WO1998007761A1 - Catalyst component for olefin polymerization, catalyst for olefin polymerization, and process for preparing polyolefin - Google Patents
Catalyst component for olefin polymerization, catalyst for olefin polymerization, and process for preparing polyolefin Download PDFInfo
- Publication number
- WO1998007761A1 WO1998007761A1 PCT/JP1997/002880 JP9702880W WO9807761A1 WO 1998007761 A1 WO1998007761 A1 WO 1998007761A1 JP 9702880 W JP9702880 W JP 9702880W WO 9807761 A1 WO9807761 A1 WO 9807761A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- bis
- substituted
- polymerization
- catalyst
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F110/00—Homopolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F110/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2420/00—Metallocene catalysts
- C08F2420/04—Cp or analog not bridged to a non-Cp X ancillary anionic donor
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65904—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with another component of C08F4/64
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65908—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an ionising compound other than alumoxane, e.g. (C6F5)4B-X+
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65912—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an organoaluminium compound
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65916—Component covered by group C08F4/64 containing a transition metal-carbon bond supported on a carrier, e.g. silica, MgCl2, polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/6592—Component covered by group C08F4/64 containing a transition metal-carbon bond containing at least one cyclopentadienyl ring, condensed or not, e.g. an indenyl or a fluorenyl ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/6592—Component covered by group C08F4/64 containing a transition metal-carbon bond containing at least one cyclopentadienyl ring, condensed or not, e.g. an indenyl or a fluorenyl ring
- C08F4/65922—Component covered by group C08F4/64 containing a transition metal-carbon bond containing at least one cyclopentadienyl ring, condensed or not, e.g. an indenyl or a fluorenyl ring containing at least two cyclopentadienyl rings, fused or not
- C08F4/65925—Component covered by group C08F4/64 containing a transition metal-carbon bond containing at least one cyclopentadienyl ring, condensed or not, e.g. an indenyl or a fluorenyl ring containing at least two cyclopentadienyl rings, fused or not two cyclopentadienyl rings being mutually non-bridged
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S526/00—Synthetic resins or natural rubbers -- part of the class 520 series
- Y10S526/943—Polymerization with metallocene catalysts
Definitions
- Olefin polymerization catalyst component Olefin polymerization catalyst
- the present invention relates to an olefin polymerization catalyst component, an olefin polymerization catalyst, and a method for producing polyolefin using the catalyst. More specifically, the present invention relates to a catalyst for polymerization of olefins, which has high activity, has a wide molecular weight distribution, is excellent in moldability, and is capable of obtaining a polyolefin having a small amount of low molecular weight components, and a method for producing polyolefin using the catalyst.
- a catalyst comprising a combination of a meta-acene compound and an organic aluminum oxy compound is widely known.
- JP-A-58-19309 European Patent No. 69951
- JP-A-60-35007 European Patent No. 129368, U.S. Pat. No. 5,324,800
- Makromol. Chem., Rapid Commun. 461 (1988) and others have reported a catalyst for polymerizing olefins using various meta-mouth compounds and linear or cyclic organoaluminum oxy compounds as catalysts.
- JP-A-7-2917 discloses an amidinato complex having a trimethylsilyl group on a nitrogen atom, specifically,
- ethylene copolymers obtained using titanium-based catalysts or chromium-based catalysts have improved melt tension, but many low-molecular-weight components with a broad molecular weight distribution (evaluated by hexane extraction). Therefore, there was a problem that smoke was generated during molding and the molded product was sticky.
- a catalyst system using a meta-oxene complex having a non-cross-linking ligand and a meta-oxene complex having a cross-linking ligand in combination with aluminoxane and using a carrier and an organoaluminum compound as necessary Methods for producing polyethylene and ethylene- a- olefin copolymer to be used are disclosed (JP-A-3-203904, JP-A-4-213305 (European Patent No. 452920)).
- JP-A-5-255436, JP-A-5-255437 (European Patent No. 515132), JP-A-5-155932, and JP-A-5-155933 have two types. Production of polyethylene and ethylene-monoolefin copolymers using a catalyst system that uses an organoaluminum compound as necessary, in combination with an aluminoxane and a meta-aluminum complex of A method is disclosed.
- a transition having at least two alkyl-substituted cyclopentajenyl ligands is disclosed.
- Manufacture of polyethylene or ethylene- ⁇ -olefin copolymer using a catalyst system using a carrier, an organoaluminum compound or a catalyst system obtained by pre-polymerizing this catalyst, if necessary, combining two metal compounds with aluminoxane A method is disclosed.
- the ethylene-based copolymer obtained by these methods has a narrow composition distribution and low low molecular weight components, which reduces smoke during molding and improves the stickiness of the molded body, but the moldability is sufficiently improved. It was a powerful thing.
- the present invention provides the following catalyst components for olefin polymerization, an olefin polymerization catalyst containing the components, and a method for producing polyolefin using the catalyst.
- M represents a transition metal atom of Group 4 of the periodic table.
- R 1 and R 2 represent an aryl group or a substituted aryl group, which may be the same or different,
- a and B represent the same or different atoms of Group 15 of the periodic table, respectively;
- D represents an atom of group 14 of the periodic table
- B is coupled by a force coordinated to M by a lone pair or by resonance when ⁇ , ⁇ , D and ⁇ resonate,
- Cp represents cyclopentagenenyl, substituted cyclopentagenenyl, indenyl, substituted indenyl, fluorenyl or substituted fluorenyl group, m is 1 or 2,
- n 1 when m is 1, 0 when m is 2,
- X 1 and X 2 may be the same or different and each represents a hydrogen atom, a halogen atom, an organic metalloid, an alkoxy, an amino, a hydrocarbon or a hetero atom-containing hydrocarbon group.
- a catalyst component for polymerization of olefins comprising a transition metal compound represented by the formula:
- a catalyst for polymerization of olefins comprising at least one compound selected from the group consisting of a compound (B-2) which reacts with a transition metal compound to form an ion pair.
- R 4 to R 13 may be the same or different and are each a hydrogen atom, a hydrocarbon group having 1 to 20 carbon atoms, a silyl group substituted with 1 to 3 alkyl groups, or 1 to 3 Represents a 4- to 6-membered carbocyclic group, wherein the force represents a germyl group substituted by two alkyl groups or two adjacent groups together with the carbon atom to which each is attached;
- a plurality of Qi may be the same or different and each may be substituted with a hydrogen atom, a halogen atom, a hydrocarbon group having 1 to 20 carbon atoms, an alkoxy group, an aryloxy group, or an alkyl group having 1 to 3 carbon atoms.
- Me represents a transition metal selected from Groups 3, 4, 5, and 6 of the periodic table; p is 0 or 1. ] (A-2a), and a general formula (4)
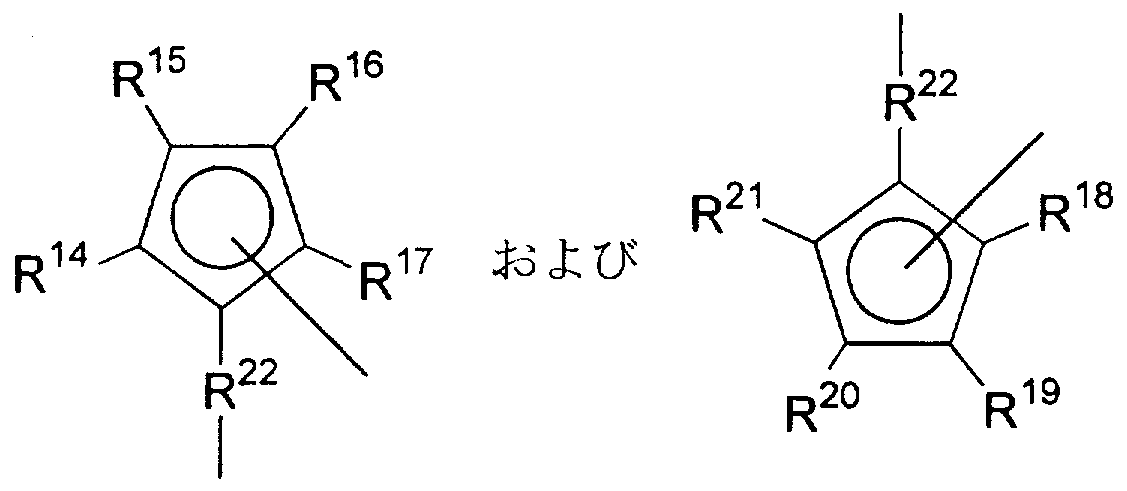
- R 14 to R 21 may be the same or different and each is a hydrogen atom, a hydrocarbon group having 1 to 20 carbon atoms, a silyl group substituted with 1 to 3 alkyl groups, or! Represents a germyl group substituted with up to 3 alkyl groups,
- R 22 is an alkylene, alkylidene, or a ring in which one of the carbon atoms forming the ring may be replaced by an oxygen atom, a cycloalkylidene, a disubstituted germylene or a disubstituted silylene group disubstituted by an alkyl group or a phenyl group. Represents
- each hydrogen atom be the same or different, a halogen atom, a hydrocarbon group having 1 to 20 carbon atoms, an alkoxy group, Ariruokishi group, substituted Ri by the to three alkyl groups ⁇ A silyloxy group or a substituted alkyl group having 1 to 30 carbon atoms and at least one carbon atom in the skeleton substituted with an element selected from Groups 13, 14 and 16 of the periodic table;
- Me represents a transition metal selected from Groups 3, 4, 5, and 6 of the periodic table.
- (B-2 ′) A catalyst for polymerization of olefins, comprising at least one compound selected from the group consisting of
- the catalyst for polymerization of olefin according to the above item 2 or 3, comprising at least one kind of an organometallic compound (C) selected from organic lithium, organomagnesium and organoaluminum, and a carrier (D).
- organometallic compound (C) selected from organic lithium, organomagnesium and organoaluminum
- D a carrier
- the olefin polymerization catalyst component the olefin polymerization catalyst containing the component, and a method for producing polyolefin using the catalyst according to the present invention will be specifically described.
- the olefin polymerization catalyst component (A-1) of the present invention is a transition metal compound represented by the following general formula (1).
- M is a transition metal of Group 4 of the periodic table (the group is based on the inorganic chemical nomenclature 1990 rules; the same applies hereinafter), and specifically means titanium, zirconium and hafnium. Preferred is zirconium.
- R 1 and R 2 may be the same or different and each represents an aryl group or a substituted aryl group.
- a and B may be the same or different and each represents an atom of Group 5 of the periodic table. Specifically, nitrogen, phosphorus, arsenic, antimony and the like can be exemplified, and nitrogen and phosphorus are preferable.
- D represents an atom of Group 14 of the periodic table, and is preferably carbon.
- B is a force that is coordinated to M by a lone pair, or resonates between M, 8, D, and B, and in some cases, combines by the resonance.
- R 3 represents a hydrogen atom, a halogen atom, an organic metalloid, an alkoxy, an amyloid hydrocarbon or a heteroatom-containing hydrocarbon group. ).
- Cp represents a cyclopentagenenyl, substituted cyclopentagenenyl, indenyl, substituted indenyl, fluorenyl or substituted fluorenyl group.
- n is 1 when m is 1; 0 when m is 2; when m is 1, it may be bridged between the L group and the Cp group. When m is 2, it may be bridged between L groups.
- X 1 and X 2 may be the same or different and each represents a hydrogen atom, a halogen atom, an organic metalloid, an alkoxy, an amino, a hydrocarbon or a hetero atom-containing hydrocarbon group.
- R 1 and R it is important for R 1 and to be an aryl group or a substituted aryl group in order to obtain a highly active and high molecular weight polyolefin.
- substituent of the aryl group include a halogen atom and an alkyl group.
- Preferred examples of R 1 and R include phenyl, fluorophenyl, trifluorophenyl, methylphenyl, 2,6-methylphenyl, naphthalenyl, fluoronaphthalenyl, anthracenyl and the like. And a phenyl group and a naphthalenyl group are particularly preferred.
- R 3 is preferably a hydrogen atom, an alkyl group or a (substituted) aryl group.
- zirconium may be replaced by hafnium, and zirconium may be replaced by titanium.
- the force is not limited to these.
- the compound (A-1) represented by the above general formula (1) can be used alone as the transition metal component, but the transition metal represented by the general formula (3) can be used as another transition metal component.
- At least one compound selected from the group consisting of the metal compound (A-2a) and the transition metal compound (A-2b) represented by the general formula (4) ( ⁇ -2) can also be used in combination.
- the symbols in the general formula (3) have the following meanings.
- R 4 to R 13 may be the same or different and each represents a hydrogen atom, a hydrocarbon group having 1 to 20 carbon atoms, a silyl group substituted with 1 to 3 alkyl groups, or 1 to 3 alkyl groups; Represents a germyl group substituted by a group, or represents a 4- to 6-membered carbocyclic group together with the carbon atom to which each two adjacent groups are attached.
- Two or three Q 1 s may be the same or different and each represents a hydrogen atom, a halogen atom, a hydrocarbon group having 1 to 20 carbon atoms, an alkoxy group, an aryloxy group, or:!
- To 3 alkyl groups Represents a silyloxy group which may be substituted by, or a substituted alkyl group having 1 to 30 carbon atoms and wherein at least one carbon atom in the skeleton is substituted with an element selected from Groups 13, 14 and 16 of the periodic table. .
- Me is a transition metal selected from Groups 3, 4, 5, and 6 of the periodic table.
- p 0 or 1.
- R '4 to R 21 are Yogu each hydrogen atom be the same or different, hydrocarbon having 1 to 20 carbon atoms Represents a hydrogen group, a silyl group substituted with 1 to 3 alkyl groups, or a germyl group substituted with 1 to 3 alkyl groups.
- R represents alkylene, alkylidene, cycloalkylidene in which one of the carbon atoms forming the ring may be replaced by an oxygen atom, disubstituted germylene or disubstituted silylene group disubstituted by an alkyl or phenyl group.
- Q 2 and Q 3 may be the same or different and each is substituted by a hydrogen atom, a halogen atom, a hydrocarbon group having 1 to 20 carbon atoms, an alkoxy group, an aryloxy group, or an alkyl group having 1 to 3 carbon atoms. Or a substituted alkyl group having 1 to 30 carbon atoms and having at least one carbon atom in the skeleton substituted by an element selected from Groups 13, 14 and 16 of the periodic table.
- Me is a transition metal selected from Groups 3, 4, 5, and 6 of the periodic table.
- Me is a transition metal element belonging to groups 3, 4, 5, and 6 of the periodic table, and preferably a transition metal element belonging to group 4 of the periodic table, that is, titanium.
- Elements selected from zirconia, zirconium and hafnium are preferred. Particularly preferred is zirconia or hafnium.
- examples of the hydrocarbon group having 1 to 20 carbon atoms represented by R 4 to R 13 include a methyl group, an ethyl group, a propyl group, a butyl group, an isobutyl group, a t-butyl group, and an ammonium group.
- Aryl groups such as alkenyl group, phenyl group and tolyl group; alkylaryl groups such as 2,6-dimethylphenyl group and 2,4,6-trimethylphenyl group; phenylmethyl group; diphenylmethyl group; triphenylmethyl group;
- An arylalkyl group can be exemplified.
- Examples of the silyl group substituted with 1 to 3 alkyl groups include a trimethylsilyl group, and examples of the germyl group substituted with 1 to 3 alkyl groups include a trimethylgermyl group.
- examples of the cyclopentagenenyl ring include an indenyl ring and a fluorenyl ring.
- Ligands having substituents as described above examples thereof include cyclopentagenenyl group, methylcyclopentagenenyl group, ethylcyclopentagenenyl group, n-butynolecyclopentagenenyl group, t-butylcyclopentagenenyl group, trimethylsilylcyclopentagenenyl group, dimethyl
- Examples thereof include an alkyl-substituted cyclopentagenyl group such as a cyclopentagenyl group and a pentamethylcyclopentagenyl group, and an indenyl group and a fluorenyl group having or not having the same substituent.
- Specific examples of the germyl group include those similar to the above R 4 to R 13 .
- Examples thereof include a cyclopentagenenyl group, a methylcyclopentagenenyl group, an ethylcyclopentagenenyl group, an n-butylcyclopentagenenyl group, a t-butylcyclopentagenenyl group, a trimethylsilylcyclopentagenenyl group, and a dimethylcyclopentadienyl group.
- Examples thereof include an alkyl-substituted cyclopentagenyl group such as a pentagenyl group and a tetramethylcyclopentagenyl group.
- examples of the alkylene group represented by R include a methylene group, an ethylene group, a propylene group, a diphenylmethylene group, and the like.
- examples of the cycloalkylidene in which one of the carbon atoms may be replaced with an oxygen atom include an ethylidene group, an isopropylidene group, a cyclopentylidene group, a cyclohexylidene group, a tetrahydropyran-141-ylidene group and the like.
- Examples of the di-substituted germylene or di-substituted silylene group di-substituted by an alkyl group or a phenyl group include a dimethylsilylene group, a diphenylsilylene group, a dimethylgermylene group, and a diphenylgermylene group.
- the hydrocarbon group include the same groups as those described above for R 4 to R l: i, and specific examples of the halogen atom include a fluorine atom, a chlorine atom, a bromine atom and an iodine atom.
- the substituted alkyl group having 1 to 30 carbon atoms in which at least one carbon atom in the skeleton is substituted with an element selected from Groups 13, 14 and 16 of the periodic table include a bis (trimethylsilinole) methyl group, Examples thereof include a methoxymethyl group and an ethoxymethyl group.
- Pentamethylcyclopentagenenyl zirconium trimethyl can be exemplified.
- transition metal compound (A-2b) represented by the general formula (4)
- a transition metal compound in which zirconium is replaced by hafnium is replaced by titanium.
- bis (n-butylcyclopentagenenyl) zirconium dichloride and dimethylsilylenebis (n-butylcyclopentagenenyl) zirconium dichloride are preferred.
- specific combinations include bis (n-butylcyclopentapentaenyl) zirconium dichloride and (pentamethylcyclopentagenenyl) (N, N'-bis (phenyl) benzamidinato) Combination with zirconium dichloride, or dimethylsilylenebis (n-butylcyclopentagenenyl) zirconium dichloride and (pentamethylcyclopentagenenyl) (N, N'-bis (phenyl) benzamidinato) zirconium dichloride Are preferred.
- the mixing ratio is the molar ratio of (A-1) to (A-2) ((A-1) / (A-2)), 99 / :! ⁇ 10/90, more preferably 95/5 ⁇ 20 80, more preferably 93/7 ⁇ 30/70, most preferably 90 ⁇ 10 ⁇ 40 ⁇ 60.
- the second component ( ⁇ ) of the olefin polymerization catalyst of the present invention comprises an organoaluminoxy compound (B-1) and a compound which forms an ion pair by reacting with the transition metal compound represented by the general formula (1).
- (II-2) at least one compound selected from the group consisting of Further, in addition to the transition metal compound ( ⁇ -1) represented by the general formula (1) as the transition metal component, a transition metal compound ( ⁇ -2) represented by the aforementioned general formula (3) or (4) is combined.
- an organoaluminoxy compound ( ⁇ -1) and a transition metal compound represented by the general formula (1), (3) or (4) are used as the second component ( ⁇ .
- At least one compound selected from the group consisting of compounds that react to form an ion pair ( ⁇ -2 ′) is used.
- an aluminoxane-based compound is usually preferably used as the organoaluminoxy compound ( ⁇ -1).
- aluminoxane is an organoaluminum compound represented by the general formula (5) or (6).
- R 23 is a hydrogen atom, a hydrocarbon group having 1 to 20 carbon atoms, an alkyl halide group or an aryl halide group.
- the hydrocarbon group include a methyl group, an ethyl group, a propyl group, a butyl group, an isobutyl group and the like, and a methyl group and an isobutyl group are preferred.
- a plurality of R 2: i in the same formula may be different from each other.For example, even when repeating units having different hydrocarbon groups are bonded in a block, they are bonded regularly or irregularly. It may be something.
- m is 1 to 100 And preferably 4 or more, particularly preferably 8 or more.
- Methods for producing this type of compound are known, such as a method in which an organoaluminum compound is added to a hydrocarbon solvent suspension of a salt having water of crystallization (copper sulfate hydrate, aluminum sulfate hydrate), A method in which solid, liquid, or gaseous water is allowed to act on an organoaluminum compound in a hydrogen solvent.
- aluminoxane two kinds of the compounds of the general formula (5) and the general formula (6) may be used.
- the organoaluminum compounds used in the production of aluminoxanes include trimethylaluminum, triethylaluminum, tripropylaluminum, triisopropylaluminum, tri-n-butynolenoreminium, triisobutynolenoreminium, and trisec.
- Organic aluminum such as quinoaluminoreminium, tri-tert-butylaluminum, tripentinoreanominium, trihexylaluminum, trioctylaluminum, tricyclohexylaluminum, dimethylaluminum chloride, getylaluminum chloride , Dialkylaluminum halides such as diisobutylaluminum dimethylchloride, dialkylaluminum alkoxides such as dimethylaluminum methoxide, Selected force among dialkyl aluminum ⁇ reel O sulfoxides such as E Chi le aluminum phenoxide.
- trialkylaluminums particularly trimethinoleanoreminium, and triisobutylaluminum are preferred.
- Hydrocarbon solvents used in the production of aluminoxane include aromatic hydrocarbons such as benzene, toluene and xylene, aliphatic hydrocarbons such as pentane, hexane, heptane, octane and decane, cyclopentane and cyclopentane. Examples thereof include alicyclic hydrocarbons such as hexane. Of these solvents, aromatic hydrocarbons are preferred.
- Examples of the compound (B-2 ′) which reacts with a metal compound to form an ion pair include Japanese Patent Publication No. 1-1501950 (International Publication No. 88/05792) and Japanese Patent Publication No. 1-502036 (International Publication No. Publication No. 88/05793), Japanese Patent Application Laid-Open No. 3-179005 (European Patent No. 427697), Japanese Patent Application Laid-Open No. 179006 (European Patent No. 427696, US Pat. No. 5,155,080, US Pat. No.
- Lewis acids include triphenylboron, tris (4-fluorophenyl) boron, tris (p-tolyl) boron, tris (o-tolyl) borane, tris (3,5-dimethylphenylinole) boron, tris ( pentafluorophenyl) boron, MgCL, Al ⁇ Si0 2 - .. A10 3 such that Ru can be exemplified.
- Examples of ionic compounds include triphenylcarbenyltetrakis (pentafluorophenyl) borate, tri-n-butylammoniumtetrakis (pentafluorophenyl) borate, and N, N-dimethylaniliniumtetrakis ( Examples thereof include (pentafluorophenyl) borate and ferrocenedium tetrakis (pentafluorophenyl) borate.
- carborane compound examples include dodecaborane, 11-carounde force borane, bis-n-butylammonium (11-carbodede force) borate, and tri-n-butylammonium (tridecahydride-17-carounde force) borate. .
- component (B-2) or component (B-2 ′) can be used by mixing two or more kinds.
- component (A) the component (A-1) or a mixture of the component (A-1) and the component (A-2) (hereinafter, collectively referred to as component (A)), and
- component (B) at least one organometallic compound (C) selected from organolithium, organomagnesium and organoaluminum can be used.
- organic lithium examples include methyllithium, ethyllithium, n-propynorium, n-butyllithium, isobutyllithium, sec-butyllithium, tert-butyllithium, n-pentyllithium, isopentyllithium, and neopentyllithium. No. Of these, n-butyllithium and tert-butyllithium are preferred.
- organomagnesium examples include n-butylethylmagnesium, g-sec-butylmagnesium, n-butyl-sec-butylmagnesium, g-tert-butynolemagnesium, dineopentylmagnesium, and di-n-hexylmagnesium. Is mentioned You. Of these, n-butylethylmagnesium, g-sec-butylmagnesium, and di-n-hexylmagnesium are preferred.
- organoaluminum examples include trimethylaluminum, triethylaluminum, tripropynoleanolinolemin, triisopropylaluminum, tri-n-butinoleanolinoleminium, triisobutylaluminum, tree sec-butylaluminum, tree tert.
- trimethylaluminum and triisobutylaluminum are preferred.
- the above component (C) can be used as a mixture of two or more kinds.
- the support (D) can be used for the catalyst of the present invention.
- the support (D) is a porous fine-particle support, and is selected from inorganic oxides, inorganic chlorides, inorganic carbonates, inorganic sulfates, and organic polymers which are preferably solid in the polymerization medium.
- the inorganic oxide for example Si_ ⁇ 2, Al 2 ⁇ 3, MgO, Zr_ ⁇ 2, Ti_ ⁇ 2, inorganic oxides of Ca_ ⁇ or Si0 2 _Al 2 0 3, Si_ ⁇ 2 - MgO, Si_ ⁇ 2 - Zr_ ⁇ 2, Si0 2 - Ti_ ⁇ 2, Si_ ⁇ 2 - CaO, A1 2 0 3 - MgO, A1 2 0 3 - Zr_ ⁇ 2, Al 2 0: i - Ti_ ⁇ 2, A1 2 0:, - CaO , Zr_ ⁇ 2 - Ti_ ⁇ 2, Zr_ ⁇ 2 - CaO, Zr0 2 - MgO, TiC ⁇ - composite oxides such Mg_ ⁇ , inorganic chlorides such as chloride magnesium ⁇ beam, magnesium carbonate, calcium carbonate, strontium carbonate, etc. And inorganic sulfates such as magnesium sulfate, calcium sulfate, and barium sulfate. Examples of the
- the carrier (D) has an average particle size of 1 to 300 ⁇ , preferably 10 to 20 Om, and more preferably 20 to 100 // m.
- the specific surface area is preferably in the range of 10 to 1000 m 2 Zg, more preferably in the range of 100 to 800 m 2 Zg, and particularly preferably in the range of 200 to 600 m 2 Zg.
- 0. S Scm ⁇ g more preferably ranges preferably instrument in the range of 0. 5 ⁇ 2. 5cm 3 Zg, particularly preferably 1. 0 ⁇ 2. 0cm 3 Zg of Range.
- Si ⁇ 2 , A Os and their composite oxides, which are preferable as the carrier (D) have different amounts of adsorbed water and surface hydroxyl groups depending on the treatment conditions.
- the water content is 5% by weight or less, and the surface hydroxyl group content is 1 or more Z (nm) 2 per surface area.
- the water content and the amount of surface hydroxyl groups can be controlled by selecting the calcination temperature and calcination time, and by treating with an organoaluminum compound or an organoboron compound.
- the olefin polymerization catalyst it is desirable that at least one of the component (A) and the component (B) is supported on the carrier (D).
- the method for example,
- the component (C) can be added.
- the loading of at least one of the component (II) and the component (B) on the carrier (D) can be carried out in an inert hydrocarbon solvent.
- aromatic hydrocarbons such as benzene, toluene and xylene, aliphatic hydrocarbons such as pentane, hexane, heptane, octane and decane, and alicyclic hydrocarbons such as cyclopentane and cyclohexane are used.
- the contacting temperature is preferably an aromatic hydrocarbon solvent, and the contacting temperature is usually 15 () to 200 C, preferably 120 to 100 C, more preferably 0 to 50 C. .
- the time between the temples to be horned is 0.05-200 B temples, preferably about 0.2-20 hours.
- the contact between the component (A) and the component (B) in the catalyst for polymerization of olefins of the present invention may be carried out in advance in the presence or absence of a monomer, or may be carried out without prior contact. You can introduce it to ⁇ .
- the order of contact may be arbitrarily selected. However, when the component (A-1) and the component (A-2) are used in combination as the transition metal component, the component (A-1) is preceded by the component (A-1).
- a preferred method is to contact component (A-2) after contacting with B)!
- This contact is performed in an inert solvent.
- solvent examples include aromatic hydrocarbons such as benzene, toluene and xylene; aliphatic hydrocarbons such as pentane, hexane, heptane, octane and decane; and lipids such as cyclopentane and cyclohexane.
- aromatic hydrocarbons such as benzene, toluene and xylene
- aliphatic hydrocarbons such as pentane, hexane, heptane, octane and decane
- lipids such as cyclopentane and cyclohexane.
- a cyclic hydrocarbon or the like can be used.
- the contact ratio between the component (A) and the component (B) is as follows. Assuming that the number is [A], the value of [A] Z [B-1] is in the range of lZl to 1/10000, more preferably 1 to 10 to 11000.
- component (B-2) or component (B-2 ')
- component (B-2) if the number of moles of the boron atom is [B-2], the value of — 1/100, more preferably in the range of SZlIZlO.
- the use ratio of the component (A) to the component (C) is preferably in a molar ratio ((A) :( C)) of from 1:10 to 1: 100,000, more preferably from 1: 100 to 1: 10000. It is.
- Component (B) and component (D) are preferably used in a weight ratio ((B-1) :( D)) of component (B-1) of 1: 0.5-1 to 1: 1. 100, more preferably 1 ::! To 1:10, and in the case of component (B-2) (or component (B-2 ′)), the weight ratio ((B-2): (D )), Preferably in the range of 1 ::! To 1: 10000, more preferably in the range of 1: 5 to 1: 100.
- the ratio of the component (A) to the component (D) is preferably in a weight ratio ((A) :( D)) of 1: 5 to 1: 10000, more preferably 1:10 to 1: 500. Range.
- each catalyst component during the polymerization reaction is carried out with aromatic hydrocarbons such as benzene, toluene and xylene, aliphatic hydrocarbons such as pentane, hexane, heptane, octane and decane, and oils such as cyclopentane and cyclohexane. It can be prepared in a cyclic hydrocarbon in the presence or absence of olefin.
- the temperature at the time of contact is -70 ° C to 200 ° C, preferably The temperature is between 20 ° C and 120 ° C, and the mixing time is 1 minute to 60 minutes.
- the contact time of each catalyst component can be arbitrarily selected.
- a method of initiating a polymerization reaction by adding a component in which component (I) and component (B) are brought into contact in advance to a reaction vessel charged with component (C) and olefin to be used for polymerization.
- the polymerization reaction may be started by charging the component (C) and the olefin to be used for the polymerization in a reaction vessel, and separately adding the component ( ⁇ ) and the component (B).
- ⁇ -olefins used in the copolymerization include propylene, 1-butene, 1-pentene, 1-hexene, 1-heptene, 1-octene,] -decene, 4-methyl-1-pentene, cyclopentene and cyclopentane It can exemplify olefins, cyclic olefins, and gens such as gen, butadiene, 1,5-hexadiene, 1,4-hexadiene, and 1,4-pentadiene. These two or more comonomers can be mixed and used for copolymerization with ethylene.
- the polymerization method used in the present invention can be any of solution polymerization, slurry polymerization, and gas phase polymerization. Preferably, it is slurry polymerization or gas phase polymerization, and multistage polymerization is also possible. It is also possible to prepolymerize the olefin.
- the amount of use Les is a polymerization catalyst in the production method of the polyolefin according to the present invention is represented by the concentration of the transition metal compound in the polymerization reaction system, usually,] 0- 8 ⁇ : 10- 2 mol / 1, Preferably, it is in the range of 10 7 to 10 3 mol Zl.
- the reaction system pressure is not particularly limited, but is preferably in the range of normal pressure to 50 kg / cm3 ⁇ 4.
- the polymerization temperature is not limited, but is preferably in the range of ⁇ 30 U to 200 ° C. Particularly preferably, it is in the range of 0 ° C to 120 ° C. More preferably, it is 50 to 90 ° C.
- the molecular weight at the time of polymerization can be controlled by known means, for example, by selecting a temperature or introducing hydrogen.
- the (co) polymer obtained by the method of the present invention has the following characteristics.
- the MFR at 19 CTC and a load of 21.6 kg depends on the type of the transition metal compound of the catalyst component, the polymerization temperature, and the amount of hydrogen introduced during the polymerization. (3 ⁇ 4 / 10 min. 190. A load of 2.16 kg with an MFR of lOOOOg / 10 min. Is obtained.
- the molecular weight distribution is broad.
- the gel permeation chromatography (GPC) force The calculated MwZMn is about 4-30.
- the amount of low molecular weight components is very small, smoke generated during molding is almost nonexistent.
- NMR was measured at 30 ° C. in a double-mouthed form using a JEOL EX-400 machine.
- MFR melting flow rate
- C measured under the condition of a load of 2.16 kg
- HLMFR high load melt opening rate
- MT melting tension
- the molecular weight (Mn, Mw, Mz) and molecular weight distribution (Mw / Mn, MzZlVlw) were measured using GPC (Waters 150C, column shodex).
- a 200-ml container sufficiently purged with argon was separately prepared, into which 1.3 g (4 mmol) of pentamethylcyclopentapentaenyl zirconium trichloride was added, and dissolved in 50 ml of dry and dry tonolene.
- pentamethylcyclopentapentaenyl zirconium trichloride was added, and dissolved in 50 ml of dry and dry tonolene.
- add the entire volume of the solution of lithium mono-N, N'-bis (phenyl) benzamidinate in toluene at room temperature stir for 5 hours, and separate the insoluble components in the reaction solution by centrifugation. did.
- pentamethylcyclopentagenylzirconium trichloride (4.5 mmol) was charged into a separately prepared 100 ml container, and dissolved in toluene (50 ml). To this was added the entire amount of the above-mentioned solution of lithium-N, N'-bis (4-fluorophenyl) benzamidinato in tonolen at room temperature, and the mixture was stirred at room temperature for 5 hours.
- reaction solution was concentrated to 10 ml. Hexane (5 ml) was added thereto, and the mixture was allowed to stand in a freezer at -20 ° C. After standing overnight, the desired (bentamethylcyclopentagenenyl) (N, N'-bis (4-fluorophenyl) benzamidinate) dinoreconidum dichloride (pale yellow crystals) was obtained.
- diphenylbenzanidine (5.0 mmol) was charged into a 100-ml container, and dissolved in toluene (60 ml).
- a toluene solution was obtained.
- Zirconium tetrachloride (2.5 mmol) was charged into a separately prepared 300 ml container under an argon atmosphere, and dissolved with toluene (60 ml). To this, the whole amount of the toluene solution of lithium-N ,, '-bis (fuunil) benzamidinato was added at room temperature, and the mixture was stirred at room temperature for 5 hours.
- reaction solution was concentrated at about 100 m. After leaving overnight in a freezer at 20 ° C, the desired bis (N, N'-bis (phenyl) benz Midinato) zirconium dichloride (yellow crystals) was obtained.
- diphenylnaphthoamidine (8.6 mmoi) was charged into a 100-ml container, and dissolved in toluene (50 ml).
- a 1.6 M solution of normal butyllithium in hexane (8.6 mmol) was slowly added dropwise under ice-cooling, and the mixture was stirred at room temperature for 3 hours to give lithium-N, N, -bis (phenyl) naphthoamidina.
- toluene solution 8.6 mmoi
- pentamethylcyclopentagenenylzirconium trichloride (8.6 mmol) was charged into a separately prepared 300 ml container, and dissolved in toluene (50 ml). To this, the whole amount of the above solution of lithium-N, N'-bis (phenyl) naphthoamidinate in Tonolen was added at room temperature, and the mixture was stirred at room temperature for 30 minutes and at 80 ° C for 2 hours. After insoluble components in the reaction solution were separated by centrifugation, the reaction solution was concentrated and left in a 12 CTC freezer (). After leaving overnight, the target (pentamethylcyclopentagenenyl) (N, N'- Bis (phenyl) naphthoamidinato) diconidum dichloride was obtained as pale yellow crystals.
- dinaphthyl benzamidine (6.7 mmol) was charged into a 100-ml container, and dissolved in toluene (50 ml).
- a 1.6 M hexane solution of normal butyl lithium (6.7 mmol) was slowly added dropwise thereto under ice-cooling, and the mixture was stirred at room temperature for 3 hours, and lithium-N, N, -bis (naphthyl) benzamidinato was added.
- a toluene solution was obtained.
- a 200 ml container sufficiently purged with argon was separately prepared, and 1.6 g (5 mmol) of pentamethylcyclopentagenyl zirconium trichloride was added thereto and dissolved in 50 ml of dry toluene. To this was added the entire amount of the lithium-N, ⁇ '-bis (2,6-dimethylphenyl) benzamidinato tonoleene solution at room temperature, and the mixture was stirred for 5 hours, and the insoluble components in the reaction solution were removed. Separated by centrifugation.
- Butylethylmagnesium (BEM) was used instead of triisobutylaluminum (TBA), and (pentamethylcyclopentagenenyl) (N, N'-bis (phenyl) benzamidinato) dinoleconidum dichloride (PPBZ) was replaced with (pentamethylcyclopentagenenyl) (N, N'-bis (phenyl) naphthoamidinate) zirco dimethyldichloride (PPNZ) prepared in Example 4.
- Polymerization was carried out as in Example 10. As a result, 76 g of polyethylene was obtained.
- the activity per complex was 7600 g polyethylene / mmol complex 'hr' atm.
- the MFR of this polyethylene at 190 ° C and a load of 2.16 kg could not be measured because no outflow of polyethylene could be observed.
- the molecular weight Mw by GPC was 343,000.
- the MFR of this polyethylene at 190 ° C and a load of 2.16 kg was 11 gZlO min, and the HLMFR at a load of 21.6 kg was 222 gZlO min.
- TSA triisobutylaluminum
- Example 1 (Penta Methylcyclopentagenyl) (N, N'-bis (phenyl) benzamidinato) zirconium dichloride (PPB Z) in toluene solution (1. Ommol / ⁇ ) was contacted with 4 ml of a mixed solution for 1 minute, The polymerization has begun. During the polymerization, a constant ethylene partial pressure in the autoclave within LOkgZcm 2 Ethylene was continuously introduced as much as possible. After performing polymerization at 70 DC for 30 minutes, the polymerization was stopped by purging. As a result, 56 g of polyethylene was obtained. The activity per complex was 2800 g polyethylene / mmol complex 'hr' atm. 190 of this polyethylene. C, MFR at a load of 2.16 kg could not be measured due to the force that could not observe the outflow of polyethylene. The molecular weight Mw by GPC was 3,620000.
- Example 16 Example 16:
- TSA triisobutylaluminum
- BEM butylethylmagnesium
- the activity per complex was 2350 g polyethylene Zmmol complex 'hr-atm.
- the MFR of this polyethylene at 190 ° C and a load of 2.16 kg could not be measured because no outflow of polyethylene could be observed.
- the molecular weight Mw by GPC was 3,500,000.
- Comparative Example 8 (Pentamethylcyclopentagenenyl) (N, N'-bis (phenyl) benzamidinato) Instead of zirconium dichloride (PPBZ), (pentamethylcyclopentagenenyl) (N, N'-bis (trimethylsilyl) Polymerization was carried out in the same manner as in Example 15 except that (benzamidinato) zirconium dichloride (PS13Z) was used.
- Example 1 Pentamethylcyclopentagenenyl) (N, N'-bis (phenyl) benzamidinato) zirco
- a mixed solution obtained by contacting 4 ml of a toluene solution (1. OmmoiZl) of dimethyl dichloride (PPBZ) for 1 minute was added to initiate polymerization.
- the ethylene partial pressure in the autoclave was continuously introduced ethylene so as to constantly maintain lOkgZcm 2. 70.
- Example 20 After performing polymerization for 30 minutes at C, the polymerization was stopped by purging. As a result, 45 g of polyethylene was obtained. The activity per complex was 2250 g polyethylene / mmol complex 'hr'atm. 190 of this polyethylene. C, MFR at a load of 2.16 kg could not be measured because no outflow of polyethylene could be observed.
- Example 20
- the MFR of this polyethylene at 190 C and a load of 2.16 kg is 0.57 g / 10 minutes, 190. C, the ratio of MFR at a load of 21.6 kg to the MFR at 190 ° C and a load of 2.16 kg (HLMFR MFR) is 91 ⁇ 3, MwZMn is 10.1, melt tension is 7.5 g, The hexane-soluble component fraction was 0.6 wt%.
- BEM butylethylmagnesium
- ⁇ triisobutylaluminum
- the MFRf of this polyethylene at 190 ° C and a load of 2.16 kg is 0.03 g / 10 minutes, and the ratio of the MFR at 190 ° C and a load of 21.6 kg to the MFR at 190 ° C and a load of 2.16 kg ( HLMFR ZMFR) was 261.2, MwZMn was 14.3, the melt tension was 7.8 g, and the cyclohexane soluble component fraction was 0.2 wt%.
- HLMFR ZMFR HLMFR ZMFR
- MFRi of this polyethylene at 190 C under a load of 2.16 kg is 0.1 l l gZ lO, 190. C, MFR at a load of 21.6 kg, 190 ° C, MFR at a load of 2.16 kg]: MORI (HLMFR / MFR) is 431 ⁇ 3, MwZMn is 13.1, and melt tension is 10. 3 g, the cyclohexane-soluble component fraction was 0.6 wt%.
- a hexane solution of a mixture of butylethylmagnesium (BEM) and triisobutylaluminum (TBA) (mixing molar ratio [Mg] / [A1] 1) was placed in a 1.5-liter SUS autoclave with sufficient nitrogen replacement. / 1, prepared by stirring in hexane at 20 ° C for 30 minutes, total concentration 0.5 mol / l) 0.9 ml, 1-hexene 5 g, isobutane 800 ml were introduced, and then the temperature was raised to 70 ° C. .
- BEM butylethylmagnesium
- TSA triisobutylaluminum
- the polymerization was initiated by injecting a three-component mixture of the solution with nitrogen at a pressure of 40 kg / cm 2 . Polymerization was carried out at a mixed gas pressure of 10 kg / cn ⁇ 70 ° C for 30 minutes to obtain 159 g of polyethylene.
- a hexane solution of a mixture of butylethylmagnesium (BEM) and triisobutylaluminum (TBA) (mixing molar ratio [Mg] Z [Al] l / l, 20 in hexane, prepared by stirring at C for 30 minutes, total concentration of 0. Smol / D O. 9 ml, 1-hexene 5 g, isobutane 800 ml were introduced, and the temperature was raised to 70 ° C.
- BEM butylethylmagnesium
- TSA triisobutylaluminum
- Example 29 Example 30
- Example 31 Example 32
- Example 33 Example 34
- Comparative Example 12 Component (A-1) PPBZ PMBZ * 5 PPBZ *-PPNZ * 6 PNBZ * 7
- B CZ * 8 Content 2.2 mg 2.3mg 4.4mg * ⁇ 2.4mg 2.6mg 1.5mg
- Ingredient (A-2) BBZ SBCZ * 9 BBZ «-
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
Description

















Claims



Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP97935830A EP0926166A4 (en) | 1996-08-21 | 1997-08-20 | CATALYST COMPONENT FOR OLEFIN POLYMERIZATION, CATALYST FOR OLEFIN POLYMERIZATION AND METHOD FOR PRODUCING POLYOLEFINES |
| US09/242,664 US6303714B1 (en) | 1996-08-21 | 1997-08-20 | Catalyst component for olefin polymerization catalyst for olefin polymerization, and process for preparing polyolefin |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP22020796A JP3773304B2 (ja) | 1996-08-21 | 1996-08-21 | オレフィン重合用触媒成分、その成分を含むオレフィン重合用触媒およびポリオレフィンの製造方法 |
| JP8/220207 | 1996-08-21 | ||
| JP14496797A JPH10330414A (ja) | 1997-06-03 | 1997-06-03 | オレフィン重合用触媒およびポリオレフィンの製造方法 |
| JP9/144967 | 1997-06-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998007761A1 true WO1998007761A1 (en) | 1998-02-26 |
Family
ID=26476239
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1997/002880 Ceased WO1998007761A1 (en) | 1996-08-21 | 1997-08-20 | Catalyst component for olefin polymerization, catalyst for olefin polymerization, and process for preparing polyolefin |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US6303714B1 (ja) |
| EP (1) | EP0926166A4 (ja) |
| KR (1) | KR20000068220A (ja) |
| WO (1) | WO1998007761A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6369176B1 (en) | 1999-08-19 | 2002-04-09 | Dupont Dow Elastomers Llc | Process for preparing in a single reactor polymer blends having a broad molecular weight distribution |
| CN100434444C (zh) * | 2001-10-19 | 2008-11-19 | 旭化成株式会社 | 烯烃聚合催化剂及使用该催化剂的烯烃聚合方法 |
| AU2003277808A1 (en) * | 2002-06-21 | 2004-01-06 | Exxonmobil Chemical Patents Inc. | Yttrium-based ethylene polymerization catalysts with bulky amidinate ancillary ligands |
| US9296836B2 (en) * | 2011-05-12 | 2016-03-29 | Dow Global Technologies Llc | Non-cyclopentadienyl-based chromium catalysts for olefin polymerization |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH093113A (ja) * | 1995-06-16 | 1997-01-07 | Showa Denko Kk | ポリオレフィンの製造方法 |
| JPH09124722A (ja) * | 1995-08-31 | 1997-05-13 | Sumitomo Chem Co Ltd | 遷移金属錯体、オレフィン重合用触媒及びオレフィン重合体の製造方法 |
| JPH09143221A (ja) * | 1995-09-18 | 1997-06-03 | Showa Denko Kk | エチレン系重合用触媒 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5690810A (en) | 1979-12-24 | 1981-07-23 | Asahi Chem Ind Co Ltd | Production of polyethylene |
| JPH06810B2 (ja) | 1983-11-14 | 1994-01-05 | 三井石油化学工業株式会社 | チタン触媒成分の改質方法 |
| JP2953521B2 (ja) | 1989-12-29 | 1999-09-27 | 三井化学株式会社 | エチレン(共)重合用触媒およびエチレンの(共)重合方法 |
| JP3110156B2 (ja) | 1991-09-17 | 2000-11-20 | 三井化学株式会社 | オレフィン重合触媒およびオレフィンの重合方法 |
| JP3110155B2 (ja) | 1992-01-16 | 2000-11-20 | 三井化学株式会社 | オレフィン重合用触媒およびオレフィンの重合方法 |
| JP3465325B2 (ja) | 1992-11-19 | 2003-11-10 | 三井化学株式会社 | オレフィン重合用触媒およびオレフィンの重合方法 |
| JP3465323B2 (ja) | 1992-11-19 | 2003-11-10 | 三井化学株式会社 | オレフィン重合用触媒およびオレフィンの重合方法 |
| JP3332051B2 (ja) * | 1992-12-18 | 2002-10-07 | 出光興産株式会社 | 重合用触媒及び該触媒系を用いる重合体の製造方法 |
| DE69602329T2 (de) * | 1995-09-14 | 1999-09-23 | Showa Denko K.K., Tokio/Tokyo | Katalysator für ethylenische Polymerisation |
-
1997
- 1997-08-20 US US09/242,664 patent/US6303714B1/en not_active Expired - Fee Related
- 1997-08-20 EP EP97935830A patent/EP0926166A4/en not_active Withdrawn
- 1997-08-20 WO PCT/JP1997/002880 patent/WO1998007761A1/ja not_active Ceased
- 1997-08-20 KR KR1019997001347A patent/KR20000068220A/ko not_active Withdrawn
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH093113A (ja) * | 1995-06-16 | 1997-01-07 | Showa Denko Kk | ポリオレフィンの製造方法 |
| JPH09124722A (ja) * | 1995-08-31 | 1997-05-13 | Sumitomo Chem Co Ltd | 遷移金属錯体、オレフィン重合用触媒及びオレフィン重合体の製造方法 |
| JPH09143221A (ja) * | 1995-09-18 | 1997-06-03 | Showa Denko Kk | エチレン系重合用触媒 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0926166A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20000068220A (ko) | 2000-11-25 |
| EP0926166A1 (en) | 1999-06-30 |
| EP0926166A4 (en) | 2000-07-19 |
| US6303714B1 (en) | 2001-10-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0570982B1 (en) | Catalysts and process for producing olefin polymers | |
| KR940006217B1 (ko) | 올레핀 중합용 촉매 및 이 올레핀 중합용 촉매를 사용한 올레핀의 중합방법 | |
| EP0586168B1 (en) | Catalyst compositions and process for preparing polyolefins | |
| EP0868445B1 (en) | High temperature olefin polymerization process | |
| EP0586167B1 (en) | Catalyst compositions and process for preparing Polyolefins | |
| EP0522581B1 (en) | Process for producing olefin based polymers and olefin polymerization catalyst | |
| JP2003513116A (ja) | 担持された触媒系の製造方法及び重合方法でのその使用 | |
| KR20010042207A (ko) | 올레핀 중합용 중합체 지지 촉매 | |
| JP4234327B2 (ja) | 改良された嵩高な配位子のメタロセン型触媒系を使用する重合方法 | |
| JP2004507589A (ja) | 触媒系および重合プロセスにおけるその使用 | |
| US6759361B2 (en) | Aluminoboronate activators for single-site olefin polymerization catalysts | |
| US6790805B2 (en) | Process for the in-situ preparation of single-site transition metal catalysts and polymerization process | |
| JP2003501523A (ja) | 担持された触媒系の製造方法及び重合方法におけるその使用 | |
| US6214953B1 (en) | Process for the preparation of olefinic polymers using supported metallocene catalyst | |
| JPH11236410A (ja) | 超高分子量エチレン系重合体の製造方法 | |
| WO1998007761A1 (en) | Catalyst component for olefin polymerization, catalyst for olefin polymerization, and process for preparing polyolefin | |
| EP0576213A2 (en) | Process for polymerizing olefins | |
| JP3572339B2 (ja) | オレフィン重合体製造用触媒、その製造方法及びそれを用いたオレフィン重合体の製造方法 | |
| WO2000015676A1 (en) | Monocyclopentadienyl metal catalyst composition for the polymerization of olefins | |
| KR100583822B1 (ko) | 비대칭성 비가교형 메탈로센 화합물 및 이를 포함하는촉매 조성물 | |
| JP2002194019A (ja) | オレフィン重合用触媒及びこれを利用した重合方法 | |
| US6239239B1 (en) | Quinolinoxy and pyridinoxy single-site catalysts containing benzyl ligands | |
| EP1246850A1 (en) | Process for making polyolefins | |
| WO2000044793A1 (en) | Catalyst for olefin polymerization and process for producing olefin polymer | |
| US6294626B1 (en) | Olefin polymerization catalysts containing modified boraaryl ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1997935830 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019997001347 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09242664 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997935830 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019997001347 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1019997001347 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1997935830 Country of ref document: EP |