WO1998051677A1 - Process for synthesizing carbapenem intermediates - Google Patents
Process for synthesizing carbapenem intermediates Download PDFInfo
- Publication number
- WO1998051677A1 WO1998051677A1 PCT/US1998/009036 US9809036W WO9851677A1 WO 1998051677 A1 WO1998051677 A1 WO 1998051677A1 US 9809036 W US9809036 W US 9809036W WO 9851677 A1 WO9851677 A1 WO 9851677A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- accordance
- produce
- reacting
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c(c1ccccc11)ccc1S(Cl)(=O)=O Chemical compound *c(c1ccccc11)ccc1S(Cl)(=O)=O 0.000 description 2
- VXOCZRRDXTUHLE-UHFFFAOYSA-N Cc(c1c2c(N3)ccc1)ccc2S3(=O)=O Chemical compound Cc(c1c2c(N3)ccc1)ccc2S3(=O)=O VXOCZRRDXTUHLE-UHFFFAOYSA-N 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D275/00—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings
- C07D275/04—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings condensed with carbocyclic rings or ring systems
- C07D275/06—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings condensed with carbocyclic rings or ring systems with hetero atoms directly attached to the ring sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- Carbapenems are antibiotics having a broad antibacterial spectrum, which includes gram positive, gram negative and anaerobic microorganisms.
- the carbapenems in which the naphthosultams of the present invention are useful contain a naphthosultam ring system in the side chain which is attached to the carbapenem nucleus at position two. Examples of carbapenems which are substituted with a naphthosultam-containing side chain at position two are found in U. S. Application No. 60/016,184 filed on April 24, 1996, the teachings of which are hereby incorporated by reference.
- the naphthosultam platform is attached to the carbapenem nucleus directly or through a linking moiety, such as a sulfur atom, a methylene group or a higher alkylene group. Also attached to the naphthosultam platform may be one or more substituent groups.
- the naphthosultam produced is an active pharmacophore when appropriately coupled to a carbapenem.
- R represents halo or Ci-6 alkyl , unsubstituted or substituted with OP, wherein P represents a protecting group, comprising:
- Alkyl refers to Ci-6 alkyl groups which may be straight or branched.
- Halogen and halo refer to chlorine, bromine and iodine, selected on an independent basis. Preferred values of halo include bromo and iodo. Most preferably, halo is bromo.
- Acid as used herein refers to strong acids such as hydrochloric, formic, sulfuric, toluene sulfonic acid and the like.
- the process described herein involves the cyclization of an appropriately substituted naphthalene sulfonamide to provide appropriately substituted 1,8-naphthosultams.
- the invention involves synthesizing a compound of formula I:
- R represents halo or Ci-6 alkyl , unsubstituted or substituted with OP, wherein P represents a protecting group, comprising:
- R represents methyl.
- R represents halo, preferably Br or I, and most preferably Br.
- one of R a and Rb represent H or Ci-6 alkyl, and the other is Ci-6 alkyl. More particularly, both represent Ci-6 alkyl, and most preferably, both represent ethyl.
- R represents halo or Ci-6 alkyl , unsubstituted or substituted with OP, wherein P represents a protecting group.
- R represents halo or Ci-6 alkyl , unsubstituted or substituted with OP, wherein P represents a protecting group; and R a and R b represent H or Ci-6 alkyl.
- an intermediate compound is included which is represented by formula XI:
- the reaction between the 1-halonaphthalene or 1-alkylnaphthalene and the chlorosulfonic acid can be conducted in an organic solvent, such as nitro substituted organics, e.g., nitromethane and nitrobenzene, and halo substituted organic solvents, such as dichlorobenzene, including ortho, meta or para substituted benzene, dichloromethane, chloroform, carbon tetrachloride,, dichloroethane and trifluoroacetic acid (TFA).
- TFA trifluoroacetic acid
- the reaction is typically conducted at a temperature of from about -40°C to as high as about 100°C, preferably about 0°C to about 25°C.
- the formation of the sulfonamide is typically conducted in a non-reactive organic solvent. Examples include secondary and tertiary alcohols, such as isopropanol, tertiary butanol, aromatics, such as toluene, and xylene, hexanes, ethers, esters, tetrahydrofuran, and many chlorinated solvents, as described above.
- the reaction is typically conducted between about -50°C and reflux temperature.
- the reaction with nitric acid is typically conducted in trifluoroacetic acid, sulfuric acid, nitrobenzene or a chlorinated organic solvent, as described above.
- the temperature range is typically from about -30°C to about 60°C, and more preferably from about 0°C to about 20°C.
- Reduction of the nitro group to form an amine and cyclization can be conducted in essentially any solvent, at a temperature ranging from about -40°C to reflux temperature.
- Reducing agents which are useful in connection with the present invention include, for example, H2/P as well as any other catalyst, Pd/C with chemical transfer reagents, such as formic acid, ammonium formate, any metal formate, any trialkylammonium formate or cyclohexene, tin chloride and the like.
- Acids which are useful for the cyclization include for example, hydrochloric, sulfuric, trifluoroacetic and methanesulfonic acids.
- hydrochloric acid is used.
- Carboxylation (chain extension) can typically be conducted in solvents such as THF and ether, e.g., diethylether, at a temperature generally in the range of about -100°C to about 70°C.
- the base that is included in the reaction is typically lithium dialkylamide, e.g., lithium diisopropylamide.
- Reduction of the carboxylic acid to form the alcohol is typically conducted in a solvent such as THF or diethylether.
- Suitable reducing agents include borane in THF and similar agents. This reaction is typically conducted at a temperature ranging from about -
- the carbapenems which are synthesized in accordance with the present invention are prepared by reacting a suitably protected, activated 2-hydroxymethyl-carbapen-2-em-3- carboxylate with a naphthosultam, modifying the thus-introduced side chain as desired, and then removing any protecting groups which are present to afford the desired final product.
- the process is illustrated by the following generic scheme:
- the naphthosultam side chain group can initially be reacted with a suitably protected carbapen-2-em-3-carboxylate having an activated hydroxymethyl group at the 2-position.
- the carbapenem nucleus having a -CH2OH substituent at position 2 can be obtained in accordance with Schmitt, S. M. et al., J. Antibiotics 41(6): 780-787 (1988), the teachings of which are incorporated herein by reference.
- the carboxylic acid group at C-3 of the carbapenem is generally protected as a carboxyl protecting group such as p-nitrobenzyl (PNB), allyl, p-methoxybenzyl, trichloroethyl, 2- trimethylsilylethyl, and the like.
- PNB p-nitrobenzyl
- allyl allyl
- p-methoxybenzyl trichloroethyl
- 2- trimethylsilylethyl 2- trimethylsilylethyl
- the hydroxyl group of the 6-(hydroxyethyl) side-chain is optionally protected with a hydroxyl protecting group such as trimethylsilyl (TMS), triethylsilyl (TES), tert- butyldimethylsilyl (TBDMS), tert-butyldiphenylsilyl (TBDPS), acetyl, allyloxycarbonyl, 2-trimethylsilylethoxy carbonyl, 2- trichloroethoxycarbonyl and the like.
- TMS trimethylsilyl
- TES triethylsilyl
- TDMS tert- butyldimethylsilyl
- TDPS tert-butyldiphenylsilyl
- acetyl acetyl, allyloxycarbonyl, 2-trimethylsilylethoxy carbonyl, 2- trichloroethoxycarbonyl and the like.
- the addition of the naphthosultam side chain group (SCG) to the carbapenem is accomplished by treating a solution of the hydroxymethyl-carbapenem and the naphthosultam side chain group in a suitable solvent such as tetrahydrofuran (THF), ether, acetonitrile, dimethylformamide (DMF), benzene, dimethylsulfoxide (DMSO), and the like with a (premixed) suitable activating reagent such as diethyl azodicarboxylate (DEAD) / triphenylphosphine, diisopropyl azodicarboxylate (DIAD) / tributylphosphine, and the like, at a temperature between about -20 °C and 35 °C for about 5 to 90 minutes.
- a suitable solvent such as tetrahydrofuran (THF), ether, acetonitrile, dimethylformamide (DMF), benzene, dimethylsul
- the naphthosultam and carbapenem can be mixed together with either the azodicarboxylate or the phosphine reagent in a suitable and the other component of the activating reagent (the phosphine or the azodicarboxylate, respectively) can be added to that mixture.
- the reaction is allowed to proceed at a temperature between about -20 °C and 35 °C for about 5 to 90 minutes.
- a positively charged substituent may be introduced into the side chain by first activating the hydroxyl group by converting it to a suitable leaving group such as a triflate, mesylate, tosylate, iodide, chloride, bromide, and the like, and then displacing the resulting leaving group with a quaternizing compound, such as N-methyl- imidazole, N-(2-hydroxyethyl)-imidazole, N-methyl-diazabicyclooctane, l-(carbamoylmethyl)-4-aza-l-azoniabicyclo-[2.2.2.]-octane, l-(3- hydroxyprop-l-yl)-4-aza-l-azoniabicyclo-[2.2.2.]-octane, pyridine, morpholine and the like which contains a nitrogen atom that can act as a nucleophile.
- a suitable leaving group such as a triflate, mesy
- the charged substituent may be incorporated in the naphthosultam side chain before addition of the naphthosultam to the carbapenem or may be introduced after deprotection. Introduction of the charged substituent before deprotection is greatly preferred.
- the conversion of the hydroxyl group to a suitable leaving group is accomplished by treating the hydroxyl substituted compound in a suitable solvent such as dichloromethane, tetrahydro- furan, ether, benzene, and the like with an activating reagent, such as trifluoromethanesulfonic anhydride, methanesulfonic anhydride, toluenesulfonic anhydride, methanesulfonyl chloride, benzenesulfonyl chloride, toluenesulfonyl chloride, and the like in the presence of a suitable base such as triethylamine, tributylamine, diisopropylethyl- amine, and the like at a temperature between about -100°C and 0°C for about 5 to 120 minutes.
- a suitable solvent such as dichloromethane, tetrahydro- furan, ether, benzene, and the like
- the intermediate thus obtained contains a leaving group, which may be converted to an alternative leaving group, iodide, by treating a solution of the intermediate in a suitable solvent such as acetone, methyl ethyl ketone, and the like at about -10°C to 50°C with an excess of sodium iodide or potassium iodide for about 0.25 to 24 hours.
- a suitable solvent such as acetone, methyl ethyl ketone, and the like
- sodium iodide or potassium iodide for about 0.25 to 24 hours.
- the iodide is obtained in sufficiently pure form that it may be used without further purification.
- the iodide if not crystalline, may be lyophilized from benzene to afford an amorphous, easily handled, solid.
- the activated hydroxyl group or iodide is displaced by reacting the activated intermediate with activating reagent.
- activation and displacement may be accomplished in a single step.
- the activating reagent is added to a solution of the hydroxyl substituted compound in the presence of a suitable base in a suitable solvent such as dichloromethane, tetrahydrofuran, ether, DMF, benzene, acetonitrile, DMSO, and the like.
- a suitable solvent such as dichloromethane, tetrahydrofuran, ether, DMF, benzene, acetonitrile, DMSO, and the like.
- the resulting activated intermediate is treated with 1-3 molar equivalents of the naphthosultam at a temperature between about -78°C and 50°C for about 15 to 120 minutes.
- the displacement may be conducted without isolation of the intermediate and, in cases where Q* is also used as a base, may even be concurrent with the formation of the activated intermediate.
- a solution of the iodide is combined with an approximately equivalent amount (0.9 - 1.05 molar equivalents) of the naphthosultam.
- a salt of a non-nucleophilic acid such as silver trifluoromethanesulfonate, silver tetrafluoroborate and the like can be added.
- the salt assists in the removal of the displaced iodide from the reaction mixture which can improve the efficiency of subsequent steps.
- the resulting mixture is then subjected to a standard work-up procedure familiar to those skilled in the art to afford a crude product which is purified, if necessary, by recrystallization or chromatography.
- An alternative method for introducing a positive charge into the side chain may be applied to side chains that contain a nitrogen atom which may be quaternized by reaction with a suitable alkylating reagent, such as methyl iodide, methyl bromide, benzyl trichloroacetimidate, methyl trif uoromethanesulfonate, triethyloxonium tetrafluoroborate, and the like. Quaternization of the nitrogen atom in the side chain is effected by treating a solution of the compound with a slight excess (1.05 to 1.2 molar equivalents) of the alkylating reagent.
- the synthesis of the target compound is typically completed by removing any protecting groups which are present in the penultimate intermediate using standard techniques which are well known to those skilled in the art.
- the deprotected final product is then purified, as necessary, using standard techniques such as ion exchange chroma- tography, HPLC on reverse phase silica gel, MPLC on reverse phase polystyrene gel, and the like or by recrystallization.
- the final product may be characterized structurally by standard techniques such as NMR, IR, MS, and UV.
- the final product if not crystalline, may be lyophilized from water to afford an amorphous, easily handled solid.
- R examples include halo, methyl, ethyl and propyl, unsubstituted or substituted with OP, wherein P represents a hydroxyl protecting group.
- protecting groups P include trimethylsilyl (TMS), triethylsilyl (TES), tert-butyldimethylsilyl (TBDMS), tert-butyldiphenylsilyl (TBDPS), acetyl, allyloxycarbonyl, 2-trimethylsilylethoxy carbonyl, 2-trichloroethoxycarbonyl and the like.
- Preferred values of R a and R b include H, methyl, ethyl and propyl. Most preferably each represents ethyl.
- NHR a Rb represents dimethylamine, diethylamine or dipropylamine.
- reducing agent is used to refer to compounds which when reacted with compound V, convert the nitro group at position 8 to an amino group.
- suitable reducing agents include H2 in combination with a metal, e.g., Pd/C, formic acid in combination with a metal, trialkylammonium formates with a metal, cyclohexene and a metal, tin chloride and the like.
- the product crystallized upon addition to water to give a white slurry.
- reaction vessel was washed with TF A/water (1: 1, 1.0L) and the wash added to the quench vessel.
- the cake was filtered on a polypropylene filter cloth and washed with water. The cake was dried in nitrogen stream overnight.
- the cake was dried in a nitrogen stream until the residual water content was ⁇ 35%w/w.
- the partially dried solid was slurried in ethyl acetate (75 mL) at 30°C for 30min. Hexane (150mL) was added over 20min and the slurry aged at 20°C for 30 min. The slurry was filtered, washed with hexane (lOOmL) and dried in a nitrogen stream overnight.
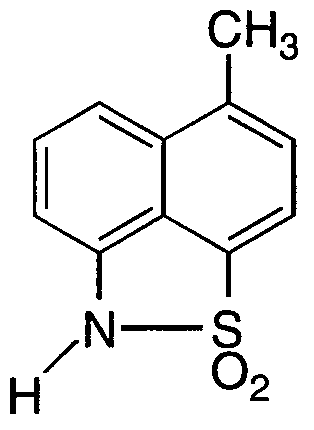
- a 72 L round bottom flask was equipped with a N2 inlet, thermocouple, and an overhead stirrer. The flask was charged with 2.20 Kg of l-methyl-4-diethylsulfonamide-5-nitronaphthalene, along with ethanol (20 L).
- Pd C 10 t% 50 wt% water wet, 0.17 Kg was charged as a slurry in water (800 ml) and rinsed down with water (80 ml) and ethanol (6 L). To the resultant slurry was added potassium formate (1.74 Kg) in one portion. The slurry was warmed to 60°C for 1 h then to reflux for 1 h. The reaction was considered complete when ⁇ 0.2% SM remained relative to the starting material as determined by HPLC.
- the resultant slurry was filtered through a pad of solka- flocTM and the cake washed with 10% hydrochloric acid in ethanol (total of 13 L).
- the combined filtrates were recharged to the cleaned 72L flask and heated to reflux (81°C) for 3-4h to achieve complete cyclization.
- the solution was cooled to 40°C and concentrated to a slurry.
- the slurry was cooled to 20°C and water (7.5L) added over 30 min.
- the slurry was cooled to 5°C and aged for 15 min.
- the slurry was filtered and washed with water (5 L).
- the crystalline solid was dried under a stream of nitrogen overnight.
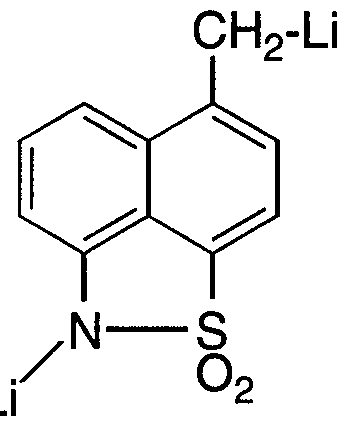
- a 20 L flask equipped with nitrogen inlet, stirrer, temperature probe and addition funnel was charged with THF (5L). Diisopropylamine(1.34L) was added. The solution was cooled to -15°C and n-butyllithium (6.0L) added dropwise maintaining the reaction temperature between -15°C and 0°C to produce lithium diisopropylamide (LDA).
- a separate 50 L flask equipped with stirrer and nitrogen inlet was charged with THF ( 10 L) and 1-Me NH naphthosultam (1.0 Kg). The solution was degassed by vacuum/purging with nitrogen and cooled to -15°C and the LDA solution added dropwise keeping the solution temperature between -15°C and -5°C to produce the dianion solution.
- the dianion solution was aged for 30 min at -15°C.
- a 50L round bottom flask was set up and equipped with a
- the slurry was aged at 15-20°C and the reaction progress monitored by HPLC.
- the slurry was cooled to 10°C and methanol (1.2L) slowly added to quench excess borane. 2N HC1 (11L ) was added slowly and aged for 30 min.
- the quenched reaction mixture was distilled at atmospheric pressure to remove volatiles.
- the resultant solution was cooled to 55°C, seeded to initiate crystallization and cooled to 15°C over lh. After aging for 30 min, the slurry was filtered, washed with 10L water and dried under a stream of nitrogen.
- Diisopropylamine(0.14 mL) was added.
- the solution was cooled to -15°C and n-butyllithium (0.66 mL) added dropwise maintaining the reaction temperature between -15°C and 0°C to produce lithium diisopropylamide (LDA).
- LDA lithium diisopropylamide
- the dianion solution was aged for 30 min at -15°C.
- a separate 20 mL flask equipped with stirrer, nitrogen inlet and temperature probe was charged with THF (2 mL) and para formaldehyde (0.12 gm) and degassed with a vacuum /nitrogen purge.
- the solution was cooled to -20°C.
- the dianion solution was then slowly added to this parafomaldehyde solution while ensuring efficient mixing.
- 10 mL of 2N HC1 was added the solution and was aged for 30 min.
- the solution was concentrated atmospherically to about 10 mL to remove THF and initiate crystallization.
- the resultant slurry was aged at 15°C for 30 min, filtered and the cake washed with water(10 mL) and dried in a nitrogen stream.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description































Claims

















Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002287931A CA2287931A1 (en) | 1997-05-13 | 1998-05-08 | Process for synthesizing carbapenem intermediates |
| AU73678/98A AU728091B2 (en) | 1997-05-13 | 1998-05-08 | Process for synthesizing carbapenem intermediates |
| JP54927898A JP2001527557A (en) | 1997-05-13 | 1998-05-08 | Synthetic method of carbapenem intermediate |
| EP98920961A EP0981522A1 (en) | 1997-05-13 | 1998-05-08 | Process for synthesizing carbapenem intermediates |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US4635797P | 1997-05-13 | 1997-05-13 | |
| US60/046,357 | 1997-05-13 | ||
| GBGB9806023.9A GB9806023D0 (en) | 1998-03-20 | 1998-03-20 | Process for synthesizing carbapenem intermediates |
| GB9806023.9 | 1998-03-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998051677A1 true WO1998051677A1 (en) | 1998-11-19 |
Family
ID=26313321
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/009036 Ceased WO1998051677A1 (en) | 1997-05-13 | 1998-05-08 | Process for synthesizing carbapenem intermediates |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP0981522A1 (en) |
| JP (1) | JP2001527557A (en) |
| AR (1) | AR011735A1 (en) |
| AU (1) | AU728091B2 (en) |
| CA (1) | CA2287931A1 (en) |
| TW (1) | TW408116B (en) |
| WO (1) | WO1998051677A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001000624A1 (en) * | 1999-06-25 | 2001-01-04 | Merck & Co., Inc. | Synthetic process for naphthosultam carbapenems |
| WO2003029230A1 (en) * | 2001-10-02 | 2003-04-10 | Pharmacia Corporation | Method for preparing benzenesulfonyl compounds |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2773765A (en) * | 1952-08-16 | 1956-12-11 | Azoplate Corp | Light sensitive material for photomechanical reproduction |
| WO1997040048A1 (en) * | 1996-04-24 | 1997-10-30 | Merck & Co., Inc. | Carbapenem antibacterial compounds, compositions containing such compounds and methods of treatment |
-
1998
- 1998-05-08 JP JP54927898A patent/JP2001527557A/en active Pending
- 1998-05-08 EP EP98920961A patent/EP0981522A1/en not_active Withdrawn
- 1998-05-08 AU AU73678/98A patent/AU728091B2/en not_active Ceased
- 1998-05-08 WO PCT/US1998/009036 patent/WO1998051677A1/en not_active Ceased
- 1998-05-08 CA CA002287931A patent/CA2287931A1/en not_active Abandoned
- 1998-05-11 TW TW087107245A patent/TW408116B/en active
- 1998-05-12 AR ARP980102207A patent/AR011735A1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2773765A (en) * | 1952-08-16 | 1956-12-11 | Azoplate Corp | Light sensitive material for photomechanical reproduction |
| WO1997040048A1 (en) * | 1996-04-24 | 1997-10-30 | Merck & Co., Inc. | Carbapenem antibacterial compounds, compositions containing such compounds and methods of treatment |
Non-Patent Citations (3)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 29, no. 4, 20 February 1935, Columbus, Ohio, US; abstract no. 1085, XP002073541 * |
| PAUL CAGNIANT ET AL: "Contribution à l'étude des hétérocycles soufrés condensés.", BULLETIN DE LA SOCIETE CHIMIQUE DE FRANCE., 1966, PARIS FR, pages 2037 - 2042, XP002073540 * |
| ROBERT E. STEINER: "Sensitivity to light of aromatic nitro compounds. V peri-dervatives of 1-nitronaphthalene", HELVETICA CHIMICA ACTA., vol. 17, 1934, BASEL CH, pages 1142 - 1157 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001000624A1 (en) * | 1999-06-25 | 2001-01-04 | Merck & Co., Inc. | Synthetic process for naphthosultam carbapenems |
| WO2003029230A1 (en) * | 2001-10-02 | 2003-04-10 | Pharmacia Corporation | Method for preparing benzenesulfonyl compounds |
| RU2284324C2 (en) * | 2001-10-02 | 2006-09-27 | Фармация Корпорейшн | Method for preparing benzenesulfonyls |
| CN1308315C (en) * | 2001-10-02 | 2007-04-04 | 法玛西雅公司 | Method for preparing benzenesulfonyl compounds |
| AU2008205429B2 (en) * | 2001-10-02 | 2008-11-06 | Pharmacia Corporation | Method for preparing benzenesulfonyl compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| TW408116B (en) | 2000-10-11 |
| EP0981522A1 (en) | 2000-03-01 |
| JP2001527557A (en) | 2001-12-25 |
| AR011735A1 (en) | 2000-08-30 |
| AU7367898A (en) | 1998-12-08 |
| CA2287931A1 (en) | 1998-11-19 |
| AU728091B2 (en) | 2001-01-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101522740B1 (en) | Process for the preparation of tauroursodeoxycholic acid | |
| CN111187269A (en) | Synthetic method of Reidesciclovir intermediate | |
| US20220024850A1 (en) | Method for preparing 2-ethyl-4-fluoro-1-nitrobenzene | |
| US20250002506A1 (en) | Improved process for the preparation of lurbinectedin and its morphs thereof | |
| CN109942593B (en) | Total synthesis method of racemic tetrandrine | |
| KR20240056726A (en) | Synthesis of nirogacestat | |
| CN104918912B (en) | Processes for the synthesis of 2-amino-4,6-dimethoxybenzamide and other benzamide compounds | |
| CN113444083B (en) | A method for preparing a small molecule tyrosine kinase inhibitor | |
| AU728091B2 (en) | Process for synthesizing carbapenem intermediates | |
| US5908936A (en) | Process for synthesizing carbapenem intermediates | |
| EP3807268A1 (en) | Process for the preparation of lifitegrast | |
| CN110551144B (en) | Preparation method of amoxicillin | |
| CN114315755B (en) | A synthetic method for key intermediates of Tubulysin and its analogs | |
| WO2003045896A1 (en) | New process | |
| CN103562207B (en) | For preparing the preparation method of 2-amino-9-((2-phenyl-1,3-dioxane-5-base epoxide) methyl)-1H-purine-6 (9H) the-one compound of valganciclovir | |
| US4960832A (en) | Polymer-supported alkyl azodicarboxylates and their use in Mitsunobu reactions | |
| CN115417816B (en) | A kind of preparation method of 3,6-dibromo-1-chloro-isoquinoline | |
| CN117088797A (en) | A kind of synthesis method of 2-bromo-1-(4-methanesulfonyl)acetophenone | |
| IL133151A (en) | Nitration process for diphenyl ethers | |
| EP0528941A4 (en) | Process for preparing cephalexin monohydrate | |
| CN111479800B (en) | Intermediate compound, preparation method thereof and solid-phase synthesis method for preparing polypeptide by using intermediate compound | |
| JPS631952B2 (en) | ||
| CN1903836A (en) | Compound containing multinitrobenzene amantadine and its synthesis method | |
| KR970005377B1 (en) | Process for preparation of 2,6-dinitroparacresol | |
| CN114478437A (en) | Method for preparing N-methyl-2-pyrrolidine ethylamine or salt thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU AZ BA BB BG BR BY CA CN CU CZ EE GE GW HU ID IL IS JP KG KR KZ LC LK LR LT LV MD MG MK MN MX NO NZ PL RO RU SG SI SK SL TJ TM TR TT UA US UZ VN YU |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2287931 Country of ref document: CA Ref country code: CA Ref document number: 2287931 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998920961 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 1998 549278 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 73678/98 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998920961 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 73678/98 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998920961 Country of ref document: EP |