WO1997023650A2 - Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of hla types - Google Patents
Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of hla types Download PDFInfo
- Publication number
- WO1997023650A2 WO1997023650A2 PCT/US1996/020202 US9620202W WO9723650A2 WO 1997023650 A2 WO1997023650 A2 WO 1997023650A2 US 9620202 W US9620202 W US 9620202W WO 9723650 A2 WO9723650 A2 WO 9723650A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- type
- sample
- chain terminating
- sequencing
- positions
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6881—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for tissue or cell typing, e.g. human leukocyte antigen [HLA] probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
- C12Q1/689—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms for bacteria
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N27/00—Investigating or analysing materials by the use of electric, electrochemical, or magnetic means
- G01N27/26—Investigating or analysing materials by the use of electric, electrochemical, or magnetic means by investigating electrochemical variables; by using electrolysis or electrophoresis
- G01N27/416—Systems
- G01N27/447—Systems using electrophoresis
- G01N27/44704—Details; Accessories
- G01N27/44717—Arrangements for investigating the separated zones, e.g. localising zones
- G01N27/44721—Arrangements for investigating the separated zones, e.g. localising zones by optical means
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/16—Primer sets for multiplex assays
Definitions
- SSCP 'single-stranded conformational polymorphism
- ddF dideoxy-fingerprinting
- the tests employed in the hierarchy may frequently be combinations of different types of molecular tests, for examples combinations of immunoassays, oligonucleotide probe hybridization tests, oligonucleotide fragment analyses, and direct nucleic acid sequencing
- This application relates to a particular type of test which can be useful alone or as part of a hierarchical testing protocol, particularly for highly polymorphic genes
- a particular example of the use of this test is its application to determining the allelic type of human HLA genes, although the test is applicable to many genes of known sequence, and the invention should not be construed as limited to HLA
- Human HLA genes are part of the major histocompatability complex (MHC), a cluster of genes associated with tissue antigens and immune responses Within the MHC genes are two groups of genes which are of substantial importance in the success of tissue and organ transplants between individuals
- MHC major histocompatability complex
- the HLA Class I genes encode transplantation antigens which are used by cytotoxic T cells to distinguish self from non-self
- the HLA class II genes, or immune response genes determine whether an individual can mount a strong response to a particular antigen
- Both classes of HLA genes are highly polymorphic, and in fact this polymorphism plays a critical role in the immune response potential of a host On the other hand, this polymorphism also places an immunological burden on the host transplanted with allogeneic tissues As a result, careful testing and matching of HLA types between tissue donor and recipient is a major factor in the success of allogeneic tissue and marrow transplants
- HLA genes have proceeded along two basic lines serological and nucleic acid-based
- serological typing antibodies have been developed which are specific for certain types of HLA proteins Panels of these tests can be performed to evaluate the type of a donor or recipient tissue
- nucleic acid based-approaches samples of the HLA genes may be hybridized with sequence-specific oligonucleotide probes to identify particular alleles or allele groups
- determination of HLA type by sequencing of the HLA gene has also been proposed Santamaria P, et al "HLA Class I Sequence-Based Typing", Human Immunology 37 39-50 (1993)
- the allelic type of a polymorphic genetic locus in a sample is identified by first combining the sample with a sequencing reaction mixture containing a template-dependent nucleic acid polymerase, A, T, G and C nucleotide feedstocks, one type of chain terminating nucleotide and a sequencing primer under conditions suitable for template dependant primer extension to form a plurality of oligonucleotide fragments of differing lengths, and then evaluating the length of the oligon
- the use of the method of the invention can increase laboratory throughput (since up to four times as many samples can be processed on the same amount of equipment) and reduce the cost per test by up to a factor of four compared to sequencing of all four bases for every sample B 1EF DESCRIPTION OF THE DRAWINGS
- Fig 1 shows the application of the invention to typing of a simple polymorphic gene
- Fig 2 illustrates an improved method for distinguishing heterozygotic alleles using the present invention
- Fig 3 illustrates a situation in which heterozygote pairs remain ambiguous even after full sequencing.
- Fig 4 illustrates the use of a control lane to evaluate the number of intervening bases in a single base sequencing reaction
- Fig 5 shows results from an automated DNA sequencing apparatus.
- Fig 6 illustrates peak-by-peak correlation of sequencing results
- Fig 7 shows a plot of the maxima of each data peak plotted against the separation from the nearest other peak
- Figs 8A-8C illustrate the application of the invention to typing of Chlamy ⁇ a trach ⁇ tis.
- Allele refers to a specific version of a nucleotide sequence at a polymorphic genetic locus
- Polymorphism means the variability found within a population at a genetic locus
- Polymorphic site means a given nucleotide location in a genetic locus which is variable within a population
- Gene or “Genetic locus” means a specific nucleotide sequence within a given genome
- the “location” or “position” of a nucleotide in a genetic locus means the number assigned to the nucleotide in the gene, generally taken from the cDNA sequence or the genomic sequence of the gene
- nucleotides Adenine, Cytosine, Guanine and Thymine are sometimes represented by their designations of A, C, G or T, respectively While it has long been apparent to persons skilled in the art that knowledge of the identity of the base at a particular location within a polymorphic genetic locus may be sufficient to determine the allelic type of that locus, this knowledge has not led to any modification of sequencing procedures Rather, the knowledge has driven development of techniques such as allele-specific hybridization assays, and allele-specific ligation assays Despite the failure of the art to recognize the possibility, however, it is not always necessary to determine the sequence of all four nucleotides of a polymorphic genetic locus in order to determine which allele is present in a specific patient sample Certain alleles of a genetic locus may be distinguishable on the basis of identification of the location of less than four, and often only one nucleotide This finding allows the development of the present method for improved allele identification at a polymorphic genetic locus
- each of the four sequencing reactions generates a plurality of primer extension products, all of which end with a specific type of dideoxy-nucleotide
- Each lane on the electrophoresis gel thus reflects the positions of one type of base in the extension product, but does not reveal the order and type of nucleotides intervening between the bases of this specific type
- the information provided by the four lanes is therefore combined in known sequencing procedures to arrive at a composite picture of the sequence as a whole
- single sequencing reactions are performed and evaluated independently to provide the number of intervening bases between each instance of a selected base and thus a precise indication of the positional location of the selected base
- a single sequencing reaction would first be performed using either dideoxy-A or dideoxy-T as the chain terminating nucleotide If the third allelic type did not exist or was unknown
- a second sequencing test could be performed using either dideoxy-C or the dideoxy-A/T not used in the first test to resolve the identity of the allelic type .
- some other test such as an allele-specific hybridization probe or an antibody test which distinguished well between allele 1 or 2 and allele 3 could be used in this case
- the method of the invention specifically identifies "known" alleles of a polymorphic locus, and is not necessarily useful for identification of new and hitherto unrecorded alleles.
- An unknown allele might be missed if it were incorrectly assumed that the single nucleotide sequence obtained from a patient sample corresponded to a unique allele, when in fact other nucleotides of the allele had been rearranged in a new fashion.
- the method is specific for distinguishing among known alleles of a polymorphic locus (though it may fortuitously come across new mutations if the right single nucleotide sequence is chosen) Databases listing known alleles must therefore be continually updated to provide greatest utility for the invention
- the second sequencing reaction performed may not yield unique patterns for all of the samples tested In this case, prior to performing a third sequencing reaction, it is desirable to combine the results of the first two sequencing reactions and evaluate these composite results for unique base patterns
- a first and second sequencing reaction may have four alleles which can be characterized as follows
- Allele 2 and Allele 4 give unique results from the T-sequence reaction alone, and can therefore be typed based upon this information Alleles 1 and 3, however have the same T- sequencing pattern. Because these two allele have different A-sequencing reaction patterns, however, they are clearly distinguishable and can be typed based upon the combined patterns without further testing.
- HLA Human Leukocyte Antigen
- the method is not limited to human polymorphisms It may be used for other animals, plants, bacteria, viruses or fungi It may be used to distinguish the allelic variants present among a mixed sample of organisms
- the method can be used to identify which subspecies of bacteria or viruses are present in a body sample This diagnosis could be essential for determining whether drug-resistant strains of pathogens are present in an individual
- the first step of the method of the invention is obtaining a suitable sample of material for testing using this methodology
- the genetic material tested using the invention may be chromosomal DNA, messenger RNA, cDNA, or any other form of nucleic acid polymer which is subject to testing to evaluate polymorphism, and may be derived from various sources including whole blood, tissue samples including tumor cells, sperm, and hair follicles
- Amplification primers for this purpose are advantageously designed to be highly selective for the genetic locus in question
- group specific and locus specific amplification primers have been disclosed in US Patent No 5,424, 184 and Cereb et al , "Locus-specific amplification of HLA class 1 genes from genomic DNA locus-specific sequences in the first and third introns of HLA-A, -B and -C alleles " Tissue Antigens 45 1-1 1 (1995) which are incorporated herein by reference
- This reaction mixture contains a template-dependent nucleic acid polymerase, A, T, G and C nucleotide feedstocks, one type of chain terminating nucleotide and a sequencing primer
- Thermo SequenaseTM a thermostable polymerase enzyme marketed by Amersham Life Sciences
- suitable enzymes include regular SequenaseTM and other enzymes used in sequencing reactions
- Selection of appropriate sequencing primers is generally done by finding a part of the gene, either in an intron or an exon, that lies near (within about 300 nt) the polymorphic region of the gene which is to be evaluated, is 5' to the polymorphic region (either on the sense or the antisense strand), and that is highly conserved among all known alleles of the gene
- a sequencing primer that will hybridize to such a region with high specificity can then be used to sequence through the polymorphic region
- Other aspects of primer quality such as lack of palindromic sequence, and preferred G/C content are identified in the US Patent No 5,545,527
- Two or more primers may be necessary to test among some sub-groups of a genetic locus In these cases it is necessary to attempt a sequencing reaction using one of the primers If hybridization is successful, and a sequencing reaction proceeds, then the results can be used to determine allele identity If no sequencing reactions occur, it may be necessary to use another one of the primers
- the sequencing reaction mixture is processed through multiple cycles during which primer is extended and then separated from the template DNA from the sample and new primer is reannealed with the template At the end of these cycles, the product oligonucleotide fragments are separated by gel electrophoresis and detected
- This process is well known in the art
- this separation is performed in an apparatus of the type described in US Patent Application No 08/353,932, the continuation in part thereof filed on December 12, 1995 as International Patent Application No PCT/US95/15951 using thin microgels as described in International Patent Application No. PCT/US9514531, all of which applications are incorporated herein by reference
- the practice of the instant invention is assisted by technically advanced methods for precisely identifying the location of nucleotides in a genetic locus using single nucleotide sequencing
- the issue is that in the technique of single nucleotide sequencing using dideoxy-sequencing/ electrophoresis analysis it is sometimes a challenge to determine how many nucleotides fall between two of the identified nucleotides A A A or A A A
- control lane indicates precisely the number of nucleotides that lie in the gaps between the identified nucleotides, as in Fig. 3
- Any sequencing format can use such a control lane, be it "manual" sequencing, using radioactively labeled oligonucleotides and autoradiograph analysis (see Chp 7, Current Protocols in Molecular Biology, Eds Ausubel, F M et al, (John Wiley & Sons, 1 95)), or automated laser fluorescence systems
- a further embodiment of the invention which may be applied in some cases, including HLA typing, to further expedite and reduce the expense of testing, involves the simultaneous use of two chain terminating nucleotides in a single reaction mixture
- a single reaction containing a mixture of ddATP and ddCTP could be performed initially
- the peaks observed on the sequencing gel are either A or C, and cannot be distinguished (unless dye-labeled terminators with different labels are used). In some cases, however, this information is sufficient to identify the nature of the allele For example, in the simple three allele case shown in Fig. 1, the sequence information would identify the T allele unambiguously.
- a second sequencing run including two chain terminating nucleotides, one being the same as one included in the first reaction and the other being different from those included in the first reaction mixture.
- wave forms may represent heterozygote mixtures.
- the database should include wave forms from all known heterozygote combinations to ensure that the matching process includes the full variety of possibilities.
- the software can be designed to inform the user of the next analytical test that should be performed to help distinguish among possible allelic members of the heterozygote
- Heterozygous polymorphic genetic loci need special consideration. Where more than one variant of the same loci exists in the patient sample, complex results are obtained when single lane sequencing begins at a commonly shared sequencing primer site. This problem is also found in traditional 4 lane sequencing (see Santamaria P, et al "HLA Class I Sequence-Based Typing” Human Immunology 37, 39-50 (1993)). However, Figure 2 illustrates an improved method for distinguishing heterozygotic alleles using the present invention.
- Fig. 2a The problem presented by a heterozygous allele is illustrated in Fig. 2a.
- the observed data from single nucleotide sequencing of the A lane can not point to the presence of a unique allele. Either the loci is heterozygous or a new allele has been found. (For well studied genetic loci, new alleles will be rare, so heterozygosity may be assumed.)
- the problem flows from a mixture of alleles in the patient sample which is analyzed. For exam ⁇ ple, the observed data may result from the additive combination of allele 1 and allele 2.
- Fig 3c shows further, that sometimes observed data may appear to be a homozygote for one allele, but in fact it may consist of a heterozygote pair, either including the suggested allele, or not
- the alleles that might lead to such confusion, by masking possible heterozygotes, can be identified in the known allele database Identification of these alleles can not be confirmed unless further tests are made which can confirm whether a heterozygote underlies the observed data
- Allelic dropout is resolved by amplifying both alleles from genomic DNA using quantitative polymerase chain reaction (see for example, Chp 15, Current Protocols in Molecular Biology, Eds Ausubel, F M et al, (John Wiley & Sons, 1995))
- the sequencing primer is used as one of a pair of PCR primers A fragment of DNA spanning the alleles in question is amplified quantitatively
- quantities of PCR products will be only half the expected amount if only one allele is being amplified
- Quantitative analysis can be made on the basis of peak heights of amplified bands observed by automated DNA sequencing instruments
- a plurality of pathogens can produce even more complex results from single nucleotide sequencing
- the complexity flows from an unlimited number of variants of the pathogen that may be present in the patient sample
- viruses, and bacteria may have variable surface antigen coding domains which allow them to evade host immune system detection
- the genetic locus selected for examination is preferably highly conserved among all variants of the pathogen, such as ribosomal DNA or functionally critical protein coding regions of DNA
- an extended series of comparisons between the observed data and the known alleles can assist the diagnosis by determining which alleles are not substantial components of the observed data
- the method of the present invention lends itself to the construction of tailored kits which provide components for the sequencing reactions As described in the examples, these components include oligonucleotide sequencing primers, enzymes for sequencing, nucleoside and dideoxynucleoside preparations, and buffers for reactions Unlike conventional kits, however, the amount of each type of dideoxynucleoside required for any given assay is not the same Thus, for an assay in which the A sequencing reaction is performed first and on all samples, the amount of dideoxy-A included in the kit may be 5 to 10 times greater than the amount of the other dideoxynucleosides
- DRB 1 is a polymorphic HLA Class II gene with at least 107 known alleles (See Bodmer et al Nomenclature for Factors of the HLA System, 1994 Hum 1mm 41 , 1 -20 ( 1994))
- the broad serological subtype of the patient sample DRB 1 allele is first determined by attempting to amplify the allele using group specific primers
- Genomic DNA is prepared from the patient sample using a standard technique such as proteinase proteolysis Allele amplification is carried out in Class II PCR buffer 10 mM T ⁇ s pH 8 4 50 mM KC1 1 5 mM MgC12 0 1% gelatin
- the 5'-primers of the above groups are terminally labelled with a fluorophore such as a fluorescein dye at the 5'- end
- reaction mixture is mixed well 2 5 units Taq Polymerase are added and mixed immediately prior to thermocycling
- reaction tubes are placed in a Robocycler Gradient 96 (Stratagene, Inc ) and subject to thermal cycling as follows
- the PCR amplification primers are a biotinylated 3'-PRIMER amp B
- the PCR product ( 10 ul) is mixed with 10 ul of washed Dynabeads M-280 (as per manufacturers recommendations, Dynal, Oslo, Norway) and incubated for 1 hr at room temperature
- the beads are washed with 50 ul of IX BW buffer ( 10 mM Tris, pH 7 5, 1 mM EDTA, 2M NaCl) followed by 50 ul of 1 X TE buffer (10 mM Tris, 1 mM EDTA)
- Lane 1 first requires alignment with the database results
- the alignment requires determination of one or more normalization coefficients (for stretching or shrinking the results of lane 1 ) to provide a high degree of overlap (i.e. maximize the intersection) with the previously aligned database results
- the alignment co-efficient(s) may be calculated using the Genetic Algorithm method of the above noted application, or another method.
- the normalization coefficients are then applied to Lane 1.
- the aligned result of Lane 1 is then systematically correlated to each of the 22 known alleles.
- the correlation takes place on a peak by peak basis as illustrated in Fig. 6
- the area under each peak is calculated within a limited radius of the peak maxima (i e. 20 data points for A L F. Sequencer results).
- a similar calculation is made for the area under the curve of the known allele at the same point
- the swath of overlapping areas is then compared. Any correlation below a threshold of reasonable variation, for example 80%, indicates that a peak is present in the patient data stream and not in the other If one peak is missing, then the known allele is rejected as a possible identifier of the sample.
- peaks in the known data stream are identified and compared, one by one, to the patient sample results Again, the presence of a peak in one data stream, that is not present in the other, eliminates the known data stream as an identifier of the sample
- lane 2 for allele DRB 1 *0405, has a peak (marked X) not found in the patient sample Peak comparison between aligned lane 1 and lane 2 will fall below threshold at the peak marked X.
- Lane 3 is for part of known allele DRB1 *0401 In this case, each peak is found to have a correlate in the other data stream. DRB 1 *0401 may therefore identify the patient sample. (The results illustrated are much shorter than the 200-300 nt usually used for comparison, so identity of the patient sample is not confirmed until the full diagnostic sequence is compared )
- Example 2 Results are obtained from the patient sample according to Example 1 , above The sample results are converted into a "text" file as follows The maxima of each peak is located and plotted against the separation from the nearest other peak (minor peaks representing noise are ignored). Fig. 7 The peaks that are closest together are assumed to represent single nucleotide separation and an narrow range for single nucleotide separation is determined.
- a series of timing tracks are proposed which attempts to locate all the peaks in terms of multiples of a possible single nucleotide separation
- the timing track that correlates best (by least mean squares analysis) with the maxima of the sample data is selected as the correct timing track
- the peak maxima are then plotted on the timing track
- the spaces between the peaks are assumed to represent other nucleotides
- a text file may now be generated which identifies the location of all nucleotides of one type and the single nucleotide steps in between.
- the text file for the patient sample is compared against all known alleles
- the known allele that best matches the patient sample identifies the sample
- Example 3 For HLA Class II DRB1 Serological group DR4, 22 alleles are known. A hierarchy of single nucleotide sequencing reactions can be used to minimize the number of reactions required for identification of which allele is present Reactions are performed according to the methods of example 1 , above If it is established from the group specific reaction that only one DRB1 allele is a DR4 subtype, then identification of that allele is made by the following steps
- Example 4 If the group specific reaction in example 1 indicates that two DR4 alleles are present in the patient sample, then from the 22 known alleles, there are 253 possible allelic pair combinations (22 homozygotes + 231 heterozygotes) Again, a hierarchy of single nucleotide sequencing reactions can be used to minimize the number of reactions required for identification of which allelic pair is present Reactions are performed according to the methods of example 1, above
- Virtually all the alleles of the HLA Class I C gene can be determined on the basis of exon 2 and 3 genomic DNA sequence alone (Cereb, N et al "Locus-specific amplification of HLA class I genes from genomic DNA locus-specific sequences in the first and third introns of HLA-A, -B and -C alleles " Tissue Antigens 45 1-1 1 ( 1995))
- the primers used amplify the polymorphic exons 2 and 3 of all C-alleles without any co- amplification of pseudogenes or B or A alleles
- These primers utilize C-specific sequences in introns 1, 2 and 3 of the C-locus
- the following primers are used to amplify the HLA Class 1 C gene exon 2
- the amplification was carried out in PCR buffer composed of 15.6 mM ammonium sulfate, 67 mM Tris-HCI (pH 8.8), 50 uM EDTA, 1 5 mM MgCI2, 0 01% gelatin, 0 2 mM of each dNTP (dATP, dCTP, dGTP and dTTP) and 0 2 mM of each amplification primer
- dNTP dATP, dCTP, dGTP and dTTP
- 0 2 mM of each amplification primer Prior to amplification 40 ng of patient sample DNA is added followed by 2 5 units of Taq Polymerase (Roche Molecular)
- the amplification cycle consisted of 1 min 96 C
- the termination reaction selected depends on whether a forward or reverse primer is chosen. Appendix 1 lists which alleles can be distinguished if a forward primer is used (i.e sequencing template is the anti-sense strand) If a reverse primer is used for sequencing, the termination reaction selected is the complementary one (A for T, C for G, and vice versa).
- the remaining 2 alleles, Cw* 12022.hla and Cw* 12021.hla can not be distinguished by nucleotide sequencing of only exons 2 and 3. Further reactions according to the invention may be performed to distinguish among these alleles If the patient sample is identified at any one step, then the following step(s) need not be performed for that sample
- Heterozygotes are analyzed on the same basis, the order of single nucleotide sequencing reactions is determined by picking which reactions will distinguish among the greatest number of samples (data not shown), and performing those reactions first
- LPL lipoprotein lipase
- Asn291 Ser is associated with reduced HDL cholesterol levels in premature atherosclerosis
- This variant has a single missense mutation of A to C at nucleotide 1 127 of the sense strand in Exon 6
- This variant can be distinguished according to the instant invention as follows
- Exon 6 of the LPL gene from a patient sample is amplified with a 5' PCR primer located in intron 5 near the 5' boundary of exon 6
- the 3' PCR primer is located in exon 6 a short distance from the Asn291 Ser mutation and labeled with biotin
- PCR amplification reactions were performed according to the methods detailed in Reymer, PWA , et al , "A lipoprotein lipase mutation (Asn291 Ser) is associated with reduced HDL cholesterol levels in premature atherosclerosis " Nature Genetics 10 28- 34 (1995) Sequencing analysis was then performed according to the Thermo SequenaseTM (Amersham) method of example 1 , using a fluorescent-labeled version of the 5' PCR primer noted above Since the deleterious allele has a C at nucleotide 1127 of the sense strand, the C termination sequencing reaction was performed The results of the reaction were recorded on an automated DNA sequencing apparatus and analyzed at the 1127 site The patient sample either carries the C at that site, or it does not If a C is present, the patient is identified as having the "unhealthy” allele If no C is present, then the "healthy" form of the allele is identified Patient reports may be prepared on this basis
- Example 7 Health care workers currently seek to distinguish among Chlamy ⁇ a trachomatis strains to determine the molecular epidemiologic association of a range of diseases with infecting genotype (See Dean, D et al "Major Outer Membrane Protein Variants of Chlamy ⁇ a trachomatis Are Associated with Severe Upper Genital Tract Infections and Histopathology in San Francisco " J Infect Dis 172 1013-22 (1995))
- the presence and genotype of pure and mixed cultures of C. trachomatis may be determined by examining the C. trachomatis ompl gene (Outer Membrane Protein
- the ompl gene has at least 4 variable sequence ("VS") domains that may be used to distinguish among the 15 known genotypes (Yuan, Y et al "Nucleotide and Deduced Amino Acid Sequences for the Four Variable Domains of the Major Outer Membrane Proteins of the 15 Chlamy ⁇ a trachomatis Serovars" Infect Immun 57 1040-1049 (1989))
- VS variable sequence
- thermocycler temperature profile was 95 degrees C for 45 sec, 55 degrees C for 1 min, and 72 degrees C for 2 min, with a final extension of 10 min at 72 degrees C after the last cycle
- One microliter of the PCR product was then used in each of two separate nested 100 microliter reactions with primer pair MF21
- flank VS1 (Variable Sequence 1 ) and VS2, and primer pair
- trachomatis serovars A-K and LI -3 including Ba, Da, la, and L2a
- a sample of each product 10 microliters was run on a 1 5% agarose gel to confirm the size of the amplification product
- All PCR products were purified (GeneClean II, Bio 101, La Jolla, CA) according to the manufacturer's instructions
- biotinylated strand was separated with Dynal beads and selected termination reactions were performed as in Example 1 using a 5' fluorescent labeled version of MF21 or MVF3
- the first 25 nt of the T termination reaction for C trachomatis VS1 can be used to distinguish among 3 groups of genotypes, as illustrated in Fig 8A
- the observed results for Sample 1 in Fig 8 A demonstrates that detectable levels of at least one of Group 1 and at least one of the Group 3 genotypes are present Group 2 is not detected
- FIG 8B illustrates possible A results
- the observed results of Sample 1 shows an A at site 257 This A could be provided by only E, F or G genotypes Since the T track has already established the absence of both F and G, then E must be among the genotypes present Further, the absence of an A at 283 indicates that neither D nor F nor G are present The presence of E and the absence of D, F and G may be reported
- Group 1 genotypes may be present in addition to E, they do not appear because their presence is effectively masked by E Other single nucleotide termination reactions can be performed to distinguish among these other possible contributors, if necessary The investigator simply determines which single nucleotide reaction will effectively distinguish among the genotypes which may be present and need to be distinguished Alternatively, Sample 2, which showed the presence of Group 1 only in the T reaction is shown to be comprised of only Ba genotype because of an absence of A at 268 This shows that both the presence and absence of nucleotides can be used to determine the presence of some genotypes in some circumstances
- the first 25 nt of C and G termination reactions for VS1 only are included in Fig 8C to show how an investigator can determine which reaction to select and perform If higher degrees of resolution are required, the termination reactions for VS2, VS3 and VS4 may be performed
- the only combinations that can not be distinguished after this point include 2 remaining alleles, Cw* 12022 and Cw* 12021 , which can not be distinguished by nucleotide sequencing of only exons 2 and 3 Further reactions according to the invention may be performed to distinguish among these alleles Note that since these alleles differ only at a silent mutation, they are identical at the amino acid level, and do not need to be distinguished in practice Sample reports can simply confirm the presence of the one allele plus either of Cw* 12022 or * 12021
- EXAMPLE 9 Analysis of the HLA-DRB1 allelic type of a sample may be performed according to Example 1 using two chain terminating nucleotides 100 ng of patient sample DNA (previously amplified as in Example 1 ) is combined with labeled sequencing primer 5' - GAGTGTCATTTCTTCAA - 3' [SEQ ID NO 18]
- the termination reaction mixture is thermal cycled in a Robocycler for 30 cycles (or fewer if found to be satisfactory) 95 C 40 sec 50 C 30 sec 68 C 60 sec
- HLA Class I C locus allele analysis on the basis of exons 2 and 3.
- 35 alleles may be combined as 35 homozygous pairs or 630 heterozygous pairs.
- Homozygous pairs mav be distinguished by single nucleotide sequencing in the following order:
- Cw*02022.hla .Cw*02022.hla)
- Cw*1503.hla (Cw*1503.hla.
- Cw*02021.hla .Cw*02021.hla.
- Cw*0303.hla .Cw*0303.hla.
- Cw*1502.hla tCw*1502.hla.
- Cw*0303.hla (Cw*0303.hla. Cw*0304.hla. Cw*0801.hl . Cw*0803.hla.
- Cw*0302.hla (Cw*0302.hla. Cw*0304.hta. Cw*0801.hla. Cw*0803.hla.
- Cw*0302.hla tCw*0302.hla. Cw*0303.hla. Cw*0801.hla. Cw*0803.hla.
- Cw*0302.hla ⁇ Cw*0302.hla. Cw*0303.hla. Cw*0304.hla. Cw*0803.hla.
- CwM402.hla CwM402.hla.
- Cw*0302.hla (Cw*0302.hla. Cw*0303.hla. Cw*0304.hla. Cw*0801.hla.
- CwM2022.hla (CwM2022.hla.
- CwM2021.hla .CwM2021.hla ⁇
- CwM2022.hla (CwM2022.hla.
- CwM2021.hla fCwM2021.hlai
- CwM403.hla rCwM403.hlal
- C M6Qi.hla (CwM6 Q i ⁇ hla)
- CWOlOl.hla 2 Cw*0102.hla 3: Cw*0201.hla 4: Cw*0301.hla 8: Cw*0501.hla 15: CwM201.hla 5: Cw*0302.hla 9: Cw*0602.hla 16: CwM203.hla 6: Cw*0401.hla 10: Cw*0702.hla 17: CwM301.hla 7: Cw*0402.hla 11 : Cw*0701.hla 18: CwM501.hla 12: Cw*0703.hla 19: CwM503.hla 13: Cw*0704.hla 20: CwM504.hla 14: Cw » 0802.hla 21 : CwM701.hla
- CwM2022.hla fCwM2022.hla.
- CwM2021.hla .CwM2021.hlal
- CwM2022.hla ⁇ CwM2022.hlai
- CwM2021.hla (CwM2021.hla1
- CwM503.hla (CwM503.hlal
- CwM502.hla (CwM502.hla.
- CwM2022.hla rCwM2022.hla.
- CwM2021.hla .CwM2021.hla.
- CwM402.hla (CwM402.hla1
- CwM505.hla (CwM505.hlai
- CwM502.hla fCwM502.hlai
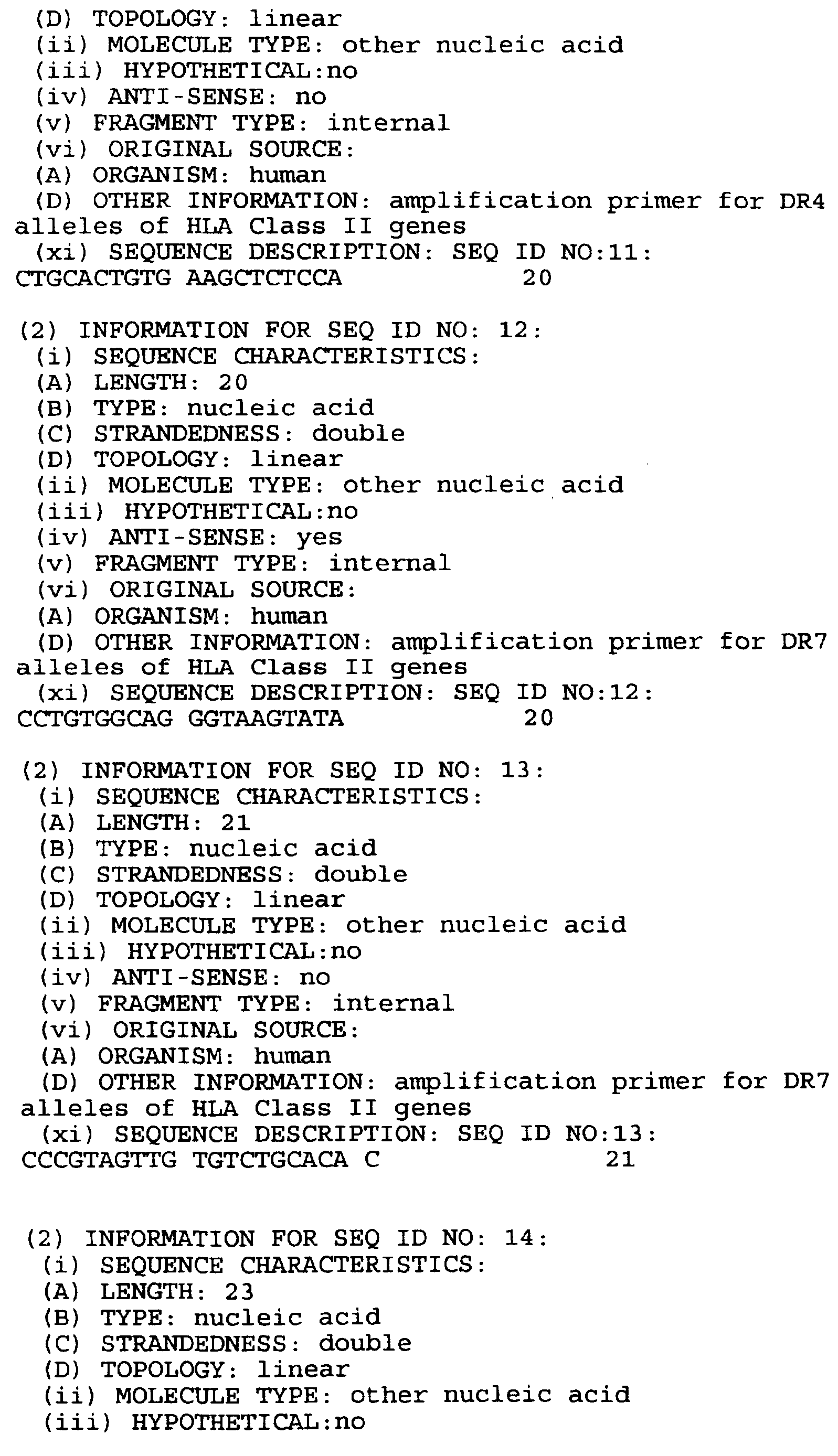
- MOLECULE TYPE other nucleic acid
- HYPOTHETICAL no
- ANTI-SENSE yes
- FRAGMENT TYPE internal
- ORIGINAL SOURCE (A) ORGANISM: human
- MOLECULE TYPE other nucleic acid
- HYPOTHETICAL no
- ANTI-SENSE yes
- ORGANISM human
- OTHER INFORMATION forward sequencing primer for HLA-C gene, exon 3
- MOLECULE TYPE other nucleic acid
- HYPOTHETICAL no
- ANTI-SENSE yes
- FRAGMENT TYPE internal
- ORIGINAL SOURCE (A) ORGANISM: human
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Analytical Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pathology (AREA)
- Cell Biology (AREA)
- Electrochemistry (AREA)
- General Physics & Mathematics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description









































Claims
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP96944459A EP0870059B1 (en) | 1995-12-22 | 1996-12-19 | Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of hla types |
| AU14262/97A AU718670B2 (en) | 1995-12-22 | 1996-12-19 | Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of HLA types |
| CA002239896A CA2239896C (en) | 1995-12-22 | 1996-12-19 | Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of hla types |
| DE69620791T DE69620791T2 (en) | 1995-12-22 | 1996-12-19 | METHOD FOR EVALUATING POLYMORPHIC GENETIC SEQUENCES, AND THEIR USE FOR DETERMINING HLA TYPES |
| JP9523796A JP2000502260A (en) | 1995-12-22 | 1996-12-19 | Methods for evaluating polymorphic gene sequences and their use in identifying HLA types |
| US08/892,003 US5981186A (en) | 1995-06-30 | 1997-07-14 | Method and apparatus for DNA-sequencing using reduced number of sequencing mixtures |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/577,858 US5834189A (en) | 1994-07-08 | 1995-12-22 | Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of HLA types |
| US08/577,858 | 1995-12-22 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US08/892,003 Continuation-In-Part US5981186A (en) | 1995-06-30 | 1997-07-14 | Method and apparatus for DNA-sequencing using reduced number of sequencing mixtures |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1997023650A2 true WO1997023650A2 (en) | 1997-07-03 |
| WO1997023650A3 WO1997023650A3 (en) | 1997-08-21 |
Family
ID=24310433
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1996/020202 Ceased WO1997023650A2 (en) | 1995-06-30 | 1996-12-19 | Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of hla types |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US5834189A (en) |
| EP (1) | EP0870059B1 (en) |
| JP (1) | JP2000502260A (en) |
| AU (1) | AU718670B2 (en) |
| CA (1) | CA2239896C (en) |
| DE (1) | DE69620791T2 (en) |
| WO (1) | WO1997023650A2 (en) |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999016910A1 (en) * | 1997-09-26 | 1999-04-08 | Visible Genetics Inc. | Method and kit for evaluation of hiv mutations |
| US6007983A (en) * | 1995-12-22 | 1999-12-28 | Visible Genetics Inc. | Method and kit for evaluation of HIV mutations |
| WO2000036142A1 (en) * | 1998-12-11 | 2000-06-22 | Visible Genetics Inc. | METHOD AND KIT FOR THE CHARACTERIZATION OF ANTIBIOTIC-RESISTANCE MUTATIONS IN $i(MYCOBACTERIUM TUBERCULOSIS) |
| US6265152B1 (en) | 1995-12-22 | 2001-07-24 | Visible Genetics Inc. | Method and kit for evaluation of HIV mutations |
| WO2000061795A3 (en) * | 1999-04-09 | 2001-08-23 | Innogenetics Nv | Method for the amplification of HLA class I alleles |
| US6830887B2 (en) | 1997-03-18 | 2004-12-14 | Bayer Healthcare Llc | Method and kit for quantitation and nucleic acid sequencing of nucleic acid analytes in a sample |
| US7169560B2 (en) | 2003-11-12 | 2007-01-30 | Helicos Biosciences Corporation | Short cycle methods for sequencing polynucleotides |
| US7220549B2 (en) | 2004-12-30 | 2007-05-22 | Helicos Biosciences Corporation | Stabilizing a nucleic acid for nucleic acid sequencing |
| US7397546B2 (en) | 2006-03-08 | 2008-07-08 | Helicos Biosciences Corporation | Systems and methods for reducing detected intensity non-uniformity in a laser beam |
| US7462449B2 (en) | 1999-06-28 | 2008-12-09 | California Institute Of Technology | Methods and apparatuses for analyzing polynucleotide sequences |
| US7476734B2 (en) | 2005-12-06 | 2009-01-13 | Helicos Biosciences Corporation | Nucleotide analogs |
| US7482120B2 (en) | 2005-01-28 | 2009-01-27 | Helicos Biosciences Corporation | Methods and compositions for improving fidelity in a nucleic acid synthesis reaction |
| US7501245B2 (en) | 1999-06-28 | 2009-03-10 | Helicos Biosciences Corp. | Methods and apparatuses for analyzing polynucleotide sequences |
| WO2007035836A3 (en) * | 2005-09-20 | 2009-06-11 | Univ Oklahoma | Accelerated class i and class ii hla dna sequence-based typing |
| US7635562B2 (en) | 2004-05-25 | 2009-12-22 | Helicos Biosciences Corporation | Methods and devices for nucleic acid sequence determination |
| US7645596B2 (en) | 1998-05-01 | 2010-01-12 | Arizona Board Of Regents | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US7666593B2 (en) | 2005-08-26 | 2010-02-23 | Helicos Biosciences Corporation | Single molecule sequencing of captured nucleic acids |
| WO2011030159A1 (en) | 2009-09-11 | 2011-03-17 | King's College London | Method using hla epitope determination |
| US7981604B2 (en) | 2004-02-19 | 2011-07-19 | California Institute Of Technology | Methods and kits for analyzing polynucleotide sequences |
| US8008002B2 (en) | 2004-03-26 | 2011-08-30 | Qiagen Gmbh | Nucleic acid sequencing |
| US8426129B2 (en) | 1998-04-20 | 2013-04-23 | Innogenetics N.V. | Method for typing HLA alleles |
| US9096898B2 (en) | 1998-05-01 | 2015-08-04 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
Families Citing this family (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5830655A (en) | 1995-05-22 | 1998-11-03 | Sri International | Oligonucleotide sizing using cleavable primers |
| GB9620209D0 (en) | 1996-09-27 | 1996-11-13 | Cemu Bioteknik Ab | Method of sequencing DNA |
| GB9626815D0 (en) | 1996-12-23 | 1997-02-12 | Cemu Bioteknik Ab | Method of sequencing DNA |
| US6379957B1 (en) * | 1998-09-21 | 2002-04-30 | Leslie A. Johnston-Dow | Methods for HIV sequencing and genotyping |
| US6703228B1 (en) | 1998-09-25 | 2004-03-09 | Massachusetts Institute Of Technology | Methods and products related to genotyping and DNA analysis |
| EP1001037A3 (en) * | 1998-09-28 | 2003-10-01 | Whitehead Institute For Biomedical Research | Pre-selection and isolation of single nucleotide polymorphisms |
| US6573047B1 (en) | 1999-04-13 | 2003-06-03 | Dna Sciences, Inc. | Detection of nucleotide sequence variation through fluorescence resonance energy transfer label generation |
| US7655443B1 (en) * | 1999-05-07 | 2010-02-02 | Siemens Healthcare Diagnostics, Inc. | Nucleic acid sequencing with simultaneous quantitation |
| US7058517B1 (en) | 1999-06-25 | 2006-06-06 | Genaissance Pharmaceuticals, Inc. | Methods for obtaining and using haplotype data |
| JP2003521024A (en) * | 1999-06-25 | 2003-07-08 | ジェネサンス・ファーマシューティカルズ・インコーポレーテッド | Methods for obtaining and using haplotype data |
| US6406854B1 (en) * | 1999-09-16 | 2002-06-18 | Mj Research | Method and compositions for evaluating resolution of nucleic acid separation systems |
| US7332275B2 (en) | 1999-10-13 | 2008-02-19 | Sequenom, Inc. | Methods for detecting methylated nucleotides |
| WO2001040520A1 (en) | 1999-12-02 | 2001-06-07 | Dna Sciences, Inc. | Methods for determining single nucleotide variations and genotyping |
| US20010049102A1 (en) * | 2000-02-24 | 2001-12-06 | Huang Xiaohua C. | Methods for determining single nucleotide variations |
| US6760668B1 (en) | 2000-03-24 | 2004-07-06 | Bayer Healthcare Llc | Method for alignment of DNA sequences with enhanced accuracy and read length |
| US6931326B1 (en) | 2000-06-26 | 2005-08-16 | Genaissance Pharmaceuticals, Inc. | Methods for obtaining and using haplotype data |
| US6958214B2 (en) | 2000-07-10 | 2005-10-25 | Sequenom, Inc. | Polymorphic kinase anchor proteins and nucleic acids encoding the same |
| US6397150B1 (en) | 2000-07-27 | 2002-05-28 | Visible Genetics Inc. | Method and apparatus for sequencing of DNA using an internal calibrant |
| GB0021286D0 (en) * | 2000-08-30 | 2000-10-18 | Gemini Genomics Ab | Identification of drug metabolic capacity |
| EP1368497A4 (en) | 2001-03-12 | 2007-08-15 | California Inst Of Techn | METHOD AND DEVICE FOR ANALYZING POLYNUCLEOTIDE SEQUENCES BY ASYNCHRONOUS BASE EXTENSION |
| CA2387277C (en) * | 2001-05-25 | 2015-03-03 | Hitachi, Ltd. | Information processing system using nucleotide sequence-related information |
| US7222059B2 (en) * | 2001-11-15 | 2007-05-22 | Siemens Medical Solutions Diagnostics | Electrophoretic trace simulator |
| US20040267458A1 (en) * | 2001-12-21 | 2004-12-30 | Judson Richard S. | Methods for obtaining and using haplotype data |
| AU2003228809A1 (en) | 2002-05-03 | 2003-11-17 | Sequenom, Inc. | Kinase anchor protein muteins, peptides thereof, and related methods |
| US20030220844A1 (en) * | 2002-05-24 | 2003-11-27 | Marnellos Georgios E. | Method and system for purchasing genetic data |
| EP1396962A1 (en) * | 2002-08-05 | 2004-03-10 | Sony International (Europe) GmbH | Bus service interface |
| EP2112229A3 (en) | 2002-11-25 | 2009-12-02 | Sequenom, Inc. | Methods for identifying risk of breast cancer and treatments thereof |
| EP1773860A4 (en) * | 2004-07-22 | 2009-05-06 | Sequenom Inc | METHODS OF ASSESSING THE RISK OF TYPE II DIABETES APPEARANCE AND RELATED TREATMENTS |
| US20070207490A1 (en) * | 2006-03-06 | 2007-09-06 | Applera Corporation | Method and system for generating sample plate layout for validation |
| EP3260556B1 (en) | 2006-05-31 | 2019-07-31 | Sequenom, Inc. | Methods for the extraction of nucleic acid from a sample |
| EP2236623A1 (en) | 2006-06-05 | 2010-10-06 | Cancer Care Ontario | Assessment of risk for colorectal cancer |
| US7902345B2 (en) | 2006-12-05 | 2011-03-08 | Sequenom, Inc. | Detection and quantification of biomolecules using mass spectrometry |
| WO2008098142A2 (en) | 2007-02-08 | 2008-08-14 | Sequenom, Inc. | Nucleic acid-based tests for rhd typing, gender determination and nucleic acid quantification |
| US20110189663A1 (en) | 2007-03-05 | 2011-08-04 | Cancer Care Ontario | Assessment of risk for colorectal cancer |
| AU2008230813B2 (en) | 2007-03-26 | 2014-01-30 | Sequenom, Inc. | Restriction endonuclease enhanced polymorphic sequence detection |
| WO2009032781A2 (en) | 2007-08-29 | 2009-03-12 | Sequenom, Inc. | Methods and compositions for universal size-specific polymerase chain reaction |
| CA2717320A1 (en) | 2008-03-11 | 2009-09-17 | Sequenom, Inc. | Nucleic acid-based tests for prenatal gender determination |
| WO2009120808A2 (en) | 2008-03-26 | 2009-10-01 | Sequenom, Inc. | Restriction endonuclease enhanced polymorphic sequence detection |
| US8962247B2 (en) | 2008-09-16 | 2015-02-24 | Sequenom, Inc. | Processes and compositions for methylation-based enrichment of fetal nucleic acid from a maternal sample useful for non invasive prenatal diagnoses |
| US8476013B2 (en) | 2008-09-16 | 2013-07-02 | Sequenom, Inc. | Processes and compositions for methylation-based acid enrichment of fetal nucleic acid from a maternal sample useful for non-invasive prenatal diagnoses |
| WO2010075188A2 (en) | 2008-12-23 | 2010-07-01 | Illumina Inc. | Multibase delivery for long reads in sequencing by synthesis protocols |
| IL313131A (en) | 2009-02-11 | 2024-07-01 | Caris Mpi Inc | Molecular profiling of tumors |
| EP3514244B1 (en) | 2009-04-03 | 2021-07-07 | Sequenom, Inc. | Nucleic acid preparation methods |
| CA2779223A1 (en) | 2009-10-27 | 2011-05-12 | Caris Mpi, Inc. | Molecular profiling for personalized medicine |
| EP3088532B1 (en) | 2009-12-22 | 2019-10-30 | Sequenom, Inc. | Processes and kits for identifying aneuploidy |
| WO2012149339A2 (en) | 2011-04-29 | 2012-11-01 | Sequenom, Inc. | Quantification of a minority nucleic acid species |
| HK1206055A1 (en) | 2012-03-02 | 2015-12-31 | Sequenom, Inc. | Methods and processes for non-invasive assessment of genetic variations |
| US9920361B2 (en) | 2012-05-21 | 2018-03-20 | Sequenom, Inc. | Methods and compositions for analyzing nucleic acid |
| US10504613B2 (en) | 2012-12-20 | 2019-12-10 | Sequenom, Inc. | Methods and processes for non-invasive assessment of genetic variations |
| US20140093873A1 (en) | 2012-07-13 | 2014-04-03 | Sequenom, Inc. | Processes and compositions for methylation-based enrichment of fetal nucleic acid from a maternal sample useful for non-invasive prenatal diagnoses |
| US9896728B2 (en) | 2013-01-29 | 2018-02-20 | Arcticrx Ltd. | Method for determining a therapeutic approach for the treatment of age-related macular degeneration (AMD) |
| HK1216655A1 (en) | 2013-03-13 | 2016-11-25 | Sequenom, Inc. | Primers for dna methylation analysis |
| CN105722977A (en) | 2013-10-30 | 2016-06-29 | 绿色生活生物技术有限责任公司 | Pathogenesis quantification systems and treatment methods for citrus greening blight |
| US11365447B2 (en) | 2014-03-13 | 2022-06-21 | Sequenom, Inc. | Methods and processes for non-invasive assessment of genetic variations |
| JP7462632B2 (en) | 2018-11-30 | 2024-04-05 | カリス エムピーアイ インコーポレイテッド | Next-generation molecular profiling |
| WO2021112918A1 (en) | 2019-12-02 | 2021-06-10 | Caris Mpi, Inc. | Pan-cancer platinum response predictor |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4582788A (en) * | 1982-01-22 | 1986-04-15 | Cetus Corporation | HLA typing method and cDNA probes used therein |
| US5171534A (en) * | 1984-01-16 | 1992-12-15 | California Institute Of Technology | Automated DNA sequencing technique |
| US5207880A (en) * | 1984-03-29 | 1993-05-04 | The Board Of Regents Of The University Of Nebraska | DNA sequencing |
| US4865968A (en) * | 1985-04-01 | 1989-09-12 | The Salk Institute For Biological Studies | DNA sequencing |
| US4811218A (en) * | 1986-06-02 | 1989-03-07 | Applied Biosystems, Inc. | Real time scanning electrophoresis apparatus for DNA sequencing |
| US4971903A (en) * | 1988-03-25 | 1990-11-20 | Edward Hyman | Pyrophosphate-based method and apparatus for sequencing nucleic acids |
| US4962020A (en) * | 1988-07-12 | 1990-10-09 | President And Fellows Of Harvard College | DNA sequencing |
| DE3841565C2 (en) * | 1988-12-09 | 1998-07-09 | Europ Lab Molekularbiolog | Methods for sequencing nucleic acids |
| US5547839A (en) * | 1989-06-07 | 1996-08-20 | Affymax Technologies N.V. | Sequencing of surface immobilized polymers utilizing microflourescence detection |
| US5119316A (en) * | 1990-06-29 | 1992-06-02 | E. I. Du Pont De Nemours And Company | Method for determining dna sequences |
| CA2105585A1 (en) * | 1991-03-06 | 1992-09-07 | Pedro Santamaria | Dna sequence-based hla typing method |
| US5424184A (en) * | 1991-05-08 | 1995-06-13 | Regents Of The University Of Minnesota | DNA sequence-based HLA class I typing method |
| US5365455A (en) * | 1991-09-20 | 1994-11-15 | Vanderbilt University | Method and apparatus for automatic nucleic acid sequence determination |
| EP0540997A1 (en) * | 1991-11-05 | 1993-05-12 | F. Hoffmann-La Roche Ag | Methods and reagents for HLA class I DNA typing |
| EP0592060B1 (en) * | 1992-10-09 | 2001-07-18 | The Board of Regents of the University of Nebraska | Digital DNA typing |
-
1995
- 1995-12-22 US US08/577,858 patent/US5834189A/en not_active Expired - Lifetime
-
1996
- 1996-12-19 AU AU14262/97A patent/AU718670B2/en not_active Ceased
- 1996-12-19 CA CA002239896A patent/CA2239896C/en not_active Expired - Fee Related
- 1996-12-19 WO PCT/US1996/020202 patent/WO1997023650A2/en not_active Ceased
- 1996-12-19 DE DE69620791T patent/DE69620791T2/en not_active Expired - Lifetime
- 1996-12-19 JP JP9523796A patent/JP2000502260A/en not_active Abandoned
- 1996-12-19 EP EP96944459A patent/EP0870059B1/en not_active Expired - Lifetime
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6007983A (en) * | 1995-12-22 | 1999-12-28 | Visible Genetics Inc. | Method and kit for evaluation of HIV mutations |
| US6265152B1 (en) | 1995-12-22 | 2001-07-24 | Visible Genetics Inc. | Method and kit for evaluation of HIV mutations |
| US6653107B2 (en) | 1995-12-22 | 2003-11-25 | Visible Genetics Inc. | Method and kit for evaluation of HIV mutations |
| US6830887B2 (en) | 1997-03-18 | 2004-12-14 | Bayer Healthcare Llc | Method and kit for quantitation and nucleic acid sequencing of nucleic acid analytes in a sample |
| WO1999016910A1 (en) * | 1997-09-26 | 1999-04-08 | Visible Genetics Inc. | Method and kit for evaluation of hiv mutations |
| AU751471B2 (en) * | 1997-09-26 | 2002-08-15 | Bayer Healthcare Llc | Method and kit for evaluation of HIV mutations |
| US8426129B2 (en) | 1998-04-20 | 2013-04-23 | Innogenetics N.V. | Method for typing HLA alleles |
| US10208341B2 (en) | 1998-05-01 | 2019-02-19 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US10214774B2 (en) | 1998-05-01 | 2019-02-26 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US9957561B2 (en) | 1998-05-01 | 2018-05-01 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US9725764B2 (en) | 1998-05-01 | 2017-08-08 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US9458500B2 (en) | 1998-05-01 | 2016-10-04 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US9212393B2 (en) | 1998-05-01 | 2015-12-15 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US9096898B2 (en) | 1998-05-01 | 2015-08-04 | Life Technologies Corporation | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| US7645596B2 (en) | 1998-05-01 | 2010-01-12 | Arizona Board Of Regents | Method of determining the nucleotide sequence of oligonucleotides and DNA molecules |
| WO2000036142A1 (en) * | 1998-12-11 | 2000-06-22 | Visible Genetics Inc. | METHOD AND KIT FOR THE CHARACTERIZATION OF ANTIBIOTIC-RESISTANCE MUTATIONS IN $i(MYCOBACTERIUM TUBERCULOSIS) |
| WO2000061795A3 (en) * | 1999-04-09 | 2001-08-23 | Innogenetics Nv | Method for the amplification of HLA class I alleles |
| EP2314715A3 (en) * | 1999-04-09 | 2012-04-18 | Innogenetics N.V. | Method for the amplification of HLA class I alleles |
| US7462449B2 (en) | 1999-06-28 | 2008-12-09 | California Institute Of Technology | Methods and apparatuses for analyzing polynucleotide sequences |
| US7501245B2 (en) | 1999-06-28 | 2009-03-10 | Helicos Biosciences Corp. | Methods and apparatuses for analyzing polynucleotide sequences |
| US7169560B2 (en) | 2003-11-12 | 2007-01-30 | Helicos Biosciences Corporation | Short cycle methods for sequencing polynucleotides |
| US9657344B2 (en) | 2003-11-12 | 2017-05-23 | Fluidigm Corporation | Short cycle methods for sequencing polynucleotides |
| US9012144B2 (en) | 2003-11-12 | 2015-04-21 | Fluidigm Corporation | Short cycle methods for sequencing polynucleotides |
| US7491498B2 (en) | 2003-11-12 | 2009-02-17 | Helicos Biosciences Corporation | Short cycle methods for sequencing polynucleotides |
| US7981604B2 (en) | 2004-02-19 | 2011-07-19 | California Institute Of Technology | Methods and kits for analyzing polynucleotide sequences |
| US8008002B2 (en) | 2004-03-26 | 2011-08-30 | Qiagen Gmbh | Nucleic acid sequencing |
| US7635562B2 (en) | 2004-05-25 | 2009-12-22 | Helicos Biosciences Corporation | Methods and devices for nucleic acid sequence determination |
| US7220549B2 (en) | 2004-12-30 | 2007-05-22 | Helicos Biosciences Corporation | Stabilizing a nucleic acid for nucleic acid sequencing |
| US7482120B2 (en) | 2005-01-28 | 2009-01-27 | Helicos Biosciences Corporation | Methods and compositions for improving fidelity in a nucleic acid synthesis reaction |
| US7666593B2 (en) | 2005-08-26 | 2010-02-23 | Helicos Biosciences Corporation | Single molecule sequencing of captured nucleic acids |
| US9868978B2 (en) | 2005-08-26 | 2018-01-16 | Fluidigm Corporation | Single molecule sequencing of captured nucleic acids |
| WO2007035836A3 (en) * | 2005-09-20 | 2009-06-11 | Univ Oklahoma | Accelerated class i and class ii hla dna sequence-based typing |
| US7476734B2 (en) | 2005-12-06 | 2009-01-13 | Helicos Biosciences Corporation | Nucleotide analogs |
| US7397546B2 (en) | 2006-03-08 | 2008-07-08 | Helicos Biosciences Corporation | Systems and methods for reducing detected intensity non-uniformity in a laser beam |
| WO2011030159A1 (en) | 2009-09-11 | 2011-03-17 | King's College London | Method using hla epitope determination |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2239896A1 (en) | 1997-07-03 |
| AU718670B2 (en) | 2000-04-20 |
| EP0870059A2 (en) | 1998-10-14 |
| WO1997023650A3 (en) | 1997-08-21 |
| JP2000502260A (en) | 2000-02-29 |
| US5834189A (en) | 1998-11-10 |
| DE69620791D1 (en) | 2002-05-23 |
| DE69620791T2 (en) | 2002-08-14 |
| AU1426297A (en) | 1997-07-17 |
| CA2239896C (en) | 2004-11-30 |
| EP0870059B1 (en) | 2002-04-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0870059B1 (en) | Method for evaluation of polymorphic genetic sequences, and the use thereof in identification of hla types | |
| EP0907752B1 (en) | Method for determination of nucleic acid sequences and diagnostic applications thereof | |
| EP0333465B1 (en) | Mutation detection by competitive oligonucleotide priming | |
| US5593830A (en) | DNA sequence-based HLA class I typing method | |
| AU2003247715B8 (en) | Methods and compositions for analyzing compromised samples using single nucleotide polymorphism panels | |
| EP0914468B1 (en) | Method for sequencing of nucleic acid polymers | |
| JP2001520020A (en) | Methods and kits for amplifying, sequencing and typing classical HLA class I genes - Patents.com | |
| AU8846898A (en) | Method and kit for hla class i typing dna | |
| KR950010188B1 (en) | Detection of Novel Gaucher Disease Mutations in Intron 2 of the Glucocerebrosidase Gene | |
| US6015675A (en) | Mutation detection by competitive oligonucleotide priming | |
| US20040014101A1 (en) | Separating and/or identifying polymorphic nucleic acids using universal bases | |
| US20020018999A1 (en) | Methods for characterizing polymorphisms | |
| US6265152B1 (en) | Method and kit for evaluation of HIV mutations | |
| US6007983A (en) | Method and kit for evaluation of HIV mutations | |
| AU751471B2 (en) | Method and kit for evaluation of HIV mutations | |
| EP0910662B1 (en) | Methods and reagents for typing hla class i genes | |
| WO1998026091A2 (en) | Method and kit for hla class i typing | |
| US6413718B1 (en) | Method for sequencing of nucleic acid polymers | |
| JP2022129868A (en) | Methods and reagents for detecting genetic factors of immune responsiveness to hepatitis B vaccine | |
| JP2010246400A (en) | Polymorphism identification method | |
| US20110117547A1 (en) | Target dna detection method and target dna detection kit | |
| GB2395787A (en) | Method and kit for evaluation of HIV mutations | |
| WO2010008809A2 (en) | Compositions and methods for early stage sex determination | |
| JP2005198525A (en) | Method for detecting glutathione s-transferase gene polymorphism and kit for detection | |
| HK1035004A (en) | Detection of sequence variation of nucleic acid by shifted termination analysis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE HU IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG |
|
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE HU IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2239896 Country of ref document: CA Ref country code: CA Ref document number: 2239896 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 1997 523796 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996944459 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996944459 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1996944459 Country of ref document: EP |