WO1999026963A1 - Hypusine peptides - Google Patents
Hypusine peptides Download PDFInfo
- Publication number
- WO1999026963A1 WO1999026963A1 PCT/US1998/017221 US9817221W WO9926963A1 WO 1999026963 A1 WO1999026963 A1 WO 1999026963A1 US 9817221 W US9817221 W US 9817221W WO 9926963 A1 WO9926963 A1 WO 9926963A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- peptide
- gly
- hypusine
- thr
- hpu
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/0606—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing heteroatoms not provided for by C07K5/06086 - C07K5/06139, e.g. Ser, Met, Cys, Thr
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
- C07K5/06147—Dipeptides with the first amino acid being heterocyclic and His-amino acid; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/081—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing O or S as heteroatoms, e.g. Cys, Ser
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0815—Tripeptides with the first amino acid being basic
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to novel hypusine-containing peptides.
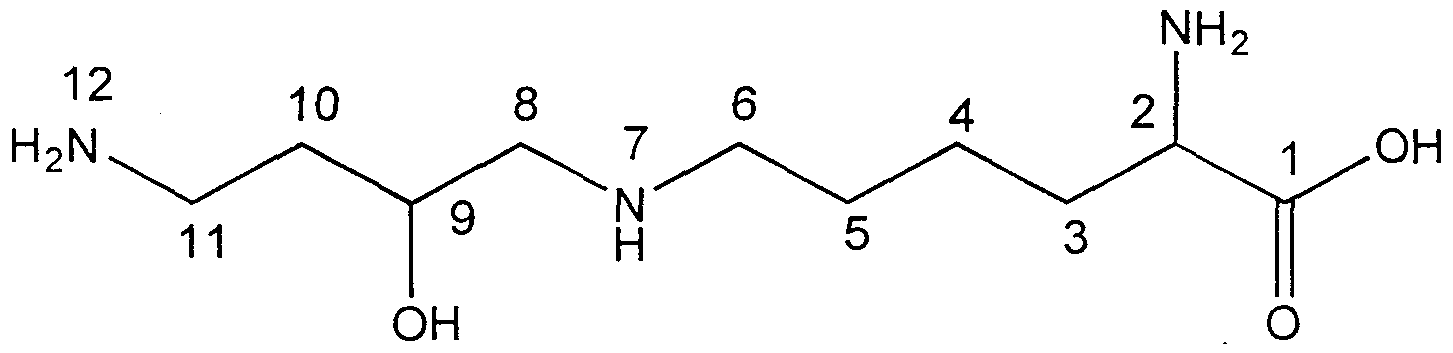
- hydroxy-7-azaundecanoic acid an unusual naturally occurring amino acid, having the structure:
- This initiation factor 5A is unique in that it is the only known cellular protein that contains the amino acid hypusine (Hpu).
- Hpu amino acid hypusine
- elF-5A was shown to stimulate ribosomal subunit joining and to enhance 80 S-bound Met-t-RNAj reactivity with puromycin [Anderson et al, FEBS Lett., Vol. 76, pages 1-10 (1977); and Kemper et al. J. Biol. Chem., Vol. 251. pages 5551-5557 (1976)1. Later, in
- the present invention relates to novel hypusine-containing peptides synthesized utilizing the hypusine reagent:
- Q ⁇ Q 2 , and Q 3 may be the same or different and are amino protective groups, provided that Q 3 is orthogonal to Qi and Q 2 ; and Z is a hydroxy protective group.
- a further embodiment of the invention relates to compounds of structure (2)
- hypusine reagent (1) which may be synthesized using hypusine reagent (1), wherein Hpu is the hypusine amino acid residue, S and T are each independently peptide residues from zero to about 12 amino acids in length.
- Hpu is the hypusine amino acid residue
- S and T are each independently peptide residues from zero to about 12 amino acids in length.
- Compounds of the invention find utility in the study of biochemical processes involving hypusine.
- Another embodiment of the invention concerns improved methods of peptide synthesis wherein the above-described hypusine reagent is employed to prepare novel hypusine-containing peptides.
- FIGs. 1 and 2 depict example reaction schemes for preparing peptides of the invention.
- Figures 1 and 2 correspond to the chemistry described in Examples 1 through 8.
- novel peptides (2) of the present invention comprise any synthetic peptide that incorporates within its structure the hypusine moiety, which is synthesized according to a method involving the use of the above-described hypusine reagent (1).
- S and T are peptide residues from zero to about 12 amino acids in length, and preferably, are peptide residues from zero to about six amino acids in length. Most preferably, S and T are peptides residues from zero to about three amino acids in length S and T may vary independently in length and in composition of amino acid residues. The terminal amino acid reside of T may be hydroxylated.
- Non-limiting examples of peptides of the invention are: L-Ser-L-Thr-L-Ser-L-Lys-L-Thr-Gly-Hpu-L-His-Gly-L-His-L-Ala-L-Lys,
- peptides of the invention find utility in the study of biochemical processes involving hypusine, such as in the study of transport mechanisms for elF5A.
- the peptides of the invention may be prepared employing conventional steps of peptide synthesis except that the above-described hypusine reagent (1) is employed to incorporate the hypusine moiety into the peptide chain.
- Conventional peptide synthesis steps are disclosed, for example, in Moroder et al., "Hormonal Receptors in Digestive Tract Physiology," G. Rosselin et al., eds., Elsevier/North- Holland Biomedical Press, Amsterdam, pages 129-135 (1979); and Moroder et al., Phvsiol. Chem., Vol. 360, pages 787-790 (1979).
- the synthesis of peptides is generally carried out through the condensation of the carboxyl group of an amino acid, and the amino group of another amino acid, to form a peptide bond.
- a sequence can be constructed by repeating the condensation of individual amino acid residues in stepwise elongation or, in some cases, by condensation between two pre-formed peptide fragments (fragment condensation).
- fragment condensation the amino and carboxy groups that are not to participate in the reaction must be blocked with protecting groups which should be readily introduced, be stable to the condensation reactions and be removed selectively from the completed peptide. If a peptide involves amino acids with side chains that may react during condensation, the problem of protection becomes increasingly difficult.
- Solid phase peptide synthesis involves attachment of a first amino acid to a solid support, such as a resin, followed by sequential addition of subsequent amino acids which results in assembly of the peptide chain on the solid support.
- Peptides can also be synthesized by related methods involving coupling peptide fragments to solid supports as discussed by Erickson et al., supra, pages 268-269. This technique involves the synthesis of small peptide segments containing a few amino acids, which segments are then coupled to each other using fragment condensation techniques to form larger peptides. Fragment condensation techniques can be combined with standard solid phase techniques wherein small peptides are attached to resins followed by sequential attachment of single amino acids or other peptide segments. Alternatively, sequential attachment of small peptides to single resin-bound amino acids can also be accomplished. The combination of the two approaches provides flexibility to synthetic schemes.
- the synthesized peptide is then removed from the resin, usually by chemical means such as treatment with hydrofluoric acid (HF).
- HF hydrofluoric acid
- the chemical treatment also removes various amino acid and peptide protecting groups, such as CBZ, t-BOC or tosyl, which mask the reactivity of amino acid functional groups during synthesis.
- the initial attachment to the resin involves the C- terminal amino acid of the peptide to be synthesized, which amino acid is covalently attached to the resin through an ester or amide linkage involving its ⁇ -carboxyl group. Synthesis then proceeds from the C- to the N-terminal. N-terminal to C- terminal peptide synthesis is less frequently used because the chemistry is more difficult and unwanted side reactions are more common.
- the first amino acid may be covalently attached to the resin, in some cases, through its functional side chain.
- Initial attachment of an amino acid to the resin by means of the side chain functional group allows the possibility of bi-directional synthesis starting with the attached amino acid. Bi-directional synthesis cannot be performed if the initial amino acid is attached through the ⁇ -COOH or ⁇ -NH 2 group.
- Side chain functional groups which have been used for attachment to resins include the sulfhydryl group of cysteine, the imidazole group of histidine, the ⁇ -amino group
- hypusine reagent described herein may be employed to access any hypusine-containing peptide
- the method of the invention will be illustrated with reference to the following syntheses. It will be understood that any conventional peptide synthesis may be modified to prepare a hypusine-containing peptide by simply utilizing the herein described hypusine reagent at any convenient stage thereof.
- the hypusine-containing pentapeptide found in eIF-5A capped at its N-terminus with L-Cys i.e., L-Cys-Thr-Gly-Hpu-His-Gly is a typical target peptide.
- L-Cys which is not contained in the natural peptide, was fixed to the sequence with the idea of being able to covalently link the peptide via a disulfide bond to a larger protein, in order to ultimately generate antibodies.
- the final hexapeptide 12 was constructed stepwise from the three aforementioned fragments, i.e., 1 , 15 and 18. Hydrogenolysis of the N ⁇ - CBZ group of 17 provided the amine HCI salt (74%) which was condensed with hypusine reagent 1 to give the di-CBZ-THP protected Hpu-His-Gly tripeptide 18 in 85% yield.
- the hypusine reagent described has been demonstrated to be a highly useful synthon in accessing the elF-5A pentapeptide sequence. While the yields are generally excellent for these kinds of systems, the most notable feature is the flexibility that this methodology offers in synthesizing related elF-5A mimics.
- the polymer-bound peptide 21 was synthesized using an Applied Biosystems 432A Synthesizer. Amino acid analysis for 21 : Gly 2.09, His 1.03, Thr 0.88. An aliquot of 21 (49 mg, 19.3 ⁇ mol), phenol (250 mg) and pentamethylbenzene (250 mg) were dissolved in degassed TFA (5.0 ml) at 0°C. Saturated HBr in acetic acid solution (0.2 ml), triisopropylsilane (0.1 ml) and 1 , 2-ethanedithiol (0.1 ml) were added under an argon atmosphere. The solution was stirred at room temperature for 1 hour and concentrated under reduced pressure.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Life Sciences & Earth Sciences (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description


Claims


Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002310027A CA2310027A1 (en) | 1997-11-21 | 1998-08-19 | Hypusine peptides |
| AU91090/98A AU9109098A (en) | 1997-11-21 | 1998-08-19 | Hypusine peptides |
| EP98943259A EP1032586A4 (en) | 1997-11-21 | 1998-08-19 | PEPTIDES CONTAINING HYPUSIN |
| JP2000522120A JP2001524488A (en) | 1997-11-21 | 1998-08-19 | Hypsin peptide |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US97565697A | 1997-11-21 | 1997-11-21 | |
| US08/975,656 | 1997-11-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999026963A1 true WO1999026963A1 (en) | 1999-06-03 |
Family
ID=25523257
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/017221 Ceased WO1999026963A1 (en) | 1997-11-21 | 1998-08-19 | Hypusine peptides |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1032586A4 (en) |
| JP (1) | JP2001524488A (en) |
| AU (1) | AU9109098A (en) |
| CA (1) | CA2310027A1 (en) |
| WO (1) | WO1999026963A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010131962A3 (en) * | 2009-05-15 | 2011-03-03 | Stichting Het Nederlands Kanker Instituut | Lysine compounds and their use in site- and chemoselective modification of peptides and proteins |
| US12099064B2 (en) | 2015-08-28 | 2024-09-24 | Genentech, Inc. | Anti-hypusine antibodies and uses thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5344846A (en) * | 1992-12-30 | 1994-09-06 | The United States Of America As Represented By The Department Of Health And Human Services | Compositions and methods for inhibiting deoxyhypusine synthase and the growth of cells |
| US5538897A (en) * | 1994-03-14 | 1996-07-23 | University Of Washington | Use of mass spectrometry fragmentation patterns of peptides to identify amino acid sequences in databases |
-
1998
- 1998-08-19 CA CA002310027A patent/CA2310027A1/en not_active Abandoned
- 1998-08-19 WO PCT/US1998/017221 patent/WO1999026963A1/en not_active Ceased
- 1998-08-19 JP JP2000522120A patent/JP2001524488A/en active Pending
- 1998-08-19 AU AU91090/98A patent/AU9109098A/en not_active Abandoned
- 1998-08-19 EP EP98943259A patent/EP1032586A4/en not_active Withdrawn
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5344846A (en) * | 1992-12-30 | 1994-09-06 | The United States Of America As Represented By The Department Of Health And Human Services | Compositions and methods for inhibiting deoxyhypusine synthase and the growth of cells |
| US5538897A (en) * | 1994-03-14 | 1996-07-23 | University Of Washington | Use of mass spectrometry fragmentation patterns of peptides to identify amino acid sequences in databases |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1032586A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010131962A3 (en) * | 2009-05-15 | 2011-03-03 | Stichting Het Nederlands Kanker Instituut | Lysine compounds and their use in site- and chemoselective modification of peptides and proteins |
| CN102459160A (en) * | 2009-05-15 | 2012-05-16 | 荷兰癌症研究所基金会 | Lysine compounds and their use in site- and chemoselective modification of peptides and proteins |
| US8729009B2 (en) | 2009-05-15 | 2014-05-20 | Stichting Het Nederlands Kanker Instituut | Lysine compounds and their use in site- and chemoselective modification of peptides and proteins |
| US12099064B2 (en) | 2015-08-28 | 2024-09-24 | Genentech, Inc. | Anti-hypusine antibodies and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1032586A4 (en) | 2000-11-15 |
| JP2001524488A (en) | 2001-12-04 |
| AU9109098A (en) | 1999-06-15 |
| EP1032586A1 (en) | 2000-09-06 |
| CA2310027A1 (en) | 1999-06-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7589170B1 (en) | Synthesis of cyclic peptides | |
| EP0561412B1 (en) | Parathyroid hormone derivatives | |
| US5635598A (en) | Selectively cleavabe linners based on iminodiacetic acid esters for solid phase peptide synthesis | |
| AP1117A (en) | Process for the preparation of resin-bound cyclic peptides. | |
| CA1339793C (en) | Cyclic analogs of atrial natriuretic peptides | |
| EP1115739B1 (en) | Auxiliary for amide bond formation | |
| MacDonald et al. | Approaches to cyclic peptide beeta turn mimics | |
| JPH06510028A (en) | Lanthionine cross-linked peptide | |
| Bergeron et al. | Development of a hypusine reagent for peptide synthesis | |
| EP1032586A1 (en) | Hypusine peptides | |
| Hu et al. | Cyclosporin analogs modified in the 3, 7, 8-positions: substituent effects on peptidylprolyl isomerase inhibition and immunosuppressive activity are nonadditive | |
| JP3436559B2 (en) | Peptide synthesis method and novel synthetic intermediate | |
| US6492489B1 (en) | Synthetic hypusine peptides | |
| CN118591551A (en) | Synthetic methods for producing modified GCC receptor agonists | |
| Spatola | Synthesis of pseudopeptides | |
| EP0370165B1 (en) | Novel calcitonin derivative and salt thereof | |
| WO2023097207A1 (en) | Synthetic process for production of modified gcc receptor agonists | |
| Brown et al. | Peptide synthesis. Part 4. Solid-phase syntheses of peptides related to gastrin | |
| EP0765341A1 (en) | Template directed cyclization | |
| AU766495B2 (en) | Synthesis of cyclic peptides | |
| US20110046348A1 (en) | Methods of preparing peptide derivatives | |
| MXPA00010056A (en) | Process for the preparation of resin-bound cyclic peptides | |
| Warnke | Studies in Peptide Synthesis | |
| Angell | The solid-phase synthesis of cyclosporin A analogues |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2310027 Country of ref document: CA Ref country code: CA Ref document number: 2310027 Kind code of ref document: A Format of ref document f/p: F |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 522120 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998943259 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998943259 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998943259 Country of ref document: EP |