WO1999019321A1 - Synthesis of 3-carbomethoxy-4,5-dimethylthiophene - Google Patents
Synthesis of 3-carbomethoxy-4,5-dimethylthiophene Download PDFInfo
- Publication number
- WO1999019321A1 WO1999019321A1 PCT/US1998/021526 US9821526W WO9919321A1 WO 1999019321 A1 WO1999019321 A1 WO 1999019321A1 US 9821526 W US9821526 W US 9821526W WO 9919321 A1 WO9919321 A1 WO 9919321A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- thiophene
- product
- group
- aryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/32—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D333/40—Thiophene-2-carboxylic acid
Definitions
- This invention relates generally to the synthesis of substituted thiophenes and, more particularly, to 3-carbomethoxy-4,5-dimethylthiophene, which is an intermediate to a family of fungicides.
- the invention relates to a novel compound, which is a precursor of the thiophene intermediates.
- One fungicide that may be made from the subject intermediate is 4,5-dimethyl- N-2-propenyl-2-(trimethylsilyl)-3-thiophenecarboxamide, which is claimed as a novel compound in U.S. Patent No. 5,486,621. Two methods of making such a compound are provided therein. Another method is described in U.S. Patent No. 5,498,630, which concerns more generally controlling Take-All disease of plants by applying a fungicide.
- One related thiophene fungicide is described in Example 271, along with the method of making such compound.
- a number of methods of making thiophene compounds have been disclosed.
- One method that is related to the method of the present invention is described, for example, in SYNLETT, Nov. 1995, p. 1 143, by G. M. Coppola, R.E. Damon, and H. Yu. That method reacts an alpha-mercaptoketone with a phosphorus-substituted acrylate in the presence of a base to form a substituted dihydrothiophene ring, from which the corresponding aromatic thiophene ring can be made. It has been believed that the presence of a phosphorus-containing moiety at the 2-position was necessary for the cycloaddition reaction to occur.
- the invention is a new method of forming substituted thiophene rings and derivatives, such as the fungicide of U.S. Patent No. 5,486,621.
- the new method reacts an alpha mercaptoketone, such as 3-mercapto-2-butanone, prepared in situ from an ⁇ -haloketone such as 3-chloro-2-butanone, with an acrylate, such as methyl-3-methoxyacrylate, in the presence of an alkoxide base, such as NaOMe, when dissolved in an aprotic solvent, e.g., toluene.
- the product of the reaction is a substituted tetrahydrothiophene, which can be converted to the aromatic thiophene by an acid treatment.
- the generic reaction can be written as follows:
- R ⁇ and R 2 are independently chosen from hydrogen, alkyl, aryl, the substituted equivalents thereof, and a ring formed from R ⁇ and R 2 having 5 to 7 atoms;
- X is a suitable leaving group such as halogen (Cl, Br, I) or methanesulfonyloxy;
- R is a member of the group of hydrogen, alkyl, aryl, and the substituted equivalents thereof;
- Z is CN or CO 2 R , wherein R 4 is chosen from the group consisting of alkyl and aryl and the substituted equivalents thereof, H, Na, Li, K, and NH + ; and A is an alkoxy group, preferably a C
- the invention is a method of making the fungicide 4,5- d ⁇ methyl-N-2-propenyl-2-(t ⁇ methyls ⁇ lyl)-3-th ⁇ ophenecarboxam ⁇ de and related compounds in which the propenyl group is replaced by C 2 to C branched or straight alkyl chain.
- the invention is the precursor compound of the thiophene intermediate, as illustrated in the schematic diagram of the reaction shown above.
- the preferred compounds will be determined by the substitutions that one desires for the thiophene in the 4- and 5-positions. In turn, these may be the substitutions desired in a fungicide or other compound for which the thiophene is an intermediate. Alternatively, Ri and R 2 could be selected to facilitate their replacements by substituents that are desired in the ultimate chemical product.
- R* and R are methyl.
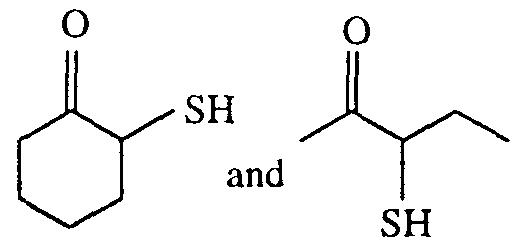
- Examples of other mercaptoketones include, but are not limited to,
- Mercaptoketones such as those used in the present invention are known in the literature. They are typically prepared by treating the corresponding haloketones (Eur. J. Med. Chem. - Chim. Ther. 1986-21, No. 6, p. 461 by R. Dubief, Y. Robbe, J.-P. Fernandez, G. Subra, A. Terol, J.-P. Chapat, H. Sentenac-Roumanou, and M. Fatome) in solvents with NaSH or other sulfur sources, or from the ketones by treatment with ammonia and elemental sulfur (Liebigs Ann. Chem. 1957, Bd. 610, p. 25 by F. Asinger, M. Thiel, and I.
- mercaptoketones such as with 3-mercapto-2-butanone
- mercaptoketones can be prepared from ⁇ - haloketones, such as 3-chloro-2-butanone, in a two-phase solvent system comprising water and a nonpolar organic solvent such as toluene or heptane, in the presence of NaSH.
- the product mercaptoketone is then recovered in high yield and purity as a solution in the organic solvent after phase separation, thereby avoiding the degradation associated with purification.
- Water can be removed from the mercaptoketone solution by various means such as by azeotropic distillation or by bringing the solution in contact with desiccants such as molecular sieves, calcium chloride, or sodium sulfate, prior to reaction with the acrylic compound.
- desiccants such as molecular sieves, calcium chloride, or sodium sulfate
- the second compound that adds to the mercaptoketone to make the substituted thiophene is an acrylic compound defined by the formula
- A is selected from the group consisting of alkoxy.
- A is methoxy.
- the reactive acrylic compound include, but are not limited to,
- the product in whole or in part, may be the corresponding tetrahydrothiophene, that is,
- the reaction of an ⁇ -haloketone with NaSH is carried out in a two-phase, water/organic solvent system.
- a solution of 1-1.25 equivalents of NaSH in water (10-30 wt%) is prepared and combined with an organic solvent such as toluene or heptane (2-4 equivalents) under an inert atmosphere with vigorous stirring.
- the mixture is maintained between about 0°C and about 30°C as the ⁇ -haloketone is added.
- the phases are allowed to separate, and the lower, aqueous phase is discarded.
- the upper, organic phase is then dried by azeotropic distillation (30-50°C, 100-600 mmHg) or by contact with a desiccant such as molecular sieves, calcium chloride, or sodium sulfate for a sufficient amount of time (15 min to 3 h).
- azeotropic distillation (30-50°C, 100-600 mmHg) or by contact with a desiccant such as molecular sieves, calcium chloride, or sodium sulfate for a sufficient amount of time (15 min to 3 h).
- the reaction of the mercaptoketone and acrylic compound is carried out in the presence of a base catalyst.
- base catalysts are alkoxides such as sodium methoxide, sodium t-amylate, potassium t-amylate, and potassium t-butoxide; strong amine bases such as diazabicycloundecene; sodium hydride; and alkali metal hydroxides.
- the base is preferably an alkoxide.
- the amount of the base catalyst will be about 0.025-0.2 equivalents relative to the amount of the mercaptoketone. Larger amounts of the base catalyst have been found to yield the thiophene, although the yields are lower.
- the product under the preferred conditions comprises a substantial proportion of tetrahydrothiophene.
- reaction will be carried out while the reactants are dissolved in a solvent, preferably, a hydrocarbon solvent such as toluene or another aprotic solvent such as chlorobenzene, heptane, or xylene.
- a solvent preferably, a hydrocarbon solvent such as toluene or another aprotic solvent such as chlorobenzene, heptane, or xylene.
- the reaction may be carried out at temperatures in the range of about 0-50°C.
- the treatment of the reaction product with acid to convert any tetrahydrothiophene to the thiophene form is preferably carried out at about 0-50°C.
- Recovery of the substituted thiophene from the reaction mixture may be carried out in at least two ways.
- the tetrahydrothiophene is recovered by separating it from the reaction mixture and then reacting it with acid to convert it to the thiophene.
- the second method reacts the tetrahydrothiophene in-situ with acid and subsequently recovers the product in the thiophene form.
- the first recovery process will include at least the following steps. Dilute sulfu ⁇ c acid will be added to the reaction mixture in order to neutralize the reaction mixture. Then, a solvent, such as ethyl acetate, will be added, and water and organic phases will be separated.
- the organic phase which includes the reaction product, will be washed with aqueous brine. After this, the washed organic phase will be dried, for example, by contact with sodium sulfate and the solvent evaporated to yield a mixture of substituted tetrahydrothiophene and substituted thiophene.
- This mixture will be treated with an aqueous acid, such as concentrated HC1, 50% H 2 SO 4 , or 50% phosphoric acid, or a non-aqueous acid such as anhydrous methanesulfonic acid, to convert the tetrahydrothiophene to the thiophene form. After that reaction, the steps described above can be repeated to separate the thiophene product.
- the substituted tetrahydrothiophene is converted in-situ to the thiophene lorm rather than being first separated.
- an aqueous acid such as concentrated HC1 or 50% H 2 S0 4 , will be added to the reaction mixture to convert the tetrahydrothiophene to the thiophene form.
- the water and organic phases will be separated and the solvent evaporated.
- the crude product may be distilled to produce the refined substituted thiophene.
- the product of the invention is a substituted thiophene, which is an intermediate for preparation of a family of thiophene-based fungicides described in
- Ri and R 2 are methyl
- R 3 is trimethylsilyl (SiMe 3 )
- R 3 which is preferably hydrogen in the acrylic starting compound, must be replaced by the trimethylsilyl moiety or another desired substituent.
- Z will be a carboxylic acid ester moiety. Therefore, hydrolysis of the carboxylic acid ester moiety to the corresponding Li salt followed by silylation as described above and subsequent conversion to the amide by standard methods would give the preferred fungicide.
- Example 1
- Aqueous NaSH (1J equivalents) was diluted with water to give a 23.5% solution by weight. This solution was combined with 3 equivalents of toluene under an inert atmosphere, stirred vigorously, and cooled to 5°C. One equivalent of 3- chloro-2-butanone was added to the mixture at a rate to keep the temperature below 10°C. This required 2 h. After the addition, the mixture was stirred at 5-10°C until
- Example 3 One equivalent of each of 3-mercapto-2-butanone and methyl-3- methoxyacrylate were dissolved in toluene, and 0.1 equivalent of NaOMe was added as a catalyst. The reaction was carried out at room temperature under a nitrogen atmosphere for 3-4 hours. Then, the mixture was poured into dilute sulfuric acid and extracted with ethyl acetate. The organic phase was washed with brine and then dried over sodium sulfate, filtered and evaporated. The product was the tetrahydrothiophene precursor described above, i.e., 2-methoxy-3-carbomethoxy-4- hydroxy-4' -methyl-5-methyl-tetrahydrothiophene.
- the tetrahydrothiophene precursor was treated with concentrated HC1 at room temperature with rapid stirring. After 5 minutes, the mixture was poured into water and extracted with ethyl acetate. The organic phase was washed with saturated NaHCO 3 , dried over NaSO 4 , filtered and evaporated at room temperature on a rotary evaporator, leaving the aromatic thiophene product, 3-carbomethoxy-4,5- dimethylthiophene.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description







Claims





Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL339914A PL205200B1 (en) | 1997-10-14 | 1998-10-13 | Method of synthesising 3-carbomethoxy-4,5-dimethylthiophene |
| EA200000415A EA003088B1 (en) | 1997-10-14 | 1998-10-13 | Synthesis of thiophene derivatives |
| DK98951049T DK1023283T3 (en) | 1997-10-14 | 1998-10-13 | Synthesis of 3-carboxymethoxy-4,5-dimethylthiophene |
| HU0003988A HU228367B1 (en) | 1997-10-14 | 1998-10-13 | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene |
| UA2000052717A UA57810C2 (en) | 1997-10-14 | 1998-10-13 | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene |
| AT98951049T ATE231853T1 (en) | 1997-10-14 | 1998-10-13 | PRODUCTION OF 3-CARBOMETHOXY-4,5-DIMETHYLTHIOPHENE |
| DE69811103T DE69811103T2 (en) | 1997-10-14 | 1998-10-13 | PREPARATION OF 3-CARBOMETHOXY-4,5-DIMETHYLTHIOPHEN |
| EP98951049A EP1023283B1 (en) | 1997-10-14 | 1998-10-13 | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene |
| CA002306910A CA2306910C (en) | 1997-10-14 | 1998-10-13 | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene |
| AU96942/98A AU9694298A (en) | 1997-10-14 | 1998-10-13 | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6197297P | 1997-10-14 | 1997-10-14 | |
| US60/061,972 | 1997-10-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999019321A1 true WO1999019321A1 (en) | 1999-04-22 |
Family
ID=22039390
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/021526 Ceased WO1999019321A1 (en) | 1997-10-14 | 1998-10-13 | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US6084108A (en) |
| EP (1) | EP1023283B1 (en) |
| CN (1) | CN1116294C (en) |
| AT (1) | ATE231853T1 (en) |
| AU (1) | AU9694298A (en) |
| CA (1) | CA2306910C (en) |
| CZ (1) | CZ297978B6 (en) |
| DE (1) | DE69811103T2 (en) |
| DK (1) | DK1023283T3 (en) |
| EA (1) | EA003088B1 (en) |
| ES (1) | ES2190610T3 (en) |
| HU (1) | HU228367B1 (en) |
| PL (1) | PL205200B1 (en) |
| UA (1) | UA57810C2 (en) |
| WO (1) | WO1999019321A1 (en) |
| ZA (1) | ZA989348B (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6992047B2 (en) * | 2001-04-11 | 2006-01-31 | Monsanto Technology Llc | Method of microencapsulating an agricultural active having a high melting point and uses for such materials |
| DE10148437A1 (en) * | 2001-10-01 | 2003-04-17 | Bayer Ag | Process for the alkylation of 3,4-dihydroxythiophene-2,5-dicarboxylic acid esters |
| CN103044479B (en) * | 2012-12-12 | 2015-09-02 | 河南农业大学 | The synthetic method of bactericide of silthiopham |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4041047A (en) * | 1973-05-18 | 1977-08-09 | Ciba-Geigy Corporation | 9-Thiaprostaglandins |
| DE3229538A1 (en) * | 1982-08-07 | 1984-02-09 | Basf Ag, 6700 Ludwigshafen | Process for the preparation of sterically uniform 3-hydroxycarboxylic acids and esters thereof |
| US5486621A (en) * | 1994-12-15 | 1996-01-23 | Monsanto Company | Fungicides for the control of take-all disease of plants |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU663299A3 (en) * | 1975-05-27 | 1979-05-15 | Ю.Ц.Б.С.А. (Фирма) | Method of obtaining n-benzil-2,3-dimethoxyacetamines |
| US4689343A (en) * | 1983-12-09 | 1987-08-25 | Bayer Aktiengesellschaft | Novel thiolane-2,4-dione-3-carboxamide fungicides |
| US5206375A (en) * | 1985-03-02 | 1993-04-27 | Basf Aktiengesellschaft | Thiophene derivatives |
| GB8609452D0 (en) * | 1986-04-17 | 1986-05-21 | Ici Plc | Fungicides |
| DE3901074A1 (en) * | 1989-01-16 | 1990-07-19 | Basf Ag | THIOPHENE-2-CARBON-SAE DERIVATIVES, METHOD FOR THE PRODUCTION THEREOF AND THEIR USE FOR CONTROLLING UNWANTED PLANT GROWTH |
| US5087288A (en) * | 1990-09-17 | 1992-02-11 | Eastman Kodak Company | Herbicidal thioparaconic acid derivatives |
| US5679801A (en) * | 1991-04-12 | 1997-10-21 | American Home Products Corporation | Tetronic and thiotetronic acid derivatives as phospholipase A2 inhibitors |
| CA2119155C (en) * | 1991-10-18 | 1999-06-15 | Dennis Paul Phillion | Fungicides for the control of take-all disease of plants |
| JP3337728B2 (en) * | 1992-11-20 | 2002-10-21 | イハラケミカル工業株式会社 | Method for producing 2-acetylbenzo [b] thiophene |
| DE69618370T2 (en) * | 1995-04-11 | 2002-09-26 | Mitsui Chemicals, Inc. | Substituted thiophene derivatives and fungicides containing them as an active ingredient for agriculture and horticulture |
-
1998
- 1998-10-13 AU AU96942/98A patent/AU9694298A/en not_active Abandoned
- 1998-10-13 EP EP98951049A patent/EP1023283B1/en not_active Expired - Lifetime
- 1998-10-13 US US09/170,441 patent/US6084108A/en not_active Expired - Lifetime
- 1998-10-13 ES ES98951049T patent/ES2190610T3/en not_active Expired - Lifetime
- 1998-10-13 CA CA002306910A patent/CA2306910C/en not_active Expired - Lifetime
- 1998-10-13 CN CN98812151A patent/CN1116294C/en not_active Expired - Lifetime
- 1998-10-13 EA EA200000415A patent/EA003088B1/en not_active IP Right Cessation
- 1998-10-13 HU HU0003988A patent/HU228367B1/en unknown
- 1998-10-13 CZ CZ20001330A patent/CZ297978B6/en not_active IP Right Cessation
- 1998-10-13 WO PCT/US1998/021526 patent/WO1999019321A1/en not_active Ceased
- 1998-10-13 AT AT98951049T patent/ATE231853T1/en active
- 1998-10-13 DK DK98951049T patent/DK1023283T3/en active
- 1998-10-13 UA UA2000052717A patent/UA57810C2/en unknown
- 1998-10-13 PL PL339914A patent/PL205200B1/en unknown
- 1998-10-13 DE DE69811103T patent/DE69811103T2/en not_active Expired - Lifetime
- 1998-10-13 ZA ZA9809348A patent/ZA989348B/en unknown
-
1999
- 1999-09-13 US US09/394,760 patent/US6037478A/en not_active Expired - Lifetime
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4041047A (en) * | 1973-05-18 | 1977-08-09 | Ciba-Geigy Corporation | 9-Thiaprostaglandins |
| DE3229538A1 (en) * | 1982-08-07 | 1984-02-09 | Basf Ag, 6700 Ludwigshafen | Process for the preparation of sterically uniform 3-hydroxycarboxylic acids and esters thereof |
| US5486621A (en) * | 1994-12-15 | 1996-01-23 | Monsanto Company | Fungicides for the control of take-all disease of plants |
Non-Patent Citations (6)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 71, no. 15, 13 October 1969, Columbus, Ohio, US; abstract no. 70433h, TILAK BAL D. ET AL.: "Synthesis of sulfur heterocyclics. II" page 300; column 1; XP002095303 * |
| CHEMICAL ABSTRACTS, vol. 94, no. 13, 30 March 1981, Columbus, Ohio, US; abstract no. 103096f, KAMBE SATOSHI ET AL.: "A simplified procedure for the preparation of 2-alkoxycarbonyl-5-aryl-4-cyano-3-hydroxy-3-phenyltetrahydrothiophenes" page 717; column 2; XP002095302 * |
| GARY M COPPOLA ET AL., SYNLETT, no. 11, November 1995 (1995-11-01), pages 1143 - 1144, XP002095300 * |
| INDIAN J. CHEM., vol. 7, no. 1, 1969, pages 9-16 * |
| REINHARD W. HOFFMANN ET AL.: "The sense of asymmetric Induction on Addition to alpha-Chiral Aldehydes", CHEMISCHE BERICHTE, vol. 118, 1985, weinheim, pages 3966 - 3979, XP002095301 * |
| SYNTHESIS, no. 10, 1980, pages 839-40 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1116294C (en) | 2003-07-30 |
| EA003088B1 (en) | 2002-12-26 |
| HUP0003988A2 (en) | 2002-01-28 |
| ATE231853T1 (en) | 2003-02-15 |
| AU9694298A (en) | 1999-05-03 |
| HUP0003988A3 (en) | 2003-01-28 |
| HU228367B1 (en) | 2013-03-28 |
| US6037478A (en) | 2000-03-14 |
| CA2306910C (en) | 2008-07-29 |
| ES2190610T3 (en) | 2003-08-01 |
| DE69811103D1 (en) | 2003-03-06 |
| DE69811103T2 (en) | 2003-10-09 |
| CZ20001330A3 (en) | 2000-09-13 |
| EP1023283B1 (en) | 2003-01-29 |
| DK1023283T3 (en) | 2003-06-23 |
| CN1281446A (en) | 2001-01-24 |
| EP1023283A1 (en) | 2000-08-02 |
| US6084108A (en) | 2000-07-04 |
| CA2306910A1 (en) | 1999-04-22 |
| ZA989348B (en) | 1999-07-01 |
| UA57810C2 (en) | 2003-07-15 |
| PL205200B1 (en) | 2010-03-31 |
| CZ297978B6 (en) | 2007-05-16 |
| EA200000415A1 (en) | 2000-10-30 |
| PL339914A1 (en) | 2001-01-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Patrocı́nio et al. | Synthesis of acylsilanes via oxidative hydrolysis of 2-silyl-1, 3-dithianes mediated by N-bromosuccinimide | |
| AU2003248280A1 (en) | New process for the industrial synthesis of tetraesters of 5-[bis(carboxymethyl)amino]-3-carboxymethyl-4-cyano-2-thiophenecarboxylic acid, and application to the synthesis of bivalent salts of ranelic acid and their hydrates | |
| US4140698A (en) | 1,2-Dihydro-3H-pyrrolo[1,2-a]pyrrole-1-nitriles | |
| EP0669924B1 (en) | Regioselective synthesis of 4-chloro-2-thiophenecarboxylic acid | |
| EP1023283B1 (en) | Synthesis of 3-carbomethoxy-4,5-dimethylthiophene | |
| CN110511193B (en) | Alpha-ketothioamide compound and synthetic method thereof | |
| JP4610740B2 (en) | Process for the preparation of 3- (substituted phenyl) -5-thienyl or furyl) -1,2,4-triazole and novel intermediates used therein | |
| CA1106850A (en) | 8h-indeno ¬ 2,1-b| thiophen-2-oxaminic acids and esters thereof | |
| US4352756A (en) | Production of furfuryl alcohols | |
| US6255540B1 (en) | Methods for producing two-substituted glycerols having various levels of protection | |
| JP4664903B2 (en) | Process for producing 4,10β-diacetoxy-2α-benzoyloxy-5β, 20-epoxy-1,13α-dihydroxy-9-oxo-19-norcyclopropa [g] taxa-11-ene | |
| CA1135271A (en) | Process for the production of 3-thienylmalonic acid | |
| CA2017911A1 (en) | 4,5,6,11-tetrahydrobenzo [6,7] -cycloocta [1,2-b] thiophen-6,11-imines and 6,11-dihydrobenzo [6,7] -cycloocta [1,2-b] thiophen-6,11 imines | |
| JPH069553A (en) | Preparation of 1-(2s-methyl-3-mercaptopropionyl)- pyrrolidine-2s-carboxylic acid | |
| JP3231207B2 (en) | Method for producing sulfenylacetic acid derivative | |
| JP4475901B2 (en) | Method for producing 3-acetylthiophenes | |
| KR100412334B1 (en) | Novel process for preparing 4-substituted-1H-pyrrole-3-carboxylic acid ester | |
| JPH06199832A (en) | Production of 2-@(3754/24)3-thienyl)ethanol derivative | |
| FR2616431A1 (en) | PROCESS FOR THE PREPARATION OF (TRANS) -4-PHENYL-L-PROLINE DERIVATIVES | |
| JPS6246546B2 (en) | ||
| JPH04149179A (en) | Production of acetylthiophene derivative | |
| JP2002503648A (en) | Methods and intermediates useful for producing antifolates | |
| WO2005002523A2 (en) | Process for preparing highly pure and free-flowing solid of 7-ethyltryptophol | |
| JPH032861B2 (en) | ||
| PL142442B1 (en) | Method of obtaining 3-methyl-2-thiophenylcarboxylamides and 3-methyl-2-thiophenylcarboxylthiamides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 98812151.4 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2306910 Country of ref document: CA Ref document number: 2306910 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2000-1330 Country of ref document: CZ |
|
| NENP | Non-entry into the national phase |
Ref country code: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998951049 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200000415 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998951049 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-1330 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998951049 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV2000-1330 Country of ref document: CZ |