WO1997026258A1 - Angiogenesis inhibiting pyridazinamines - Google Patents
Angiogenesis inhibiting pyridazinamines Download PDFInfo
- Publication number
- WO1997026258A1 WO1997026258A1 PCT/EP1997/000201 EP9700201W WO9726258A1 WO 1997026258 A1 WO1997026258 A1 WO 1997026258A1 EP 9700201 W EP9700201 W EP 9700201W WO 9726258 A1 WO9726258 A1 WO 9726258A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- alk
- phenyl
- 6alkyl
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *CN(C*)c1c(*)c(*)c(-c2nc(*)n[s]2)nn1 Chemical compound *CN(C*)c1c(*)c(*)c(-c2nc(*)n[s]2)nn1 0.000 description 3
- NWPWRAWAUYIELB-UHFFFAOYSA-N CCOC(c1ccc(C)cc1)=O Chemical compound CCOC(c1ccc(C)cc1)=O NWPWRAWAUYIELB-UHFFFAOYSA-N 0.000 description 1
- ZWULKULQMNOJHS-UHFFFAOYSA-N CCc1n[o]c(-c2ccc(C)cc2)n1 Chemical compound CCc1n[o]c(-c2ccc(C)cc2)n1 ZWULKULQMNOJHS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- This invention concerns 3-(3-substituted-l,2,4-thiadiazol-5-yl)pyridazinamines acting as angiogenesis inhibitors, and their preparation; it further relates to compositions comprising them, as well as their use as a medicine.
- Angiogenesis i.e. the formation of new vessels by endothelial cells, plays an important role in a variety of physiologic and pathophysiologic processes.
- the development of a vascular supply is essential for the growth, maturation and maintenance of normal tissues. It is also required for wound healing.
- angiogenesis is also critical for solid tumor growth and metastasis and is involved in a variety of other pathological conditions such as neovascular glaucoma, diabetic retinopathy, psoriasis and rheumatoid arthritis.
- pathological states are characterized by augmented angiogenesis during which normally quiescent endothelial cells become activated, degrade extracellular matrix barriers, proliferate, and migrate to form new vessels.
- compounds with angiogenesis inhibitory properties would be very useful.
- hydrocortisone is a well known angiogenesis inhibitor (Folkman et al., Science 230:1375, 1985' "A new class of steroids inhibits angiogenesis in the presence of heparin or a heparin fragment”; Folkman et al., Science 221 :719, 1983, "Angiogenesis inhibition and tumor regression caused by heparin or a heparin fragment in the presence of cortisone").
- pyridazinamines are described having antipicornaviral activity.
- the compounds of the present invention differ from the cited pyridazinamines by the fact that they are invariably substituted with a thiadiazolyl moiety and particularly by the fact that unexpectedly these compounds have angiogenesis inhibiting properties.
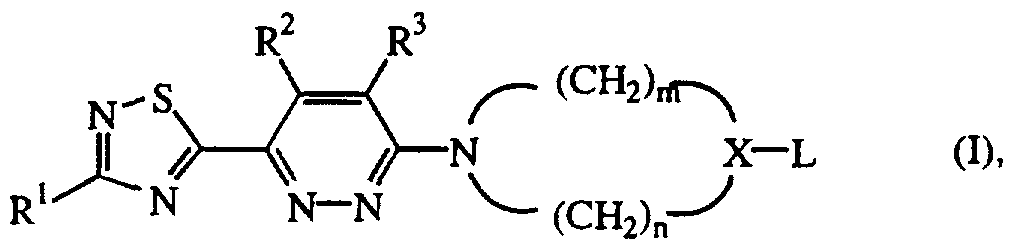
- This invention concerns compounds of formula
- ⁇ -(CH 2 ) n ⁇ also contain one double bond, may be substituted with Chalky!, amino, aminocarbonyl, mono- or di(C ⁇ _6alkyl)amino, C ⁇ _6alkyloxycarbonyl,
- R 1 is hydrogen, Chalky!, Ci ⁇ alkyloxy, C ⁇ .6alkylthio, amino, mono- or di(Ci-6alkyl)amino, Ar, Ar ⁇ H-, C3_6cycloalkyl, hydroxymethyl or benzyloxymethyl;
- R 2 and R 3 are hydrogen, or taken together may form a bivalent radical of formula
- L 1 is Ar-C ⁇ _6alkyloxy, Ar-oxy, Ar-thio, Ar-carbonylamino, di-Ar-methyloxy-,
- L 3 is Ci-6alkyl substituted with one or two radicals selected from Ar, Ar-oxy, or
- Ar-piperidinyl or Ar-NR 4 - Alk-; R 4 and R 5 are each independently selected from hydrogen or C ⁇ _6alkyl; each Ar is independently selected from phenyl; phenyl substituted with 1, 2 or 3 substituents each independently selected from halo, amino, nitro, Cj. 6 alkyl, trihalomethyl, C ⁇ .
- heterocycles in the definition of Het are preferably connected to the rest of the molecule by a carbon atom.
- 2-Benzimidazolinonyl is preferably connected to the rest of the molecule by a nitrogen atom.
- the Ar group is situated on the nitrogen atom of the piperazinyl or homopiperazinyl moiety.
- halo is generic to fluoro, chloro, bromo and iodo
- C ⁇ _4alkyl defines straight and branched chain saturated hydrocarbon radicals having from 1 to 4 carbon atoms such as, for example, methyl, ethyl, propyl, butyl, 1-methylethyl, 2-methylpropyl and the like
- Ci ⁇ alkyl is meant to include Ci-4alkyl and the higher homologues thereof having 5 to 6 carbon atoms such as, for example, pentyl, 2-methylbutyl, hexyl, 2-methylpentyl and the like
- C 2 -4alkanediyl defines bivalent straight and branched chain saturated hydrocarbon radicals having from 2 to 4 carbon atoms such as, for example, 1,2-ethanediyl, 1,3-propanediyl,
- Ci- ⁇ alkanediyl defines bivalent straight and branched chain saturated hydrocarbon radicals having from 1 to 6 carbon atoms such as, for example, methylene, 1,2-ethanediyl, 1,3-propanediyl, 1,4-butanediyl, 1,5-pentanediyl, 1,6-hexanediyl and the like.
- C3-6cycloalkyl comprises cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- the pharmaceutically acceptable acid addition salts as mentioned hereinabove are meant to comprise the therapeutically active non-toxic acid addition salt forms which the compounds of formula (I) are able to form.
- Said salts can conveniently be obtained by treating the base form of the compounds of formula (I) with appropriate acids such as, for example, inorganic acids such as hydrohalic acids, e.g.
- hydrochloric or hydrobromic acid sulfuric; nitric; phosphoric and the like acids; or organic acids such as, for example, acetic, propanoic, hydroxyacetic, lactic, pyruvic, oxalic, malonic, succinic, maleic, fumaric, malic, tartaric, citric, methanesulfonic, ethanesulfonic, benzenesulfonic, p-toluenesulfonic, cyclamic, salicylic, p-aminosalicylic, pamoic and the like acids.
- organic acids such as, for example, acetic, propanoic, hydroxyacetic, lactic, pyruvic, oxalic, malonic, succinic, maleic, fumaric, malic, tartaric, citric, methanesulfonic, ethanesulfonic, benzenesulfonic, p-toluenesulfonic, cycl
- salt forms can be converted by treatment with an appropriate base into the free base form.
- acid addition salt as used hereinabove also comprises the solvates which the compounds of formula (I) as well as the salts thereof, are able to form.
- solvates are for example hydrates, alcoholates and the like.
- stereochemically isomeric forms as used hereinbefore defines all the possible isomeric as well as conformational forms which the compounds of formula (I) may possess.
- chemical designation of compounds denotes the mixture, more in particular the racemic mixture, of all possible stereochemically isomeric forms, said mixtures containing all diastereomers and/or enantiomers of the basic molecular structure.
- stereogenic centers may have the R- or S-configuration; substituents on bivalent cyclic saturated radicals may have either the cis- or tr ⁇ ns-configuration.
- the relative stereodesc ⁇ ptors R and S are used in accordance with the Chemical Abstracts rules (Chemical Substance Name Selection Manual (CA), 1982 Edition, Vol. HI, Chapter 20). All stereochemically isomeric forms of the compounds of formula (I) both in pure form or mixtures thereof are intended to be embraced within the scope of the present invention.
- the N-oxide forms of the compounds of formula (I) are meant to comprise those compounds of formula (I) wherein one or several nitrogen atoms are oxidized to the so-called N-oxide, particularly those N-oxides wherein one or more of the piperidine-, piperazine or pyridazinyl-nitrogens are N-oxidized.
- X is CH or ⁇ , and the CH2 groups of the — ⁇ X — moiety are unsubstituted.
- Another group of interesting compounds consists of those compounds of formula (I) wherein R 1 is C ⁇ _6alkyl, X is CH and m is 2 and n is 2.
- Still another group of interesting compounds consists of those compounds of formula
- a particular group of compounds are those compounds of formula (I) wherein R 1 is hydrogen or di(C ⁇ _6alkyl)amino.
- Preferred compounds are those compounds of formula (I) wherein R 1 is methyl, R 2 and R 3 are hydrogen, X is CH or ⁇ and L is Ar-piperidinyl or a phenyl substituted with 1, 2 or 3 substituents each independently selected from halo or trifluoromethyl.
- More particular compounds are those compounds of formula (I) wherein R 1 is hydrogen or di(C ⁇ _6alkyl)amino, R 2 and R 3 are hydrogen, X is CH or ⁇ , m is 2, n is 2.
- the compounds of the present invention can generally be prepared by reacting a pyridazine of formula (II) with an amine of formula (HI), following art-known N-alkylation procedures.
- W represents an appropriate reactive leaving group such as, for example, halo, e.g. fluoro, chloro, bromo, iodo, or in some instances W may also be a sulfonyloxy group, e.g. methanesulfonyloxy, benzenesulfonyloxy, trifluoromethanesulfonyloxy and the like reactive leaving groups.
- Said reaction is performed following art-known procedures such as for instance stirring and heating both reactants together in a reaction-inert solvent , e.g. dimethyiformamide, preferably in the presence of a base, e.g. sodiumcarbonate.
- said N-alkylation reaction may be carried out by applying art-known conditions of phase transfer catalysis reactions.
- Said conditions comprise stirring the reactants, with an appropriate base and optionally under an inert atmosphere such as, for example, oxygen-free argon or nitrogen gas, in the presence of a suitable phase transfer catalyst such as, for example, a trialkylphenylmethylammonium, tetraalkylammonium, tetraalkylphosphonium, tetraarylphosphonium halide, hydroxide and the like catalysts.
- phase transfer catalyst such as, for example, a trialkylphenylmethylammonium, tetraalkylammonium, tetraalkylphosphonium, tetraarylphosphonium halide, hydroxide and the like catalysts.
- the compounds of formula (I) wherein X is ⁇ and L is L 3 , said compounds being represented by formula (I-a) can also be prepared by N-alkylating a substituted piperazine of formula (IV) with a intermediate of formula (V), following similar procedures as described hereinbefore for the preparation of (I) starting from (II) and (HI).
- Said reductive N-alkylation may be performed in a reaction-inert solvent such as, for example, dichloromethane, ethanol, toluene or a mixture thereof, and in the presence of a reducing agent such as, for example, a borohydride, e.g. sodium borohydride, sodium cyanoborohydride or triacetoxy borohydride.
- a reducing agent such as, for example, a borohydride, e.g. sodium borohydride, sodium cyanoborohydride or triacetoxy borohydride.
- hydrogen as a reducing agent in combination with a suitable catalyst such as, for example, palladium-on-charcoal or platinum-on-charcoal.
- it may be advantageous to add a dehydrating agent to the reaction mixture such as, for example, aluminium rt-butoxide.
- an appropriate catalyst-poison to the reaction mixture, e.g., thiophene or quinoline-sulphur. Stirring and optionally elevated temperatures and/or pressure may enhance the rate of the reaction.
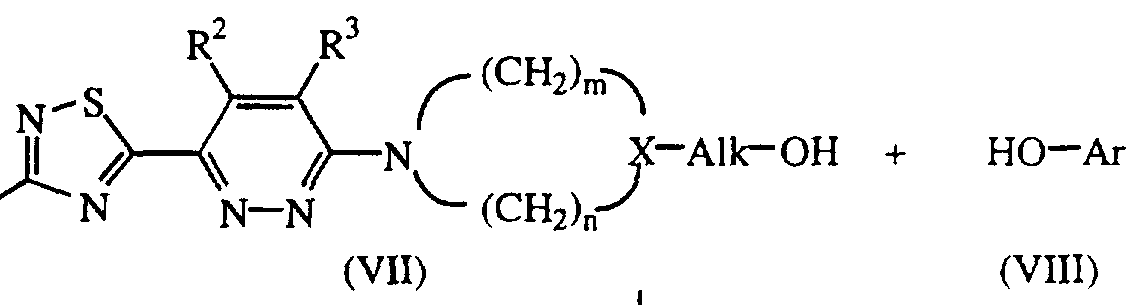
- the compounds of formula (I) wherein L is Ar-O-Ci-6alkyl-, said compounds being represented by formula (I-b), may also be prepared by condensing a phenol of formula (V ⁇ i) and an intermediate of formula (VII), e.g. by using the Mitsunobu reaction (Synthesis, 1, 1981).
- the compounds of formula (I-b) can also be prepared following art-known O-alkylation reactions by alkylating a phenol of formula (VHI) with a pyridazinamine derivative of formula (IX).
- Said O-alkylation reaction can conveniently be carried out by mixing the reactants, optionally in a reaction- inert solvent such as, for example, water; an aromatic solvent, e.g. benzene, methylbenzene and the like; a C ⁇ _6alkanol, e.g. methanol, ethanol and the like; a ketone, e.g. 2-propanone, 4-methyl-2-pentanone and the like; an ester, e.g. ethylacetate, ⁇ -butyrolactone and the like; an ether, e.g.
- a reaction- inert solvent such as, for example, water; an aromatic solvent, e.g. benzene, methylbenzene and the like; a C ⁇ _6alkanol, e.g. methanol, ethanol and the like; a ketone, e.g. 2-propanone, 4-methyl-2-pentanone and the like; an ester, e.
- a base such as, e.g. sodium carbonate, sodium hydrogen carbonate, sodium hydroxide and the like, or an organic base such as, for example, a tertiary amine, e.g.
- NN-diethylethanamine, N-ethyl-N-(l -methyl ethyl)-2-propanamine and the like may optionally be used to pick up the acid which is formed during the course of the reaction.
- a suitable salt form thereof such as, for example, an alkali or earth alkaline metal salt
- Stirring and somewhat elevated temperatures may enhance the rate of the reaction; more in particular the reaction may be conducted at the reflux temperature of the reaction mixture.
- said O-alkylation reaction may be carried out by applying art-known conditions of phase transfer catalysis reactions as described hereinbefore.
- Appropriate solvents are, for example, hydrocarbons, e.g. hexane, heptane, cyclo- hexane and the like; ethers, e.g. diethyl ether, tetrahydrofuran, 1 ,2-dimethoxyethane and the like; dipolar aprotic solvents, e.g. dimethylsulfoxide, hexamethyl-phosphor triamide, and the like.
- hydrocarbons e.g. hexane, heptane, cyclo- hexane and the like
- ethers e.g. diethyl ether, tetrahydrofuran, 1 ,2-dimethoxyethane and the like
- dipolar aprotic solvents e.g. dimethylsulfoxide, hexamethyl-phosphor triamide, and the like.
- the unsaturated intermediates (XII) can be reduced following an appropriate reduction procedure, for example, by stirring and, if desired, heating the unsaturated intermediates in a suitable reaction-inert solvent in the presence in the presence of a reducing agent such as, for example, a borohydride, e.g. sodium borohydride, sodium cyanoborohydride or triacetoxy borohydride.
- a reducing agent such as, for example, a borohydride, e.g. sodium borohydride, sodium cyanoborohydride or triacetoxy borohydride.
- Suitable solvents are alkanols, e.g. methanol, ethanol and the like, and carboxylic acids, e.g. acetic acid and the like.
- the intermediate ylides of formulae (XI) can be obtained by treating a phosphonium salt or a phosphonate with an appropriate base such as, for example, potassium tert- butoxide, methyllithium, butyllithium, sodium amide, sodium hydride, sodium alkoxide and the like bases under an inert atmosphere and in a reaction-inert solvent such as, for example, an ether, e.g. tetrahydrofuran, 1,4-dioxane and the like.
- an appropriate base such as, for example, potassium tert- butoxide, methyllithium, butyllithium, sodium amide, sodium hydride, sodium alkoxide and the like bases under an inert atmosphere and in a reaction-inert solvent such as, for example, an ether, e.g. tetrahydrofuran, 1,4-dioxane and the like.
- the compounds of formula (I) may further be prepared by converting compounds of formula (I) into each other according to art-known group transformation reactions. For instance, compounds of formula (I) bearing a phenyl moiety may be submitted to a bromination reaction with N-bromosuccinimide in a reaction-inert solvent to introduce a bromine atom on the phenyl moiety. Also, compounds of formula (I) may be hydrolysed under acidic conditions.
- the compounds of formula (I) may also be converted to the corresponding N-oxide forms following art-known procedures for converting a trivalent nitrogen into its N-oxide form.
- Said N-oxidation reaction may generally be carried out by reacting the starting material of formula (I) with an appropriate organic or inorganic peroxide.
- Appropriate inorganic peroxides comprise, for example, hydrogen peroxide, alkali metal or earth alkaline metal peroxides, e.g. sodium peroxide, potassium peroxide;
- appropriate organic peroxides may comprise peroxy acids such as, for example, benzenecarboperoxoic acid or halo substituted benzenecarboperoxoic acid, e.g.
- 3-chlorobenzenecarboperoxoic acid peroxoalkanoic acids, e.g. peroxoacetic acid, alkylhydroperoxides, e.g. tert-butyl hydroperoxide.
- Suitable solvents are, for example, water, lower alkanols, e.g. ethanol and the like, hydrocarbons, e.g. toluene, ketones, e.g. 2-butanone, halogenated hydrocarbons, e.g. dichloromethane, and mixtures of such solvents.
- Intermediates of formula (II) may be prepared by reacting compounds of formula (XIII), wherein W is an appropriate leaving group as defined above, with an intermediate of formula (XIV), optionally added as its acid addition salt.
- intermediates of formula (VTI) may be prepared in an analogous way as compounds of formula (I) by reacting an intermediate of formula (II), wherein W is appropriate leaving group as defined above, with an intermediate of formula (XV),
- reaction-inert solvent e.g. dimethyiformamide
- base e.g. sodiumcarbonate
- Compounds of formula (I) and some of the intermediates may have one or more stereogenic centers in their structure, present in a R or a S configuration.
- the compounds of formula (I) as prepared in the hereinabove described processes may be synthesized in the form of racemic mixtures of enantiomers which can be separated from one another following art-known resolution procedures.
- the racemic compounds of formula (I) may be converted into the corresponding diastereomeric salt forms by reaction with a suitable chiral acid. Said diastereomeric salt forms are subsequently separated, for example, by selective or fractional crystallization and the enantiomers are liberated therefrom by alkali.
- An alternative manner of separating the enantiomeric forms of the compounds of formula (I) involves liquid chromatography using a chiral stationary phase.
- Said pure stereochemically isomeric forms may also be derived from the corresponding pure stereochemically isomeric forms of the appropriate starting materials, provided that the reaction occurs stereospecifically.
- said compound will be synthesized by stereospecific methods of preparation. These methods will advantageously employ enantiomerically pure starting materials.
- the compounds of formula (I) have valuable pharmacological properties in that they inhibit angiogenesis, both in vivo and in vitro.
- angiogenesis inhibitors are useful to control or treat angiogenesis dependent disorders such as, e.g. ocular neovascular diseases, neovascular glaucoma, diabetic retinopathy, retrolental fibroplasia, hemangiomas, angiofibromas, psoriasis and rheumatoid arthritis.
- angiogenesis inhibitors are useful to control solid tumor growth, such as, e.g. breast, prostate, melanoma, renal, colon, cervival cancer and the like; and metastasis.
- the present invention thus also relates to compounds of formula (I) as defined hereinabove for use as a medicine.
- the present invention provides a method of treating warm-blooded animals suffering from such disorders, said method comprising the systemic administration of a therapeutic effective amount of a compound of formula (I), a N-oxide or a pharmaceutically acceptable acid addition salt thereof.
- the subject compounds may be formulated into various pharmaceutical forms for administration purposes.
- compositions of this invention an effective amount of a particular compound, in base or acid addition salt form, as the active ingredient is combined in intimate admixture with a pharmaceutically acceptable carrier, which carrier may take a wide variety of forms depending on the form of preparation desired for administration.
- a pharmaceutically acceptable carrier which carrier may take a wide variety of forms depending on the form of preparation desired for administration.
- These pharmaceutical compositions are desirably in unitary dosage form suitable, preferably, for administration orally, rectally, percutaneously, or by parenteral injection.
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols and the like in the case of oral liquid preparations such as suspensions, syrups, elixirs and solutions; or solid carriers such as starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like in the case of powders, pills, capsules and tablets. Because of their ease in administration, tablets and capsules represent the most advantageous oral dosage unit form, in which case solid pharmaceutical carriers are obviously employed.
- the carrier will usually comprise sterile water, at least in large part, though other ingredients, to aid solubility for example, may be included.
- Injectable solutions may be prepared in which the carrier comprises saline solution, glucose solution or a mixture of saline and glucose solution. Injectable suspensions may also be prepared in which case appropriate liquid carriers, suspending agents and the like may be employed.
- the carrier optionally comprises a penetration enhancing agent and/or a suitable wetting agent, optionally combined with suitable additives of any nature in minor proportions, which additives do not cause a significant deleterious effect to the skin. Said additives may facilitate the administration to the skin and/or may be helpful for preparing the desired compositions.
- These compositions may be administered in various ways, e.g., as a transdermal patch, as a spot-on, as an ointment.
- Dosage unit form as used in the specification and claims herein refers to physically discrete units suitable as unitary dosages, each unit containing a predetermined quantity of active ingredient calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- dosage unit forms are tablets (including scored or coated tablets), capsules, pills, powder packets, wafers, injectable solutions or suspensions, teaspoonfuls, tablespoonfuls and the like, and segregated multiples thereof.
- an effective amount would be from lxlO -5 mg/kg to 10 mg/kg body weight, and in particular from 0.001 mg/kg to 1 mg/kg body weight. It may be appropriate to administer the required dose as two, three, four or more sub-doses at appropriate intervals throughout the day. Said sub-doses may be formulated as unit dosage forms, for example, containing 0.001 to 500 mg, and in particular 0.01 mg to 200 mg of active ingredient per unit dosage form.
- DMF means NN-dimethylformamide
- DCM dichloromethane
- DIPE diisopropylether
- THF tetrahydrofuran
- Example A.1 A mixture of 3-chloro-6-methylpyridazine (12.9 g) and thionyl chloride (119 g) was stirred for 1 night at reflux temperature. The mixture was evaporated, 200 ml of DCM was added and the mixture was cooled till -5°C. 1-Imino-ethanamine hydrochloride (9.5 g) was added portionwise and the whole was stirred for 15 minutes. Then, sodium hydroxide (50%) (25 ml) was added dropwise (temp. ⁇ 5°C). The mixture was stirred till room temperature was reached. After stirring for 30 minutes, the layers were separated. The organic layer was dried, filtered and evaporated.
- Example A.2 a) A mixture of 3-chloro-6-methylpyridazine (19.3 g) in thionyl chloride (160 ml) was stirred and refluxed overnight. The mixture was evaporated, toluene (100 ml) was added and evaporated again, yielding 40 g (100%) of ⁇ , ⁇ ,3-trichloro-6-pyridazine- methanesulfenyl chloride (intermediate 2-a). b) A mixture of intermediate (2-a) (79.2 g) in DCM (600 ml) was stirred at 0°C. 2-(Phenylmethoxy)-e_hanimidamide (40.1 g) was added. NaOH (50%, 60ml) was added dropwise at 0°C. The mixture was cooled and stirred at 0°C for 1 hour. Water
- Table 1 lists intermediates that were prepared analogously.
- Tables 2 to 1 1 list the compounds that were prepared according to one of the above examples and table 12 lists both the experimental (column heading “exp.”) and theoretical (column heading “theor.”) elemental analysis values for carbon, hydrogen and nitrogen of the compounds as prepared in the experimental part hereinabove..
- Angiogenesis inhibitory activity was measured in vitro using the rat aortic ring model of angiogenesis as described by Nicosia, R.F. and Ottinetti in "Laboratory Investigation", vol. 63, p. 115, 1990.
- the ability of compounds to inhibit microvessel formation was compared to vehicle-treated control rings.
- Quantitation (microvessel area) following eight days in culture was performed using an image analysis system, consisting of a light microscope, a CCD camera and an automated, custom-designed image analysis program as described by Nissanov, J., Tuman, R.W., Gruver, L.M., and Fortunato, J.M. in "Laboratory Investigation", vol 73 (#5), p. 734, 1995.
- Compounds were tested at several concentrations for determination of inhibitory potency (IC50's). Several compounds, as listed in table 13, have an IC50 value lower than 1 nM. Table 13 :
- the in vivo angiogenesis inhibitory activity was measured using the Matrigel model, as described in US-5, 382,514. Briefly, a liquid, containing extracts of murine basement membrane and an angiogenic growth factor (e.g. VEGF, bFGF, aFGF), was injected in a warm-blooded animal where it forms a gel matrix. After a period of time, the gel is recovered from the animal and angiogenesis is quantitated. The test compounds were administered orally at a dose of 0.1 mg kg. Compounds 2, 30, 31, 47, 53, 54, 74, 75, 82, 88, 134, 138 and 177 were found to inhibit angiogenesis by more than 70%.
- an angiogenic growth factor e.g. VEGF, bFGF, aFGF
- Active ingredient as used throughout these examples relates to a compound of formula (I), a N-oxide form, a pharmaceutically acceptable acid addition salt or a stereochemically isomeric form thereof.
- the resulting solution is filled in suitable containers.
- Example D.3 Film-coated tablets
- a mixture of 100 g of the A.L, 570 g lactose and 200 g starch is mixed well and thereafter humidified with a solution of 5 g sodium dodecyl sulfate and 10 g polyvinyl- pyrrolidone in about 200 ml of water.
- the wet powder mixture is sieved, dried and sieved again.
- 100 g microcrystalline cellulose and 15 g hydrogenated vegetable oil The whole is mixed well and compressed into tablets, giving 10.000 tablets, each comprising 10 mg of the active ingredient.
- Example D.4 Injectable solution 1.8 g methyl 4-hydroxybenzoate and 0.2 g propyl 4-hydroxybenzoate were dissolved in about 0.5 1 of boiling water for injection. After cooling to about 50°C there were added while stirring 4 g lactic acid, 0.05 g propylene glycol and 4 g of the A.L The solution was cooled to room temperature and supplemented with water for injection q.s. ad 1 1 volume, giving a solution of 4 mg/ml of A.L The solution was sterilized by filtration and filled in sterile containers.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Cardiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Heart & Thoracic Surgery (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Abstract
Description








































Claims









Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU14439/97A AU717744B2 (en) | 1996-01-15 | 1997-01-14 | Angiogenesis inhibiting pyridazinamines |
| SI9730191T SI0876366T1 (en) | 1996-01-15 | 1997-01-14 | Angiogenesis inhibiting pyridazinamines |
| AT97901059T ATE203534T1 (en) | 1996-01-15 | 1997-01-14 | ANGIOGENESIS INHIBITING PYRIDAZINAMINES |
| JP52465697A JP4169368B2 (en) | 1996-01-15 | 1997-01-14 | Angiogenesis inhibitory pyridazineamine |
| DE69705819T DE69705819T2 (en) | 1996-01-15 | 1997-01-14 | ANGIOGENESIS INHIBITING PYRIDAZINE AMINES |
| DK97901059T DK0876366T3 (en) | 1996-01-15 | 1997-01-14 | Angiogenesis-inhibiting pyridazine amines |
| NZ326354A NZ326354A (en) | 1996-01-15 | 1997-01-14 | 3-(substituted-1-piperazinyl)-6-(substituted-1,2,4-thiadiazol-5 -yl)-pyridazine derivatives and medicaments |
| EP97901059A EP0876366B1 (en) | 1996-01-15 | 1997-01-14 | Angiogenesis inhibiting pyridazinamines |
| IL12446197A IL124461A (en) | 1996-01-15 | 1997-01-14 | Angiogenesis inhibiting 3-(1,2,4-thiadiazol-5-yl)piperazine pyridazine derivatives their preparation and pharmaceutical compositions containing them |
| CA002237273A CA2237273C (en) | 1996-01-15 | 1997-01-14 | Angiogenesis inhibiting pyridazinamines |
| NO982037A NO309653B1 (en) | 1996-01-15 | 1998-05-05 | Autiogenesis inhibiting pyridazine amines |
| GR20010401770T GR3036900T3 (en) | 1996-01-15 | 2001-10-16 | Angiogenesis inhibiting pyridazinamines |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP96200085.7 | 1996-01-15 | ||
| EP96200085 | 1996-01-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997026258A1 true WO1997026258A1 (en) | 1997-07-24 |
Family
ID=8223585
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1997/000201 Ceased WO1997026258A1 (en) | 1996-01-15 | 1997-01-14 | Angiogenesis inhibiting pyridazinamines |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US5985878A (en) |
| EP (1) | EP0876366B1 (en) |
| JP (1) | JP4169368B2 (en) |
| KR (1) | KR100443893B1 (en) |
| CN (1) | CN1104430C (en) |
| AT (1) | ATE203534T1 (en) |
| AU (1) | AU717744B2 (en) |
| CA (1) | CA2237273C (en) |
| DE (1) | DE69705819T2 (en) |
| DK (1) | DK0876366T3 (en) |
| ES (1) | ES2162235T3 (en) |
| GR (1) | GR3036900T3 (en) |
| IL (1) | IL124461A (en) |
| MY (1) | MY117098A (en) |
| NO (1) | NO309653B1 (en) |
| NZ (1) | NZ326354A (en) |
| PT (1) | PT876366E (en) |
| SI (1) | SI0876366T1 (en) |
| TW (1) | TW480256B (en) |
| WO (1) | WO1997026258A1 (en) |
| ZA (1) | ZA97288B (en) |
Cited By (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998035958A1 (en) * | 1997-02-13 | 1998-08-20 | Novartis Ag | Phthalazines with angiogenesis inhibiting activity |
| WO1998058919A1 (en) * | 1997-06-24 | 1998-12-30 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting 5-substituted-1,2,4-thiadiazolyl derivatives |
| WO1998058929A1 (en) * | 1997-06-24 | 1998-12-30 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting thiadiazolyl pyridazine derivatives |
| WO1999020606A3 (en) * | 1997-10-23 | 1999-07-29 | Uriach & Cia Sa J | Piperidines and piperazines as platelet aggregation inhibitors |
| WO1999043658A1 (en) * | 1998-02-27 | 1999-09-02 | Warner-Lambert Company | Heterocyclic substituted aniline calcium channel blockers |
| US6162804A (en) * | 1997-09-26 | 2000-12-19 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| US6228871B1 (en) | 1995-07-10 | 2001-05-08 | Merck & Co., Inc. | Angiogenesis inhibitors |
| US6265407B1 (en) | 1997-06-24 | 2001-07-24 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting thiadiazolyl pyridazine derivatives |
| WO2001053258A1 (en) * | 2000-01-20 | 2001-07-26 | Eisai Co., Ltd. | Nitrogenous cyclic compounds and pharmaceutical compositions containing the same |
| JP2001521025A (en) * | 1997-10-27 | 2001-11-06 | ニューロサーチ、アクティーゼルスカブ | Heteroaryldiazacycloalkanes as cholinergic ligands at the nicotinic acetylcholine receptor |
| US6465484B1 (en) | 1997-09-26 | 2002-10-15 | Merck & Co., Inc. | Angiogenesis inhibitors |
| US6548699B1 (en) | 1997-05-14 | 2003-04-15 | Atherogenics, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| JP2003523460A (en) * | 2000-01-25 | 2003-08-05 | シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ | A derivative of partially desulfated glycosaminoglycan having anti-angiogenic activity and no anticoagulant effect |
| US6670398B2 (en) | 1997-05-14 | 2003-12-30 | Atherogenics, Inc. | Compounds and methods for treating transplant rejection |
| US6833369B2 (en) | 1997-06-24 | 2004-12-21 | Janssen Pharmaceutica, Nv | Angiogenesis inhibiting 5-substituted-1,2,4,-thiadiazolyl derivatives |
| US6852878B2 (en) | 1998-05-14 | 2005-02-08 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of VCAM-1 |
| US6881860B2 (en) | 2000-04-11 | 2005-04-19 | Atherogenics, Inc. | Compounds and methods to increase plasma HDL cholesterol levels and improve HDL functionality |
| US6887870B1 (en) * | 1999-10-12 | 2005-05-03 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
| WO2005070925A1 (en) * | 2004-01-25 | 2005-08-04 | Sanofi-Aventis Deutschland Gmbh | Aryl substituted heterocycles, method for production and use thereof as medicaments |
| WO2006034440A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| WO2006101521A2 (en) | 2004-09-20 | 2006-09-28 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| WO2007009236A1 (en) * | 2005-07-20 | 2007-01-25 | Merck Frosst Canada Ltd. | Heteroaromatic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| US7208503B2 (en) | 2000-04-21 | 2007-04-24 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting 5-substituted-1,2,4-thiadiazolyl derivatives |
| WO2007046867A3 (en) * | 2005-05-19 | 2007-07-05 | Xenon Pharmaceuticals Inc | Piperidine derivatives and their uses as therapeutic agents |
| US7265115B2 (en) | 1999-01-29 | 2007-09-04 | Abbott Laboratories | Diazabicyclic CNS active agents |
| US7319108B2 (en) | 2004-01-25 | 2008-01-15 | Sanofi-Aventis Deutschland Gmbh | Aryl-substituted heterocycles, process for their preparation and their use as medicaments |
| US7335658B2 (en) | 2003-07-30 | 2008-02-26 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| US7390813B1 (en) | 2001-12-21 | 2008-06-24 | Xenon Pharmaceuticals Inc. | Pyridylpiperazines and aminonicotinamides and their use as therapeutic agents |
| US7399761B2 (en) | 2001-05-04 | 2008-07-15 | Novartis Ag | Phthalazine derivatives with angiogenesis inhibiting activity |
| US7582633B2 (en) | 2007-01-26 | 2009-09-01 | Merck Frosst Canada L.L.C. | Azacycloalkane derivatives as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| US7635698B2 (en) | 2004-12-29 | 2009-12-22 | Millennium Pharmaceuticals, Inc. | Compounds useful as chemokine receptor antagonists |
| US7777036B2 (en) | 2004-09-20 | 2010-08-17 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as therapeutic agents |
| US7829712B2 (en) | 2004-09-20 | 2010-11-09 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives for inhibiting human stearoyl-CoA-desaturase |
| WO2011002067A1 (en) * | 2009-07-02 | 2011-01-06 | 武田薬品工業株式会社 | Heterocyclic compound and use thereof |
| US7880002B2 (en) | 2004-12-29 | 2011-02-01 | Millennium Pharmaceuticals, Inc. | Substituted piperazinyl-pyrrolidine compounds useful as chemokine receptor antagonists |
| US7919496B2 (en) | 2004-09-20 | 2011-04-05 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives for the treatment of diseases mediated by stearoyl-CoA desaturase enzymes |
| US7951805B2 (en) | 2004-09-20 | 2011-05-31 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as mediators of stearoyl-CoA desaturase |
| US7981892B2 (en) | 2008-04-29 | 2011-07-19 | Eli Lilly And Company | Disubstituted phthalazine hedgehog pathway antagonists |
| US8012956B2 (en) | 2007-10-25 | 2011-09-06 | Exelixis, Inc. | Tropane compounds |
| US8063224B2 (en) | 2006-12-01 | 2011-11-22 | Merck Canada Inc. | Azacycloalkane derivatives as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| US8071603B2 (en) | 2004-09-20 | 2011-12-06 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-CoA desaturase inhibitors |
| US8252840B2 (en) | 2007-03-26 | 2012-08-28 | Salutria Pharmaceuticals Llc | Methods of derivatives of probucol for the treatment of type II diabetes |
| US8273742B2 (en) | 2009-06-19 | 2012-09-25 | Eli Lilly And Company | Disubstituted phthalazine hedgehog pathway antagonists |
| US8404687B2 (en) | 2008-11-03 | 2013-03-26 | Eli Lilly And Company | Disubstituted phthalazine hedgehog pathway antagonists |
| US8445493B2 (en) | 2008-11-17 | 2013-05-21 | Eli Lilly And Company | Tetrasubstituted pyridazines hedgehog pathway antagonists |
| WO2013085957A1 (en) * | 2011-12-06 | 2013-06-13 | Janssen Pharmaceutica Nv | Substituted piperidinyl-pyridazinyl derivatives useful as scd 1 inhibitors |
| US8507490B2 (en) | 2008-11-17 | 2013-08-13 | Eli Lilly And Company | Tetrasubstituted pyridazine hedgehog pathway antagonists |
| US8541457B2 (en) | 2005-06-03 | 2013-09-24 | Xenon Pharmaceuticals Inc. | Aminothiazole derivatives as human stearoyl-CoA desaturase inhibitors |
| WO2014191737A1 (en) * | 2013-05-28 | 2014-12-04 | Redx Pharma Limited | Heterocyclic compounds as hedgehog signaling pathway inhibitors |
| WO2015001348A1 (en) * | 2013-07-03 | 2015-01-08 | Redx Pharma Limited | Pyridazine derivatives as hedgehog pathway inhibitors |
| US9238658B2 (en) | 2011-12-06 | 2016-01-19 | Janssen Pharmaceutica Nv | Substituted piperidinyl-carboxamide derivatives useful as SCD 1 inhibitors |
| CN108912116A (en) * | 2018-08-15 | 2018-11-30 | 翟学旭 | A kind of nitrogen-containing heterocycle analog derivative and its application in retinal disease |
| US11046658B2 (en) | 2018-07-02 | 2021-06-29 | Incyte Corporation | Aminopyrazine derivatives as PI3K-γ inhibitors |
| WO2023034836A1 (en) * | 2021-08-30 | 2023-03-09 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| US11926616B2 (en) | 2018-03-08 | 2024-03-12 | Incyte Corporation | Aminopyrazine diol compounds as PI3K-γ inhibitors |
| US12168011B2 (en) | 2022-02-21 | 2024-12-17 | Verona Pharma Plc | Formulation production process |
| US12213974B2 (en) | 2020-10-13 | 2025-02-04 | Endeavor Biomedicines, Inc. | Methods of treating fibrosis |
| US12251384B1 (en) | 2023-06-26 | 2025-03-18 | Verona Pharma Plc | Particulate composition |
| US12447148B2 (en) | 2024-03-05 | 2025-10-21 | Endeavor Biomedicines, Inc. | Methods of improving lung function |
| US12605377B2 (en) | 2020-10-13 | 2026-04-21 | Endeavor Biomedicines, Inc. | Methods of treating fibrosis |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6833370B1 (en) * | 1999-05-21 | 2004-12-21 | Abbott Laboratories | Heterocycle substituted aminoazacycles useful as central nervous system agents |
| MXPA02011319A (en) * | 2000-05-15 | 2003-06-06 | Pharma Mar Sa | Antitumoral analogs of et 743. |
| US20050119251A1 (en) * | 2001-12-21 | 2005-06-02 | Jian-Min Fu | Nicotinamide derivatives and their use as therapeutic agents |
| US7754711B2 (en) * | 2003-07-30 | 2010-07-13 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| US20050065178A1 (en) * | 2003-09-19 | 2005-03-24 | Anwer Basha | Substituted diazabicycloakane derivatives |
| TW200533356A (en) * | 2004-02-24 | 2005-10-16 | Mitsubishi Pharma Corp | Fused pyridazine derivatives |
| EP1827438B2 (en) * | 2004-09-20 | 2014-12-10 | Xenon Pharmaceuticals Inc. | Piperazin derivatives for inhibiting human stearoyl-coa-desaturase |
| CA2598456A1 (en) * | 2005-02-16 | 2006-08-24 | Schering Corporation | Heterocyclic substituted piperazines with cxcr3 antagonist activity |
| EP1966183A4 (en) * | 2005-12-20 | 2010-12-29 | Merck Frosst Canada Ltd | HETEROAROMATIC COMPOUNDS AS DELTA-9 DEATURASE STÉAROYL-COENZYME INHIBITORS |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0398427A1 (en) * | 1989-05-15 | 1990-11-22 | Janssen Pharmaceutica N.V. | Antirhinoviral pyridazinamines |
| EP0429344A1 (en) * | 1989-11-17 | 1991-05-29 | Sanofi | Pyridazine derivatives, process for their preparation and pharmaceutical compositions containing them |
| EP0435381A1 (en) * | 1989-12-26 | 1991-07-03 | Janssen Pharmaceutica N.V. | Antipicornaviral pyridazinamines |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5001125A (en) * | 1984-03-26 | 1991-03-19 | Janssen Pharmaceutica N.V. | Anti-virally active pyridazinamines |
| US4992433A (en) * | 1987-11-23 | 1991-02-12 | Janssen Pharmaceutica N.V. | Novel pyridazinamine derivatives |
| US5461053A (en) * | 1989-02-07 | 1995-10-24 | Sanofi | Pyridazine derivatives |
| GB8911158D0 (en) * | 1989-05-16 | 1989-07-05 | Janssen Pharmaceutica Nv | Antiviral pyridazinamines |
| ZA9010357B (en) * | 1989-12-26 | 1992-08-26 | Janssen Pharmaceutica Nv | Antipicornaviral pyridazinamines |
| US5100893A (en) * | 1990-04-18 | 1992-03-31 | Janssen Pharmaceutica N.V. | Antipicornaviral pyridazinamines |
| US5242924A (en) * | 1992-07-02 | 1993-09-07 | Sterling Winthrop Inc. | Tetrazolyl-(phenoxy and phenoxyalkyl)-piperidinylpyridazines as antiviral agents |
-
1997
- 1997-01-14 AT AT97901059T patent/ATE203534T1/en not_active IP Right Cessation
- 1997-01-14 PT PT97901059T patent/PT876366E/en unknown
- 1997-01-14 MY MYPI97000130A patent/MY117098A/en unknown
- 1997-01-14 CN CN97191705A patent/CN1104430C/en not_active Expired - Lifetime
- 1997-01-14 KR KR10-1998-0703682A patent/KR100443893B1/en not_active Expired - Fee Related
- 1997-01-14 EP EP97901059A patent/EP0876366B1/en not_active Expired - Lifetime
- 1997-01-14 SI SI9730191T patent/SI0876366T1/en unknown
- 1997-01-14 AU AU14439/97A patent/AU717744B2/en not_active Expired
- 1997-01-14 NZ NZ326354A patent/NZ326354A/en unknown
- 1997-01-14 WO PCT/EP1997/000201 patent/WO1997026258A1/en not_active Ceased
- 1997-01-14 ZA ZA97288A patent/ZA97288B/en unknown
- 1997-01-14 DE DE69705819T patent/DE69705819T2/en not_active Expired - Lifetime
- 1997-01-14 IL IL12446197A patent/IL124461A/en not_active IP Right Cessation
- 1997-01-14 DK DK97901059T patent/DK0876366T3/en active
- 1997-01-14 JP JP52465697A patent/JP4169368B2/en not_active Expired - Lifetime
- 1997-01-14 ES ES97901059T patent/ES2162235T3/en not_active Expired - Lifetime
- 1997-01-14 CA CA002237273A patent/CA2237273C/en not_active Expired - Lifetime
- 1997-01-23 TW TW086100703A patent/TW480256B/en not_active IP Right Cessation
-
1998
- 1998-05-05 NO NO982037A patent/NO309653B1/en unknown
- 1998-07-09 US US09/119,075 patent/US5985878A/en not_active Expired - Lifetime
-
2001
- 2001-10-16 GR GR20010401770T patent/GR3036900T3/en not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0398427A1 (en) * | 1989-05-15 | 1990-11-22 | Janssen Pharmaceutica N.V. | Antirhinoviral pyridazinamines |
| EP0429344A1 (en) * | 1989-11-17 | 1991-05-29 | Sanofi | Pyridazine derivatives, process for their preparation and pharmaceutical compositions containing them |
| EP0435381A1 (en) * | 1989-12-26 | 1991-07-03 | Janssen Pharmaceutica N.V. | Antipicornaviral pyridazinamines |
Cited By (97)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6228871B1 (en) | 1995-07-10 | 2001-05-08 | Merck & Co., Inc. | Angiogenesis inhibitors |
| CZ297534B6 (en) * | 1997-02-13 | 2007-01-03 | Novartis Ag | Pyridazine derivatives exhibiting inhibiting activity to angiogenesis, process of their preparation, their use and pharmaceutical compositions in which the derivatives are comprised |
| WO1998035958A1 (en) * | 1997-02-13 | 1998-08-20 | Novartis Ag | Phthalazines with angiogenesis inhibiting activity |
| US6514974B2 (en) | 1997-02-13 | 2003-02-04 | Novartis Ag | Pyrido-, pyrimido-, pyridazo- and pyrazo- pyridazines having angiogenesis inhibiting activity |
| US6258812B1 (en) | 1997-02-13 | 2001-07-10 | Novartis Ag | Phthalazines with angiogenesis inhibiting activity |
| US6617352B2 (en) | 1997-05-14 | 2003-09-09 | Atherogenics, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| US6548699B1 (en) | 1997-05-14 | 2003-04-15 | Atherogenics, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| US6828447B2 (en) | 1997-05-14 | 2004-12-07 | Atherogenics, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| US6670398B2 (en) | 1997-05-14 | 2003-12-30 | Atherogenics, Inc. | Compounds and methods for treating transplant rejection |
| US7087645B2 (en) | 1997-05-14 | 2006-08-08 | Atherogenics, Inc. | Compounds and methods for treating transplant rejection |
| US7375252B2 (en) | 1997-05-14 | 2008-05-20 | Atherogenics, Inc. | Compounds and method for the inhibition of the expression of VCAM-1 |
| US6602914B2 (en) | 1997-05-14 | 2003-08-05 | Atherogenics, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| US7189870B2 (en) | 1997-05-14 | 2007-03-13 | Atherogenic, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| US6602873B2 (en) | 1997-06-24 | 2003-08-05 | Janssen Pharamaceutica, N.V. | Angiogenesis inhibiting thiadiazolyl pyridazine derivatives |
| US6833369B2 (en) | 1997-06-24 | 2004-12-21 | Janssen Pharmaceutica, Nv | Angiogenesis inhibiting 5-substituted-1,2,4,-thiadiazolyl derivatives |
| US6265407B1 (en) | 1997-06-24 | 2001-07-24 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting thiadiazolyl pyridazine derivatives |
| WO1998058929A1 (en) * | 1997-06-24 | 1998-12-30 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting thiadiazolyl pyridazine derivatives |
| WO1998058919A1 (en) * | 1997-06-24 | 1998-12-30 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting 5-substituted-1,2,4-thiadiazolyl derivatives |
| US6465484B1 (en) | 1997-09-26 | 2002-10-15 | Merck & Co., Inc. | Angiogenesis inhibitors |
| US6162804A (en) * | 1997-09-26 | 2000-12-19 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| WO1999020606A3 (en) * | 1997-10-23 | 1999-07-29 | Uriach & Cia Sa J | Piperidines and piperazines as platelet aggregation inhibitors |
| JP2001521025A (en) * | 1997-10-27 | 2001-11-06 | ニューロサーチ、アクティーゼルスカブ | Heteroaryldiazacycloalkanes as cholinergic ligands at the nicotinic acetylcholine receptor |
| WO1999043658A1 (en) * | 1998-02-27 | 1999-09-02 | Warner-Lambert Company | Heterocyclic substituted aniline calcium channel blockers |
| US6251919B1 (en) * | 1998-02-27 | 2001-06-26 | Warner-Lambert | Heterocyclic substituted aniline calcium channel blockers |
| US6852878B2 (en) | 1998-05-14 | 2005-02-08 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of VCAM-1 |
| US7265115B2 (en) | 1999-01-29 | 2007-09-04 | Abbott Laboratories | Diazabicyclic CNS active agents |
| US6887870B1 (en) * | 1999-10-12 | 2005-05-03 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
| WO2001053258A1 (en) * | 2000-01-20 | 2001-07-26 | Eisai Co., Ltd. | Nitrogenous cyclic compounds and pharmaceutical compositions containing the same |
| EP1818326A1 (en) * | 2000-01-20 | 2007-08-15 | Eisai R&D Management Co., Ltd. | Nitrogen-containing heterocyclic compounds and pharmaceutical composition containing the compounds |
| US6906072B1 (en) | 2000-01-20 | 2005-06-14 | Eisai Co., Ltd. | Piperazine compound and pharmaceutical composition containing the compound |
| JP2003523460A (en) * | 2000-01-25 | 2003-08-05 | シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ | A derivative of partially desulfated glycosaminoglycan having anti-angiogenic activity and no anticoagulant effect |
| US8222231B2 (en) | 2000-01-25 | 2012-07-17 | Sigma-Tau Industrie Farmaceutiche Riunite Spa | Derivatives of partially desulphated glycosaminoglycans endowed with antiangiogenic activity and devoid of anticoagulating effect |
| US6881860B2 (en) | 2000-04-11 | 2005-04-19 | Atherogenics, Inc. | Compounds and methods to increase plasma HDL cholesterol levels and improve HDL functionality |
| US7183317B2 (en) | 2000-04-11 | 2007-02-27 | Atherogenics, Inc. | Compounds and methods to increase plasma HDL cholesterol levels and improve HDL functionality |
| US7208503B2 (en) | 2000-04-21 | 2007-04-24 | Janssen Pharmaceutica N.V. | Angiogenesis inhibiting 5-substituted-1,2,4-thiadiazolyl derivatives |
| US8034814B2 (en) | 2001-05-04 | 2011-10-11 | Novartis Ag | Phthalazine derivatives with angiogenesis inhibiting activity |
| US7399761B2 (en) | 2001-05-04 | 2008-07-15 | Novartis Ag | Phthalazine derivatives with angiogenesis inhibiting activity |
| US7390813B1 (en) | 2001-12-21 | 2008-06-24 | Xenon Pharmaceuticals Inc. | Pyridylpiperazines and aminonicotinamides and their use as therapeutic agents |
| EP3042895A1 (en) * | 2003-07-30 | 2016-07-13 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| US7335658B2 (en) | 2003-07-30 | 2008-02-26 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| WO2005070925A1 (en) * | 2004-01-25 | 2005-08-04 | Sanofi-Aventis Deutschland Gmbh | Aryl substituted heterocycles, method for production and use thereof as medicaments |
| US8299104B2 (en) | 2004-01-25 | 2012-10-30 | Sanofi-Aventis Deutschland Gmbh | Aryl-substituted heterocycles, process for their preparation and their use as medicaments |
| US7319108B2 (en) | 2004-01-25 | 2008-01-15 | Sanofi-Aventis Deutschland Gmbh | Aryl-substituted heterocycles, process for their preparation and their use as medicaments |
| US7777036B2 (en) | 2004-09-20 | 2010-08-17 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as therapeutic agents |
| US7951805B2 (en) | 2004-09-20 | 2011-05-31 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as mediators of stearoyl-CoA desaturase |
| WO2006034440A3 (en) * | 2004-09-20 | 2006-08-10 | Xenon Pharmaceuticals Inc | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| US7592343B2 (en) | 2004-09-20 | 2009-09-22 | Xenon Pharmaceuticals Inc. | Pyridazine-piperazine compounds and their use as stearoyl-CoA desaturase inhibitors |
| WO2006101521A2 (en) | 2004-09-20 | 2006-09-28 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| US7767677B2 (en) | 2004-09-20 | 2010-08-03 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-CoA desaturase inhibitors |
| US8071603B2 (en) | 2004-09-20 | 2011-12-06 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-CoA desaturase inhibitors |
| US7829712B2 (en) | 2004-09-20 | 2010-11-09 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives for inhibiting human stearoyl-CoA-desaturase |
| WO2006034440A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| WO2006101521A3 (en) * | 2004-09-20 | 2006-12-28 | Xenon Pharmaceuticals Inc | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| EP2266569A3 (en) * | 2004-09-20 | 2011-03-09 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| EP2269610A3 (en) * | 2004-09-20 | 2011-03-09 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| US7919496B2 (en) | 2004-09-20 | 2011-04-05 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives for the treatment of diseases mediated by stearoyl-CoA desaturase enzymes |
| US8026360B2 (en) | 2004-09-20 | 2011-09-27 | Xenon Pharmaceuticals Inc. | Substituted pyridazines as stearoyl-CoA desaturase inhibitors |
| US8168788B2 (en) | 2004-12-29 | 2012-05-01 | Millennium Pharmaceuticals, Inc. | Substituted piperazinyl-pyrrolidine compounds useful as chemokine receptor antagonists |
| US8648197B2 (en) | 2004-12-29 | 2014-02-11 | Millennium Pharmaceuticals, Inc. | Substituted piperazinyl-pyrrolidine compounds useful as chemokine receptor antagonists |
| US7880002B2 (en) | 2004-12-29 | 2011-02-01 | Millennium Pharmaceuticals, Inc. | Substituted piperazinyl-pyrrolidine compounds useful as chemokine receptor antagonists |
| US7635698B2 (en) | 2004-12-29 | 2009-12-22 | Millennium Pharmaceuticals, Inc. | Compounds useful as chemokine receptor antagonists |
| US8399455B2 (en) | 2004-12-29 | 2013-03-19 | Millennium Pharmaceuticals, Inc. | Compounds useful as chemokine receptor antagonists |
| WO2007046867A3 (en) * | 2005-05-19 | 2007-07-05 | Xenon Pharmaceuticals Inc | Piperidine derivatives and their uses as therapeutic agents |
| US8541457B2 (en) | 2005-06-03 | 2013-09-24 | Xenon Pharmaceuticals Inc. | Aminothiazole derivatives as human stearoyl-CoA desaturase inhibitors |
| WO2007009236A1 (en) * | 2005-07-20 | 2007-01-25 | Merck Frosst Canada Ltd. | Heteroaromatic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| US8063224B2 (en) | 2006-12-01 | 2011-11-22 | Merck Canada Inc. | Azacycloalkane derivatives as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| US7582633B2 (en) | 2007-01-26 | 2009-09-01 | Merck Frosst Canada L.L.C. | Azacycloalkane derivatives as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| US8252840B2 (en) | 2007-03-26 | 2012-08-28 | Salutria Pharmaceuticals Llc | Methods of derivatives of probucol for the treatment of type II diabetes |
| US8012956B2 (en) | 2007-10-25 | 2011-09-06 | Exelixis, Inc. | Tropane compounds |
| US7981892B2 (en) | 2008-04-29 | 2011-07-19 | Eli Lilly And Company | Disubstituted phthalazine hedgehog pathway antagonists |
| US8404687B2 (en) | 2008-11-03 | 2013-03-26 | Eli Lilly And Company | Disubstituted phthalazine hedgehog pathway antagonists |
| US8445493B2 (en) | 2008-11-17 | 2013-05-21 | Eli Lilly And Company | Tetrasubstituted pyridazines hedgehog pathway antagonists |
| US8507490B2 (en) | 2008-11-17 | 2013-08-13 | Eli Lilly And Company | Tetrasubstituted pyridazine hedgehog pathway antagonists |
| US8273742B2 (en) | 2009-06-19 | 2012-09-25 | Eli Lilly And Company | Disubstituted phthalazine hedgehog pathway antagonists |
| US9000023B2 (en) | 2009-06-19 | 2015-04-07 | Eli Lilly And Company | Disubstituted phthalazine hedgehog pathway antagonists |
| WO2011002067A1 (en) * | 2009-07-02 | 2011-01-06 | 武田薬品工業株式会社 | Heterocyclic compound and use thereof |
| WO2013085957A1 (en) * | 2011-12-06 | 2013-06-13 | Janssen Pharmaceutica Nv | Substituted piperidinyl-pyridazinyl derivatives useful as scd 1 inhibitors |
| US9102669B2 (en) | 2011-12-06 | 2015-08-11 | Janssen Pharmaceutica Nv | Substituted piperidinyl-pyridazinyl derivatives useful as SCD 1 inhibitors |
| US9238658B2 (en) | 2011-12-06 | 2016-01-19 | Janssen Pharmaceutica Nv | Substituted piperidinyl-carboxamide derivatives useful as SCD 1 inhibitors |
| WO2014191737A1 (en) * | 2013-05-28 | 2014-12-04 | Redx Pharma Limited | Heterocyclic compounds as hedgehog signaling pathway inhibitors |
| US9579319B2 (en) | 2013-05-28 | 2017-02-28 | Redx Pharma Plc | Heterocyclic compounds as hedgehog signaling pathway inhibitors |
| WO2015001348A1 (en) * | 2013-07-03 | 2015-01-08 | Redx Pharma Limited | Pyridazine derivatives as hedgehog pathway inhibitors |
| US12365668B2 (en) | 2018-03-08 | 2025-07-22 | Incyte Corporation | Aminopyrazine diol compounds as PI3K-y inhibitors |
| US11926616B2 (en) | 2018-03-08 | 2024-03-12 | Incyte Corporation | Aminopyrazine diol compounds as PI3K-γ inhibitors |
| US11046658B2 (en) | 2018-07-02 | 2021-06-29 | Incyte Corporation | Aminopyrazine derivatives as PI3K-γ inhibitors |
| US12421197B2 (en) | 2018-07-02 | 2025-09-23 | Incyte Corporation | Aminopyrazine derivatives as PI3K-γ inhibitors |
| CN108912116A (en) * | 2018-08-15 | 2018-11-30 | 翟学旭 | A kind of nitrogen-containing heterocycle analog derivative and its application in retinal disease |
| US12295954B2 (en) | 2020-10-13 | 2025-05-13 | Endeavor Biomedicines, Inc. | Methods of treating fibrosis |
| US12213974B2 (en) | 2020-10-13 | 2025-02-04 | Endeavor Biomedicines, Inc. | Methods of treating fibrosis |
| US12605377B2 (en) | 2020-10-13 | 2026-04-21 | Endeavor Biomedicines, Inc. | Methods of treating fibrosis |
| WO2023034836A1 (en) * | 2021-08-30 | 2023-03-09 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| US12168011B2 (en) | 2022-02-21 | 2024-12-17 | Verona Pharma Plc | Formulation production process |
| US12409180B2 (en) | 2022-02-21 | 2025-09-09 | Verona Pharma Plc | Formulation production process |
| US12251384B1 (en) | 2023-06-26 | 2025-03-18 | Verona Pharma Plc | Particulate composition |
| US12447148B2 (en) | 2024-03-05 | 2025-10-21 | Endeavor Biomedicines, Inc. | Methods of improving lung function |
| US12605380B2 (en) | 2024-03-05 | 2026-04-21 | Endeavor Biomedicines, Inc. | Methods of improving lung function |
| US12605379B2 (en) | 2024-03-05 | 2026-04-21 | Endeavor Biomedicines, Inc. | Methods of improving lung function |
Also Published As
| Publication number | Publication date |
|---|---|
| GR3036900T3 (en) | 2002-01-31 |
| JP4169368B2 (en) | 2008-10-22 |
| NO982037D0 (en) | 1998-05-05 |
| ATE203534T1 (en) | 2001-08-15 |
| US5985878A (en) | 1999-11-16 |
| ES2162235T3 (en) | 2001-12-16 |
| JP2000503014A (en) | 2000-03-14 |
| NZ326354A (en) | 1999-05-28 |
| DE69705819T2 (en) | 2002-04-11 |
| PT876366E (en) | 2002-01-30 |
| CN1104430C (en) | 2003-04-02 |
| IL124461A (en) | 2000-07-26 |
| AU1443997A (en) | 1997-08-11 |
| NO982037L (en) | 1998-09-15 |
| DE69705819D1 (en) | 2001-08-30 |
| NO309653B1 (en) | 2001-03-05 |
| DK0876366T3 (en) | 2001-11-05 |
| MY117098A (en) | 2004-05-31 |
| CA2237273A1 (en) | 1997-07-24 |
| TW480256B (en) | 2002-03-21 |
| KR100443893B1 (en) | 2004-10-15 |
| CN1208415A (en) | 1999-02-17 |
| EP0876366B1 (en) | 2001-07-25 |
| EP0876366A2 (en) | 1998-11-11 |
| AU717744B2 (en) | 2000-03-30 |
| SI0876366T1 (en) | 2001-12-31 |
| IL124461A0 (en) | 1998-12-06 |
| CA2237273C (en) | 2009-01-13 |
| ZA97288B (en) | 1998-07-14 |
| KR19990067654A (en) | 1999-08-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0876366B1 (en) | Angiogenesis inhibiting pyridazinamines | |
| US6159982A (en) | 2,4-diaminopyrimidine derivates as dopamine D4 receptor antagonist | |
| US5929075A (en) | Apolipoprotein-B synthesis inhibitors | |
| US6103725A (en) | Alkylaminobenzothiazole and -benzoxazole derivatives | |
| US8530474B2 (en) | Substituted 6-(1-piperazinyl)-pyridazines as 5-HT6 receptor antagonists | |
| US5100893A (en) | Antipicornaviral pyridazinamines | |
| JP2001519413A (en) | Heterocyclic compounds and their intermediates useful for the treatment of benign prostatic hyperplasia | |
| EP0993452B1 (en) | Angiogenesis inhibiting 5-substituted-1,2,4-thiadiazolyl derivatives | |
| US4988699A (en) | Substituted tetrahydrobenzothiazoles as dopaminergic agents | |
| EP0991649A1 (en) | Angiogenesis inhibiting thiadiazolyl pyridazine derivatives | |
| US4665187A (en) | Certain 1,2,3,6-tetrahydro-pyridyl-N-lower-alkanoyl-pyridines as intermediates | |
| US7208503B2 (en) | Angiogenesis inhibiting 5-substituted-1,2,4-thiadiazolyl derivatives | |
| US5106973A (en) | Pyridzainamine derivatives | |
| US5196535A (en) | Intermediates for producing antipicornaviral pyridazinamines | |
| US6265407B1 (en) | Angiogenesis inhibiting thiadiazolyl pyridazine derivatives | |
| CA2525849A1 (en) | 4-arylsulphonylpiperidine derivatives for antagonism of the 5-ht2a receptor | |
| US6833369B2 (en) | Angiogenesis inhibiting 5-substituted-1,2,4,-thiadiazolyl derivatives | |
| US4906642A (en) | Pyridine derivatives | |
| AU708344C (en) | 2,4-diaminopyrimidine derivates as dopamine D4 receptor antagonists | |
| EP3212636A1 (en) | New pyridinyloxy- and phenyloxy-pyrazolyl compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 97191705.1 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU BA BB BG BR CA CN CU CZ EE GE HU IL IS JP KG KR LC LK LR LT LV MD MG MK MN MX NO NZ PL RO SG SI SK TR TT UA US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2237273 Country of ref document: CA Ref document number: 2237273 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 326354 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1997901059 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019980703682 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/A/1998/004671 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09119075 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997901059 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019980703682 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1997901059 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1019980703682 Country of ref document: KR |