WO1999012931A1 - Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase - Google Patents
Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase Download PDFInfo
- Publication number
- WO1999012931A1 WO1999012931A1 PCT/EP1998/005604 EP9805604W WO9912931A1 WO 1999012931 A1 WO1999012931 A1 WO 1999012931A1 EP 9805604 W EP9805604 W EP 9805604W WO 9912931 A1 WO9912931 A1 WO 9912931A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- isopropyl
- pyrrolo
- hexahydro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C=C[C@@](CCN(*)P)N(C(c1ccccc11)=O)C1=O Chemical compound *C=C[C@@](CCN(*)P)N(C(c1ccccc11)=O)C1=O 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/08—Plasma substitutes; Perfusion solutions; Dialytics or haemodialytics; Drugs for electrolytic or acid-base disorders, e.g. hypovolemic shock
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to therapeutically active bicyclic compounds, processes for their manufacture, pharmaceutical formulations containing them and their use in chemotherapy.
- novel bicyclic compounds which are effective in treating inflammatory diseases.
- Inflammation is a primary response to tissue injury or microbial invasion and is characterised by circulating leukocytes binding to and extravasation through vascular endothelium. Circulating leukocytes include neutrophils, eosinophils, basophils, monocytes and lymphocytes. Different forms of inflammation involve different types of infiltrating leukocytes.
- the inflammatory process can be triggered in a number of ways, including by infection, tissue damage and autoimmune reactions.
- neutrophils move from the bloodstream into the tissue at the site of tissue lesion.
- the neutrophils contain large numbers of different intracellular granules and when activated at the site of inflammation the contents of these granules are secreted into the tissue.
- the different granules contain a variety of enzymes and other proteins, many of which have antibacterial properties.
- neutrophil elastase One of the enzymes found in the azurophilic granules is neutrophil elastase.
- Neutrophil elastase has a wide spectrum of activities in the body. For example, within the lung the enzyme increases mucus production and changes the cellular composition of the epithelium The enzyme also causes vascular permeability changes within the microcirculation of many tissues and it is a potent destructive agent against a number of connective tissue components.
- elastase activity has been implicated in the pathogenesis of a number of disease states including inflammatory diseases of the airways, the joints and the skin.
- the enzyme is also responsible for some or most of the symptoms of acute respiratory distress syndrome (ARDS) and other acute inflammatory states brought about by trauma and/or sepsis.
- ARDS acute respiratory distress syndrome
- R represents C 1-6 alkyl
- R 2 represents C ⁇ alkyl or C ⁇ alkenyl
- a represents 1 or 2
- R 3 represents C 1-8 alkyl or (CH 2 ) n Ar
- n represents 1 or 2;
- Ar represents optionally substituted phenyl; and salts and solvates thereof (hereinafter “compounds of the invention”).
- Formula (I) shows the relative stereochemistry of the chiral centres.
- the invention embraces compounds of the invention in racemic form as well as in a form in which one enantiomer predominates or is present exclusively.
- the present invention also covers the physiologically acceptable salts of the compounds of formula (I).
- physiologically acceptable salts of the compounds of formula (I) include inorganic and organic acid salts such as hydrochloride and tartrate.
- Examples of groups by with the phenyl of Ar may be substituted include halogen, C 1-4 alkyl, C ⁇ alkoxy, nitro, hydroxy, amine (optionally substituted by one or two C 1-4 alkyi groups), CF 3 , COOH, COOC ⁇ alkyl and CONH 2 (optionally substituted by one or two C ⁇ alkyl groups).
- alkyl includes branched as well as straight chain alkyl and may also include cycloalkyl when 3 or more carbon atoms are present.
- Suitable R 1 alkyl groups include methyl, ethyl and propyl.
- Suitable R 3 C 1-8 alkyl groups include n-butyl, cyclopropyl and t-butyl.
- R 1 to represent methyl or ethyl, especially methyl.
- R 2 to represent isopropyl or propyl, especially isopropyl.
- R 3 to represent C ⁇ alkyl or CH 2 Ar.
- Preferred Ar includes phenyl and phenyl substituted by one or more halogen groups.
- Intracellular elastase is prepared from neutrophils collected by lavage and from femoral bone marrow . This is achieved by sonication of the neutrophils and centrifugation to yield intracellular granules. These are disrupted by freeze/thawing and sonication. Elastase and myeloperoxidase assays are then performed on these samples to assess the efficacy of the compounds and to normalise for neutrophil recovery.
- the wash is with saline (300 ⁇ l), followed by centrifugation at 100g for 10 minutes at 4°C. Pellets are washed twice more, before resuspension of the final cell pellet in buffer (200 ⁇ l of 100mM Tris, 300mM NaCI, 1% (w/v) HTAB, pH 8.6). Samples are stored at -20 °C. After freeze-thawing of the samples four times, elastase activity is determined by a colorimetric assay in 50mM Tris, 150mM NaCI, 0.6mM MeO-Succ-Ala-Ala-Ala-Pro-Val-pNA at pH 8.6, measuring the rate of increase in absorbance at 405nm.
- the compounds of the invention are of potential therapeutic benefit in the treatment and amelioration of symptoms of diseases where elastase activity is implicated.
- diseases particularly include bronchitis, including chronic bronchitis. Also any chronic obstructive pulmonary disease (COPD).
- COPD chronic obstructive pulmonary disease
- Examples of disease states in which the compounds of the invention have potentially beneficial effects include inflammatory diseases of the respiratory tract such as bronchitis (including chronic bronchitis), bronchiectasis, asthma and hyper-reactivity states of the lung, acute respiratory distress syndrome and septic shock, inflammatory or destructive conditions of the lung such as emphysema and cystic fibrosis and inflammatory or destructive conditions of external tissue such as skin diseases (e.g. lupus and psoriasis) and periodontal disease including gingivitis.
- inflammatory diseases of the respiratory tract such as bronchitis (including chronic bronchitis), bronchiectasis, asthma and hyper-reactivity states of the lung, acute respiratory distress syndrome and septic shock, inflammatory or destructive conditions of the lung such as emphysema and cystic fibrosis and inflammatory or destructive conditions of external tissue such as skin diseases (e.g. lupus and psoriasis) and periodontal disease including ging
- cardiovascular diseases such as myocardial infarction and stroke, peripheral vascular disease including intermittent claudication, atherosclerosis, reperfusion injury, cardiovascular changes occurring during cardiopulmonary bypass surgery and septicemia.
- Compounds of the invention may also be useful in the treatment of connective tissue disorders such as rheumatoid arthritis, osteoarthritis and spondylitis and inflammatory conditions of the kidney such as glomerulonephritis.
- leukemias including acute myelogenous leukemia, acute myelomonocytic leukemia and the chronic monocytic leukemias and in prevention or inhibition of metastasis of solid tumours e.g. lung, breast, prostate and stomach cancers and melanomas.
- a particular aspect of the present invention is the use of compounds of formula (I) in the treatment of chronic bronchitis.
- Chronic bronchitis is a condition which results from the exposure of the airway surface to noxious chemicals or agents or is secondary to another disease. The symptoms of the condition are caused by the excessive secretion of mucus onto the surface of the airways. This excess mucus cannot be cleared effectively and the result is reduced gas exchange within the lungs resulting in laboured breathing and hypoxemia, recurrent microbial infections and persistent cough associated with the expectoration of mucoid material.
- the proposed mechanism for the excessive secretion of mucus involves the recruitment of neutrophils into the airways following the exposure of the epithelium to irritant materials; the neutrophils secrete elastase onto the surface of the airways and the enzyme brings about both an increase in the amount of mucus secreted onto the airway surfaces and a dramatic change in the cellular composition of the airway epithelium.
- Inhibition of elastase activity by the administration of compounds of this invention is therefore an approach to the treatment of chronic bronchitis.
- Reduced lung function in COPD eg in chronic bronchitics with airflow obstruction
- an elastase inhibitor will improve lung function.
- compounds of the invention are useful in human or veterinary medicine, in particular as inhibitors of the enzyme neutrophil elastase.
- a compound of formula (I) or a physiologically acceptable salt or solvate thereof for use in human or veterinary medicine, particularly in the treatment of conditions where elastase activity is implicated such as chronic bronchitis.
- references herein to treatment extend to prophylaxis as well as the treatment of established conditions.
- a compound of formula (I) or a physiologically acceptable salt or solvate thereof for the manufacture of a medicament for the treatment of conditions where elastase activity is implicated, particularly in chronic bronchitis.
- a method for the treatment of a human or animal subject with a condition caused or mediated by elastase activity comprises administering to said human or animal subject an effective amount of a compound of formula (I) or a physiologically acceptable salt or solvate thereof.
- the compounds according to the invention may be formulated for administration in any convenient way, and the invention therefore also includes within its scope pharmaceutical compositions for use in therapy, comprising a compound of formula (I) or a physiologically acceptable salt or solvate thereof in admixture with one or more physiologically acceptable diluents or carriers.
- a process for preparation of such a pharmaceutical composition which comprises mixing the ingredients.
- the compounds according to the invention may, for example, be formulated for oral, buccal, parenteral, topical or rectal administration.
- Tablets and capsules for oral administration may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, mucilage of starch or polyvinyl pyrrolidone; fillers, for example, lactose, microcrystalline cellulose, sugar, maize- starch, calcium phosphate or sorbitol; lubricants, for example, magnesium stearate, stearic acid, talc, polyethylene glycol or silica; disinteg rants, for example, potato starch, croscarmellose sodium or sodium starch glycollate; or wetting agents such as sodium lauryl sulphate.
- the tablets may be coated according to methods well known in the art.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for constitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents, for example, sorbitol syrup, methyl cellulose, glucose/sugar syrup, gelatin, hydroxymethyl cellulose, carboxymethyl cellulose, aluminium stearate gel or hydrogenated edible fats; emulsifying agents, for example, lecithin, sorbitan mono-oleate or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters, propylene glycol or ethyl alcohol; or preservatives, for example, methyl or propyl p_- hydroxybenzoates or sorbic acid.
- the preparations may also contain buffer salts, flavouring, colouring and/or sweeten
- the compounds may also be formulated as suppositories, e.g. containing conventional suppository bases such as cocoa butter or other glycerides.
- the compounds according to the invention may also be formulated for parenteral administration by bolus injection or continuous infusion and may be presented in unit dose form, for instance as ampoules, vials, small volume infusions or pre-filled syringes, or in multi-dose containers with an added preservative.
- the compositions may take such forms as solutions, suspensions, or emulsions in aqueous or non-aqueous vehicles, and may contain formulatory agents such as anti-oxidants, buffers, antimicrobial agents and/or toxicity adjusting agents.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g. sterile, pyrogen-free water, before use.
- the dry solid presentation may be prepared by filling a sterile powder aseptically into individual sterile containers or by filling a sterile solution aseptically into each container and freeze-drying.
- topical administration as used herein, we include administration by insufflation and inhalation.
- preparation for topical administration include ointments, creams, lotions, powders, pessaries, sprays, aerosols, capsules or cartridges for use in an inhaler or insufflator or drops (e.g. eye or nose drops).
- Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents and/or solvents.
- bases may thus, for example, include water and/or an oil such as liquid paraffin or a vegetable oil such as arachis oil or castor oil or a solvent such as a polyethylene glycol.
- Thickening agents which may be used include soft paraffin, aluminium stearate, cetostearyl alcohol, polyethylene giycols, microcrystalline wax and beeswax.
- Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents or thickening agents.
- Powders for external application may be formed with the aid of any suitable powder base, for example, talc, lactose or starch. Drops may be formulated with an aqueous or non-aqueous base also comprising one or more dispersing agents, solubilising agents or suspending agents.
- Spray compositions may be formulated, for example, as aqueous solutions or suspensions or as aerosols delivered from pressurised packs, with the use of a suitable propellant, e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafiuoroethane, 1 ,1 ,1 ,2,3,3,3-heptafluoropropane, 1 ,1 ,1 ,2- tetrafluorethane, carbon dioxide or other suitable gas.
- a suitable propellant e.g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafiuoroethane, 1 ,1 ,1 ,2,3,3,3-heptafluoropropane, 1 ,1 ,1 ,2- tetrafluorethane, carbon dioxide or other suitable gas.
- Capsules and cartridges for use in an inhaler or insufflator, of for example gelatin may be formulated containing a powder mix of a compound of the invention and a suitable powder base such as lactose or starch.
- compositions according to the invention may also be used in combination with other therapeutic agents, for example anti-inflammatory agents such as corticosteroids or NSAIDs, bronchodilators such as beta-2 adrenergic agonists and xanthines (e.g. theophylline), mucolytic agents, anti- muscarinics, anti-leukotrienes, inhibitors of cell adhesion (e.g. ICAM antagonists), anti-oxidants (eg N-acetylcysteine), lung surfactants and/or antimicrobial and anti-viral agents.
- anti-inflammatory agents such as corticosteroids or NSAIDs
- bronchodilators such as beta-2 adrenergic agonists and xanthines (e.g. theophylline)
- mucolytic agents e.g. theophylline
- anti-muscarinics e.g. theophylline
- anti-leukotrienes e.g. the
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a physiologically acceptable salt or solvate thereof together with another therapeutically active agent.
- compositions comprising a combination as defined above together with a pharmaceutically acceptable carrier thereof represent a further aspect of the invention.
- the compound of the invention may conveniently be administered in amounts of, for example, 0.01 to 50mg/kg body weight, suitably 0.05 to 25mg/kg body weight orally, one or more times a day.
- the precise dose will of course depend on the age and condition of the patient, the particular route of administration chosen, and the disease being treated.
- the compound is preferably administered orally for the treatment of bronchitis. Other routes of administration may be needed for other indications, for instance i.v. for ARDS.
- the compounds of the invention have useful duration of action.
- the compounds of formula (I) and salts and solvates thereof may be prepared by the methodology described hereinafter, constituting a further aspect of this invention.
- a process according to the invention for preparing a compound of formula (I) comprises: (i) condensation of a compound of formula (II):
- This condensation reaction is suitably carried out in the presence of a coupling agent such as EDC, HATU or TBTU, preferably also in the presence of HOBT, and a solvent such as dichloromethane, DMF, MeCN or tetrahydrofuran at a temperature of suitably between O °C and ambient.
- a coupling agent such as EDC, HATU or TBTU
- HOBT hydroxybenzoic acid
- a solvent such as dichloromethane, DMF, MeCN or tetrahydrofuran
- the sulphonylation reaction is suitably carried out in the presence of LHMDS or NaH, in a solvent such as tetrahydrofuran at a temperature of suitably between -78 ° C and O °C.
- the cyclisation reaction is suitably carried out in the presence of 2-chloro-1- methylpyridinium iodide or EDC in a solvent such as dichloromethane, at a temperature of suitably 0 °C - reflux.
- Acid derivatives including acid halides (eg acid chlorides) (preferably in the presence of a base), anhydrides, thioesters may also be used in this reaction.
- This oxidation reaction may be carried out in conventional manner such as by peracid oxidation.
- Preferred leaving groups include halogen (such as chlorine, bromine or iodine), mesylate and tosylate.
- the reaction may be performed by combining the reactants optionally in the presence of a base such as triethylamine or potassium carbonate in an inert aprotic solvent such as DMF or MeCN.
- Purification of a single enantiomer may be achieved by conventional methods such as chiral chromatography (e.g. chiral HPLC) and crystallisation using a homochiral acid (e.g. tartaric acid).
- chiral chromatography e.g. chiral HPLC
- crystallisation using a homochiral acid e.g. tartaric acid
- Physiologically acceptable acid salts of the compound of formula (I) such as the hydrochloride or tartrate may be prepared by treating a basic compound of formula (I) with the desired acid.
- the compound of formula (VI) may first be treated with a corresponding aldehyde, followed by reduction with a reducing agent such as triacetoxyborohydride typically in the presence of acetic acid in an inert solvent such as DCM.
- a reducing agent such as triacetoxyborohydride typically in the presence of acetic acid in an inert solvent such as DCM.
- BOC may be performed by reacting with (BOC) 2 O in the presence of base (e.g.
- This conversion may be performed on treatment with ammonium bicarbonate in the presence of a suitable solvent such as pyridine/DMF and in the presence of
- CBZ may be performed by reaction with nBuLi followed by CBZ-CI in the presence of an inert solvent such as THF below -50 °C.
- This reaction may be performed by treatment with RX where RX is a compound
- R e.g. Mel, benzyl iodide or Me 2 SO 4
- a suitable solvent e.g. propanone or acetonitrile.
- R will represent alkyl or aralkyi and X will represent halide especially iodide or sulphate. Protection of the amide is convenient, although not essential, for this reaction.
- Step (e) This ring closure reaction may be performed by treatment with Dowex 2X8 400 mesh OH " resin in a suitable solvent, e.g. MeCN. Alternatively, the ring closure may be performed by treatment with potassium carbonate in a suitable solvent e.g. MeCN. Step (f)
- Deprotection may be performed in a conventional manner, for example, a BOC protecting group may be removed by treatment with HCI, e.g. in dioxan.
- This reaction may be performed by treatment with a trifluoroacetic acid alkyl ester (e.g. the methyl ester) or trifluoroacetic anhydride in the presence of a suitable base e.g. N-methylmorpholine.
- a trifluoroacetic acid alkyl ester e.g. the methyl ester
- a suitable base e.g. N-methylmorpholine.
- This conversion will take place on treating the compound of formula (XIII) with a reducing agent e.g. lithium borohydride, followed by treatment with concentrated sulphuric acid in the presence of an alkyl alcohol e.g. ethanol solvent.
- a reducing agent e.g. lithium borohydride
- concentrated sulphuric acid in the presence of an alkyl alcohol e.g. ethanol solvent.
- the reaction of compounds of formula (XIV) and (XV) takes place in the presence of a Lewis acid e.g. boron trifluoride dietherate and an inert solvent e.g. dichloromethane.
- the group "alkyl" in Oalkyl and OSi(alkyl) 3 generally represents C 1-6 alkyl.
- suitable alkyl groups in the silyl alkyl moiety include methyl, isopropyl and t-butyl.
- Preferred Oalkyl is OEt and preferred OSi(alkyl) 3 is OSi(i-Pr) 3 or OSi(Me) 2 (t-Bu).
- the use of variants of compounds of formula (XV) in which Oalkyl is replaced by OSi(alkyl) 3 is also envisaged.
- Compounds of formula (XV) may be prepared by treatment of the corresponding carboxylic acid ester (R 2 CH 2 COOEt or another alkyl ester, which compounds are either known or may be prepared by known methods) with a strong base (eg LHMDS) followed by a trialkylsilylchloride (such as trimethylsilylchloride) or a trialkylsilyltriflate.
- a strong base eg LHMDS
- a trialkylsilylchloride such as trimethylsilylchloride
- a trialkylsilyltriflate Typically the reaction will be performed at low temperature (less than 0°C) in an inert solvent (such as THF) in the presence of DMPU.
- Step G This deprotection reaction will take place on treatment with base, such as potassium carbonate.
- This ring closure reaction may be performed on treatment with an alkyl Grignard reagent (eg t-butylmagnesium choride) in an inert solvent such as THF in the presence of tetramethylethylenediamine at a temperature of -20°C to 25°C.
- an alkyl Grignard reagent eg t-butylmagnesium choride
- an inert solvent such as THF
- Step (m) This is an N-deprotection reaction, which can suitably be carried out in conventional manner.
- P 1 is CBZ
- it is suitably carried out by hydrogenation over Pd (OH) 2 catalyst in solvents such as ethyl acetate or THF.
- R 13 is suitably a C 1-6 alkyl group, preferably methyl.
- the cyclisation reaction will take place on stirring in water with Dowex 2X8 (preferably 400 mesh).
- the TFA protected amine is formed by treating the compound of formula (XXII) with methyl trifluoroacetate in a polar protic solvent, eg MeOH. Step (d)
- Suitable protecting groups P. include CBZ.
- the compound of formula (XXIII) may be treated with a strong base such as LHMDS or nBuLi in an inert solvent such as THF, followed by treatment with CBZ-CI.
- the compounds of formula (XXIV) are either known compounds or may be made in analogous manner to known compounds.
- P ⁇ is an N-protecting group, preferably CBZ.
- Step (a) is a further N-protection reaction.
- P 2 in formula (XXV) is a different N-protecting group, preferably BOC. When P 2 is BOC, the reaction is suitably carried out using BOC 2 O.
- reaction is carried out in the presence of a base such as triethylamine or 4-dimethylaminopyridine in a solvent such as ethyl acetate, at temperature of suitably 0°-25° C.
- a base such as triethylamine or 4-dimethylaminopyridine
- a solvent such as ethyl acetate
- Step (b) This conversion is suitably carried out with pyridinium p-toluenesulfonate, in a solvent such as acetone/water, at a temperature suitably between 25°-75° C.
- Step (c) This is a condensation rearrangement reaction suitably carried out using a 2- phenylsulfinyl acetic acid ester (PhSOCH 2 CO 2 R 13 ) and piperidine, in a solvent such as acetonitriie, suitably at ambient temperature.
- a 2- phenylsulfinyl acetic acid ester PhSOCH 2 CO 2 R 13
- piperidine a solvent such as acetonitriie
- R 13 is suitably a C 1-6 alkyl group, preferably methyl.
- THF at a temperature of suitably 0 °-40 °C.
- This is a deprotection reaction, preferably using a strong acid such as TFA in a solvent such as DCM, at a temperature of suitably 0 °-40 °C.
- R 13 is suitably C 1-6 alkyl, preferably ethyl.
- a cyclisation reaction suitably carried out as an intramolecular Michael reaction.
- NaH is used, in a solvent such as THF, at a temperature such as 0 ° - 25 °C.
- N-deprotection and re-protection two reactions occur: N-deprotection and re-protection.
- the phthalimido group is removed suitably with hydrazine hydrate in a solvent such as ethanol at a temperature between 0 °C and reflux.
- Protecting group P 3 is incorporated in a conventional manner. When P 3 is BOC, this is suitably achieved with BOC 2 O.
- the R 2 side chain may be introduced by alkylation, using as reactant R 2 -Y, wherein Y is a reactive group such as bromo or iodo.
- R 2 -Y wherein Y is a reactive group such as bromo or iodo.
- the reaction is carried out using a base, preferably a strong base such as LHMDS.
- LHMDS suitably a cosolvent DMPU in THF is used.
- Suitable reaction temperatures are -78 ° to 50 °C. Under these conditions the reaction generally takes place with good stereochemical control.
- the former is carried out in a conventional manner, for example by using KOH in aqueous ethanol, at a temperature of suitably 25°-80°C .
- the latter is carried out in a conventional manner, for example by using HCI in dioxan, at a temperature of suitably 0 °-50 °C or, if the protecting group is trifluoroacetate by treatment with base,
- a suitable base such as N, N-diisopropyl ethylamine
- a solvent such as dichloromethane
- Step (a) The compounds of formula (XXXIV) are either known compounds or may be prepared in analogous manner to known compounds.
- P 3 is a protecting group as discussed above, and is suitably BOC.
- the reaction is suitably carried out using PIFA (phenyl iodosylbis(trifluoroacetate)) and a base such as pyridine in an aqueous solvent, such as aqueous THF, dioxan or acetonitrile. This is the method of Stansfield, C.F. Organic Preparations and Procedures Int., 1990, 22(5), 593-603.
- Step (b) P 1 is a protecting group eg CBZ.
- This protection reaction may be carried out in a conventional manner. For instance it is suitably carried out in a water miscible solvent such as THF, DMF or dioxan using N-(benzyloxycarbonyloxy) succinamide, benzyloxycarbonyl chloride, or any suitable source of the benzyloxycarbonyl group, with pH adjustment to alkaline with sodium carbonate.
- the compound of formula (XXXVI) can be prepared in conventional manner from diaminobutyric acid.
- This reaction is suitably carried out in two stages.
- the first stage involves reacting the compound of formula (XXXVI) at reduced temperature with N- methylmorphoiine and then an alkyl chloroformate such as ethyl chloroformate, in an organic solvent such as DCM, dioxan or THF.
- the second stage the product is reduced, suitably with sodium borohydride at a reduced temperature, such as -20 ° to 10 °C, in a solvent such as THF.
- This oxidation reaction may be carried out in any suitable manner, for instance using oxalyl chloride in DMSO and DCM under nitrogen at reduced temperature, such as -30 ° to -70 °C, followed by triethylamine.
- the intermediate (XXXVIII) suitably is not isolated.
- a phosphonate in a Wadsworth-Emmons reaction.
- Step (f) This Michael addition reaction is suitably carried out using LHMDS or other suitable strong base in a suitable organic solvent such as THF, ether or toluene, and preferably a complexing agent such as TMEDA is also present.
- a suitable organic solvent such as THF, ether or toluene, and preferably a complexing agent such as TMEDA is also present.
- the intermediate compounds of formula (III) may be prepared by reacting a deprotected compound of formula (XVIII) from Scheme 1 with a compound of formula (XXXX)
- an acid derivative thereof such as an acid halide (e.g. the acid choride) in the manner described above in relation to main process (i) above.
- an acid halide e.g. the acid choride
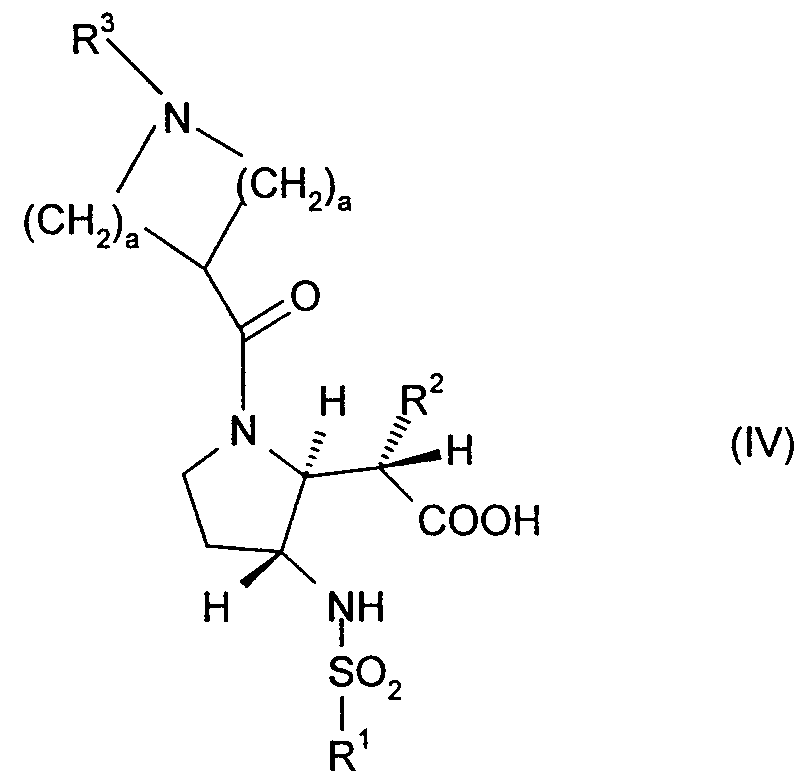
- the intermediate compounds of formula (IV) may be prepared from a compound of formula (XVII) (with the primary amine suitably protected) in an analogous manner to that described above for preparing a compound of formula (III) from a compound of formula (XVIII) together with main process (ii) above.
- Compounds of formula (V) wherein X a represents S may be prepared by reaction of a corresponding compound of formula (III) with a compound of formula R 1 SSR 1 under standard conditions for nucleophilic displacement.
- Compounds of formula (V) wherein X a represents SO may be prepared by peracid oxidation of a corresponding compound wherein X a represents S.
- Compounds of formula (VI) may be prepared by a process comprising reacting a compound of formula (II) with a compound of formula (XXXXA)
- compounds of formula (XXXX) or (XXXXA) may be prepared by hydrolysis of a corresponding nitrile compound, which may be prepared from the corresponding hydroxy compound.
- This chemistry is illustrated for compounds in which a represents 1 in Anderson and Lok (1972), J Org. Chem. 37 (24) 3953-3955 and references mentioned therein.
- Schemes 1, 2, 3 and 4 may be modified to produce homochiral products by using homochiral starting materials (e.g. S-methionine in Scheme 1 or S-diaminobutyric acid in Scheme 4) or by performing an additional chiral resolution step.
- homochiral starting materials e.g. S-methionine in Scheme 1 or S-diaminobutyric acid in Scheme 4
- the isomers of the compounds of formula (XII) may be resolved by a dynamic resolution procedure.
- a racemic compound of formula (XII) may be treated with homochiral di-p-toluoyl tartaric acid in the presence of 3,5-dichloro-2-hydroxybenzaldehyde as catalyst in an inert solvent, e.g. THF.
- a homochiral salt of the compound of formula (XII) results.
- a compound of formula (XIII) may then be produced by subsequent treatment with trifluoroacetic acid methyl ester in the presence of N- methylmorph
- Both enantiomers of the compound of formula (XII) may also be produced from a synthesis based on S-methionine following a similar procedure.
- Novel chiral intermediates in the above described chiral and resolution sections also form an important aspect of this invention.
- the compounds of the invention have the advantage that they may be more efficacious, show greater selectivity, have fewer side effects, have a longer duration of action, be more bioavailable by the preferred route, have more attractive pharmacodynamic or pharmacokinetic properties or have other more desirable properties than similar known compounds.
- Example 1 rel-(3R,3aR,6aS)-4-(1-Cyclopropylmethyl-azetidine-3-carbonyl)-3- isopropyl-1-methanesulfonyl-hexahydro-pyrrolo[3,2-b]pyrrol-2-one hydrochloride
- Intermediate 12 (0.019g), acetic acid (1 drop), sodium triacetoxyborohydride (0.021 g), cyclopropanecarboxaldehyde (0.075ml) and dichloromethane (1ml) were stirred at room temperature for 3 days. The mixture was diluted with dichloromethane (5ml), stirred with 8% sodium bicarbonate solution (5ml) and separated.
- Example 2 rel-(3R,3aR,6aS)-4-(1-Benzyl-azetidine-3-carbonyl)-3-isopropyl-1- methanesulfonyl-hexahydro-pyrrolo[3,2-b]pyrrol-2-one hydrochloride
- Example 2 was similarly prepared to Example 1 from Intermediate 12 and benzaldehyde (0.011 ml) to afford the title compound as a white solid (0.009g).
- T.l.c SiO 2 Dichloromethane:ethanol:ammonia [100:8:1], Rf 0.46 Mass spec MH + (found) 420, MH + (calc) 420
- Example 3 rel-(3R,3aR,6aS)-4-[1 -(2,2-Dimethyl-propyl)-azetidine-3-carbonyl]-3- isopropyl-1-methanesulfonyl-hexahydro-pyrrolo[3,2-b]pyrrol-2-one hydrochloride
- Example 3 was similarly prepared to Example 1 from Intermediate 12 and pivaldehyde (0.011ml) to afford the title compound as a white solid (0.01 Og).
- T.l.c SiO 2 Dichloromethane:ethanol:ammonia [100:8:1] Rf 0.44 Mass spec MH + (found) 400, MH + (calc) 400
- Example 4 rel-(3R,3aR,6aS)-4-[1-(2,6-Dichloro-benzyl)-azetidine-3-carbonyl]-3- isopropyl-1-methanesulfonyl-hexahydro-pyrrolo[3,2-b]pyrrol-2-one hydrochloride
- Example 4 was prepared similarly to Example 1 from Intermediate 12 and 2,6- dichlorobenzaldehyde (0.017g) to afford after trituration with ether (3x1 ml) the title compound as a white solid (0.003g).
- Example 6 (3S, 3aS, 6aR)-4-[1-(2,2-Dimethyl-propyl)-azetidine-3-carbonyl]-3- isopropyl-1-methanesufonyl-hexahydro-pyrrolo[3,2-b]pyrrol-2-one
- acetic acid 9 drops - ca 100 ⁇ L
- sodium triacetoxyborohydride 0.80g
- pivaldehyde (0.24mL). After 18h, the mixture was concentrated in vacuo and the residue treated with ethyl acetate (90mL).
- Example 3 The compound of Example 3 was tested in the in vivo hamster IL-8 test described earlier in the description and showed a duration of action of at least 6 hours at a dose of less than 10 mg/kg.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Dermatology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
Description


















Claims









Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HU0003475A HUP0003475A3 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| KR1020007002505A KR20010023830A (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone Derivatives as Inhibitors of Neutrophil Elastase |
| IL13493598A IL134935A0 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| EP98948905A EP1019406A1 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| APAP/P/2000/001760A AP2000001760A0 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase. |
| HR20000136A HRP20000136A2 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| EA200000205A EA200000205A1 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrlonone derivatives as ELASTASIS INHIBITORS NEUTROPHILS |
| AU95360/98A AU9536098A (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| EEP200000137A EE200000137A (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| CA002303249A CA2303249A1 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| US09/508,326 US6228853B1 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| PL98339186A PL339186A1 (en) | 1997-09-09 | 1998-09-07 | Derivatives of pyrrolopyrrolone as inhibitors of neutrophilic elastase |
| SK324-2000A SK3242000A3 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
| JP2000510738A JP2001515902A (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolones as neutrophil elastase inhibitors |
| BR9811651-7A BR9811651A (en) | 1997-09-09 | 1998-09-07 | Compound, use of the same, process to prepare it, single enantiomer purified, pharmaceutical composition, and, processes for the treatment of chronic bronchitis or chronic obstructive pulmonary disease in a human or animal individual, and for treatment of asthma in a human or animal |
| IS5394A IS5394A (en) | 1997-09-09 | 2000-03-03 | Pyrrolopyrrolone derivatives that inhibit the elastase neutralization |
| NO20001200A NO20001200L (en) | 1997-09-09 | 2000-03-08 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9719187.8A GB9719187D0 (en) | 1997-09-09 | 1997-09-09 | Compounds |
| GB9719187.8 | 1997-09-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999012931A1 true WO1999012931A1 (en) | 1999-03-18 |
Family
ID=10818820
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/005604 Ceased WO1999012931A1 (en) | 1997-09-09 | 1998-09-07 | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
Country Status (24)
| Country | Link |
|---|---|
| US (1) | US6228853B1 (en) |
| EP (1) | EP1019406A1 (en) |
| JP (1) | JP2001515902A (en) |
| KR (1) | KR20010023830A (en) |
| CN (1) | CN1278821A (en) |
| AP (1) | AP2000001760A0 (en) |
| AU (1) | AU9536098A (en) |
| BR (1) | BR9811651A (en) |
| CA (1) | CA2303249A1 (en) |
| EA (1) | EA200000205A1 (en) |
| EE (1) | EE200000137A (en) |
| GB (1) | GB9719187D0 (en) |
| HR (1) | HRP20000136A2 (en) |
| HU (1) | HUP0003475A3 (en) |
| ID (1) | ID24364A (en) |
| IL (1) | IL134935A0 (en) |
| IS (1) | IS5394A (en) |
| MA (1) | MA26544A1 (en) |
| NO (1) | NO20001200L (en) |
| PL (1) | PL339186A1 (en) |
| SK (1) | SK3242000A3 (en) |
| TR (1) | TR200001026T2 (en) |
| WO (1) | WO1999012931A1 (en) |
| ZA (1) | ZA988162B (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003525268A (en) * | 2000-03-03 | 2003-08-26 | アベンテイス・フアルマ・ソシエテ・アノニム | Azetidine derivatives, their preparation and pharmaceutical compositions containing them |
| WO2021053058A1 (en) | 2019-09-17 | 2021-03-25 | Mereo Biopharma 4 Limited | Alvelestat for use in the treatment of graft rejection, bronchiolitis obliterans syndrome and graft versus host disease |
| WO2021209740A1 (en) | 2020-04-16 | 2021-10-21 | Mereo Biopharma 4 Limited | Methods involving neutrophil elastase inhibitor alvelestat for treating coronavirus infection |
| WO2023067103A1 (en) | 2021-10-20 | 2023-04-27 | Mereo Biopharma 4 Limited | Neutrophil elastase inhibitors for use in the treatment of fibrosis |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7745226B2 (en) | 2005-04-06 | 2010-06-29 | Quest Diagnostics Investments Incorporated | Methods for detecting vitamin D metabolites |
| US7972868B2 (en) | 2007-11-28 | 2011-07-05 | Quest Diagnostics Investments Incorporated | Methods for detecting dihydroxyvitamin D metabolites by mass spectrometry |
| US7977117B2 (en) | 2009-12-03 | 2011-07-12 | Quest Diagnostics Investments Incorprated | Vitamin D metabolite determination utilizing mass spectrometry following derivatization |
| CN102812356B (en) | 2009-12-11 | 2017-08-08 | 奎斯特诊断投资公司 | Mass Spectrometry of Steroids in Multiplexed Samples |
| US20110240840A1 (en) | 2009-12-11 | 2011-10-06 | Quest Diagnostics Investments Incorporated | Mass spectrometric determination of cookson-derivatized, non-metabolized vitamin d |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3983245A (en) * | 1975-02-06 | 1976-09-28 | Smithkline Corporation | Certain 4-(3-azacycloalkoxy or azacycloalkylmethoxy)benzoylbenzofurans or benzothiophenes |
| EP0201957A2 (en) * | 1985-05-14 | 1986-11-20 | Shell Internationale Researchmaatschappij B.V. | Process for the preparation of a carboxylic acid salt |
| WO1993024519A1 (en) * | 1992-06-04 | 1993-12-09 | Zeneca Limited | Lactam dipeptides having hle inhibiting activity |
| WO1995021855A1 (en) * | 1994-02-11 | 1995-08-17 | Zeneca Limited | Diastereomeric pure trifluoromethyl ketone peptide derivatives as inhibitors of human leukocyte elastase |
| WO1997025309A1 (en) * | 1996-01-03 | 1997-07-17 | Smithkline Beecham Plc | Carbamoyloxy derivatives of mutiline and their use as antibacterials |
| WO1997036903A1 (en) * | 1996-03-28 | 1997-10-09 | Glaxo Group Limited | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
-
1997
- 1997-09-09 GB GBGB9719187.8A patent/GB9719187D0/en not_active Ceased
-
1998
- 1998-09-07 MA MA25244A patent/MA26544A1/en unknown
- 1998-09-07 IL IL13493598A patent/IL134935A0/en unknown
- 1998-09-07 EA EA200000205A patent/EA200000205A1/en unknown
- 1998-09-07 SK SK324-2000A patent/SK3242000A3/en unknown
- 1998-09-07 AP APAP/P/2000/001760A patent/AP2000001760A0/en unknown
- 1998-09-07 HR HR20000136A patent/HRP20000136A2/en not_active Application Discontinuation
- 1998-09-07 AU AU95360/98A patent/AU9536098A/en not_active Abandoned
- 1998-09-07 HU HU0003475A patent/HUP0003475A3/en unknown
- 1998-09-07 CN CN98810967A patent/CN1278821A/en active Pending
- 1998-09-07 ID IDW20000665A patent/ID24364A/en unknown
- 1998-09-07 KR KR1020007002505A patent/KR20010023830A/en not_active Withdrawn
- 1998-09-07 EE EEP200000137A patent/EE200000137A/en unknown
- 1998-09-07 JP JP2000510738A patent/JP2001515902A/en active Pending
- 1998-09-07 US US09/508,326 patent/US6228853B1/en not_active Expired - Fee Related
- 1998-09-07 TR TR2000/01026T patent/TR200001026T2/en unknown
- 1998-09-07 EP EP98948905A patent/EP1019406A1/en not_active Withdrawn
- 1998-09-07 ZA ZA9808162A patent/ZA988162B/en unknown
- 1998-09-07 CA CA002303249A patent/CA2303249A1/en not_active Abandoned
- 1998-09-07 BR BR9811651-7A patent/BR9811651A/en not_active Application Discontinuation
- 1998-09-07 PL PL98339186A patent/PL339186A1/en not_active Application Discontinuation
- 1998-09-07 WO PCT/EP1998/005604 patent/WO1999012931A1/en not_active Ceased
-
2000
- 2000-03-03 IS IS5394A patent/IS5394A/en unknown
- 2000-03-08 NO NO20001200A patent/NO20001200L/en not_active Application Discontinuation
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3983245A (en) * | 1975-02-06 | 1976-09-28 | Smithkline Corporation | Certain 4-(3-azacycloalkoxy or azacycloalkylmethoxy)benzoylbenzofurans or benzothiophenes |
| EP0201957A2 (en) * | 1985-05-14 | 1986-11-20 | Shell Internationale Researchmaatschappij B.V. | Process for the preparation of a carboxylic acid salt |
| WO1993024519A1 (en) * | 1992-06-04 | 1993-12-09 | Zeneca Limited | Lactam dipeptides having hle inhibiting activity |
| WO1995021855A1 (en) * | 1994-02-11 | 1995-08-17 | Zeneca Limited | Diastereomeric pure trifluoromethyl ketone peptide derivatives as inhibitors of human leukocyte elastase |
| WO1997025309A1 (en) * | 1996-01-03 | 1997-07-17 | Smithkline Beecham Plc | Carbamoyloxy derivatives of mutiline and their use as antibacterials |
| WO1997036903A1 (en) * | 1996-03-28 | 1997-10-09 | Glaxo Group Limited | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
Non-Patent Citations (1)
| Title |
|---|
| D.L. LEE ET AL.: "alpha-Methylenelactam rearrangement", JOURNAL OF ORGANIC CHEMISTRY., vol. 39, no. 7, 1974, EASTON US, pages 893 - 902, XP002090864 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003525268A (en) * | 2000-03-03 | 2003-08-26 | アベンテイス・フアルマ・ソシエテ・アノニム | Azetidine derivatives, their preparation and pharmaceutical compositions containing them |
| WO2021053058A1 (en) | 2019-09-17 | 2021-03-25 | Mereo Biopharma 4 Limited | Alvelestat for use in the treatment of graft rejection, bronchiolitis obliterans syndrome and graft versus host disease |
| WO2021209740A1 (en) | 2020-04-16 | 2021-10-21 | Mereo Biopharma 4 Limited | Methods involving neutrophil elastase inhibitor alvelestat for treating coronavirus infection |
| WO2023067103A1 (en) | 2021-10-20 | 2023-04-27 | Mereo Biopharma 4 Limited | Neutrophil elastase inhibitors for use in the treatment of fibrosis |
Also Published As
| Publication number | Publication date |
|---|---|
| GB9719187D0 (en) | 1997-11-12 |
| CN1278821A (en) | 2001-01-03 |
| HRP20000136A2 (en) | 2000-08-31 |
| CA2303249A1 (en) | 1999-03-18 |
| JP2001515902A (en) | 2001-09-25 |
| NO20001200D0 (en) | 2000-03-08 |
| EP1019406A1 (en) | 2000-07-19 |
| AP2000001760A0 (en) | 2000-03-31 |
| EA200000205A1 (en) | 2000-10-30 |
| AU9536098A (en) | 1999-03-29 |
| ID24364A (en) | 2000-07-13 |
| MA26544A1 (en) | 2004-12-20 |
| NO20001200L (en) | 2000-05-08 |
| BR9811651A (en) | 2000-08-08 |
| TR200001026T2 (en) | 2000-09-21 |
| KR20010023830A (en) | 2001-03-26 |
| PL339186A1 (en) | 2000-12-04 |
| US6228853B1 (en) | 2001-05-08 |
| HUP0003475A2 (en) | 2001-09-28 |
| SK3242000A3 (en) | 2001-03-12 |
| IL134935A0 (en) | 2001-05-20 |
| IS5394A (en) | 2000-03-03 |
| ZA988162B (en) | 2000-03-22 |
| HUP0003475A3 (en) | 2002-01-28 |
| EE200000137A (en) | 2001-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6057457A (en) | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase | |
| EP0946561B1 (en) | Triazolo(4,5-d)pyrimidinyl derivatives and their use as medicaments | |
| US6228853B1 (en) | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase | |
| WO1999012933A2 (en) | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase | |
| JPS6140235B2 (en) | ||
| EP3515915B1 (en) | Process for preparing beta-lactamase inhibitor hydroxylurea intermediates | |
| US6177425B1 (en) | Pyrrolopyrrolone derivatives | |
| WO2000018770A1 (en) | Pyrrolopyrrolone derivatives as antiviral agents | |
| MXPA00002370A (en) | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase | |
| CZ2000878A3 (en) | Pyrrolopyrrolone derivatives | |
| NO150040B (en) | ANALOGY PROCEDURE FOR THE PREPARATION OF THERAPEUTIC ACTIVE 2-AZETIDINONES | |
| WO1999012932A1 (en) | Method of inhibiting serine protease enzymes | |
| MXPA00002325A (en) | Pyrrolopyrrolone derivatives as inhibitors of neutrophil elastase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 134935 Country of ref document: IL Ref document number: P-142/00 Country of ref document: YU Ref document number: 98810967.0 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1998948905 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2303249 Country of ref document: CA Ref document number: 2303249 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3242000 Country of ref document: SK Ref document number: 95360/98 Country of ref document: AU Ref document number: 200000205 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2000/002370 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20000136A Country of ref document: HR Ref document number: PV2000-878 Country of ref document: CZ Ref document number: 1020007002505 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000/01026 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09508326 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998948905 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-878 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007002505 Country of ref document: KR |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV2000-878 Country of ref document: CZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998948905 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1020007002505 Country of ref document: KR |