WO1997027177A2 - Dihydropyridine-, pyridine-, benzopyran-4-one- and triazoloquinazoline derivatives, their preparation and their use as adenosine receptor antagonists - Google Patents
Dihydropyridine-, pyridine-, benzopyran-4-one- and triazoloquinazoline derivatives, their preparation and their use as adenosine receptor antagonists Download PDFInfo
- Publication number
- WO1997027177A2 WO1997027177A2 PCT/US1997/001252 US9701252W WO9727177A2 WO 1997027177 A2 WO1997027177 A2 WO 1997027177A2 US 9701252 W US9701252 W US 9701252W WO 9727177 A2 WO9727177 A2 WO 9727177A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- phenyl
- alkyloxy
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*)(C=C1)C=CC(ON2)=C1C2=O Chemical compound CC(*)(C=C1)C=CC(ON2)=C1C2=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/80—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D211/84—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen directly attached to ring carbon atoms
- C07D211/90—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/22—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/22—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4
- C07D311/26—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3
- C07D311/28—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3 with aromatic rings attached in position 2 only
- C07D311/30—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3 with aromatic rings attached in position 2 only not hydrogenated in the hetero ring, e.g. flavones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/22—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4
- C07D311/26—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3
- C07D311/28—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3 with aromatic rings attached in position 2 only
- C07D311/32—2,3-Dihydro derivatives, e.g. flavanones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/70—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with ring systems containing two or more relevant rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
Definitions
- the present invention relates to adenosine receptor antagonists, pharmaceutical compositions, and methods of selectively blocking adenosine receptors in a mammal.
- the present invention also relates to methods of treating various medical disorders with adenosine receptor antagonists .
- caffeine and other alkylxanthines as physiological stimulants is well known.
- the principle mechanism by which caffeine and other alkylxanthines act as physiological stimulants is by blocking the effects of the ubiquitous neuromodulator adenosine.
- Mechanism of action of caffeine in Caffeine, Coffee and Health, (S. Garattini, ed) , Chapter 4, pp. 97-150 (1993) .
- Adenosine is produced locally in response to increased activity or stress to the system. This feedback mechanism allows the organ to compensate for the stress by decreasing energy demand (depressant activity) and increasing oxygen supply (e.g., by vasodilation) .
- energy demand depressant activity
- oxygen supply e.g., by vasodilation
- Adenosine plays several key physiological roles. In addition to its role in intermediary metabolism, adenosine displays a number of receptor-mediated physiological actions, including dilation of coronary vessels, inhibition of platelet aggregation, and inhibition of lipolysis. Bruns et al . , Proc. Nat. Acad. Sci. U.S.A.. 77, 5547-5551 (1980) .
- Adenosine receptors belonging to the superfamily of the G protein-coupled receptors, are generally divided into two major subclasses, A. and A_, , on the basis of the differential affinities of a number of adenosine receptor agonists and antagonists for the receptors, their primary structures, and the secondary messenger systems to which they couple.
- A- receptors which can be further subdivided into A- a and A_ b , stimulate adenylate cyclase
- a 1 receptors may couple to a variety of secondary messenger systems, including those involved in the inhibition of adenylate cyclase, the inhibition or stimulation of phosphoinositol turnover, the activation of guanylate cyclase, the activation of potassium influx, and the inhibition of calcium influx (van Galen et al . , Med. Res. Rev. , 12. 423- 471 (1992) ; Jacobson et al . , J. Med. Chem.. 35. 407-422 (1992) ) .
- a novel adenosine receptor was identified on the basis of its primary structure and cloned from rat brain (Zhou et al. , Proc. Natl. Acad. Sci. U.S.A., 89, 7432-7436 (1992)) and rat testes (Meyerhof et al. , FEBS Lett . , 284, 155-160 (1991) ) .
- the putative transmembrane domains of the novel adenosine receptor which has been designated the A 3 receptor, show 58% identity with the canine A x receptor and 57% with the canine A_ a receptor.
- the A 3 receptor is negatively coupled to adenylate cyclase (Zhou et al.) .
- a 3 receptor The distribution of the A 3 receptor is found primarily in the central nervous system (CNS) (Zhou et al . ) , brain, testes (Meyerhof et al . ) , and immune system, where it appears to be involved in the modulation of release from mast cells of mediators of the immediate hypersensitivity reaction (Ramkumar et al . , J. Biol. Chem.. 268, 16887- 16890 (1993)) . It is believed that A 3 -selective compounds will have utility in the therapeutic and/or prophylactic treatment of cardiac disease, infertility, kidney disease, and CNS disorders.
- a 3 -adenosine receptor antagonists should serve as cerebroprotective, anti-asthmatic, or anti-inflammatory agents. Beaven et al., Trends Pharmacol . Sci .. 15, 13-4 (1994); Jacobson et al., Drugs of the Future, 20, 689-699 (1995) ; von Lubitz et al., Eur. J. Pharmacol., 275, 23-29 (1995) .
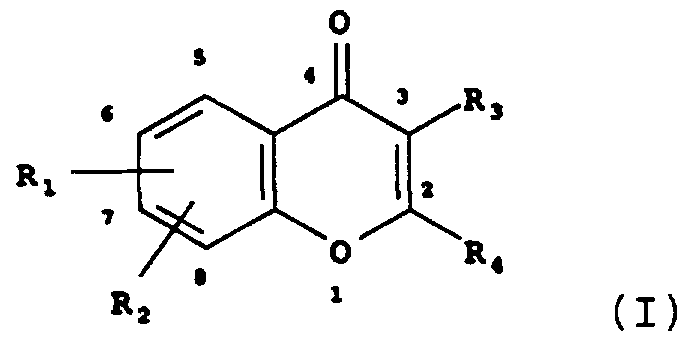
- the present invention provides a compound of the formula I
- R. and R 3 are selected from the group consisting of hydrogen, hydroxy, C.-C 3 alkyloxy, and alkylcarbonyloxy;
- R 2 is selected from the group consisting of hydrogen, hydroxy, Cj-Cg alkyloxy, and C 2 -C 6 alkenyloxy, said alkenyloxy together with the carbon atom of the phenyl ring forming an oxygen heterocycle;
- the present invention further provides a compound of the formula II
- R ⁇ is selected from the group consisting of hydroxy1 and C.-C 6 alkoxy
- R 2 is selected from the group consisting of styryl and phenylacetylenyl.
- R 2 is a C ⁇ C g alkyl
- R 6 is selected from the group consisting C.-C 6 alkyl, haloalkyl, and phenyl which may be further substituted with C.-C 6 alkyl, halo, nitro, furyl, or thienyl
- R 3 is selected from the group consisting of C.- C 6 alkyl, C- .
- R 4 is selected from the group consisting of C--C 6 alkyl, aryl C 2 -C 6 alkenyl, C.- C 6 alkylamino, alkyl silyl C.-C 6 alkyloxy, aryl, heterocyclic, aryl C.-C 6 alkyl, phenylacetylenyl which may be further substituted with nitro, C--C 6 alkyl, hydroxy, halo, amino, carboxy, C.-C 6 alkoxy, haloalkyl, or C.-C
- the present invention further provides a compound of the formula IV
- R 2 is selected from the group consisting of hydrogen and C.-C 6 alkyl
- R 3 is selected from the group consisting of hydrogen and C--C 6 alkyloxycarbonyl
- R 4 is selected from the group consisting of alkyl, phenyl C 2 -C 6 alkenyl, phenyl C 2 -C 6 alkynyl, aryl, and aryl substituted with one or more substituents selected from the group consisting of nitro and C.-Cg alkyloxy
- R 5 is selected from the group consisting of hydrogen, C ⁇ -C 6 alkyloxycarbonyl, and aryl C j -C 8 alkyloxy carbonyl
- the present invention further provides a compound of the formula V
- R x is selected from the group consisting of alkylcarbonyl, C--C 6 alkyloxycarbonyl, amino C.-C 6 alkylcarbonyl, and arylcarbonyl wherein the aryl may be further substituted with halo, nitro, hydroxy, amino or cyano; and R 2 is hydrogen or halogen.
- the present invention further provides pharmaceutical compositions comprising any of the aforesaid compounds and a method of treating a mammal comprising selectively blocking one or more of the adenosine receptors of the mammal by administering to the mammal at least one compound of formulas I-V.
- Figure 1 depicts a method of synthesis of alkylated derivatives of galangin and morin.
- Alkyl iodide was used as the alkylating agent for the preparation of 2, 3a, 3b, 4, and 6.
- Ethyl bromide was used as the alkylating agent for the preparation of 7
- alkyl sulfate was used as the alkylating agent for the preparation of 8.
- Figure 2 depicts a method of synthesis of the 2-phenylacetylenyl and 2-cinnamyl derivatives, 10, 15, and 16, starting from 4-methoxyfuranochromone.
- Figure 3 depicts a method of synthesis of certain 2- substituted flavonoid derivatives, 19-24, starting from visnagin.
- Figure 4 depicts the structures of flavonoid derivatives 41-44.
- Figure 5 depicts the structures of flavonoid derivatives 45-51.
- Figure 6 depicts a method of synthesis of certain 1,4-dihydropyridine derivatives.
- Figure 7 depicts a method of oxidation of certain 1, 4-dihydropyridine derivatives.
- Figure 8 depicts a representative set of competition curves for inhibition binding of [ 125 I]AB-MECA by compounds 12 (triangles) , 14 (circles) , and 18 (diamonds) , at human brain A 3 receptors expressed in human HEK-293 cells at 25°C.
- Figure 9A depicts a part of the reaction scheme for the synthesis of a diastereomeric pair of a 5-ester substituted 1,4-dihydropyridine.
- Figure 9B depicts another part of the reaction scheme for the synthesis of a diastereomeric pair of a 5-ester substituted 1,4-dihydropyridine.
- Figure 10A depicts the percentage of HL-60 apoptotic cells (vertical axis) as a function of the concentration (horizontal axis) of A 3 adenosine receptor agonists IB-MECA (°) and Cl-IB-MECA (•) , as determined by fluorescent cell sorting (flow cytometric DNA analysis) .
- Figure 10B depicts the percentage of U-937 apoptotic cells (vertical axis) as a function of the concentration (horizontal axis) of A 3 adenosine receptor agonists IB-MECA
- Figure 11A depicts the number of living HL-60 cells (vertical axis) as a function of the concentration (horizontal axis) of A 3 denosine receptor antagonist and a low concentration of Cl-IB-MECA.
- the antagonist was compound 101.
- Figures 11A-D for each set of curves, control (--) , antagonist alone ( ⁇ ) , or antagonist in the presence of lOnM (°) or 1 ⁇ M (•) Cl-IB-MECA are shown.
- Figure 11B depicts the number of living HL-60 cells (vertical axis) as a function of the concentration (horizontal axis) of the A 3 adenosine receptor antagonist and low concentration of Cl-IB-MECA.
- the antagonist was compound L-249313(6-carboxymethyl-5,9-dihydro-9-methyl-2- phenyl- [1,2,4] triazolo [5, 1-a] [2,7] -naphthyridine) .
- Figure 11C depicts the number of living U-937 cells (vertical axis) as a function of the concentration (horizontal axis) of the A 3 adenosine receptor antagonist and a low concentration of Cl-IB-MECA.
- the antagonist was compound 101.
- Figure 11D depicts the number of living U-937 cells (vertical axis) as a function of the concentration (horizontal axis) of the A 3 adenosine receptor antagonist and a low concentration of Cl-IB-MECA.
- the antagonist was compound L-249313.
- Figure 12 depicts the percent change in dynamic compliance (C dyn , vertical axis) of rabbit lung as a function of the adenosine agonist concentration using Cl-
- IB-MECA (o) APNEA ( ) , or IB-MECA ( ⁇ ) .
- (•) represents percent change in C dyn as a result of exposure to compound
- the present invention provides certain derivatives of flavonoids, dihydropyridines, pyridines, and triazoloquinazolines, suitable for blocking one or more of the adenosine receptors of a mammal such as human, as set forth herebelow in greater detail.
- the present invention provides a compound of the formula I
- R. and R 3 are selected from the group consisting of hydrogen, hydroxy, C--C 6 alkyloxy, and alkylcarbonyloxy;
- R 2 is selected from the group consisting of hydrogen, hydroxy, ⁇ - ⁇ alkyloxy, and C 2 -C 6 alkenyloxy, said alkenyloxy together with the carbon atom of the phenyl ring forming an oxygen heterocycle;
- R 2 are neither hydroxy nor alkoxy; when R-, R 2 , and R 3 are hydrogen, R 4 is neither phenyl nor alkyloxyphenyl; when R3 is hydrogen and R 4 is phenyl, neither R. nor R 2 is alkylcarbonyloxy; and when R 3 is hydroxy or alkyloxy, R. and R 2 are not dihydroxy.
- Particular embodiments include a compound of formula I wherein (a) R 4 is phenyl, (b) R 4 is phenyl and R 3 is a C ⁇ C 3 alkyloxy, (c) R 4 is phenyl, R 3 is a C.-C 3 alkyloxy, and R 2 and R 2 are 5,7-di(C--C 3 alkyloxy) , (d) R 4 is a 2,4-di(C--C 3 alkyloxy)phenyl, (e) R 3 is a C.-C 3 alkyloxy and R 4 is a 2,4- di(C.-C 3 alkyloxy)phenyl, (f) R 3 is a C.-C 3 alkyloxy, R 4 is a 2,4-di(C 1 -C 3 alkyloxy)phenyl, and R- and R 2 are the same and are selected from the group consisting of methoxy and ethoxy, (g) Rj is 5-hydroxy and R 2 is one of
- the present invention further provides a compound of formula I, wherein the compound is a 4-(C 1 -C 6 alkyloxy) -7- styrylvisnagin.
- the compound is a 4-(C 1 -C 6 alkyloxy) -7- styrylvisnagin.
- Examples of such compounds include 4- methoxy-7- trans-styrylvisnagin, 4-ethoxy-7-tra ⁇ s- styrylvisnagin, and 4-propoxy-7 -trans-styrylvisnagin.
- Other particular embodiments include compounds of formula I, wherein the compound is a C.-C 6 alkyloxy-7- phenylbutadienylvisnagin. Examples of such compounds include 4-methoxy-7-phenylbutadienylvisnaginand 4-ethoxy- 7-phenylbutadienylvisnagin.
- the present invention further provides a compound of formula I, wherein the compound is a C j -Cg alkyloxy-7-
- the present invention further provides a compound of formula I, wherein the compound is selected from the group consisting of 3, 5, 7-triacetoxyflavone, 3,5,7- trimethoxyflavone, 3, 5, 7-triethoxyflavone, 3 , 7-diethoxy-5- hydroxyflavone, 3 , 5, 7-tripropoxyflavone, 3, 4', 5, 7- tetramethoxyflavone, 2 ⁇ , 3, 4 ' , 7-tetraethoxy-5- hydroxyflavone, 2 ' , 3 , ' , 5, 7-pentamethoxyflavone, 2 ' , 3,4 ' , 5, 7-pentaethoxyflavone, hexamethylmyricetin, and 3-hydroxy-4 ' -phenylacetylenyl-6-methoxyflavone.
- the present invention further provides a compound of the formula II
- R x is selected from the group consisting of hydroxyl and C.-C 6 alkoxy
- R 2 is selected from the group consisting of styryl and phenylacetylenyl.
- Examples of compounds of formula II include 2- phenylacetylenyl-3-hydroxy-6-methoxyflavone, trans-2- styryl-3-hydroxy-6-methoxyflavone, and trans-2- phenylacetylenyl-3-hydroxy-6-methoxyflavone.
- the present invention further provides a compound of the formula III
- R 2 is a C.-C 6 alkyl
- R 6 is selected from the group consisting
- R 3 is selected from the group consisting of C-- C 6 alkyl, C j -Cg alkyloxycarbonyl, aryl C--C 6 alkyloxycarbonyl, C.-C 6 alkylthiocarbonyl, C--C 6 alkylaminocarbonyl, and C.-C 6 alkyloxy C--C 6 alkylcarbonyl, or R 3 together with R 2 forms a ring having 2-4 methylene groups, and C.-C 6 alkenyloxycarbonyl; R 4 is selected from the group consisting of C.-C 6 alkyl, aryl C 2 -C 6 alkenyl, C-- C 6 alkylamino, C.-C 6 alkyl silyl Cj-Cg al
- Particular embodiments include compounds of formula III, wherein (a) R 2 is methyl, (b) R 2 is methyl and R 3 is selected from the group consisting of methoxycarbonyl and ethoxycarbonyl, (c) R 2 is methyl, R 3 is selected from the group consisting of methoxycarbonyl and ethoxycarbonyl, and R 6 is selected from the group consisting of C.-C 4 alkyl and phenyl, (d) R 2 is methyl, R 3 is selected from the group consisting of methoxycarbonyl and ethoxycarbonyl, R 6 is selected from the group consisting of C.-C 4 alkyl and phenyl, and R 4 is selected from the group consisting of C x - C 3 alkyl, (e) R 2 is methyl, R 3 is selected from the group consisting of methoxycarbonyl and ethoxycarbonyl, R 6 is selected from the group consisting of C.-C 4 alkyl and phenyl, R 4 is
- R 2 is selected from the group consisting of hydrogen and C--C 6 alkyl
- R 3 is selected from the group consisting of hydrogen and Ci-Cg alkyloxycarbonyl
- R 4 is selected from the group consisting of C.-C 6 alkyl, phenyl C 2 -C 6 alkenyl, phenyl C 2 -C 6 alkynyl, aryl, and aryl substituted with one or more substituents selected from the group consisting of nitro and C.-Cg alkyloxy
- R s is selected from the group consisting of hydrogen, Cj-Cg alkyloxycarbonyl, and aryl C.-Cg alkyloxy carbonyl
- Particular embodiments include compounds of formula IV, wherein (a) R 2 is selected from the group consisting of hydrogen and methyl, (b) R 2 is selected from the group consisting of hydrogen and methyl, and R 3 and R 5 are same or different and selected from the group consisting of hydrogen, methoxycarbonyl, and ethoxycarbonyl, (c) R 2 is selected from the group consisting of hydrogen and methyl, R 3 and R 5 are same or different and selected from the group consisting of hydrogen, methoxycarbonyl, and ethoxycarbonyl, and R 4 is selected from the group consisting of methyl, o-nitrophenyl, and p-methoxyphenyl, and (d) R 2 is selected from the group consisting of hydrogen and methyl, R 3 and R B are same or different and selected from the group consisting of hydrogen, methoxycarbonyl, and ethoxycarbonyl, R 4 is selected from the group consisting of methyl, o-nitrophenyl, and p- methoxyphenyl
- the present invention further provides a compound of the formula V
- R x is selected from the group consisting of C--C 6 alkylcarbonyl, aryl C.-C 6 alkylcarbonyl, aryl C 2 -C 6 alkenylcarbonyl, C j -Cg alkyloxycarbonyl, amino C--C 6 alkylcarbonyl, and arylcarbonyl, wherein said aryl may be further substituted with halo, nitro, hydroxy, amino or cyano; and R 2 is hydrogen or halogen.
- Aryl includes phenyl, naphthyl, and aromatic moieties having 3 or 4 rings.
- Particular embodiments include a compound of formula V wherein R 2 is hydrogen, and R j is ethylcarbonyl, benzoyl phenylethylcarbonyl, styrylcarbonyl, 4- nitrobenzylcarbonyl, 4-aminobenzylcarbonyl, or 3-iodo-4- aminobenzylcarbonyl .
- Aryl in this application refers to phenyl, naphthyl, and aromatic groups with 3 or more rings, and preferably phenyl, unless otherwise described. All of the aforesaid compounds of the present invention can be used as is or in the form of a pharmaceutical composition comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of the compound.
- the present invention further provides a method of treating a mammal comprising selectively blocking one or more of the adenosine receptors of the mammal by administering to the mammal at least one compound of formulas I-V.
- the present invention provides a method of treating a mammal comprising selectively blocking the adenosine receptors of a mammal by administering to said mammal at least one compound of the formula I
- R. and R 3 are selected from the group consisting of hydrogen, hydroxy, C.-C 6 alkoxy, and C ⁇ - C 6 alkylcarbonyloxy;
- R 2 is selected from the group consisting of hydroxy, C.-C 6 alkoxy, and C 2 -C 6 alkenoxy, said alkenoxy together with the carbon atom of the phenyl ring forming an oxygen heterocycle;
- R 2 are neither hydroxy nor alkoxy; when R-, R 2 , and R 3 are hydrogen, R 4 is neither phenyl nor alkyloxyphenyl; and when R 3 is hydrogen and R 4 is phenyl, neither R. nor R 2 is alkylcarbonyloxy.
- Selected compounds of formula I are set forth in Tables 3-5. These compounds have affinity for adenosine receptors in general. Among these compounds, certain compounds have greater affinity for one type of adenosine receptors, e.g., A 3 , than other types of adenosine receptors. Therefore these compounds can be used to selectively block that type of adenosine receptors for which they have greater affinity. Thus, for instance, compounds 3b, 4, 6, 7, 8, 10, lib, lie, lid, 16, 20, 21 and 24 can be used to selectively block the A 3 adenosine receptors in a mammal .
- the present invention further provides a method of treating a mammal comprising selectively blocking the adenosine receptors of a mammal by administering to said mammal at least one compound selected from the group consisting of genistein, (+)dihydrogenistein, sakuranetin, ⁇ -naphthoflavone, ⁇ -naphthoflavone, amaryllidaceae, oxogalanthine lactam, acetylhaemanthine methiodide, 2,3- methylenedioxy-fluorene-9-one, hematoxylin, and arborinine .
- the present invention further provides a method of treating a mammal comprising selectively blocking one or more of the adenosine receptors of a mammal by administering to said mammal at least one compound of the formula II
- R. is selected from the group consisting of hydroxyl and C 1 -C 6 alkoxy
- R 2 is selected from the group consisting of styryl and phenylacetylenyl.
- Selected compounds of formula II are set forth in Tables 6-7. These compounds have affinity for adenosine receptors in general. Among these compounds, certain compounds have greater affinity for one type of adenosine receptors, e.g., A 3 , than other types adenosine of receptors. Therefore these compounds can be used to selectively block that type of adenosine receptors for which they have greater affinity. Thus, for instance, compound 38 can be used to selectively block the A 3 adenosine receptors in a mammal.
- Selected compounds of formula III are set forth in Tables 10 and 18. These compounds have affinity for adenosine receptors in general. Among these compounds, certain compounds have greater affinity for one type of receptors, e.g., A lf than other types adenosine of receptors. Therefore, these compounds can be used to selectively block that type of receptors for which they have greater affinity. Thus, for instance, compounds 52, 58, 60, 61, 62, 63, 64, 65, 68, 69, 70, 71, 75, 76, and 77, which have greater affinity to A- adenosine receptors than A- a receptors, can be used to selectively block A x adenosine receptors.
- compounds 63, 64, 65, 74, 75, 76, 79, 87, 90, 93-95, 98-101, 105-107, 109, 115-126, 129-130b, 133, and 136 which have greater affinity for A 3 adenosine receptors than A. or A-. a adenosine receptors, can be used to selectively block A 3 adenosine receptors.
- the present invention further provides a method of treating a mammal comprising selectively blocking one or more of the adenosine receptors of a mammal by administering to said mammal at least one compound of the formula IV
- R 2 is selected from the group consisting of hydrogen and C ⁇ C g alkyl
- R 3 is selected from the group consisting of hydrogen and C.-Cg alkyloxycarbonyl
- R 4 is selected from the group consisting of C--C 6 alkyl, phenyl C 2 -C 6 alkenyl, phenyl C 2 -C 6 alkynyl, aryl, and aryl substituted with one or more substituents selected from the group consisting of nitro and C.-Cg alkyloxy
- R s is selected from the group consisting of hydrogen, C.-Cg alkyloxy carbonyl, and aryl C.-Cg alkyloxycarbonyl
- R 6 is selected from the group consisting of hydrogen, aryl, and alkyl.
- pharmaceutically acceptable carriers described herein for example, vehicles, adjuvants, excipients, or diluents, are well-known to those who are skilled in the art and are readily available to the public. It is preferred that the pharmaceutically acceptable carrier be one which is chemically inert to the active compounds and one which has no detrimental side effects or toxicity under the conditions of use.
- compositions of the present invention are merely exemplary and are in no way limiting.
- Formulations suitable for oral administration can consist of (a) liquid solutions, such as an effective amount of the compound dissolved in diluents, such as water, saline, or orange juice; (b) capsules, sachets, tablets, lozenges, and troches, each containing a predetermined amount of the active ingredient, as solids or granules; (c) powders; (d) suspensions in an appropriate liquid; and (e) suitable emulsions.
- Liquid formulations may include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent.
- Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and corn starch.
- Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid, microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible carriers.
- Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the active ingredient, such carriers as are known in the art.
- a flavor usually sucrose and acacia or tragacanth
- pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the active ingredient, such carriers as are known in the art.
- the compounds of the present invention can be made into aerosol formulations to be administered via inhalation.
- aerosol formulations can be placed into pressurized acceptable propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like. They also may be formulated as pharmaceuticals for non- pressured preparations, such as in a nebulizer or an atomizer.
- Formulations suitable for parenteral administration include aqueous and non-aqueous, isotonic sterile injection solutions, which can contain anti-oxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and non-aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives.
- the compound can be administered in a physiologically acceptable diluent in a pharmaceutical carrier, such as a sterile liquid or mixture of liquids, including water, saline, aqueous dextrose and related sugar solutions, an alcohol, such as ethanol, isopropanol, or hexadecyl alcohol, glycols, such as propylene glycol or polyethylene glycol, glycerol ketals, such as 2, 2-dimethyl-l, 3- dioxolane-4-methanol, ethers, such as poly(ethyleneglycol) 400, an oil, a fatty acid, a fatty acid ester or glyceride, or an acetylated fatty acid glyceride with or without the addition of a pharmaceutically acceptable surfactant, such as a soap or a detergent, suspending agent, such as pectin, carbomers, methylcellulose, hydroxypropylmethylcellulose, or carboxymethylcellulose, or emulsifying agents and other pharmaceutical adju
- oils include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral.
- Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
- Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts
- suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides, (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and polyoxyethylenepolypropylene copolymers, (d) amphoteric detergents such as, for example, alkyl- ⁇ -aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
- the parenteral formulations will typically contain from about 0.5 to about 25% by weight of the active ingredient in solution. Suitable preservatives and buffers can be used in such formulations. In order to minimize or eliminate irritation at the site of injection, such compositions may contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17. The quantity of surfactant in such formulations ranges from about 5 to about 15% by weight. Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol .
- HLB hydrophile-lipophile balance
- parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example, water, for injections, immediately prior to use.
- sterile liquid carrier for example, water
- Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described.
- the compounds of the present invention may be made into injectable formulations.
- the requirements for effective pharmaceutical carriers for injectable compositions are well known to those of ordinary skill in the art. See Pharmaceutics and Pharmacy Practice. J.B. Lippincott Co., Philadelphia, PA, Banker and Chalmers, eds., pages 238-250 (1982) , and ASHP Handbook on Injectable Drugs, Toissel, 4th ed., pages 622-630 (1986) .
- the compounds of the present invention may be made into suppositories by mixing with a variety of bases, such as emulsifying bases or water-soluble bases.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams, or spray formulas containing, in addition to the active ingredient, such carriers as are known in the art
- the compounds of the present invention can be used in the treatment of any disease state or condition involving the release of inositol-1,4, 5-triphosphate (IP3) , diacylglycerol (DAG) , and free radicals and subsequent arachidonic acid cascades.
- IP3 inositol-1,4, 5-triphosphate
- DAG diacylglycerol
- free radicals and subsequent arachidonic acid cascades can be treated in accordance with the present inventive method, wherein one of the above-described compounds is acutely administered, e.g., within about a few minutes to about an hour of the onset or realization of symptoms.
- the method also has utility in the treatment of chronic disease states and conditions, in particular those conditions and disease states wherein chronic prophylactic or therapeutic administration of one of the above-described compounds will prevent the onset of symptoms or will reduce recovery time.
- diseases states and conditions include inflammatory disorders, such as vascular inflammation and arthritis, allergies, Crohn's disease, asthma, wound healing, stroke, cardiac failure, acute spinal cord injury, acute head injury or trauma, seizure, neonatal hypoxia (cerebral palsy; prophylactic treatment involves chronic exposure through placental circulation) , chronic hypoxia due to arteriovenous malformations and occlusive cerebral artery disease, severe neurological disorders related to excitotoxicity, Parkinson's disease, Huntington's chorea, and other diseases of the central nervous system (CNS) , cardiac disease, kidney disease, and contraception.
- CNS central nervous system
- these compounds can be significant cerebral protectants.
- the above compounds can be used to treat and/or protect against a variety of disorders, including, for example, seizures, transient ischemic shock, strokes, focal ischemia originating from thrombus or cerebral hemorrhage, global ischemia originating from cardiac arrest, trauma, neonatal palsy, hypovolemic shock, and hyperglycemia and associated neuropathies.
- the above method is applicable, for example, where a mammal has or is at risk of having a condition, disorder, or disease state associated with the cellular release of inositol- 1,4, 5-triphosphate or diacylglycerol.
- the method is also applicable when said mammal has or is at risk for hyperactivity and said compound in binding to said A 3 adenosine receptors functions as a locomotor depressant.
- the present inventive method is also applicable when said mammal has or is at risk for hypertension and said compound in binding to said A 3 adenosine receptors functions as a hypotensive agent.
- the method is also applicable when said mammal has or is at risk for anxiety and said compound in binding to said A 3 adenosine receptors functions as an anxiolytic agent.
- the method is furthermore applicable when said mammal has or is at risk for cerebral ischemia and said compound in binding to said
- a 3 adenosine receptors functions as a cerebroprotectant .
- the method is also applicable when said mammal has or is at risk for seizures and said compound in binding to said A 3 adenosine receptors functions as an antiseizure agent .
- the present inventive method can be administered chronically as well as acutely.
- the present inventive method includes the administration to an animal, such as a mammal, particularly a human, in need of the desired adenosine receptor-dependent response of an effective amount, e.g., a therapeutically effective amount, of one or more of the aforementioned present inventive compounds or pharmaceutically acceptable salts or derivatives thereof, alone or in combination with one or more other pharmaceutically active compounds. Any suitable pharmaceutically acceptable salts can be used.
- Example of suitable salts include carbonate, bicarbonate, sulfate, bisulfate, nitrate, halides, phosphates, oxalate, acetate, formate, citrates, and amino acid salts.
- Some of the compounds of the present invention can be utilized as functionalized congeners for coupling to other molecules, such as amines and peptides.
- the use of such congeners provide for increased potency, prolonged duration of action, specificity of action, and prodrugs. Water solubility is also enhanced, which allows for reduction, if not complete elimination, of undesirable binding to plasma proteins and partition into lipids. Accordingly, improved pharmacokinetics can be realized.
- the dose administered to an animal, particularly a human, in the context of the present invention should be sufficient to effect a prophylactic or other therapeutic response in the animal over a reasonable time frame.
- dosage will depend upon a variety of factors including the strength of the particular compound employed, the age, species, condition, and body weight of the animal, as well as the severity/stage of the disease or condition.
- the size of the dose will also be determined by the route, timing and frequency of administration as well as the existence, nature, and extent of any adverse side-effects that might accompany the administration of a particular compound and the desired physiological effect. It will be appreciated by one of skill in the art that various conditions or disease states, in particular chronic conditions or disease states, may require prolonged treatment involving multiple administrations.
- Suitable doses and dosage regimens can be determined by conventional range-finding techniques known to those of ordinary skill in the art. Generally, treatment is initiated with smaller dosages, which are less than the optimum dose of the compound. Thereafter, the dosage is increased by small increments until the optimum effect under the circumstances is reached. For convenience, the total daily dosage may be divided and administered in portions during the day if desired. In proper doses and with suitable administration of certain compounds, the present invention provides for a wide range of selective adenosine receptor-dependent responses. Exemplary dosages range from about 0.01 to about 100 mg/kg body weight of the animal being treated/day. Preferred dosages range from about 0.1 to about 10 mg/kg body weight/day.
- Bay K 8422 l,4-dihydro-2,6-dimethyl-5-nitro-4- [2- (trifluoromethyl)-ph enyl] -3-pyridine carboxylic acid methyl ester
- Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer and spectra were taken in d6-DMSO. Electron-impact mass spectrometry was performed with a VG7070F mass spectrometer at 6 kV. Elemental analysis was performed by Atlantic Microlabs, Inc. (Norcross, GA) .
- EXAMPLE 2 This Example illustrates the procedure for determining the affinity of the present inventive compounds for adenosine receptors.
- K. values at A x and A 2a receptors were determined in radioligand binding assays in brain membranes vs. [3H] PIA or [3H.CGS 21680) , respectively.
- Schwabe et al . Naunvn-Schmiedeber ' s Arch. Pharmacol. , 313. 179-187
- the flavone derivatives can be accomplished following the teachings in literature and the procedures set forth herein. Wollenweber et al . , J.B. Harborne. (Chapman and Hall, eds.) , London, 259-335 (1993) ; Cushman et al. , J. Med. Chem.. 37, 3373-3382 (1994) ; Cunningham et al. , Anti-Cancer Drug Design, 7_, 365-384 (1992) ; Lee et al . , Tetrahedron. 51. 4909-4922 (1995) . As shown in Figure 2, the flavone ring system could be formed via a condensation of a
- the coupling constant for the 2- and 3-position protons for compounds 15 - 16 is -12 Hz, which is characteristic of trans vicinal coupling.
- Williams et al . Spectroscopic Methods in Organic Chemistry. 4th ed. , McGraw Hill, London (1989) .
- An alternate approach to providing olefinic substitution at the 2-position (Figure 2) was to condense a 2-methylchromone (such as the natural product visnagin, 17)27 with an aldehyde, Hafez, Czech. Chem. Commun., 59, (1994) , under basic conditions. This method was used to prepare the 2-cinnamyl derivative, Ji et al., J. Med.
- EXAMPLE 4 This Example illustrates the alkylation of galangin, 1 and orin, 5a.
- Galangin (27 mg, 0.1 mmol) was dissolved in dried acetone (20 mL) , solid potassium carbonate (0.5 g) and dimethyl sulfate (1 mL) were added, and the mixture was refluxed for 4 hrs, then cooled to room temperature. The solution was filtered and evaporated, water (20 mL) and concentrated ammonium hydroxide (2 mL) were added and the solution was extracted with ethyl acetate (15 mL x 2) . The solvent was evaporated and the residue was recrystallized from methanol/water to give 21 mg product (67%) .
- Galangin (30 mg, 0.11 mmole) was dissolved in dried acetone (45 mL) , solid potassium carbonate (0.5 g) and iodoethane were added, and the mixture was refluxed for overnight. The solution was filtered and evaporated, water (20 mL) and concentrated ammonia (1 mL) were added, and the solution was extracted with ethyl acetate. The ethyl acetate was evaporated, and the crude mass was purified on a preparative silica TLC plate to give 12 mg
- Galangin (30 mg, 0.11 mmole) was dissolved in dried acetone (45 mL) , solid potassium carbonate (0.5 g) and 1-iodopropane were added and the mixture was refluxed overnight. The solution was filtered and evaporated, water (20 mL) and concentrated ammonia (1 mL) were added, and the solution was extracted with ethyl acetate. The ethyl acetate was evaporated, and the crude mass was purified on a preparative TLC plate (silica) to give 25 mg
- EXAMPLE 8 This Example illustrates the synthesis of 4-methyloxy-7-trans-styryl-visnagin (19) and 4-ethyloxy-7-trans-styryl-visnagin (20) .
- EXAMPLE 10 This Example illustrates the synthesis of 4-methoxy- 7-phenylbutadienyl visnagin (22) and 4-ethoxy-7- phenylbutadienyl visnagin (23) .
- Compound 22 was prepared by dissolving visnagin and cinnamaldehyde in ethanol in the presence of sodium ethoxide according to the above procedure for preparing compounds 19 and 20 to yield compound 22, mp 162-165°C.
- Mass (El) m/z 344 (M+, base) .
- Galangin (9.0 mg, 33 ⁇ mol) was dissolved in 1 mL DMF and treated with acetic anhydride (0.2 mL) and 4,4-dimethylaminopyridine (3 mg) . After stirring for 10 min, 2 mL of 1 N NaH 2 P0 4 was added. A white precipitate was removed by filtration, and recrystallized from methanol/water to yield 11.4 mg of a solid (87%) , which was homogeneous by TLC (Rf 0.56, silica, chloroform: methanol: acetic acid, 95:4:1) . Mass spec: 414
- EXAMPLE 13 This Example illustrates another method of synthesis of 2' , 3 , 4 ' , 5, 7-pentamethyloxyflavone (38, also designated as compound 6 in Example 4) .
- Morin hydrate (25 mg, 82 ⁇ mol, Aldrich) was dissolved in dry acetone (20 mL) , in which was suspended potassium carbonate (1.0 g) . Dimethyl sulfate (1.0 mL, 11 mmol) was added, and the mixture was refluxed for four h under nitrogen. After cooling in an ice bath, 2 mL concentrated ammonium hydroxide were added in aliquots followed by 20 mL water. The solution was extracted with ethyl acetate.
- EXAMPLE 14 This Example illustrates the synthesis of compound 45.
- Compound 45 was synthesized by a modification of a literature procedure. Fales et al . J. Amer. Chem. Soc.. 77, 5885-5890 (1955) . 6-Bromo-3 ,4-dimethoxybenzoic acid (6-bromoveratric acid, 2.0 g, 7.7 mmol, Spectrum Chem. Corp., New Brunswick, NJ) was dissolved in 50% EtOH/H20 (100 mL) , and treated with resorcinol (0.85 g, 7.7 mmol) , 50 mg copper powder, 50 mg cupric acetate, and sodium hydroxide (0.31 g, 7.7 mmol) .
- This Example illustrates the synthesis of 2, 3-methylenedioxy-fluorene-9-one (49) .
- EXAMPLE 16 This Example illustrates the efficacy of some the flavonoid derivatives of the present invention in blocking adenosine receptors.
- the affinities of the flavonoid derivatives 1-lld were determined using radioligand binding assays, and the results thereof are set forth in Table 3.
- EXAMPLE 17 This Example further illustrates the efficacy of the compounds of the present invention in blocking adenosine receptors.
- the affinities of the flavonoid derivatives 12- 16 were determined using radioligand binding assays, and the results thereof are set forth in Table 4.
- d Displacement of ⁇ 10 % of specific binding at the indicated concentration in M. 10 e 2, 3-trans
- EXAMPLE 18 This Example further illustrates the efficacy of the compounds of the present invention in blocking adenosine receptors.
- the affinities of the flavonoid derivatives 17- 24 were determined using radioligand binding assays, and the results thereof are set forth in Table 5.
- CH CH- d (10 ⁇ 4 ) d (10 "4 ) d dO "4 )
- CH CH-Ph 23 OC 2 H 5
- CH CH- d (10- 4 ) 167 45.5 ⁇ 10.3 >4
- CH CH-Ph 24 OCH
- CH NH-Ph d (lo "4 ) d (10 4 ) 9.18+2.56 >20
- EXAMPLE 19 This Example further illustrates the efficacy of the compounds of the present invention in blocking adenosine receptors.
- the affinities of the flavonoid derivatives 25- 39 were determined using radioligand binding assays, and the results thereof are set forth in Table 6.
- EXAMPLE 20 This Example further illustrates the efficacy of the compounds of the present invention in blocking adenosine receptors.
- the affinities of the flavonoid derivatives 40- 51 were determined using radioligand binding assays, and the results thereof are set forth in Table 7.
- EXAMPLE 21 This Example further illustrates the efficacy of the compounds of the present invention in blocking adenosine receptors.
- the effects of flavonoid derivatives 3a, 15, and 20 on the A 3 agonist-elicited inhibition of adenylyl cyclase were determined, and the results thereof are set forth in Table 8.
- Compounds 1, 11a, 12, and 17 were obtained from Fluka, Ronkonoma, NY or from Aldrich, St. Louis, MO.
- Compounds 13 and 14 were obtained from Apin Chemicals, Ltd. , Oxon, UK.
- Compound 5a was obtained from K+K Laboratories, Jamaica, NY.
- Compounds 4, 27-34, 43, and 44 were obtained from Fluka, Ronkonoma, NY or from Aldrich, St. Louis, MO.
- Compound 36 was obtained from Apin Chemicals, Ltd., Oxon, UK.
- Compounds 25, 26, 27, and 42 were obtained from K+K Laboratories, Jamaica, NY.
- Compound 40 was obtained from
- This Example illustrates a procedure for the oxidation (see Figure 7) of 1, 4-dihydropyridine-3 , 5-dicarboxylate esters .
- EXAMPLE 24 This Example illustrates a procedure for the preparation (see Figure 6) of 1, 4-dihydropyridine- 3, 5-dicarboxylate esters.
- Triethylamine (20 mg) was added to a mixture of compound 135 (90 mg, 0.2 mmol) , diphenyl phosphoryl azide (56 mg, 0.2 mmol) , and ethanethiol (20 mg, 0.3 mmol) in DMF (1 mL) with stirring and ice cooling. The mixture was stirred at room temperature for 3 hr, diluted with dichloromethane (20 mL) , washed with water (10 mL x 2) , dried with sodium sulfate. The solvent was evaporated and the residue was carried out for deprotection with 1NHC1 to give 15 mg of product.
- TBAF (hydrate, 208 mg, 0.8 mmol) was added to a solution of 138 (115 mg, 0.21 mmol) in DMF (1 mL) .
- the mixture was stirred under argon at room temperature for 2h, diluted with ethyl acetate (20 mL) , washed with IN HCI (5 mL) , H 2 0 (20m mL x 2) and brine (20 mL x 2) , dried with magnesium sulfate.
- the solvent was evaporated and residue was separated with preparative TLC plates to give 80 mg of product.
- EXAMPLE 25 This Example illustrates the affinities of certain dihydropyridine derivatives. The affinities were determined in radioligand binding assays, and the results thereof are set forth in Table .
- EXAMPLE 26 This Example illustrates the affinities of certain pyridine derivatives. The affinities were determined in radioligand binding assays, and the results thereof are set forth in Table 10.
- EXAMPLE 27 This Example illustrates the affinities of certain dihydropyridine and pyridine derivatives. The affinities were determined in radioligand binding assays, and the results thereof are set forth in Tables 11-12.
- R 2 Methyl, Ki ( ⁇ M) or % inhibition'
- Ph-C C- 25 ⁇ 3%(10 4 )
- EXAMPLE 28 This Example illustrates a method of synthesis and resolution of diastereomers of certain 1,4-dihydropyridines .
- the chemical synthesis and resolution are outlined in Figures 9A-9B. Characterization of the 1,4-dihydropyridines derivatives is set forth in Table 13.
- each of the isolated isomers was shown to be a pure diastereomer and with the peaks of the other isomer not visible in the spectrum.
- EuFOD 4 mg/mL
- the resonances of the S,S isomer were shifted downfield to a greater degree in the presence of the NMR shift reagent.
- the signals from the 4-H were shifted by EuFOD by >0.7 ppm in the S,S isomer and by only 0.3 ppm in the R,S isomer.
- the 2,2-dimethyl-l, 3-dioxolane moiety also served as a protected form of a diol, 211a obtained following deprotection in HCl/THF (Fig. 9B) .
- This diol showed a selective reactivity vs the 5-ethyl ester in basic transesterfication reactions.
- the 3-ethyl ester, 214 was obtained in this manner using sodium hydroxide in 95% ethanol.
- the resolved diastereomers 209a and 210a were also deprotected separately, to give 212a and 213a. These diols were then tested for biological activity. The results obtained are set forth in Table 14.
- Table 13 Chemical shifts of diastereomeric 1,4- dihydropyridine derivatives in proton NMR.
- CGS15943 refers to compound 144 wherein R. and R 2 are hydrogen.
- EXAMPLE 30 This Example illustrates the affinities of certain triazoloquinazolines .
- the affinities were determined in radioligand binding assays, and the results are set forth in Tables 15-16.
- c Displacement of specific [ 125 I]AB-MECA binding at human A 3 receptors expressed in HEK cells, in membranes, expressed as
- Table 16 Affinities of triazoloquinazoline derivatives in radioligand binding assays at A 1# A 2a , and A 3 receptors. a'c
- EXAMPLE 31 This Example illustrates the utility of the adenosine receptor antagonists of the present invention in the killing of cancer cells. Reagents:
- HL-60 and U-937 cells were obtained from the ATCC (Bethesda, MD) .
- RPMI 1640 medium and fetal bovine serum were obtained from Gibco BRL (Gaithersburg, MD) .
- IB-MECA (2- [4- [92-carboxyethyl)phenyl]ethylamino] -5' -N- ethylcarboxamidoadenosine
- Cl-IB-MECA N € - (3-iodobenzyl) - 2-chloro-adenosine-5' -N-methyluronamide
- the A 3 selective adenosine antagonists 101 and 153 were synthesized using procedures described earlier.
- L-249313 (6- carboxymethyl-5, 9-dihydro-9-methyl-2-phenyl-
- the HL-60 cells were maintained in RPMI 1640 supplmented with 10% fetal calf serum, 100 units/ml penicillin, 100 mg/ml streptomycin and 2 mM L-glutamine. The cells were split every third day, and 2 days before each experiment cultures were diluted to 2 X 10 5 cells/ml. For analysis of DNA content, aliquots of 2 ml were placed into 12-well flat-bottomed plates (Costar, Cambridge, MA, USA) containing 2 ⁇ 6 ⁇ l test-compound solutions at defined concentrations or 6 ⁇ l DMSO (diluting medium) . Live cell counting was carried out using 0.1% trypan blue.
- DNA content analysis by flow cytometry Cells were fixed by adding ⁇ 10 7 cells suspended in 1 ml of PBS of 1 ml 80% ethanol at -20°C and stored for 48-120 hrs. After the cells were washed twice with PBS, the cells were stained with 20 mg/ml chromomycin A3 dissolved in PBS containing 2 mM MgCl 2 by incubation in subdued light (30 min; 4 °C) . The cells were then analyzed using a FACScan flow cytometer (Becton Dickinson, Mountain View, CA) .
- FACScan flow cytometer Becton Dickinson, Mountain View, CA
- the cells were restained by propidium iodide l ⁇ g/ml in PBS for 15 min. After every step, the cells were rinsed 2-3 times with fresh PBS for 5 min each. Finally, the glass slides were sealed with nail polish. The slides were stored at 4 °C in the dark.
- Flow cytometry data obtained are set forth in Figures 10A and 10B and indicate that high concentrations (_> ⁇ M) of the A 3 receptor agonists, IB-MECA and Cl-IB-MECA, caused apoptosis in HL-60 promyelocytic leukemia cells. A similar response was observed in U-937 histiocytic lymphoma cells.
- the percent of apoptotic cells was estimated from the percent cells having hypodiploid DNA content in a DNA frequency histogram.
- Figures 10A-B or general cell death, as determined using trypan blue staining ( Figures 11A-D) .
- Figures 11A-D cell death was studied in HL-60 cells ( Figures 11A-B) and in U-937 cells ( Figures 11C-D) over a time course of 9 days. Cells in culture receiving no drug treatment proliferated geometrically until nearly the end of the experiment. In the presence of 0.01 or 1.0 ⁇ M Cl-IB-MECA alone, no deviation from this growth curve was observed.
- Cl-IB-MECA at a concentration of 20 or 40 ⁇ M caused death of >90% of HL-60 cells in culture after 4 days followed by a very slow increase in cell number.
- U-937 cells were slightly less sensitive to this agonist, with 40 ⁇ M Cl-IB-MECA required to obtain a similar effect.
- this A 3 agonist at sublethal concentrations, including the very low concentration of 10 nM, reversed the impairment of cell proliferation induced by either of the selective antagonists in both HL-60 cells ( Figures 11A-B) and U-937 cells ( Figures 11C-D) .
- bak protein was also induced in both HL-60 cells and U-937 cells using the triazoloquinazoline A 3 antagonist 153 (10 nM) .
- Cl- IB-MECA at 30 ⁇ M but not concentrations ⁇ 1 ⁇ M, induced a significant level of expression of bak in both HL-60 cells and U-937 cells.
- Various other cell lines were investigated for the expression of bak in response to the A 3 agonist .
- Cl-IB-MECA at 10 ⁇ M was found to induce the expression of bak in MCF7 breast adenocarcinoma cells and 1321N astrocytoma cells, but not in U373 astrocytoma cells, so the upregulation of bak expression by A 3 agonists appears to be a widespread but not universal phenomenon.
- a 3 agonists and antagonists by virtue of regulating programmed cell death, may have usefulness in treating diseases either in which cytotoxicity is undesirable, such as neurodegeneration, or desirable, such as cancer and inflammation.
- the level of agonist or antagonist should be carefully balanced to obtain the desired effect on the cells, e.g., death or protection.
- Table 17 Percentage of apoptotic cell in HL60 and U937 after 48 hour treatment with A 3 antagonists
- EXAMPLE 32 This Example illustrates the utility of the present inventive compounds in preserving neurons in stroke suffered by an animal. Female Mongolian gerbils received bilateral 10 min. carotid occlusion, followed by injection 15 min. later with
- EXAMPLE 33 This Example illustrates the bronchoconstricting effect of A 3 adenosine receptor agonists and the utility of A 3 adenosine receptor antagonists in combating the bronchconstricting effect of A 3 adenosine receptor agonists.
- Adenosine receptors have been implicated in the bronchoconstriction (BC) in allergic asthma. It was found that after selectively blocking A- receptors by diphenyl cyclopentyl xanthine, IB-MECA, an A 3 agonist, induced a dose-dependent BC in allergic rabbits.
- a 3 adenosine receptor antagonists can combat the BC induced by the A 3 agonist.
- New Zealand white pasteurella free rabbit litter mates were exposed to A 3 adenosine agonist APNEA, IB-MECA, or Cl- IB-MECA and the percent change in dynamic compliance (C dyn ) was measured as a function of the adenosine dose using known procedures. Ali et al. , J. Pharmacol. & Exper. Therap. , 268, 1328-1334 (1993) . Allergy was induced in the rabbits using dust mites.
- Fig. 12 The results obtained are set forth in Fig. 12 (open symbols) and show that the adenosine agonist causes the Cdyn to decrease, which is an indication of bronchoconstriction.
- an adenosine antagonist compound 101
- the antagonist was administered at a constant dose of 10 "5 M through a 2 minute aerosilization, followed by a 15 minute wait before the measurement of dynamic compliance.
- the results confirm that the A 3 adenosine receptor antagonist is effective in combating inflammatory disorders such as asthma.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Hydrogenated Pyridines (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
























































Claims






Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU22466/97A AU709190B2 (en) | 1996-01-29 | 1997-01-29 | Dihydropyridine-, pyridine-, benzopyran- one- and triazoloquinazoline derivative, their preparation and their use as adenosine receptor antagonists |
| US09/117,598 US6066642A (en) | 1996-01-29 | 1997-01-29 | Dihydropyridine-, pyridine-, benzopyran-4-one- and triazoloquinazoline derivative, their preparation and their use as adenosine receptor antagonists |
| EP97905627A EP0885192B8 (en) | 1996-01-29 | 1997-01-29 | Dihydropyridine-, pyridine-, benzopyran- one- and triazoloquinazoline derivative, their preparation and their use as adenosine receptor antagonists |
| JP52706597A JP4431638B2 (en) | 1996-01-29 | 1997-01-29 | Dihydropyridine-, pyridine-, benzopyran-one- and triazoloquinazoline derivatives, their preparation and their use as adenosine receptor antagonists |
| AT97905627T ATE548351T1 (en) | 1996-01-29 | 1997-01-29 | DIHYDROPYRIDINE-PYRIDINE, BENZOPYRANONE AND TRIAZOLOQUINAZOLINE DERIVATIVES, THEIR PREPARATION AND USE AS ADENOSINE RECEPTOR ANTAGONISTS |
| CA002244774A CA2244774C (en) | 1996-01-29 | 1997-01-29 | Dihydropyridine derivatives, their preparation and their use as adenosine receptor antagonists |
| HK99102653.6A HK1017676B (en) | 1996-01-29 | 1997-01-29 | Dihydropyridine-, pyridine-, benzopyran- one- and triazoloquinazoline derivative, their preparation and their use as adenosine receptor antagonists |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US1073796P | 1996-01-29 | 1996-01-29 | |
| US60/010,737 | 1996-01-29 | ||
| US2119196P | 1996-07-03 | 1996-07-03 | |
| US60/021,191 | 1996-07-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1997027177A2 true WO1997027177A2 (en) | 1997-07-31 |
| WO1997027177A3 WO1997027177A3 (en) | 1997-11-13 |
Family
ID=26681538
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1997/001252 Ceased WO1997027177A2 (en) | 1996-01-29 | 1997-01-29 | Dihydropyridine-, pyridine-, benzopyran-4-one- and triazoloquinazoline derivatives, their preparation and their use as adenosine receptor antagonists |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6066642A (en) |
| EP (2) | EP2311806A3 (en) |
| JP (1) | JP4431638B2 (en) |
| AT (1) | ATE548351T1 (en) |
| AU (1) | AU709190B2 (en) |
| CA (1) | CA2244774C (en) |
| WO (1) | WO1997027177A2 (en) |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000002861A1 (en) * | 1998-07-10 | 2000-01-20 | The United States Of America, Represented By Secretary, Department Of Health And Human Services | A3 adenosine receptor antagonists |
| WO2000003741A3 (en) * | 1998-07-16 | 2000-09-28 | Univ Pennsylvania | Methods for reducing intraocular pressure using a3-adenosine antagonists |
| EP1108710A4 (en) * | 1998-07-23 | 2001-11-21 | Chen Ing Jun | Guaiacoxypropanolamines with alpha/beta-adrenergic blocking activity |
| US6358964B1 (en) * | 2000-07-26 | 2002-03-19 | King Pharmaceuticals Research And Development, Inc. | Adenosine, A3 receptor modulators |
| WO2002070520A1 (en) * | 2001-03-07 | 2002-09-12 | Bayer Aktiengesellschaft | Substituted 2,6-diamino-3,5-dicyano-4-aryl-pyridines and their use as adenosine receptor-selective ligands |
| WO2002079196A1 (en) * | 2001-03-30 | 2002-10-10 | Bayer Aktiengesellschaft | Substituted 2-carba-3,5-dicyano-4-aryl-6-aminopyridines and the use of the same as selective ligands of the adenosine receptor |
| US6482841B1 (en) * | 1997-10-09 | 2002-11-19 | Cermol S.A. | Pyridyl compounds and pharmaceutical compositions containing them |
| WO2002009699A3 (en) * | 2000-07-28 | 2003-01-03 | Immupharm Aps | Method of treating symptoms of common cold, allergic rhinitis and infections relating to the respiratory tract |
| WO2003031430A3 (en) * | 2001-10-04 | 2004-04-08 | Brane Tech S R L | Flavonoid compounds and their pharmaceutical uses |
| JP2005518384A (en) * | 2001-12-21 | 2005-06-23 | サノフィ−アベンティス | Triazolo-quinoline derivatives useful as adenosine receptor ligands |
| US7087589B2 (en) | 2000-01-14 | 2006-08-08 | The United States Of America As Represented By The Department Of Health And Human Services | Methanocarba cycloakyl nucleoside analogues |
| WO2011029956A1 (en) * | 2009-09-14 | 2011-03-17 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Flavones and flavanones derivates as dna methyltransferases inhibitors |
| US8304412B2 (en) | 2006-12-01 | 2012-11-06 | Bayer Intellectual Property Gmbh | Cyclically substituted 3,5-dicyano-2-thiopyridines and use thereof |
| US8420825B2 (en) | 2009-01-29 | 2013-04-16 | Bayer Intellectual Property Gmbh | Alkylamino-substituted dicyanopyridines and their amino acid ester prodrugs |
| US8426602B2 (en) | 2008-03-11 | 2013-04-23 | Bayer Intellectual Property Gmbh | Heteroaryl-substituted dicyanopyridines and their use |
| US8440700B2 (en) | 2007-07-27 | 2013-05-14 | Bayer Intellectual Property Gmbh | Substituted aryloxazoles and their use |
| US8518957B2 (en) | 2009-12-02 | 2013-08-27 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methanocarba adenosine derivatives, pharmaceutical compositions, and method of reducing intraocular pressure |
| US8609686B2 (en) | 2007-12-20 | 2013-12-17 | Bayer Intellectual Property Gmbh | Substituted azabicyclic compounds and the use thereof |
| US8618119B2 (en) | 2007-12-20 | 2013-12-31 | Bayer Intellectual Property Gmbh | Fused cyanopyridines and the use thereof |
| US8653109B2 (en) | 2006-09-08 | 2014-02-18 | Bayer Intellectual Property Gmbh | Substituted bipyridine derivatives and their use as adenosine receptor ligands |
| US8703696B2 (en) | 2007-08-01 | 2014-04-22 | Bayer Intellectual Property Gmbh | Dipeptoid prodrugs and the use thereof |
| US8703934B2 (en) | 2006-12-01 | 2014-04-22 | Bayer Intellectual Property Gmbh | Substituted 4-amino-3,5-dicyano-2-thiopyridines and use thereof |
| US8741834B2 (en) | 2008-12-16 | 2014-06-03 | Bayer Intellectual Property Gmbh | Dipeptoid prodrugs and the use thereof |
| CN103864741A (en) * | 2014-03-06 | 2014-06-18 | 陈朝银 | Preparation and application of walnut shell flavone lipid-decreasing active ingredient |
| WO2014101113A1 (en) * | 2012-12-28 | 2014-07-03 | Merck Sharp & Dohme Corp. | Piperazine-substituted 7-methoxy-[1,2,4]triazolo[1,5-c]quinazolin-5-amine compounds with a2a antagonist properties |
| US8791146B2 (en) | 2008-05-29 | 2014-07-29 | Bayer Intellectual Property Gmbh | 2-alkoxy-substituted dicyanopyridines and their use |
| US9040566B2 (en) | 2010-09-02 | 2015-05-26 | Bayer Intellectual Property Gmbh | Adenosine A1 agonists for the treatment of glaucoma and ocular hypertension |
| US9187428B2 (en) | 2010-06-30 | 2015-11-17 | Bayer Intellectual Property Gmbh | Substituted dicyanopyridines and use thereof |
| EP3189837A4 (en) * | 2014-09-05 | 2018-01-17 | Oneness Biotech Co., Ltd | Use of flavonoid compound in preparation of composition for healing wound |
Families Citing this family (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HN1998000125A (en) * | 1997-08-28 | 1999-02-09 | Pfizer Prod Inc | 2-AMINOPYRIDIDS WITH BRANCHED ALCOXY SUBSTITUTES |
| US6448253B1 (en) * | 1998-09-16 | 2002-09-10 | King Pharmaceuticals Research And Development, Inc. | Adenosine A3 receptor modulators |
| US6921825B2 (en) | 1998-09-16 | 2005-07-26 | King Pharmaceuticuals Research & Development, Inc. | Adenosine A3 receptor modulators |
| JP3662519B2 (en) * | 2000-07-13 | 2005-06-22 | シャープ株式会社 | Optical pickup |
| US6869956B2 (en) * | 2000-10-03 | 2005-03-22 | Bristol-Myers Squibb Company | Methods of treating inflammatory and immune diseases using inhibitors of IκB kinase (IKK) |
| EP1465634B1 (en) | 2001-12-12 | 2014-10-22 | The Government of the United States of America, as represented by the Secretary Department of Health and Human Services | Methods for using adenosine receptor inhibitors to enhance immune response and inflammation |
| WO2004000224A2 (en) * | 2002-06-24 | 2003-12-31 | King Pharmaceuticals Research & Development, Inc. | Enhancing treatment of mdr cancer with adenosine a3 antagonists |
| MXPA04001731A (en) * | 2002-06-24 | 2004-05-31 | King Pharmaceuticals Res & Dev | Enhancing treatment of mdr cancer with adenosine a3 antagonists. |
| ATE411996T1 (en) * | 2002-09-30 | 2008-11-15 | Bayer Healthcare Ag | CONDENSED AZOLPYRIMIDINE DERIVATIVES |
| EP1663227A2 (en) * | 2003-09-10 | 2006-06-07 | Synta Pharmaceuticals Corporation | Dihydropyridine compounds for treating or preventing metabolic disorders |
| US7473884B2 (en) * | 2005-04-21 | 2009-01-06 | Avago Technologies Ecbu Ip (Singapore) Pte. Ltd. | Orientation determination utilizing a cordless device |
| EP2258372B8 (en) * | 2005-06-07 | 2012-12-19 | Kyowa Hakko Kirin Co., Ltd. | A2A antagonists for use in the treatment of motor disorders |
| JP2010505848A (en) * | 2006-10-06 | 2010-02-25 | ザ トラスティーズ オヴ ザ ユニヴァーシティー オヴ ペンシルバニア | Effective delivery of species-crossed A3 adenosine receptor antagonists to reduce intraocular pressure |
| US8916570B2 (en) | 2008-03-31 | 2014-12-23 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | A3 adenosine receptor agonists and antagonists |
| AU2009231978C1 (en) * | 2008-03-31 | 2014-01-30 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Purine derivatives as A3 adenosine receptor- selective agonists |
| EP2331542B1 (en) * | 2008-08-01 | 2016-07-27 | The United States of America, as Represented by The Secretary, Department of Health and Human Services | A3 adenosine receptor antagonists and partial agonists |
| US9181253B2 (en) | 2008-08-01 | 2015-11-10 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Adenosine receptor agonists, partial agonists, and antagonists |
| WO2010055384A1 (en) * | 2008-11-17 | 2010-05-20 | Glenmark Pharmaceuticals S.A. | Chromenone derivatives as trpv3 antagonists |
| WO2010132671A1 (en) * | 2009-05-15 | 2010-11-18 | The University Of Kentucky Research Foundation | Treatment of mci and alzheimer's disease |
| US9968574B2 (en) * | 2009-05-15 | 2018-05-15 | The University Of Kentucky Research Foundation | Treatment of MCI and Alzheimer's disease |
| WO2011123518A1 (en) | 2010-03-31 | 2011-10-06 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Adenosine receptor agonists for the treatment and prevention of vascular or joint capsule calcification disorders |
| AU2011282776B2 (en) | 2010-07-27 | 2014-06-12 | Boston Medical Center Corporation | Aryl hydrocarbon receptor (AhR) modifiers as novel cancer therapeutics |
| WO2012068539A1 (en) * | 2010-11-19 | 2012-05-24 | Limerick Biopharma, Inc. | Use and composition of quercetin-3'-o-sulfate for therapeutic treatment |
| JP2014524426A (en) | 2011-08-12 | 2014-09-22 | ソーク インスティテュート フォー バイオロジカル スタディーズ | Neuroprotective polyphenol analog |
| WO2013025484A1 (en) * | 2011-08-12 | 2013-02-21 | Lapchak Paul A | Polyphenol analogs to treat ischemia |
| US9227979B2 (en) * | 2012-01-25 | 2016-01-05 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Fluorescent antagonists of the A3 adenosine receptor |
| CN104270945B (en) * | 2012-03-19 | 2017-03-29 | 巴克老龄化研究所 | APP-specific BACE inhibitors (ASBI) and uses thereof |
| HK1254892A1 (en) | 2016-01-14 | 2019-08-02 | 韩德株式会社 | Compounds antagonizing a3 adenosine receptor, method for preparing them, and medical-use thereof |
| CN110248678A (en) | 2016-12-03 | 2019-09-17 | 朱诺治疗学股份有限公司 | The method for adjusting CAR-T cell |
| BR112019011065A2 (en) | 2016-12-03 | 2019-10-01 | Juno Therapeutics Inc | methods for determining car cell dosage |
| KR20190098747A (en) | 2016-12-05 | 2019-08-22 | 주노 쎄러퓨티크스 인코퍼레이티드 | Method of manufacturing engineered cells for adoptive cell therapy |
| AU2018275894B2 (en) | 2017-06-02 | 2025-04-24 | Juno Therapeutics, Inc. | Articles of manufacture and methods for treatment using adoptive cell therapy |
| JP2020526194A (en) | 2017-06-29 | 2020-08-31 | ジュノー セラピューティクス インコーポレイテッド | Mouse model for assessing toxicity associated with immunotherapeutic agents |
| PT3703750T (en) | 2017-11-01 | 2025-01-17 | Memorial Sloan Kettering Cancer Center | Chimeric antigen receptors specific for b-cell maturation antigen and encoding polynucleotides |
| SG11202003501XA (en) | 2017-11-01 | 2020-05-28 | Juno Therapeutics Inc | Antibodies and chimeric antigen receptors specific for b-cell maturation antigen |
| WO2019089858A2 (en) | 2017-11-01 | 2019-05-09 | Juno Therapeutics, Inc. | Methods of assessing or monitoring a response to a cell therapy |
| MA51210A (en) | 2017-12-01 | 2020-10-07 | Juno Therapeutics Inc | METHODS FOR DOSING AND MODULATING GENETICALLY MODIFIED CELLS |
| MA51184A (en) | 2017-12-15 | 2020-10-21 | Juno Therapeutics Inc | ANTI-CCT5 BINDING MOLECULES AND RELATED METHODS OF USE |
| MA54078A (en) | 2018-11-01 | 2021-09-15 | Juno Therapeutics Inc | METHODS FOR THE TREATMENT WITH CHEMERA ANTIGEN RECEPTORS SPECIFIC FOR B LYMPHOCYTE MATURATION ANTIGEN |
| EP3873937A2 (en) | 2018-11-01 | 2021-09-08 | Juno Therapeutics, Inc. | Chimeric antigen receptors specific for g protein-coupled receptor class c group 5 member d (gprc5d) |
| IL283218B2 (en) | 2018-11-16 | 2025-11-01 | Juno Therapeutics Inc | Methods of dosing engineered t cells for the treatment of b cell malignancies |
| PT3886875T (en) | 2018-11-30 | 2024-06-27 | Juno Therapeutics Inc | METHODS FOR TREATMENT USING ADOPTIVE CELL THERAPY |
| SG11202107976SA (en) | 2019-01-29 | 2021-08-30 | Juno Therapeutics Inc | Antibodies and chimeric antigen receptors specific for receptor tyrosine kinase like orphan receptor 1 (ror1) |
| JP2023522857A (en) | 2020-04-10 | 2023-06-01 | ジュノー セラピューティクス インコーポレイテッド | Methods and Uses for Chimeric Antigen Receptor Engineered Cell Therapy Targeting B Cell Maturation Antigens |
| EP4543923A1 (en) | 2022-06-22 | 2025-04-30 | Juno Therapeutics, Inc. | Treatment methods for second line therapy of cd19-targeted car t cells |
| JP2025525937A (en) | 2022-08-05 | 2025-08-07 | ジュノー セラピューティクス インコーポレイテッド | Chimeric antigen receptor specific for GPRC5D and BCMA |
| KR20250135354A (en) | 2022-12-13 | 2025-09-12 | 주노 쎄러퓨티크스 인코퍼레이티드 | Chimeric antigen receptor specific for BAFF-R and CD19 and methods and uses thereof |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2005116C3 (en) * | 1970-02-05 | 1980-02-14 | Bayer Ag, 5090 Leverkusen | Symmetrical 1,4-dihydropyridine-3,5-dicarboxylic acid esters |
| DE2117571C3 (en) * | 1971-04-10 | 1979-10-11 | Bayer Ag, 5090 Leverkusen | Asymmetrical 1,4-dihydropyridine-33-dicarboxylic acid esters, process for their preparation and their use as pharmaceuticals |
| US3932645A (en) * | 1971-04-10 | 1976-01-13 | Farbenfabriken Bayer Ag | Pharmaceutical compositions containing unsymmetrical esters of 1,4-dihydropyridine 3,5-dicarboxylic acid |
| DE2228377A1 (en) * | 1972-06-10 | 1974-01-03 | Bayer Ag | DIHYDROPYRIDINE CARBONIC ACID AMIDES, THE METHOD FOR THEIR MANUFACTURING AND THEIR USE AS A MEDICINAL PRODUCT |
| DE3022030A1 (en) * | 1980-06-12 | 1981-12-17 | Bayer Ag, 5090 Leverkusen | 4-THIAZOLE or 4-IMIDAZOLE-SUBSTITUTED, 1,4-DIHYDROPYRIDINE, METHOD FOR THE PRODUCTION THEREOF AND THE MEDICINAL PRODUCTS CONTAINING THEM |
| NZ201395A (en) * | 1981-07-30 | 1987-02-20 | Bayer Ag | Pharmaceutical compositions containing 1,4-dihydropyridines and certain of these dihydropyridines |
| US4672068A (en) * | 1984-05-04 | 1987-06-09 | Fujirebio Kabushiki Kaisha | Antihypertensive 1,4-dihydropyridines having a conjugated ester |
| JPS61227567A (en) * | 1985-04-01 | 1986-10-09 | Eisai Co Ltd | 1,4-dihydropyridine derivative |
| IT1215298B (en) * | 1985-08-06 | 1990-01-31 | Boehringer Biochemia Srl | 2- (ETEROALCHIL) -1,4-DIHYDROPYRIDINE AND A PROCESS FOR THEIR PRODUCTION. |
| US4659717A (en) * | 1985-08-21 | 1987-04-21 | Eli Lilly And Company | Dihydropyridines useful in the treatment of angina and stroke |
| US4771057A (en) * | 1986-02-03 | 1988-09-13 | University Of Alberta | Reduced pyridyl derivatives with cardiovascular regulating properties |
| GB8905526D0 (en) * | 1989-03-10 | 1989-04-19 | Fujisawa Pharmaceutical Co | N-containing heterocyclic compounds,processes for the preparation thereof and composition comprising the same |
| DE4236707A1 (en) * | 1992-10-30 | 1994-05-05 | Bayer Ag | 4-heterocyclyl substituted dihydropyridines |
-
1997
- 1997-01-29 CA CA002244774A patent/CA2244774C/en not_active Expired - Fee Related
- 1997-01-29 AU AU22466/97A patent/AU709190B2/en not_active Ceased
- 1997-01-29 AT AT97905627T patent/ATE548351T1/en active
- 1997-01-29 JP JP52706597A patent/JP4431638B2/en not_active Expired - Fee Related
- 1997-01-29 EP EP10011436A patent/EP2311806A3/en not_active Withdrawn
- 1997-01-29 EP EP97905627A patent/EP0885192B8/en not_active Expired - Lifetime
- 1997-01-29 US US09/117,598 patent/US6066642A/en not_active Expired - Lifetime
- 1997-01-29 WO PCT/US1997/001252 patent/WO1997027177A2/en not_active Ceased
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6482841B1 (en) * | 1997-10-09 | 2002-11-19 | Cermol S.A. | Pyridyl compounds and pharmaceutical compositions containing them |
| US6376521B1 (en) | 1998-07-10 | 2002-04-23 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | A3 adenosine receptor antagonists |
| WO2000002861A1 (en) * | 1998-07-10 | 2000-01-20 | The United States Of America, Represented By Secretary, Department Of Health And Human Services | A3 adenosine receptor antagonists |
| AU765423B2 (en) * | 1998-07-16 | 2003-09-18 | Government Of The United States Of America, The | Methods for reducing intraocular pressure |
| WO2000003741A3 (en) * | 1998-07-16 | 2000-09-28 | Univ Pennsylvania | Methods for reducing intraocular pressure using a3-adenosine antagonists |
| US6528516B1 (en) | 1998-07-16 | 2003-03-04 | Trustees Of The University Of Pennsylvania, The Center For Technology Transfer | Methods for reducing intraocular pressure using A3 adenosine receptor antagonists |
| EP1108710A4 (en) * | 1998-07-23 | 2001-11-21 | Chen Ing Jun | Guaiacoxypropanolamines with alpha/beta-adrenergic blocking activity |
| US7790735B2 (en) | 2000-01-14 | 2010-09-07 | The United States Of America As Represented By The Department Of Health And Human Services | Methanocarba cycloalkyl nucleoside analogues |
| US7087589B2 (en) | 2000-01-14 | 2006-08-08 | The United States Of America As Represented By The Department Of Health And Human Services | Methanocarba cycloakyl nucleoside analogues |
| US6358964B1 (en) * | 2000-07-26 | 2002-03-19 | King Pharmaceuticals Research And Development, Inc. | Adenosine, A3 receptor modulators |
| US8003688B2 (en) | 2000-07-28 | 2011-08-23 | Immupharm Aps | Method of treating symptoms of common cold, allergic rhinitis and infections relating to the respiratory tract |
| WO2002009699A3 (en) * | 2000-07-28 | 2003-01-03 | Immupharm Aps | Method of treating symptoms of common cold, allergic rhinitis and infections relating to the respiratory tract |
| EA008612B1 (en) * | 2000-07-28 | 2007-06-29 | Иммуфарм Апс | Method of treating symptoms of common cold, allergic rhinitis and infections relating to the respiratory tract |
| WO2002070520A1 (en) * | 2001-03-07 | 2002-09-12 | Bayer Aktiengesellschaft | Substituted 2,6-diamino-3,5-dicyano-4-aryl-pyridines and their use as adenosine receptor-selective ligands |
| US7129255B2 (en) | 2001-03-30 | 2006-10-31 | Bayer Aktiengesellschaft | Substituted 2-carba-3,5-dicyano-4-aryl-6-aminopyridines and the use of the same as selective ligands of the adenosine receptor |
| WO2002079196A1 (en) * | 2001-03-30 | 2002-10-10 | Bayer Aktiengesellschaft | Substituted 2-carba-3,5-dicyano-4-aryl-6-aminopyridines and the use of the same as selective ligands of the adenosine receptor |
| WO2003031430A3 (en) * | 2001-10-04 | 2004-04-08 | Brane Tech S R L | Flavonoid compounds and their pharmaceutical uses |
| JP2005518384A (en) * | 2001-12-21 | 2005-06-23 | サノフィ−アベンティス | Triazolo-quinoline derivatives useful as adenosine receptor ligands |
| US8653109B2 (en) | 2006-09-08 | 2014-02-18 | Bayer Intellectual Property Gmbh | Substituted bipyridine derivatives and their use as adenosine receptor ligands |
| US8304412B2 (en) | 2006-12-01 | 2012-11-06 | Bayer Intellectual Property Gmbh | Cyclically substituted 3,5-dicyano-2-thiopyridines and use thereof |
| US8703934B2 (en) | 2006-12-01 | 2014-04-22 | Bayer Intellectual Property Gmbh | Substituted 4-amino-3,5-dicyano-2-thiopyridines and use thereof |
| US8440700B2 (en) | 2007-07-27 | 2013-05-14 | Bayer Intellectual Property Gmbh | Substituted aryloxazoles and their use |
| US9095582B2 (en) | 2007-07-27 | 2015-08-04 | Bayer Intellectual Property Gmbh | Substituted aryloxazoles and their use |
| US8703696B2 (en) | 2007-08-01 | 2014-04-22 | Bayer Intellectual Property Gmbh | Dipeptoid prodrugs and the use thereof |
| US8609686B2 (en) | 2007-12-20 | 2013-12-17 | Bayer Intellectual Property Gmbh | Substituted azabicyclic compounds and the use thereof |
| US8618119B2 (en) | 2007-12-20 | 2013-12-31 | Bayer Intellectual Property Gmbh | Fused cyanopyridines and the use thereof |
| US8426602B2 (en) | 2008-03-11 | 2013-04-23 | Bayer Intellectual Property Gmbh | Heteroaryl-substituted dicyanopyridines and their use |
| US8791146B2 (en) | 2008-05-29 | 2014-07-29 | Bayer Intellectual Property Gmbh | 2-alkoxy-substituted dicyanopyridines and their use |
| US8741834B2 (en) | 2008-12-16 | 2014-06-03 | Bayer Intellectual Property Gmbh | Dipeptoid prodrugs and the use thereof |
| US8420825B2 (en) | 2009-01-29 | 2013-04-16 | Bayer Intellectual Property Gmbh | Alkylamino-substituted dicyanopyridines and their amino acid ester prodrugs |
| WO2011029956A1 (en) * | 2009-09-14 | 2011-03-17 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Flavones and flavanones derivates as dna methyltransferases inhibitors |
| US8518957B2 (en) | 2009-12-02 | 2013-08-27 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methanocarba adenosine derivatives, pharmaceutical compositions, and method of reducing intraocular pressure |
| US9187428B2 (en) | 2010-06-30 | 2015-11-17 | Bayer Intellectual Property Gmbh | Substituted dicyanopyridines and use thereof |
| US9040566B2 (en) | 2010-09-02 | 2015-05-26 | Bayer Intellectual Property Gmbh | Adenosine A1 agonists for the treatment of glaucoma and ocular hypertension |
| WO2014101113A1 (en) * | 2012-12-28 | 2014-07-03 | Merck Sharp & Dohme Corp. | Piperazine-substituted 7-methoxy-[1,2,4]triazolo[1,5-c]quinazolin-5-amine compounds with a2a antagonist properties |
| CN103864741A (en) * | 2014-03-06 | 2014-06-18 | 陈朝银 | Preparation and application of walnut shell flavone lipid-decreasing active ingredient |
| EP3189837A4 (en) * | 2014-09-05 | 2018-01-17 | Oneness Biotech Co., Ltd | Use of flavonoid compound in preparation of composition for healing wound |
Also Published As
| Publication number | Publication date |
|---|---|
| US6066642A (en) | 2000-05-23 |
| EP2311806A3 (en) | 2011-08-10 |
| ATE548351T1 (en) | 2012-03-15 |
| CA2244774C (en) | 2006-10-17 |
| JP2000516910A (en) | 2000-12-19 |
| AU2246697A (en) | 1997-08-20 |
| CA2244774A1 (en) | 1997-07-31 |
| EP0885192A1 (en) | 1998-12-23 |
| EP2311806A2 (en) | 2011-04-20 |
| WO1997027177A3 (en) | 1997-11-13 |
| EP0885192B8 (en) | 2012-05-16 |
| AU709190B2 (en) | 1999-08-26 |
| EP0885192B1 (en) | 2012-03-07 |
| JP4431638B2 (en) | 2010-03-17 |
| HK1017676A1 (en) | 1999-11-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6066642A (en) | Dihydropyridine-, pyridine-, benzopyran-4-one- and triazoloquinazoline derivative, their preparation and their use as adenosine receptor antagonists | |
| HUT71558A (en) | Pyran-2-one- and 5,6-dihydro-pyran-2-one derivatives for treating hiv- and other retrovirus infections | |
| JPH0561273B2 (en) | ||
| EP2364298B1 (en) | Quinolone compound and pharmaceutical composition | |
| EP0116769B1 (en) | Dihydropyridines | |
| TW200934765A (en) | Amino-heteroaryl-containing prokineticin 1 receptor antagonists | |
| EP0217530B1 (en) | Dihydropyridines | |
| BG63012B1 (en) | 5,6-Dihydropyrone derivatives as protease inhibitors and antiviral agents | |
| EP0164247B1 (en) | Dihydropyridines | |
| KR100821107B1 (en) | Compounds for the Treatment of Damaged Gastric Hypertrophy | |
| WO2014020101A1 (en) | Griseofulvin derivatives | |
| CZ33196A3 (en) | Derivatives of benzopyran per se, for the use as medicinal preparations and for treating diseases, process and intermediates for their preparation, pharmaceutical compositions based thereon and process of their preparation | |
| AU2007304881B2 (en) | Novel chromenone potassium channel blockers and uses thereof | |
| EP0161917B1 (en) | Dihydropyridines | |
| AU755525B2 (en) | Pyridine derivatives, their preparation and their use as Adenosine receptor antagonist | |
| EP0222598A2 (en) | Dihydropyridine anti-ischaemic and antihypertensive agents | |
| EP0257616A2 (en) | Dihydropyridine derivates and pharmaceutical composition thereof | |
| EP0460418A2 (en) | Novel Dihydropyridine derivative | |
| EP0163238B1 (en) | Substituted and bridged pyridines useful as pharmaceutical agents | |
| Ghoorbannejad et al. | Synthesis and assessment of the cytotoxic effect of some of 1, 4-dihydropyridine derivatives which contain azole moiety | |
| US4957930A (en) | Dihydropyridine derivatives | |
| WO1997000875A1 (en) | Benzimidazole derivatives containing fused pyridine | |
| EA052895B1 (en) | AGENTS FOR KLF2 INDUCTION AND METHODS OF THEIR USE | |
| EP0165032A2 (en) | Dihydropyridine anti-ischaemic and antihypertensive agents | |
| HU194170B (en) | Process for production of derivatives of dihydropiridin with positive inotrop influence and medical compounds containing thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AL AM AT AU AZ BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU IL IS JP KE KG KP KR KZ LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG US US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AL AM AT AU AZ BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU IL IS JP KE KG KP KR KZ LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK TJ TM TR TT UA UG US US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM |
|
| ENP | Entry into the national phase |
Ref document number: 2244774 Country of ref document: CA Ref country code: CA Ref document number: 2244774 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1997905627 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09117598 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997905627 Country of ref document: EP |