WO1996022968A1 - Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles - Google Patents
Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles Download PDFInfo
- Publication number
- WO1996022968A1 WO1996022968A1 PCT/US1996/000548 US9600548W WO9622968A1 WO 1996022968 A1 WO1996022968 A1 WO 1996022968A1 US 9600548 W US9600548 W US 9600548W WO 9622968 A1 WO9622968 A1 WO 9622968A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- carbon atoms
- improved process
- independentiy
- straight chain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(CCC=C1)=C1OP(Oc1ccccc1*)Oc(c(*)cc1ccccc11)c1-c(c(cccc1)c1cc1*)c1OC1C2Oc(cccc3)c3-c(cccc3)c3OC12 Chemical compound *C(CCC=C1)=C1OP(Oc1ccccc1*)Oc(c(*)cc1ccccc11)c1-c(c(cccc1)c1cc1*)c1OC1C2Oc(cccc3)c3-c(cccc3)c3OC12 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6571—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and oxygen atoms as the only ring hetero atoms
- C07F9/6574—Esters of oxyacids of phosphorus
- C07F9/65746—Esters of oxyacids of phosphorus the molecule containing more than one cyclic phosphorus atom
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/18—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms
- B01J31/1845—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms the ligands containing phosphorus
- B01J31/185—Phosphites ((RO)3P), their isomeric phosphonates (R(RO)2P=O) and RO-substitution derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/08—Preparation of carboxylic acid nitriles by addition of hydrogen cyanide or salts thereof to unsaturated compounds
- C07C253/10—Preparation of carboxylic acid nitriles by addition of hydrogen cyanide or salts thereof to unsaturated compounds to compounds containing carbon-to-carbon double bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/141—Esters of phosphorous acids
- C07F9/1411—Esters of phosphorous acids with hydroxyalkyl compounds with further substituents on alkyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/141—Esters of phosphorous acids
- C07F9/1412—Polyol derivatives esterified at least twice by phosphorous acid groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/141—Esters of phosphorous acids
- C07F9/145—Esters of phosphorous acids with hydroxyaryl compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/141—Esters of phosphorous acids
- C07F9/146—Esters of phosphorous acids containing P-halide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/22—Amides of acids of phosphorus
- C07F9/24—Esteramides
- C07F9/2404—Esteramides the ester moiety containing a substituent or a structure which is considered as characteristic
- C07F9/242—Esteramides the ester moiety containing a substituent or a structure which is considered as characteristic of hydroxyaryl compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/655—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having oxygen atoms, with or without sulfur, selenium, or tellurium atoms, as the only ring hetero atoms
- C07F9/65515—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having oxygen atoms, with or without sulfur, selenium, or tellurium atoms, as the only ring hetero atoms the oxygen atom being part of a five-membered ring
- C07F9/65517—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having oxygen atoms, with or without sulfur, selenium, or tellurium atoms, as the only ring hetero atoms the oxygen atom being part of a five-membered ring condensed with carbocyclic rings or carbocyclic ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6571—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and oxygen atoms as the only ring hetero atoms
- C07F9/6574—Esters of oxyacids of phosphorus
- C07F9/65744—Esters of oxyacids of phosphorus condensed with carbocyclic or heterocyclic rings or ring systems
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/30—Addition reactions at carbon centres, i.e. to either C-C or C-X multiple bonds
- B01J2231/32—Addition reactions to C=C or C-C triple bonds
- B01J2231/323—Hydrometalation, e.g. bor-, alumin-, silyl-, zirconation or analoguous reactions like carbometalation, hydrocarbation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/50—Redistribution or isomerisation reactions of C-C, C=C or C-C triple bonds
- B01J2231/52—Isomerisation reactions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/02—Compositional aspects of complexes used, e.g. polynuclearity
- B01J2531/0261—Complexes comprising ligands with non-tetrahedral chirality
- B01J2531/0266—Axially chiral or atropisomeric ligands, e.g. bulky biaryls such as donor-substituted binaphthalenes, e.g. "BINAP" or "BINOL"
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/80—Complexes comprising metals of Group VIII as the central metal
- B01J2531/84—Metals of the iron group
- B01J2531/847—Nickel
Definitions
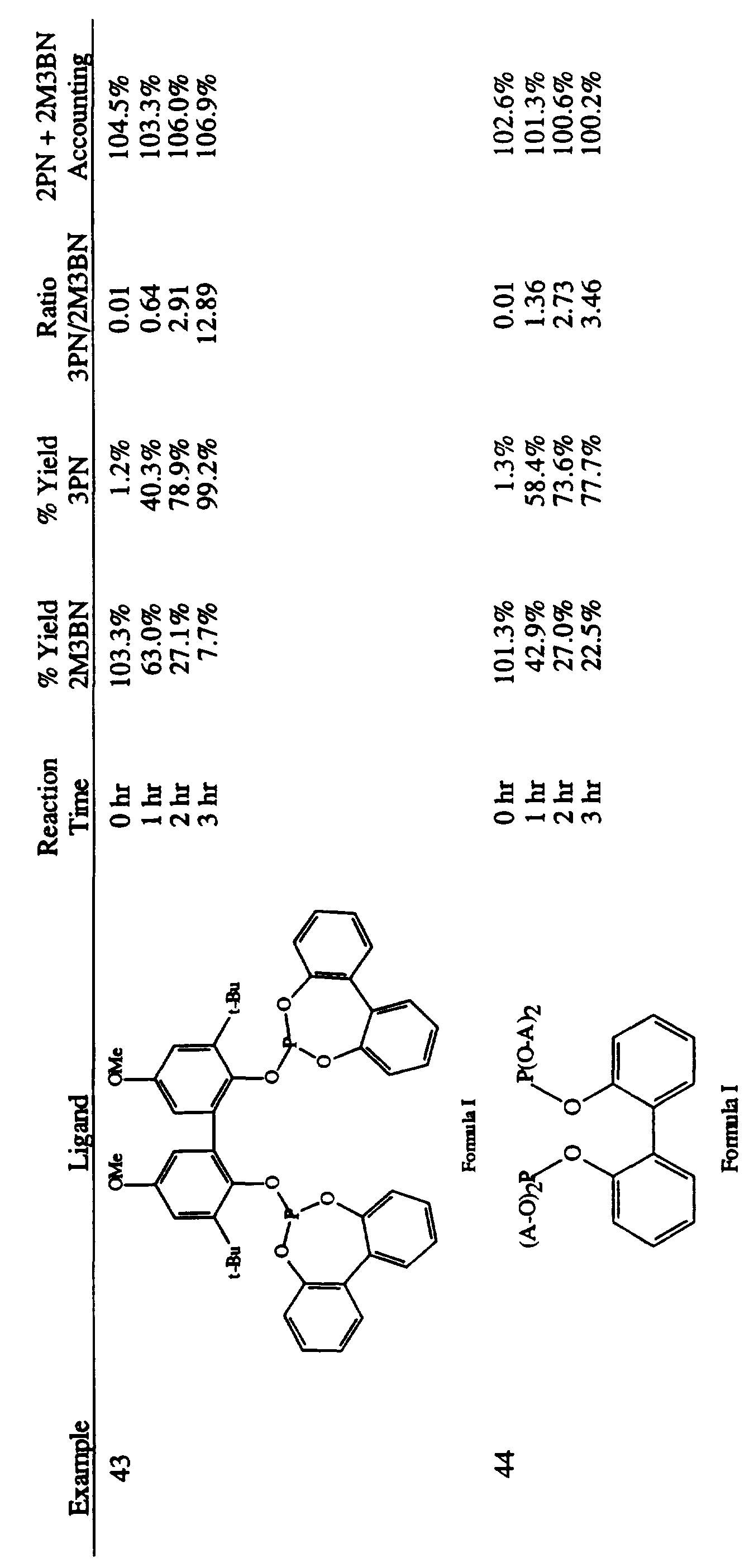
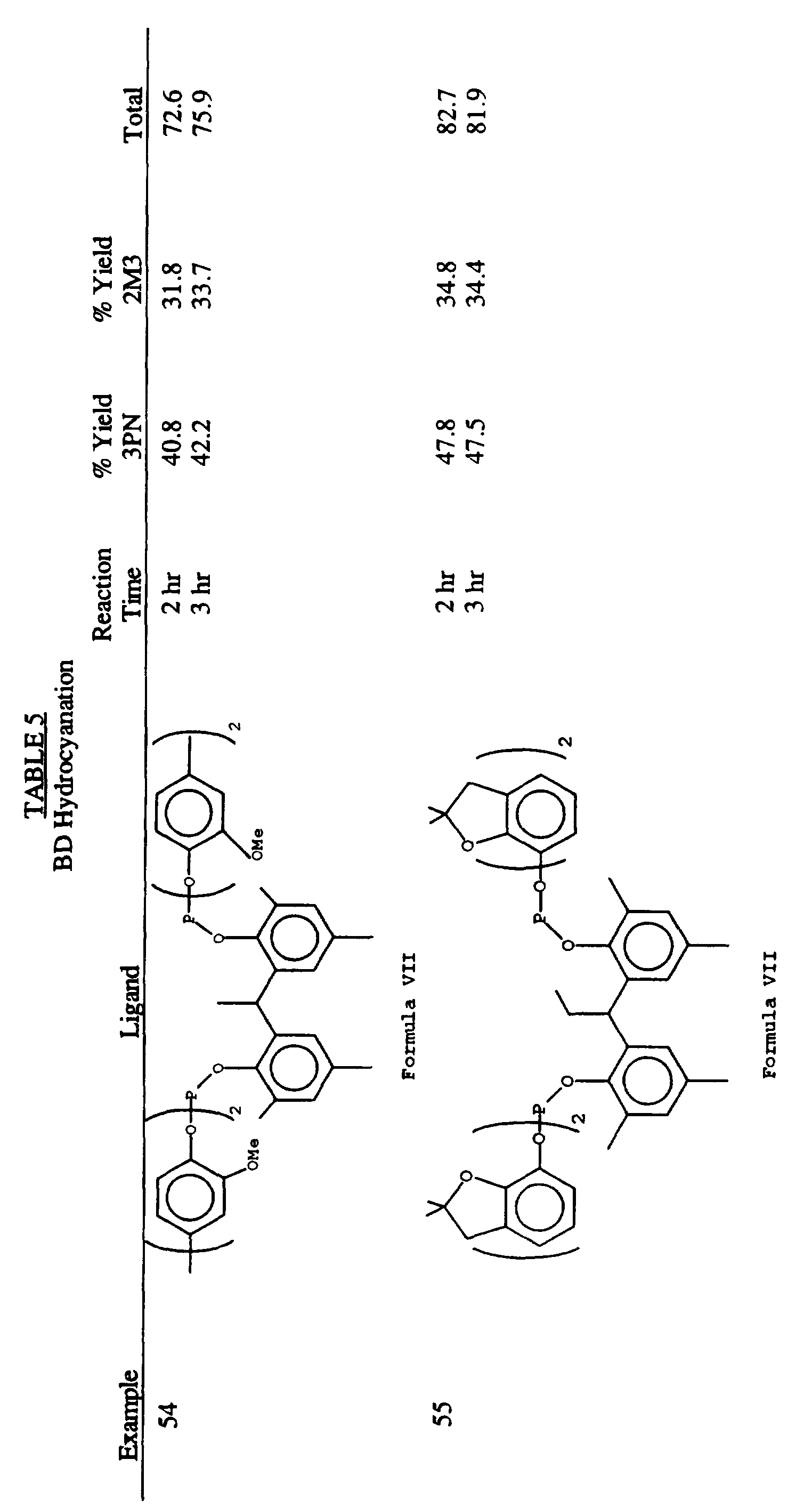
- the present invention provides an improved process for the liquid phase hydrocyanation of diolefinic compounds and isomerization of the resulting nonconjugated acyclic nitriles comprising, reacting an acyclic aliphatic diolefinic compound, preferably butadiene, with a source of HCN, wherein the improvement comprises conducting the hydrocyanation and/or isomerzation in the presence of a catalyst precursor composition comprising zero-valent nickel and at least one multidentate phosphite ligand selected from the group consisting of compounds represented by Formulas I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, and XV as set forth below:
- each R 1 is independently a branched or straight chain alkyl of up to 12 carbon
- each R 2' is independentiy H or a primary, secondary or tertiary hydrocarbyl of 1 to 12 carbon atoms at either the meta or para position to the oxygen; or CN, CO 2 R 4 or OR 4" wherein R 4 is a C 1 to C 6 alkyl at either the meta or para position to the oxygen of the phenoxy ring;
- the reactions are most conveniently performed continuously from hydrocyanation of the starting diolefin to the final 3- and/or 4-monoalkene linear nitriles.
- the processes can be conducted stepwise, i.e., the
- ligands may be prepared by a variety of methods known in the art, for example, see descriptions in WO 93,03839, U.S. 4,769,498; U.S. 4,688,651, J. Amer. Chem. So c., 1993, 115, 2066.
- the reaction of 2,2'-biphenol with phosphorus trichloride gives 1,1'-biphenyl-2,2'-diyl phosphorochloridite.
- the zero-valent nickel can be prepared or generated according to techniques known in the art (U.S. 3,496,217; 3,631,191; 3,846,461; 3,847,959; and 3,903,120 which are incorporated herein by reference).
- Zero-valent nickel compounds that contain ligands which can be displaced by the organophosphorus ligand are a preferred source of zero-valent nickel.
- Two such preferred zero-valent nickel compounds are Ni(COD) 2 (COD is 1,5-cyclooctadiene) and
- the reaction mixture is agitated, such as by stirring or shaking.
- the cyanated product can be recovered by conventional techniques such as crystallization of the product from solution or by distillation.
- the process is usually carried out "neat", i.e., without an added diluent or solvent.
- Any solvent or diluent that is nondestructive of the catalyst can be used, however.
- Suitable solvents include aliphatic or aromatic hydrocarbons (hexane, cyclohexane, benzene), ethers (diethyl ether, tetrahydrofuran, dioxane, glycol dimethyl ether, anisole), esters (ethyl acetate, methyl benzoate), nitriles
- Ligand B Formula VI where R 2 is the cyclic group, -O-C(CH 3 ) 2 -CH 2 -
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Inorganic Chemistry (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Toxicology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Polyesters Or Polycarbonates (AREA)
- Lubricants (AREA)
- Luminescent Compositions (AREA)
- Polyurethanes Or Polyureas (AREA)
Abstract
Description
























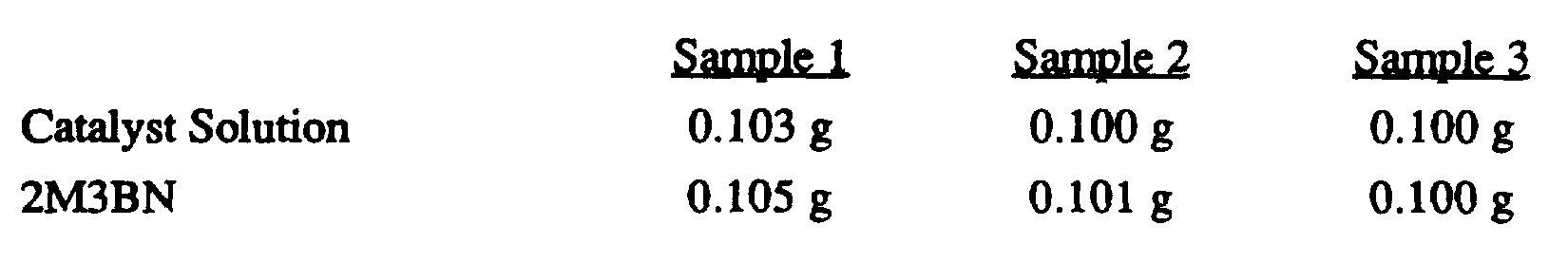








































Claims






























Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69605398T DE69605398T2 (en) | 1995-01-27 | 1996-01-17 | CYAN HYDROGEN ADDITION TO DIOLEFINS AND ISOMERIZATION OF NON-CONJUGATED 2-LKYL-3-MONOALKENNITRILES |
| EP96902689A EP0804412B1 (en) | 1995-01-27 | 1996-01-17 | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| JP52290296A JP3959111B2 (en) | 1995-01-27 | 1996-01-17 | Diolefin hydrocyanation and non-conjugated 2-alkyl-3-monoalkenenitrile isomerization |
| BRPI9606718-7A BR9606718B1 (en) | 1995-01-27 | 1996-01-17 | improved processes for liquid phase hydrocyanation, improved process for isomerization, multidentate phosphite binder and composition. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37942995A | 1995-01-27 | 1995-01-27 | |
| US08/379,429 | 1995-11-28 | ||
| US08/563,718 US5821378A (en) | 1995-01-27 | 1995-11-28 | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| US08/563,718 | 1995-11-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996022968A1 true WO1996022968A1 (en) | 1996-08-01 |
Family
ID=27008622
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1996/000548 Ceased WO1996022968A1 (en) | 1995-01-27 | 1996-01-17 | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US5696280A (en) |
| EP (1) | EP0804412B1 (en) |
| JP (1) | JP3959111B2 (en) |
| CN (2) | CN1076342C (en) |
| AT (2) | ATE236172T1 (en) |
| BR (1) | BR9606718B1 (en) |
| CA (1) | CA2208040A1 (en) |
| DE (2) | DE69627207T2 (en) |
| ES (2) | ES2141474T3 (en) |
| ID (1) | ID24513A (en) |
| IN (1) | IN187044B (en) |
| WO (1) | WO1996022968A1 (en) |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997033892A1 (en) * | 1996-03-15 | 1997-09-18 | Dsm N.V. | Process to prepare a multidentate phosphite compound |
| WO1998027054A1 (en) * | 1996-12-16 | 1998-06-25 | Basf Aktiengesellschaft | Monoolefinic c5-mononitriles, method for the production and the use thereof |
| WO1999006418A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Selective synthesis of organodiphosphite compounds |
| WO1999006355A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| WO1999006356A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Hydrocyanation processes and multidentate phosphite ligand and nickel catalyst compositions therefor |
| WO1999006359A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Improved hydrocyanation process |
| WO1999006357A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alykl-3-monoalkenenitriles |
| WO1999013983A1 (en) * | 1997-09-12 | 1999-03-25 | Basf Aktiengesellschaft | Catalyst comprising at least one phosphonite ligand based nickel (o) complex and method for the production of nitriles |
| US5910600A (en) * | 1996-04-30 | 1999-06-08 | Mitsubishi Chemical Corporation | Bisphosphite compound, process for its production and hydroformylation process employing the bisphosphite compound |
| WO1999064155A1 (en) * | 1998-06-05 | 1999-12-16 | Basf Aktiengesellschaft | Catalyst comprising a complex of a metal from subgroup viii based on a bidentate phosphonite ligand, and method for producing nitriles |
| WO2001021579A1 (en) * | 1999-09-20 | 2001-03-29 | E.I. Du Pont De Nemours And Company | Multidentate phosphite ligands, catalytic compositions containing such ligands and catalytic processes utilizing such catalytic compositions |
| WO2001021580A1 (en) * | 1999-09-20 | 2001-03-29 | E.I. Du Pont De Nemours And Company | Multidentate phosphite ligands, catalytic compositions containing such ligands and catalytic processes utilizing such catalytic compositions |
| WO2002013964A3 (en) * | 2000-08-02 | 2002-07-18 | Basf Ag | Suitable catalyst for producing nitriles |
| US6486359B1 (en) | 1998-06-18 | 2002-11-26 | Basf Aktiengesellschaft | Catalyst comprising a complex of a metal of subgroup viii based on a phosphinite ligand, and a method for hydroformylation |
| US6515161B1 (en) | 1999-09-20 | 2003-02-04 | E. I. Du Pont De Nemours And Company | Hydroformylation process utilizing multidentate phosphite ligands |
| US6660876B2 (en) | 2001-11-26 | 2003-12-09 | E. I. Du Pont De Nemours And Company | Phosphorus-containing compositions and their use in hydrocyanation, isomerization and hydroformylation reactions |
| KR100714324B1 (en) * | 1999-09-20 | 2007-05-04 | 이 아이 듀폰 디 네모아 앤드 캄파니 | Multidentate phosphite ligands, and catalyst compositions containing them |
| WO2008008928A3 (en) * | 2006-07-14 | 2008-02-28 | Invista Tech Sarl | Process for making 3-pentenenitrile by hydrocyanation of butadiene |
| WO2008008929A3 (en) * | 2006-07-14 | 2008-02-28 | Invista Tech Sarl | Process for making 3-pentenenitrile by hydrocyanation of butadiene |
| WO2009091771A3 (en) * | 2008-01-15 | 2009-09-24 | Invista Technologies S.A R.L | Process for making and refining 3-pentenenitrile, and for refining 2-methyl-3-butenenitrile |
| US7629484B2 (en) | 2006-03-17 | 2009-12-08 | Invista North America S.A.R.L. | Method for the purification of triorganophosphites by treatment with a basic additive |
| WO2010046226A1 (en) | 2008-10-21 | 2010-04-29 | Rhodia Operations | Method for producing compounds including nitrile functions |
| WO2010086246A1 (en) | 2009-01-29 | 2010-08-05 | Rhodia Operations | Method for producing compounds including nitrile functions |
| EP2239248A2 (en) | 2002-12-02 | 2010-10-13 | INVISTA Technologies S.à.r.l. | A selective synthesis of organophosphites |
| US7897801B2 (en) | 2003-05-12 | 2011-03-01 | Invista North America S.A R.L. | Process for the preparation of dinitriles |
| US7919646B2 (en) | 2006-07-14 | 2011-04-05 | Invista North America S.A R.L. | Hydrocyanation of 2-pentenenitrile |
| US7973174B2 (en) | 2005-10-18 | 2011-07-05 | Invista North America S.A.R.L. | Process of making 3-aminopentanenitrile |
| US8088943B2 (en) | 2008-01-15 | 2012-01-03 | Invista North America S.A R.L. | Hydrocyanation of pentenenitriles |
| US8101790B2 (en) | 2007-06-13 | 2012-01-24 | Invista North America S.A.R.L. | Process for improving adiponitrile quality |
| US8247621B2 (en) | 2008-10-14 | 2012-08-21 | Invista North America S.A.R.L. | Process for making 2-secondary-alkyl-4,5-di-(normal-alkyl)phenols |
| US8338636B2 (en) | 2009-08-07 | 2012-12-25 | Invista North America S.A R.L. | Hydrogenation and esterification to form diesters |
| US8373001B2 (en) | 2003-02-10 | 2013-02-12 | Invista North America S.A R.L. | Method of producing dinitrile compounds |
| WO2013076569A1 (en) | 2011-10-26 | 2013-05-30 | Invista Technologies S.A R.L. | Methods for producing organodi phosphites from phosphorochloridites characterized by measuring side- product levels to determine further additions |
| US8609901B2 (en) | 2009-04-21 | 2013-12-17 | Invista North America S.A R.L. | Highly selective process for producing organodiphosphites |
| US9061970B2 (en) | 2008-01-25 | 2015-06-23 | Invista North America S.A.R.L. | Production of compounds comprising nitrile functional groups |
| US9233917B2 (en) | 2008-06-17 | 2016-01-12 | Invista North America S.A R.L. | Preparation of nitriles from ethylenically unsaturated compounds |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3812095B2 (en) | 1997-10-28 | 2006-08-23 | 三菱化学株式会社 | Method for producing aldehydes and bisphosphite used therefor |
| CA2328866A1 (en) * | 1998-04-16 | 1999-10-21 | E.I. Du Pont De Nemours And Company | Hydrocyanation of olefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| DE10136488A1 (en) * | 2001-07-27 | 2003-02-06 | Basf Ag | Catalyst system comprising Ni(0) and phosphorous ligands is useful for the production of nitrile and dinitrile compounds |
| US6489517B1 (en) * | 2001-11-26 | 2002-12-03 | E. I. Du Pont De Nemours And Company | Process for preparing 3,3′,6,6′-tetraalkyl-2,2′-biphenols and 3,3′,6,6′-tetraalkyl-5,5′-dihalo-2,2′-biphenols |
| EP1344770A1 (en) * | 2002-03-12 | 2003-09-17 | E.I. du Pont de Nemours and Company | Process for the hydrocyanation of butadiene |
| WO2004078766A1 (en) * | 2003-02-27 | 2004-09-16 | Mitsubishi Chemical Corporation | Optically active phosphites and phosphoramides bearing biphenol skeletons with axial chirality, and their use in catalytic asymmetric reactions |
| FR2854892B1 (en) | 2003-05-12 | 2005-06-24 | Rhodia Polyamide Intermediates | PROCESS FOR PRODUCING DINITRILES |
| DE102004004720A1 (en) * | 2004-01-29 | 2005-08-18 | Basf Ag | Process for the preparation of 3-pentenenitrile |
| DE102004004724A1 (en) * | 2004-01-29 | 2005-08-18 | Basf Ag | Preparation of 3-pentenenitrile from 1,3-butadiene |
| CN1309728C (en) * | 2004-07-30 | 2007-04-11 | 中国科学院上海有机化学研究所 | Chiral organic, inorganic polymer assembled catalyst, synthesis method and use |
| US7287477B2 (en) * | 2004-10-13 | 2007-10-30 | Foster Wheeler Energy Corporation | Cyclone bypass for a circulating fluidized bed reactor |
| BRPI0619982A2 (en) * | 2005-12-15 | 2011-10-25 | The Penn State Research Foundation | tetraphosphorus binders for catalytic hydroformylation and related reactions |
| US7709674B2 (en) | 2006-07-14 | 2010-05-04 | Invista North America S.A R.L | Hydrocyanation process with reduced yield losses |
| WO2008017626A1 (en) * | 2006-08-08 | 2008-02-14 | Basf Se | Method for producing 3-pentenenitrile by means of the hydrocyanation of 1,3-butadiene |
| CN101687658B (en) | 2007-05-14 | 2013-07-24 | 因温斯特技术公司 | High efficiency reactor and process |
| FR2932476B1 (en) * | 2008-06-17 | 2010-07-30 | Rhodia Operations | PROCESS FOR THE PRODUCTION OF NITRILIC COMPOUNDS FROM ETHYLENE-UNSATURATED COMPOUNDS |
| CN105753906A (en) * | 2014-12-18 | 2016-07-13 | 中国科学院兰州化学物理研究所 | Chiral bidentate phosphite ligand derived from cyclohexanediol and preparation method and application of ligand |
| EP4074720B1 (en) * | 2021-04-16 | 2023-07-19 | Evonik Operations GmbH | Mixture of bisphosphites with an open and a closed volatile building block and its use as catalyst mixture in hydroformylation |
| CN113912516B (en) * | 2021-10-15 | 2023-06-27 | 浙江新和成股份有限公司 | Application of multidentate phosphite ligand in catalytic synthesis of adiponitrile |
| CN116947696A (en) * | 2023-08-07 | 2023-10-27 | 刘凯凯 | A method for in-situ catalytic synthesis of adiponitrile |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2934564A (en) * | 1957-11-08 | 1960-04-26 | American Potash & Chem Corp | Organohalophosphines |
| EP0005500A1 (en) * | 1978-05-18 | 1979-11-28 | Ciba-Geigy Ag | Dioxaphosphepines, their preparation and use as stabilisers for organic materials |
| WO1993003839A1 (en) * | 1991-08-21 | 1993-03-04 | Union Carbide Chemicals & Plastics Technology Corporation | Asymmetric syntheses |
| US5288918A (en) * | 1992-09-29 | 1994-02-22 | Union Carbide Chemicals & Plastics Technology Corporation | Hydroformylation process |
Family Cites Families (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1084599A (en) * | 1964-10-15 | 1967-09-27 | Asahi Chemical Ind | Process for the preparation of unsaturated aliphatic nitriles |
| US3853948A (en) * | 1965-11-23 | 1974-12-10 | Du Pont | Catalytic isomerization of 2-methyl-3-butenenitrile to linear pentene-nitriles |
| US3496215A (en) * | 1965-11-23 | 1970-02-17 | Du Pont | Hydrocyanation of olefins using selected nickel phosphite catalysts |
| US3536748A (en) * | 1965-11-23 | 1970-10-27 | Du Pont | Catalytic isomerization of 2-methyl-3-butenenitrile to linear pentenenitriles |
| GB1112539A (en) * | 1965-11-26 | 1968-05-08 | Du Pont | Preparation of organic nitriles |
| US3485917A (en) * | 1966-04-14 | 1969-12-23 | Janssen Pharmaceutica Nv | Composition and method for combating fungus with imidazole carboxylates |
| US3496210A (en) * | 1966-07-28 | 1970-02-17 | Du Pont | Hydrocyanation of terminal alkynes |
| US3496217A (en) * | 1967-05-23 | 1970-02-17 | Du Pont | Hydrocyanation of olefins |
| US3578695A (en) * | 1967-07-26 | 1971-05-11 | Standard Oil Co Ohio | Olefinic nitriles by the catalytic oxidation of olefins using hydrogen cyanide |
| US3775461A (en) * | 1967-11-06 | 1973-11-27 | Du Pont | Hydrocyanation of olefins |
| US3869500A (en) * | 1968-03-23 | 1975-03-04 | Asahi Chemical Ind | Process for the production of unsaturated aliphatic nitriles |
| US3584029A (en) * | 1968-04-09 | 1971-06-08 | Asahi Chemical Ind | Process for the production of lower saturated aliphatic nitriles |
| US3655723A (en) * | 1969-10-31 | 1972-04-11 | Du Pont | Hydrocyanation of olefins |
| NL6916495A (en) * | 1969-11-01 | 1971-05-04 | Pivalonitrile from isobutene and hcn | |
| US3631191A (en) * | 1970-04-08 | 1971-12-28 | Du Pont | Synthesis of zero valent nickel-tetrakis triaryl phosphite complexes |
| JPS4928495B1 (en) * | 1970-06-16 | 1974-07-26 | ||
| US3676481A (en) * | 1970-06-29 | 1972-07-11 | Du Pont | Catalytic isomerization of 2-methyl-3-butenenitrile to linear pentenenitriles in the presence of certain metal salt and/or tri(hydrocarbyl)boron promoters |
| US3739011A (en) * | 1971-04-30 | 1973-06-12 | Du Pont | Catalytic isomerization of 2-methyl-3-butenenitrile to linear pentenenitriles |
| US3798256A (en) * | 1971-08-02 | 1974-03-19 | Du Pont | Hydrocyanation of olefins |
| US3925445A (en) * | 1971-08-02 | 1975-12-09 | Du Pont | Hydrocyanation of olefins |
| US3766231A (en) * | 1971-08-02 | 1973-10-16 | Du Pont | Compounds of zero valent nickel containing n bonded nitriles |
| US3766237A (en) * | 1972-01-25 | 1973-10-16 | Du Pont | Hydrocyanation of olefins |
| GB1417554A (en) * | 1972-02-07 | 1975-12-10 | Ici Ltd | Organo metal complexes |
| US3773809A (en) * | 1972-06-28 | 1973-11-20 | Du Pont | Separation of organic phosphorus compounds and their metal complexes from organic nitriles in the hydrocyanation of olefins |
| US3846461A (en) * | 1972-10-25 | 1974-11-05 | Du Pont | Process of preparing a zerovalent nickel complex with organic phosphorus compounds |
| US3847959A (en) * | 1972-10-25 | 1974-11-12 | Du Pont | Process of preparing a zerovalent nickel complex with organic phosphorus compounds |
| US3903120A (en) * | 1973-06-19 | 1975-09-02 | Du Pont | Preparation of zerovalent nickel complexes from elemental nickel |
| US3852325A (en) * | 1973-08-29 | 1974-12-03 | Du Pont | Selective isomerization of pentenenitriles |
| US3852328A (en) * | 1973-09-26 | 1974-12-03 | Du Pont | Catalytic isomerization of 2-methyl-3-butenenitrile to a linear pentenenitrile |
| US3852329A (en) * | 1973-10-02 | 1974-12-03 | Du Pont | Process for isomerization of 2-methyl-3-butene-nitrile to a linear pentenenitrile |
| US4298546A (en) * | 1980-08-05 | 1981-11-03 | E. I. Du Pont De Nemours And Company | Isomerization of 2-methyl-3-butenenitrile |
| US4371474A (en) * | 1982-01-13 | 1983-02-01 | E. I. Du Pont De Nemours And Company | Hydrocyanation of olefins |
| US4599206A (en) * | 1984-02-17 | 1986-07-08 | Union Carbide Corporation | Transition metal complex catalyzed reactions |
| US4737588A (en) * | 1984-12-28 | 1988-04-12 | Union Carbide Corporation | Transition metal complex catalyzed reactions |
| US4668651A (en) * | 1985-09-05 | 1987-05-26 | Union Carbide Corporation | Transition metal complex catalyzed processes |
| US4774353A (en) * | 1986-06-05 | 1988-09-27 | E. I. Du Pont De Nemours And Company | Triorganotin catalyst promoters for hydrocyanation |
| US4739000A (en) * | 1986-07-22 | 1988-04-19 | Ethyl Corporation | Antioxidant aromatic tetraphosphites |
| US4705881A (en) * | 1986-11-17 | 1987-11-10 | E. I. Du Pont De Nemours And Company | Continuous hydrocyanation process using zinc halide promoter |
| US4714773A (en) * | 1987-01-09 | 1987-12-22 | E. I. Du Pont De Nemours And Company | Hydrocyanation of butadiene |
| US4783546A (en) * | 1987-06-02 | 1988-11-08 | E. I. Du Pont De Nemours And Company | Preparation of 4-pentenenitrile by isomerization |
| US4874884A (en) * | 1988-03-31 | 1989-10-17 | E. I. Du Pont De Nemours And Company | Promoter synergism in pentenenitrile hydrocyanation |
| DE4026406A1 (en) * | 1990-08-21 | 1992-02-27 | Basf Ag | RHODIUM HYDROFORMYLATION CATALYSTS WITH BIS-PHOSPHITE LIGANDS |
| TW213465B (en) * | 1991-06-11 | 1993-09-21 | Mitsubishi Chemicals Co Ltd | |
| JP3416956B2 (en) * | 1991-06-11 | 2003-06-16 | 三菱化学株式会社 | Hydroformylation method and bisphosphite compound |
| US5175335A (en) * | 1991-11-12 | 1992-12-29 | E. I. Du Pont De Nemours And Company | Enantioselective hydrocyanation of aromatic vinyl compounds |
| US5292785A (en) * | 1992-05-05 | 1994-03-08 | Ciba-Geigy Corporation | Bis-phosphite stabilized compositions |
| EP0730574B1 (en) * | 1993-11-23 | 1998-08-19 | E.I. Du Pont De Nemours And Company | Processes and catalyst compositions for hydrocyanation of monoolefins |
| US5512695A (en) * | 1994-04-14 | 1996-04-30 | E. I. Du Pont De Nemours And Company | Bidentate phosphite and nickel catalyst compositions for hydrocyanation of monoolefins |
| US5543536A (en) * | 1994-04-26 | 1996-08-06 | E. I. Du Pont De Nemours And Company | Monodentate phosphite and nickel catalyst composition for monoolefin hydrocyanation |
| US5512696A (en) * | 1995-07-21 | 1996-04-30 | E. I. Du Pont De Nemours And Company | Hydrocyanation process and multidentate phosphite and nickel catalyst composition therefor |
| US5440067A (en) * | 1994-11-18 | 1995-08-08 | E. I. Dupont De Nemours And Company | Catalyzed gas phase isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| US5449807A (en) * | 1994-11-18 | 1995-09-12 | E. I. Du Pont De Nemours And Company | Catalyzed vapor phase hydrocyanation of diolefinic compounds |
-
1996
- 1996-01-08 IN IN36CA1996 patent/IN187044B/en unknown
- 1996-01-17 WO PCT/US1996/000548 patent/WO1996022968A1/en not_active Ceased
- 1996-01-17 BR BRPI9606718-7A patent/BR9606718B1/en not_active IP Right Cessation
- 1996-01-17 ES ES96902689T patent/ES2141474T3/en not_active Expired - Lifetime
- 1996-01-17 JP JP52290296A patent/JP3959111B2/en not_active Expired - Fee Related
- 1996-01-17 DE DE69627207T patent/DE69627207T2/en not_active Expired - Lifetime
- 1996-01-17 CA CA002208040A patent/CA2208040A1/en not_active Abandoned
- 1996-01-17 ES ES98203601T patent/ES2196481T3/en not_active Expired - Lifetime
- 1996-01-17 EP EP96902689A patent/EP0804412B1/en not_active Expired - Lifetime
- 1996-01-17 CN CN96191601A patent/CN1076342C/en not_active Expired - Lifetime
- 1996-01-17 AT AT98203601T patent/ATE236172T1/en not_active IP Right Cessation
- 1996-01-17 CN CNB011208791A patent/CN100427495C/en not_active Expired - Lifetime
- 1996-01-17 DE DE69605398T patent/DE69605398T2/en not_active Expired - Lifetime
- 1996-01-17 AT AT96902689T patent/ATE187161T1/en not_active IP Right Cessation
- 1996-01-26 ID IDP20000245A patent/ID24513A/en unknown
- 1996-10-11 US US08/729,457 patent/US5696280A/en not_active Expired - Lifetime
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2934564A (en) * | 1957-11-08 | 1960-04-26 | American Potash & Chem Corp | Organohalophosphines |
| EP0005500A1 (en) * | 1978-05-18 | 1979-11-28 | Ciba-Geigy Ag | Dioxaphosphepines, their preparation and use as stabilisers for organic materials |
| WO1993003839A1 (en) * | 1991-08-21 | 1993-03-04 | Union Carbide Chemicals & Plastics Technology Corporation | Asymmetric syntheses |
| US5288918A (en) * | 1992-09-29 | 1994-02-22 | Union Carbide Chemicals & Plastics Technology Corporation | Hydroformylation process |
Non-Patent Citations (5)
| Title |
|---|
| G. MÄRKL ET AL.: "Zur Chemie des Cyclooctatetraenyldilithiums", JOURNAL OF ORGANOMETALLIC CHEMISTRY, vol. 273, 1984, LAUSANNE CH, pages 1 - 29, XP002002593 * |
| J. HEINICKE ET AL.: "1,3-Carbanionische Umlagerungen, Synthese von Bis(o-hydroxyaryl)-Phosphorverbindungen", PHOSPHORUS, SULFUR SILICON RELAT. ELEM., vol. 44, no. 3-4, 1989, pages 209 - 16, XP000570204 * |
| M. J. BAKER ET AL.: "Chelating diphosphite complexes of nickel(0) and platinum(0): Their remarkable stability and hydrocyanation activity", JOURNAL OF THE CHEMICAL SOCIETY, CHEMICAL COMMUNICATIONS, 1991, LETCHWORTH GB, pages 803 - 4, XP002002591 * |
| M. J. BARKER ET AL.: "Chiral aryl diphosphites: a new class of ligands for hydrocyanation catalysis", JOURNAL OF THE CHEMICAL SOCIETY, CHEMICAL COMMUNICATIONS, 1991, LETCHWORTH GB, pages 1292 - 3, XP002002592 * |
| ZH. M. IVANOVA: "Phosphoramidous Difluorides", J. GEN. CHEM. USSR (ENGL. TRANSL.), vol. 35, 1965, pages 165 - 7, XP000570216 * |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997033892A1 (en) * | 1996-03-15 | 1997-09-18 | Dsm N.V. | Process to prepare a multidentate phosphite compound |
| US5910600A (en) * | 1996-04-30 | 1999-06-08 | Mitsubishi Chemical Corporation | Bisphosphite compound, process for its production and hydroformylation process employing the bisphosphite compound |
| WO1998027054A1 (en) * | 1996-12-16 | 1998-06-25 | Basf Aktiengesellschaft | Monoolefinic c5-mononitriles, method for the production and the use thereof |
| RU2217416C2 (en) * | 1996-12-16 | 2003-11-27 | Басф Акциенгезелльшафт | Method for preparing mixtures of monoolefinic c5-mononitriles and method for preparing adipodinitrile based on thereof |
| WO1999006359A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Improved hydrocyanation process |
| US6020516A (en) * | 1997-07-29 | 2000-02-01 | E. I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| JP2002509550A (en) * | 1997-07-29 | 2002-03-26 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | Hydrocyanation of diolefins and isomerization of non-conjugated 2-alkyl-3-monoalkenenitrile |
| WO1999006357A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alykl-3-monoalkenenitriles |
| WO1999006418A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Selective synthesis of organodiphosphite compounds |
| WO1999006416A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Selective synthesis of organodiphosphite compounds |
| US5959135A (en) * | 1997-07-29 | 1999-09-28 | E. I. Du Pont De Nemours And Company | Hydrocyanation processes and multidentate phosphite ligand and nickel catalyst compositions thereof |
| US5981772A (en) * | 1997-07-29 | 1999-11-09 | E. I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| WO1999006355A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| WO1999006356A1 (en) * | 1997-07-29 | 1999-02-11 | E.I. Du Pont De Nemours And Company | Hydrocyanation processes and multidentate phosphite ligand and nickel catalyst compositions therefor |
| US6031120A (en) * | 1997-07-29 | 2000-02-29 | E. I. Du Pont De Nemours And Company | Selective synthesis of organodiphosphite compounds |
| US6069267A (en) * | 1997-07-29 | 2000-05-30 | E. I. Du Pont De Nemours And Company | Selective synthesis of organodiphosphite compounds |
| US6120700A (en) * | 1997-07-29 | 2000-09-19 | E. I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles |
| US6171997B1 (en) | 1997-07-29 | 2001-01-09 | E. I. Du Pont De Nemours And Company | Hydrocyanation of diolefins and isomerization of nonconjugated 2-aklyl-3-monoalkenenitriles |
| WO1999013983A1 (en) * | 1997-09-12 | 1999-03-25 | Basf Aktiengesellschaft | Catalyst comprising at least one phosphonite ligand based nickel (o) complex and method for the production of nitriles |
| US6242633B1 (en) | 1997-09-12 | 2001-06-05 | Basf Aktiengesellschaft | Catalyst comprising at least one phosphonite ligand based nickel (O) complex and method for the production of nitriles |
| US6355833B2 (en) | 1997-09-12 | 2002-03-12 | Basf Aktiengesellschaft | Catalyst comprising at least one nickel(0) complex based on a phosphonite ligand, and the preparation of nitriles |
| WO1999064155A1 (en) * | 1998-06-05 | 1999-12-16 | Basf Aktiengesellschaft | Catalyst comprising a complex of a metal from subgroup viii based on a bidentate phosphonite ligand, and method for producing nitriles |
| US6521778B1 (en) | 1998-06-05 | 2003-02-18 | Basf Aktiengesellschaft | Catalyst comprising a complex of a metal from subgroup VIII based on a bidentate phosphonite ligand, and method for producing nitriles |
| US6486359B1 (en) | 1998-06-18 | 2002-11-26 | Basf Aktiengesellschaft | Catalyst comprising a complex of a metal of subgroup viii based on a phosphinite ligand, and a method for hydroformylation |
| KR100714323B1 (en) * | 1999-09-20 | 2007-05-04 | 이 아이 듀폰 디 네모아 앤드 캄파니 | Catalytic Process Using Catalyst Compositions Containing Multidentate Phosphite Ligands |
| KR100714324B1 (en) * | 1999-09-20 | 2007-05-04 | 이 아이 듀폰 디 네모아 앤드 캄파니 | Multidentate phosphite ligands, and catalyst compositions containing them |
| US6515161B1 (en) | 1999-09-20 | 2003-02-04 | E. I. Du Pont De Nemours And Company | Hydroformylation process utilizing multidentate phosphite ligands |
| US6380421B1 (en) | 1999-09-20 | 2002-04-30 | E. I. Du Pont De Nemours And Company | Multidentate phosphite ligands, catalytic compositions containing such ligands and catalytic processes utilizing such catalytic compositions |
| WO2001021580A1 (en) * | 1999-09-20 | 2001-03-29 | E.I. Du Pont De Nemours And Company | Multidentate phosphite ligands, catalytic compositions containing such ligands and catalytic processes utilizing such catalytic compositions |
| US6812352B2 (en) | 1999-09-20 | 2004-11-02 | Invista North America S.A.R.L. | Multidentate phosphite ligands, catalytic compositions containing such ligands, and catalytic processes utilizing such catalytic compositions |
| WO2001021579A1 (en) * | 1999-09-20 | 2001-03-29 | E.I. Du Pont De Nemours And Company | Multidentate phosphite ligands, catalytic compositions containing such ligands and catalytic processes utilizing such catalytic compositions |
| WO2002013964A3 (en) * | 2000-08-02 | 2002-07-18 | Basf Ag | Suitable catalyst for producing nitriles |
| US6660876B2 (en) | 2001-11-26 | 2003-12-09 | E. I. Du Pont De Nemours And Company | Phosphorus-containing compositions and their use in hydrocyanation, isomerization and hydroformylation reactions |
| US6924345B2 (en) | 2001-11-26 | 2005-08-02 | Invista North America S.A R.L. | Phosphorus-containing compositions and their use in hydrocyanation, isomerization and hydroformylation reactions |
| EP1905511A2 (en) | 2001-11-26 | 2008-04-02 | INVISTA Technologies S.à.r.l. | Phosphorus-containing compositions and their use in hydrocyanation, isomerization and hydroformylation reactions |
| EP2277623A2 (en) | 2001-11-26 | 2011-01-26 | INVISTA Technologies S.à.r.l. | Polymeric phosphorus-containing compositions and their use in hydrocyanation, unsaturated nitrile isomerization and hydroformylation reactions |
| EP2243763A2 (en) | 2002-12-02 | 2010-10-27 | Invista Technologies S.à.r.l. | A selective synthesis of organophosphites |
| EP2239248A2 (en) | 2002-12-02 | 2010-10-13 | INVISTA Technologies S.à.r.l. | A selective synthesis of organophosphites |
| US8373001B2 (en) | 2003-02-10 | 2013-02-12 | Invista North America S.A R.L. | Method of producing dinitrile compounds |
| US7897801B2 (en) | 2003-05-12 | 2011-03-01 | Invista North America S.A R.L. | Process for the preparation of dinitriles |
| US7973174B2 (en) | 2005-10-18 | 2011-07-05 | Invista North America S.A.R.L. | Process of making 3-aminopentanenitrile |
| US8178711B2 (en) | 2006-03-17 | 2012-05-15 | Invista North America S.A R.L. | Method for the purification of triorganophosphites by treatment with a basic additive |
| US7629484B2 (en) | 2006-03-17 | 2009-12-08 | Invista North America S.A.R.L. | Method for the purification of triorganophosphites by treatment with a basic additive |
| WO2008008929A3 (en) * | 2006-07-14 | 2008-02-28 | Invista Tech Sarl | Process for making 3-pentenenitrile by hydrocyanation of butadiene |
| US7880028B2 (en) | 2006-07-14 | 2011-02-01 | Invista North America S.A R.L. | Process for making 3-pentenenitrile by hydrocyanation of butadiene |
| US7709673B2 (en) | 2006-07-14 | 2010-05-04 | Invista North America S.A R.L. | Process for making 3-pentenenitrile by hydrocyanation of butadiene |
| US7919646B2 (en) | 2006-07-14 | 2011-04-05 | Invista North America S.A R.L. | Hydrocyanation of 2-pentenenitrile |
| US8394981B2 (en) | 2006-07-14 | 2013-03-12 | Invista North America S.A R.L. | Hydrocyanation of 2-pentenenitrile |
| WO2008008928A3 (en) * | 2006-07-14 | 2008-02-28 | Invista Tech Sarl | Process for making 3-pentenenitrile by hydrocyanation of butadiene |
| US8101790B2 (en) | 2007-06-13 | 2012-01-24 | Invista North America S.A.R.L. | Process for improving adiponitrile quality |
| US8088943B2 (en) | 2008-01-15 | 2012-01-03 | Invista North America S.A R.L. | Hydrocyanation of pentenenitriles |
| WO2009091771A3 (en) * | 2008-01-15 | 2009-09-24 | Invista Technologies S.A R.L | Process for making and refining 3-pentenenitrile, and for refining 2-methyl-3-butenenitrile |
| US7977502B2 (en) | 2008-01-15 | 2011-07-12 | Invista North America S.A R.L. | Process for making and refining 3-pentenenitrile, and for refining 2-methyl-3-butenenitrile |
| US9061970B2 (en) | 2008-01-25 | 2015-06-23 | Invista North America S.A.R.L. | Production of compounds comprising nitrile functional groups |
| US9233917B2 (en) | 2008-06-17 | 2016-01-12 | Invista North America S.A R.L. | Preparation of nitriles from ethylenically unsaturated compounds |
| US8247621B2 (en) | 2008-10-14 | 2012-08-21 | Invista North America S.A.R.L. | Process for making 2-secondary-alkyl-4,5-di-(normal-alkyl)phenols |
| WO2010046226A1 (en) | 2008-10-21 | 2010-04-29 | Rhodia Operations | Method for producing compounds including nitrile functions |
| US9174207B2 (en) | 2008-10-21 | 2015-11-03 | Invista North America S.A.R.L. | Process for producing compounds comprising nitrile functions |
| WO2010086246A1 (en) | 2009-01-29 | 2010-08-05 | Rhodia Operations | Method for producing compounds including nitrile functions |
| US8772549B2 (en) | 2009-04-21 | 2014-07-08 | INVISTA North America S.à r.l. | Highly selective process for producing organodiphosphites |
| US8609901B2 (en) | 2009-04-21 | 2013-12-17 | Invista North America S.A R.L. | Highly selective process for producing organodiphosphites |
| US8338636B2 (en) | 2009-08-07 | 2012-12-25 | Invista North America S.A R.L. | Hydrogenation and esterification to form diesters |
| KR20140093218A (en) * | 2011-10-26 | 2014-07-25 | 인비스타 테크놀러지스 에스.에이 알.엘. | Methods for producing organodi phosphites from phosphorochloridites characterized by measuring side- product levels to determine further additions |
| WO2013076569A1 (en) | 2011-10-26 | 2013-05-30 | Invista Technologies S.A R.L. | Methods for producing organodi phosphites from phosphorochloridites characterized by measuring side- product levels to determine further additions |
| US9221852B2 (en) | 2011-10-26 | 2015-12-29 | Invista North America S.A.R.L. | Method for making organodiphosphites from phosphorochloridites characterized by measuring side-product levels to determine further additions |
| KR101890976B1 (en) | 2011-10-26 | 2018-08-22 | 인비스타 텍스타일스 (유.케이.) 리미티드 | Methods for producing organodi phosphites from phosphorochloridites characterized by measuring side- product levels to determine further additions |
Also Published As
| Publication number | Publication date |
|---|---|
| ATE236172T1 (en) | 2003-04-15 |
| DE69605398D1 (en) | 2000-01-05 |
| DE69627207D1 (en) | 2003-05-08 |
| CN1169143A (en) | 1997-12-31 |
| ES2196481T3 (en) | 2003-12-16 |
| CN1356335A (en) | 2002-07-03 |
| CA2208040A1 (en) | 1996-08-01 |
| EP0804412B1 (en) | 1999-12-01 |
| IN187044B (en) | 2002-01-05 |
| CN1076342C (en) | 2001-12-19 |
| US5696280A (en) | 1997-12-09 |
| DE69605398T2 (en) | 2000-07-27 |
| BR9606718B1 (en) | 2009-01-13 |
| JP3959111B2 (en) | 2007-08-15 |
| ID24513A (en) | 1996-07-25 |
| ATE187161T1 (en) | 1999-12-15 |
| JPH10512879A (en) | 1998-12-08 |
| EP0804412A1 (en) | 1997-11-05 |
| CN100427495C (en) | 2008-10-22 |
| BR9606718A (en) | 1998-01-13 |
| DE69627207T2 (en) | 2004-01-15 |
| ES2141474T3 (en) | 2000-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0804412B1 (en) | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles | |
| EP0911339B1 (en) | Method of making phosphorochloridites | |
| EP1000021B1 (en) | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alykl-3-monoalkenenitriles | |
| EP1000020B1 (en) | Hydrocyanation of diolefins and isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles | |
| JP3535172B2 (en) | Hydrocyanation process and polydentate phosphite and nickel catalyst compositions therefor | |
| EP1000022B1 (en) | Hydrocyanation processes and multidentate phosphite ligand and nickel catalyst compositions therefor | |
| US5847191A (en) | Process for the hydrocyanation of monoolefins using bidentate phosphite ligands and zero-valent nickel | |
| EP1191018B1 (en) | Process for the isomerization of nonconjugated 2-alkyl-3-monoalkenenitriles | |
| US5543536A (en) | Monodentate phosphite and nickel catalyst composition for monoolefin hydrocyanation | |
| JP3519410B2 (en) | Composition of bidentate phosphite and nickel catalyst for hydrocyanation of monoolefins | |
| EP1073520B1 (en) | Hydrocyanation of olefins and isomerisation of nonconjugated 2-alkyl-3-monoalkenenitriles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 96191601.X Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): BR CA CN JP KR SG |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2208040 Country of ref document: CA Ref document number: 2208040 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996902689 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019970704750 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 1996 522902 Country of ref document: JP Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996902689 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019970704750 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1996902689 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1019970704750 Country of ref document: KR |