WO1998049157A1 - Farnesyltransferase inhibiting quinazolinones - Google Patents
Farnesyltransferase inhibiting quinazolinones Download PDFInfo
- Publication number
- WO1998049157A1 WO1998049157A1 PCT/EP1998/002357 EP9802357W WO9849157A1 WO 1998049157 A1 WO1998049157 A1 WO 1998049157A1 EP 9802357 W EP9802357 W EP 9802357W WO 9849157 A1 WO9849157 A1 WO 9849157A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- hydrogen
- 6alkyl
- compound
- halo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c(ccc(C(C1)(c2ccccc2)O*1N=C)c1)c1N=I Chemical compound *c(ccc(C(C1)(c2ccccc2)O*1N=C)c1)c1N=I 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/78—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 2
- C07D239/80—Oxygen atoms
- C07D239/82—Oxygen atoms with an aryl radical attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention is concerned with novel quinazolinone derivatives, the preparation thereof, pharmaceutical compositions comprising said novel compounds and the use of these compounds as a medicine as well as methods of treatment by administering said compounds.
- ras genes that have been identified in mammals, birds, insects, mollusks, plants, fungi and yeasts.
- the family of mammalian ras genes consists of three major members ("isoforms") : H-ras , K-ras and N-ras genes. These ras genes code for highly related proteins generically known as p21 ra,y . These p2 ⁇ ras proteins comprise a family of proteins that regulate cell growth when bound to the inner surface of the plasma membrane.
- farnesyl transferase inhibitors can be very useful as anticancer agents for tumors in which ras contributes to transformation.
- EP-0,371,564 discloses (lH-azol-1-ylmethyl) substituted quinoline, quinazoline and quinoxaline derivatives which suppress the plasma elimination of retinoic acids. Some of these compounds also have the ability to inhibit the formation of androgens from progestines and/or inhibit the action of the aromatase enzyme complex.
- the present invention concerns compounds of formula
- R 1 and R 2 each independently are hydrogen, hydroxy, halo, cyano, Ci-galkyl, trihalomethyl, trihalomethoxy, C2-6alkenyl, Ci- ⁇ alkyloxy, hydroxyCi-6alkyloxy, Ci-6alkyloxyC ⁇ -6alkyloxy, C]-6alkyloxycarbonyl, aminoC ⁇ - 6 alkyloxy, mono- or di(Ci-6alkyl)aminoCi-6alkyloxy, Ar 1 , R 3 and R 4 each independently are hydrogen, halo, cyano, Ci-galkyl, -galkyloxy, A ⁇ oxy, Ci-galkylthio, di(Ci-6alkyl)amino, trihalomethyl or trihalomethoxy; R 5 is hydrogen, halo, Ci- ⁇ alkyl, cyano, haloCi-6alkyl, hydroxyC]-6alkyl, cyanoCi-6alkyl, aminoCi-6alkyl, Ci-6alky
- R 10 is hydrogen, Ci-6alkyl, Ci- 6 alkylcarbonyl, Ar 1 ,
- Ci-6alkyloxycarbonylCi-6alkyl or a radical of formula -Alk-OR 13 or -Alk-NR 14 R 15 ;
- R 11 is hydrogen, Ci-6alkyl,
- Ar 1 or R 12 is hydrogen, Ci-6alkyl, C]-6alkylcarbonyl, Ci-6alkyloxycarbonyl,
- Alk is C ⁇ _6alkanediyl
- R 13 is hydrogen, C]-6alkyl, C ⁇ - 6 alkylcarbonyl, hydroxyCi- 6 alkyl, Ar 1 or Ar ⁇ Ci- ⁇ alkyl;
- R 14 is hydrogen, Ci- 6 alkyl, Ar 1 or
- R 15 is hydrogen
- R 6 is a radical of formula
- R 16 is hydrogen, halo, Ar 1 , C ⁇ -6alkyl, hydroxyCi-galkyl,
- Ci-6alkyloxyC ⁇ -6alkyl Ci-6alkyloxy, Ci-6alkylthio, amino, Ci-6alkyloxycarbonyl, Ci-6a__ylthioCi-6alkyl, Cj-6alkylS(O)C ⁇ _6alkyl or Ci- 6 alkylS(O)2C ⁇ - 6 alkyl;
- R 17 is hydrogen, Ci- ⁇ alkyl or di(C].4alkyl)aminosulfonyl;
- R 7 is hydrogen or Ci.galkyl provided that the dotted line does not represent a bond;
- R 8 is hydrogen, Ci- ⁇ alkyl or Ar 2 CH2 or Het ] CH2;
- R 9 is hydrogen, Ci-6alkyl , Ci- ⁇ alkyloxy or halo; or R 8 and R 9 taken together to form a bivalent radical of formula
- Ar 1 is phenyl; or phenyl substituted with 1 or 2 substituents each independently selected from halo.
- Ci-6alkyl C ⁇ -6alkyloxy or trifluoromethyl;
- Ar 2 is phenyl: or phenyl substituted with 1 or 2 substituents each independently selected from halo.
- halo is generic to fluoro, chloro, bromo and iodo
- C ⁇ _ 2 alkyl defines methyl or ethyl
- Ci-4alkyl includes Ci-2alkyl and the higher homologues thereof having 3 to 4 carbon atoms such as, e.g. propyl, butyl,
- C ⁇ .6al yl includes C ⁇ _4alkyl and the higher homologues thereof having 5 to 6 carbon atoms such as, for example, pentyl, 2-methyl- butyl, hexyl, 2-methylpentyl and the like;
- C2- 6 alkenyl defines straight and branched chain hydrocarbon radicals containing one double bond and having from 2 to 6 carbon atoms such as, for example, ethenyl, 2-propenyl, 3-butenyl, 2-pentenyl, 3-pentenyl, 3-methyl-2- butenyl, and the like;
- Ci. ⁇ alkanediyl defines bivalent straight and branched chained saturated hydrocarbon radicals having from 1 to 6 carbon atoms, such as, for example, methylene, 1 ,2-ethanediyl.
- the pharmaceutically acceptable acid addition salts as mentioned hcreinabove are meant to comprise the therapeutically active non-toxic acid addition salt forms which the compounds of formula (I) are able to form.
- the compounds of formula (I) which have basic properties can be converted in their pharmaceutically acceptable acid acid addition salts by treating said base form with an appropriate acid.
- Appropriate acids comprise, for example, inorganic acids such as hydrohalic acids, e.g. hydrochloric or hydrobromic acid; sulfuric; nitric; phosphoric and the like acids; or organic acids such as, for example, acetic, propanoic, hydroxyacetic, lactic, pyruvic, oxalic, malonic, succinic ⁇ i.e.
- butane- dioic acid maleic, fuma ⁇ c, malic, tartaric. citric, methanesulfonic, ethanesulfonic, benzenesulfonic. /?-toluenesulfonic, cycla ic, salicylic, /? -amino-salicylic, pamoic and the like acids.
- acid addition salts also comprises the hydrates and the solvent addition forms which the compounds of formula (I) are able to form. Examples of such forms are e.g. hydrates, alcoholates and the like.
- stereochemically isomeric forms of compounds of formula (I), as used hereinbefore, defines all possible compounds made up of the same atoms bonded by the same sequence of bonds but having different three-dimensional structures which are not interchangeable, which the compounds of formula (I) may possess. Unless otherwise mentioned or indicated, the chemical designation of a compound encompasses the mixture of all possible stereochemically isomeric forms which said compound may possess. Said mixture may contain all diastereomers and/or enantiomers of the basic molecular structure of said compound. All stereochemically isomeric forms of the compounds of formula (I) both in pure form or in admixture with each other are intended to be embraced within the scope of the present invention.
- R 8 and R 9 are taken together to fo ⁇ n a bivalent radical of formula (c-4) or (c-5), the CH2 moiety in said bivalent radical is preferably connected to the nitrogen atom of the 2-quinazolinone moiety of the compounds of formula (I).
- a group of interesting compounds consists of those compounds of f ormula (I) wherein one or more of the following restrictions apply : a) R 1 and R 2 are each independently selected from hydrogen, halo, Ci-6alkyl, C ⁇ _6alkyloxy or trihalomethyl; in particular hydrogen, halo or C ⁇ _4alkyl; b) R 3 and R 4 are each independently selected from hydrogen, halo, Ci-6alkyl, Ci-6alkyloxy or trihalomethyl; in particular hydrogen, halo or C ⁇ _4alkyl; c) R 5 is is hydrogen, hydroxy, haloC ⁇ .6alkyl, hydroxyCi-6alkyl, cyanoCi-6alkyl, Ci-6alkyloxycarbonylCi-6alkyl, or a radical of formula -NR 1 :l R 12 wherein R 11 is hydrogen or Ci-6alkyl and R 12 is hydrogen, Ci.galkyl, Ci- ⁇ alkyloxy, C ⁇ _6alkyloxy-
- Ci-6alkylcarbonyl in particular R 5 is hydrogen, hydroxy, halo or amino; d) R 6 is a radical of formula (b-1) or (b-2) wherein R 16 is hydrogen or Ci- ⁇ alkyl and R 17 is C ⁇ . 6 alkyl; e) R 7 is hydrogen or C ⁇ .6alkyl in case the dotted line docs not represent a bond; 1) R 8 is hydrogen, Ci- ⁇ alkyl or Het ] CH2; g) R 9 is hydrogen.
- a particular group of compounds consists of those compounds of formula (I) wherein X is oxygen, R 1 and R 2 are each independently selected from hydrogen, halo or Ci-4alkyl; R 3 and R 4 are each independently selected from hydrogen, halo or Ci-4a__yl; R 5 is hydrogen, hydroxy, halo or a amino; R 6 is a radical of formula (b-1) or (b-2) wherein R 16 is hydrogen or Ci-4alkyl and R 17 is C ⁇ _ ⁇ alkyl; R 7 is hydrogen or C ⁇ 4 alkyl in case the dotted line does not represent a bond; R 8 is hydrogen; Ci-4alkyl or Het 1 CH2; and R 9 is hydrogen.
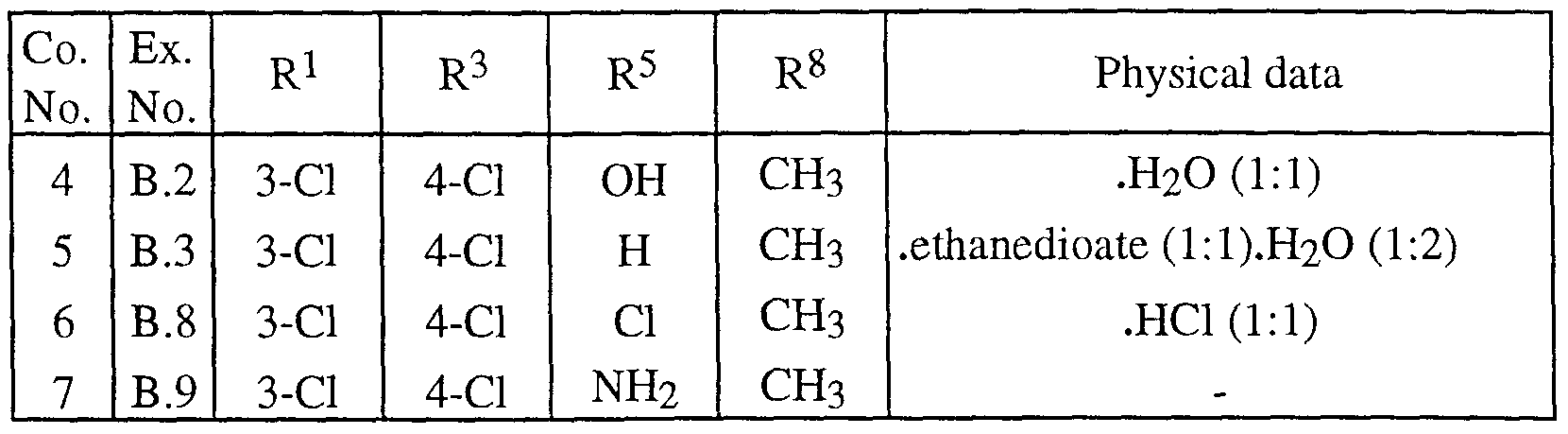
- Preferred compounds are those compounds of formula (I) wherein X is oxygen, R 1 is 3-chloro, R 2 is hydrogen, R 3 is 4-chloro, R 4 is hydrogen, R 5 is hydrogen, Ci-2alkyl, halo or amino; R 6 is a radical of formula (b-1) or (b-2) wherein R 16 is hydrogen and R 17 is Ci-2alkyl; and R 7 is hydrogen or Ci-2alkyl in case the dotted line does not represent a bond; R 8 is hydrogen; C ⁇ _2alkyl or Het 1 CH 2 ; and R 9 is hydrogen.
- the compounds of formula (I) wherein R 6 is a radical of formula (b-1), represented by compounds of formula (I-a), can generally be prepared by N-alkylating an intermediate of formula (III), with an intermediate of formula (II), wherein W is an appropriate leaving group such as, for example, chloro, bromo, methanesulfonyloxy or benzenesulfonyloxy.
- the reaction can be performed in a reaction-inert solvent such as, for example, acetonitrile, and optionally in the presence of a suitable base such as, for example, sodium carbonate, potassium carbonate or triethylamine. Stirring may enhance the rate of the reaction.
- the reaction may conveniently be carried out at a temperature ranging between room temperature and reflux temperature. -7-
- compounds of formula (I-a) can be prepared by reacting an intermediate of formula (IV) with an intermediate of formula (V), wherein Y is carbon or sulfur, such as, for example, a l,l'-carbonyldiimidazole.
- reaction may conveniently be conducted in a reaction-inert solvent, such as, e.g. tetrahydrofuran, optionally in the presence of a base, such as sodium hydride, and at a temperature ranging between room temperature and the reflux temperature of the reaction mixture.
- a reaction-inert solvent such as, e.g. tetrahydrofuran
- a base such as sodium hydride
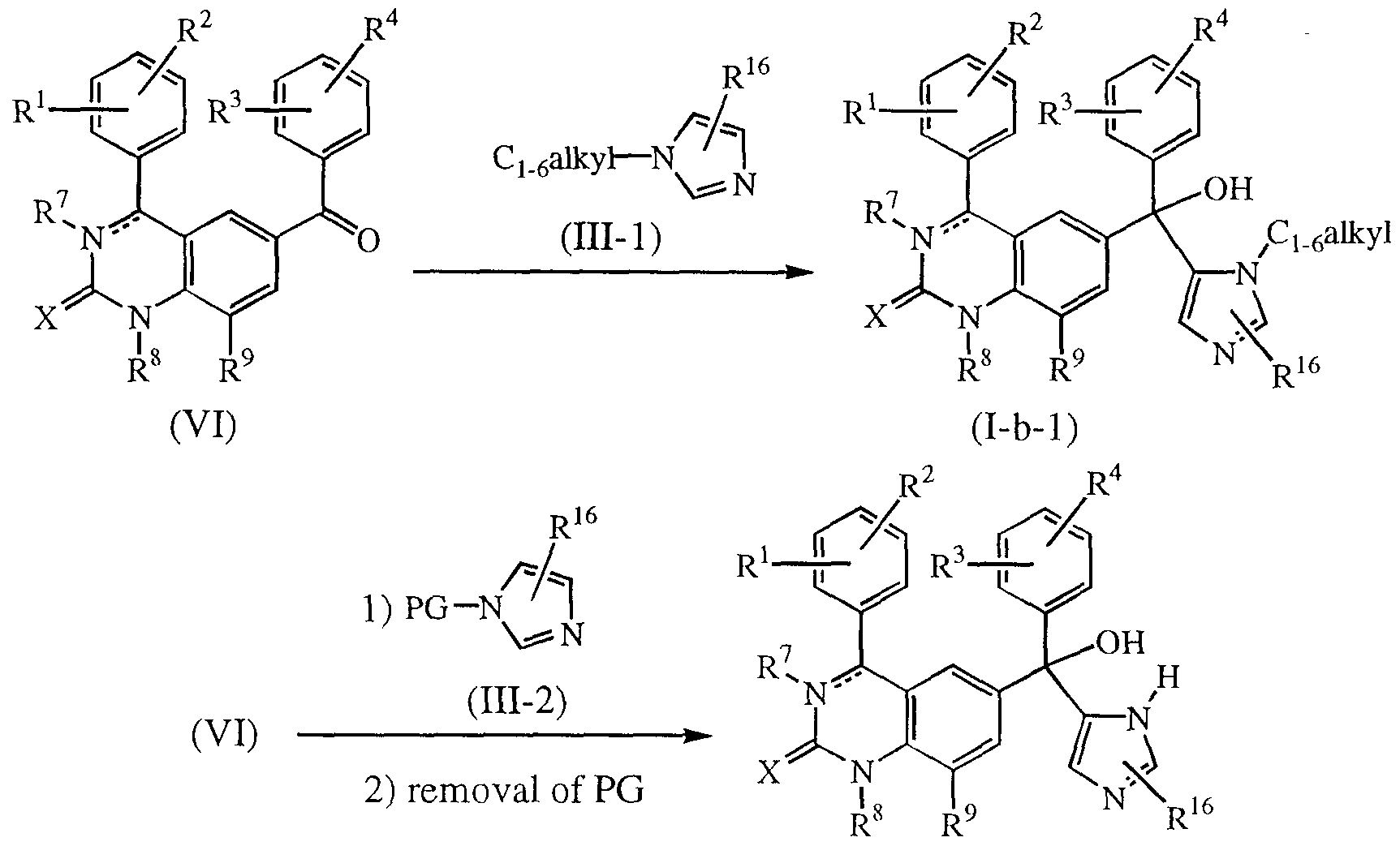
- the compounds of formula (I) wherein R 6 represents a radical of formula (b-2), R 5 is hydroxy and R 17 is Ci-6alkyl, said compounds being referred to as compounds of formula (I-b-1) may be prepared by reacting an intermediate ketone of formula (VI) with an intermediate of formula (IITl). Said reaction requires the presence of a suitable strong base, such as, for example, butyl lithium in an appropriate solvent, such as, for example, tetrahydrofuran, and the presence of an appropriate silanederivative, such as, for example, triethylchlorosilane. During the work-up procedure an intermediate silane derivative is hydrolyzed. Other procedures with protective groups analogous to silanederivatives can also be applied. (I-b-2)
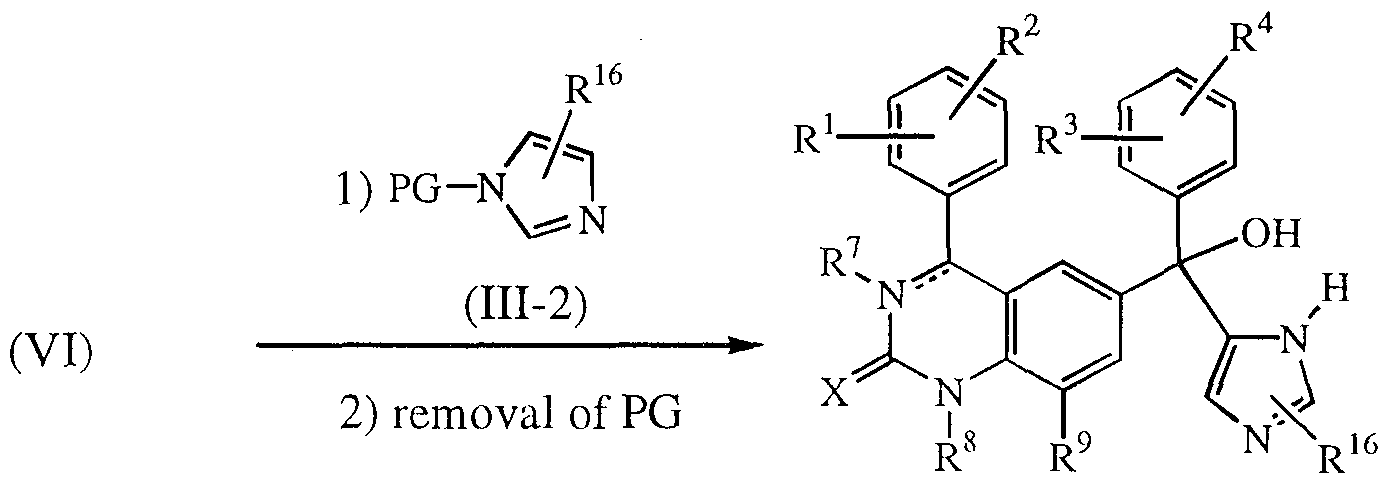
- the compounds of formula (I), wherein R 6 is a radical of formula (b-2), R 5 is hydroxy and R 17 is hydrogen, said compounds being referred to as compounds of formula (I-b-2) may be prepared by reacting an intermediate ketone of formula (VI) with a intermediate of formula (111-2), wherein PG is a protective group such as, for example, a sulfonyl group, e.g. a dimethylamino sulfonyl group, which can be removed after the addition reaction. Said reaction is conducted analogously as for the preparation of compounds of formula (I-b-1), followed by removal of the protecting group PG, yielding compounds of formula (I-b-2).
- compounds of formula (I-d) can be converted to compounds of formula (I-c) using art-known reduction procedures such as, e.g. treatment with sodiumborohydride in a suitable solvent, e.g. methanol.
- compounds of formula (I-c) can be converted to compounds of formula (I-c-1) by treating compounds (I-c) with a reagent of formula R 7 -W 1 , wherein W 1 is an appropriate leaving group such as, for example, chloro, bromo, methanesulfonyloxy or benzenesulfonyloxy, using the above-described N-alkylation procedure.
- W 1 is an appropriate leaving group such as, for example, chloro, bromo, methanesulfonyloxy or benzenesulfonyloxy
- the compounds of formula (I-b) can be converted to compounds of formula (I-e), defined as a compound of formula (I) wherein R 6 is a radical of formula (b-2) and R 5 is hydrogen, by submitting the compounds of formula (I-b) to appropriate reducing conditions, such as, e.g. stirring in acetic acid in the presence of formamide. -10-
- compounds of formula (I-b) can be converted to compounds of formula (I-f) wherein R 5 is halo, by reacting the compounds of formula (I-b) with a suitable halogenating agent, such as, e.g. thionyl chloride or phosphorus tribromide. Successively, the compounds of formula (I-f) can be treated with a reagent of formula H-NR ⁇ R 12 in a reaction-inert solvent, thereby yielding compounds of formula (I-g).
- a suitable halogenating agent such as, e.g. thionyl chloride or phosphorus tribromide.
- a compound of formula (I-i), defined as a compound of formula (I) wherein X is sulfur, may be prepared by reacting the corresponding compound of formula (I-h), defined as a compound of formula (I) wherein X is oxygen, with a reagent like phosphorus pentasulfide or Lawesson's reagent in a suitable solvent such as, for example, pyridine.
- An intermediate of formula (Il-a), being an intermediate of formula (II) wherein X is oxygen and R 7 and R 8 are hydrogen, can be prepared starting from an intermediate of formula (VII).
- Said intermediate (VII), wherein n is 2 or 3, is conveniently prepared by protecting the corresponding art-known ketone as a ketal.
- An intermediate of formula (VII) is reacted with an intermediate of formula (VIII) in the presence of a base such as sodium hydroxide, in an appropriate solvent, e.g. methanol.
- a base such as sodium hydroxide
- an appropriate solvent e.g. methanol.
- the thus obtained intermediate of formula (IX) undergoes ring opening of the isoxazole moiety by hydrogenation of intermediate (IX) in the presence of a suitable catalyst such as, e.g. Raney Nickel.
- the hydroxy group of intermediates of formula (X ⁇ ) is converted to a leaving group W by treating inte ⁇ nediates (XII) with a suitable reagent such as, e.g. methanesulfonyloxy chloride, or a halogenating reagent such as, e.g. POCI3 or SOCI2, yielding intermediates of formula (Il-a).
- a suitable reagent such as, e.g. methanesulfonyloxy chloride, or a halogenating reagent such as, e.g. POCI3 or SOCI2, yielding intermediates of formula (Il-a).
- Intermediates of formula (ITb), defined as intermediates of formula (II) wherein X is O and R 7 is hydrogen, can be prepared by reacting intermediates of formula (XI) with R 8 -W 1 , wherein W 1 is a suitable leaving group such as, e.g. chloro, bromo, methanesulfonyloxy or benzenesulfonyloxy; using the above-described N-alkylation procedure. Subsequent reduction with e.g. sodiumborohydride in a suitable solvent, e.g. methanol, and hydrolysis under acidic conditions, yields intermediates of formula (XIV). Convertion of the hydroxy group of intermediates (XIV) into leaving group W, e.g. by treatment with methanesulfonyloxy chloride or a halogenating reagent such as, e.g. SOCI2, POCI3, gives intermediates of formula (Il-b).
- W 1 is a suitable leaving group such as, e.g
- Intermediates of formula (Vl-a), defined as intermediates of formula (VI) wherein X is O and the dotted line does not represent a bond, can be prepared by submitting intermediates of formula (XIII) to art-known reduction procedures, such as, e.g. treatment with sodium borohydride in a reaction-inert solvent e.g. methanol, thereby •13-
- Inte ⁇ nediates (XV) are N-alkylated with R 7 -W 1 , wherein W 1 is a leaving group as above-described, and subsequently hydrolysed under acidic conditions to inte ⁇ nediates of formula (Vl-a).
- intermediates of formula (Vl-b) can be prepared by hydrolysis of the intermediate of formula (IX) with an acid, such as for example, TiCl3, in the presence of water.
- an acid such as for example, TiCl3, in the presence of water.
- acylation with a reactive carboxylic acid derivative such as, e.g. trichloroacetyl chloride, yields an intermediate of formula (XVII), which undergoes ring closure in the presence of an ammonium salt, e.g. ammonium acetate, and an appropriate base such as, e.g. hexamethylphosphorous triamide (HMPT), thereby yielding an intermediate of formula (Vl-b).
- an ammonium salt e.g. ammonium acetate
- an appropriate base such as, e.g. hexamethylphosphorous triamide (HMPT)
- the compounds of formula (I) and some of the intermediates have at least one stereogenic center in their structure.
- This stereogenic center may be present in a R or a S configuration.
- the compounds of formula (I) as prepared in the hereinabove described processes are generally racemic mixtures of enantiomers which can be separated from one another following art-known resolution procedures.
- the racemic compounds of formula (I) may be converted into the corresponding diastereomeric salt forms by reaction with a suitable chiral acid. Said diastereomeric salt forms are subsequently separated, for example, by selective or fractional crystallization and the enantiomers are liberated therefrom by alkali.
- An alternative manner of separating the enantiomeric forms of the compounds of formula (I) involves liquid chromatography using a chiral stationary phase.
- Said pure stereochemically isomeric forms may also be derived from the corresponding pure stereochemically isomeric forms of the appropriate starting materials, provided that the reaction occurs stereospeciiically.
- Specific stereoisomers can be synthesized by stereospecific methods of preparation. These methods will advantageously employ enantiomerically pure starting materials.
- This invention provides a method for inhibiting the abnormal growth of cells, including transfo ⁇ ned cells, by administering an effective amount of a compound of the invention.
- Abnormal growth of cells refers to cell growth independent of normal regulatory mechanisms (e.g. loss of contact inhibition). This includes the abnormal growth of : (1) tumor cells (tumors) expressing an activated ras oncogene; (2) tumor cells in which the ras protein is activated as a result of oncogenic mutation of another gene; (3) benign and malignant cells of other proliferative diseases in which aberrant ras activation occurs.
- ras oncogenes not only contribute to the growth of of tumors in vivo by a direct effect on tumor cell growth but also indirectly, i.e. by facilitating tumor-induced angiogenesis (Rak. J. et al, Cancer Research, 55, 4575-4580, 1995).
- pharmacologically targetting mutant ras oncogenes could conceivably suppress solid tumor growth in vivo, in part, by inhibiting tumor-induced angiogenesis.
- This invention also provides a method for inhibiting tumor growth by administering an effective amount of a compound of the present invention, to a subject, e.g. a mammal (and more particularly a human) in need of such treatment.
- this invention provides a method for inhibiting the growth of tumors expressing an activated ras oncogene by the administration of an effective amount of the compounds of the present invention.
- tumors which may be inhibited, but are not limited to, lung cancer (e.g. adenocarcinoma), pancreatic cancers (e.g. pancreatic carcinoma such as, for example exocrine pancreatic carcinoma), colon cancers (e.g.
- colorectal carcinomas such as, for example, colon adenocarcinoma and colon adenoma
- hematopoietic tumors of lymphoid lineage e.g. acute lymphocytic leukemia, B-cell lymphoma, Burkitt's lymphoma
- myeloid leukemias for example, acute myelogenous leukemia (AML)
- thyroid follicular cancer myelodysplastic syndrome (MDS)
- tumors of mesenchymal origin e.g. fibrosarcomas and rhabdomyosarcomas
- melanomas teratocarcinomas
- neuroblastomas gliomas
- benign tumor of the skin e.g. keratoacanthomas
- breast carcinoma e.g. keratoacanthomas
- kidney carninoma ovary carcinoma
- bladder carcinoma e.g. keratoacanthomas
- This invention may also provide a method for inhibiting proliferative diseases, both benign and malignant, wherein ras proteins are aberrantly activated as a result of oncogenic mutation in genes, i.e. the ras gene itself is not activated by mutation to an oncogenic form, with said inhibition being accomplished by the administration of an effective amount of the compounds described herein, to a subject in need of " such a treatment.
- the benign proliferative disorder neurofibromatosis, or tumors in which ras is activated due to mutation or overexpression of tyrosine kinase oncogenes may be inhibited by the compounds of this invention.
- the present invention discloses the compounds of formula (I) for use as a medicine as well as the use of these compounds of formula (I) for the manufacture of a medicament for treating one or more of the above mentioned conditions.
- the subject compounds may be formulated into various pharmaceutical forms for administration purposes.
- compositions of " this invention an effective amount of a particular compound, in base or acid addition salt form, as the active ingredient is combined in intimate admixture with a pharmaceutically acceptable carrier, which carrier may take a wide variety of forms depending on the form of preparation desired for administration.
- a pharmaceutically acceptable carrier which carrier may take a wide variety of forms depending on the form of preparation desired for administration.
- These pharmaceutical compositions are desirably in unitary dosage form suitable, preferably, for administration orally, rectally, percutaneously, or by parenteral injection.
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols and the like in the case of oral liquid preparations such as suspensions, syrups, elixirs and solutions; or solid carriers such as starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like in the case of powders, pills, capsules and tablets. Because of their ease in administration, tablets and capsules represent the most advantageous oral dosage unit form, in which case solid pharmaceutical carriers are obviously employed.
- the carrier will usually comprise sterile water, at least in large part, though other ingredients, to aid solubility for example, may be included.
- Injectable solutions may be prepared in which the carrier comprises saline solution, glucose solution or a mixture of saline and glucose solution. Injectable suspensions may also be prepared in which case appropriate liquid carriers, suspending agents and the like may be employed.
- the carrier optionally comprises a penetration enhancing agent and/or a suitable wetting agent, optionally combined with suitable additives of any nature in minor proportions, which additives do not cause a significant deleterious effect to the skin. Said additives may facilitate the administration to the skin and/or may be helpful for preparing the desired compositions.
- These compositions may be administered in various ways, e.g., as a transdermal patch, as a spot-on, as an ointment.
- Dosage unit form as used in the specification and claims herein refers to physically discrete units suitable as unitary dosages, each unit containing a predetermined quantity of active ingredient calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- dosage unit forms are tablets (including scored or coated tablets), capsules, pills, powder packets, wafers, injectable solutions or suspensions, teaspoonfuls. tablespoonfuls and the like, and segregated multiples thereof.
- an effective amount would be from 0.01 mg/kg to 100 mg/kg body weight, and in particular from 0.05 mg/kg to 10 mg/kg body weight. It may be appropriate to administer the required dose as two, three, four or more sub-doses at appropriate intervals throughout the day. Said sub-doses may be formulated as unit dosage forms, for example, containing 0.05 to 500 mg, and in particular 0.1 mg to 200 mg of active ingredient per unit dosage form. The following examples are provided for purposes of illustration.
- THF tetrahydrofuran
- DIPE diisopropylether
- DCM dichloromethane
- DMF N,N-dimethylformamide
- AC ⁇ means acetonitrile
- Example B.5 A dispersion of sodium hydride in mineral oil (60%)(0.0047 mol) was added portionwise to a mixture of compound (9) (0.0043 mol) in DMF (40 ml) under N2 flow. The mixture was stirred for 30 minutes at room temperature. A solution of iodomethane (0.0047 mol) in DMF (10 ml) was added dropwise and the resulting reaction mixture was stirred overnight at room temperature. The reaction mixture was poured out into water (200 ml) and this mixture was extracted with toluene (3 x 100 ml). The separated organic layer was dried, filtered and the solvent evaporated.
- a dispersion of sodium hydride in mineral oil (60%) (0.01122 mol) was added portionwise to a mixture of compound (1) (0.0051 mol) in DMF (25 ml) under N 2 flow. The mixture was stirred for 30 minutes at room temperature.
- a solution of 4-(chloromethyl)pyridine hydrochloride (0.00561 mol) in DMF (5 ml) was added dropwise and the resulting reaction mixture was stirred over the weekend at room temperature. The reaction mixture was poured out into water and this mixture was extracted with toluene. The separated organic layer was dried, filtered and the solvent evaporated.
- the residue was purified by column chromatography over silica gel (eluent : CH 2 Cl2/CH 3 OH/(CH3OH/NH3) 90/5/5). The desired fractions were collected and the solvent was evaporated. This fraction was repurified by high-performance liquid chromatography over Kromasil RP-18 (100 A, 10 ⁇ m, 5 cm DAC; eluent: (0.5% NH 4 OAc in H 2 O)/CH 3 OH/CH3CN 47/25/28 v/v). The pure fractions were collected and the organic solvent was evaporated. The aqueous residue was extracted with DCM.
- Example B 7 A mixture of compound (4) (0.0069 mol) in formamide (34 ml) and acetic acid (68 ml) was stirred at 160°C for 24 hours, then poured out into ice water and alkalised with a concentrated N ⁇ 3 (aq.) solution. The precipitate was filtered off " , washed with water and taken up in DCM. The organic layer was separated, dried, filtered and the solvent was evaporated till dryness. The residue was purified by column chromatography over silica gel (eluent: CH 2 CI 2 /CH3OH/NH4OH 96/4/0.2). The pure fractions were collected and the solvent was evaporated. The residue was crystallized from 2-propanone/DIPE.
- Tables F- 1 to F-4 list the compounds that were prepared according to one of the above Examples.
- Compounds are screened in tissue culture in NIH 3T3 cells transi ' onned by the T24 activated human -ras gene.
- Cells are seeded at an initial density of 200,000 cells per well (9.6 cm 2 surface area) in six-well cluster tissue culture plates.
- Test compounds are immediately added to 3.0 ml cell growth medium in a 3.0 ⁇ l volume of DMSO, with a final concentration of DMSO in the cell growth medium of 0.1 %.
- the test compounds are run at concentrations of 5, 10, 50, 100, and 500 nM along with a DMSO treated vehicle control. (In case a high activity is observed at 5 nM, the test compound is tested at even lower concentrations.)
- the cells are allowed to proliferate for 72 hours. Then the cells are detached in 1.0 ml trypsin-EDTA cell dissociation medium and counted on a Coulter particle counter.
- Control cell counts [cell counts from cells incubated with DMSO vehicle - 200,000]
- Test compound cell counts [cell counts from cells incubated with test compound
- test compound cell counts ⁇ rk ⁇ / Test compound %inh ⁇ b ⁇ t ⁇ on [l - cQntrol ⁇ CQ _ nts J x 100%. Compounds 5, 7, 14 and 15 had an IC50 less than 500 nM.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Medicines Containing Plant Substances (AREA)
- Silver Salt Photography Or Processing Solution Therefor (AREA)
- Cosmetics (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
Description





















Claims







Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002288140A CA2288140C (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones |
| NZ336233A NZ336233A (en) | 1997-04-25 | 1998-04-17 | Phenyl substituted quinazolines on 4-position and 2-quinazolinone moiety bearing a carbon or nitrogen-linked imidazolyl moiety |
| SK1461-99A SK146199A3 (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones |
| JP54656198A JP4308919B2 (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase-inhibiting quinazolinones |
| AU76460/98A AU738628B2 (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones |
| IL13036398A IL130363A (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones, their preparation and pharmaceutical compositions comprising them |
| BR9809398-3A BR9809398A (en) | 1997-04-25 | 1998-04-17 | Quinazolinones that inhibit farnesyl transferase |
| US09/403,705 US6177432B1 (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones |
| HU0001122A HUP0001122A3 (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones |
| EP98924161A EP0977750B1 (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones |
| PL336468A PL190944B1 (en) | 1997-04-25 | 1998-04-17 | A derivative of quinazolinone and method for its manufacture as well as pharmaceutical compound and method for its manufacture |
| DE69838025T DE69838025T2 (en) | 1997-04-25 | 1998-04-17 | CHINAZOLINONE HEMMEN THE FARNESYLTRANSFERASE |
| KR10-1999-7005507A KR100520401B1 (en) | 1997-04-25 | 1998-04-17 | Farnesyl transferase inhibiting quinazolinones |
| NO19995169A NO317576B1 (en) | 1997-04-25 | 1999-10-22 | Farnesyltransferase inhibiting quinazolinones |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP97201259.5 | 1997-04-25 | ||
| EP97201259 | 1997-04-25 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/380,856 A-371-Of-International US6187786B1 (en) | 1997-03-10 | 1998-03-03 | Farnesyl transferase inhibiting 1,8-annelated quinolinone derivatives substituted with N- or C-linked imidazoles |
| US09/687,153 Continuation US6358961B1 (en) | 1997-04-25 | 2000-10-13 | Farnesyltransferase inhibiting quinazolinones |
| US09/725,391 Division US6444812B1 (en) | 1997-03-10 | 2000-11-29 | Intermediate compounds in the preparation of farnnesyl transferase inhibiting 1,8-annelated quinolinone derivatives substituted with N-or C linked imidazoles |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998049157A1 true WO1998049157A1 (en) | 1998-11-05 |
Family
ID=8228267
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/002357 Ceased WO1998049157A1 (en) | 1997-04-25 | 1998-04-17 | Farnesyltransferase inhibiting quinazolinones |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US6177432B1 (en) |
| EP (1) | EP0977750B1 (en) |
| JP (1) | JP4308919B2 (en) |
| KR (1) | KR100520401B1 (en) |
| CN (1) | CN1094937C (en) |
| AT (1) | ATE366250T1 (en) |
| AU (1) | AU738628B2 (en) |
| BR (1) | BR9809398A (en) |
| CA (1) | CA2288140C (en) |
| CZ (1) | CZ296959B6 (en) |
| DE (1) | DE69838025T2 (en) |
| ES (1) | ES2289783T3 (en) |
| HU (1) | HUP0001122A3 (en) |
| IL (1) | IL130363A (en) |
| NO (1) | NO317576B1 (en) |
| NZ (1) | NZ336233A (en) |
| PL (1) | PL190944B1 (en) |
| RU (1) | RU2205831C2 (en) |
| SK (1) | SK146199A3 (en) |
| TR (1) | TR199902606T2 (en) |
| WO (1) | WO1998049157A1 (en) |
| ZA (1) | ZA983504B (en) |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000039082A3 (en) * | 1998-12-23 | 2000-10-26 | Janssen Pharmaceutica Nv | 1,2-annelated quinoline derivatives |
| KR20010077400A (en) * | 2000-02-02 | 2001-08-17 | 성재갑 | Anticancer agents by combination of Ftase inhibitor(LB42908) and other anticancer drugs |
| US6284755B1 (en) | 1998-12-08 | 2001-09-04 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| US6316436B1 (en) | 1998-12-08 | 2001-11-13 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| WO2001064199A3 (en) * | 2000-02-29 | 2001-12-27 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with taxane compounds |
| WO2001098302A1 (en) * | 2000-06-22 | 2001-12-27 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 1,2-annelated quinoline enantiomer |
| WO2001064246A3 (en) * | 2000-02-29 | 2002-02-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with an her2 antibody |
| WO2001064226A3 (en) * | 2000-02-29 | 2002-03-07 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with platinum compounds |
| WO2001064194A3 (en) * | 2000-02-29 | 2002-03-07 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with camptothecin compounds |
| WO2001064198A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with anti-tumor podophyllotoxin derivatives |
| WO2001064252A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with further anti-cancer agents |
| WO2001064195A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with anti-tumor nucleoside derivatives |
| WO2001064196A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with vinca alkaloids |
| WO2001064197A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with anti-tumor anthracycline derivatives |
| WO2001064218A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations |
| WO2002024683A1 (en) * | 2000-09-25 | 2002-03-28 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 6-[(substituted phenyl)methyl]-quinoline and quinazoline derivatives |
| WO2001064217A3 (en) * | 2000-02-29 | 2002-03-28 | Janssen Pharmaceutica Nv | Combinations of a farnesyl protein transferase inhibitor with nitrogen mustard or nitrosourea alkylating agents |
| WO2002024682A1 (en) * | 2000-09-25 | 2002-03-28 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting quinoline and quinazoline derivatives as farnesyl transferase inhibitors |
| WO2002024687A1 (en) * | 2000-09-25 | 2002-03-28 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinolinone derivatives |
| WO2002042296A1 (en) * | 2000-11-21 | 2002-05-30 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting benzoheterocyclic derivatives |
| WO2002043733A1 (en) * | 2000-11-28 | 2002-06-06 | Janssen Pharmaceutica N.V. | Farnesyl protein transferase inhibitors for the treatment of inflammatory bowel disease |
| WO2002024686A3 (en) * | 2000-09-25 | 2002-06-13 | Janssen Pharmaceutica Nv | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinoline and quinazoline derivatives |
| WO2002051835A1 (en) * | 2000-12-27 | 2002-07-04 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 4-substituted quinoline and quinazoline derivatives |
| WO2002064142A1 (en) * | 2001-02-15 | 2002-08-22 | Janssen Pharmaceutica N.V. | Farnesyl protein transferase inhibitor combinations with antiestrogen agents |
| US6451812B1 (en) | 1998-07-06 | 2002-09-17 | Janssen Pharmaceutica N.V. | Farnesyl protein transferase inhibitors for treating arthropathies |
| WO2003051880A1 (en) * | 2001-12-19 | 2003-06-26 | Janssen Pharmaceutica N.V. | 1,8-annelated quinoline derivatives substituted with carbon-linked triazoles as farnesyl transferase inhibitors |
| JP2003521509A (en) * | 2000-02-04 | 2003-07-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Farnesyl protein transferase inhibitors for the treatment of breast cancer |
| FR2837201A1 (en) * | 2002-03-18 | 2003-09-19 | Servier Lab | NOVEL COMPOUNDS DERIVED FROM QUINAZOLINE, THEIR PREPARATION PROCESS AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| US6838467B2 (en) | 2000-02-24 | 2005-01-04 | Janssen Pharmaceutica N. V. | Dosing regimen |
| US6844439B2 (en) | 2001-03-12 | 2005-01-18 | Jansen Pharmaceutica, Nv | Process for the preparation of imidazole compounds |
| US7241777B2 (en) | 2002-03-22 | 2007-07-10 | Janssen Pharmaceutica N.V. | Benzylimidazolyl substituted 2-quinoline and quinazoline derivatives for use as farnesyl transferase inhibitors |
| US7511138B2 (en) | 2002-04-15 | 2009-03-31 | Janssen Pharmaceutica Nv | Farnesyl transferase inhibiting tricyclic quinazoline derivatives substituted with carbon-linked imidazoles or triazoles |
| EP2362218A2 (en) | 2004-11-05 | 2011-08-31 | Janssen Pharmaceutica N.V. | Methods of monitoring the efficacy of farnesyltransferase inhibitors |
| US8481564B2 (en) | 2006-04-20 | 2013-07-09 | Janssen Pharmaceutica, N.V. | Inhibitors of c-fms kinase |
| US8497376B2 (en) | 2007-10-17 | 2013-07-30 | Janssen Pharmaceutica N.V. | Inhibitors of c-fms kinase |
| US8557847B2 (en) | 2005-06-10 | 2013-10-15 | Janssen Pharmaceutica, N.V. | Synergistic modulation of FLT3 kinase using a FLT3 inhibitor and a farnesyl transferase inhibitor |
| US8697716B2 (en) | 2006-04-20 | 2014-04-15 | Janssen Pharmaceutica Nv | Method of inhibiting C-KIT kinase |
| US8859602B2 (en) | 2006-04-20 | 2014-10-14 | Janssen Pharmaceutica Nv | Inhibitors of c-fms kinase |
| US9029352B2 (en) | 2012-08-07 | 2015-05-12 | Janssen Pharmaceutica Nv | Process for the preparation of C-FMS kinase inhibitors |
| US9303046B2 (en) | 2012-08-07 | 2016-04-05 | Janssen Pharmaceutica Nv | Process for the preparation of heterocyclic ester derivatives |
| EP2445502B1 (en) * | 2009-06-25 | 2017-06-21 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| US11273158B2 (en) | 2018-03-05 | 2022-03-15 | Alkermes Pharma Ireland Limited | Aripiprazole dosing strategy |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2205831C2 (en) * | 1997-04-25 | 2003-06-10 | Янссен Фармацевтика Н.В. | Quinazolinones inhibiting farnesyltransferase activity |
| US20020177575A1 (en) * | 2001-05-04 | 2002-11-28 | Ward Wakeland | Identification of farnesyl-protein transferase as a target for systemic lupus erythematosus therapies |
| US20030125268A1 (en) * | 2002-08-28 | 2003-07-03 | Rybak Mary Ellen Margaret | Farnesyl protein transferase inhibitor combinations with anti-tumor anthracycline derivatives |
| US20050272068A1 (en) * | 2004-03-18 | 2005-12-08 | The Brigham And Women's Hospital, Inc. | UCH-L1 expression and cancer therapy |
| US20060106060A1 (en) * | 2004-03-18 | 2006-05-18 | The Brigham And Women's Hospital, Inc. | Methods for the treatment of synucleinopathies (Lansbury) |
| US20050272722A1 (en) * | 2004-03-18 | 2005-12-08 | The Brigham And Women's Hospital, Inc. | Methods for the treatment of synucleinopathies |
| JP2007529555A (en) * | 2004-03-18 | 2007-10-25 | ザ ブライハム アンド ウイメンズ ホスピタル, インコーポレイテッド | How to treat synucleinopathy |
| CA2559285A1 (en) * | 2004-03-18 | 2005-09-29 | Brigham And Women's Hospital, Inc. | Methods for the treatment of synucleinopathies |
| US20070293539A1 (en) * | 2004-03-18 | 2007-12-20 | Lansbury Peter T | Methods for the treatment of synucleinopathies |
| EP1655289A1 (en) * | 2004-11-04 | 2006-05-10 | Embl | Quinazoline derivatives, process for their preparation, their use as antimitotics and pharmaceutical compositions comprising said derivatives |
| US20060194821A1 (en) * | 2005-02-18 | 2006-08-31 | The Brigham And Women's Hospital, Inc. | Compounds inhibiting the aggregation of superoxide dismutase-1 |
| EP1871347B1 (en) | 2005-04-19 | 2016-08-03 | Novartis AG | Pharmaceutical composition |
| CA2634598A1 (en) | 2005-12-23 | 2007-07-05 | Link Medicine Corporation | Treatment of synucleinopathies |
| CN101600698B (en) | 2006-09-11 | 2012-01-11 | 欧加农股份有限公司;药典有限责任公司 | Quinazolinone and isoquinolinone acetamide derivatives |
| US7932036B1 (en) | 2008-03-12 | 2011-04-26 | Veridex, Llc | Methods of determining acute myeloid leukemia response to treatment with farnesyltransferase |
| WO2009151683A2 (en) * | 2008-03-12 | 2009-12-17 | Link Medicine Corporation | Quinolinone farnesyl transferase inhibitors for the treatment of synucleinopathies and other indications |
| US20110060005A1 (en) * | 2008-11-13 | 2011-03-10 | Link Medicine Corporation | Treatment of mitochondrial disorders using a farnesyl transferase inhibitor |
| NZ593090A (en) * | 2008-11-13 | 2013-06-28 | Link Medicine Corp | Azaquinolinone derivatives and uses thereof |
| US20100331363A1 (en) * | 2008-11-13 | 2010-12-30 | Link Medicine Corporation | Treatment of mitochondrial disorders using a farnesyl transferase inhibitor |
| ES2705719T3 (en) | 2010-07-28 | 2019-03-26 | Janssen Diagnostics Llc | Methods for determining the response of acute myeloid leukemia to treatment with farnesyltransferase inhibitors |
| CN103275085B (en) * | 2013-05-30 | 2015-04-08 | 温州大学 | Quinazoline and quinazolinone compound, its synthesis method and application |
| SMT202100712T1 (en) | 2015-08-17 | 2022-01-10 | Kura Oncology Inc | Methods of treating cancer patients with farnesyl transferase inhibitors |
| TW201818965A (en) | 2016-11-03 | 2018-06-01 | 美商庫拉腫瘤技術股份有限公司 | Method for treating cancer patients by using FARNESYL TRANSFERASE inhibitor |
| WO2019113269A1 (en) | 2017-12-08 | 2019-06-13 | Kura Oncology, Inc. | Methods of treating cancer patients with farnesyltransferase inhibitors |
| US20220143006A1 (en) | 2019-03-15 | 2022-05-12 | Kura Oncology, Inc. | Methods of treating cancer with farnesyltransferase inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0371564A2 (en) * | 1988-11-29 | 1990-06-06 | Janssen Pharmaceutica N.V. | (1H-azol-1-ylmethyl)substituted quinoline, quinazoline or quinoxaline derivatives |
| EP0664128A1 (en) * | 1992-10-07 | 1995-07-26 | Sumitomo Pharmaceuticals Company, Limited | Pharmaceutical composition for inhibiting tumor necrosis factor production |
| WO1996015118A1 (en) * | 1994-11-12 | 1996-05-23 | Zeneca Limited | Aniline derivatives |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5430148A (en) * | 1992-03-31 | 1995-07-04 | Agouron Pharmaceuticals, Inc. | Antiproliferative quinazolines |
| RU2205831C2 (en) * | 1997-04-25 | 2003-06-10 | Янссен Фармацевтика Н.В. | Quinazolinones inhibiting farnesyltransferase activity |
-
1998
- 1998-04-17 RU RU99124815/04A patent/RU2205831C2/en active
- 1998-04-17 HU HU0001122A patent/HUP0001122A3/en unknown
- 1998-04-17 ES ES98924161T patent/ES2289783T3/en not_active Expired - Lifetime
- 1998-04-17 AU AU76460/98A patent/AU738628B2/en not_active Expired
- 1998-04-17 IL IL13036398A patent/IL130363A/en not_active IP Right Cessation
- 1998-04-17 CZ CZ0371799A patent/CZ296959B6/en not_active IP Right Cessation
- 1998-04-17 CA CA002288140A patent/CA2288140C/en not_active Expired - Lifetime
- 1998-04-17 KR KR10-1999-7005507A patent/KR100520401B1/en not_active Expired - Fee Related
- 1998-04-17 BR BR9809398-3A patent/BR9809398A/en not_active Application Discontinuation
- 1998-04-17 DE DE69838025T patent/DE69838025T2/en not_active Expired - Lifetime
- 1998-04-17 US US09/403,705 patent/US6177432B1/en not_active Expired - Lifetime
- 1998-04-17 EP EP98924161A patent/EP0977750B1/en not_active Expired - Lifetime
- 1998-04-17 TR TR1999/02606T patent/TR199902606T2/en unknown
- 1998-04-17 JP JP54656198A patent/JP4308919B2/en not_active Expired - Lifetime
- 1998-04-17 PL PL336468A patent/PL190944B1/en unknown
- 1998-04-17 AT AT98924161T patent/ATE366250T1/en not_active IP Right Cessation
- 1998-04-17 WO PCT/EP1998/002357 patent/WO1998049157A1/en not_active Ceased
- 1998-04-17 NZ NZ336233A patent/NZ336233A/en unknown
- 1998-04-17 SK SK1461-99A patent/SK146199A3/en unknown
- 1998-04-17 CN CN98804366A patent/CN1094937C/en not_active Expired - Lifetime
- 1998-04-24 ZA ZA9803504A patent/ZA983504B/en unknown
-
1999
- 1999-10-22 NO NO19995169A patent/NO317576B1/en unknown
-
2000
- 2000-10-13 US US09/687,153 patent/US6358961B1/en not_active Expired - Lifetime
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0371564A2 (en) * | 1988-11-29 | 1990-06-06 | Janssen Pharmaceutica N.V. | (1H-azol-1-ylmethyl)substituted quinoline, quinazoline or quinoxaline derivatives |
| EP0664128A1 (en) * | 1992-10-07 | 1995-07-26 | Sumitomo Pharmaceuticals Company, Limited | Pharmaceutical composition for inhibiting tumor necrosis factor production |
| WO1996015118A1 (en) * | 1994-11-12 | 1996-05-23 | Zeneca Limited | Aniline derivatives |
Cited By (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6451812B1 (en) | 1998-07-06 | 2002-09-17 | Janssen Pharmaceutica N.V. | Farnesyl protein transferase inhibitors for treating arthropathies |
| US6284755B1 (en) | 1998-12-08 | 2001-09-04 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| US6316436B1 (en) | 1998-12-08 | 2001-11-13 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| CZ302374B6 (en) * | 1998-12-23 | 2011-04-20 | Janssen Pharmaceutica N.V. | 1,2-Anellated quinoline derivative, process and intermediate product for preparation thereof and pharmaceutical composition containing thereof |
| US6914066B2 (en) | 1998-12-23 | 2005-07-05 | Janssen Pharmaceutica N.V. | 1,2-annelated quinoline derivatives |
| BG65124B1 (en) * | 1998-12-23 | 2007-03-30 | Janssen Pharmaceutica N.V. | 1,2-annelated quinoline derivatives |
| KR100712226B1 (en) * | 1998-12-23 | 2007-04-27 | 얀센 파마슈티카 엔.브이. | 1,2-annealed quinoline derivatives |
| WO2000039082A3 (en) * | 1998-12-23 | 2000-10-26 | Janssen Pharmaceutica Nv | 1,2-annelated quinoline derivatives |
| US6458800B1 (en) | 1998-12-23 | 2002-10-01 | Janssen Pharmaceutica N.V. | 1,2-annelated quinoline derivatives |
| KR20010077400A (en) * | 2000-02-02 | 2001-08-17 | 성재갑 | Anticancer agents by combination of Ftase inhibitor(LB42908) and other anticancer drugs |
| JP2003521509A (en) * | 2000-02-04 | 2003-07-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Farnesyl protein transferase inhibitors for the treatment of breast cancer |
| US6838467B2 (en) | 2000-02-24 | 2005-01-04 | Janssen Pharmaceutica N. V. | Dosing regimen |
| WO2001064198A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with anti-tumor podophyllotoxin derivatives |
| WO2001064196A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with vinca alkaloids |
| WO2001064218A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations |
| WO2001064199A3 (en) * | 2000-02-29 | 2001-12-27 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with taxane compounds |
| WO2001064217A3 (en) * | 2000-02-29 | 2002-03-28 | Janssen Pharmaceutica Nv | Combinations of a farnesyl protein transferase inhibitor with nitrogen mustard or nitrosourea alkylating agents |
| WO2001064246A3 (en) * | 2000-02-29 | 2002-02-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with an her2 antibody |
| WO2001064226A3 (en) * | 2000-02-29 | 2002-03-07 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with platinum compounds |
| WO2001064197A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with anti-tumor anthracycline derivatives |
| WO2001064194A3 (en) * | 2000-02-29 | 2002-03-07 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with camptothecin compounds |
| WO2001064252A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with further anti-cancer agents |
| WO2001064195A3 (en) * | 2000-02-29 | 2002-03-21 | Janssen Pharmaceutica Nv | Farnesyl protein transferase inhibitor combinations with anti-tumor nucleoside derivatives |
| US8329714B2 (en) | 2000-06-22 | 2012-12-11 | Janssen Pharmaceutica Nv | Farnesyl transferase inhibiting 1,2-annelated quinoline enantiomer |
| HRP20020989B1 (en) * | 2000-06-22 | 2011-05-31 | Janssen Pharmaceutica N.V. | ENANTIOMER 1,2-ANELIRANOG KINOLINA KOJI INHIBIRA FARNEZIL TRANSFERAZU |
| WO2001098302A1 (en) * | 2000-06-22 | 2001-12-27 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 1,2-annelated quinoline enantiomer |
| BG65894B1 (en) * | 2000-06-22 | 2010-04-30 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 1,2-annelated quinoline enantiomer |
| US8318753B2 (en) | 2000-06-22 | 2012-11-27 | Janssen Pharmaceutica Nv | Farnesyl transferase inhibiting 1,2-annelated quinoline enantiomer |
| JP2004509887A (en) * | 2000-09-25 | 2004-04-02 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | 6-Heterocyclylmethylquinoline and quinazoline derivatives inhibiting farnesyltransferase |
| WO2002024682A1 (en) * | 2000-09-25 | 2002-03-28 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting quinoline and quinazoline derivatives as farnesyl transferase inhibitors |
| WO2002024687A1 (en) * | 2000-09-25 | 2002-03-28 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinolinone derivatives |
| JP2004509884A (en) * | 2000-09-25 | 2004-04-02 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | 6-[(Substituted phenyl) methyl] -quinoline and quinazoline derivatives that inhibit farnesyltransferase |
| US7196094B2 (en) | 2000-09-25 | 2007-03-27 | Janssen Pharmaceutica, N.V. | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinoline and quinazoline derivatives |
| JP2004521863A (en) * | 2000-09-25 | 2004-07-22 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | 6-Heterocyclylmethylquinolinone derivatives inhibiting farnesyltransferase |
| WO2002024686A3 (en) * | 2000-09-25 | 2002-06-13 | Janssen Pharmaceutica Nv | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinoline and quinazoline derivatives |
| US7173040B2 (en) | 2000-09-25 | 2007-02-06 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 6-[(substituted phenyl)methyl]-quinoline and quinazoline derinazoline derivatives |
| WO2002024683A1 (en) * | 2000-09-25 | 2002-03-28 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 6-[(substituted phenyl)methyl]-quinoline and quinazoline derivatives |
| US7053105B2 (en) | 2000-09-25 | 2006-05-30 | Janssen Pharmaceutica, N.V. | Farnesyl transferase inhibiting quinoline and quinazoline derivatives as farnesyl transferase inhibitors |
| US7067531B2 (en) | 2000-09-25 | 2006-06-27 | Angibaud Patrick Rene | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinolinone derivatives |
| US7153958B2 (en) | 2000-11-21 | 2006-12-26 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting benzoheterocyclic derivatives |
| WO2002042296A1 (en) * | 2000-11-21 | 2002-05-30 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting benzoheterocyclic derivatives |
| WO2002043733A1 (en) * | 2000-11-28 | 2002-06-06 | Janssen Pharmaceutica N.V. | Farnesyl protein transferase inhibitors for the treatment of inflammatory bowel disease |
| US7129356B2 (en) | 2000-12-27 | 2006-10-31 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 4-substituted quinoline and quinazoline derivatives |
| WO2002051835A1 (en) * | 2000-12-27 | 2002-07-04 | Janssen Pharmaceutica N.V. | Farnesyl transferase inhibiting 4-substituted quinoline and quinazoline derivatives |
| JP2004517960A (en) * | 2001-02-15 | 2004-06-17 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Combination of antiestrogens and farnesyl protein transferase inhibitors |
| WO2002064142A1 (en) * | 2001-02-15 | 2002-08-22 | Janssen Pharmaceutica N.V. | Farnesyl protein transferase inhibitor combinations with antiestrogen agents |
| US6844439B2 (en) | 2001-03-12 | 2005-01-18 | Jansen Pharmaceutica, Nv | Process for the preparation of imidazole compounds |
| CZ300622B6 (en) * | 2001-03-12 | 2009-07-01 | Janssen Pharmaceutica N. V. | Process for preparing 4-(3-chlorophenyl)-6-[(4-chlorophenyl)hydroxy(1-methyl-1 H -imidazol-5-yl)methyl]-1-methyl-2(1 H )-quinolinone |
| WO2003051880A1 (en) * | 2001-12-19 | 2003-06-26 | Janssen Pharmaceutica N.V. | 1,8-annelated quinoline derivatives substituted with carbon-linked triazoles as farnesyl transferase inhibitors |
| US7408063B2 (en) | 2001-12-19 | 2008-08-05 | Janssen Pharmaceutica, N.V. | 1,8-annelated quinoline derivatives substituted with carbon-linked triazoles as farnesyl transferase inhibitors |
| FR2837201A1 (en) * | 2002-03-18 | 2003-09-19 | Servier Lab | NOVEL COMPOUNDS DERIVED FROM QUINAZOLINE, THEIR PREPARATION PROCESS AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| EP1346992A1 (en) * | 2002-03-18 | 2003-09-24 | Les Laboratoires Servier | Quinazoline derivatives, process for their preparation and pharmaceutical compositions containing them |
| US7943635B2 (en) | 2002-03-22 | 2011-05-17 | Janssen Pharmaceutica Nv | Benzylimidazolyl substituted 2-quinoline and quinazoline derivatives for use as farnesyl transferase inhibitors |
| US7241777B2 (en) | 2002-03-22 | 2007-07-10 | Janssen Pharmaceutica N.V. | Benzylimidazolyl substituted 2-quinoline and quinazoline derivatives for use as farnesyl transferase inhibitors |
| US7655654B2 (en) | 2002-04-15 | 2010-02-02 | Janssen Pharmaceutica Nv | Farnesyl transferase inhibiting tricyclic quinazoline derivatives substituted with carbon-linked imidazoles or triazoles |
| US7511138B2 (en) | 2002-04-15 | 2009-03-31 | Janssen Pharmaceutica Nv | Farnesyl transferase inhibiting tricyclic quinazoline derivatives substituted with carbon-linked imidazoles or triazoles |
| EP2362218A2 (en) | 2004-11-05 | 2011-08-31 | Janssen Pharmaceutica N.V. | Methods of monitoring the efficacy of farnesyltransferase inhibitors |
| US8557847B2 (en) | 2005-06-10 | 2013-10-15 | Janssen Pharmaceutica, N.V. | Synergistic modulation of FLT3 kinase using a FLT3 inhibitor and a farnesyl transferase inhibitor |
| US8697716B2 (en) | 2006-04-20 | 2014-04-15 | Janssen Pharmaceutica Nv | Method of inhibiting C-KIT kinase |
| US9394289B2 (en) | 2006-04-20 | 2016-07-19 | Janssen Pharmaceutica Nv | Inhibitors of c-fms kinase |
| US8481564B2 (en) | 2006-04-20 | 2013-07-09 | Janssen Pharmaceutica, N.V. | Inhibitors of c-fms kinase |
| US8759347B2 (en) | 2006-04-20 | 2014-06-24 | Janssen Pharmaceutica Nv | Inhibitors of C-FMS kinase |
| US8859602B2 (en) | 2006-04-20 | 2014-10-14 | Janssen Pharmaceutica Nv | Inhibitors of c-fms kinase |
| US8895584B2 (en) | 2006-04-20 | 2014-11-25 | Janssen Pharmaceutica Nv | Inhibitors of c-fms kinase |
| US8933091B2 (en) | 2006-04-20 | 2015-01-13 | Janssen Pharmaceutica Nv | Method of inhibiting C-KIT kinase |
| US9526731B2 (en) | 2006-04-20 | 2016-12-27 | Janssen Pharmaceutica Nv | Method of inhibiting C-KIT kinase |
| US9266866B2 (en) | 2006-04-20 | 2016-02-23 | Janssen Pharmaceutica Nv | Inhibitors of C-FMS kinase |
| US9296726B2 (en) | 2006-04-20 | 2016-03-29 | Janssen Pharmaceutica Nv | Inhibitors of c-fms kinase |
| US9403804B2 (en) | 2006-04-20 | 2016-08-02 | Janssen Pharmaceutica Nv | Inhibitors of c-fms kinase |
| US8497376B2 (en) | 2007-10-17 | 2013-07-30 | Janssen Pharmaceutica N.V. | Inhibitors of c-fms kinase |
| US10112903B2 (en) | 2009-06-25 | 2018-10-30 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| EP2445502B1 (en) * | 2009-06-25 | 2017-06-21 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| EP3309151A1 (en) * | 2009-06-25 | 2018-04-18 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| US10023537B2 (en) | 2009-06-25 | 2018-07-17 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| US10351529B2 (en) | 2009-06-25 | 2019-07-16 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| US10822306B2 (en) | 2009-06-25 | 2020-11-03 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| US11518745B2 (en) | 2009-06-25 | 2022-12-06 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| US12180164B2 (en) | 2009-06-25 | 2024-12-31 | Alkermes Pharma Ireland Limited | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| US9029352B2 (en) | 2012-08-07 | 2015-05-12 | Janssen Pharmaceutica Nv | Process for the preparation of C-FMS kinase inhibitors |
| US9303046B2 (en) | 2012-08-07 | 2016-04-05 | Janssen Pharmaceutica Nv | Process for the preparation of heterocyclic ester derivatives |
| US11273158B2 (en) | 2018-03-05 | 2022-03-15 | Alkermes Pharma Ireland Limited | Aripiprazole dosing strategy |
| US12251381B2 (en) | 2018-03-05 | 2025-03-18 | Alkermes Pharma Ireland Limited | Aripiprazole dosing strategy |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6177432B1 (en) | Farnesyltransferase inhibiting quinazolinones | |
| US6187786B1 (en) | Farnesyl transferase inhibiting 1,8-annelated quinolinone derivatives substituted with N- or C-linked imidazoles | |
| EP1106610B1 (en) | Farnesyl transferase inhibiting 2-quinolone derivatives | |
| US6458800B1 (en) | 1,2-annelated quinoline derivatives | |
| AP1108A (en) | Farnesyl protein transferase inhibiting (imidazol-5-Y1) Methyl-2-Quinoline derivatives. | |
| EP1322650B1 (en) | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinoline and quinazoline derivatives | |
| US7067531B2 (en) | Farnesyl transferase inhibiting 6-heterocyclylmethyl quinolinone derivatives | |
| US20080255191A1 (en) | Benzylimidazolyl substituted 2-quinoline and quinazoline derivatives for use as farnesyl transferase inhibitors | |
| EP1347966A1 (en) | Farnesyl transferase inhibiting 4-substituted quinoline and quinazoline derivatives | |
| MXPA99009763A (en) | Farnesyltransferase inhibiting quinazolinones | |
| HK1036064B (en) | Farnesyl transferase inhibiting 2-quinolone derivatives | |
| HK1027576B (en) | Farnesyl transferase inhibiting 2-quinolone derivatives | |
| HK1024689B (en) | Farnesyl transferase inhibiting 1,8-annelated quinolinone derivatives substituted with n- or c-linked imidazoles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 98804366.1 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM GW HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 336233 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019997005507 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998924161 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 1998 546561 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2288140 Country of ref document: CA Ref document number: 2288140 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999/02606 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1999-3717 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09403705 Country of ref document: US Ref document number: PA/a/1999/009763 Country of ref document: MX Ref document number: 146199 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 76460/98 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998924161 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1999-3717 Country of ref document: CZ |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019997005507 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 76460/98 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1019997005507 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV1999-3717 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998924161 Country of ref document: EP |