WO1998055497A1 - Betulinol derivatives - Google Patents
Betulinol derivatives Download PDFInfo
- Publication number
- WO1998055497A1 WO1998055497A1 PCT/US1998/011456 US9811456W WO9855497A1 WO 1998055497 A1 WO1998055497 A1 WO 1998055497A1 US 9811456 W US9811456 W US 9811456W WO 9855497 A1 WO9855497 A1 WO 9855497A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- betulinol
- formula
- antibody
- group
- peptide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(CCC1C(C)=C)(CC2)C1C(CC1)C2(C)C(C)(CC2)C1C1(C)C2C(C)(C)C(*)CC1 Chemical compound CC(CCC1C(C)=C)(CC2)C1C(CC1)C2(C)C(C)(CC2)C1C1(C)C2C(C)(C)C(*)CC1 0.000 description 5
- FVWJYYTZTCVBKE-UHFFFAOYSA-N CC(C)(C(CC1)C(C)(CC2)C(CC3)C1(C)C1(C)C3C(C(CC3)C(C)=C)C3(CO)CC1)C2O Chemical compound CC(C)(C(CC1)C(C)(CC2)C(CC3)C1(C)C1(C)C3C(C(CC3)C(C)=C)C3(CO)CC1)C2O FVWJYYTZTCVBKE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J53/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by condensation with a carbocyclic rings or by formation of an additional ring by means of a direct link between two ring carbon atoms, including carboxyclic rings fused to the cyclopenta(a)hydrophenanthrene skeleton are included in this class
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
Definitions
- the present invention relates generally to betulinol derivatives and, in particular, to betulinol-antibody conjugates and to methods for making and using these betulinol derivatives and betulinol-antibody conjugates.
- Betulinol is one of the more plentiful triterpenes, constituting up to 24 per cent of the outer bark of the white birch (Betula alba) and as much as 35 per cent of the outer bark and about 5 per cent of the inner bark of the Manchurian white birch (Betula platvphylla (Hirota et al., J.S.C.I. Japan. 47:922 (1944)). It also occurs in the free state in the barks of the following trees: the yellow and black birch (Steiner, Mikrochemie. Molisch-Festschrift. p. 405 (1936)), Corylus avellana. Carpinus betulus (Feinberg et al., Monatsh.
- Birch tree cortex-extracted betulinol was first mentioned as an antiseptic in 1899. Subsequently, compounds singled out from extracts of Hvptis emorv and Alnus oregonu. identified as pentacyclic styrenes and their derivatives, were shown to inhibit carcino sarcoma growth (Sheth et al, J. Pharm. Sci.. 61 :1819 (1972) and Sheth et al., J. Pharm. Sci.. 62:139-140 (1973)). It has been suggested that betulinic acid is the main anti-tumor agent in the mixture of terpenoids (Tomas et al., Planta Medicina.
- chemotherapeutic agents act not only on malignant cells but have adverse effects on non-target cells as well, particularly on the rapidly proliferating cells of the gastrointestinal tract and bone marrow.
- these cytotoxic drugs give rise to undesirable and frequently severe side effects.
- site-directed chemotherapy is quite old, only a small number of anti-neoplastic drugs and toxins have been successfully coupled to monoclonal and polyclonal antibodies.
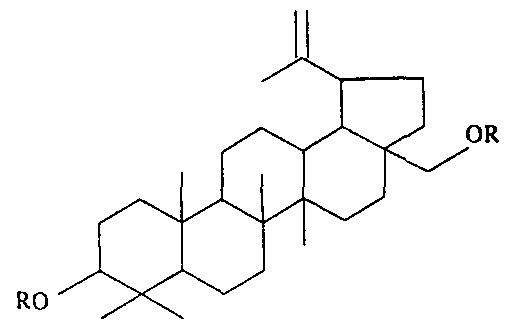
- the present invention relates to a diether having the formula:
- R is an alkyl group
- the present invention also relates to a method for preparing the diether.
- the method includes alkylating a dialcohol having the formula:
- the present invention further relates to a method of preparing betulonic aldehyde.
- the method includes oxidizing betulinol with chromium anhydride in acetone in the presence of sulfuric acid under conditions effective to produce betulonic aldehyde.
- the present invention relates to a compound having the formula:
- X or Y is a -peptide-Q moiety and the other of X and Y is a hydroxy group, an alkoxy group, an alkanoyloxy group, or a -peptide-Q moiety;
- Q is a hydroxy group, a -NHNH 2 moiety, an -NHNH-C(O)CH 2 Hal moiety, an -antibody-OH moiety, or an -NHNH-C(O)-antibody-OH moiety;
- Hal is a halogen
- the present invention is also related to a method of producing a betulinol- antibody conjugate having the formula:
- Y is a hydroxy group, an alkoxy group, or an alkanoyloxy group
- the method includes converting a haloacetylhydrazide having the formula: -peptide-NHNH-C (0) -CH 2 Hal
- Hal is a halogen
- the present invention in yet another aspect thereof, relates to a betulinol- antibody conjugate having the formula:
- A are independently selected from the group consisting of a -CHO moiety and a moiety having the formula:
- Y is a hydroxy group, an alkoxy group, or an alkanoyloxy group.
- the present invention also relates to a method of producing the betulinol- antibody conjugate described in the preceding paragraph. The method includes converting a carrier molecule having the formula:
- the present invention relates to a betulinol-antibody conjugate having the formula:
- A is a moiety having the formula:
- Y is a hydroxy group, an alkoxy group, or an alkanoyloxy group; and n is an integer from 1 to 100.
- the present invention also relates to a method of producing the betulinol- antibody conjugate described in the preceding paragraph.
- a crosslinker having a first reactive terminus and one or more second reactive termini is provided.
- An antibody is reacted with the first reactive terminus, and a hydrazide having the formula:
- the compounds, diethers, and betulinol-antibody conjugates of the present invention can be used to treat patients suffering from cancer.
- the present invention relates to method of treating cancer.
- the method includes administering to a cancer patient an effective amount of a compound.
- the compound is selected from the group consisting of betulonic aldehyde and compounds having the formulae:
- A is a moiety having the formula:
- a 2 is a moiety having the formula:
- n is an integer from 1 to 100;
- X and Y 1 are each independently selected from the group consisting of a hydroxy group, an alkoxy group, an alkanoyloxy group, and a -peptide-NHNH-C(O)-antibody-OH moiety;
- Y 2 is selected from the group consisting of a hydroxy group, an alkoxy group, and an alkanoyloxy group; and HO-antibody-H is an antibody targeted to a site to be treated in the patient.
- Figure 1 is a schematic flow diagram depicting a process for producing betulinol.
- the present invention relates to a diether having the formula:
- R is an alkyl group.
- R can be an unsubstituted alkyl, or it can be substituted with any number and combination of known substituents, such as sulfo, carboxy, cyano, halogen (e.g., fluoro, chloro), hydroxyl, alkenyl (e.g., allyl, 2-carboxy- allyl), alkoxy (e.g., methoxy, ethoxy), aryl (e.g., phenyl, p-sulfophenyl), aryloxy (e.g., phenyloxy), carboxylate (e.g., methoxycarbonyl, ethoxycarbonyl), acyloxy (e.g., acetyloxy), acyl (e.g., acetyl, propionyl), amino (including unsubstituted- monosubstituted-, and disubstituted-amino as well as cyclic
- the alkyl group can be linear, branched, or cyclic.
- suitable alkyl groups include, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, neopentyl, cyclopentyl, n-hexyl, and cyclohexyl.
- R is methyl, in which case the diether is betulinol dimethyl ether (also designated as "cornelon").
- the diether of the present invention has a number of optically active carbon atoms. It is preferred that the diether be optically pure, and it is yet more preferred that each of the chiral centers in the diether have the conformation of that of naturally occurring betulinol as shown in the formula below:
- the diether can be prepared in a variety of ways.
- One particularly preferred preparative method, a method to which the present invention also relates, includes alkylating a dialcohol having the formula:
- the dialcohol starting material for this reaction can be betulinol, such as betulinol isolated from natural products.
- betulinol can be isolated from the outer layer of the bark of the white birch tree Betula alba by sublimation (Lowitz, Crell's Annalen, 1 :312 (1788) and Mason, Silliman's Am. J.. 20:282 (1831), which are hereby incorporated by reference) or by extraction with an alcohol, such as ethanol (Hunefeld, J. Prakt. Chem., 7:53 (1836) and Hess, Poggendorff s Annalen.
- betulinol is isolated from the non-saponifiable substance of floral soap, such as, for example, by the method depicted in Figure 1.
- the crushed initial leaf wood and components of a sulfate boiling procedure (NaOH, Na 2 SO 4 , Na 2 S 2 O 3 , Na 2 SO 3 ) are lodged to a boiling pot in a batch or continuous process.
- lignin the component of wood
- Crude cellulose is derived from the pulping liquor which is composed of lignin, cellulose, and black buck.
- Black buck is a composition of black buck with salts of tall acid and non-saponifiable substances.
- the crude cellulose is used in paper production, whereas the sulfate soap is separated from the black buck by centrifugation or by a settling process. Treatment of the sulfate soap with sulfuric acid produces tall oil.
- the non-saponifiable substances are separated as crude betulinol. Recrystallization of the crude betulinol, such as from acetone, ethyl acetate, isopropanol, butanol, ethanol, and the like, yields pure betulinol.
- the black buck residue present after centrifugation or settling can be advantageously recycled as shown in Figure 1.
- betulinol used as the starting material in the synthesis of the diether is not critical to the practice of the present invention, it is preferred that betulinol having a purity of at least 92-94% and a melting point of 241-243 °C be used. Betulinol having these properties can be obtained using the preferred isolation and purification methods described above.
- the dialcohol is alkylated with a nitrile having the formula R-C ⁇ N.
- the identity of the nitrile used depends on the identity of the R groups desired in the diether. For example, where betulinol dimethyl ether is desired, the nitrile is acetonitrile.
- Other nitriles suitable for use in preparing other diethers include propionyl nitrile, butyryl nitrile, pentanoyl nitrile, hexanoyl nitrile, benzylacetonitrile, and the like.
- the dialcohol and nitrile are present in at least a 1 :2 molar ratio, more preferably, in a molar ratio of from about 1 :20 to 1 :60, and, most preferably, in a molar ratio of about 1 :40.
- the reaction can be carried out without the use of a solvent in the case where the nitrile is a liquid in which the dialcohol is soluble, such as is the case where the nitrile is acetonitrile.
- the reaction can be carried out in a reaction solvent, preferably one in which both the dialcohol and the nitrile are appreciably soluble and with which neither reacts.
- Suitable solvents include, for example, ketone solvents, such as acetone, and chlorinated hydrocarbon solvents, such as methylene chloride and chloroform.
- the reaction can be carried out at a temperature from about room temperature to about the reflux temperature of the nitrile or the reaction solvent, preferably, from about 30°C to about 70°C, and, more preferably, at about 50°C.
- the duration of the reaction depends, in large measure, on the reactivity of the nitrile, the concentration of the reactants, and other factors. Typically, the reaction is carried out for a period of time from about 5 minutes to about 12 hours, preferably, from about 5 minutes to about 1 hour, and, more preferably, about 20 minutes.
- the diether is isolated.
- isolation is best carried out by filtering the precipitated diether, preferably after cooling the reaction mixture.
- the diether can be separated, as an oil or as a precipitate, by addition of a solvent to the reaction mixture of a solvent in which the diether lacks appreciable solubility, typically an alkane, such as petroleum ether, or an ether, such as diethyl ether.
- the present invention also relates to a method for preparing betulonic aldehyde.
- the method starts with betulinol, provided, for example, by the methods described above in connection with preparing the diether of the present invention. Betulinol is then oxidized with chromium anhydride in acetone in the presence of sulfuric acid.
- betulinol is first dissolved in acetone, preferably in a weight ratio of from about 1 :50 to about 1 :200, and more preferably in a weight ratio of from about 1 : 100 to about 1:110.
- the reaction is allowed to proceed at a temperature of from about room temperature to about the reflux temperature of the acetone, preferably at the reflux temperature of the acetone, for from about 15 minutes to about 24 hours, preferably from about 1 hour to about 4 hours, more preferably from about 2.5 to about 3 hours.
- the reaction can then be worked up by standard procedures to isolate the resulting betulonic aldehyde product.
- the reaction mixture is cooled, water is added to it to produce a sediment containing the product, and the sediment is filtered and washed with water to remove residual sulfuric acid.
- the filtered betulonic aldehyde can be purified, such as by recrystallization or chromatography, preferably by recrystallization from an alcohol, such as ethanol, isopropanol, or butanol.
- the present invention also relates to a compound having the formula:
- One of X or Y is a -peptide-Q moiety and the other of X and Y is a hydroxy group, an alkoxy group, an alkanoyloxy group, or a -peptide-Q moiety.
- Alkoxy groups have the general formula -OR, where R is an alkyl group, defined and illustrated as it was above in connection with the diether of the present invention.
- suitable alkoxy groups include methoxy, ethoxy, propoxy (including n-propoxy and iso-propoxy), butoxy, pentoxy, and hexoxy (including n- hexoxy and cyclohexoxy).
- Alkanoyloxy group include those having the general formula -OC(O)R, where R is an alkyl group, defined and illustrated as it was above in connection with the diether of the present invention.
- suitable alkoxy groups include acetoxy, propionyloxy (including n-propionyloxy and iso-propionyloxy), butanoyloxy, pentanoyloxy, and hexanoyloxy (including n-hexanoyloxy and cyclohexanoyloxy).
- -peptide- means the diradical of a peptide having the formula H-peptide-OH, where -H denotes the peptide's amino terminus and -OH denotes the peptide's carboxy terminus. The peptide is bonded to the -CH 2 group of the betulinol ring structure through its amino terminus.
- the peptide can be made of any amino acid sequence and any number of amino acid residues, it is preferred that the peptide be a tetrapeptide, particularly -Leu-Ala-Leu-Ala-, or a pentapeptide, particularly -Gly-Ala-Leu-Gly-Leu-.
- Q can be a hydroxy group, an -NHNH 2 moiety, an -NHNH-C(O)CH 2 Hal moiety, an -antibody-OH moiety, or an -NHNH-C(O)-antibody-OH moiety.
- -antibody-OH is a radical form of an antibody having the formula H-antibody-OH, where the H- denotes the amino terminus and the -OH denotes the carboxy terminus of the antibody.
- the antibody is bound to the -peptide- moiety or to the -peptide-NHNHC(O)- moiety through its amino terminus.
- the preferred type of antibody for use in the invention is an immuno globulin which is a ga maglobulin.
- immunoglobulin which is a ga maglobulin.
- IgG, IgA, IgE, and IgM subclasses are particularly preferred.
- Some representative immunoglobulins are monoclonal or polyclonal antibodies to human or animal tumor associated antigens; human B- and T-cell antigens; human la antigens; viral, fungal and bacterial antigens; and cells involved in human inflammatory or allergic reactions.
- Preferred antibodies to human or animal tumor associated antigensin include:
- Ig from goats or sheep immunized with carcinoembryonic antigen Ig from rabbit antiacute lymphoblastic leukemia serum; Ig from various primate antisera raised against acute lymphoblastic leukemia, acute myleoblastic leukemia, chronic lymphoblastic leukemia and chronic granulocytic leukemia; Ig from goats or sheep immunized with lung carcinoma cells, or cellular fractions; monoclonal Ig from mouse hybridomas secreting anti-human colorectal carcinoma antibodies; monoclonal Ig from mouse hybridomas secreting anti-human melanoma antibodies; monoclonal Ig from mouse hybridomas that secrete antibodies reacting with human leukemia cells; monoclonal Ig from mouse hybridomas secreting antibodies reacting with human neuroblastoma cells; monoclonal Ig from mouse hybridomas secreting antibodies reacting with human breast cancer antigens; monoclonal Ig from mouse hybridomas secreting antibodies reacting with human ovarian carcinoma cells; monoclon
- Antibody is also meant to include immunoglobulin fragments Ig', referred to also as Fab, Fab', F(ab') 2 , and IgM monomer derived from an antibody, for example, by proteolytic enzyme digestion with, for example, pepsin or papain, or by reductive alkylation. Procedures for preparing these antibody fragments are described in Parham, J. Immunology. 131 :2895 (1983V Lamovi et al. J. Immunological Methods, 56:235 (1983); Parham, J. Immunological Methods. 53:133 (1982); and Matthew et al., Immunological Methods. 50:239 (1982), which are hereby incorporated by reference.
- Preferred conjugates are those prepared from monoclonal antibodies, especially those which recognize human cancer cells such as adenocarcinoma, squamous cell carcinoma, transitional cell carcinoma, melanoma, neuroblastoma, small cell carcinoma, leukemia, lymphoma, and sarcoma.
- Methods for preparing antibodies and monoclonal antibodies to particular haptenic or antigenic target substrates are described in Goding, Monoclonal Antibodies: Principles and Practice. 2nd. ed., New York: Academic Press, (1986); Kennett et al., Monoclonal Antibodies. New York:Plenum Press (1980); U.S. Patent. No. 4,423,147 to Secher et al.; U.S. Patent No.
- the present invention relates to a betulinol-antibody conjugate having the formula:
- Y is a hydroxy group, an alkoxy group, or an alkanoyloxy group.
- the betulinol-antibody conjugate described in the preceding paragraph is not limited by its method of preparation.
- One particularly preferred method for preparing the betulinol-antibody conjugate described in the preceding paragraph includes converting a betulinol-peptide derivative having the formula:
- reaction is carried out under conditions effective for formation of a covalent peptide bond between the amino terminus of the antibody and the carboxy terminus of the peptide.
- reaction is carried out using a betulinol-peptide derivative:antibody molar ratio of from about 1:1 to about 100:1; in an inert solvent, such as dimethylformamide; and under mild conditions, such as by gently stirring the reaction mixture at a reduced temperature, preferably from about 0°C to about 10°C, more preferably about 4°C, for from about 1 hour to about 10 days, preferably for about 3 days.
- Catalysts typically used in peptide bond formation reactions such as N,N'- dicyclohexylcarbodiimide (“DCC”), can be used, preferably in a molar amount approximately equal to that of the betulinol-peptide present.
- DCC N,N'- dicyclohexylcarbodiimide
- the crude betulinol-antibody conjugate is isolated, such as by adding water to the reaction mixture to precipitate the product and then filtering the precipitate. Purification of the precipitate can be effected, for example, by chromatography using an appropriate stationary phase, such as silica gel, and a suitable solvent, such as a 4:1 to 1 :1 (volume ratio) mixture of chloroform and methanol.
- the betulinol-peptide derivative used to produce the betulinol-antibody conjugate can be prepared, for example, from a compound having the formula:
- the reaction is carried out under conditions effective for formation of a covalent bond between the amino terminus of the peptide and the betulinol hydroxy group.
- the reaction is carried out using a betulinol compound:antibody molar ratio of about 50:1; in an inert solvent, such as dimethylformamide; and under mild conditions, such as by gently stirring the reaction mixture at a reduced temperature, preferably from about 0°C to about 10°C, more preferably about 4°C, for from about 1 hour to about 10 days, preferably for about 3 days.
- Catalysts typically used in peptide bond formation reactions, such as DCC can be used, preferably in a molar amount approximately equal to the molar amount of betulinol compound present.
- the crude betulinol-peptide can be isolated, for example, by adding water to the reaction mixture to precipitate the product and then filtering the precipitate. Purification of the precipitate can be effected, for example, by chromatography using an appropriate stationary phase, such as silica gel, and a suitable solvent, such as a 4: 1 to 1 : 1 (volume ratio) mixture of chloroform and methanol.
- the present invention also relates to a betulinol-antibody conjugate having the formula:
- Y is a hydroxy group, an alkoxy group, or an alkanoyloxy group.
- the betulinol-antibody conjugate described in the preceding paragraph is not limited by its method of preparation. However, in one particularly preferred method for preparing the betulinol-antibody conjugate, a haloacetylhydrazide having the formula:
- Hal denotes a halogen atom, such as a chlorine, a bromine, or, preferably, an iodine.
- the reaction is carried out under conditions effective for formation of a covalent bond between the amino terminus of the antibody and the NHNHC(O)- moiety.
- the reaction is carried out by mixing the haloacetylhydrazide, dissolved in an appropriate solvent, such as DMF, with the antibody, dissolved in an appropriate solvent, such as aqueous buffer.
- an appropriate solvent such as DMF
- One particularly useful buffer for dissolving the antibody is 0.1 M Tris-HCl buffer, adjusted to a pH of 8 and containing 0.1 M NaCl.
- the concentrations of the haloacetylhydrazide and antibody in their respective solvents and the amounts of the two solutions employed are selected so that a haloacetylhydrazide:antibody molar ratio of about 1:1 is achieved upon mixing of the two solutions.
- the conversion can be carried out under mild conditions, such as by gently stirring the reaction mixture at a temperature from about 0°C to about 70°C, preferably from about 20°C to about 30°C, more preferably at about 4°C, for from about 1 hour to about 10 days, preferably for about 17 hours.
- the crude betulinol-antibody conjugate is isolated, such as by dialyzing the crude betulinol-antibody conjugate against phosphate buffer, preferably 10 mM, pH 7.2 phosphate buffer containing 0.14 M NaCl. Purification of the isolated betulinol-antibody conjugate can be effected, for example, by chromatography.
- haloacetylhydrazide used to produce the betulinol-antibody conjugate can be prepared by providing a hydrazide having the formula:
- Nitrophenyl haloacetates suitable for use in this preparative method include p-nitrophenyl chloroacetate, p-nitrophenyl bromoacetate, and, preferably, p-nitrophenyl iodoacetate.
- the reaction is preferably carried out in a reaction solvent in which both the hydrazide and the p-nitrophenyl haloacetate are soluble, such as DMF or a chlorinated hydrocarbon, such as chloroform.
- the hydrazide and the p-nitrophenyl haloacetate are mixed in the reaction solvent, preferably in a hydrazide :p- nitrophenyl haloacetate molar ratio of from about 2:1 to about 1:2, more preferably about 1:1.
- the reaction mixture is stirred gently, preferably in the dark, at a temperature from about 10°C to about 100°C, preferably at about room temperature, for a period of time ranging from 1 hour to 3 days, preferably for about 19 hours.
- the crude haloacetylhydrazide is precipitated, preferably after cooling, such as by addition of a solvent, such as ethyl acetate, which reduces the solubility of the haloacetylhydrazide product in the reaction mixture.
- the precipitate can be collected by any suitable means, such as by filtration or centrifugation. Purification of the precipitate can be effected, for example, by chromatography or recrystallization, but is generally not necessary.
- the hydrazide used in the above-described preparation of haloacetylhydrazide can be produced by providing a betulinol-peptide having the formula:
- the hydrazide preparative method further includes converting the betulinol-peptide with hydrazine hydrate under conditions effective to produce the hydrazide.
- the reaction is preferably carried out in a reaction solvent in which both hydrazine hydrate and the betulinol-peptide are soluble, such as DMF or a chlorinated hydrocarbon, such as chloroform.
- a reaction solvent in which both hydrazine hydrate and the betulinol-peptide are soluble, such as DMF or a chlorinated hydrocarbon, such as chloroform.
- the hydrazine hydrate and the betulinol-peptide are mixed in the reaction solvent, preferably in a hydrazine hydrate: betulinol-peptide molar ratio of from about 2:1 to about 1 :2, more preferably about 1:1.
- the reaction mixture is stirred, preferably at a temperature from about 10°C to about 100°C, more preferably at about room temperature, for a period of time ranging from 12 hours to about 10 days, preferably for about 5 days.
- the crude hydrazide is precipitated, preferably after cooling, such as by addition of a solvent, such as ethyl alcohol, which reduces the solubility of the hydrazide product in the reaction mixture.
- the precipitate can be collected by any suitable means, such as by filtration or centrifugation. Purification of the precipitate can be effected, for example, by chromatography or recrystallization, but is generally not necessary.
- the present invention also provides a betulinol-antibody conjugate having the formula: H 2 NOCH 2 CO-(Gly). Lys Gl y-ant ibody-OH
- A are independently selected from an aldehyde group or a moiety having the formula:
- Y can be a hydroxy group, an alkoxy group, or an alkanoyloxy group.
- betulinol-antibody conjugate described in the preceding paragraph can be made by providing a carrier molecule having the formula:
- the carrier molecule is then converted with a hydrazide having the formula:
- the carrier molecule can be reacted with the antibody under conditions effective to produce an antibody-bound carrier molecule having the formula:
- the antibody-bound carrier molecule is then reacted with the hydrazide under conditions effective to produce the betulinol-antibody conjugate.
- the carrier molecule can be reacted with the hydrazide under conditions effective to produce a betulinol-bound carrier molecule having the formula:
- the betulinol-bound carrier molecule is then reacted with the antibody under conditions effective to produce the betulinol-antibody conjugate.
- reaction of the antibody with the Gly residue of the carrier molecule or betulinol-bound carrier molecule can be carried out under conditions effective for formation of a covalent peptide bond between the amino terminus of the antibody and the carboxy group of the Gly residue.
- the reaction is carried out using a carrier molecule:antibody molar ratio of from about 1 : 1 to about 10: 1 ; in an inert solvent, such as dimethylformamide; and under mild conditions, such as by gently stining the reaction mixture at a reduced temperature, preferably from about 0°C to about 10°C, more preferably about 4°C, for from about 1 hour to about 10 days, preferably for about 3 days.
- Catalysts typically used in peptide bond formation reactions, such as DCC can be used, preferably in a molar amount approximately equal to that of the antibody present.
- the crude antibody-bound carrier molecule product or the crude betulinol-antibody conjugate product is isolated, such as by adding water to the reaction mixture to precipitate the product and then filtering the precipitate.
- Purification of the precipitate can be effected, for example, chromatographically using an appropriate stationary phase, such as silica gel, and solvent such as a 4:1 to 1 :1 (volume ratio) mixture of chloroform and methanol.
- Reaction of the aldehyde group on the carrier molecule or on the antibody- bound carrier molecule with the hydrazide is best canied out under conditions which are conducive for the formation of hydrazone bonds.
- the number of aldehyde residues which react with hydrazide depends primarily on the molar ratio of hydrazide to carrier molecule. Suitable hydrazide: carrier molecule molar ratios range from about 1 : 1 to about 20: 1.
- the reaction is carried out in an inert solvent, such as DMF by stining the reaction mixture at a temperature from about 15°C to about 35°C, preferably about 25°C, for from about 10 hours to about 6 days, preferably for about 5 days. Further details regarding this reaction are described, for example, in Vilaseca et al., Bioconiugate Chem., 4:515-520 (1993) ("Vilaseca”), which is hereby incorporated by reference.
- an inert solvent such as DMF
- the crude betulinol-bound canier molecule product or the crude betulinol-antibody conjugate product is isolated, such as by precipitation with butanol followed by centrifugation or filtration. Purification of the precipitate can be effected, for example, by chromatography.
- the canier molecule can be prepared by the method described in Vilaseca, which is hereby incorporated by reference.
- the resin-bound protected nonapeptide Fmoc-Gly 3 -[Lys(t-Boc)] 5 -Gly-OCH 2 -PAM resin is synthesized by standard fluorenylmethyloxycarbonyl ("Fmoc") techniques from a t-Boc-Gly-OCH 2 -PAM ((phenylacetaimido)methyl resin) on an automated peptide synthesizer.
- Fmoc fluorenylmethyloxycarbonyl
- the protected nonapeptide is then deprotected with trifluoroacetic acid ("TFA") and reacted with t-Boc- Ser(Bzl)-OSu to produce Fmoc-Gly 3 -[Lys(t-Boc-Ser(Bzl))] 5 -Gly-OCH 2 -PAM.
- TFA trifluoroacetic acid
- Deprotection with piperidine in DMF followed by deprotection with TFA and reaction with t-Boc-NHOCH 2 COOSu yields t-Boc-NHOCH 2 CO-Gly 3 -[Lys(t-Boc-Ser(Bzl))] 5 -Gly- OCH 2 -P AM.
- Treatment of this material with TFA and then with a mixture of TFA and trifluoromethanesulfonic acid produces the canier molecule.
- the present invention also relates to a betulinol-antibody conjugate having the formula:
- A is a moiety having the formula:
- Y is a hydroxy group, an alkoxy group, or an alkanoyloxy group, suitable examples of which include those described above, and n is an integer from 1 to 100, preferably from 30 to 50.
- the spacer moiety is functionalized with groups capable of bonding with the -NHNH- group of the A moiety.
- Each of the moieties A are attached to the spacer.
- Each A can be incorporated into the backbone of the spacer, or, alternatively, each A can be attached to the spacer backbone as a pendant group.
- the -spacer-(A) n moiety can have the formula: [C (O) NHCH 2 CH 2 CH 2 CH 2 - C- NHC(0)O- ( CH 2 CH 2 O) a ] b -
- the spacer can be a diamine derivative of polyethylene glycol having 2-(pyridyldithio)- propionyl and N-hydroxysuccinimide ester groups bonded thereto, such as those described in Haselgrubler et al., Bioconiueate Chem.. 6:242-248 (1995) ("Haselgrubler”), which is hereby incorporated by reference.
- Another spacer suitable for use in practicing the present invention is a branched form of polyethylene glycol propionic acid N- hydroxysuccinimide ester, such as a monomethoxypoly(ethylene glycol)-propionic acid N-hydroxysuccinimide ester.
- This and other branched forms of polyethylene glycol propionic acid N-hydroxysuccinimide ester are described in Senter et al., Bioconiueate Chem., 6:389-394 (1995) ("Senter”), which is hereby incorporated by reference.
- the betulinol-antibody conjugates described in the preceding paragraphs can be prepared by providing a crosslinker having a first reactive terminus and one or more second reactive termini.
- the first reactive terminus is reacted with an antibody, and one or more of the one or more second reactive termini is reacted with a hydrazide having the formula:
- the hydrazide can be prepared by the methods described above.
- Suitable crosslinkers for the practice of the present invention include molecules which contain functional groups capable of forming covalent bonds with an antibody and with the hydrazide.
- the first terminus is typically an amino group (capable of reacting with the antibody's carboxy terminus) or a hydroxyl, an aldehyde, or a carboxylic acid group (capable of reacting with the antibody's amino terminus).
- the crosslinker can be a polymer containing pendant groups having the required reactivity.
- One suitable crosslinker is poly(polyethylene glycol-lysine), which has the formula:
- poly(polyethylene glycol-lysine) can be prepared from lysine and polyethylene glycol by the methods described in Vilaseca; Poiani et al., Biocongugate Chem., 5:621- 630 (1994); Nathan et al., Biocongugate Chem.. 4:54-62 (1993); Nathan et al.,
- crosslinkers are diamine derivatives of polyethylene glycol having 2-(pyridyldithio)-propionyl and N-hydroxysuccinimide ester groups bonded thereto, such as those described in Haselgrubler, which is hereby incorporated by reference.
- Another crosslinker suitable for use in practicing the present invention is a branched form of polyethylene glycol propionic acid N-hydroxysuccinimide ester, such as a monomethoxypoly(ethylene glycol)-propionic acid N-hydroxysuccinimide ester. Further description of and methods for preparing these polyethylene glycol propionic acid N-hydroxysuccinimide esters is provided in Senter, which is hereby incorporated by reference.
- the hydrazide can be prepared by the methods described above. Reaction of the first reactive terminus of the crosslinker with the antibody is typically carried out in an inert solvent, such as dimethyl sulfoxide ("DMSO"), by stining the reaction mixture at a temperature from about 19 °C to about 25 °C, preferably about 19°C, for from about 60 minutes to about 120 minutes, preferably for about 70 minutes. Suitable antibodyxrosslinker molecule molar ratios range from about 1 : 1.5 to about 1 :12. Further details regarding this reaction are described, for example, in Haselgrubler, which is hereby incorporated by reference.
- DMSO dimethyl sulfoxide
- Reaction of the second reactive terminus of the crosslinker with the hydrazide is typically canied out in an inert solvent, such as DMSO, by stirring the reaction mixture at a temperature from about 19 °C to about 25 °C, preferably about 19 ° C for from about 60 minutes to about 120 minutes, preferably for about 70 minutes.
- an inert solvent such as DMSO
- the number of hydrazide moieties which react with each crosslinker molecule depends primarily on the molar ratio of hydrazide to crosslinker. Suitable hydrazidexrosslinker molecule molar ratios range from about 1:1.5 to about 1:12.
- reaction can be canied out in any order.
- the first reactive terminus can be coupled to the antibody, and the second reactive termini of the resulting antibody-crosslinker product can then be reacted with the hydrazide.
- the hydrazide can be reacted with the second reactive termini of the crosslinker, and the first reactive terminus of the resulting hydrazide-crosslinker complex can then be reacted with the antibody.
- a simultaneous, one pot reaction of the crosslinker, antibody, and hydrazide is also contemplated, although purification of the betulinol-antibody conjugate may be more difficult.
- the crude hydrazide-crosslinker intermediate or the crude antibody-crosslinker intermediate can be isolated, such as by precipitation with butanol.
- the intermediate hydrazide-crosslinker or antibody-crosslinker can also be purified, for example, by chromatography.
- the crude hydrazide- crosslinker intermediate or the crude antibody-crosslinker intermediate can be reacted with the antibody or hydrazide, respectively, without purification.
- the crude betulinol-antibody conjugate produced by the above-described process can be isolated such as by precipitation with butanol.
- the isolated betulinol-antibody conjugate can also be purified, for example, by chromatography.
- the present invention also relates to a method of treating cancer by administering to a cancer patient an effective amount of a betulinol derivative.
- Suitable betulinol derivatives include betulonic aldehyde. They also include compounds having the formulae:
- X and Y 1 are each independently selected from the group consisting of a hydroxy group, an alkoxy group, an alkanoyloxy group, a -peptide-antibody-OH moiety, and -peptide-NHNH-C(O)-antibody-OH moiety, such as betulinol dimethyl ether and betulinic acid diacetate.
- Other suitable betulinol derivatives include the betulinol- antibody conjugates of the present invention.
- HO- antibody-H is an antibody targeted to a site to be treated in the patient. Suitable antibodies include those cited above.
- the betulinol derivatives can be administered orally, parenterally, subcutaneously, intravenously, intramuscularly, intraperitoneally, by intranasal instillation, by intracavitary or intravesical instillation, intraocularly, intraarterially, intralesionally, or by application to mucous membranes, such as, that of the nose, throat, and bronchial tubes.
- They may be administered alone or with pharmaceutically or physiologically acceptable carriers, excipients, or stabilizers, and can be in solid or liquid form, such as tablets, capsules, powders, solutions, suspensions, or emulsions.
- the solid unit dosage forms can be of the conventional type.
- the solid form can be a capsule, such as an ordinary gelatin type containing the betulinol derivative and a canier, for example, lubricants and inert fillers, such as lactose, sucrose, or cornstarch.
- these betulinol derivatives can be tableted with conventional tablet bases, such as lactose, sucrose, or cornstarch, in combination with binders, like acacia, cornstarch, or gelatin, disintegrating agents, such as cornstarch, potato starch, or alginic acid, and lubricants, like stearic acid or magnesium stearate.
- the betulinol derivatives may also be administered in injectable dosages by solution or suspension of these materials in a physiologically acceptable diluent with a pharmaceutical carrier.
- a pharmaceutical carrier include sterile liquids, such as water and oils, with or without the addition of a surfactants, adjuvants, excipients, or stabilizers.
- Illustrative oils are those of petroleum, animal, vegetable, or synthetic origin, for example, peanut oil, soybean oil, or mineral oil.
- water, saline, aqueous dextrose and related sugar solutions, and glycols, such as propylene glycol or polyethylene glycol are prefened liquid caniers, particularly for injectable solutions.
- the betulinol derivative in solution or suspension may be packaged in a pressurized aerosol container together with suitable propellants, for example, hydrocarbon propellants like propane, butane, or isobutane, and with conventional adjuvants.
- suitable propellants for example, hydrocarbon propellants like propane, butane, or isobutane, and with conventional adjuvants.
- the betulinol derivatives can also be administered in a non- pressurized form, such as in a nebulizer or atomizer.
- Betulinol was placed in a reactor, and acetic anhydride was then added with stining. After the addition, the molar ratio of betulinol to acetic anhydride was between 1 :20 and 1:50. The reaction mixture was then heated with stining to 139-141°C, maintained at this temperature for between 60 and 90 min, and then cooled to 110-120°C. Hot water, in an amount equal to 6-10 times the initial mass of betulinol, was then added with stining. A crystalline sediment formed, and the sediment was removed by filtration and flushed repeatedly with hot water until the pH of the filtrate reached 6.8-7.0.
- the sediment of betulinol biacetate was then dried at 60-70°C and purified by recrystallization in an organic solvent (acetone, ethyl acetate, isopropyl alcohol, or butyl alcohol). Briefly, recrystallization was canied out by diluting the betulinol biacetate in the solvent with heating, boiling the betulinol acetate solution for from 0.5 to 1 hour, and cooling the betulinol biacetate solution to 10-15°C. A sediment formed, was filtered, and, was dried. The resulting product was 97-98% betulinol biacetate and had a melting temperature of 215°C. The yield of purified betulinol biacetate was 90%.
- an organic solvent acetone, ethyl acetate, isopropyl alcohol, or butyl alcohol.
- butanol was selected as a solvent.
- the BBA sample had a melting temperature of 217°C and BBA mass content of 97.0%.
- Betulinol was placed in a thermostated reactor, and acetone, in an amount of 100-110 ml per gram of betulinol, was added with stining.
- An oxidizing mixture of CrO 3 /H 2 SO 4 (molar ratio of 2:3, respectively) was then slowly poured into the reactor with stining.
- the reaction mixture was brought to reflux and maintained at reflux for 2.5-3 hrs.
- the reaction mixture was then cooled and water was added, resulting in the formation of a sediment.
- the sediment was filtered and recrystallized from ethanol.
- the resulting solid contained 93-95% of betulonic aldehyde and melted at a temperature from 154-156°C.
- the yield of purified betulonic aldehyde was 65%.
- Example 3 Preparation of Cornelon
- Betulinol was dissolved in acetonitrile in a betulinol-to-acetonitrile mole ratio of 1 :40. The solution was heated to 50°C and stined for 20 minutes. The crystalline residue, designated as "Cornelon", was washed with acetonitrile, filtered, and dried at 60° C. Cornelon was obtained in a 80-95% yield and was analyzed by HPLC.
- Example 4 Anti-carcinogenic Activity of Betulinol Biacetate and Betulonic Aldehyde The anti-carcinogenic activity of betulinol biacetate and betulonic aldehyde was assessed using 117-120 gram white rats implanted with worker's carcinoma and 21-23 gram white mice with artificially transplanted Ehrlich's tumor.
- the volume of the tumor was measured and calculated as a multiple of the quadrate of its similar diameter by the greater diameter.
- the effect was expressed as a ratio ("E/C"), in percent, of the tumor volume in rats treated with betulinol acetate or betulonic aldehyde ("E") and the tumor volume in control rats ("C”).
- E/C ratio
- the effect of betulinol acetate or betulonic aldehyde on ALL was evaluated the same way.
- N,N'-dicyclohexylcarbodiimide (206 mg) and a pentapeptide, gly-ala-leu-gly-leu, (360 mg) will be dissolved in 30 ml of DMF.
- the DMF solution will then be added to betulinol (499 mg).
- the mixture will be stined for 3 days at 4°C. Two ml of ice water will be added, and the precipitate which forms will be removed by filtration.
- the filtrate will be evaporated and subjected to chromatography on a silica gel column (3x30 cm, YMC-Gel, SIL60, 350/250 mesh) with chloroform:methanol (4:1), to isolate the pentapeptide ester derivative of betulinol (expected yield:500 mg).
- the product will be analyzed by preparative think layer chromatography on silica gel-coated glass plates eluted with a mixture of chloroform-methanol-water (120:20:1, v/v/v), and by amino acid analysis.
- the major fraction containing the betulinol-peptide conjugate will be utilized for further linking with an antibody.
- Example 6 Hypothetical Preparation of Betulinol- Antibody Conjugate
- the monoclonal antibody will be succinylated as follows. 10 mg of the antibody will be dissolved in 0.1 ml of water, and the pH will be adjusted to 7.5.
- Succinic anhydride (0.068 mmole) will be added while maintaining the pH at 7.5.
- the mixture will be extensively dialyzed against 10 mM phosphate buffer, pH 7.2, containing 0.15 M NaCl, to give a betulinol- pentapeptide-antibody conjugate.
- the conjugate will be purified by HPLC and characterized for its homogeneity and properties.
- the conjugates will be further characterized by nuclear magnetic resonance and by sodium dodecyl sulfate polyacrylamide gel electrophoresis ("SDS-PAGE").
- SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
- DCC (206 mg) will be added to a solution of betulinol (499 mg) and tetrapeptide (360 mg) in 30 mg of DMF.
- the mixture will be stined at 4°C for 3 days. Ice-water (2 ml) will then be added to generate a precipitate, which is then removed by filtration. The filtrate will then be evaporated, and the residue will be subjected to medium pressure chromatography on a silica gel column (3x30 cm, YME-GEL, SIL60, 350/250 mesh) with chloroform-methanol (4:1 to 1 :1).
- the tetrapeptide ester derivative of betulinol is isolated as a powder (500 mg) by evaporation of the solvent, treatment with ethyl acetate and filtration. Hydrazine hydrate (500 mg) will be mixed with a solution of the tetrapeptide ester derivative of betulinol (418mg) in 20 ml of DMF, and the mixture will be stined at room temperature for 5 days. A small amount of precipitate which forms will be removed by centrifugation, and the mixture will be treated with 50 ml of ethanol. The resulting precipitate (a belulinol tetrapeptide hydrazide derivative will be collected by filtration and dried under reduced pressure.
- p-Nitrophenyl iodoacetate (61 mg) will be added to a solution of the belulinol tetrapeptide hydrazide derivative (100 mg) in DMF (2 ml), and the mixture will be stined at room temperature for 19 hr. in the dark. The mixture is then treated with ethyl acetate (30 ml), which causes a precipitate to form. The precipitate will be collected by centrifugation and dried under reduced pressure to give the iodoacetylhydrazide derivative of the betulinol tetrapeptide (105 mg).
- a solution of the iodoacetylhydrazide derivative of the betulinol tetrapeptide in DMF (48.8 mg/ml) will be added to a solution of antibody in 0.1 M Tris- HC1 buffer (pH 8.0, containing 0.1 M NaCl) (8.27 mg/ml), and the mixture will be allowed stand at 25°C for 17 hr. The mixture will then extensively dialyzed against 10 mM PBS to give the betulinol-antibody conjugate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Steroid Compounds (AREA)
Abstract
Description












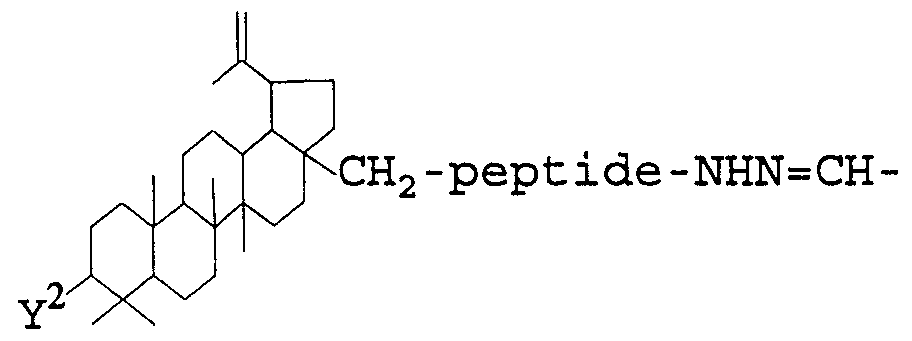













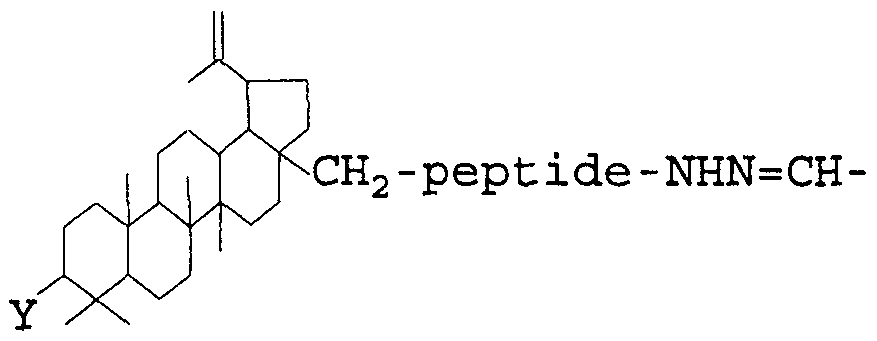










Claims























Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69825446T DE69825446T2 (en) | 1997-06-04 | 1998-06-03 | BETULINOLDERIVATE |
| CA002293502A CA2293502A1 (en) | 1997-06-04 | 1998-06-03 | Betulinol derivatives |
| EP98926258A EP0988311B9 (en) | 1997-06-04 | 1998-06-03 | Betulinol derivatives |
| AU78135/98A AU7813598A (en) | 1997-06-04 | 1998-06-03 | Betulinol derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US4862197P | 1997-06-04 | 1997-06-04 | |
| US60/048,621 | 1997-06-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998055497A1 true WO1998055497A1 (en) | 1998-12-10 |
Family
ID=21955543
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/011456 Ceased WO1998055497A1 (en) | 1997-06-04 | 1998-06-03 | Betulinol derivatives |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US6890533B2 (en) |
| EP (1) | EP0988311B9 (en) |
| AU (1) | AU7813598A (en) |
| CA (1) | CA2293502A1 (en) |
| DE (1) | DE69825446T2 (en) |
| WO (1) | WO1998055497A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008086759A3 (en) * | 2007-01-15 | 2009-02-26 | Univerzita Karlova V Praze Pri | Method of preparation and isolation of betulin diacetate from birch bark from paper mills and its optional processing to betulin |
| JP2010090385A (en) * | 2009-11-30 | 2010-04-22 | Kyokuto Kobunshi Kk | Polymer obtained from betuline, and method of manufacturing the same |
| JP2013082929A (en) * | 2012-12-14 | 2013-05-09 | Masayoshi Tabata | Polymer obtained from betulin and method of producing the same |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020155177A1 (en) * | 2000-09-29 | 2002-10-24 | Krasutsky Pavel A. | Process for extracting compounds from plants |
| US6951833B2 (en) * | 2002-09-17 | 2005-10-04 | O'neil Deborah | Anti-microbial compositions |
| US20040057983A1 (en) | 2002-09-25 | 2004-03-25 | David Schmidt | Biomolecular wearable apparatus |
| WO2006031756A2 (en) | 2004-09-10 | 2006-03-23 | Cornell Research Foundation, Inc | Betulinol derivatives as anti-cancer agents |
| TW200628161A (en) * | 2004-11-12 | 2006-08-16 | Panacos Pharmaceuticals Inc | Novel betulin derivatives, preparation thereof and use thereof |
| US20080039428A1 (en) * | 2006-06-29 | 2008-02-14 | Panacos Pharmaceuticals, Inc. | Antiretroviral combination therapy |
| WO2008070347A2 (en) * | 2006-10-27 | 2008-06-12 | Regents Of The University Of Minnesota | Betulin-peptide conjugates |
| US8602961B2 (en) | 2008-05-15 | 2013-12-10 | Lifewave Products Llc | Apparatus and method of stimulating elevation of glutathione levels in a subject |
| PT2326766E (en) * | 2008-08-15 | 2012-08-01 | Hercules Inc | Pulping additives for a reduction of resin from kraft pulp |
| US9808011B2 (en) | 2014-12-15 | 2017-11-07 | Biovectra Inc. | Pentacyclic triterpene compounds and uses thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5034223A (en) * | 1986-10-09 | 1991-07-23 | Neorx Corporation | Methods for improved targeting of antibody, antibody fragments, hormones and other targeting agents, and conjugates thereof |
| US5639656A (en) * | 1994-03-31 | 1997-06-17 | Medical College Of Hampton Road | Antibodies reactive with biological markers of benign prostate hyperplasia |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4122166A (en) | 1976-08-17 | 1978-10-24 | Philips Roxane, Inc. | Injectable contraceptive and method |
| FR2378044A1 (en) | 1977-01-20 | 1978-08-18 | Sarget Lab | NEW PLANT EXTRACT FROM CHRYSANTHELLUM VARIETIES |
| US4671958A (en) | 1982-03-09 | 1987-06-09 | Cytogen Corporation | Antibody conjugates for the delivery of compounds to target sites |
| US5144010A (en) | 1984-08-27 | 1992-09-01 | The Trustees Of Columbia University In The City Of New York | Method of producing monoclonal auto-anti-idiotypic antibodies |
| NZ213974A (en) | 1985-10-29 | 1989-08-29 | Nz Ministry Agriculture & Fisheries | Milk protein/4-androstene-3,17-dione conjugates and veterinary compositions |
| US4801688A (en) | 1986-05-27 | 1989-01-31 | Eli Lilly And Company | Hydrazone immunoglobulin conjugates |
| US5064823A (en) | 1988-08-24 | 1991-11-12 | Research Triangle Institute | Pentacyclic triterpenoid compounds as topoisomerase inhibitors or cell differentiation inducers |
| US5162218A (en) | 1988-11-18 | 1992-11-10 | The Regents Of The University Of California | Conjugated polypeptides and methods for their preparation |
| US5013547A (en) | 1989-02-07 | 1991-05-07 | Erbamont, Inc. | Anticancer drug - antibody conjugates and method for preparing same |
| US5328840A (en) | 1989-08-15 | 1994-07-12 | The Research Foundation Of The State University Of New York | Method for preparing targeted carrier erythrocytes |
| US5166319A (en) | 1989-10-10 | 1992-11-24 | Brunswick Corporation | Interfacial condensation of bioactive compounds and the site-specific compounds and conjugates thereof |
| US5272253A (en) | 1991-07-01 | 1993-12-21 | Eli Lilly And Company | Cluster conjugates of drugs with antibodies |
| FR2683531B1 (en) | 1991-11-13 | 1993-12-31 | Rhone Poulenc Rorer Sa | NEW LUPANE DERIVATIVES, THEIR PREPARATION AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| DE4239429A1 (en) | 1992-11-24 | 1994-05-26 | Merck Patent Gmbh | Process for the preparation of immunoconjugates |
| US5643884A (en) * | 1993-08-09 | 1997-07-01 | Glycomed Incorporated | Lupane triterpenoid derivatives |
| US5658947A (en) * | 1995-03-21 | 1997-08-19 | Board Of Trustees Of The University Of Illinois | Method and composition for selectively inhibiting melanoma using betalinic acid |
| US5679828A (en) * | 1995-06-05 | 1997-10-21 | Biotech Research Labs, Inc. | Betulinic acid and dihydrobetulinic acid derivatives and uses therefor |
-
1998
- 1998-06-03 EP EP98926258A patent/EP0988311B9/en not_active Expired - Lifetime
- 1998-06-03 US US09/089,894 patent/US6890533B2/en not_active Expired - Fee Related
- 1998-06-03 WO PCT/US1998/011456 patent/WO1998055497A1/en not_active Ceased
- 1998-06-03 AU AU78135/98A patent/AU7813598A/en not_active Abandoned
- 1998-06-03 DE DE69825446T patent/DE69825446T2/en not_active Expired - Fee Related
- 1998-06-03 CA CA002293502A patent/CA2293502A1/en not_active Abandoned
-
2002
- 2002-08-02 US US10/212,576 patent/US20030036540A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5034223A (en) * | 1986-10-09 | 1991-07-23 | Neorx Corporation | Methods for improved targeting of antibody, antibody fragments, hormones and other targeting agents, and conjugates thereof |
| US5639656A (en) * | 1994-03-31 | 1997-06-17 | Medical College Of Hampton Road | Antibodies reactive with biological markers of benign prostate hyperplasia |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0988311A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008086759A3 (en) * | 2007-01-15 | 2009-02-26 | Univerzita Karlova V Praze Pri | Method of preparation and isolation of betulin diacetate from birch bark from paper mills and its optional processing to betulin |
| CZ301038B6 (en) * | 2007-01-15 | 2009-10-21 | Univerzita Karlova v Praze, Prírodovedecká fakulta | Process for preparing betulin diacetate from birch bark from paper mills |
| JP2010090385A (en) * | 2009-11-30 | 2010-04-22 | Kyokuto Kobunshi Kk | Polymer obtained from betuline, and method of manufacturing the same |
| JP2013082929A (en) * | 2012-12-14 | 2013-05-09 | Masayoshi Tabata | Polymer obtained from betulin and method of producing the same |
Also Published As
| Publication number | Publication date |
|---|---|
| AU7813598A (en) | 1998-12-21 |
| US20030036540A1 (en) | 2003-02-20 |
| DE69825446T2 (en) | 2005-09-29 |
| DE69825446D1 (en) | 2004-09-09 |
| CA2293502A1 (en) | 1998-12-10 |
| EP0988311B9 (en) | 2004-12-15 |
| EP0988311B1 (en) | 2004-08-04 |
| EP0988311A4 (en) | 2003-02-05 |
| US6890533B2 (en) | 2005-05-10 |
| EP0988311A1 (en) | 2000-03-29 |
| US20040001837A1 (en) | 2004-01-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA1336119C (en) | Vinca derivative conjugates containing a c-3 detergent chain | |
| US6890533B2 (en) | Betulinol derivatives | |
| CA2975383C (en) | Antibody drug conjugates comprising dolastatin derivatives | |
| US4315851A (en) | Pharmaceutical composition having antitumor activity | |
| US5006652A (en) | Intermediates for antibody-vinca drug conjugates | |
| US20100029758A1 (en) | Derivatives of isoflavones | |
| EP4277629A1 (en) | Camptothecine antibody-drug conjugates and methods of use thereof | |
| NO169958B (en) | PROCEDURE FOR PREPARING A CONJUGATE OF ANTITUMUM MEDICINE | |
| NO315162B1 (en) | Compounds for use as lysosomal, enzyme cleavable anti-tumor drug conjugates and process for their preparation | |
| WO2001079302A1 (en) | A complex between folic acid and polysaccharides, its preparation method and a pharmaceutical composition comprising said complex as active component | |
| CN106467575A (en) | Cysteine engineered Antibody-toxin conjugate | |
| JP2006514981A (en) | GP120-specific antigen, conjugate thereof; method for its preparation and use | |
| EP1473301A2 (en) | Betulinol derivatives | |
| KR970001532B1 (en) | Novel estradiol derivative chlorambucil conjugate, process for preparing the same and pharmaceutical composition | |
| Schneider et al. | Drug targeting in human cancer chemotherapy | |
| CN101084234A (en) | Betulinol derivatives as anti-cancer agents | |
| EP1802648A2 (en) | Betulinol derivatives as anti-cancer agents | |
| Hermentin et al. | Attachment of rhodosaminylanthracyclinone-type anthracyclines to the hinge-region of monoclonal antibodies | |
| US4354977A (en) | Phosphonothioate immunogens | |
| Zhang et al. | Synthesis of a targeting drug for antifibrosis of liver; a conjugate for delivering glycyrrhetin to hepatic stellate cells | |
| WO2002053165A1 (en) | Natural anti-angiogenic composition, method of preparation thereof and its application | |
| IL143591A (en) | Palladium-substituted bacteriochlorophyll derivatives, their preparation and pharmaceutical compositions comprising them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM GW HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2293502 Country of ref document: CA Ref country code: CA Ref document number: 2293502 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998926258 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: JP Ref document number: 1999502835 Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998926258 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998926258 Country of ref document: EP |