WO1999009014A1 - Hiv nuclear localization inhibitors - Google Patents
Hiv nuclear localization inhibitors Download PDFInfo
- Publication number
- WO1999009014A1 WO1999009014A1 PCT/US1998/016814 US9816814W WO9909014A1 WO 1999009014 A1 WO1999009014 A1 WO 1999009014A1 US 9816814 W US9816814 W US 9816814W WO 9909014 A1 WO9909014 A1 WO 9909014A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- hiv
- branched
- straight
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/47—One nitrogen atom and one oxygen or sulfur atom, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
Definitions
- the present invention provides a genus of compounds and pharmaceutical compositions that have anti-HIV anti-infective therapeutic activity and that inhibit nuclear localization of the HIV preintegration complex.
- HIV-1 human immunodeficiency virus-type 1
- Human immunodeficiency virus type-1 and other lentiviruses infect non-dividing terminally differentiated cells, such as primary macrophages (Gendelman et al., J. Virol. 58:67- 74, 1986: Gartner et al. Science 233:215-219. 1986). primary blood dendritic cells (Langhoff et al., Proc. Natl Acad. Sci. USA 88:998-8002. 1991), and epidermal Langerhan ' s cells (Ramazzotti et al., Immunology 85:94-98, 1995).
- HIV-1 preintegration complex PIC
- PIC HIV-1 preintegration complex
- the PIC is composed of the gag-derived matrix antigenprotein (MA), nucleocapsid protein (NC), reverse transcriptase (RT), integrase (IN), and viral protein "r” (vpr). Reverse transcription and production of the nascent cDNA is completed in context of the PIC in the cytoplasm of the infected target cell, prior to nuclear entry. It was recently shown (Gallay et al.. J. Virol. 70:1027-1032, 1996; and Popov et al., Proc. Natl. Acad. Sci. USA 93:11859-1 1864. 1996) that the PIC of HIV-1 associates with karyopherins.
- MA gag-derived matrix antigenprotein
- NC nucleocapsid protein
- RT reverse transcriptase
- integrase integrase
- vpr viral protein "r”
- Karyopherin ⁇ binds to target proteins via their nuclear localization sequence (NLS), while karyopherin ⁇ mediates docking of the karyopherin ⁇ - target protein complex to nuclear pore structures (Radu et al., Proc. Natl. Acad. Sci. USA 92:1769-1773, 1995: Moroianu et al, Proc. Natl. Acad. Sci USA 92:2008-2011, 1995: G ⁇ rlich et al., Nature (London) 377:246-248, 1995; Adam and Gerace, Cell 66:837-847, 1991 : G ⁇ rlich and Mattaj. Science 271:1513-1518. 1996; and Hurt. Cell 84:509-515. 1996).
- HIV-1 matrix antigen protein contains one defined (K 26 KKYK) and one putative (K 110 SKKK) NLS, and represents a major karyophilic structure within the PIC (Bukrinsky et al., Nature 365:666-669. 1993; von Schwedler et al., Proc. Natl. Acad. Sci. USA 91:6992-6996, 1994; Gallay et al.. J. Virol. 70:1027-1032. 1996; and Bukrinsky et al. Proc. Natl. Acad. Sci. USA 90:6125-6129, 1993).
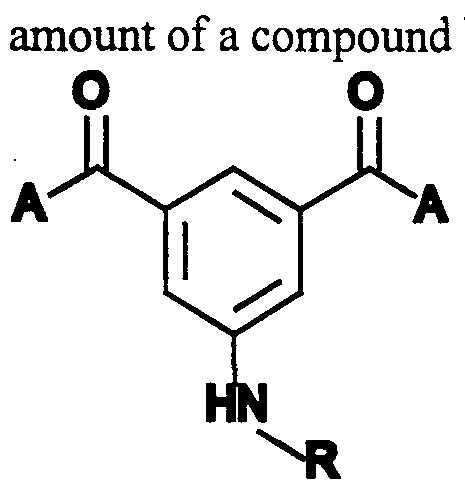
- the present invention provides a compound having the formula I:
- A is independently a straight or branched C 1-6 alkyl, a straight or branched C 2 - 6 alkenyl or a C ⁇ - 6 alkoxy;
- Y is -S-A wherein A is independently defined above: and Z and X are independently H, -(CH 2 ) n -NH wherein n is an integer from 0 to 6.
- n is an integer from 0 to 6.
- A is methyl
- X is -NH 2
- Z is H or amino.
- the present invention further provides a pharmaceutical composition comprising a compound from formula I in a pharmaceutically acceptable carrier.
- the invention further provides a process for synthesizing a compound of formula I, comprising the steps of:
- the substituted 6-halogen-methylmercaptopyrimidine is 4-amino-6-chloro-2- methylmercaptopyrimidine and the 3.5-dialkylaniline is 3,5-diacetylaniline.
- Figure 1 shows a graph comparing, in an assay of anti-HIV activity in macrophage cultures.
- anti-HIV therapeutic activity of inventive compound 53 (2-methylmercapto-4-amino- 6-(3',5 * -diacetylphenyl) amino-pyrimidine) with a structurally similar compound (“cni- h0294") that differs from compound 53 of the present invention by having a positive charge in the pyrimidine moiety and lacking a stipulated sulfur group substituted to the pyrimidine moiety.
- the assay measures reverse transcriptase activity in the infected macrophage culture supematants as a measure of virus production. These data can be directly correlated to efficacy treating HIV infection. These data show that inventive compound 53 was more efficacious that structurally similar compound 2.
- Figure 2 shows a graph comparing anti-HIV therapeutic activity, in an assay of anti- HIV activity in macrophage cultures, of inventive compound 62 (2-methylmercapto-6-(3',5'- diacetylphenyl) amino-pyrimidine) with a structurally similar compound (“cni-h0294 " ) that differs from compound 62 of the present invention by having a positive charge in the pyrimidine moiety and lacking a stipulated sulfur group substituted to the pyrimidine moiety.
- the assay measures reverse transcriptase activity in the infected macrophage culture supematants as a measure of vims production.
- Figure 3 shows a further analysis of therapeutic efficacy of compound 62 in activated (anti-CD3 and anti-CD28 monoclonal antibodies) peripheral blood mononuclear cell (PBMC) cultures infected with HIV-1 vims and treated with different concentrations of compound 62 ( ⁇ M).
- PBMC peripheral blood mononuclear cell
- the assay measures p24 as an index of viral replication and can be directly correlated to efficacy in treating HIV infection.
- Figure 4 shows that compound 62 also inhibited vims replication in PBMC from a HIV-1 infected individual when the PBMCs were activated in vitro with anti-CD3 mAb.
- PBMCs from a seropositive individual were collected and depleted of CD8 + T lymphocytes as described above. Cells were suspended in culture medium and activated with anti-CD3 mAb (l ⁇ g/ml). After 6-10 days vims production was evaluated by measuring levels of p24 in the culture supematants and comparing treated to untreated cultures.
- Figure 4 shows a dose- response relationship for compound 62 ("cni-h6297”) under the foregoing experimental conditions in this predictive assay of HIV anti-infective properties.
- A is independently a straight or branched C ⁇ - alkyl, a straight or branched C 2-6 alkenyl or a C ⁇ - 6 alkoxy;
- Y is -S-A wherein A is independently defined above: and Z and X are independently H, -(CH 2 ) n -NH wherein n is an integer from 0 to 6, a straight or branched C ⁇ -6 alkyl, a straight or branched C -6 alkenyl or a C ⁇ -6 alkoxy.
- A is methyl
- X is -NH
- Z is H or amino.
- the present invention further provides a pharmaceutical composition comprising a compound from formula I in a pharmaceutically acceptable carrier.
- the invention further provides a process for synthesizing a compound of formula I, comprising the steps of:
- the substituted 6-halogen-methylmercaptopyrimidine is 4-amino-6-chloro-2- methylmercaptopyrimidine and the 3,5-dialkylaniline is 3.5-diacetylaniline.
- the present invention provides an improvement in the design of small organic molecules that are effective for inhibiting HIV infection by creating an integral sulfur- containing substituent (see "Y" in formula I).
- the presence of this substituent, not disclosed or suggested in alternative HIV inhibitors, has provided compound characteristics of improved cellular absorption and. as a result, improved potency not disclosed or suggested by the structures of the alternative HIV preintegration complex inhibitors.
- the inventive compounds were active to inhibit receptor-mediated nuclear importation in the infection of peripheral blood mononuclear cell (PBMC) cultures.
- PBMC peripheral blood mononuclear cell
- the compound, cni- h0294. is thought to interact with the HIV-1 PIC (preintegration complex) by forming partial Schiff bases with adjacent lysine residues in the MA NLS (Popov et al., Proc. Natl. Acad. Sci. USA 93:11859-11864. 1996; Dubrovsky et al, Molec. Med. 1:217-230. 1995), contains a positive charge in its substituted pyrimidine moiety that may serve to limit its cellular bioavailability and it oral absorption characteristics.
- the exemplary compound, 2-methylmercapto-4-amino-6-(3 ' .5 ' -diacetylphenyl) amino- pyrimidine was synthesized. There are two methods for synthesizing compound 53. The first method starts by adding acetyl chloride (0.8 ml, 11 mmol) to 60 ml absolute ethanol. The mixture was stirred for 15 minutes to form a hydrochloric ethanol solution. Then. 1.75 g (10 mmol) of 4-amino-6-chloro-2-methylmercaptopyrimidine (Aldrich) and 3.5-diacetylaniline (1.8 g, 10 mmol) (Aldrich) were added in sequence.
- Illustrative compound 62 (2-methylmercapto-6-(3',5 * -diacetylphenyl) amino- pyrimidine) was synthesized by mixing 803 mg (5 mmol) 2-methylmercapto-4- chloropyrimidine and 886 mg (5 mmol) 3,5-diacetylaniline in 20 ml of water. In addition, 0.42 ml concentrated HC1 was added. This reaction mixture was heated at 90-100 °C for 4 hours, and then cooled down in an ice bath. The cooled mixture had 5 ml of IN KOH added to neutralize the acid pH from the HC1. The mixture was stirred for 10 minutes in an ice bath to form a precipitate.
- inventive pharmaceutical complex or inventive pharmaceutical combination can be administered to a patient either by itself (complex or combination) or in pharmaceutical compositions where it is mixed with suitable carriers and excipients.
- inventive compound or pharmaceutical composition can be administered parenterally, such as by intravenous injection or infusion, intraperitoneal injection, subcutaneous injection, or intramuscular injection.
- inventive compound or pharmaceutical composition can be administered orally or rectally through appropriate formulation with carriers and excipients to form tablets, pills, capsules, liquids, gels, syrups, slurries, suspensions and the like.
- inventive compound or pharmaceutical composition can be administered topically, such as by skin patch, to achieve consistent systemic levels of active agent.
- inventive compound or pharmaceutical composition is formulated into topical creams, skin or mucosal patches, liquids or gels suitable to topical application to skin or mucosal membrane surfaces.
- inventive compound or pharmaceutical composition can be administered by inhaler to the respiratory tract for local or systemic treatment of HIV infection.
- the dosage of the inventive compound or pharmaceutical composition suitable for use with the present invention can be determined by those skilled in the art from this disclosure.
- the pharmaceutical composition will contain an effective dosage (depending upon the route of administration and pharmacokinetics of the active agent) of the inventive compound or pharmaceutical composition and suitable pharmaceutical carriers and excipients. which are suitable for the particular route of administration of the formulation (i.e.. oral, parenteral, topical or by inhalation).
- the active compound is mixed into the pharmaceutical formulation by means of mixing, dissolving, granulating, dragee-making, emulsifying, encapsulating, entrapping or lyophilizing processes.
- the pharmaceutical formulations for parenteral administration include aqueous solutions of the active complex or combination in water- soluble form.
- suspensions of the active compound may be prepared as oily injection suspensions.
- Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes.
- Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxvmethyl cellulose, sorbitol. or dextran.
- the suspension may optionally contain stabilizers or agents to increase the solubility of the complex or combination to allow for more concentrated solutions.
- compositions for oral administration can be obtained by combining the active compound with solid excipients, such as sugars (e.g., lactose, sucrose, mannitol or sorbitol).
- solid excipients such as sugars (e.g., lactose, sucrose, mannitol or sorbitol).
- cellulose preparations e.g., starch, methyl cellulose, hydroxypropylmethyl cellulose, and sodium carboxymethyl cellulose
- gelatin e.g., methyl cellulose, hydroxypropylmethyl cellulose, and sodium carboxymethyl cellulose
- polyvinylpyrrolidone e.g., polyvinylpyrrolidone
- a desintegrating agent may be added, and a stabilizer may be added.
- Example 1 This example illustrates several in vitro experiments in predictive models of treatment of HIV infection to show the therapeutic utility of the inventive compounds. Macrophages, isolated and purified as described by Schumandtmanesalla et al. (Virology, 1997)) were infected with HIV-1 at a multiplicity adjusted according to p24 content (10 ng p24 per 10 6 cells). Compound (53 in Figure 1 or 62 in Figure 2) was added at different concentrations. In addition, positive control compound 2 (called “CNI-H294" in Figure 1 and "cni-h0294" in Figure 2) was added at the concentrations indicated. After a two hour incubation for viral adsorption, excess vimses were washed away, and the cells were incubated for additional indicated periods prior to analysis. RT, or reverse transcriptase activity, was measured by standard techniques in 7-11 days.
- Figure 1 shows a graph comparing anti-HIV therapeutic activity of inventive compound 53 with a structurally similar compound ("compound 2”) having a positive charge in the pyrimidine moiety and lacking a required sulfur group substituted to the pyrimidine moiety in an assay of anti-HIV activity in H9 cell cultures.
- the assay measures reverse transcriptase activity in the infected macrophage culture supematants as a measure of vims production.
- Figure 2 shows a graph comparing anti-HIV therapeutic activity of inventive compound 62 with a structurally similar compound (“cni-h0294") having a positive charge in the pyrimidine moiety and lacking a required sulfur group substituted to the pyrimidine moiety in an assay of anti-HIV activity in macrophage cultures.
- the assay measures reverse transcriptase activity in the infected H9 cell culture supematants as a measure of vims production.
- Example 2 This example illustrates that compound 62 inhibited HIV-1 vims replication in acutely infected PBMC cultures activated with anti-CD3 and anti-CD28 monoclonal antibodies (Figure 3).
- Peripheral blood mononuclear cells were isolated from an uninfected individual and depleted of CD8 + T lymphocytes using a CD8-specific monoclonal antibody, according to the procedure described by Smithgall et al., J. Immunol. 156:2324-2330, 1996. Briefly, the procedure substitutes separation with magnetic beads for complement-mediated lysis of antibody-bound cells. The remaining PBMC fractions were suspended in RPMI culture medium supplemented with 10% heat-inactivated human serum at 2x10 6 cells/200 ⁇ l.
- Cells were activated with anti-CD3 mAb (l ⁇ g/ml) together with anti-CD28 mAb (l ⁇ g/ml) in the presence of various concentrations of compound 62. This form of cell activation specifically targets D T lymphocytes in the population.
- Compound 62 also inhibited vims replication in PBMC from an HIV-1 infected individual when the PBMCs were activated in vitro with anti-CD3 mAb.
- PBMCs from a seropositive individual were collected and depleted of CD8 + T lymphocytes as described above. Cells were suspended in culture medium and activated with anti-CD3 mAb ( 1 ⁇ g/ml). After 6-10 days vims production was evaluated by measuring levels of p24 in the culture supematants and comparing treated to untreated cultures.
- Figure 4 shows a dose-response relationship for compound 62 ("CNI-H6297”) under the foregoing experimental conditions in this predictive assay of HIV anti-infective properties.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Tropical Medicine & Parasitology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- AIDS & HIV (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description


Claims


Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP98940874A EP1012146B1 (en) | 1997-08-15 | 1998-08-13 | Hiv nuclear localization inhibitors |
| AU89054/98A AU758464B2 (en) | 1997-08-15 | 1998-08-13 | HIV nuclear localization inhibitors |
| DE69816835T DE69816835T2 (en) | 1997-08-15 | 1998-08-13 | INHIBITORS OF NUCLEAR LOCALIZATION OF HIV |
| JP2000509697A JP4475487B2 (en) | 1997-08-15 | 1998-08-13 | Nuclear localization inhibitor of HIV |
| CA002300424A CA2300424C (en) | 1997-08-15 | 1998-08-13 | Hiv nuclear localization inhibitors |
| AT98940874T ATE245983T1 (en) | 1997-08-15 | 1998-08-13 | INHIBITORS OF NUCLEAR LOCALIZATION OF HIV |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/912,076 | 1997-08-15 | ||
| US08/912,076 US5808068A (en) | 1997-08-15 | 1997-08-15 | HIV nuclear localization inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999009014A1 true WO1999009014A1 (en) | 1999-02-25 |
Family
ID=25431342
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/016814 Ceased WO1999009014A1 (en) | 1997-08-15 | 1998-08-13 | Hiv nuclear localization inhibitors |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US5808068A (en) |
| EP (1) | EP1012146B1 (en) |
| JP (1) | JP4475487B2 (en) |
| AT (1) | ATE245983T1 (en) |
| AU (1) | AU758464B2 (en) |
| CA (1) | CA2300424C (en) |
| DE (1) | DE69816835T2 (en) |
| WO (1) | WO1999009014A1 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6297253B1 (en) * | 1996-10-15 | 2001-10-02 | The Picower Institute For Medical Research | Compounds and methods of use to treat infectious diseases |
| CA2686618A1 (en) * | 2003-07-17 | 2005-01-17 | At&T Corp. | Method and apparatus for windowing in entropy encoding |
| US20050203150A1 (en) * | 2004-03-09 | 2005-09-15 | Haffar Omar K. | Compounds, compositions and methods for inhibiting or treating HIV-1 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0673022A (en) * | 1992-08-27 | 1994-03-15 | Kumiai Chem Ind Co Ltd | Pyrimidine or triazine derivative and herbicide |
-
1997
- 1997-08-15 US US08/912,076 patent/US5808068A/en not_active Expired - Lifetime
-
1998
- 1998-08-13 WO PCT/US1998/016814 patent/WO1999009014A1/en not_active Ceased
- 1998-08-13 AT AT98940874T patent/ATE245983T1/en not_active IP Right Cessation
- 1998-08-13 JP JP2000509697A patent/JP4475487B2/en not_active Expired - Fee Related
- 1998-08-13 CA CA002300424A patent/CA2300424C/en not_active Expired - Fee Related
- 1998-08-13 DE DE69816835T patent/DE69816835T2/en not_active Expired - Fee Related
- 1998-08-13 AU AU89054/98A patent/AU758464B2/en not_active Ceased
- 1998-08-13 EP EP98940874A patent/EP1012146B1/en not_active Expired - Lifetime
Non-Patent Citations (2)
| Title |
|---|
| Chemical Abstracts Service (C A S); 1 January 1900 (1900-01-01), XP002914913, Database accession no. 123-25160 * |
| POPOV S.,: "CRITICAL ROLE OF REVERSE TRANSCRIPTASE IN THE INHIBITORY MECHANISM OF CNI-H0294 ON HIV-1 NUCLEAR TRANSLOCATION.", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, NATIONAL ACADEMY OF SCIENCES, US, vol. 93., 15 October 1996 (1996-10-15), US, pages 11859 - 11864., XP002914912, ISSN: 0027-8424, DOI: 10.1073/pnas.93.21.11859 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4475487B2 (en) | 2010-06-09 |
| CA2300424C (en) | 2009-04-07 |
| EP1012146A1 (en) | 2000-06-28 |
| EP1012146A4 (en) | 2002-12-04 |
| EP1012146B1 (en) | 2003-07-30 |
| DE69816835T2 (en) | 2004-04-22 |
| DE69816835D1 (en) | 2003-09-04 |
| ATE245983T1 (en) | 2003-08-15 |
| JP2001515069A (en) | 2001-09-18 |
| US5808068A (en) | 1998-09-15 |
| AU758464B2 (en) | 2003-03-20 |
| AU8905498A (en) | 1999-03-08 |
| CA2300424A1 (en) | 1999-02-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103402516B (en) | Compound, composition and application method as antiviral drugs | |
| CN101355939A (en) | Composition and synthesis of new substances for inhibiting HIV replication | |
| JPH04300834A (en) | Synergistic action of hiv reverse transcriptase inhibitor | |
| JPH04230280A (en) | Aminobenzodiazepine and drug containing same | |
| US5703086A (en) | Compounds and methods of use to derivatize neighboring lysine residues in proteins under physiological conditions | |
| AU759210B2 (en) | HIV matrix protein tyrosine position 29 pocket binders | |
| EP1012146B1 (en) | Hiv nuclear localization inhibitors | |
| JP2021500401A (en) | Wide spectrum antiviral compositions and methods | |
| US6001871A (en) | Hypoestoxides, derivatives and agonists thereof for use as antiviral agents | |
| JPH0714875B2 (en) | Virus-inhibiting pharmaceutical composition | |
| EP2196453A1 (en) | Novel substituted aryl derivatives, their process of preparation and their therapeutical uses as anti-HIV agents | |
| EP0843663B9 (en) | 4-aryl-thio-pyridin-2(1h)-ones, drugs containing same, and uses thereof for treating hiv-related diseases | |
| US20170267726A1 (en) | Non-immunosuppressive cyclosporin derivatives as antiviral agents | |
| US5574040A (en) | Pyrimidine compounds and methods of use to derivatize neighboring lysine residues in proteins under physiologic conditions | |
| Michne et al. | Keto/enol epoxy steroids: A new structural class of hiv-1 tat inhibitors | |
| WO2003049695A2 (en) | Compounds to treat hiv infection and aids | |
| JP3728028B2 (en) | Pyrimidine derivatives | |
| WO2009145839A1 (en) | Polyamine compounds that bind tar rna of hiv and methods of treating viral disorders | |
| EP2163553A1 (en) | Arene connected polyamine macroring derivatives, preparation methods and pharmaceutical uses thereof | |
| US6414004B1 (en) | 3-substituted 5-aryl-4-isoxazolecarbonitriles having antiviral activity | |
| CN115403625A (en) | S-DACOs non-nucleoside reverse transcriptase inhibitor derivative and application thereof | |
| US20060264475A1 (en) | Compounds, compositions and methods for inhibiting or treating hiv-1 | |
| Kasralikar et al. | Month 2018 Design and Synthesis of Novel 1, 2, 3-triazolyl-pyrimidinone Hybrids as Potential Anti-HIV-1 NNRT Inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA IL JP NZ |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2300424 Country of ref document: CA Ref country code: CA Ref document number: 2300424 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 509697 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 89054/98 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998940874 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998940874 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 89054/98 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998940874 Country of ref document: EP |