinated hydrocarbons, preferably diethylether, chloroform or methylene chloride.
- the amine compounds may be sulfonylated by reaction with a suitably substituted sulfonyl halide of the formula, Zi-S0 2 -halide in an aprotic solvent at a temperature from about -30"C to about 25 * C in an aprotic solvent such as tetrahydrofuran.
- the amine reactant is generally employed in equimolar proportions relative to the sulfonyl halide reactant, and preferably in the presence of an acyl transfer catalyst.
- a preferred acyl transfer catalyst for this reaction is N-methyl-morpholine (NMM) .
- R 2Z is a carboxy-protecting group.
- the carboxy-protecting group is then removed and the resultant compound is reacted with a suitably substituted amine reactant of the formula, H-NR2aR2b, substantially in accordance with the procedure detailed in Reaction I.
- a preferred solvent for this reaction is a mixture of tetrahydrofuran and dimethylformamide.
- a preferred coupling reagent for this reaction is DCC.
- a preferred promoting agent is H0BT-H 2 0.
- SUBSTITUTE SHEET (RULE 261 co pounds may be acylated by reaction with a suitable acyl halide, isocyanate or chloroformate, preferably in the presence of an acid scavenger such as a tertiary amine, preferably triethylamine.
- the reaction is carried out at a temperature of from about -20"C to about 25"C.
- Typical solvents for this reaction include ethers and chlorinated hydrocarbons, preferably diethyl ether, chloroform or methylene chloride.
- the amine compounds may be sulfonylated by reaction with a suitably substituted sulfonyl halide of the formula, Z -S0 2 -halide as described above.
- a compound of formula (I) wherein R2 is - (CH 2 ) y -W-R2a, where y and R2a are as defined above; and W is -S(O)- or -S(0) 2 - may be prepared by oxidizing an intermediate compound of formula IB
- R2 is -(CH 2 ) y -W-R2a, where y, Z and R 2a are as defined above;
- W is -S-. ; under standard reaction conditions known in the art.
- the intermediate compound using X is -S- may be combined with an oxidizing agent in an aqueous or organic solvent at a temperature of from about -78"C to 25"C.
- Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- Typical oxidizing agents include oxone®, m-chloroperoxybenzoic acid.
- a preferred oxidizing agent is oxone®.
- a , A2 , and R 4 are as defined above for formula (I);
- Bi is -CH 2 - or -CH 2 CH 2 - may be prepared by deprotectmg a compound of formula (IA) and then acylating the resultant amine with a carboxylic acid reactant of the formula:
- the reaction is carried out in the presence or absence of a promoting agent, preferably in the presence of a promoting agent, and in the presence of a coupling reagent.
- a promoting agent preferably in the presence of a promoting agent
- Typical aprotic solvents for this reaction are tetrahydrofuran and dimethylformamide, or a mixture of such solvents.
- the reaction is carried out at a temperature from about -30'C to about 25"C.
- the amine reactant is generally employed in equimolar proportions relative to the carboxylic acid reactant, in the presence of an equimolar quantity to a slight excess of the coupling reagent.
- Typical coupling reagents include the carbodiimides such as dicyclohexylcarbodiimide (DCC) and N,N'-diethylcarbodiimide; the imidazoles such as carbonyldiimidazole; as well as reagents such as bis(2-oxo-3-oxazolidinyl)phosphinic chloride (B0P-C1) or N-ethoxycarbonyl-2-ethoxy-l,2-dihydroquinoline (EEDQ) .
- a preferred coupling reagent for this reaction is DCC.
- a promoting agent is preferably included for this reaction; a preferred promoting agent is hydroxybenzotriazole hydrate (HOBT-H 2 0) .
- a 1 , A 2 , and R 4 are as defined above for formula (I);
- B is -0- , -S- , -NH- , or -N (CH 3 ) - ; may be prepared by activating an appropriately substituted compound of the formula :
- a coupling reagent specifically, an activated carbonate (Bi is -0-) , an activated carbamate (where Bi is -N- or -N(CH 3 )-) and an activated thiocarbonate (where Bi is -S-) ) .
- the coupling reagent is then reacted with an amine reactant (obtained by deprotecting a compound of formula (IA) ) , preferably in the presence of an acid scavenger such as a tertiary amine, preferably triethylamine.
- an acid scavenger such as a tertiary amine, preferably triethylamine.
- the reaction is carried out at a temperature of from about -20 C C to about 25 * C.
- Typical solvents for this reaction include ethers and chlorinated hydrocarbons, preferably diethylether, chloroform or methylene chloride.
- Ai, A 2 , -and R 4 are as defined above for formula (I);
- Bi is -NH-; may be prepared by reacting an appropriately substituted amino compound of the formula:
- the amino reactant may be reacted with triphosgene in a mutual inert solvent.
- the reactants are generally employed in an amount ranging from about equimolar proportions to about a two molar excess, preferably in about a one molar excess relative to the triphosgene.
- a base for example a trialkylamine such as triethylamine or diisopropylethylamme and the like, may be added to promote the raction.
- Typical solvents suitable for use in this reaction include any organic solvent such as toluene. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- the reaction is generally substantially complete after about 6 to 24 hours when conducted at a temperature in the range of from about 25 * C to the reflux temperature of the reaction mixture.
- the reaction is preferably conducted at a temperature in the range of from about 80 * C to the reflux temperature of the reaction mixture for about 8 hours to 12 hours.
- the isocyanate compound is then reacted with a deprotected amino compound of formula (IA) in a mutual inert solvent.
- copper salts for example copper (I) iodide or copper (I) chloride.
- the isocyanate compound is generally employed in an amount ranging from about equimolar proportions to about a one molar excess.
- Typical solvents suitable for use in this reaction include any organic solvent such as dimethylformamide. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- the reaction is generally substantially complete after about 15 minutes to 3 hours when conducted at a temperature in the range of from -about 10"C to about 40'C.
- the reaction is preferably conducted at a temperature in the range of from about 15"C to about 30 * C for about 15 minutes to 2 hours.
- R 4 , R3, RO, Ri, R5, x, R6, Y, R7 a , b, c, d, and e are as defined above for formula (I) ;
- RA is an amino-protecting group;
- reaction Scheme I represents the presence of double bonds between, for example, R a and R e , R a and R a , or R e and R e and the like, where b, b or d is 0, respectively.
- Reaction Scheme I is accomplished by carrying out reactions 1-3 (or 1-5) in sequential order.
- the intermediate compound may be isolated, if desired by procedures known in the art, for example, the compound may be crystallized and then collected by filtration, or the reaction solvent may be removed by extraction, evaporation or decantation.
- the intermediate compound may be further purified, if desired, by common techniques such as crystallization or chromatography over solid supports such as silica gel or alumina, before carrying out the next step of the reaction scheme.
- Reaction I.1 is typically carried out by activating the carboxylic acid moiety using, for example, DCC, or a mixed anhydride such as isobutyl, followed by reaction with a primary or secondary amine having the formula NRORI where RO and Ri are as defined above for formula (I) .
- the reaction is typically carried out in a nonpolar aprotic solvent or mixture of solvents in the presence or absence of an acid scavenger at a temperature of from about -20'C to about 25'C to afford the corresponding amide.
- Suitable solvents for this reaction include ethers and chlorinated hydrocarbons, preferably diethyl ether, trichloroethane, or methylene chloride.
- this reaction is preferably carried out in the presence of an acid scavenger such as a tertiary amine, preferably triethylamine.
- an acid scavenger such as a tertiary amine, preferably triethylamine.
- the amide afforded by this reaction may be isolated or further reacted as shown in Reaction 2.
- Reaction 1.2 is typically carried out by reacting the compound obtained from Reaction 1.1 using the procedures detailed in Comprehensive Organic Synthesis.
- Heteroatom Manipulation Barry M. Trost, ed. , volume 6, pages 736-746, (1991) .
- an appropriately substituted monocyclic ring is reacted with an aldehyde, such as formaldehyde or trichloroacetaldehyde in the presence of an acid.
- the acid may be used as a solvent.
- Typical acids include hydrochloric acid, hydrobromic acid, sulfuric acid, acetic acid, trifluoroacetic acid, and the like.
- a co-solvent may optionally be added to the reaction mixture. The co-solvent choice is not critical so long as the co-solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- Typical solvents for this reaction include halogenated solvents such as methylene chloride, trichloroethane, carbontetrachloride, and the like.
- halogenated solvents such as methylene chloride, trichloroethane, carbontetrachloride, and the like.
- an aldehyde equivalent may be used, for example, dimethoxymethane and a suitable acid.
- reaction 1.3 the compound isolated from reaction 1.2 is reduced to provide a saturated heterocyclic compound as depicted above.
- Catalytic hydrogenation is a preferred method of reduction.
- Typical catalysts include palladium catalysts, rhodium catalysts (for example rhodium on alumina), rhenium catalysts and the like.
- Preferred catalysts include palladium-on-carbon.
- Suitable solvents for this reaction include the C ⁇ . -C 4 alcohols, tetrahydrofuran, acetic acid in alcohol, ethyl acetate and the like.
- a preferred solvent is ethanol.
- the reaction is typically carried out under an atmosphere of hydrogen from about 500 to about 4000 psi at a temperature of from about 25 * C to about 150"C.
- the reaction is carried out under an atmosphere of hydrogen from about 2000 to about 3000 psi at a temperature of from about 50"C to 100"C.
- the catalyst is generally employed in an amount ranging from about equivalent proportions to about a twelve-fold excess (by weight) of the reactant, preferably in about a six- to ten-fold excess (by weight) of the catalyst relative to the substrate.
- Reactions 1.4 and 1.5 may be used to prepare compounds of formula (I) where
- Reaction 1.4 is a standard amino deprotection reaction using procedures and methods known in the art to afford the corresponding amine which is then used in Reaction 1.5, above.
- Chemical deprotection procedures are preferred.
- the compound isolated from 1.3 may be deprotected using trimethylsilyliodide (TMSI) in an aprotic solvent or mixture of solvents at a temperature of from about 10"C to 60'C, preferably at a temperature of from about 20"C to 40"C.
- Typical solvents include methylene chloride, acetonitrile, trichloroethane, and the like.
- reaction 1.5 the epoxide prepared in Reaction A.5, below, is reacted with compound isolated from Reaction I.4 in an alcoholic solvent at a temperature of from about 20"C to 100'C.
- Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- Typical solvents for this reaction include the alcohols, preferably isopropanol or ethanol.
- the reaction is preferably carried out at a temperature of about 65-C.
- the compound of formula (IA) isolated from reaction 1.5, may be deprotected and then coupled or reacted as described above to provide a compound of formula
- Reaction Scheme A The epoxide used in Reaction 1. 5 may be synthesized using Reaction Scheme A. Reaction Scheme A
- R is an amino-protecting group
- R3 is as defined above for formula (I)
- G is halo.
- Reaction Scheme A is accomplished by carrying out reactions 1-5 in sequential order.
- the intermediate compound may be isolated, if desired by procedures known in the art, for example, the compound may be crystallized and then collected by filtration, or the reaction solvent may be removed by extraction, evaporation or decantation.
- the intermediate compound may be further purified, if desired, by common techniques such as crystallization or chromatography over solid supports such as silica gel or alumina, before carrying out the next step of the reaction scheme.
- Reaction A.l is carried out by activating, that is, converting, an amino-protected carboxylic acid reactant having the structure:
- the amino-protected carboxylic acid reactant may be reacted with a C ! -C 6 alkylchloroformate, such as isobutylchloroformate preferably in the presence of an acid scavenger.
- Preferred acid scavengers are the trialkylamines, preferably triethylamine.
- the reaction is typically carried out in an aprotic solvent such as ethyl acetate. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- the resulting mixed anhydride reactant is preferably used in Reaction A.2 without further isolation or purification.
- Reaction A.2 is accomplished in two steps. First, a solution of sodium hydroxide, covered with a layer of an ether solvent, preferably diethyl ether, is reacted with a large excess of N-methyl-N-nitro-N-nitrosoguanidine to form a diazomethane reactant.
- the sodium hydroxide is preferably used as an aqueous solution having about four to six mol/liter of sodium hydroxide.
- the organic layer is dried over a dessicant such as potassium hydroxide.
- This solution is then reacted with the mixed anhydride from Reaction A.l, above, to form the corresponding ⁇ -diazo carbonyl compound.
- the diazomethane reactant is preferably used in this reaction without isolation or purification.
- the reaction is typically carried out at a temperature of from about -50"C to about -10°C, preferably about -20'C.
- Reaction A.3 the ⁇ -diazo carbonyl compound prepared in Reaction A.2 is reacted with an acid of the formula H-G where G is halo, in an aprotic solvent such as diethylether to form an ⁇ -halo carbonyl compound.
- a preferred acid reactant is hydrochloric acid which provides the corresponding ⁇ -chloro carbonyl compound.
- the reaction is typically carried out at a temperature from about -30 * C to about 0 * C. Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- the acid reactant is typically added in the form of an anhydrous gas in small increments until the reaction appears substantially complete. The reaction can be monitored by thin layer chromatography.
- Reaction A.4 the carbonyl moiety on the compound prepared in Reaction A.3 is reduced using standard conditions known in the art to form the corresponding oc -chloro hydroxy compound.
- the compound prepared in Reaction A.3 is combined with a reducing agent in a mixture of solvents.
- Typical reducing agents include sodium borohydride, lithium borohydride, zinc borohydride, diisobutylaluminum hydride, and sodium bis (2-methoxy-ethoxy)aluminum hydride.
- a preferred reducing agent is sodium borohydride.
- Typical solvent mixtures include a protic and aprotic mixture such as tetrahydrofuran/water.
- Solvent choice is not critical so long as the solvent employed is inert to the ongoing reaction and the reactants are sufficiently solubilized to effect the desired reaction.
- the reaction is typically carried out at a temperature from about -10"C to about 10 * C, preferably about O'C.
- the ⁇ -chloro hydroxy compound prepared in Reaction A.4 is treated with a strong base to form the corresponding epoxide (which is used above in Reaction 1.5) under standard conditions known in the art.
- the ⁇ -chloro hydroxy compound may be reacted with a potassium hydroxide/ethanol mixture in an alcoholic solvent such as ethanol.
- the reaction is typically carried out at a temperature from about O'C to about the reflux temperature of the solvent. Preferably the reaction is carried out at room temperature.
- R 3 and R A are as defined above.
- reaction scheme described in Vederas et al. , J.Am.Chem. Soc, .102, 7105-7109 (1985) .
- this reaction scheme is carried out by first reacting amino-protected serine with triphenylphosphine, dimethylazodicarboxylate (DMAD) or diethylazodicarboxylate (DEAD) in an aprotic solvent at a temperature of from about -80"C to O'C to form the corresponding ⁇ -lactone.
- DMAD dimethylazodicarboxylate
- DEAD diethylazodicarboxylate
- the reaction is typically carried out in an ether, such as tetrahydrofuran at a temperature of from about -80'C to -50"C.
- the lactone ring is opened with an appropriately substituted thio anion, -S-Ra, where R 3a is as defined above.
- the thio anion compound is preferably formed by reacting the corresponding thiol with a strong base, such as sodium hydride or potassium hydride. This reaction is typically carried out in an aprotic solvent at a temperature from about O'C to about 40"C and under an inert atmosphere, such as nitrogen. Typical solvents for this reaction include ethers, preferably tetrahydrofuran.
- the compounds of formula IA, where R 3 is -S-aryl may be prepared using the procedures detailed in Photaki, J. Am. Chem. Soc, 85 .
- the compounds may be prepared by reacting doubly protected serine (carboxy-protected and amino-protected) with toluenesulfonyl chloride in the presence of dimethylaminopyridine (DMAP) and an acid scavenger such as pyridine in an aprotic solvent such as methylene chloride to form the corresponding toluenesulfonate which may then be reacted with an appropriately substituted thioanion having the structure, -S-aryl.
- DMAP dimethylaminopyridine
- an acid scavenger such as pyridine
- an aprotic solvent such as methylene chloride
- the thioanion compound is preferably formed by reacting the corresponding thiol with a , strong base as described above.
- the carboxy-protecting group may be removed from the resulting doubly protected arylthioalanine using conditions known in the art.
- isomers may be prepared from their respective precursors by the procedures described above, by resolving the racemic mixtures or by separating diastereomers. The resolution can be carried out in the presence of a resolving agent, by chromatography, or by repeated crystallization or by some combination of these techniques which are known in the art. Further details regarding resolutions can be found in Jacques et al. , Enantiomers, Racemates, and Resolutions, John Wiley & Sons 1981.
- the various diastereomeric forms of these compounds may be isolated in amounts greater than or equal to 96%, preferably greater than or equal to 98%.
- the isomers of the carboxylic acid reactant may be separated and the individual reactants then reacted with the amine compound as described above.
- the various diastereomeric forms of the carboxylic acid reactants may be separated to provide a pair of enantiomers (cis and trans) each of which may then be separated to provide the individual carboxylic acid reactant in an amount greater than or equal to 85%, preferably greater than or equal to 90% ee.
- the enantiomeric forms of the carboxylic acid reagent may be isolated using enzymatic resolution.
- carboxylic acid reagents where Bi is -0- may be obtained by enzymatic resolution of an hydroxy-substituted heterocyclic compound which is then converted to the desired carboxylic acid reactant.
- the cis and trans forms of hydroxy-substituted heterocyclic compound are first separated using chromatographic means according to procedures known in the art. The individual cis and trans enantiomers may then be isolated, respectively, using enzymatic resolution.
- the cis enantiomers of the hydroxy-substituted heterocyclic compound may be reacted with acetic anhydride in the presence of a base such as DMAP to provide the corresponding acetate-substituted heterocyclic compound.
- a base such as DMAP
- This R,R acetate is then selectively hydrolyzed using a catalytic amount of lipase (for example, lipase PS-800, Pseudomonas fluorescens. Fluka) in the presence of a surfactant such as Triton® X-100 in a buffered solution (pH 6-8, preferably pH 7) to provide the R,R hydroxy-substituted heterocyclic compound.
- a surfactant such as Triton® X-100 in a buffered solution (pH 6-8, preferably pH 7)
- the remaining S,S acetate-substituted heterocyclic compound may be hydrolyzed using procedures known in the art to provide the desired S,S hydroxy-substituted heterocyclic compound.
- the cis enantiomers of the hydroxy-substituted heterocyclic compound may be reacted with vinyl acetate in the presence of lipase SAM II, (Pseudomonas fluorescens. Fluka) to provide a mixture of the R,R acetate-substituted heterocyclic compound and the S,S hydroxy-substituted heterocyclic compound which may then be separated using skills known in the art.
- these hydroxy compounds may be activated to form a coupling reagent (i.e., an activated carbonate) which is then reacted with an amine reactant (obtained by deprotecting a compound of formula (IA) ) to provide the corresponding diastereomer (compound of formula
- the pharmaceutically acceptable salts of the invention are typically formed by reacting a compound of formula (I) with an equimolar or excess amount of acid or base.
- the reactants are generally combined in a mutual solvent such as diethyl ether or benzene, for acid addition salts, or water or alcohols for base addition salts.
- the salts normally precipitate out of solution within about one hour to about ten days and can be isolated by filtration or other conventional methods.
- the terms melting point, nuclear magnetic resonance spectra, electron impact mass spectra, field desorption mass spectra, fast atom bombardment mass spectra, infrared spectra, ultraviolet spectra, elemental analysis, high performance liquid chromatography, and thin layer chromatography are abbreviated “m.p.”, “NMR”, “EIMS”, “MS(FD)”, “MS(FAB)”, “IR”, “UV” , “Analysis”, “HPLC”, and “TLC” , respectively.
- the absorption maxima listed for the IR spectra are only those of interest and not all of the maxima observed.
- MS(FD) spectra were taken on a Varian-MAT 731 Spectrometer using carbon dendrite emitters.
- EIMS spectra were obtained on a CEC 21-110 instrument from Consolidated Electrodynamics Corporation.
- MS(FAB) spectra were obtained on a VG ZAB-3 Spectrometer.
- IR spectra were obtained on a Perkin-Elmer 281 instrument.
- UV spectra were obtained on a Cary 118 instrument.
- TLC was carried out on E. Merck silica gel plates. Melting points are uncorrected.
- a preformed catalyst was formed by adding 3.92 g of (+) -2, 3-0-isopropylidene-2,3-dihydroxy-l,4-bis (diphenyl- phosphine)butane to 1.76 g of chloro(1, 5-cyclooctadiene) - rhodium I dimer in 800 mL of toluene, under argon. The resultant mixture was stirred under argon for approximately twenty minutes. Meanwhile, 152 g (720 mmol) of the subtitled compound of Example IB in 2500 mL of ethanol was shaken under 20 psi of hydrogen gas in a hydrogenation vessel for approximately twenty minutes. The preformed catalyst was then added the hydrogenation vessel, under nitrogen.
- the resultant reaction mixture was allowed to react at 50'C, under twenty psi of hydrogen gas for approximately sixteen hours.
- the reaction mixture was concentrated under reduced pressure to provide a brown solid which was treated with 700 mL of water containing 100 g of potassium carbonate.
- the resultant mixture was filtered through celite, washed with water and the filtrate was combined with diethyl ether.
- the resultant layers were separated and the aqueous layer was acidified with 5N hydrochloric acid, layered with 1000 mL of methylene chloride.
- reaction mixture was concentrated, then washed with 100 mL of diethyl ether.
- the result-ant layers were separated and the aqueous layer was acidified to pH ⁇ 2 with 5N hydrochloric acid and extracted with methylene chloride which was then dried with sodium sulfate, filtered and concentrated under reduced pressure to provide 10.85 g of the titled compound.
- Example 2 N- (Benzyloxycarbonyl) S-thienyl-D,L-alanine To a mixture of 3.0 g (16.9 mmol) of ⁇ -thienyl-D,L-alanine in 75 mL of water and 60 mL of dioxane, was added 5.6 g (40.6 mmol) of anhydrous potassium carbonate, followed by 2.85 mL (20 mmol) of carbobenzyloxychloride. The resulting mixture was stirred rapidly for approximately one hour. When the reaction was substantially complete, as indicated by thin layer chromatography (21/7/7/9, ethyl acetate/acetic acid/acetonitrile/water) , the reaction mixture was concentrated.
- This oil was redissolved in methylene chloride and purified using flash chromatography (Si0 2 ; eluent of 2% methanol in methylene chloride (on a smaller scale, an eluent of 1:2 diethyl ether in hexane containing 2% methanol) ) to provide the desired cis isomer (major) comes through with a small amount of a minor isomer.
- This mixture was recrystallized from a mixture of 1.5 mL methanol/20 mL diethyl ether/120
- the titled compound was prepared substantially in accordance with the procedure detailed in Example 3A-D, using the titled compound from Example 2.
- Example 4 in 3 mL of ethanol was heated to 65"C and reacted for approximately twenty hours.
- the resultant mixture was concentrated under reduced pressure to provide a crude material.
- This material was purified using radial chromatography (2000 micron plate; eluent of 1% methanol in chloroform) which separated the diastereomers to provide 98 mg of the titled compound (36%) .
- Example 3D using 85 mg (0.158 mmol) of the titled compound of Example 5, 101 ⁇ L (71 mmol) of trimethylsilyliodide in a 1:1 acetonitrile/methylene chloride mixture to provide 64 mg of a white solid (quantitative) .
- iH NMR 300 MHz, CDCl 3 ) : ⁇ 7.28 (m, 5H) ; 6.38 (s, IH) ;
- the desired titled compound was prepared substantially in accordance with the procedure detailed in Example 3D, using 1.8 g (3.15 mmol) of the subtitled compound of Example 7, 2.1 mL of trimethylsilyliodide in 20 mL of a 1:1 acetonitrile/methylene chloride mixture to provide 1.18 g of a white solid (86%) .
- the subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9B, using 102 mg (0.233 mmol) of the titled compound from Example 8, 67 ⁇ L (0.48 mmol) of triethylamine and 77 mg (0.24 mmol) of the subtitled compound from Example 10A in 2 mL of methylene chloride to provide the subtitled compounds. Yield: 52 mg.
- Example 11 A ( ⁇ ) -3-Tetrahydrothiophene succinimidyl carbonate To a solution of 0.91 g (8.58 mmol) of ( ⁇ ) -hydroxy-tetrahydrothiophene in 10 mL of anhydrous acetonitrile, was added 3.6 mL of triethylamine followed by 3.3 g (12.9 mmol) of N,N-disuccinimidyl carbonate.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 10A, using 0.88 g (3.59 mmol) of the subtitled compound from Example IIA and 2.4 g (7.54 mmol) of 55%
- reaction mixture was warmed to room temperature and allowed to react for approximately thirty hours.
- the reaction mixture was diluted with 2 mL of diethyl ether and then filtered through a small cotton plug.
- the resultant filtrate was concentrated under reduced pressure and then purified using flash chromatography (gradient eluent of 3-4% methanol in methylene chloride) to provide 50 mg of a white foam (79%) which comprised a mixture of the titled compounds.
- iH NMR 300 MHz, CDC1 3
- Example 14 Ouinaldic acid pentafluorophenyl ester To a mixture of 15.0 g (86.6 mmol) of quinaldic acid and 20.8 g (113 mmol) of pentafluorophenol in 200 mL of tetrahydrofuran, was added 18.3 g (95.3 mmol) ethylene dichloride in one portion. The reaction mixture was stirred vigorously at room temperature for approximately two hours, resulting in the formation of a gummy precipitate. The solution was decanted from the gum, and the gum was washed with methylene chloride.
- the desired compound was prepared substantially in accordance with the procedure detailed in Example 14B, using 60 mg (0.137 mmol) of the titled compound from
- Example 8 33 mg (0.148 mmol) of the subtitled compound from Example 15A and 20 mg (0.148 mmol) of HOBT-H 2 0 and 29 mg (0.142 mmol) of DCC in 2 mL of tetrahydrofuran containing 0.2 mL of dimethylformamide.
- the crude material was purified using radial chromatography (1000 micron plate; eluent of 8% methanol in ethyl acetate) to provide 59 mg of the desired subtitled compound.
- Example 12 using 61 mg (0.15 mmol) of the titled compound from Example 6, 58 mg (0.153 mmol) of 2S-N(ethanoyl)amino- 3-naphth-2-ylsulfonyl propanoic acid, 21 mg (0.53 mmol) of
- the titled compound was prepared substantially in accordance with the procedure detailed in Example 16, using 6 mg (0.15 mmol) of the titled compound of Example 6, 0.53 g (0.15 mmol) of 2S-N(t-butoxycarbonyl) amino-3-p- fluoro-phenylsulfonyl propanoic acid, 21 mg (0.15 mmol) of HOBT-H 2 0, and 31 mg (0.15 mmol) of DCC in 2 mL of tetrahydrofuran to provide a crude material. This material was purified using radial chromatography (2000 micron plate; gradient eluent of 3-4% methanol in methylene chloride) to provide 90 mg of the desired compound (82%) .
- the reaction mixture was purified using radial chromatography (1000 micron plate; eluent of 2% methanol in chloroform) to provide 20 mg of the desired subtitled compound.
- the titled compound was prepared substantially in accordance with the procedure detailed in Example 16, using 80 mg (0.183 mmol) of the titled compound of Example 8, 64 mg (0.183 mmol) of 2S-N(t-butoxycarbonyl)amino-3-p- fluoro-phenylsulfonyl propanoic acid, 25 mg (0.183 mmol) of HOBT-H 2 0, and 38 mg (0.183 mmol) of DCC in 2.5 mL of tetrahydrofuran to provide a crude material. This material was purified using flash chromatography (gradient eluent of 3-4% methanol in methylene chloride) to provide 130 mg of a white foam (93%) . MS(FD) : 768, 666 (100) .
- Example 21 Example 21
- Example 19A using 0.13 g (0.169 mmol) of the titled compound of Example 20 and 1 mL of trifluoroacetic acid in 1 mL of methylene chloride to provide 100 mg of a solid that was used without further purification.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 19B, using 0.10 g (0.150 mmol) of the subtitled compound of Example 21A, 44 ⁇ L of triethylamine and 12 ⁇ L of acetyl chloride in 1 mL of methylene chloride.
- the crude material was purified using flash chromatography (Si0 2 ; eluent of 2% methanol in methylene chloride) to provide 44 mg of a white solid (40%) .
- Example 19B using 75 mg (0.118 mmol) of the subtitled compound of Example 19A, 21 ⁇ L (0.118 mmol) of triethylamine and 8.4 ⁇ L (0.118 mmol) of acetyl chloride in 2 mL of methylene chloride to provide 70 mg of a colorless glassy material.
- This material was purified using radial chromatography (gradient eluent of 2.5-5% methanol in ethyl acetate) to provide 64 mg of a white foam.
- This foam was further purified using HPLC (eluent of 30% water in methanol) to provide the desired titled compound (purity >
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 12, using 290 mg (0.72 mmol) of the titled compound from Example 6, 274 mg (0.72 mmol) of 2S-N(t- butoxycarbonyl)amino-3-quinolin-8-ylsulfonyl propanoic acid, 98 mg (0.72 mmol) of HOBT-H 2 0, and 149 mg (0.72 mmol) of DCC in 7 mL of tetrahydrofuran to provide a brownish foam. This foam was purified using radial chromatography (eluent of 2.5% methanol in ethyl acetate) to provide 380 mg of a white glassy material. Yield: 75%. Analysis for C 39 H 53 N 5 0 S 2 : Calcd: C, 60.99; H, 6.96; N, 9.12;
- the desired titled compound was prepared substantially in accordance with the procedure detailed in Example 25, by first reacting 51 mg (0.066 mmol) of the subtitled compound of Example 24A and 1 mL of trifluoroacetic acid in 1 mL of methylene chloride to provide a residue. This residue was then redissolved in 1 mL of methylene chloride and combined with 18.4 ⁇ L (0.132 mmol) of triethylamine and 5.6 ⁇ L (0.072 mmol) of methanesulfonylchloride to provide a crude material. This material was purified using radial chromatography (eluent of 3% methanol in methylene chloride containing 0.1% ammonium hydroxide) to provide 18 mg of a white powder.
- Example 27 A. N-t-Butyl-octahvdro-5 r2R-hydroxy-3S-phenylmethyl-4- aza-5-oxo-6S-N(t-butoxycarbonyl)amino-7-naphth-2- ylsulfonyl-heptyll - (3aR,7aS) -thieno 3 ,2-clpyridine- (6S) - carboxamide
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 12, using 220 mg (0.72 mmol) of the titled compound from Example 6, 274 mg (0.72 mmol) of 2S-N(t- butoxycarbonyl)amino-3-naphth-2-ylsulfonyl propanoic acid, 98 mg (0.72 mmol) of HOBT-H 2 0, and 149 mg (0.72 mmol) of DCC in 7 mL of tetrahydrofuran to provide crude material
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 24B. First, 154 mg (0.2 mmol) of the subtitled compound of Example 27A and 3 mL of trifluoroacetic acid were reacted in 3 mL of methylene chloride to provide a residue. This residue was then dissolved in 2 mL of acetonitrile and 32 ⁇ L (0.25 mmol) of S-ethyl trifluorothioacetate was added. The resultant reaction mixture was monitored by TLC. After approximately twelve hours, an additional 3 drops of S-ethyl trifluorothioacetate was added to the reaction mixture. When the reaction was substantially complete, the crude material was isolated and purified using radial chromatography (eluent of 2% methanol in methylene chloride) to provide 96 mg of a white glassy solid.
- radial chromatography eluent of 2% methanol in methylene chloride
- the desired titled compound was prepared substantially in accordance with the procedure detailed in Example 25.
- This residue was then redissolved in 1 mL of methylene chloride and reacted with 11.7 ⁇ L (0.165 mmol) of acetyl chloride in the presence of 23 ⁇ L (0.165 mmol) of triethylamine to provide an oil.
- This oil was purified using radial chromatography (silica, eluent of 3% methanol in methylene chloride) to provide 66 mg of a white powder. .Analysis for C 37 H 8 N 4 0 6 S 2 :
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 23, using 0.140 mg (0.210 mmol) of the subtitled compound of Example 21A and 70 ⁇ L of S-ethyl trifluorothioacetate in 1 mL of methylene chloride to provide crude material. This material was purified using radial chromatography (eluent of 2% methanol in methylene chloride) to provide 65 mg of the subtitled compound. Yield: 41%.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 12, using 120 mg (0.275 mmol) of the titled compound from Example 8, 104 mg (0.275 mmol) of 2S-N(t- butoxycarbonyl)amino-3-naphth-2-ylsulfonyl propanoic acid, 37 mg (0.275 mmol) of HOBT-H 2 0, -and 57 mg (0.275 mmol) of DCC in tetrahydrofuran to provide crude material. This material was purified using radial chromatography (eluent of 2% methanol in methylene chloride) to provide 169 mg of the subtitled compound. Yield: 77%.
- the reaction mixture was filtered (using ethyl acetate to wash) to provide approximately 1 g of a white solid.
- the resulting filtrate was concentrated under reduced pressure to provide 2.3 g of an orange oil.
- This oil was purified using radial chromatography (4000 micron plate; eluent of 1% ethyl acetate in methylene chloride) to provide mixed fractions: 1.6 g (2:1 cis, trans) and 90 mg (4:1 cis, trans) which were used in Example 31C without further purification.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 10A, using 0.75 g (2.65 mmol) of the mixture of compounds isolated in Example 3IB and 1.7 g (5.4) of MCPBA in 5 mL of methylene chloride to provide 0.88 g of crude material. This material was purified using flash chromatography (eluent of 1:1 ethyl acetate and cyclohexane) to provide the desired compounds. Analysis for C 12 H 13 N0 7 S: Calcd: C, 45.71; H, 4.16; N, 4.44; Found: C, 45.50; H, 4.26; N, 4.71. Yield (trans isomers) : 0.21 g (25%) .
- the subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9B, using 0.10 g (0.23 mmol) of the titled compound from Example 8, 64 ⁇ L (0.46 mmol) of triethylamine and 72 mg (0.23 mmol) of the ( ⁇ ) cis-subtitled compounds isolated in Example 31C in 3 mL of methylene chloride to provide the subtitled compounds.
- the crude material was purified using radial chromatography (1000 micron plate; eluent of 3% methanol in methylene chloride) to provide two fractions. Analysis for C 8 H 43 N 3 0 6 S 3 :
- the desired cis and trans compounds were separated from 0.68 g of the cis/trans mixture obtained in Example 31A, using HPLC (steel column, Rainin Dynamax CN, on a Waters 4000) .
- HPLC steel column, Rainin Dynamax CN, on a Waters 4000
- the desired compounds were eluted using an eluent of 5% diethyl ether in pentane (at 40 mL/min.) .
- the subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9B, using 119 mg (0.271 mmol) of the titled compound from
- Example 8 75 ⁇ L (0.542 mmol) of triethylamine and 90 mg (0.286 mmol) of the ( ⁇ ) trans subtitled compounds isolated in Example 31C in 3 mL of methylene chloride to provide the subtitled compounds.
- the crude material was purified using radial chromatography (2000 micron plate; eluent of 2% methanol in methylene chloride) to provide two fractions.
- This oil was purified using HPLC (steel column, Rainin Dynamax CN on a Waters 4000, eluent of 30% diethyl ether in pentane, 45 mL/min.) to provide 0.44 g of a colorless oil.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9A, using 0.29 g (2.5 mmol) of the subtitled compound of Example 36B, 1.02 mL (7.4 mmol) of triethylamine, and 0.94 g (3.7 mmol) of N,N'-disuccinimidyl carbonate in 5 mL of acetonitrile to provide 0.72 g of a light brown oil (51%) .
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9B, using 0.48 g (1.1 mmol) of the titled compound from Example 8, 0.32 g (1.1 mmol) of the subtitled compound from Example 37B, and 0.32 mL (2.31 mmol) of triethylamine in 4 mL of methylene chloride to provide 0.8 g of a crude white solid.
- This solid was purified using radial chromatography (4000 micron plate; eluent of 3% methanol in methylene chloride) to provide 0.50 g of a white solid. Yield: 74%.
- MS (FD) 613.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9A, using 0.30 g (2.5 mmol) of the subtitled compound of Example 33B, 1.06 g (7.62 mmol) of triethylamine, and 0.98 g (3.8 mmol) of N,N'-disuccinimidyl carbonate in 5 mL of acetonitrile to provide 0.70 g of a light brown oil (51%) which was used without further purification.
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9B, using 0.44 g (1.0 mmol) of the titled compound from Example 8, 0.32 g (1.1 mmol) of the subtitled compound from Example 38B, and 0.28 mL (2.0 mmol) of triethylamine in 3 mL of methylene chloride to provide 0.8 g of a crude white solid.
- This solid was purified using flash chromatography (silica, eluent of 3.5% methanol in methylene chloride) to provide 0.39 g of a white solid. Yield: 64%.
- Example 38C in 5 mL of methylene chloride, was slowly added
- Example 39A To a mixture of 3.0 g of the subtitled compounds of Example 39A in 48 mL of a 0.05M phosphate buffer (pH 7), was added 0.24 mL of Triton® X-100 (Fluka) and 0.12 g of lipase (SAM II) from Pseudomonas fluorescens (Fluka) . The resultant reaction mixture was heated to 37-39 * C and allowed to react overnight. The reaction mixture was then diluted with 50 mL of diethyl ether, the resultant layers were separated and the aqueous layer was extracted twice with 40 mL of diethyl ether.
- Triton® X-100 Triton® X-100
- SAM II lipase
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9A, using 600 mg (4.1 mmol) of the subtitled compound of Example 39B, 1.71 mL (12.3 mmol) of triethylamine, and 1.58 mg (6.15 mmol) of N,N'- disuccinimidyl carbonate in acetonitrile to provide a crude material. This material was purified using column chromatography (50 g Si0 2 ; gradient eluent of 30-50% ethyl acetate in hexane) to provide 1.0 g of the desired subtitled compound (85%) .
- iH NMR 300 MHz, CDC1 3 ) : ⁇ 5.42 (m, IH) ; 3.24 (m, IH) ;
- the desired subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9A, using 1.0 g (3.48 mmol) of the subtitled compound from Example 40A and 1.83 g (7.1 mmol) of 85% m-chloroperoxybenzoic acid (MCPBA) in 50 mL of methylene chloride to provide 1.1 g (100%) which was used without further purification.
- MCPBA m-chloroperoxybenzoic acid
- Example 10B (Alternate preparation of Example 10B (52 m ⁇ fraction)
- the subtitled compound was prepared substantially in accordance with the procedure detailed in Example 9B, using 100 mg (0.230 mmol) of the titled compound from Example 8, 66 ⁇ L (0.473 mmol) of triethylamine and 77 mg (0.24 mmol) of the subtitled compound from Example 40B in 1.5 mL of methylene chloride to provide crude material.
- Example 40C in 12 mL of a 2:1 mixture of methylene chloride/acetonitrile, was slowly added 85.8 ⁇ L (1.33 mmol) of methanesulfonic acid. The resultant reaction mixture was concentrated under reduced pressure to provide a residue. This residue was redissolved in diethyl ether and hexane and concentrated under reduced pressure (three times) . The resultant solid was sonicated with hexane and concentrated under reduced pressure to provide 965 mg of the desired subtitled compound (99.5%) .
- the following compounds may be prepared in accordance with the procedures detailed above.
- N-t-butyl-octahydro-5 [2R-hydroxy-3R-N(l', l'-dioxo- 2'-methyl-tetrahydrothiophen-3'S-yloxycarbonyl)amino-4- phenylthio) -butyl] - (3aR,7aS) -thieno[3,2-c] pyridine- (6S) -carboxamide
- N-t-butyl-octahydro-5 [2R-hydroxy-3R-N(l', l'-dioxo- tetrahydrothiophen-2'R-ylmethylcarbonyl)amino-4- phenylthio-butyl] - (3aR,7aS) -thieno[3,2-c] pyridine- (6S) -carboxamide;
- the compounds of the present invention are useful for inhibiting HIV protease, which is an enzyme associated with viral component production and assembly.
- An embodiment of the present invention is a method of treating or preventing HIV infection comprising administering to a primate in need thereof an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- Another embodiment of the present invention is a method of treating or preventing AIDS comprising administering to a primate in need thereof an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- a further embodiment of the present invention is a method of inhibiting HIV replication comprising administering to an HIV infected cell, a cell susceptible to HIV infection or a primate in need thereof, an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the term "effective amount” as used herein, means an amount of a compound of formula (I) which is capable of inhibiting the HIV protease mediated viral component production and assembly.
- the HIV protease inhibition contemplated by the present method includes either therapeutic or prophylactic treatment, as appropriate.
- the specific dose of compound administered according to this invention to obtain therapeutic or prophylactic effects will, of course, be determined by the particular circumstances surrounding the case, including, for example, the compound administered, the route of administration, the condition being treated and the individual being treated.
- a typical daily dose (administered in single or divided doses) will contain a dosage level of from about 0.01 mg/kg to about 50 mg/kg of body weight of an active compound of this invention.
- Preferred daily doses generally will be from about 0.05 mg/kg to about 20 mg/kg and ideally from about 0.1 mg/kg to about 10 mg/kg.
- the compounds can be administered by a variety of routes including oral, rectal, transdermal, subcutaneous, intravenous, intramuscular and intranasal .
- the compounds of the present invention are preferably formulated prior to administration. Therefore, another embodiment of the present invention is a pharmaceutical formulation comprising an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier, diluent or excipient therefor.
- the active ingredient in such. formulations comprises from 0.1% to 99.9% by weight of the formulation.
- pharmaceutically acceptable it is meant that the carrier, diluent or excipient is compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- the present pharmaceutical formulations can be prepared by known procedures using known and readily available ingredients.
- the active ingredient will usually be admixed with a carrier, or diluted by a carrier, or enclosed within a carrier which may be in the form of a capsule, sachet, paper or other container.
- a carrier which may be in the form of a capsule, sachet, paper or other container.
- the carrier serves as a diluent, it may be a solid, semi-solid or liquid material which acts as a vehicle, excipient or medium for the active ingredient.
- compositions can be in the form of tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols, (as a solid or in a liquid medium) , ointments containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, sterile injectable solutions, sterile packaged powders and the like.
- active ingredient represents a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- Hard gelatin capsules are prepared using the following ingredients:
- Formulation 2 A tablet is prepared using the ingredients below:
- the components are blended and compressed to form tablets each weighing 665 mg.
- Formulation 3 An aerosol solution is prepared containing the following components:
- the active compound is mixed with ethanol and the mixture added to a portion of the propellant 22, cooled to -30"C and transferred to a filling device. The required amount is then fed to a stainless steel container and diluted with the remainder of the propellant. The valve units are then fitted to the container.
- Formulation 4 Tablets each containing 60 mg of active ingredient, are made as follows: Quantity
- the active ingredient, starch and cellulose are passed through a No. 45 mesh U.S. sieve and mixed thoroughly.
- the aqueous solution containing polyvinylpyrrolidone is mixed with the resultant powder, and the mixture then is passed through a No. 14 mesh U.S. sieve.
- the granules so produced are dried at 50 * C and passed through a No. 18 mesh U.S. sieve.
- the sodium carboxymethyl starch, magnesium stearate and talc, previously passed through a No. 60 mesh U.S. sieve, are then added to the granules which, after mixing, are compressed on a tablet machine to yield tablets each weighing 150 mg.
- Capsules each containing 80 mg of active ingredient, are made as follows:
- the active ingredient, cellulose, starch and magnesium stearate are blended, passed through a No. 45 mesh U.S. sieve, and filled into hard gelatin capsules in 200 mg quantities.
- Formulation 6 Suppositories, each containing 225 mg of active ingredient, are made as follows:
- the active ingredient is passed through a No. 60 mesh U.S. sieve and suspended in the saturated fatty acid glycerides previously melted using the minimum heat necessary. The mixture is then poured into a suppository mold of nominal 2 g capacity and allowed to cool.
- Suspensions each containing 50 mg of active ingredient per 5 mL dose, are made as follows:
- the active ingredient is passed through a No. 45 mesh U.S. sieve and mixed with the sodium carboxymethyl cellulose and syrup to form a smooth paste.
- the benzoic acid solution, flavor and color are diluted with a portion of the water and added, with stirring. Sufficient water is then added to produce the required volume.
- Formulation 8 An intravenous formulation may be prepared as follows:
- the solution of the above ingredients generally is administered intravenously to a subject at a rate of 1 mL per minute.
- the compounds of formula (I) are useful as HIV protease inhibitors. These compounds may be assayed using two assays: (1) an enzyme inhibition assay, and (2) an antiviral cell culture assay. These assays and the resultant data are provided below.
- a Fluorescence HIV-1 Protease Inhibitor Assay was carried out to demonstrate the ability of the compounds of the present invention to inhibit HIV protease. This assay is described in detail in published European Patent Application (EPA) 0 526 009, herein incorporated by reference. Using this assay, a number of the compounds, prepared above, were assayed for HIV protease inhibitory activity.
- the concentration of the tested compound that inhibits 50% of the enzyme (IC 50 ) is reported below in Table 1.
- the assay is unable to test compounds at less than a concentration of 0.16 ng/mL.
- some values are provided as % inhibition at [the stated] concentration (i.e. IC 7 (0.16) represents the ability of the compound to inhibit 74% of the enzyme at 0.16 ng/mL) .
- This assay compares the viability of HIV-infected cells in a cell culture medium relative to the viability of such cells in the presence of a test compound. The concentration of test compound necessary to inhibit 90% of the virus is then measured using the viability of the cells as an indicator of viral inhibition. This assay is carried out using cell lines that are sensitive to the lytic effects of HIV infection.
- this assay utilizes the metabolic reduction of a tetrazolium reagent
Landscapes
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description












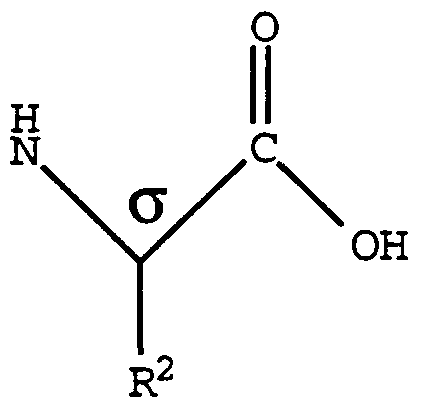

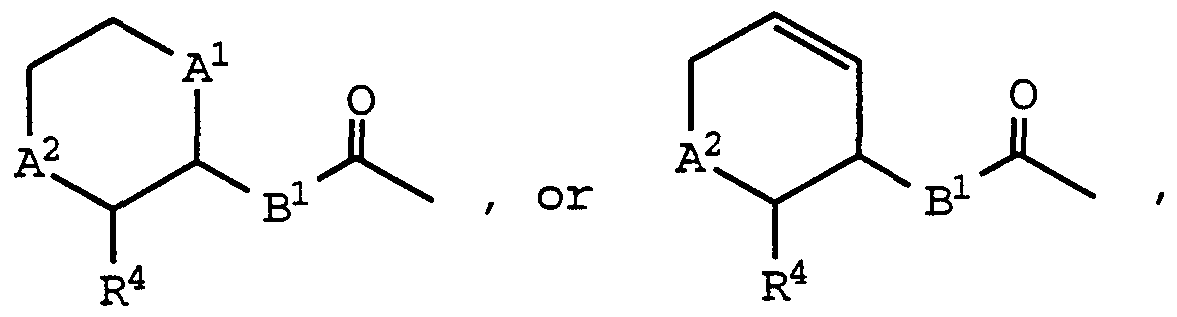





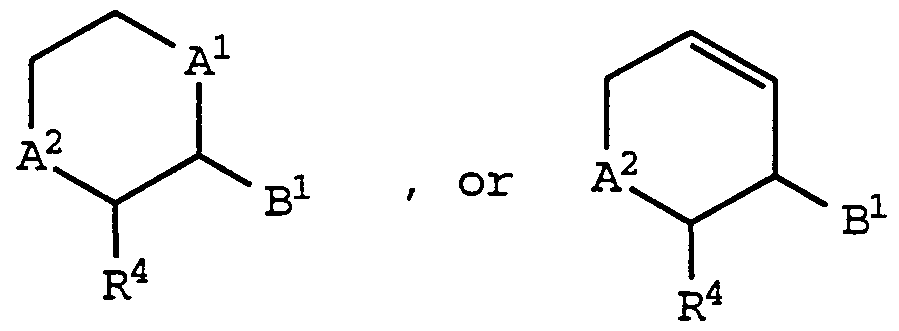











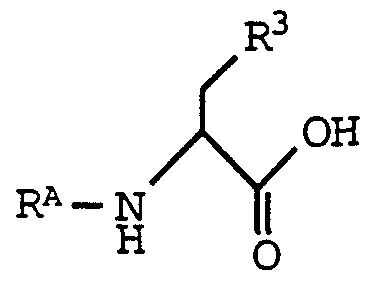








Claims


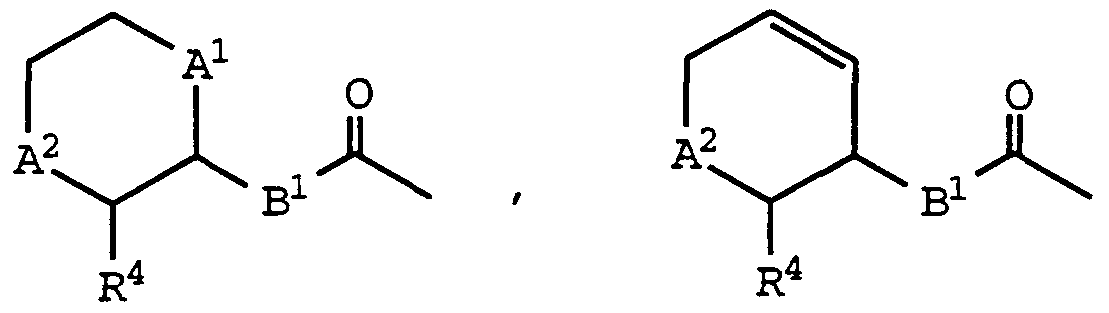






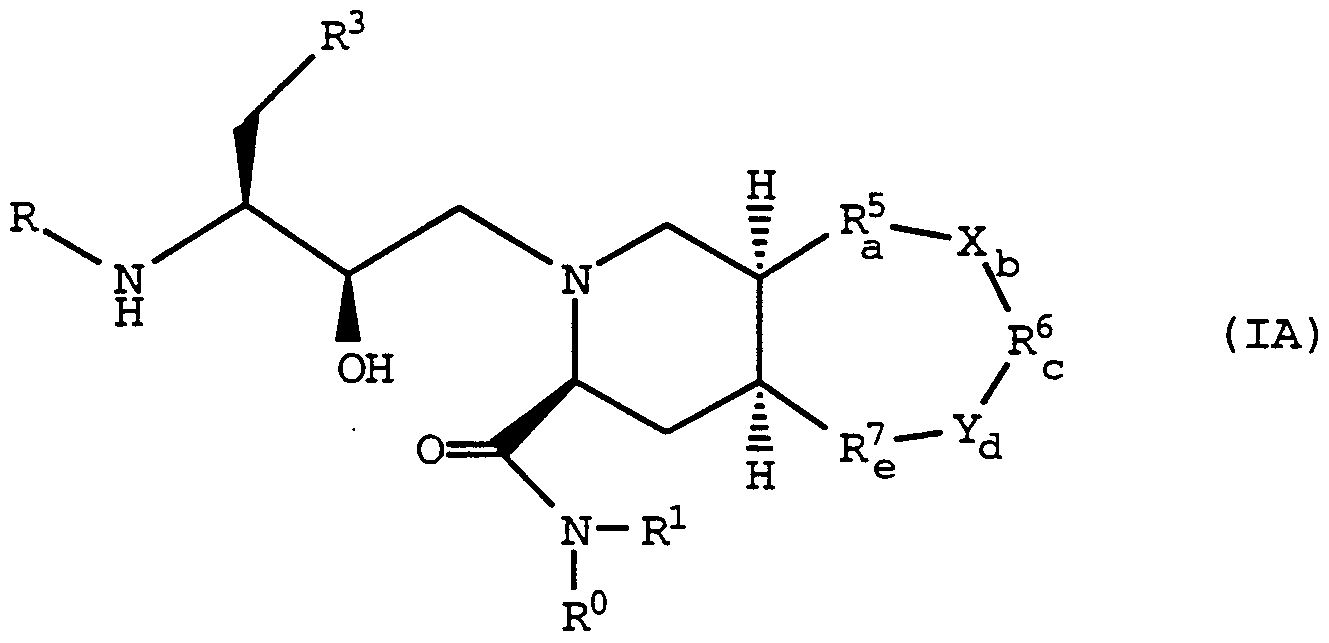

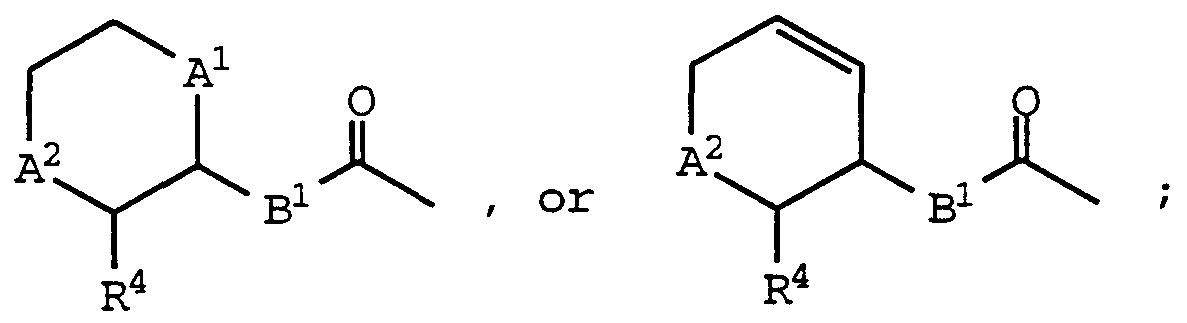



Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP94930062A EP0744948A4 (en) | 1994-02-02 | 1994-10-06 | Protease inhibitors |
| AU79302/94A AU700417B2 (en) | 1994-02-02 | 1994-10-06 | Protease inhibitors |
| JP7520583A JPH09509155A (en) | 1994-02-02 | 1994-10-06 | Protease inhibitor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US19081094A | 1994-02-02 | 1994-02-02 | |
| US08/190,810 | 1994-02-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995020962A1 true WO1995020962A1 (en) | 1995-08-10 |
Family
ID=22702885
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1994/011350 Ceased WO1995020962A1 (en) | 1994-02-02 | 1994-10-06 | Protease inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US5480887A (en) |
| EP (1) | EP0744948A4 (en) |
| JP (1) | JPH09509155A (en) |
| CN (1) | CN1145587A (en) |
| AU (1) | AU700417B2 (en) |
| CA (1) | CA2182192A1 (en) |
| WO (1) | WO1995020962A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0823424A1 (en) * | 1996-08-09 | 1998-02-11 | F. Hoffmann-La Roche Ag | Process for the preparation of quinargine |
| US5914404A (en) * | 1996-08-09 | 1999-06-22 | Hoffmann-La Roche Inc. | Process for the preparation of quinargine |
| WO2000002862A1 (en) * | 1998-07-08 | 2000-01-20 | G.D. Searle & Co. | Retroviral protease inhibitors |
| US6632826B1 (en) | 2001-09-28 | 2003-10-14 | Hoffmann-La Roche Inc. | Carbocyclic HIV protease inhibitors |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0560268B1 (en) * | 1992-03-13 | 1995-01-04 | Bio-Mega/Boehringer Ingelheim Research Inc. | Substituted pipecolinic acid derivatives as HIV protease inhibitors |
| US5484926A (en) * | 1993-10-07 | 1996-01-16 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| WO1995020965A1 (en) * | 1994-02-02 | 1995-08-10 | Eli Lilly And Company | Hiv protease inhibitors and intermediates |
| AU712120B2 (en) * | 1996-05-02 | 1999-10-28 | Merck & Co., Inc. | HIV protease inhibitors useful for the treatment of AIDS |
| JP4626896B2 (en) | 1998-02-13 | 2011-02-09 | ダイセル化学工業株式会社 | Acylating agent, acylation method using the same, and adamantane derivative |
| AU1930900A (en) * | 1998-12-04 | 2000-06-26 | University Of Maryland Biotechnology Institute | Use of protease inhibitors to modulate cellular pathways, immunity and therapiesassociated therewith |
| US6583299B1 (en) * | 1999-05-20 | 2003-06-24 | G.D. Searle & Co. | α-amino-β-sulfonyl hydroxamic acid compounds |
| ATE504572T1 (en) * | 1999-09-02 | 2011-04-15 | Shionogi & Co | INTEGRAS INHIBITORS CONTAINING DERIVATIVES OF AROMATIC HETEROCYCLES |
| US8754114B2 (en) | 2010-12-22 | 2014-06-17 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| ES2704744T3 (en) | 2012-06-13 | 2019-03-19 | Incyte Holdings Corp | Substituted tricyclic compounds as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| EA035095B1 (en) | 2013-04-19 | 2020-04-27 | Инсайт Холдингс Корпорейшн | Bicyclic heterocycles as fgfr inhibitors |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| MX373169B (en) | 2015-02-20 | 2020-04-24 | Incyte Holdings Corp | Bicyclic heterocycles as fgfr inhibitors |
| WO2016134294A1 (en) | 2015-02-20 | 2016-08-25 | Incyte Corporation | Bicyclic heterocycles as fgfr4 inhibitors |
| MA41551A (en) | 2015-02-20 | 2017-12-26 | Incyte Corp | BICYCLIC HETEROCYCLES USED AS FGFR4 INHIBITORS |
| AR111960A1 (en) | 2017-05-26 | 2019-09-04 | Incyte Corp | CRYSTALLINE FORMS OF A FGFR INHIBITOR AND PROCESSES FOR ITS PREPARATION |
| CR20200590A (en) | 2018-05-04 | 2021-04-26 | Incyte Corp | Solid forms of an fgfr inhibitor and processes for preparing the same |
| MA52493A (en) | 2018-05-04 | 2021-03-10 | Incyte Corp | FGFR INHIBITOR SALTS |
| WO2020185532A1 (en) | 2019-03-08 | 2020-09-17 | Incyte Corporation | Methods of treating cancer with an fgfr inhibitor |
| WO2021007269A1 (en) | 2019-07-09 | 2021-01-14 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| WO2021067374A1 (en) | 2019-10-01 | 2021-04-08 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| TWI891666B (en) | 2019-10-14 | 2025-08-01 | 美商英塞特公司 | Bicyclic heterocycles as fgfr inhibitors |
| WO2021076728A1 (en) | 2019-10-16 | 2021-04-22 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| BR112022010664A2 (en) | 2019-12-04 | 2022-08-16 | Incyte Corp | DERIVATIVES OF A FGFR INHIBITOR |
| EP4069696A1 (en) | 2019-12-04 | 2022-10-12 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
| WO2021146424A1 (en) | 2020-01-15 | 2021-07-22 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| TW202304459A (en) | 2021-04-12 | 2023-02-01 | 美商英塞特公司 | Combination therapy comprising an fgfr inhibitor and a nectin-4 targeting agent |
| WO2022261159A1 (en) | 2021-06-09 | 2022-12-15 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
| EP4352059A1 (en) | 2021-06-09 | 2024-04-17 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5142056A (en) * | 1989-05-23 | 1992-08-25 | Abbott Laboratories | Retroviral protease inhibiting compounds |
| IL89900A0 (en) * | 1988-04-12 | 1989-12-15 | Merck & Co Inc | Hiv protease inhibitors useful for the treatment of aids and pharmaceutical compositions containing them |
| CA1340588C (en) * | 1988-06-13 | 1999-06-08 | Balraj Krishan Handa | Amino acid derivatives |
| EP0361341A3 (en) * | 1988-09-28 | 1991-07-03 | Miles Inc. | Therapeutics for aids based on inhibitors of hiv protease |
| EP0813868B1 (en) * | 1990-11-19 | 2005-06-01 | Monsanto Company | Retroviral protease inhibitors |
| ZA92913B (en) * | 1991-02-08 | 1993-05-06 | Sankyo Co | New beta-amino-alpha hydroxycarboxylic acids and ttheir use |
| CN1071930A (en) * | 1991-07-10 | 1993-05-12 | 伊莱利利公司 | Inhibitors of human immunodeficiency virus protease for the treatment of AIDS |
| WO1993008184A1 (en) * | 1991-10-23 | 1993-04-29 | Merck & Co., Inc. | Hiv protease inhibitors |
| WO1993013066A1 (en) * | 1991-12-20 | 1993-07-08 | Syntex (U.S.A.) Inc. | Cyclic amides of 3-amino-2-hydroxy-carboxylic acids as hiv-protease inhibitors |
| US5484926A (en) * | 1993-10-07 | 1996-01-16 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| WO1995020965A1 (en) * | 1994-02-02 | 1995-08-10 | Eli Lilly And Company | Hiv protease inhibitors and intermediates |
-
1994
- 1994-08-31 US US08/299,186 patent/US5480887A/en not_active Expired - Fee Related
- 1994-10-06 JP JP7520583A patent/JPH09509155A/en active Pending
- 1994-10-06 CA CA002182192A patent/CA2182192A1/en not_active Abandoned
- 1994-10-06 EP EP94930062A patent/EP0744948A4/en not_active Ceased
- 1994-10-06 CN CN94195067A patent/CN1145587A/en active Pending
- 1994-10-06 WO PCT/US1994/011350 patent/WO1995020962A1/en not_active Ceased
- 1994-10-06 AU AU79302/94A patent/AU700417B2/en not_active Ceased
-
1995
- 1995-08-16 US US08/515,875 patent/US5624934A/en not_active Expired - Fee Related
Non-Patent Citations (2)
| Title |
|---|
| JOURNAL OF MEDICINAL CHEMISTRY, Volume 30, issued 1987, ROSENBERG et al., "Novel Renin Inhibitors Containing Analogues of Statine Retro-Inverted at the C-Termini Specificity at the P Histidine Site", see pages 1224-1228. * |
| See also references of EP0744948A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0823424A1 (en) * | 1996-08-09 | 1998-02-11 | F. Hoffmann-La Roche Ag | Process for the preparation of quinargine |
| US5914404A (en) * | 1996-08-09 | 1999-06-22 | Hoffmann-La Roche Inc. | Process for the preparation of quinargine |
| WO2000002862A1 (en) * | 1998-07-08 | 2000-01-20 | G.D. Searle & Co. | Retroviral protease inhibitors |
| US6632826B1 (en) | 2001-09-28 | 2003-10-14 | Hoffmann-La Roche Inc. | Carbocyclic HIV protease inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1145587A (en) | 1997-03-19 |
| US5624934A (en) | 1997-04-29 |
| EP0744948A1 (en) | 1996-12-04 |
| JPH09509155A (en) | 1997-09-16 |
| EP0744948A4 (en) | 1997-06-11 |
| AU700417B2 (en) | 1999-01-07 |
| US5480887A (en) | 1996-01-02 |
| CA2182192A1 (en) | 1995-08-10 |
| AU7930294A (en) | 1995-08-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU700417B2 (en) | Protease inhibitors | |
| DE69324120T2 (en) | HIV protease inhibitors and their use in the treatment of AIDS | |
| US5502061A (en) | Peptidyl substituted benzamides and naphthamies | |
| EP0746320B1 (en) | Hiv protease inhibitors and intermediates | |
| US5719287A (en) | Intermediates for inhibitors of HIV protease and method of preparation thereof | |
| US5434265A (en) | Inhibitors of HIV protease | |
| EP0604184B1 (en) | Inhibitors of HIV protease useful for the treatment of aids | |
| JPH06256277A (en) | HIV-protease inhibitors useful for the treatment of AIDS | |
| US5578608A (en) | Symmetrical diaryl and diheteroanyl cis epoxy alkanes antiviral compounds | |
| US5508407A (en) | Retroviral protease inhibitors | |
| JPH09509657A (en) | Intermediates and methods for manufacturing | |
| JPH06234716A (en) | HIV-protease inhibitors useful in the treatment of AIDS | |
| US5475136A (en) | Inhibitors of HIV protease useful for the treatment of AIDS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 94195067.0 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AM AT AU BB BG BR BY CA CH CN CZ DE DK ES FI GB GE HU JP KE KG KP KR KZ LK LT LU LV MD MG MN MW NL NO NZ PL PT RO RU SD SE SI SK TJ TT UA UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE MW SD SZ AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2182192 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994930062 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994930062 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1994930062 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994930062 Country of ref document: EP |