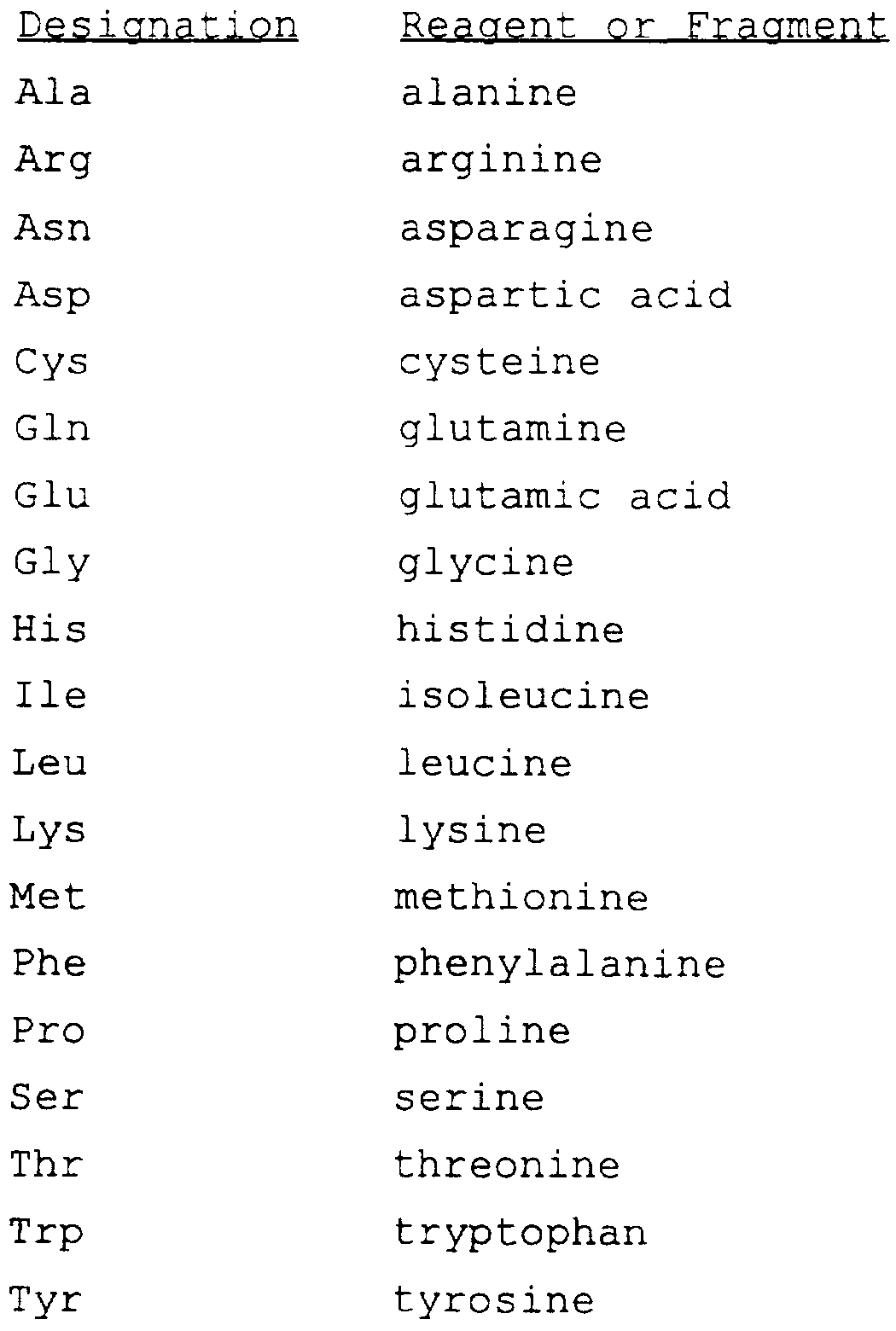
WO1997022618A1 - INHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYME - Google Patents
INHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYME Download PDFInfo
- Publication number
- WO1997022618A1 WO1997022618A1 PCT/US1996/020370 US9620370W WO9722618A1 WO 1997022618 A1 WO1997022618 A1 WO 1997022618A1 US 9620370 W US9620370 W US 9620370W WO 9722618 A1 WO9722618 A1 WO 9722618A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- disease
- straight
- optionally substituted
- branched alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*)C(C(C)(C)N(*)CC(C)=C)=O Chemical compound CC(*)C(C(C)(C)N(*)CC(C)=C)=O 0.000 description 9
- BJFSXHQWELIWQR-UHFFFAOYSA-N CCOC(CC(Cc1cccc(NC(N)=N)c1)C(C(C(C)C)NC(c1ccccc1)=O)=O)=O Chemical compound CCOC(CC(Cc1cccc(NC(N)=N)c1)C(C(C(C)C)NC(c1ccccc1)=O)=O)=O BJFSXHQWELIWQR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0205—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-(X)3-C(=0)-, e.g. statine or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0202—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-X-X-C(=0)-, X being an optionally substituted carbon atom or a heteroatom, e.g. beta-amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to novel classes of compounds which are inhibitors of terleukm-l ⁇ converting enzyme ("ICE") .
- This invention also relates to pharmaceutical compositions comprising these compounds .
- the compounds and pharmaceutical compositions of th s invention are particularly well suited for inhibiting ICE activity and consequently, may be advantageously used as agents against mterleukm-l- ("IL-1") ana apoptosis-mediated diseases, including inflammatory diseases, autoimmune diseases, proliferative disorders, infectious c seases, and degenerative diseases.
- IL-1 mterleukm-l-
- This invention also relates to methods for inhibiting ICE activity and methods for treating mterleukm-l- and apoptosis-mediated diseases using the compounds and compositions of this invention.
- Interleukin 1 is a pro- inflammatory and immunoregulatory protein that stimulates fibroblast differentiation and proliferation, the production of prostaglandms, collagenase and phospholipase by synovial cells and chondrocytes, basophil and eosmophil degranulation and neutrophil activation.
- IL-1 Interleukin 1
- Oppenhei J.H. et al, Immunology Today, 7, pp. 45-56 (1986) . As such, it is involved the pathogenesis of chronic and acute inflammatory and autoimmune diseases.
- IL-1 is both a mediator of inflammatory symptoms and of the destruction of the cartilage proteoglycan in afflicted joints.
- IL-1 is also a highly potent bone resorption agent. It is reiterately referred to as "osteoclast activating factor" in destructive bone diseases such as osteoarthritis and multiple myeloma. Batailie, R. et al., Int. J. Clm. Lab. Res.. 21, p. 283 (1992) .
- IL-1 can promote tumor cell growth and adhesion.
- IL-1 also stimulates production of other cytokines such as IL-6, which can modulate tumor development. Tartour et al . , Cancer Res. 54, 6243 (1994) .
- IL-1 is predominantly produced by peripheral blood monocytes as part of the inflammatory response and exists in two distinct agonist forms, IL-l ⁇ and IL-
- IL-l ⁇ is synthesized as a biologically inactive precursor, pIL-l ⁇ .
- pIL-l ⁇ lacks a conventional leader sequence and is not processed by a signal peptidase. March, C.J., Nature. 315, pp. 641-647 (1985) .
- pIL-l ⁇ is cleaved by interleukin-l ⁇ converting enzyme ("ICE") between Asp- 116 and Ala-117 to produce the biologically active C-termmal fragment found human serum and synovial fluid.
- ICE interleukin-l ⁇ converting enzyme
- ICE is a cysteme protease localized primarily m monocytes . It converts precursor IL-l ⁇ to the mature form. Black, R.A. et al . , FEBS Lett . , 247, pp. 386-390 (1989); Kostura, M.J. et al . , Pro . Natl. Acad. Sci. USA. 86, pp. 5227-5231 (1989) . Processing by ICE is also necessary for the transport of mature
- ICE or its homologues, also appears to be involved in the regulation of cell death or apoptosis. Yuan, J. et al., Cell. 75, pp. 641-652 (1993) ; Miura, M. et al . , Cell. 75, pp. 653-660 (1993) ; Nett-Fiordalisi, M.A. et al., J. Cell Biochem.. 17B, p. 117 (1993) . In particular, ICE or ICE homologues are thought to be associated with the regulation of apoptosis neurogenerative diseases, such as Alzheimer's ana
- Parkinson's disease Marx, J. and M. Baringa, Science, 259, pp. 760-762 (1993) ; Gagliardim, V. et al., Science. 263, pp. 826-828 (1994) .
- ICE has been demonstrated to mediate apoptosis (programmed cell death) m certain tissue types.
- McGr, H. Science. 267, p. 1445 (1995) ; Whyte, M. and Evan, G., Nature, 376, p. 17 (1995) ; Martin, S.J. and Green, D.R. , Cell, 82, p. 349 (1995) ; Alnemri, E.S., et al. , J. Biol . Chem., 270, p. 4312 (1995); Yuan, J. Curr. Qpin. Cell Biol., 7, p. 211 (1995) .
- Therapeutic applications for inhibition of apoptosis may include treatment of Alzheimer's disease, Parkinson's disease, stroke, mycardial infarction, spinal atrophy, and aging.
- a transgenic mouse with a disruption of the ICE gene is deficient in Fas-mediated apoptosis.
- Kuida, et al . (1995) This activity of ICE is distinct from its role as the processing enzyme for pro-IL-l ⁇ . It is conceivable that m certain tissue types, inhibition of ICE may not affect secretion of mature IL-l ⁇ , but may inhibit apoptosis.
- ICE has been previously described as a heterodimer composed of two subunits, p20 and plO (20kDa and lOkDa molecular weight, respectively) .
- subunits are derived from a 45kDa proenzyme (p45) by way of a p30 form, through an activation mechanism that is autocatalytic.
- Thornberry N.A. et al., Nature, 356, pp. 768-774 (1992) .
- the ICE proenzyme has been divided into several functional domains: a prodomam (pl4), a p22/20 subunit, a polypeptide linker and a plO subunit.
- a prodomam pl4
- p22/20 subunit a polypeptide linker
- plO subunit a prodomam
- Thornberry et al .. supra Casano et al., Genomics, 20, pp. 474-481 (1994) .
- Full length p45 has been characterized by its cDNA and ammo acid sequences.
- the p20 and plO cDNA and ammo acid sequences are also known.
- Murme and rat ICE have also been sequenced and cloned. They have high am o acid and nucleic acid sequence homology to human ICE. Miller, D.K. et al., Ann. N.Y. Acad. S ⁇ .. 696, pp. 133-148 (1993) ; Molmeaux, S.M. et al . , Proc. Nat. Acad. Sci., 90, pp.
- ICE has been determined at atomic resolution by X-ray crystallography. Wilson, K.P., et al., Nature, 370, pp. 270-275 (1995) .
- the active enzyme exists as a tetramer of two p20 and two plO subunits .
- human homologs of ICE with sequence similarities m the active site regions of the enzymes. Such homologs include TX (or ICE re l - II or ICH-2) (Faucheu, et al . , EMBO J.. 14, p. 1914 (1995) ; Kamens J., et al . , J. Biol.
- Each of these ICE homologs, as well as ICE itself, is capable of inducing apoptosis when overexpressed in transfected cell lines. Inhibition of one or more of these homologs with the peptidyl ICE inhibitor Tyr-Val- Ala-Asp-chloromethylketone results in inhibition of apoptosis primary cells or cell lines. Lazebnik et al., Nature, 371, p. 346 (1994) .
- the compounds described herein are also capable of inhibiting one or more homologs of ICE (see example) . Therefore, one can envisage using these compounds to inhibit apoptosis tissue types that contain ICE homologs, but which do not contain active ICE or produce mature IL-1 ⁇ .
- ICE inhibitors represent a class of compounds useful for the control of inflammation or apoptosis or both.
- Peptide and peptidyl inhibitors of ICE have been described.
- Such peptidyl inhibitors of ICE have been observed to block the production of mature IL-l ⁇ a mouse model of inflammation (Ku, et al. or vide mfra) and to suppress growth of leukemia cells i n vi tro (Estrov, et al., Blood, 84, p. 380a (1994) ) .
- the present invention provides novel classes of compounds, and pharmaceutically acceptable derivatives thereof, that are useful as inhibitors of ICE. These compounds can be used alone or in combination with other therapeutic or prophylactic agents, such as antibiotics, immunomodulators or other anti-inflammatory agents, for the treatment or prophylaxis of diseases mediated by IL-1 or by apoptosis. According to a preferred embodiment, the compounds of this invention are capable of binding to the active site of ICE and inhibiting the activity of that enzyme.
- active site refers to any or all of the following sites m ICE: the substrate binding site, the site where an inhibitor binds and the site where the cleavage of substrate occurs.
- alkenyl refers to a straight-chain or branched-cham alkenyl radical containing from 2 to 10, carbon atoms.
- examples of such radicals include, but are not limited to, ethenyl, E- and Z-propenyl, isopropenyl, E- and Z- butenyl, E- and Z- isobutenyl, E- and Z-pentenyl, decenyl and the like.
- alkynyl refers to a straight-chain or branched-cham alkynyl radical containing from 2 to 10, carbon atoms.
- examples of such radicals include, but are not limited to, ethynyl (acetylenyl) , propynyl, propargyl, butynyl, hexynyl, decynyl and the like.
- substitute refers to the replacement of a hydrogen atom in a compound with a substituent group.
- K 1 refers to a numerical measure of the effectiveness of a compound in inhibiting the activity of a target enzyme such as ICE. Lower values of K-_ reflect higher effectiveness.
- the K-_ value is a derived by fitting experimentally determined rate data to standard enzyme kinetic equations (see I. H. Segel, Enzyme Kinetics, Wiley-Interscience, 1975) .
- patient as used in this application refers to any mammal, especially humans.
- pharmaceutically effective amount refers to an amount effective in treating or ameliorating an IL-1- or apoptosis-mediated disease in a patient.
- prophylactically effective amount refers to an amount effective in preventing or substantially lessening IL-1- or apoptosis-mediated disease in a patient.
- pharmaceutically acceptable carrier or adjuvant refers to a non-toxic carrier or ad uvant that may be administered to a patient, together with a compound of this invention, and which does not destroy the pnarmacological activity thereof.
- pharmaceutically acceptable derivative means any pharmaceutically acceptable salt, ester, or salt of such ester, of a compound of this invention or any other compound which, upon administration to a recipient, is capable of providing (directly or indirectly) a compound of this invention or an anti-ICE active metabolite or residue thereof.
- Pharmaceutically acceptable salts of the compounds of this invention include, for example, those derived from pharmaceutically acceptable inorganic and organic acids and bases.
- suitable acids include hydrochloric, hydrobromic, sulfuric, nitric, perchloric, fumaric, aleic, phosphoric, glycolic, lactic, salicylic, succinic, toluene-p-sulfonic, tartaric, acetic, citric, methanesulfonic, formic, benzoic, malonic, naphthalene-2-sulfon ⁇ c and benzenesulfonic acids.
- Salts derived from appropriate bases include alkali metal (e.g., sodium), alkaline earth metal (e.g., magnesium), ammonium and N- (C ] __ alkyl) 4 salts.
- This invention also envisions the "quaternization" of any basic nitrogen-containing groups of the compounds disclosed herein.
- the basic nitrogen can be quaternized with any agents known to those of ordinary skill m the art including, for example, lower alkyl halides, such as methyl, ethyl, propyl and butyl chloride, bromides and iodides; dialkyl sulfates including dimethyl, diethyl, dibutyl and diamyl sulfates; long chain halides such as decyl, lauryl, my ⁇ styl and stearyl chlorides, bromides and iodides; and aralkyl halides including benzyl and phenethyl bromides. Water or oil-soluble or dispersible products may be obtained by such quaternization.
- the ICE inhibitors of this invention may contain one or more "asymmetric" carbon atoms and thus may occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. All such lsomeric forms of these compounds are expressly included m the present invention.
- Each stereogenic carbon may be of the R or S configuration.
- specific compounds and scaffolds exemplified in this application may be depicted in a particular stereochemical configuration, compounds and scaffolds having either the opposite stereochemistry at any given chiral center or mixtures thereof are also envisioned.
- the ICE inhibitors of this invention may comprise structures which may optionally be substituted at carbon, nitrogen or other atoms by various substituents. Such structures may be singly or multiply substituted.
- the structures contain between 0 and 3 substituents.
- each substituent may be picked independently of any other substituent as long as the combination of substituents results m the formation of a stable compound.
- Combinations of substituents and variables envisioned by this invention are only those that result in the formation of stable compounds.
- stable refers to compounds which possess stability sufficient to allow manufacture and administration to a mammal by methods known in the art. Typically, such compounds are stable at a temperature of 40°C or less, m the absence of moisture or other chemically reactive conditions, for at least a week.
- ICE inhibitors of one embodiment (A) of this invention are those of formula ( ) :
- n 1 or 2;
- R ] _2 and R 13 are independently selected from the group consisting of -R , -C(0)-R 7 , and -C (0) -N (H) -R 7 , or R 12 and R 13 taken together form a 4-8-membered saturated cyclic group;
- R 2 is -H or a -C 1 _ 6 straight or branched alkyl group optionally substituted with Ar, -OH, -OR 7 , -C(0)-0H, C(0)-NH 2 , or -OR 5 ;
- R 7 is selected from the group consisting of -Ar, a -C ;] __g straight or branched alkyl group optionally substituted with -Ar, a -C 1 _ 6 straight or branched alkenyl group optionally substituted with Ar, and a ⁇ c 2- 6 straight or branched alkynyl group optionally substituted with Ar;
- R 5 is selected from the group consisting of: -C(0)-R 7 , -C(0) -0R 9 , -C(0)-N(R 9 ) (R 10 ) ,
- each Ar is a cyclic group independently selected from the set consisting of phenyl, 1-naphthyl, 2- naphthyl, mdenyl, azulenyl, fluorenyl and anthracenyl and a heterocyclic aromatic group selected from the group consisting of 2-furyl, 3-furyl, 2-th ⁇ enyl, 3- thienyl, 2-py ⁇ dyl, 3-py ⁇ dyl, 4-pyr ⁇ dyl, pyrrolyl, oxazolyl, thiazolyl, lmidazolyl, pyraxolyl, 2- pyrazolmyl, pyrazolidmyl, isoxazolyl, isotriazolyl, 1, 2, 3-oxad ⁇ azolyl, 1, 2, 3-tr ⁇ azolyl, 1, 3, 4-th ⁇ ad ⁇ azolyl, pyndazmyl, pyri idinyl, pyrazmyl, 1, 3, 5-t ⁇ a
- each R 9 and R 10 is independently selected from the group consisting of -H, -Ar, and a ⁇ _- ⁇ straight or branched alkyl group optionally substituted with -Ar;
- each Rq is a -C ] __ 5 straight or branched alkyl group optionally substituted with -Ar or -W;
- W is -ORg, -SR 9 , -N(H)C(NR 9 )N(R 9 ) (R 10 ) , -C(0)-OR 9 , or -N(R 9 ) (R 10 ) ;
- R 3 is -CH 2 Ar or a 5 to 15-membered non-aromatic cyclic group which contains between 1 and 3 rings, and which optionally contains between 0 and 2 endocyclic oxygen atoms, sulfur atoms, or nitrogen atoms, and wherein the cyclic group is optionally fused with Ar;
- R 5 is -C(0)-R 7 or -C (0) C (0) -R 7 ;
- each R 4 is a C 1 _ 5 straight or branched alky., group optionally substituted with Ar;
- n 1;
- R? is -CH Ar or cC
- E is CH or N
- each D is independently N or C, wherein C is optionally substituted with -0R 14 , -F, -Cl, -Br, -I, -N0 2 , -S(0) 2 -N(R 9 ) (R 10 ) , -C(0)-N(R 9 ) (R 10 ) , -N(H)-C ⁇ 0)- N(R 9 ) (R 10 ), -N(R 9 ) (R 10 ), -C(0)-0R 9 , -CF 3 , -0CF 3 , a C ⁇ ⁇ straight or branched alkyl group, 1, 2-methylened ⁇ oxy, -CN, or -N(H)C(NR 9 )N(R 9 ) (R 10 ) ;
- each R 9 and R 10 is independently selected from the group consisting of -H, -Ar, and a -C 1 _ 5 straight or branched alkyl group optionally substituted with -Ar.
- the ICE inhibitors of another embodiment of this invention are those of formula ( ⁇ ) :
- R- j _ is selected from the group consisting of
- X is selected from the group consisting of 0, S, S (0) , and S (0) 2 ;
- R 6 is independently selected from the group consisting of: -H,
- R 7 is selected from the group consisting of -Ar, _c l- 6 straight or branched alkyl group optionally substituted with -Ar, a -C 1 _ 6 straight or branched alkenyl group optionally substituted with Ar, and a -C 2 _ 6 straight or branched alkynyl group optionally substituted with Ar;
- R 3 is selected from the following group, m which any ring may optionally be singly or multiply substituted by -NH 2 , -C(0)-OH, -F, -Cl, -Br, -I, -OH, -N0 2 , -CN, -perfluoroalkyl C 1 _ 3 alkyl, -R 5 , -0R 5 , -OR 7 , -N(H)-R 5 , -N(H)-R 7 , 1, 2-methylened ⁇ oxy, and -SR 7 :
- Y is independently selected from the group consisting of 0 and S;
- each Ar is a cyclic group independently selected from the set consisting of a carbocyclic aromatic group selected from the group consisting of phenyl, 1- naphthyl, 2-naphthyl, mdenyl, azulenyl, fluorenyl and anthracenyl and a heterocyclic aromatic group selected from the group consisting of 2-furyl, 3-furyl, 2- thienyl, 3-thienyl, 2-py ⁇ dyl, 3-py ⁇ dyl, 4-pyr ⁇ dyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyraxolyl, 2-pyrazolinyl, pyrazolidinyl, isoxazolyl, isotriazolyl, 1, 2, 3-oxadiazolyl, 1, 2, 3-tr ⁇ azolyl, 1, 3, 4-th ⁇ ad ⁇ azolyl, pyridazinyl, pyrimidinyl, pyra
- each R 9 and R 10 are independently selected from the group consisting of -H, -Ar, and a -C 1 _5 straight or branched alkyl group optionally substituted with Ar;
- each R 14 is -H or a C 1 _ 6 straight or branched alkyl group
- R 5 is selected from the group consisting of: -C(0)-R 7 ,
- R 4 is a -C ⁇ -,5 straight or branched alkyl group optionally substituted with Ar or W;
- W is -0R 9 , -SR 9 , -N(H)C(NR 9 )N(R 9 ) (R 10 ) , -C(0)-OR 9 , and -NR 9 , (R 10 ) ;
- R 3 is -CH 2 Ar or a 5 to 15-membered non-aromatic cyclic group which contains between 1 and 3 rings, and which optionally contains between 0 and 2 endocyclic oxygen atoms, sulfur atoms, or nitrogen atoms, and wherein the cyclic group is optionally fused with Ar;
- R 2 is -H, or a C ] __g straight or branched alkyl group, wherein the alkyl group is optionally substituted with Ar, -OH, -0R 7 , -C(0)-OH, C(0)-NH 2 , or -OR 5 ;
- R ⁇ is -C(0)-H
- R 5 is -C(0)-R 7 or -C (0) C (0) -R 7 ;
- R 4 is a -C ] __5 straight or branched alkyl group optionally substituted by -Ar;
- n 1;
- n 1;
- each D is independently N or C, wherein C is optionally substituted with -OR 14 , -F, -Cl, -Br, -I, -N0 2 , -S(0) 2 -N(R 9 ) (R 10 ), -C(0)-N(R 9 ) (R 10 ) , -N(H)-C(0)- N(R 9 ) (R 10 ), -N(R 9 ) (R 10 ), -C(0)-0R 9 , -CF 3 , -OCF 3 , a C 1 _ 6 straight or branched alkyl group, 1, 2-methylened ⁇ oxy, -CN, or -N ⁇ H)C(NR 9 )N(R 9 ) (R 10 ) ;
- each Rg and R 10 is independently selected from the group consisting of -H, -Ar, and a -C- j __ 5 straight or branched alkyl group optionally substituted with -Ar.
- R 5 is -C(0)-R 7 or -C (0) C (0) -R 7 ;
- R 4 is a _ C ] __5 straight or branched alkyl group optionally substituted by -Ar;
- n 1;
- n 1;
- each D is independently N or C, wherein C is optionally substituted with -0R 14 , -F, -Cl, -Br, -I,
- ICE inhibitors of this invention may be synthesized using conventional techniques.
- these compounds are conveniently synthesized from readily available starting materials.
- the compounds of this invention are among the most readily synthesized ICE inhibitors known.
- Previously described ICE inhibitors often contain four or more chiral centers and numerous peptide linkages. The relative ease with which the compounds of this invention can be synthesized represents an advantage m the large scale production of these compounds.
- the compounds of this invention may exist m various equilibrium forms, depending on conditions including choice of solvent, pH, and others known to the practitioner skilled in the art. All such forms of these compounds are expressly included m the present invention.
- R 1 is -(CO)-H
- compounds of this invention may also take acyloxv ketal, acyloxy acetal, ketal or acetal form:
- the compounds of this invention may be modified by appropriate functionalities to enhance selective biological properties. Such modifications are known the art and include those which increase biological penetration into a given biological system (e.g., blood, lymphatic system, central nervous system) , increase oral availability, increase solubility to allow administration by injection, alter metabolism and alter rate of excretion.
- the compounds may be altered to pro-drug form such that the desired compound is created m the body of the patient as the result of the action of metabolic or other biochemical processes on the pro-drug.
- pro-drug forms typically demonstrate little or no activity in m vi tro assays.
- pro-drug forms include ketal, acetal, oxime, and hydrazone forms of compounds which contain ketone or aldehyde groups, especially where they occur m the R- j _ group of the compounds of this invention.
- Other examples of pro-drug forms include the hemi- ketal, hemi-acetal, acyloxy ketal, acyloxy acetal, ketal, and acetal forms that are described in EQ1 ana EQ2.
- the compounds of this invention are excellent ligands for ICE. Accordingly, these compounds are capable of targeting and inhibiting events m IL-1- and apoptosis-mediated diseases and, thus, the ultimate activity of that protein inflammatory diseases, autoimmune diseases, proliferative disorders, infectious diseases, and degenerative diseases.
- the compounds of this invention innibit the conversion of precursor IL-l ⁇ to mature IL-l ⁇ by inhibiting ICE. Because ICE is essential for the production of mature IL-l ⁇ , inhibition of that enzyme effectively blocks initiation of IL-1 mediated physiological effects and symptoms, such as inflammation, by inhibiting the production of mature IL-1.
- the compounds of this invention effectively function as IL-1 inhibitors.
- the compounds of this invention may be employed in a conventional manner for the treatment of diseases which are mediated by IL-1 or apoptosis. Such methods of treatment, their dosage levels and requirements may be selected by those of ordinary skill m the art from available methods and techniques.
- a compound of this invention may be combined with a pharmaceutically acceptable adjuvant for administration to a patient suffering from an IL-1- or apoptosis-mediated disease m a pharmaceutically acceptable manner and in an amount effective to lessen the severity of that disease.
- the compounds of this invention may be used m compositions and methods for treating or protecting individuals against IL-I- or apoptosis-mediated diseases over extended periods of time.
- the compounds may be employed m such compositions either alone or together with other compounds of this invention a manner consistent with the conventional utilization of ICE inhibitors in pharmaceutical compositions.
- a compound of this invention may be combined with pharmaceutically acceptable adjuvants conventionally employed m vaccines and administered prophylactically effective amounts to protect individuals over an extended period time against IL-1- or apoptosis-mediated diseases.
- the compounds of this invention may also be co-admmistered with other ICE inhibitors to increase the effect of therapy or prophylaxis against various IL-1- or apoptosis-mediated diseases.
- the compounds of this invention may be used m combination either conventional anti- inflammatory agents or with matrix metalloprotease inhibitors, lipoxygenase inhibitors and antagonists of cyto ines other than IL-l ⁇ .
- the compounds of this invention can also be administered in combination with lmmunomodulators (e.g., bropi ⁇ mme, anti-human alpha mterferon antibody, IL-2, GM-CSF, methion e enkephalm, mterferon alpha, diethyldithiocarbamate, tumor necrosis factor, naltrexone and rEPO) or with prostaglandms, to prevent or combat IL-1- or apoptosis-mediated disease symptoms such as inflammation.
- lmmunomodulators e.g., bropi ⁇ mme, anti-human alpha mterferon antibody, IL-2, GM-CSF, methion e enkephalm, mterferon alpha, diethyldithiocarbamate, tumor necrosis factor, naltrexone and rEPO
- prostaglandms e.g., IL-1- or apoptosis-mediated disease symptoms
- compositions according to this invention may be comprised of a combination of an ICE inhibitor of this invention and another therapeutic or prophylactic agent.
- compositions of this invention comprise any of the compounds of the present invention, and pharmaceutically acceptable salts thereof, with any pharmaceutically acceptable carrier, adjuvant or vehicle.
- Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used m the pharmaceutical compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycme, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamme sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvmyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxy ethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glyco
- compositions of this invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vagmally or via an implanted reservoir. We prefer oral administration.
- the pharmaceutical compositions of this invention may contain any conventional non-toxic pharmaceutically-acceptable carriers, adjuvants or vehicles.
- parenteral as used herein includes subcutaneous, mtracutaneous, intravenous, intramuscular, tra-articular, mtrasynovial, mtrasternal, mtrathecal, mtralesional and mtracranial injection or infusion techniques.
- the pharmaceutical compositions may be m the form of a sterile mjectable preparation, for example, as a sterile mjectable aqueous or oleaginous suspension.
- This suspension may be formulated according to techniques known the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents.
- the sterile mjectable preparation may also be a sterile mjectable solution or suspension m a non-toxic parenterally- acceptable diluent or solvent, for example, as a solution m 1, 3-butaned ⁇ ol.
- suitable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and lsotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides .
- Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of mjectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially m their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant such as Ph. Helv or a similar alcohol.
- compositions of this invention may be orally administered m any orally acceptable dosage form including, but not limited to, capsules, tablets, and aqueous suspensions and solutions.
- carriers which are commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried corn starch.
- aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
- compositions of this invention may also be administered in the form of suppositories for rectal administration.
- These compositions can be prepared by mixing a compound of this invention with a suitable non-irritatmg excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components.
- suitable non-irritatmg excipient include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
- Topical administration of the pharmaceutical compositions of this invention is especially useful when the desired treatment involves areas or organs readily accessible by topical application.
- the pharmaceutical composition For application topically to the skin, the pharmaceutical composition should be formulated with a suitable ointment containing the active components suspended or dissolved m a carrier.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petroleum, white petroleum, propylene glycol, polyoxy- ethylene polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutical composition can be formulated with a suitable lotion or cream containing the active compound suspended or dissolved in a carrier.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the pharmaceutical compositions of this invention may also be topically applied to the lower intestinal tract by rectal suppository formulation or a suitable enema formulation. Topically-transdermal patches are also included m this invention.
- compositions of this invention may be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions m saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubiliz g or dispersing agents known the art.
- Dosage levels of between about 0.01 and about 100 mg/kg body weight per day, preferably between about 1 and 50 mg/kg body weight per day of the active ingredient compound are useful in the prevention and treatment of IL-1- and apoptosis-mediated diseases, including inflammatory diseases, autoimmune diseases, destructive bone disorders, proliferative disorders, infectious diseases, degenerative diseases, osteoarthritis, pancreatitis, asthma, adult respiratory distress syndrome, glomeralonephritis, rheumatoid arthritis, systemic lupus erythematosus, scleroderma, chronic thyroiditis, Graves' disease, autoimmune gastritis, insulin-dependent diabetes mellitus (Type I), autoimmune hemolytic anemia, autoimmune neutropenia, thrombocytopema, chronic active hepatitis, myasthenia gravis, inflammatory bowel disease, Crohn' s disease, psoriasis, graft vs.
- diseases including inflammatory diseases, autoimmune diseases,
- osteoporosis multiple myeloma-related bone disorder
- acute myelogenous leukemia chronic myelogenous leukemia
- metastatic melanoma metastatic melanoma
- Kaposi's sarcoma multiple myeloma sepsis
- septic shock
- compositions of this invention will be administered from about 1 to 5 times per day or alternatively, as a continuous infusion. Such administration can be used as a chronic or acute therapy.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- a typical preparation will contain from about 5% to about 95% active compound (w/w) .
- such preparations contain from about 20. to about 80 active compound.
- a maintenance dose of a compound, composition or combination of this invention may be administered, if necessary. Subsequently, the dosage or frequency o administration, or both, may be reduced, as a function of the symptoms, to a level at which the improved condition is retained when the symptoms have been alleviated to the desired level, treatment should cease. Patients may, however, require intermittent treatment on a long-term basis upon any recurrence or disease symptoms. As the skilled artisan will appreciate, lower or higher doses than those recited above may be required.
- IL-1 or apoptosis-mediated diseases which may be treated or prevented by the compounds of this invention include, but are not limited to, inflammatory diseases, autoimmune diseases, proliferative disorders, infectious diseases, degenerative, and necrotic diseases.
- Inflammatory diseases which may be treated or prevented include, but are not limited to osteoarthritis, acute pancreatitis, chronic pancreatitis, asthma, and adult respiratory distress syndrome.
- the inflammatory disease is osteoarthritis or acute pancreatitis.
- Autoimmune diseases which may be treated or prevented include, but are not limited to, glomeralonephritis, rheumatoid arthritis, systemic lupus erythematosus, scleroderma, chronic thyroiditis, Graves' disease, autoimmune gastritis, msulm- dependent diabetes mellitus (Type I), autoimmune hemolytic anemia, autoimmune neutropema, thrombocytopenia, chronic active hepatitis, myasthenia gravis, inflammatory bowel disease, Crohn' s disease, psoriasis, and graft vs. host disease.
- the autoimmune disease is rheumatoid arthritis, inflammatory bowel disease, Crohn' s disease, or psoriasis .
- Bone destructive disorders which may be treated or prevented include, but are not limited to, osteoporosis and multiple myeloma-related bone disorder.
- Proliferative diseases which may be treated or prevented include, but are not limited to, acute myelogenous leukemia, chronic myelogenous leukemia, metastatic melanoma, Kaposi's sarcoma, and multiple myeloma.
- Infectious diseases which may be treated or prevented include, but are not limited to, sepsis, septic shock, and Shigellosis.
- the IL-1-med ⁇ ated degenerative or necrotic diseases which may be treated or prevented by the compounds of this invention include, but are not limited to, Alzheimer's disease, Parkinson's disease, cerebral ischemia, and myocardial ischemia.
- the degenerative disease is Alzheimer' s disease.
- apoptosis-mediated degenerative diseases which may be treated or prevented by the compounds of this invention include, but are not limited to,
- this invention focuses on the use of the compounds disclosed herein for preventing and treating IL-1 and apoptosis-mediated diseases, the compounds of this invention can also be used as inhibitory agents for other cysteine proteases.
- the compounds of this invention are also useful as commercial reagents which effectively bind to ICE or other cysteine proteases.
- the compounds of this invention, and their derivatives may be used to block proteolysis of a target peptide m biochemical or cellular assays for ICE and ICE homologs or may be derivatized to bind to a stable resin as a tethered substrate for affinity ehromatography applications.
- Ala-Asp-pNitroanilide substrate Synthesis of analogous substrates is described by L. A. Reiter (Int. J. Peptide Protein Res. 13 . , 87-96 (1994) ; .
- the assay mixture contains :
- the visible ICE assay is run m a 96-well microtiter plate. Buffer, ICE and DMSO (if mnioitor is present) are added to the wells m the order _ ⁇ sted. The components are left to incubate at room temperature for 15 minutes starting at the time that all components are present m all wells. The microtiter plate reader is set to incubate at 37 °C. After the 15 minute incubation, substrate is added directly to tne wells and the reaction is monitored by following the release of the chromophore (pNA) at 405 - 603 nm at 37 °C for 20 minutes. A linear fit of the data is performed and the rate is calculated m mOD/mm. DMSO is only present during experiments involving inhibitors, buffer is used to make up the volume to 100 ⁇ l m the other experiments .
- pNA chromophore
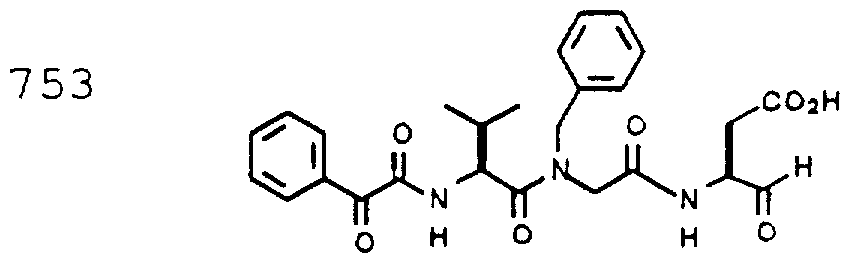
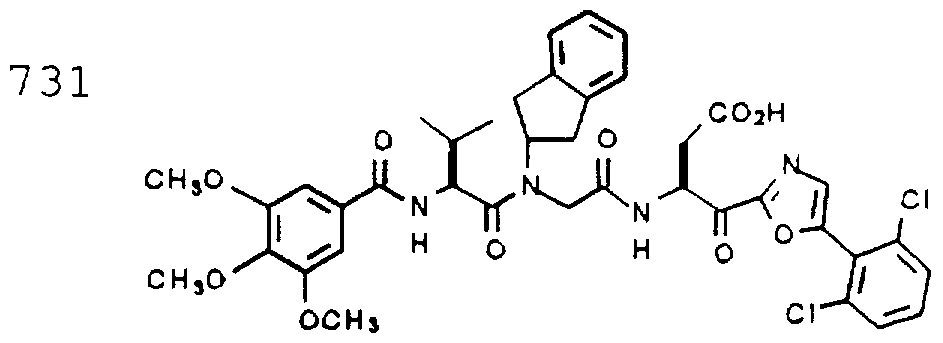
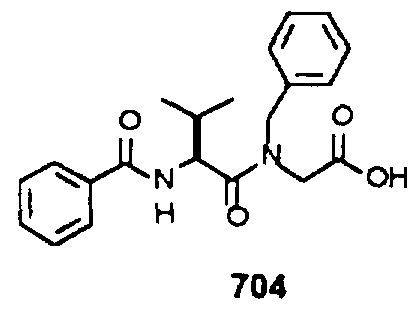
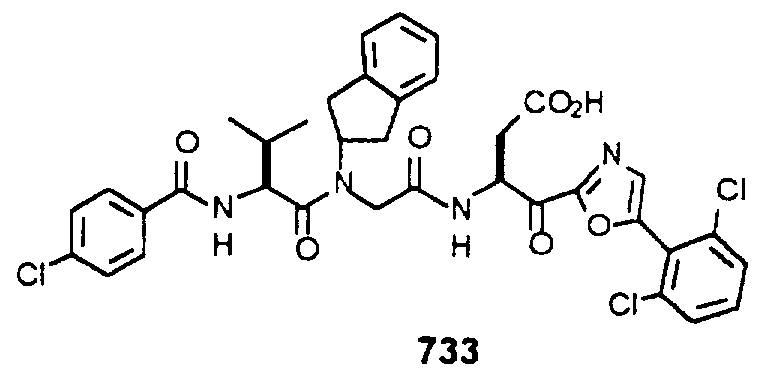
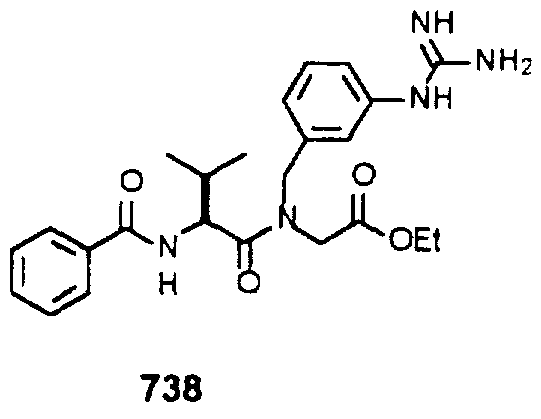
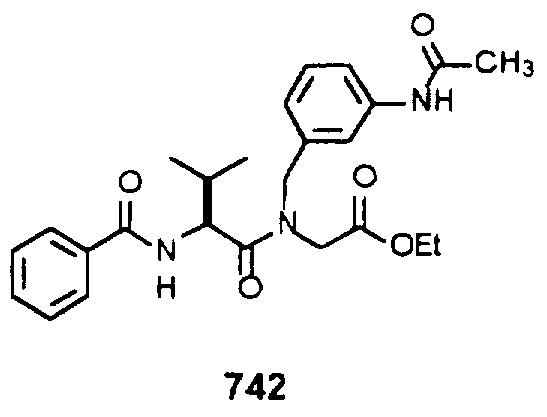
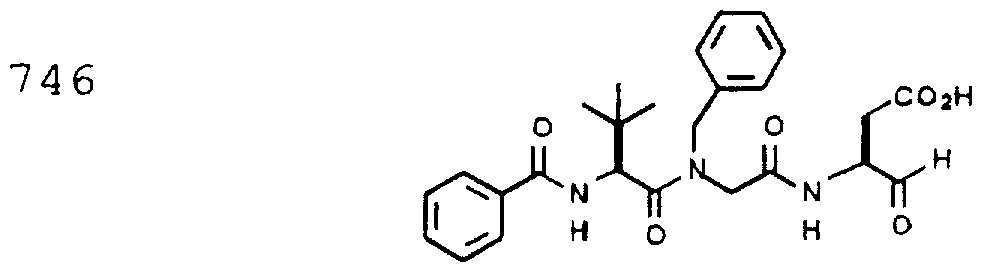
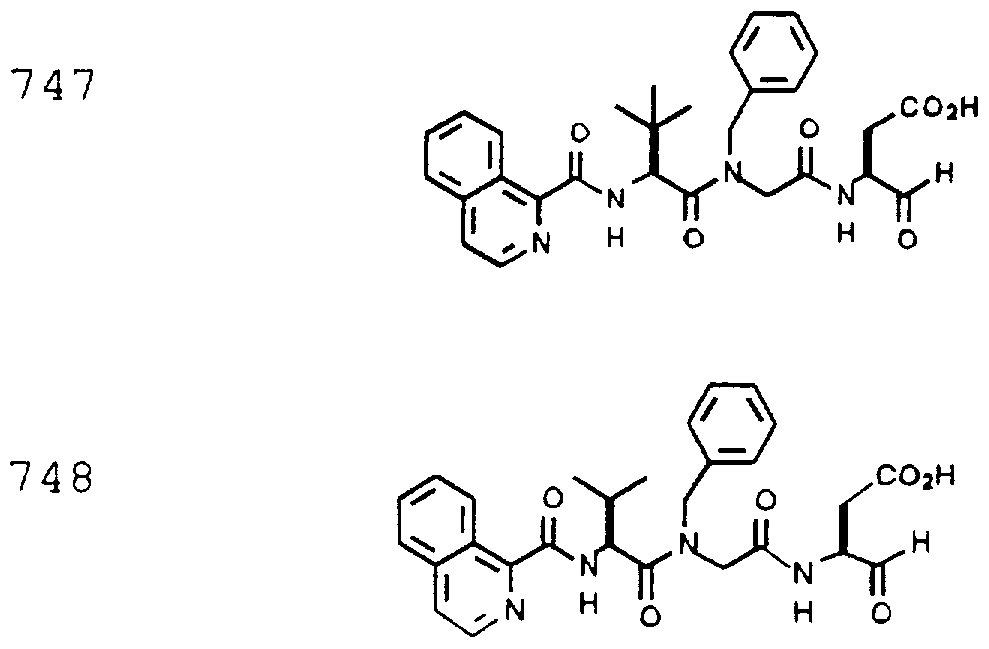
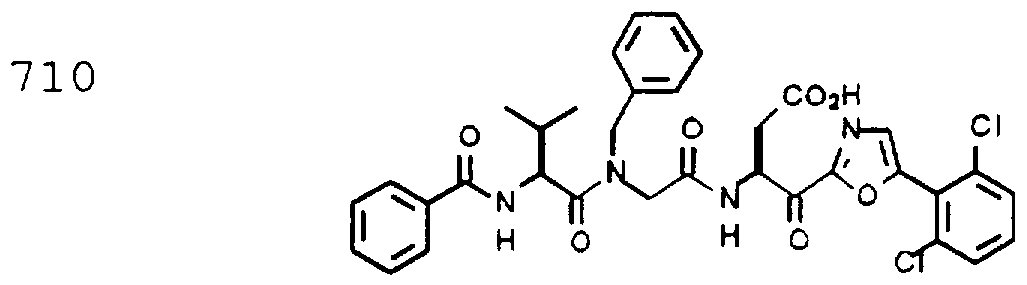
- Example 2 The following K j _ values were determined for compounds 706, 710, 719, 720, 725-727, 729, 731, 733, 743, 745, and 747-757 using the assay described Example 1. The structures of the compounds of Example 2 are shown in the following Table and in Example 3.
- Type 1 Type 2
- Example 3 Compounds of Example 2 were synthesized as follows:
- N-Benzylglycine Ethyl Ester (701) To a solution of benzaldehyde (14.0 g, 0.132 mol) in absolute EtOH (500 mL) was added glycine ethyl ester hydrochloride (37.0 g, 0.256 mol), NaOAc (32.5 g, 0.396 mol) and sodium cyanoborohydride (9.8 g, 0.158 mol) , and the resulting mixture heated to reflux. After 1 hr at reflux, the reaction was cooled and concentrated in vacuo . The residue was taken up into IN NaOH and EtOAc.
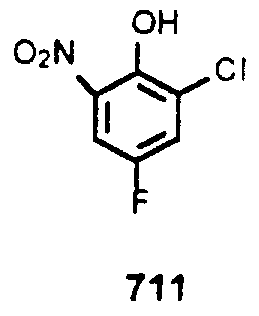
- 2-Chloro-4-fluoro-6-n ⁇ trophenol (711) To a mixture of 2-Chloro-4-fluorophenol (25 g, 0.171 mol) in H 2 0 (100 mL) and Et 0 (300 mL) at 0 °C was added dropwise concentrated nitric acid (25 mL) . After the addition was complete the reaction was warmed to rt and stirred for 3 hr. The layers were separated and the organic phase washed with 1:1 brme:H 2 0, brme, dried over MgS0 4 , filtered and concentrated m vacuo to a slurry. The slurry was diluted with hexane and the yellow solid collected and dried to provide 23.6 g of compound 711.
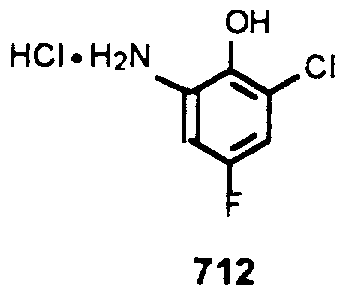
- 6-Chloro-4-fluorobenzoxazole (713) A mixture of compound 712 (17.0 g, 86.3 mmol) and trimethylorthoformate (18.9 mL, 0.173 mol) in abso-ute MeOH (90 mL) was heated to reflux upon which a solution formed. After stirring at reflux for 24 hr, the reaction was cooled and concentrated to provide an orange solid. The solid was dissolved into Et 2 0, washed with IN NaOH, brine, dried over MgS0 4 , filtered and concentrated to provide a yellow orange solid. Recyrstallization from hot aqueous EtOH with rapid cooling and filtration provided 10.0 g of compound 713 as white needles. Note that prolong standing m aqueous EtOH causes decomposition of compound 713.
- 4-D ⁇ fluoro-6-ammophenol Hydrochloride (715) A mixture of 2, 4-D ⁇ fluoro-6-n ⁇ trophenol (28.4 g, 0.162 mol; prepared by a similar method as 711 except replacing 2-chloro-4-fluorophenol with 2, 4-d ⁇ fluorophenol) and 10% palladium on carbon (3.5 g) m absolute MeOH (120 mL) was placed under 1 atm of H and stirred until complete reduction had occurred. The H 2 was replaced with nitrogen and the reaction was filtered through Celite. Gaseous HCl was bubbled through the filtrate and the resulting solution concentrated.
- N-Indan-2-ylglycine t-Butyl Ester (721) To a suspension of 2-ammoindane hydrochloride (5.0g, 29.5 mmol) and powdered K 2 C0 3 (8.3 g, 60.0 mmol) m absolute EtOH (30 mL) was added tert-butyl bromoacetate (4.4 mL, 29.5 mmol) . After stirring for 10 mm at rt the reaction was heated to 45 °C and stirred for 2 hr . The reaction was cooled to rt, diluted with EtOAc, filtered and concentrated. ehromatography of the residue on silica gel (elution with 20% EtOAc:hexane) provided 4.7g of compound 721 as a white crystalline solid.
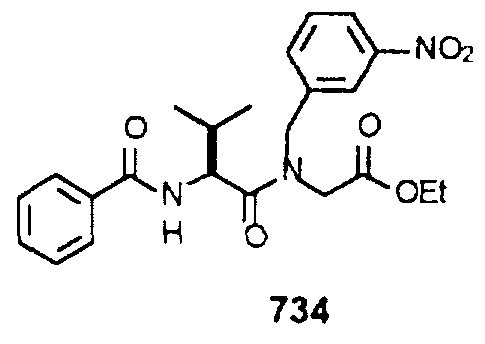
- Bocammobenzyl) amino) acetic Acid Ethyl Ester (703): To a solution of compound 735 (1.45 g, 3.5 mmol) and DIEA (740 ⁇ l, 4.25 mmol) in CH 2 C1 2 (7.0 mL) containing a catalytic amount of N,N-dimethylammopyridme, was added di-tert-butyldicarbonate (850 mg, 3.9 mmol) . After 1 hr, the reaction was diluted with EtOAc, washed with H 2 0, sat. aq. KHS0 4 , brine, dried over MgS0 4 , filtered and concentrated in vacuo to provide 1.78 g of compound 736.
- Step A Synthesis of 401 a/b.
- TentaGel SS NH 2 resm (0.16 mmol/g, 10.0 g) was placed in a sintered glass funnel and washed with DMF (3 X 50 mL) , 10 ⁇ (v/v) DIEA in DMF (2 X 50 mL) and finally with DMF (4 X 50 mL) .
- Sufficient DMF was added to the resm to obtain a slurry followed by 713a (1.42 g, 2.4 mmol, prepared from either (3S) 3-
- HOBT HOBT-H 2 0; 0.367 g 2.4 mmol
- HBTU 0- benzotriazole-N, N,N,N' - tetramethyluronium hexafluorophosphate
- Method 1 Synthesis of 761a/b and 762a.
- Resms 761a and 762a were prepared from resm 401a (0.24 g, 0.038 mmol) and Fmoc-Val e or Fmoc-t-Leucme, respectively, while resm 761b was prepared from resm 401b and Fmoc-Valme using an Advanced ChemTech 396 Multiple Peptide synthesizer.
- the automated cycles consisted of a resm wash with DMF (3 X 1 L) , deprotection with 25% (v/v) pipe ⁇ dme m DMF (1 mL) for 3 mm followed by fresh reagent (1 mL) for 10 mm.
- the resm was washed with DMF (3 X 1 mL) and N- methypyrrolidone (3 X 1 mL) .
- the res was then acylated with a solution of either 0.4M Fmoc-1-Valme or Fmoc-t-Leucme and 0.4M HOBT m N-methypyrrolidone (1 mL) , a solution of 0. M HBTU in N-methypyrrolidone (0.5 mL) and a solution of 1.6M DIEA m N- methypyrrolidone (0.35 mL) and the reaction was shaken for 2 hr at rt. The acylation step was repeated. Finally, the resins were washed with DMF (3 X 1 mL) .
- the appropriate carboxylic acid (0.4 M in 0.4 M HOBt/NMP) was coupled to the resm as described in Step B.
- the aldehyde was cleaved from the resm and globally deprotected by treatment with 95% TFA/ 5% H 2 0 (v/v, 1.5 mL) for 30 mm at rt. After washing the resin with cleavage reagent (1 mL) , the combined filtrates were added to cold 1:1 Et 2 0:pentane (12 mL) and the resulting precipitate was isolated by centrifugation and decantation.
- the resulting pellet was dissolved in 10% CH 3 CN/90% H 2 0/0.1% TFA (15 mL) and lyophilized to obtain the crude product as a white powder.
- the compound was purified by semi-prep RP-HPLC with a Rainin MicrosorbTM C18 column (5 u, 21.4 X 250 mm) elutmg with a linear CH 3 CN gradient (10 ⁇ - 60%) containing 0.1% TFA (v/v) over 45 mm at 12 mL/min. Fractions containing the desired product were pooled and lyophilized.
- Step C Method 1A. Synthesis of 751. Following a similar procedure as method 1, resin 761a was acylated with 4-(l- fluorenylmethoxycarbonylamino) benzoic acid and repeated. The Fmoc group was removed as described in Step C and the free amine was acetylated with 20% (v/v) acetic anhydride in DMF (1 mL) and 1.6M DIEA m N- methylpyrrolidone (0.35 mL) for 2 hr at rt. The acetylation step was repeated. Cleavage of the aldehyde from the resm gave 751.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Physical Education & Sports Medicine (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Neurosurgery (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Rheumatology (AREA)
- Biochemistry (AREA)
- Biomedical Technology (AREA)
- Oncology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Immunology (AREA)
- Communicable Diseases (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Virology (AREA)
- Hospice & Palliative Care (AREA)
Abstract
Description


























































































Claims































Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP52300897A JP4009320B2 (en) | 1995-12-20 | 1996-12-20 | Inhibitor of interleukin-1β converting enzyme |
| KR1019980704611A KR20000064454A (en) | 1995-12-20 | 1996-12-20 | Inhibitors of Interleukin-1beta Converting Enzyme |
| HK99101441.5A HK1016611B (en) | 1995-12-20 | 1996-12-20 | Inhibitors of interleukin-1beta converting enzyme |
| BR9612191A BR9612191A (en) | 1995-12-20 | 1996-12-20 | Interleukin-1beta converting enzyme inhibitors |
| AU14658/97A AU722936B2 (en) | 1995-12-20 | 1996-12-20 | Inhibitors of interleukin-1beta converting enzyme |
| EP96945237A EP0876395B1 (en) | 1995-12-20 | 1996-12-20 | Inhibitors of interleukin-1beta converting enzyme |
| NZ326555A NZ326555A (en) | 1995-12-20 | 1996-12-20 | Inhibitors of interleukin-1beta converting enzyme (ICE) |
| IL12495496A IL124954A (en) | 1995-12-20 | 1996-12-20 | Inhibitors of interleukin-1betaconverting enzyme and pharmaceutical compositions containing the same |
| AT96945237T ATE310011T1 (en) | 1995-12-20 | 1996-12-20 | INHIBITORS OF THE INTERLEUKIN-1 BETA CONVERTING ENZYME |
| DE69635458T DE69635458T2 (en) | 1995-12-20 | 1996-12-20 | INHIBITORS OF INTERLEUKIN-1-BETA CONVERTING ENZYME |
| PL96327333A PL188813B1 (en) | 1995-12-20 | 1996-12-20 | Inhibitors of interleukin- 1beta converting enzyme |
| NO982774A NO982774L (en) | 1995-12-20 | 1998-06-16 | Inhibitors of Interleukin-1 <beta> -converting enzyme |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/575,648 US5843904A (en) | 1995-12-20 | 1995-12-20 | Inhibitors of interleukin-1βconverting enzyme |
| US08/575,648 | 1995-12-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997022618A1 true WO1997022618A1 (en) | 1997-06-26 |
Family
ID=24301162
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1996/020370 Ceased WO1997022618A1 (en) | 1995-12-20 | 1996-12-20 | INHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYME |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US5843904A (en) |
| EP (1) | EP0876395B1 (en) |
| JP (3) | JP4009320B2 (en) |
| KR (1) | KR20000064454A (en) |
| CN (2) | CN1127511C (en) |
| AT (1) | ATE310011T1 (en) |
| AU (1) | AU722936B2 (en) |
| BR (1) | BR9612191A (en) |
| CA (1) | CA2240489A1 (en) |
| CZ (1) | CZ292633B6 (en) |
| DE (1) | DE69635458T2 (en) |
| HU (1) | HUP9902254A3 (en) |
| IL (1) | IL124954A (en) |
| NO (1) | NO982774L (en) |
| NZ (1) | NZ326555A (en) |
| PL (1) | PL188813B1 (en) |
| RU (1) | RU2213096C2 (en) |
| TR (1) | TR199801168T2 (en) |
| WO (1) | WO1997022618A1 (en) |
| ZA (1) | ZA9610797B (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998011129A1 (en) * | 1996-09-12 | 1998-03-19 | Idun Pharmaceuticals, Incorporated | C-TERMINAL MODIFIED (N-SUBSTITUTED)-2-INDOLYL DIPEPTIDES AS INHIBITORS OF THE ICE/ced-3 FAMILY OF CYSTEINE PROTEASES |
| US5869519A (en) * | 1996-12-16 | 1999-02-09 | Idun Pharmaceuticals, Inc. | C-terminal modified (n-substituted)-2-indolyl dipeptides as inhibitors of the ICE/ced-3 family of cysteine proteases |
| US5877197A (en) * | 1996-12-16 | 1999-03-02 | Karanewsky; Donald S. | C-terminal modified (N-substituted)-2-indolyl dipeptides as inhibitors of the ICE/ced-3 family of cysteine proteases |
| WO1999033859A3 (en) * | 1997-12-24 | 1999-12-23 | Stanford Res Inst Int | Novel anti-estrogenic steroids, and associated pharmaceutical compositions and methods of use |
| WO2000023421A1 (en) * | 1998-10-22 | 2000-04-27 | Idun Pharmaceuticals, Inc. | (SUBSTITUTED)ACYL DIPEPTIDYL INHIBITORS OF THE ICE/ced-3 FAMILY OF CYSTEINE PROTEASES |
| US6184244B1 (en) * | 1996-12-16 | 2001-02-06 | Idun Pharmaceuticals, Inc. | C-terminal modified (N-substituted)-2-indolyl dipeptides as inhibitors of the ICE/ced-3 family of cysteine proteases |
| WO2001092210A1 (en) * | 2000-05-30 | 2001-12-06 | Trans Tech Pharma | Compounds for modulating the rage receptor |
| WO2002066063A1 (en) * | 2001-02-23 | 2002-08-29 | Nippon Organon K.K. | Remedies for metabolic bone diseases |
| JP2002539201A (en) * | 1999-03-15 | 2002-11-19 | アクシス・ファーマシューティカルズ・インコーポレイテッド | Amine derivatives as protease inhibitors |
| US6525024B1 (en) | 2000-04-17 | 2003-02-25 | Idun Pharmaceuticals, Inc. | Inhibitors of the ICE/ced-3 family of cysteine proteases |
| US6525025B2 (en) | 2000-09-08 | 2003-02-25 | Merck Frosst Canada & Co. | Gamma-ketoacid dipeptides as inhibitors of caspase-3 |
| US6730671B2 (en) | 1999-03-02 | 2004-05-04 | Boehringer Ingelheim Pharmaceuticals, Inc. | Compounds useful as reversible inhibitors of cathespin S |
| US6756372B2 (en) | 1999-09-13 | 2004-06-29 | Boehringer Ingelheim Pharmaceuticals, Inc. | Compounds useful as reversible inhibitors of cysteine proteases |
| US7087632B2 (en) | 2001-03-05 | 2006-08-08 | Transtech Pharma, Inc. | Benzimidazole derivatives as therapeutic agents |
| US7087604B2 (en) | 2002-03-08 | 2006-08-08 | Bristol-Myers Squibb Company | Cyclic derivatives as modulators of chemokine receptor activity |
| US7157430B2 (en) | 1998-10-22 | 2007-01-02 | Idun Pharmaceuticals, Inc. | (Substituted)acyl dipeptidyl inhibitors of the ICE/CED-3 family of cysteine proteases |
| US7361678B2 (en) | 2002-03-05 | 2008-04-22 | Transtech Pharma, Inc. | Azole derivatives and fused bicyclic azole derivatives as therapeutic agents |
| US7423177B2 (en) | 2001-03-05 | 2008-09-09 | Transtech Pharma, Inc. | Carboxamide derivatives as therapeutic agents |
| US8580833B2 (en) | 2009-09-30 | 2013-11-12 | Transtech Pharma, Inc. | Substituted imidazole derivatives and methods of use thereof |
| US9352010B2 (en) | 2011-07-22 | 2016-05-31 | The J. David Gladstone Institutes | Treatment of HIV-1 infection and AIDS |
| EP3603634A1 (en) | 2004-05-18 | 2020-02-05 | Novartis AG | Pharmaceutical composition containing glycopyrrolate and a beta2 adrenoceptor agonist |
| WO2022238507A1 (en) * | 2021-05-11 | 2022-11-17 | Awakn Ls Europe Holdings Limited | Therapeutic aminoindane compounds and compositions |
| WO2026082873A1 (en) | 2024-10-17 | 2026-04-23 | Institut National de la Santé et de la Recherche Médicale | Use of caspase-1 inhibitors for treating ineffective erythropoiesis in patients suffering from sickle cell disease |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5843904A (en) * | 1995-12-20 | 1998-12-01 | Vertex Pharmaceuticals, Inc. | Inhibitors of interleukin-1βconverting enzyme |
| US6093742A (en) * | 1997-06-27 | 2000-07-25 | Vertex Pharmaceuticals, Inc. | Inhibitors of p38 |
| US6184210B1 (en) | 1997-10-10 | 2001-02-06 | Cytovia, Inc. | Dipeptide apoptosis inhibitors and the use thereof |
| KR20010041905A (en) * | 1998-03-16 | 2001-05-25 | 시토비아 인크. | Dipeptide caspase inhibitors and the use thereof |
| AU3876600A (en) | 1999-03-16 | 2000-10-04 | Cytovia, Inc. | Substituted 2-aminobenzamide caspase inhibitors and the use thereof |
| US6355618B1 (en) | 1999-04-09 | 2002-03-12 | Cytovia, Inc. | Caspase inhibitors and the use thereof |
| IL147970A0 (en) * | 1999-08-09 | 2002-09-12 | Tripep Ab | Pharmaceutical compositions containing tripeptides |
| US6495522B1 (en) | 1999-08-27 | 2002-12-17 | Cytovia, Inc. | Substituted alpha-hydroxy acid caspase inhibitors and the use thereof |
| US6566338B1 (en) | 1999-10-12 | 2003-05-20 | Cytovia, Inc. | Caspase inhibitors for the treatment and prevention of chemotherapy and radiation therapy induced cell death |
| EP1712239A3 (en) * | 2000-05-12 | 2007-08-22 | Immunex Corporation | Interleukin-1 inhibitors in the treatment of diseases |
| PE20011350A1 (en) | 2000-05-19 | 2002-01-15 | Vertex Pharma | PROPHARMAC OF AN INHIBITOR OF INTERLEUKIN-1ß CONVERTER ENZYME (ICE) |
| CA2418720A1 (en) * | 2000-09-13 | 2002-03-21 | Vertex Pharmaceuticals Incorporated | Caspase inhibitors and uses thereof |
| US6706712B2 (en) | 2000-12-20 | 2004-03-16 | Bristol-Myers Squibb Pharma Company | Cyclic derivatives as modulators of chemokine receptor activity |
| WO2002050019A2 (en) * | 2000-12-20 | 2002-06-27 | Bristol-Myers Squibb Pharma Co. | Diamines as modulators of chemokine receptor activity |
| US20030049255A1 (en) * | 2001-08-07 | 2003-03-13 | Sims John E. | Interleukin-1 receptors in the treatment of diseases |
| US6593455B2 (en) * | 2001-08-24 | 2003-07-15 | Tripep Ab | Tripeptide amides that block viral infectivity and methods of use thereof |
| WO2003024995A1 (en) * | 2001-09-19 | 2003-03-27 | Tripep Ab | Molecules that block viral infectivity and methods of use thereof |
| US6916813B2 (en) * | 2001-12-10 | 2005-07-12 | Bristol-Myers Squibb Co. | (1-phenyl-2-heteoaryl)ethyl-guanidine compounds as inhibitors of mitochondrial F1F0 ATP hydrolase |
| AU2003231012B2 (en) * | 2002-04-22 | 2008-05-08 | University Of Maryland, Baltimore County | Antiviral inhibitions of capsid proteins |
| KR20050101221A (en) * | 2003-02-21 | 2005-10-20 | 트리펩 아베 | Glycinamide derivative for inhibiting hiv replication |
| US20050096319A1 (en) * | 2003-02-21 | 2005-05-05 | Balzarini Jan M.R. | Identification of compounds that inhibit replication of human immunodeficiency virus |
| GB0329572D0 (en) * | 2003-12-20 | 2004-01-28 | Astrazeneca Ab | Amide derivatives |
| EP2399916B1 (en) * | 2004-03-12 | 2014-12-10 | Vertex Pharmaceuticals Incorporated | Process and intermediates for the preparation of aspartic acetal caspase ihnhibitors |
| WO2005115362A1 (en) * | 2004-05-15 | 2005-12-08 | Vertex Pharmaceuticals Incorporated | Treating seizures using ice inhibitors |
| CN101268084A (en) | 2005-07-28 | 2008-09-17 | 沃泰克斯药物股份有限公司 | Caspase inhibitor prodrugs |
| CN104650016A (en) * | 2015-01-29 | 2015-05-27 | 武汉大学 | Tripeptide analog compound and preparation method thereof |
| CN112979636A (en) * | 2021-02-06 | 2021-06-18 | 绍兴文理学院 | Thiazole compound containing phenylsulfide structure and preparation method and application thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0644198A1 (en) * | 1993-06-03 | 1995-03-22 | Sterling Winthrop Inc. | Alpha-heteroaryloxymethyl ketones as interleukin-1beta converting enzyme inhibitors |
| WO1995035308A1 (en) * | 1994-06-17 | 1995-12-28 | Vertex Pharmaceuticals Incorporated | INHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYME |
Family Cites Families (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4276298A (en) * | 1978-03-24 | 1981-06-30 | Merck & Co., Inc. | 2-Aryl-1,2-benzisothiazolinone-1,1-dioxides and their use as selective protease inhibitors |
| US4369183A (en) * | 1979-09-06 | 1983-01-18 | Merck & Co., Inc. | 2-Pyridyl-1,2-benzisothiazolinone-1,1-dioxides and their use as selective protease inhibitors |
| US4499295A (en) * | 1983-05-09 | 1985-02-12 | G. D. Searle & Co. | Protease inhibitors |
| US4584397A (en) * | 1983-05-09 | 1986-04-22 | G. D. Searle & Co. | Protease inhibitors |
| US4551279A (en) * | 1984-01-09 | 1985-11-05 | G. D. Searle & Co. | Protease inhibitors |
| US5055451A (en) * | 1986-12-22 | 1991-10-08 | Syntex Inc. | Aryloxy and arylacyloxy methyl ketones as thiol protease inhibitors |
| US5158936A (en) * | 1986-12-22 | 1992-10-27 | Syntex (U.S.A.) Inc. | Aryloxy and arylacyloxy methyl ketones as thiol protease inhibitors |
| NZ223148A (en) * | 1987-01-16 | 1989-10-27 | Merrell Dow Pharma | Peptide derivatives having peptidase inhibition activity |
| WO1989004838A1 (en) * | 1987-11-25 | 1989-06-01 | Immunex Corporation | Interleukin-1 receptors |
| US5081228A (en) * | 1988-02-25 | 1992-01-14 | Immunex Corporation | Interleukin-1 receptors |
| US4968607A (en) * | 1987-11-25 | 1990-11-06 | Immunex Corporation | Interleukin-1 receptors |
| US5008245A (en) * | 1988-10-27 | 1991-04-16 | University Of Kentucky Research Foundation | Novel peptidyl carbamate inhibitors of the enzyme elastase |
| KR0183397B1 (en) * | 1989-05-04 | 1999-05-01 | 존 엠. 스피나토 | Saccharin derivatives useful as protease inhibitors and methods for their preparation |
| US5527882A (en) * | 1989-07-07 | 1996-06-18 | The Regents Of The University Of California | Polypeptide having an amino acid replaced with N-benzylglycine |
| ZA905737B (en) * | 1989-07-26 | 1991-05-29 | Merrell Dow Pharma | Novel peptidase inhibitors |
| NZ235155A (en) * | 1989-09-11 | 1993-04-28 | Merrell Dow Pharma | Peptidase substrates in which the carboxy terminal group has been replaced by a tricarbonyl radical |
| WO1991015577A1 (en) * | 1990-04-04 | 1991-10-17 | Black, Roy, A. | INTERLEUKIN 1'beta' PROTEASE |
| US5416013A (en) * | 1990-04-04 | 1995-05-16 | Sterling Winthrop Inc. | Interleukin 1β protease and interleukin 1β protease inhibitors |
| IL99527A (en) * | 1990-09-28 | 1997-08-14 | Lilly Co Eli | Tripeptide antithrombotic agents |
| EP0504938A3 (en) * | 1991-03-22 | 1993-04-14 | Suntory Limited | Prophylactic and therapeutic agent for bone diseases comprising di- or tripeptide derivative as active ingredient |
| DE69226820T2 (en) * | 1991-06-21 | 1999-05-12 | Merck & Co., Inc., Rahway, N.J. | Peptidyl derivatives as inhibitors of interleukin-1B converting enzymes |
| JP3190431B2 (en) * | 1991-07-01 | 2001-07-23 | 三菱化学株式会社 | Ketone derivatives |
| US5278061A (en) * | 1991-08-16 | 1994-01-11 | Merck & Co., Inc. | Affinity chromatography matrix useful in purifying interleukin-1β converting enzyme |
| DE69229252T2 (en) * | 1991-08-16 | 1999-12-16 | Merck & Co., Inc. | DNA encoding the interleukin-1B precursor converting enzyme |
| US6348570B1 (en) * | 1991-08-16 | 2002-02-19 | Merck & Co., Inc. | Chromophore containing compounds and their use in determining interleukin-1β convertase activity |
| EP0533226A3 (en) * | 1991-08-16 | 1993-08-18 | Merck & Co. Inc. | Novel chromophore containing compounds |
| AU657701B2 (en) * | 1991-08-30 | 1995-03-23 | Vertex Pharmaceuticals Incorporated | Interleukin 1beta protease and interleukin 1beta protease inhibitors |
| GB9123326D0 (en) * | 1991-11-04 | 1991-12-18 | Sandoz Ltd | Improvements in or relating to organic compounds |
| AU3479593A (en) * | 1992-01-31 | 1993-09-01 | Merck & Co., Inc. | Peptidyl derivatives as inhibitors of interleukin-1beta converting enzyme |
| AU3666893A (en) * | 1992-02-21 | 1993-09-13 | Merck & Co., Inc. | Peptidyl derivatives as inhibitors of interleukin-1beta converting enzyme |
| WO1993025683A1 (en) * | 1992-06-12 | 1993-12-23 | Massachusetts Institute Of Technology | A gene which prevents programmed cell death |
| JPH08500482A (en) * | 1992-06-12 | 1996-01-23 | マサチューセッツ インスティテュート オブ テクノロジー | Inhibitors of Ced-3 and related proteins |
| WO1993025685A1 (en) * | 1992-06-12 | 1993-12-23 | Massachusetts Institute Of Technology | Cloning and characterization of the cell death genes ced-3 and ced-4 |
| AU4634993A (en) * | 1992-06-24 | 1994-01-24 | Merck & Co., Inc. | Dna encoding precursor interleukin 1beta converting enzyme |
| JP2602181B2 (en) * | 1992-07-31 | 1997-04-23 | ファイザー・インク. | Peptidyl 4-amino-2,2-difluoro-3-oxo-1,6-hexanedioic acid derivative as anti-inflammatory agent |
| US5374623A (en) * | 1992-08-20 | 1994-12-20 | Prototek, Inc. | Cysteine protease inhibitors effective for in vivo use |
| CA2109646C (en) * | 1992-11-24 | 2000-03-07 | Gaston O. Daumy | Para-nitroanilide peptides |
| EP0618223A3 (en) * | 1993-03-08 | 1996-06-12 | Sandoz Ltd | Peptides inhibit the release of interleukin 1-beta useful as anti-inflammatory agents. |
| US5462939A (en) * | 1993-05-07 | 1995-10-31 | Sterling Winthrop Inc. | Peptidic ketones as interleukin-1β-converting enzyme inhibitors |
| US5411985A (en) * | 1993-05-17 | 1995-05-02 | Merck & Co., Inc. | Gamma-pyrone-3-acetic acid as an inhibitor or interleukin-1 β inventory enzyme |
| ATE170868T1 (en) * | 1993-06-04 | 1998-09-15 | Vertex Pharma | PEPTIDE PHOSPHINYLOXYMETHYL KETONES AS INHIBITORS OF INTERLEUKIN-1 BETA-CONVERTING ENZYMES |
| ES2114654T3 (en) * | 1993-06-08 | 1998-06-01 | Vertex Pharma | PIRIDAZINES AS INHIBITORS OF THE INTERLEUKIN-1BETA CONVERSION ENZYME. |
| WO1995000160A1 (en) * | 1993-06-24 | 1995-01-05 | The General Hospital Corporation | Programmed cell death genes and proteins |
| US5866545A (en) * | 1993-08-13 | 1999-02-02 | Merck & Co., Inc. | Substituted ketone derivatives as inhibitors of interleukin-1β converting enzyme |
| US5486623A (en) * | 1993-12-08 | 1996-01-23 | Prototek, Inc. | Cysteine protease inhibitors containing heterocyclic leaving groups |
| US5508262A (en) * | 1993-12-15 | 1996-04-16 | University Of South Florida | Interleukin-1 receptor antagonist decreases severity of acute pancreatitis |
| CN1504462A (en) * | 1994-03-31 | 2004-06-16 | ������˹ҩƷ��˾ | Pyrimidinyl derivatives as interleukin inhibitors |
| FI964342A7 (en) * | 1994-04-29 | 1996-10-28 | Vertex Pharma | Halogenomethylamides as IL-1beta protease inhibitors |
| US5552400A (en) * | 1994-06-08 | 1996-09-03 | Sterling Winthrop Inc. | Fused-bicyclic lactams as interleukin-1β converting enzyme inhibitors |
| US5565430A (en) * | 1994-08-02 | 1996-10-15 | Sterling Winthrop Inc. | Azaaspartic acid analogs as interleukin-1β converting enzyme inhibitors |
| US5498616A (en) * | 1994-11-04 | 1996-03-12 | Cephalon, Inc. | Cysteine protease and serine protease inhibitors |
| US5843904A (en) * | 1995-12-20 | 1998-12-01 | Vertex Pharmaceuticals, Inc. | Inhibitors of interleukin-1βconverting enzyme |
-
1995
- 1995-12-20 US US08/575,648 patent/US5843904A/en not_active Expired - Lifetime
-
1996
- 1996-12-20 BR BR9612191A patent/BR9612191A/en not_active Application Discontinuation
- 1996-12-20 CA CA002240489A patent/CA2240489A1/en not_active Abandoned
- 1996-12-20 KR KR1019980704611A patent/KR20000064454A/en not_active Ceased
- 1996-12-20 CN CN96199733A patent/CN1127511C/en not_active Expired - Fee Related
- 1996-12-20 IL IL12495496A patent/IL124954A/en not_active IP Right Cessation
- 1996-12-20 WO PCT/US1996/020370 patent/WO1997022618A1/en not_active Ceased
- 1996-12-20 NZ NZ326555A patent/NZ326555A/en unknown
- 1996-12-20 ZA ZA9610797A patent/ZA9610797B/en unknown
- 1996-12-20 HU HU9902254A patent/HUP9902254A3/en unknown
- 1996-12-20 PL PL96327333A patent/PL188813B1/en not_active IP Right Cessation
- 1996-12-20 CZ CZ19981905A patent/CZ292633B6/en not_active IP Right Cessation
- 1996-12-20 EP EP96945237A patent/EP0876395B1/en not_active Expired - Lifetime
- 1996-12-20 TR TR1998/01168T patent/TR199801168T2/en unknown
- 1996-12-20 DE DE69635458T patent/DE69635458T2/en not_active Expired - Lifetime
- 1996-12-20 AT AT96945237T patent/ATE310011T1/en not_active IP Right Cessation
- 1996-12-20 JP JP52300897A patent/JP4009320B2/en not_active Expired - Fee Related
- 1996-12-20 CN CNA031589928A patent/CN1500781A/en active Pending
- 1996-12-20 AU AU14658/97A patent/AU722936B2/en not_active Ceased
- 1996-12-20 RU RU98113932/04K patent/RU2213096C2/en not_active IP Right Cessation
-
1998
- 1998-02-17 US US09/024,537 patent/US6162790A/en not_active Expired - Lifetime
- 1998-06-16 NO NO982774A patent/NO982774L/en not_active Application Discontinuation
-
2003
- 2003-12-05 JP JP2003408331A patent/JP2004143182A/en not_active Withdrawn
-
2007
- 2007-11-09 JP JP2007292640A patent/JP2008101008A/en not_active Withdrawn
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0644198A1 (en) * | 1993-06-03 | 1995-03-22 | Sterling Winthrop Inc. | Alpha-heteroaryloxymethyl ketones as interleukin-1beta converting enzyme inhibitors |
| WO1995035308A1 (en) * | 1994-06-17 | 1995-12-28 | Vertex Pharmaceuticals Incorporated | INHIBITORS OF INTERLEUKIN-1β CONVERTING ENZYME |
Non-Patent Citations (3)
| Title |
|---|
| DOLLE R.E. ET AL.: "P1 Aspartate-based peptide alpha-(2,6-dichlorobenzoyl)oxy)methyl ketones as potent time-dependent inhibitors of interleukin 1-beta-converting enzyme", JOURNAL OF MEDICINAL CHEMISTRY, vol. 37, no. 5, 1994, WASHINGTON US, pages 563 - 564, XP002029379 * |
| DOLLE, ROLAND E. ET AL: "Aspartyl.alpha.-((1-Phenyl-3-(trifluoromethyl)- pyrazol-5-yl)oxy)methyl Ketones as Interleukin-1.beta. Converting Enzyme Inhibitors. Significance of the P1 and P3 Amido Nitrogens for Enzyme-Peptide Inhibitor Binding", JOURNAL OF MEDICINAL CHEMISTRY, vol. 37, no. 23, 11 November 1994 (1994-11-11), WASHINGTON US, pages 3863 - 6, XP000652064 * |
| MULLICAN, MICHAEL D. ET AL: "The synthesis and evaluation of peptidyl aspartyl aldehydes as inhibitors of ICE", BIOORG. MED. CHEM. LETT., vol. 4, no. 19, 1994, pages 2359 - 2364, XP000653042 * |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998011129A1 (en) * | 1996-09-12 | 1998-03-19 | Idun Pharmaceuticals, Incorporated | C-TERMINAL MODIFIED (N-SUBSTITUTED)-2-INDOLYL DIPEPTIDES AS INHIBITORS OF THE ICE/ced-3 FAMILY OF CYSTEINE PROTEASES |
| US6184244B1 (en) * | 1996-12-16 | 2001-02-06 | Idun Pharmaceuticals, Inc. | C-terminal modified (N-substituted)-2-indolyl dipeptides as inhibitors of the ICE/ced-3 family of cysteine proteases |
| US5869519A (en) * | 1996-12-16 | 1999-02-09 | Idun Pharmaceuticals, Inc. | C-terminal modified (n-substituted)-2-indolyl dipeptides as inhibitors of the ICE/ced-3 family of cysteine proteases |
| US5877197A (en) * | 1996-12-16 | 1999-03-02 | Karanewsky; Donald S. | C-terminal modified (N-substituted)-2-indolyl dipeptides as inhibitors of the ICE/ced-3 family of cysteine proteases |
| WO1999033859A3 (en) * | 1997-12-24 | 1999-12-23 | Stanford Res Inst Int | Novel anti-estrogenic steroids, and associated pharmaceutical compositions and methods of use |
| US6242422B1 (en) | 1998-10-22 | 2001-06-05 | Idun Pharmacueticals, Inc. | (Substituted)Acyl dipeptidyl inhibitors of the ice/ced-3 family of cysteine proteases |
| WO2000023421A1 (en) * | 1998-10-22 | 2000-04-27 | Idun Pharmaceuticals, Inc. | (SUBSTITUTED)ACYL DIPEPTIDYL INHIBITORS OF THE ICE/ced-3 FAMILY OF CYSTEINE PROTEASES |
| US7157430B2 (en) | 1998-10-22 | 2007-01-02 | Idun Pharmaceuticals, Inc. | (Substituted)acyl dipeptidyl inhibitors of the ICE/CED-3 family of cysteine proteases |
| US6730671B2 (en) | 1999-03-02 | 2004-05-04 | Boehringer Ingelheim Pharmaceuticals, Inc. | Compounds useful as reversible inhibitors of cathespin S |
| JP2002539201A (en) * | 1999-03-15 | 2002-11-19 | アクシス・ファーマシューティカルズ・インコーポレイテッド | Amine derivatives as protease inhibitors |
| US6982272B2 (en) | 1999-09-13 | 2006-01-03 | Boehringer Ingelheim Pharmaceuticals, Inc. | Compounds useful as reversible inhibitors of cysteine proteases |
| US7279472B2 (en) | 1999-09-13 | 2007-10-09 | Boehringer Ingelheim Pharmaceuticals Inc. | Compounds useful as reversible inhibitors of cysteine proteases |
| US7265132B2 (en) | 1999-09-13 | 2007-09-04 | Boehringer Ingelheim Pharmaceuticals Inc. | Compounds useful as reversible inhibitors of cysteine proteases |
| US7056915B2 (en) | 1999-09-13 | 2006-06-06 | Boehringer Ingelheim Pharmaceuticals, Inc. | Compounds useful as reversible inhibitors of cysteine proteases |
| US6756372B2 (en) | 1999-09-13 | 2004-06-29 | Boehringer Ingelheim Pharmaceuticals, Inc. | Compounds useful as reversible inhibitors of cysteine proteases |
| US6525024B1 (en) | 2000-04-17 | 2003-02-25 | Idun Pharmaceuticals, Inc. | Inhibitors of the ICE/ced-3 family of cysteine proteases |
| US6969703B2 (en) | 2000-04-17 | 2005-11-29 | Idun Pharmaceuticals, Inc. | Inhibitors of the ICE/ced-3 family of cysteine proteases |
| EP1642888A1 (en) | 2000-05-30 | 2006-04-05 | TransTech Pharma Inc. | compounds for modulating the rage receptor |
| US6613801B2 (en) | 2000-05-30 | 2003-09-02 | Transtech Pharma, Inc. | Method for the synthesis of compounds of formula I and their uses thereof |
| US7067554B2 (en) | 2000-05-30 | 2006-06-27 | Transtech Pharma, Inc. | Method for the synthesis of compounds of formula I and their uses thereof |
| AU780368B2 (en) * | 2000-05-30 | 2005-03-17 | Transtech Pharma | Compounds for modulating the rage receptor |
| WO2001092210A1 (en) * | 2000-05-30 | 2001-12-06 | Trans Tech Pharma | Compounds for modulating the rage receptor |
| US6525025B2 (en) | 2000-09-08 | 2003-02-25 | Merck Frosst Canada & Co. | Gamma-ketoacid dipeptides as inhibitors of caspase-3 |
| US6858623B2 (en) | 2000-09-08 | 2005-02-22 | Boehringer Ingelheim Pharmaceuticals, Inc. | Compounds useful as reversible inhibitors of cysteine proteases |
| WO2002066063A1 (en) * | 2001-02-23 | 2002-08-29 | Nippon Organon K.K. | Remedies for metabolic bone diseases |
| US7423177B2 (en) | 2001-03-05 | 2008-09-09 | Transtech Pharma, Inc. | Carboxamide derivatives as therapeutic agents |
| US7776919B2 (en) | 2001-03-05 | 2010-08-17 | TransTech Pharm, Inc. | Carboxamide derivatives as therapeutic agents |
| US7329684B2 (en) | 2001-03-05 | 2008-02-12 | Transtech Pharma, Inc. | Benzimidazole derivatives as therapeutic agents |
| US7087632B2 (en) | 2001-03-05 | 2006-08-08 | Transtech Pharma, Inc. | Benzimidazole derivatives as therapeutic agents |
| US7361678B2 (en) | 2002-03-05 | 2008-04-22 | Transtech Pharma, Inc. | Azole derivatives and fused bicyclic azole derivatives as therapeutic agents |
| US7714013B2 (en) | 2002-03-05 | 2010-05-11 | Transtech Pharma, Inc. | Azole derivatives and fused bicyclic azole derivatives as therapeutic agents |
| US7737285B2 (en) | 2002-03-05 | 2010-06-15 | Transtech Pharma, Inc. | Azole derivatives and fused bicyclic azole derivatives as therapeutic agents |
| US7776884B2 (en) | 2002-03-08 | 2010-08-17 | Bristol-Myers Squibb Company | Cyclic derivatives as modulators of chemokine receptors activity |
| US7087604B2 (en) | 2002-03-08 | 2006-08-08 | Bristol-Myers Squibb Company | Cyclic derivatives as modulators of chemokine receptor activity |
| EP3603634A1 (en) | 2004-05-18 | 2020-02-05 | Novartis AG | Pharmaceutical composition containing glycopyrrolate and a beta2 adrenoceptor agonist |
| US8580833B2 (en) | 2009-09-30 | 2013-11-12 | Transtech Pharma, Inc. | Substituted imidazole derivatives and methods of use thereof |
| US9598375B2 (en) | 2009-09-30 | 2017-03-21 | Vtv Therapeutics Llc | Substituted imidazole derivatives and methods of use thereof |
| US10363241B2 (en) | 2009-09-30 | 2019-07-30 | Vtv Therapeutics Llc | Substituted imidazole derivatives and methods of use thereof |
| US9352010B2 (en) | 2011-07-22 | 2016-05-31 | The J. David Gladstone Institutes | Treatment of HIV-1 infection and AIDS |
| US9956260B1 (en) | 2011-07-22 | 2018-05-01 | The J. David Gladstone Institutes | Treatment of HIV-1 infection and AIDS |
| WO2022238507A1 (en) * | 2021-05-11 | 2022-11-17 | Awakn Ls Europe Holdings Limited | Therapeutic aminoindane compounds and compositions |
| WO2026082873A1 (en) | 2024-10-17 | 2026-04-23 | Institut National de la Santé et de la Recherche Médicale | Use of caspase-1 inhibitors for treating ineffective erythropoiesis in patients suffering from sickle cell disease |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU722936C (en) | Inhibitors of interleukin-1beta converting enzyme | |
| EP0942925B1 (en) | Inhibitors of interleukin-1 beta converting enzyme | |
| JP5285130B2 (en) | Intermediate of prodrug of ICE inhibitor | |
| US5874424A (en) | Inhibitors of interleukin-1β converting enzyme | |
| KR100561504B1 (en) | Inhibitors of Interleukin-1 Beta Converting Enzyme | |
| EP0944645B1 (en) | INHIBITORS OF INTERLEUKIN-1beta CONVERTING ENZYME | |
| JP2000503635A (en) | Inhibitor of interleukin-1β converting enzyme | |
| US6008217A (en) | Inhibitors of interleukin-1β converting enzyme | |
| MXPA98005015A (en) | Inhibitors of the enzyme that becomes the interleucine | |
| HK1016611B (en) | Inhibitors of interleukin-1beta converting enzyme | |
| RU2249598C2 (en) | Interleikyn-1beta converting enzyme inhibitors | |
| KR100856767B1 (en) | Inhibitors of interleukin-1beta converting enzyme | |
| EP1466921A1 (en) | Inhibitors of interleukin-1 beta converting enzyme | |
| AU7612201A (en) | Inhibitors of interleukin-1beta converting enzyme |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 96199733.8 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE HU IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1199800508 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 326555 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2240489 Country of ref document: CA Ref document number: 2240489 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1998-1905 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019980704611 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1998/005015 Country of ref document: MX Ref document number: 1998/01168 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996945237 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996945237 Country of ref document: EP Ref document number: PV1998-1905 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019980704611 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV1998-1905 Country of ref document: CZ |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1019980704611 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1996945237 Country of ref document: EP |