WO1997005270A1 - Inhibitors of farnesyl-protein transferase - Google Patents
Inhibitors of farnesyl-protein transferase Download PDFInfo
- Publication number
- WO1997005270A1 WO1997005270A1 PCT/US1996/012114 US9612114W WO9705270A1 WO 1997005270 A1 WO1997005270 A1 WO 1997005270A1 US 9612114 W US9612114 W US 9612114W WO 9705270 A1 WO9705270 A1 WO 9705270A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- preparation
- recited
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 COC(C1=CC=COC(Oc2c3c(OC)cc(*)c2)=C1C3=O)=O Chemical compound COC(C1=CC=COC(Oc2c3c(OC)cc(*)c2)=C1C3=O)=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/18—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms containing at least two hetero rings condensed among themselves or condensed with a common carbocyclic ring system, e.g. rifamycin
- C12P17/181—Heterocyclic compounds containing oxygen atoms as the only ring heteroatoms in the condensed system, e.g. Salinomycin, Septamycin
Definitions
- Ras gene is found activated in many human cancers, including colorectal carcinoma, exocrine pancreatic carcinoma, and myeloid leukemias. Biological and biochemical studies of Ras action indicate that Ras functions like a G-regulatory protein, since Ras must be localized in the plasma membrane and must bind with GTP in order to transform cells (Gibbs, J. et al., Microbiol. Rev. 53: 171 -286 ( 1989)). Forms of Ras in cancer cells have mutations that distinguish the protein from Ras in normal cells.
- Ras C-terminus contains a sequence motif termed a "CAAX” or "Cys-Aaa 1 -Aaa 2 -Xaa” box (Aaa is an aliphatic amino acid, the Xaa is any amino acid) (Willumsen et al., Nature 370:583-586 (1984)).
- Other proteins having this motif include the Ras-related GTP-binding proteins such as Rho, fungal mating factors, the nuclear lamins, and the gamma subunit of transducin.
- Ha-Ras and N-Ras are palmitoylated via formation of a thioester on a Cys residue near a C-terminal Cys farnesyl acceptor (Gutierrez et al., EMBO J. 5:1093- 1098 ( 1989); Hancock et al., Cell 57:1 167-1171 (1989)). Ki-Ras lacks the palmitate acceptor Cys. The last 3 amino acids at the Ras C-terminal end are removed proteolytically, and methyl esterification occurs at the new C-terminus (Hancock et al., ibid). Fungal mating factor and mammalian nuclear lamins undergo identical modification steps (Anderegg et al., J. Biol. Chem.
- Inhibition of farnesyl-protein transferase and, thereby, of famesylation of the Ras protein blocks the ability of Ras to transform normal cells to cancer cells.
- the compounds of the invention inhibit Ras famesylation and, thereby, generate soluble Ras which, as indicated infra, can act as a dominant negative inhibitor of Ras function. While soluble Ras in cancer cells can become a dominant negative inhibitor, soluble Ras in normal cells would not be an inhibitor.
- a cytosol-localized (no Cys-Aaa 1 -Aaa 2 -Xaa box membrane domain present) and activated (impaired GTPase activity, staying bound to GTP) form of Ras acts as a dominant negative Ras inhibitor of membrane-bound Ras function (Gibbs et al., Proc. Natl. Acad. Sci. USA 56:6630-6634(1989)). Cytosollocalized forms of Ras with normal GTPase activity do not act as inhibitors. Gibbs et al., ibid, showed this effect in Xenopus oocytes and in mammalian cells.
- cytosolic pool of Ras In tumor cells having activated Ras, the cytosolic pool acts as another antagonist of membrane-bound Ras function. In normal cells having normal Ras, the cytosolic pool of Ras does not act as an antagonist. In the absence of complete inhibition of famesylation, other farnesylated proteins are able to continue with their functions.
- Farnesyl-protein transferase activity may be reduced or completely inhibited by adjusting the compounds dose. Reduction of farnesyl-protein transferase enzyme activity by adjusting the compounds dose.
- Farnesyl-protein transferase utilizes farnesyl pyrophosphate to covalently modify the Cys thiol group of the Ras CAAX box with a farnesyl group.
- Inhibition of farnesyl pyrophosphate biosynthesis by inhibiting HMG-CoA reductase blocks Ras membrane localization in vivo and inhibits Ras function.
- CA 1 A 2 X-type FPTase inhibitors contain acyclic amino acids in the second position. Incorporation of proline in the Al position in such inhibitors has been shown to be the least well tolerated amino acid substitution in that position (Reiss et al., PNAS (1991)).
- Such inhibitors may inhibit while serving as alternate substrates for the Ras farnesyl-transferase enzyme, or may be purely competitive inhibitors (U.S. Patent 5,141 ,851 , University of Texas).
- Inhibitors of farnesyl protein transferase which are citraconic acid derivatives have been isolated as fermentation products from a strain of Chaetomella acutiseta (U.S. Pat. No. 5,260,479 and EP-547671 -A). Synthetic analogs of those compounds have also been described (U.S. Pat. Nos. 5,245,061 and 5,260,479).
- Non-peptide compounds that are inhibitors of Ras farnesyl-protein transferase have been isolated from a strain of Cylindocarpon lucidum (U.S. Pat. No. 5,420,334).
- the present invention relates to compounds which inhibit farnesyl-protein transferase and the famesylation of the oncogene protein Ras, chemotherapeutic compositions containing the compounds of this invention, and methods for producing the compounds of this invention.
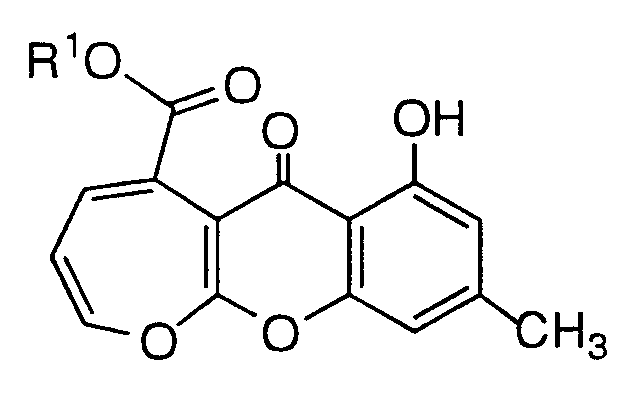
- R 1 is H, or C 1 -C 4 alkyl
- R 3 is C 1 -C 4 alkyl ; or the pharmaceutically acceptable salt, hydrate or prodrug thereof.
- R 1 is H, or C 1 -C 4 alkyl; or the pharmaceutically acceptable salt, hydrate or prodrug.
- R 3 is C 1 -C 4 alkyl ; or the pharmaceutically acceptable salt, hydrate or prodrug thereof.
- Compound A has the formula:
- Compound B has the formula:
- Compound H has the formula:
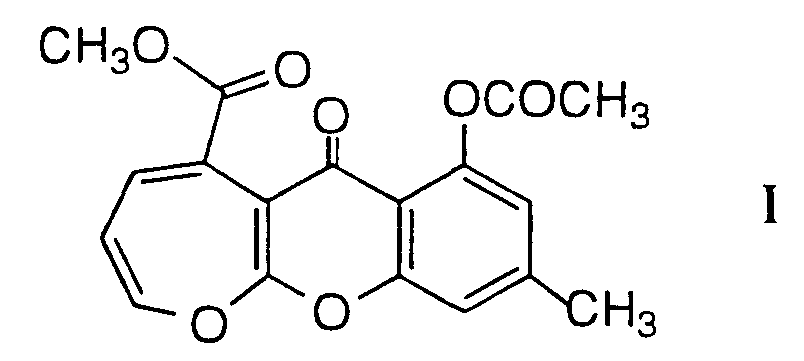
- Compound I has the formula:
- alkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- alkyl includes methyl, ethyl, propyl, iso-propyl, butyl, sec-butyl, tert-butyl and the like.
- mutants of the above described organism may also be capable of producing the compounds of this invention.
- the culture MF 61 18 is that of a fungus, Phoma sp., isolated from leaf litter of the desert shrub, Zygophyllum staffii, collected in Omdel, Thailand. This culture has been deposited with the American Type Culture Collection at 12301 Parklawn Drive, Rockville, MD 20852 as ATCC 74347.
- Conidiogenous cells ampulliform, phialidic.
- Conidia predominately elliptical, sometimes obovate, dumbbell shaped or cylindrical, hyaline, guttules at each end, 4 - 5 ⁇ 1 - 2 ⁇ m.
- Phoma Coelomycetes, Deuteromycotina
- MF 6118 exhibits all of the aforementioned generic characteristics but, in culture, does not exhibit any distinguishing characteristics to confidently place this fungus in a particular species. Therefore, it is designated as Phoma sp.
- Nutrient media may also contain
- the preferred sources of carbon in the nutrient medium are sucrose or fructose.
- the preferred sources of carbon in the nutrient medium are sucrose or fructose.
- trace metals such as manganese, iron,
- the fermentation production medium is incubated for about 3 to 30 days, preferably about 12 to 21 days.
- the fermentation is conducted aerobically at temperatures ranging from about 20 to 30°C. To obtain optimum results, the temperatures are in the range of about 22 to 28°C, most preferably about 22 to 25°C.
- fermentation flasks are harvested and the active compound isolated.
- the compounds is useful as a pharmaceutical agent for mammals, especially for humans. These compounds may be administered to patients for use in the treatment of cancer. Examples of the type of cancer which may be treated with the compounds of this
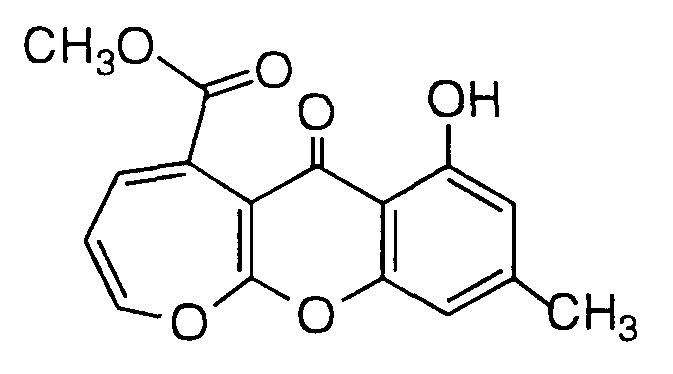
- Step C Isolation and Purification of 7-hydroxy-9-methyl-6-oxo6H-oxepino[2,3-b][1]benzopyran-5-carboxylic acid methyl ester
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Microbiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Public Health (AREA)
- General Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description









Claims



















Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP9507688A JPH11510388A (en) | 1995-07-25 | 1996-07-22 | Inhibitors of farnesyl-protein transferase |
| EP96925425A EP0842291A4 (en) | 1995-07-25 | 1996-07-22 | INHIBITORS OF FARNESYL PROTEIN TRANSFERASE |
| AU65940/96A AU706341B2 (en) | 1995-07-25 | 1996-07-22 | Inhibitors of farnesyl-protein transferase |
| CA002227369A CA2227369A1 (en) | 1995-07-25 | 1996-07-22 | Inhibitors of farnesyl-protein transferase |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US141495P | 1995-07-25 | 1995-07-25 | |
| US60/001,414 | 1995-07-25 | ||
| GB9606502.4 | 1996-03-28 | ||
| GBGB9606502.4A GB9606502D0 (en) | 1996-03-28 | 1996-03-28 | Inhibitors of farnesyl-protein transferase |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997005270A1 true WO1997005270A1 (en) | 1997-02-13 |
Family
ID=26309006
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1996/012114 Ceased WO1997005270A1 (en) | 1995-07-25 | 1996-07-22 | Inhibitors of farnesyl-protein transferase |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0842291A4 (en) |
| JP (1) | JPH11510388A (en) |
| AU (1) | AU706341B2 (en) |
| CA (1) | CA2227369A1 (en) |
| WO (1) | WO1997005270A1 (en) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5082489A (en) * | 1990-02-06 | 1992-01-21 | The Royal Institution For The Advancement Of Learning (Mcgill University) | Composition for biocontrol of wild buckwheat |
| US5151529A (en) * | 1988-11-28 | 1992-09-29 | Sankyo Company, Limited | Platelet activating factor antagonists, named "the phomactins" their preparation and use |
| US5254727A (en) * | 1992-10-19 | 1993-10-19 | Merck & Co., Inc. | Acyclic tricarboxylic acid compounds |
| US5310949A (en) * | 1992-09-02 | 1994-05-10 | Merck & Co., Inc. | Cholesterol lowering compounds |
| US5364948A (en) * | 1991-08-02 | 1994-11-15 | Merck & Co., Inc. | Biologically active compounds isolated from aerobic fermentation of Trichoderma viride |
| US5498627A (en) * | 1994-04-15 | 1996-03-12 | Takeda Chemical Industries, Ltd. | Octahydro-2-naphthalenecarboxylic acid derivative, its production and use |
-
1996
- 1996-07-22 CA CA002227369A patent/CA2227369A1/en not_active Abandoned
- 1996-07-22 AU AU65940/96A patent/AU706341B2/en not_active Ceased
- 1996-07-22 WO PCT/US1996/012114 patent/WO1997005270A1/en not_active Ceased
- 1996-07-22 EP EP96925425A patent/EP0842291A4/en not_active Withdrawn
- 1996-07-22 JP JP9507688A patent/JPH11510388A/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5151529A (en) * | 1988-11-28 | 1992-09-29 | Sankyo Company, Limited | Platelet activating factor antagonists, named "the phomactins" their preparation and use |
| US5082489A (en) * | 1990-02-06 | 1992-01-21 | The Royal Institution For The Advancement Of Learning (Mcgill University) | Composition for biocontrol of wild buckwheat |
| US5364948A (en) * | 1991-08-02 | 1994-11-15 | Merck & Co., Inc. | Biologically active compounds isolated from aerobic fermentation of Trichoderma viride |
| US5310949A (en) * | 1992-09-02 | 1994-05-10 | Merck & Co., Inc. | Cholesterol lowering compounds |
| US5254727A (en) * | 1992-10-19 | 1993-10-19 | Merck & Co., Inc. | Acyclic tricarboxylic acid compounds |
| US5498627A (en) * | 1994-04-15 | 1996-03-12 | Takeda Chemical Industries, Ltd. | Octahydro-2-naphthalenecarboxylic acid derivative, its production and use |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0842291A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0842291A1 (en) | 1998-05-20 |
| CA2227369A1 (en) | 1997-02-13 |
| AU6594096A (en) | 1997-02-26 |
| AU706341B2 (en) | 1999-06-17 |
| JPH11510388A (en) | 1999-09-14 |
| EP0842291A4 (en) | 1998-11-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5283256A (en) | Cholesterol-lowering agents | |
| AU680847B2 (en) | Inhibitors of farnesyl-protein transferase | |
| US5286895A (en) | Cholesterol lowering compounds | |
| US5260465A (en) | Inhibitors of farnesyl protein transferase | |
| WO1993016065A1 (en) | Cholesterol lowering compounds | |
| US5420334A (en) | Inhibitors of farnesyl-protein transferase | |
| US5364948A (en) | Biologically active compounds isolated from aerobic fermentation of Trichoderma viride | |
| EP0526936A2 (en) | Cholesterol-lowering agents | |
| US5420157A (en) | Inhibitors of farnesyl protein transferase or prodrugs thereof | |
| US5663193A (en) | Inhibitors of farnesyl-protein transferase | |
| AU706341B2 (en) | Inhibitors of farnesyl-protein transferase | |
| US5627057A (en) | Inhibitor compounds of farnesyl-protein transferase and chemotherapeutic compositions containing the same, produced by strain ATCC 55532 | |
| GB2261373A (en) | Inhibitors of farnesyl protein transferase as anti-cancer agents | |
| US5436263A (en) | Inhibitors of farnesyl-protein transferase | |
| EP0582267A1 (en) | Substances and microorganisms which produce them | |
| US5703067A (en) | Inhibitors of farnesyl-protein transferase | |
| GB2261375A (en) | Inhibitors of farnesyl protein transferase as anti-cancer agents | |
| US5270332A (en) | Cholesteral lowering agents | |
| GB2261374A (en) | Inhibitors of farnesyl protein transferase as anti-cancer agents | |
| JPH0394692A (en) | Bioactive substance be-18257's | |
| US5294627A (en) | Directed biosynthesis of biologically active compounds | |
| US4902781A (en) | Novel tripetide derivatives | |
| US4725621A (en) | CL-1957E antibiotic compound and its production | |
| US5447717A (en) | Cholesterol-lowering agents | |
| US5789438A (en) | Inhibitors of farnesyl-protein transferase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU AZ BB BG BR BY CA CN CU CZ EE GE HU IL IS JP KG KR KZ LK LR LT LV MD MG MK MN MX NO NZ PL RO RU SG SI SK TJ TM TR TT UA US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1996925425 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2227369 Country of ref document: CA Ref country code: CA Ref document number: 2227369 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 1997 507688 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996925425 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1996925425 Country of ref document: EP |