WO1999035156A1 - Novel macrolides - Google Patents
Novel macrolides Download PDFInfo
- Publication number
- WO1999035156A1 WO1999035156A1 PCT/IB1998/002099 IB9802099W WO9935156A1 WO 1999035156 A1 WO1999035156 A1 WO 1999035156A1 IB 9802099 W IB9802099 W IB 9802099W WO 9935156 A1 WO9935156 A1 WO 9935156A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- groups
- substituted
- optionally
- alkenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCC*(*)C(C)C(C(C)(*)C(O*(C(C)C(C(*)*(C(C)(*)*)OC(C1O)OC(C)C*C1N(C)C)O*)=O)I)O Chemical compound CCC*(*)C(C)C(C(C)(*)C(O*(C(C)C(C(*)*(C(C)(*)*)OC(C1O)OC(C)C*C1N(C)C)O*)=O)I)O 0.000 description 10
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
Definitions
- This invention relates to novel erythromycins and azalides that are useful as antibacterial agents and antiprotozoa agents and other applications (e.g , anticancer, atherosclerosis, gastric mo lity reduction, etc.) in mammals, including man, as well as in fish and birds.
- This invention also relates to pharmaceutical compo ⁇ itions containing the novel compounds and to methods of treating bacterial infections and protozoa infections and in mammals, fish and birds by administering the novel compounds to mammals, fish and birds requiring such treatment
- Macrolide antibiotics are known to be useful In the treatment of a broad sprectrum of bacterial infections and protozoa infections in mammals, fish and birds.
- Such antibiotics include various derivatives of erythromycin A such as azithromycin which is commercially available and is referred to in United States patents 4,474,758 and 4,517,359, both of which are incorporated herein by reference in their entirety.
- Additional macrolides are referred to in U.S. patent application serial number 60/063676, filed October 29, 1597 (Yong-Jin Wu), U.S appiication serial number 60/063161, filed October 29, 1997 (Yong-Jin Wu), U.S.
- PCT/GB97/01810 filed July 4, 1997 (Peter Francis Leadlay, James Staunton, Jesus Cortes and Michael Stephen Pacey), International Application No. PCT/GB97/01819 filed July 4, 1997 (Peter Francis Leadlay, James Staunton, and Jecuc Cort ⁇ e), U.S. application serial no, 60/070343, filed January 2, 1998, (Diriam), U.S. application serial no, 60/070358, filed January 2, 1998 (Yong-Jin Wu) and U.S. application serial no. 60 ⁇ 097075, filed August 19, 1998 (He ⁇ grniao Cheng, Michael A. Letavic, Carl B. Ziegier, Jason K, Dutra, Bnan S.
- novel macrolide compounds of the present invention possess potent activity against various bacterial infections and protozoa infections as described below. Summary of the Invention The present invention relates to compounds of the formula
- Y is H, d-Cio alkyl, c 2 -C-
- m is an inteoer ranging from 0 to 4, and wherein the alkyl, alkenyl, aryl, heteroaryl and alkynyl groups are optionally substituted by 1 to 3 sub ⁇ tituents independently selected from halo, cyano, nitro, trifluoromethyl, azido, -C(0)R Z1 , -OC(0)R 21 , - NR 21 C(0)R 22 , -C(0)NR 21 R 22 , -NR 21 R 22 , hydroxy, C,-C 6 alkyl, C r C ⁇ alkoxy, C 6 -C 10 aryl, and 5- 10 membered heteroaryl;
- R 1 is an alpha-branched Cj-C 8 alkyl, alkenyl, alkynyl, alkoxyalkyl or alkylthioalkyl group any of which may optionally be substituted by one or more hydroxyl groups; a Cs-C ⁇ cycloalkylalkyl group wherein the alkyl group is an alpha-branched C C 5 alkyl group; a C,-C 3 cycloalkyl or C 5 -C profession cycloalke ⁇ yl group, either of which may optionally be substituted by methyl or one or more hydroxyl or one or more C,-C alkyl groups or halo atoms: or a 3 to 6 membered oxygen or sulphur containing heterocyclic ring which may be saturated, or fully or partially unsaturated and which may optionally be substituted by one or more 0,-0-, alkyl groups or halo atoms; or R 1 is phenyl which may be optionally substituted with at least one substitu
- X is O, S or -CH 2 -, a, b, c, and d are each independently an integer ranging from 0 to 2 and a + b ⁇ c + d ⁇ 5; or R 1 is CH 2 R 2 *, wherein R 24 is H, C.-C a alkyl, C 2 -C 8 alkenyl, C 2 -C ⁇ alkynyl, alkoxyalkyl or alkylihloalkyl containing from 1 to s carbon aioms In each alkyl or alkoxy group wherein any of said alkyl. alkoxy.
- alkenyl or alkynyl groups may be substituted by one or more hydroxyl groups or by one or more halo atoms; or a ( VC ⁇ cycloalkyl or C 3 -C B cycloalkenyl either or which may be optionally substituted by methyl or one or more C,-C 4 alkyl groups or halo atoms; or a 3 to 6 membered oxygen or sulphur containing heterocyclic ring which may be saturated or fully or partially unsaturated and which may optionally be substituted by one or more d-C ⁇ alkyl groups or halo atoms; or a group of the formula SR 43 wherein R 23 is C n - C 8 alkyl, C 2 -C Threadalkenyl, C 2 -C 8 a
- R 2 is H or OH
- R 9 is H, OH, or OCH 3 ;
- R 4 is H, -C(0)R 9 , -C(0)OR 9 , -C(0)NR 8 R 10 or a hydroxy protecting group;
- R 5 is -SR 8 , -(CH 2 ) n C(0)R ⁇ wherein n is 0 or 1 , C,-C 10 alkyl, C 2 -C 10 alkenyl, C 2 -
- each R 6 and R 7 is independently H, hydroxy, C r C s alkoxy, C r C, alkyl, C 2 -C ⁇ alkenyl, C 2 -C 6 alkynyl, -(CH 2 ) m (C 6 -C 10 aryl), or -(CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4;
- each R B is independently H, C r C 10 alkyl, C2-C 10 alkenyl, C 2 -C 10 alkynyl, -(CH 2 ) e( CR 11 R 1z (CH 2 ) l
- a 4-7 membered saturated ring is formed that optionally includes 1 or 2 carbon-carbon double or triple bonds; or R S and R 14 are taken together to form a 4-10 membered monocyclic or poiycyclic saturated ring or a 5-10 membered heteroaryl ring, wherein 3aid saturated and heteroaryl rings optionally include 1 or 2 heteroatoms selected from O, S and -N(R B )-, in addition to the nitrogen to which R 13 and R 14 are attached, said saturated ring optionally includes 1 or 2 carbon-carbon double or triple bonds, and said saturated and heteroaryl rings are optionally substituted by 1 to 3 R 1s groups;
- R 1S is H. C ⁇ -C, 6 alkyl, C a -C 10 alkenyl. Or C z -C,_ alkynyl, wherein the foregoing R 1S groups are optionally substituted by 1 to 3 substituents independently selected from halo and - OR 9 ; each R 1B is independently selected from halo, cyano, nitro, trifluoromethyl, azido, - C(0)R 17 , -C(0)OR 17 , -C(0)OR 17 , -OC(0)OR 17 , -NR 6 C(0)R 7 , -C(0)NR 8 R 7 , -NR 6 R 7 , hydroxy, C-rCg alkyl, C
- each R 17 is independently selected from H, C C 10 alkyl, C 2 -C 10 alkenyl, C 2 -C 10 alkynyl, -(CH 2 ) m (C e -C 10 aryl), and -(CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, provided that R a is not H where R 1 ⁇ is -CH 2 S(0) n R e ; R 1, is OH;
- R 9 is C r C,o alkyl, C 2 -C 10 alkenyl, C 2 -C, 0 alkynyl, cyano, -CHjS(O) 0 R 8 wherein n is an integer ranging from 0 to 2, -CH 2 OR e , -CH 2 N(OR 9 )R e , -CH 2 NR*R 15 , -(CH 2 ) m (C e -C, Q aryl), or - (CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, and wherein the foregoing R 10 groups are optionally substituted by i to 3 R 1 ⁇ groups; or R 1 ⁇ and R 19 are taken together to form an oxazolyl ring as shown below
- each R 21 and R 22 is independently H, hydroxy, C r C ⁇ alkoxy, C,-C ⁇ alkyl, C 2 -C ⁇ alkenyl, (CH z ) m (C 6 -C 1D ) aryl, (CH 2 ) m (5-1 ⁇ membered heteroaryl), wherein m Is an integer ranging from 0 to 4, or C 2 -C
- the present invention further relates to compounds of the formula
- Y is H, C t -C 10 alkyl, C 2 -C 10 alkenyl, C z -C l0 alkynyl, -(CH 2 ) m C 6 -C l0 aryl, -(CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, and wherein the alkyl, alkenyl, aryl, heteroaryl and alkynyl groups are optionally substituted by 1 to 3 substituents independently selected from halo, cyano, nitro, trifluoromethyl, azido, -C(0)R 2 ⁇ -OC(0)R 21 , - NR 21 C(0)R 22 , -CfOJN ⁇ R 22 , -NR 2l R a hydroxy, C,-C 6 alkyl, C,-C 6 alkoxy, C B -C, 0 aryl, and 5- 10 membered heteroaryl; R 1 Is an
- X is O, S or -CH 2 -, a, b, c, and d are each independently 0-2 and a + b + c + d ⁇ 5; or R 1 is CH t R 24 , wherein R 24 is H, C,-C B alkyl, d-C ⁇ alkenyl, C C B alkynyl, alkoxyalkyl or alkylthioalkyl containing from 1 to 6 carbon atoms in each alkyl or alkoxy group wherein any of said alkyl, alkoxy, alkenyl or alkynyl groups may be substituted by one or more hydroxyl groups or by one or more halo atoms; or a C 3 -C ⁇ cycloalkyl or C 5 -C B cycloalkenyl either or which may be optionally substituted by methyl or one or more C r C 4 alkyl groups or halo atoms; or a 3 to 6 membered oxygen or
- R z is H or OH;
- R 3 is H, OH, or OCH 3 ;
- R 4 is H, -C(0)R 9 , -C(0)OR B , -C(0)NR 9 R 10 or a hydroxy protecting group;
- each R 9 and R 19 is independently H or d-d alkyl;
- R" is OH;
- each R 21 and R 22 is independently H, hydroxy, C r C Pain alkoxy, C r C s alkyl, C 2 -C 6 alkenyl, (CH 2 ) m (C 8 -C,o) aryl, (CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, or C 2 -C l0 al ytyl.
- the compounds of formula I are exemplified by the compounds of formulas 5 and 6, described below, wherein Y m H and CH 3 , respectively.
- R 1 is isopropyl, R 2 is H and R 1 ⁇ is OH;
- R 1 is isopropyl, R 2 is OH and R 19 is OH;
- R 1 is cyclopropyl, R 2 is H and R 19 is OH;
- R 1 is cyclopropyl, R z is OH and R 1 ⁇ is OH;
- R 1 is sec-butyl, R 2 is H and R 1 ⁇ is OH;
- R 1 is sec-butyl, R 2 is OH and R 19 is OH;
- R 1 Is cyclobutyl, R 2 Is H and R 19 Is OH;
- R 1 is cyclobutyl, R 2 is OH and R 19 is OH;
- R 1 is cyclopentyl, R 2 is H and R 19 is OH;
- R 1 is cyclopentyl, R 2 is OH and R 1 ⁇ is OH; R 1 is methylthioethyl, R z is H and R 19 is OH;
- R 1 is methylthioethyl, R 2 is OH and R 1 ⁇ is OH;
- R 1 is 3-furyl
- R 2 is H
- R 19 is OH and or
- R 1 is 3-furyl
- R 2 is OH
- R 19 is OH
- the invention further relates to compounds of the 2, 2a, 3, 3a, 4, 5, 6, 7, 8, 9, 10, 11. 12. H. H. J!, .16. 17 and 18.
- the invention further relates to a compound of the formula
- R 1 is an alpha-branched d-d alkyl, alkenyl, alkynyl, alkoxyalkyl or alkylthioalkyl group any of which may optionally be substituted by one or more hydroxyl groups; a C 5 -C 3 cycloalkylalkyl group wherein the alkyl group is an alpha-branched C 2 -C 3 alkyl group; a C 3 -C 3 cycloalkyl or C 5 -C.
- R 1 is phenyl which may be optionally substituted with at least one substituent selected from C r C 4 alkyl, C,-C 4 alkoxy and C,-C 4 alkylthio groups, halogen atoms, hydroxyl groups, trifluoromethyl, and cyano; or R may be with a formula (a) as shown below:
- R 1 is CH 2 R 24 , wherein R* 4 is H, C,-C B alkyl, C 2 -C B alkenyl, C 2 -C 8 alkynyl, alkoxyalkyl or alkylthioalkyl containing from 1 to 6 carbon atoms in each alkyl or alkoxy group wherein any of said alkyl, alkoxy, alkenyl or alkynyl groups may be substituted by one or more hydroxyl groups or by one or more halo atoms; or a C 3 -C 8 cycloalkyl or C r C 8 cycloalkenyl either or which may be optionally substituted by methyl or one or more C,-C 4 alkyl groups or halo atoms; or a 3 to 6 membered oxygen or s
- R 2 is H or OH.
- the invention further relates to a compound of the formula
- R 1 is an alpha-branched C 3 -C B alkyl, alkenyl, alkynyl, alkoxyalkyl or alkylthioalkyl group any of which may optionally be substituted by one or more hydroxyl groups; a C 3 -C 8 cycloalkylalkyl group wherein the alkyl group is an alpha-branched C 2 -C 3 alkyl group; a C 3 -C 8 cycloalkyl or d-C, cycloalkenyl group, either of which may optionally be substituted by methyl or one or more hydroxyl or one or more d-d alkyl groups or halo atoms; or a 3 to 6 membered oxygen or sulphur containing heterocyclic ring which may be saturated, or fully or partially unsaturated and which may optionally be substituted by one or more C,-d alkyl groups or halo atoms; or R' is phenyl, alkynyl, alk
- X is O, S or -CH Z -, a, b, o, and d are each independently 0-2 and a + b + c * d ⁇ 6; or R 1 is CHj-R 24 , wherein R 24 is H, C,-C 8 alkyl, C 2 -C 8 alkenyl, C 2 -C B alkynyl, alkoxyalkyl or alkylthioalkyl containing from 1 to 6 carbon atoms in each alkyl or alkoxy group wherein any of said alkyl, alkoxy, alkenyl or alkynyl groups may be substituted by one or more hydroxyl groups or by one or more halo atoms; or a Ca-C ⁇ cycloalkyl or C 3 -C 8 cycloalkenyl either or which may be optionally substituted by methyl or one or more C,-C 4 alkyl groups or halo atoms; or a 3 to 6 membered oxygen or s
- the invention also relates to a pharmaceutical composition for the treatment of a bacterial infection or a protozoa infection in a mammal, fish, or bird which comprises a therapeutlcally effective amount of a compound of formulas I, II, 8, or 9, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the invention also relates to a method of treating a bacterial infection or a protozoa infection in a mammal, fish, or bird which comprises administering to said mammal, fish or bird a therapeutically effective amount of a compound of formula I, II, 8, or 9, or a pharmaceutically acceptable salt thereof.
- the invention also relates to a process for preparing a compound of the formula
- R 1 and R 2 are as defined for the compound of formula I, which comprises treating a compound of the formula
- R 1 and R 2 are as defined for the compound of formula I, with a reducing agent
- the invention also relates to the above process wherein the reducing agent is NaBH 4 or platinum oxide.
- the invention also relates to a process for preparing a compound of the formula
- R 1 and R 2 are as defined for the compound of formula I, which comprises treating a compound of the formula
- R* and R 2 are as defined for the compound of formula I, with a methylating agent.
- Trie Invention also relates to the above process wherein the methylating agent Is formaldehyde.
- the invention also relates to a process for preparing a compound of the formula
- R 1 and R 2 are as defined for the compound of formula I, which comprises treating a compound of the formula
- R 1 and R 2 are as defined for the compound of formula I, with a reducing agent.
- the invention also relates to the above process wherein the reducing agent is NaBH 4 or platinum oxide.
- the invention also relates to a process for preparing a compound of the formula
- R 1 and R 2 are as defined for the compound of formula I, which comprises treating a compound of the formula
- R 1 and R 2 are as defined for the compound of formula I, with a methylating agent.
- the Invention also relates to tha above process wherein the methylating agent is formaldehyde.
- the invention also relates to a compound of the formula
- Y is H, d-C . o alkyl, C 2 -C 10 alkenyl, C 2 -C 10 alkynyl, -(CH 2 ) m C 6 -C 10 aryl, -(CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, and wherein the alkyl, alkenyl, aryl, heteroaryl and alkynyl groups are optionally substituted by 1 to 3 substituents independently selected from halo, cyano, nitro, trifluoromethyl, azido, -C(0)R 21 , -OC(0)R 21 , - NR 21 C(0)R 22 , -C(0)NR 21 R 2Z , -NR 1 R 22 , hydroxy, d-C 3 alkyl, d- alkoxy, C 6 -C 1Q aryl, and 5- 10 membered heteroaryl;
- R 1 is an alpha-branched C 3 -C Conduct alkyl. alkenyl, alkynyl, alkoxyalkyl or alkylthioalkyl group any of which may optionally be substituted by one or more hydroxyl groups; a C S -C B cycloalkylalkyl group wherein the alkyl group is an alpha-branched d-d alkyl group; a d-d cycloalkyl or C 3 -C 8 cycloalkenyl group, either of which may optionally be substituted by methyl or one or more hydroxyl or one or more C r C 4 alkyl groups or halo atoms; or a 3 to 6 membered oxygen or sulphur containing heterocyclic ring which may be saturated, or fully or partially unsaturated and which may optionally be substituted by one or more C alkyl groups or halo atoms; or R 1 is phenyl which may be optionally substituted with at least one substituent selected
- R 2 is H or OH
- R 3 is H, OH, or OCH 3 ;
- R 4 is H, -C(0)R 9 , -C(0)OR*, -C(0)NR* R 1 ⁇ or a hydroxy protecting group
- R 6 is -SR 8 , -(CH 2 ) friendshipC(0)R 8 wherein n is 0 or 1, C,-C 10 alkyl, C 2 -C 10 alkenyl, C 2 - do alkynyl, -(CH 2 ) m (C ⁇ -C,o aryl), or -(CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, and wherein the foregoing R s groups are optionally substituted by 1 to 3 R 18 groups; each R a ⁇ d R 7 is independently H, hydroxy, 0,-0 8 alkoxy, 0-,-C ⁇ alkyl, C 2 -C 6 alkenyl, C 2 -C classroom alkynyl, -(CH 2 ) r ⁇ (C 6 -C 10 aryl
- R 1S is H, d-C 1p alkyl, C 2 -C 10 alkenyl, or C 2 -C 10 alkynyl, wherein the foregoing R lS groups are optionally substituted by 1 to 3 substituents independently selected from halo and - OR ⁇ ; each R 1S is independently selected from halo, cyano, nitro, trifluoromethyl, azido, - C(0)R 17 , -C(0)OR 17 , -C(0)OR 17 , -OC(0)OR 17 , -NR 6 C(0)R 7 , -C(0)NR 6 R 7 , -NR 6 R 7 , hydroxy, C,-C 6 alkyl, CrC 6 alkoxy, -(CH 2 ) m (C 6 -C 10 aryl), and -(CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, and wherein said aryl and heteroaryl subsituent
- each R 17 is independently selected from H, C C 0 alkyl, C j -C 1D alkenyl, Ci-C 1 ⁇ alkynyl, -(CH 2 ) m (d-do aryl), and -(CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, provided that R ⁇ is not H where R 19 is -CH 2 S(0) n R B ; R 1B is OH;
- R 19 is d-C 10 alkyl, C r C 10 alkenyl, C 2 -C 1Q alkynyl, cyano, -CH 2 S(0) n R B wherein ⁇ is an integer ranging from 0 to 2, -CH 2 0R 8 , -CH 2 N(OR ⁇ )R ⁇ , -CH 2 NR B R 15 , -(CH a ) -do aryl), or - (CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, and wherein the foregoing R 1B groups are optionally substituted by 1 to 3 R 16 groups; or R 1a and R 19 are taken together to form an oxazolyl ring as shown below
- each R 21 and R 22 is independently H, hydroxy, d-C B alkoxy, C,-C 6 alkyl, C 2 -C 6 alkenyl, (CH 2 ) m (C 6 -C 0 ) aryl, (CH 2 ) m (5-10 membered heteroaryl), wherein m is an integer ranging from 0 to 4, or C 2 -C 10 alkylyl.
- treatment includes the treatment or prevention of a bacterial infection or protozoa infection as provided in the method of the present invention.
- bacterial infection(s) or "protozoa infections; includes bacterial infections and protozoa infections that occur in mammals, fish and birds as well as disorders related to bacterial infections and protozoa infections that may be treated or prevented by administering antibiotics such as the compounds of the present invention.
- Such bacterial infections and protozoa infections and disorders related to such infections include the following; pneumonia, otitis media, sinusitus, bronchitis, tonsillitis, and mastoiditis related to infection by Streptococcus pneumoniae, Haemophilus influenzae, Moraxalla catarrhalis, Staphylococcus aureus, or P ⁇ ptostreptococcus spp.; pharynigitis, rheumatic fever, and glomerulonephritis related to infection by Streptococcus pyogenes, Groups C and G streptococci, Clostridium diptheria ⁇ , or Actinobacillus ha ⁇ molytlcum; respiratory tract infections related to infection by Mycoplasma pneumoniae, Legionella pneumophila, Streptococcus pneumoniae, Haemophilus influenzae, or Chlamydia pneumoniae; uncomplicated skin and soft tissue infections, abscesses and osteomyelitis, and puerper
- aureus food poisoning and Toxic shock syndrome
- Groups A, B, and C streptococci ulcers related to infection by H ⁇ llcobact ⁇ r pylori: systemic febrile syndromes related to WO 99/35156 _ ⁇ g _ PCT/IB98/02099
- influenzae or List ⁇ ria spp.
- disseminated Mycobact ⁇ rium avium complex (MAC) disease related to infection by Mycobacterium avium, or Mycobacterium intracellulare
- gastroenteritis related to infection by Cdmpyl b ⁇ ct ⁇ r j ⁇ j ⁇ ni
- intestinal protozoa related to infection by Cryptospo ⁇ dium spp.
- odontogenic infection related to infection by viridans streptococci
- persistent cough related to infection by Bordetella pertussis
- gas gangrene related to infection by Clostridium perf ⁇ ngens or Bacteroides spp.
- atherosclerosis related to infection by Helicobacter pylon or Chlamydia pneumoniae.
- Bacterial infections and protozoa infections and disorders related to such infections that may be treated or prevented in animals include the following: bovine respiratory disease related to infection by P. ha ⁇ m., P. multocida, Mycoplasma bows, or Bordetella spp.; cow enteric disease related to infection by E. coll or protozoa (i.e., coccidia, cryptosporidia, etc.); dairy cow mastitis related to infection by Sfap ⁇ .
- aureus swine respiratory disease related to infection by A. pleuro., P. multocida, or Mycoplasma spp.
- swine enteric disease related to infection by E. coli, Lawsonia intracellularis, Salmonella, or Serpulina hyodyisinteriae
- cow footrot related to infection by Fusobacterium spp.
- cow hairy warts related to infection by Fusobacterium necrophorum or Bacteroides nodosus cow pink-eye related to infection by Moraxalla bo ⁇ is; cow premature abortion related to infection by protozoa (i.e. neosporium); urinary tract infection in dogs and cats related to infection by £ coli; skin and soft tissue infections in dogs and cats related to infection by Staph. epidermidis, Staph. int ⁇ rmedius, coagulas ⁇ n ⁇ g. Staph. or P.
- the present invention also relates to a method of preparing the above compounds of formulas I, II, 8, or 9.
- the compounds used in the preparation of the compounds of formulas I, II, 8, and 9 can prepared using the methods described in International Application No. PCT/GB97/01810 filed July 4, 1997 (Peter Francis Leadlay, James Staunton, Jesus Cortes and Michael Stephen Pacey), and International Application No. PCT/GB97/01819 tiled July 4, 1997 (Peter Francis Leadlay, James Staunton, and Jesus Cortes), both of which are is incorporated herein by reference in their entirety.
- the present Invention also relates to the compounds of formulas 2, 2a, 3 ⁇ 3a, and 4 to
- hydroxy protecting group includes acetyl, benzyloxycarbonyl, and various hydroxy protecting groups familiar to those skilled in the art include the groups referred to in T. W. Greene, P. Q. M. Wuts, "Protective Groups In Organic Synthesis,” (J. Wiley & Sons, 1991),
- halo as used herein, unless otherwise indicated, includes fluoro, chloro, bromo or iodo.
- alkyr as used herein, unless otherwise Indicated, includes saturated monovalent hydrocarbon radicals having straight, cyclic or branched moieties, or mixtures thereof. It is to be understood that where cyclic moieties are intended, at least three carbons in said alkyl must be present Such cyclic moieties include cyclopropyl, cyclobutyl and cyclopentyl.
- alkoxy as used herein, unless otherwise indicated, includes -O-alkyl groups wherein alkyl is as defined above.
- aryl as used herein, unless otherwise indicated, includes an organic radical derived from an aromatic hydrocarbon by removal of one hydrogen, such as phenyl or naphthyl.
- 5-10 membered heteroaryl includes aromatic heterocyclic groups containing one or more heteroatoms each selected from O, S and N, wherein each heterocyclic group has from 5 to 10 atoms in its ring system.
- Suitable 5-10 membered heteroaryl groups include pyridinyl, imidazolyl, pyrimidinyl, pyrazolyl, (1 ,2,3,)- and (1 ,2,4)-triazolyl, pyra-zinyl, tatrazolyl, furyl, thionyl, isoxazolyl, oxazolyl, pyrrolyl and thiazolyl,
- pharmaceutically acceptable salt(s) includes salts of acidic or basic groups which may be present in the compounds of the present invention.
- the compounds of the present invention that are basic in nature are capable of forming a wide variety of salts with various inorganic and organic acids.
- acids that may be used to prepare pharmaceutically acceptable acid addition salts of such basic compounds of the present invention are those that form non-toxic acid addition salts, Le., salts containing pharmacologically acceptable anlons, such as the hydrochlorlde, hydrobromlde, hydrolodlde, nitrate, sulfate, bisulfate, phosphate, acid phosphate, isonicotinate, acetate, lactate, salicylate, citrate, acid citrate, tartrate, pa ⁇ tothenate, bitartrate, ascorbate, succi ⁇ ate, maleate, gentisinate, fumarate, gluconate, glucaronat ⁇ , saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfo ⁇ ate, p-toluenesulfonate and pamoate [i.e., l ,l'-methylene-bis-(2- hydroxy
- Those compounds of the present invention that are acidic in nature are capable of forming base salts with various pharmacologically acceptable cations.
- Examples of such salts include the alkali metal or alkaline earth metal salts and, particulariy, the calcium, magnesium, sodium and potassium salts of the compounds of the present invention.
- Certain compounds of the present invention may have asymmetric centers and therefore exist in different enantiomeric and diastereomeric forms.
- This invention relates to the use of all optical Isomers and stereolsomers of the compounds of the present invention, and mixtures thereof, and to all pharmaceutical compositions and methods of treatment that may employ or contain them.
- the present invention includes the compounds of the present invention, and the pharmaceutically acceptable salts thereof, wherein one or more hydrogen, carbon or other atoms are replaced by isotopes thereof.
- Such compounds may be U3eful as research and diagnostic tools in metabolism pharmacokinetic studies and in binding assays.
- the compounds of the present invention are readily prepared.
- the compounds used in the preparation of the compounds of formulas I, II, 8, and 9 can prepared using the methods described in International Appiication No. PCT/GB97/01810 filed July 4, 1997 (Peter Francis Leadlay, James Staunton, Jesus Cortes and Michael Stephen Pacey), and International Application No. PCT/GB97/01819 filed July 4, 1997 (Peter Francis Leadlay, James Staunton, and Jesus Cortes), both of which are Is Incorporated herein by reference in their entirety.
- novel polyketides and methods and means for preparing them, and specifically the novel macrolides that are useful in the preparation of the compounds of the present invention are prepared by fermenting suitable organisms in the presence of where R 1 is as defined in claim 1 or claim 2.
- a preferred organism is Saccharopolyspora erythraea preferably containing an integrated plasmid capable of directing synthesis of desired compounds,
- polyketide biosynthetic genes or portions of them, which may be derived from different polyketide biosynthetic gene clusters are manipulated to allow the production of novel erythromycins,
- Polyketides are a large and structurally diverse class of natural products that includes many compounds possessing antibiotic or other pharmacological properties, such as erythromycj ⁇ , tetracyclines, rapamycin, avermectin, polyether ionophores, and FK5D6.
- antibiotic or other pharmacological properties such as erythromycj ⁇ , tetracyclines, rapamycin, avermectin, polyether ionophores, and FK5D6.
- polyketides are abundantly produced by Streptomyces and related actinomycete bacteria.
- PKS polyketide synthase
- Naturally module refers to the set of contiguous domains, from a ⁇ -ketoacylsynthase (“KS”) gene to the next acyl carrier protein (“ACP”) gene, which accomplishes one cycle of polyketide chain extension.
- KS ⁇ -ketoacylsynthase
- ACP acyl carrier protein
- combinatorial module is used to refer to any group of contiguous domains (and domain parts), extending from a first point in a first natural module, to a second equivalent point in a second natural module.
- the first and second points will generally be in core domains which are present in all modules, i.e., both at equivalent points of respective KS, AT (acyl transferase), ACP domains, or in linker regions between domains.
- T e organisation of the erythromycin producing PKS, (also known as ⁇ - deoxyerythronolide B synthase, DEBS) genes contains three open reading frames encode the DEBS polypeptides. The genes are organised in six repeated units designated modules.
- the first open reading frame encodes the first multi-enzyme or cassette (DEBS1) which consists of three modules: the loading module (ery_-load) and two extension modules (modules 1 and 2).
- the loading module comprises an acyl transferase and an acyl carrier protein.
- the complex polyketides produced by modular Type I PKS's are particularly valuable, in that they include compounds with known utility as anthelminthics, insecticides, immunosuppressants, antifungal, and/or antibacterial agents. Because of their structural complexity, such novel polyketides are not readily obtainable by total chemical synthesis, or by chemical modifications of known polyketides.
- the Type I PKS gene assembly encodes a loading module which is followed by extension modules. It is particularly useful to provide a hybrid PKS gene assembly in which the loading module is heterologous to the extension modules and is such as to lead to a polyketide having an altered starter unit.
- W093/13863 refers to altering PKS genes by inactivating a single function (i.e. a single enzyme) or affecting "an entire module" by deletion, insertion, or replacement thereof.
- the loading assembly in their terms, is not a module.
- the hybrid gene assembly can be used to produce many different polyketide ⁇ .
- a hybrid gene assembly may employ nucleic acid encoding an avr loading module with ery extender modules.
- a loading module may accept unnatural acid units and derivatives thereof, the avr loading module is particulariy useful in this regard (Dutto ⁇ et al,, (1991) J. Antibiot., 44:357-365).
- Methyl and other substituents can be added or removed by acyltransferase domain replacement or total module replacement. Consequently, it also becomes apparent to those skilled in the art that it is possible to combine the use of the relaxed substrate specificity of the erythromycin loading module with extension module replacement and hybrid leading module substitution with extension module replacement as a mechanism to produce a wide range of novel erythromycins.
- International Application PCT/GB97/01810 describes the production of novel erythromycins by non-transformed organisms and also such gene assemblies, vectors containing such gene assemblies, and transformant organisms that can express them to produce novel erythromycins in transformed organisms. Transformant organisms may harbour recombinant plasmids, or the plasmids may integrate.
- a plasmid with an int sequence will integrate into a specific attachment site (att) of a host's chromosome.
- Transformant organisms may be capable of modifying the initial products, e.g., by carrying out all or some of the biosynthetic modifications normal in the production of erythromycins. However, use may be made of mutant organisms such that some of the normal pathways are blocked, e.g., to produce products without one or more "natural" hydroxy-groups or sugar groups, for instance as described in WO 91/16334 or in Weber et al. (1985) J. Bacterlol. 164:425-433 hich are Incorporated herein by reference in their entirety.
- the DNA is highly conserved here between all modular PKS's, and this may aid in the construction of hybrids that can be transcribed. It may also assist in maintaining the spacing of the active sites of the encoded enzymes, which may be important
- the ery module together with a small amount of the following ketosynthase (KS) domain was removed.
- the start of the KS domain (well spaced from the active site) is highly conserved and therefore provides a suitable splicing site as an alternative to the linker region between the loading domain and the start of the KS domain.
- the excised er ⁇ module was then replaced by an avr loading module.
- acyl transferase AT
- ACP acyl carrier protein
- KS acyl carrier protein
- the excised loading module would have provided a propionate starter, and the replacement is intended to provide one or more different starters.
- Propionate may feed into the KS of the extension module from a propionate pool in the host cell, leading to dilution of the desired products. This can be largely prevented by substituting an extended loading module including all or most of the KS domain.
- the splice site may be in the end region of the KS gene, or early in the following AT gene, or the linker region between them.
- a “combinatorial module” to be excised and/or replaced and/or inserted may extend from the corresponding domain of two natural-type modules, e.g., from the AT of one module to the AT of the next, or from KS to KS.
- the splice sites will be in corresponding conserved marginal regions or in linker regions.
- a combinatorial module can also be a 'double' or larger multiple, for adding 2 or more modules at a time.
- An erythromycin analogue (being a macrolide compound with a 14-membered ring) in which a substituent R, on the C-13 position, bears a side-chain other than ethyl, generally a straight chain C3-C6 alkyl group, a branched C 3 -C 8 alkyl group, a C3-C 8 cycloalkyi or cycloalkenyl group (optionally substituted, e.g., with one or more hydroxy, C-)_ alkyl or alkoxy groups or halogen atoms), or a 3-6 membered heterocycle containing O or S, saturated or fully or partially unsaturated, optionally substituted (as for cycloalkyi), or R is phenyl which may be optionally substituted with at least one substituent selected from C,-C 4 alkyl, C 1 -C alkoxy and C,-C 4 alkylthio groups, halogen atoms, trifluoromethyl
- Preferred candidate ⁇ for the C-13 substituent R are the groups of carboxylate units RCOOR', usable as substrates by an avr starter module, or rapamycin starter variants.
- Preferred substrates are the carboxylic acids RCOOH.
- Alternative substrates that can be effectively used are carboxylic acid salts, carboxylic acid esters, or amides.
- Preferred esters are N- acetyl-cysteami ⁇ e thioesters which can readily be utilised as substrates by the avr starter module as illustrated by Dutton et al.
- amides are N-acyl imidazoles.
- Other alternative substrates that may be used are derivatives which are oxidative precursors for the carboxylic acids; thus, for example suitable substrates would be amino acids of the formula RCH(NH 2 )COOH, glyoxylic acids of the formula RCOCOOH, methylamine derivatives of the formula RCH a NH a , methanol derivatives of the formula RCH 2 OH, aldehydes of the formula RCHO or substituted alkanoie acids of the formula R(CH 2 ) n COOH wherein n is 2, 4, or 6.
- examples of preferred substrates include isobutyrate (Risi-Pr) and 2-methylbutyrate (Ris1-methylpropyl).
- Other possibilities include n-butyrate, cyclopropyl carboxylate, cyclobutyl carboxylate, cyclopentyl carboxylate cyclohexyl carboxylate, cycloheptanyl carboxylate, cyclohexenyl carboxylates, cycloheptenyl carboxylates, and ring-methylated variants of the cyclic carboxylates and the aforementioned derivatives thereof,
- the erythromycin analogue may correspond to the initial product of a PKS (6- daoxyerythranolide) or the product after one or more of the normal biosynthetic steps. These comprise: 6-hydroxylation; 3-0-glycosylation; 5-0-glycosylation; 12-hydro ⁇ ylation; and specific sugar ethylation.
- the analogues may include those corresponding to 6-deoxyerythronoiide B, erythromycin A, and various intermediates and alternatives thereof.
- Erythromycin analogues differing from the corresponding 'natural' in the oxidation state of one or more of the ketjde units (i.e. selection of alternatives from the group: -CO-, - CH(OH)-. IsCH-. and -CH -).
- PKS enzymes e.g., one or more of hydroxylation, epoxidation, glycosylation, and methylation.
- unnatural starter units preferably, but not restricted to the carboxylic acid analogues or the unnatural starter units
- a preferred approach involves introduction of the starter unit into fermentation broths of the erythromycin-producing organism, an approach which is more effective for transformed organisms capable of producing erythromycins.
- the starter unit analogue can also be introduced to alternative preparations of the erythromycin- producing organisms, for example, fractionated or unfractionated broken-cell preparations. Again, this approach is equally effective for transformed organisms capable of producing erythromycins.
- one or more segments of DNA encoding individual modules or domains within a heterologous Type I PKS have been used to replace the DNA encoding, respectively, individual modules or domains within the DEBS genes of an erythromycin-producing organism.
- Loading modules and extension modules drawn from any natural or non-natural Type I PKS are suitable for this "donor” PKS but particularly suitable for this purpose are the components of Type I PKS's for the biosynthesis of erythromycin, rapamycin, avermectin, tetronasin, oleandomycin, monensin, amphotericin, and rifamycin, for which the gene and modular organisation is known through gene sequence analysis, at least in part.
- the loading modules of the donor PKS are those loading modules showing a relaxed specificity, for example, the loading module of the avermectin (avr)-producing PKS of Streptomyces avermitilis; or those loading modules possessing an unusual specificity, for example, the loading modules of the rapamycin-, FK5D6- and ascomycin-producing PKS's. all of which naturally accept a shikimate-derived starter unit.
- both the untransformed and genetically engineered erythromycin-producing organisms when cultured under suitable conditions have been found to produce non-natural erythromycins, and where appropriate, the products are found to undergo the same processing as the natural erythromycin.
- the novel erythromycin products contain a starter unit typical of those used by the avr PKS.
- Saccharopolyspora erythraea strains containing such hybrid PKS are found to produce 14-membered macrolides containing starter units typically used by the avr PKS,
- erythromycin PKS placed under the control of a Type II PKS promoter and activator gene.
- desired genes present on an SCP2*-derived plasmid are placed under the control of the bidirectional actl promoter derived from the actinorhodin biosynthetic gene cluster of Streptomyces coelicolor, and in which the vector also contains the structural gene encoding the specific activator protein Act ll-orf 4.
- the recombinant plasmid is introduced into Saccharopolyspora erythraea, under conditions where either the introduced PKS genes, or PKS genes already present in the host strain, are expressed under the control of the actl promoter.
- Such strains produce the desired erythromycin product and the activator gene requires only the presence of the specific promoter in order to enhance transcriptional efficiency from the promoter. This is particularly surprising in that activators of the Actll-orf4 family do not belong to a recognised class of DNA-binding proteins. Therefore it would be expected that additional proteins or other control elements would be required for activation to occur in a heterologous host not known to produce actinorhodin or a related isochromanequinone pigment.
- the recombinant strains can produce more than ten-fold erythromycin product than when the same PKS genes are under the control of the natural promoter, and the specific erythromycin product is also produced precociously in growing culture, rather than only during the transition from growth to stationary phase, Such erythromycins are useful as antibiotics and for many other purposes in human and veterinary medicine.
- the activator and promoter are derived from the actinorhodin PKS gene cluster and the actl/actll-orf4-regulated ery PKS gene cluster is housed in the chromosome, following the site-specific integration of a low copy number plasmid vector, culturing of these cells under suitable conditions can produce more than ten-fold total 14-membered macrolide product than in a comparable strain not under such heterologous control.
- the activator and promoter are derived from the actinorhodin PKS gene cluster and the actl/actll-orf4-regulated ery PKS gene cluster is housed in the chromosome, following the site-specific integration of a low copy number plasmid vector, culturing of these cells under suitable conditions can produce more than ten-fold total 14-membered macrolide product than in a comparable strain not under such heterologous control.
- the PKS genes under this heterologous control are hybrid Type I PKS genes whose construction is described herein, more than ten-fold hybrid polyketide product can be obtained compared to the same hybrid Type I PKS genes not under ⁇ uch control.
- the hybrid Type I PKS genes are the ery PKS genes in which the loader module is replaced by the avr loading module, a ten-fold increase is found in the total amounts of novel 14-membered macrolides produced by the genetically engineered cells when cultured under suitable conditions as described in PCT/GB97/01810.
- Erythromycin analogus described in International Application PCT/GB97/01810 are produced by fermentation of an untransformed or transformed organism capable of producing erythromycins, including but not limited to Saccharopolyspora species, Streptomyces griseoplanus, Nocardia sp., Micromonospora sp., Arthobacter sp., and Streptomyces antibioticus, but excluding S. coelicolor.
- Particularly suitable in this regard are untransformed and transformed strains of Saccharopolyspora erythraea, for example NR L 2338, 18643, 21484.
- Particularly preferred transformed strains are those in which the erythromycin loading module has been replaced with the loading module from the avermectin producer, Strepbmyces avermitilis, or the rapamycin producer, Streptomyces hygroscopicus.
- the preferred method of producing compounds of the current invention is by fermentation of the appropriate organism in the presence of the appropriate carboxylic acid of the formula R.,COOH, wherein R, is as defined in formulae 1_ or 2 of International Application PCT/GB97/01810 or is R 1 of the compounds of the present invention, or a salt, ester (particularly preferable being the N-acetylcysteamine thioester), or amide thereof or oxidative precursor thereof.
- the acid or derivative thereof is added to the fermentation either at the time of inoculation or at intervals during the fermentation.
- Production of the compounds of this invention may be monitored by removing samples from the fermentation, extracting with an organic solvent and following the appearance of the compounds of this invention by chromatography, for example using high pressure liquid chromatography. incubation Is continued untB the yield of the compound of formulae 1_ or 2 has been maximised, generally for a period of 4 to 10 days.
- a preferred level of each addition of the carboxylic acid or derivative thereof is between 0.05 and 4.0 g/L.
- the best yields of the compounds from formulae 1_ or 2 are generally by gradually adding the acid or derivative to the fermentation, for example by daily addition over a period of several days.
- the medium used for the fermentation may be a conventional complex medium containing assimilable BOU ⁇ C ⁇ S of carbon, nitrogen and trace elements.
- step 1 of Scheme 1 the C-9 carbonyl, of formula 1_, is converted to the corresponding oxim ⁇ at the C-9 position by reacting the macrolide with hydroxylamine or preferably, a hydroxylamine salt such as a hydrochloride.
- At least one molar equivalent is usually employed in a weakly basic, tertiary amine (preferably pyridine) as a solvent; at a temperature range 20-80°C.
- a protic solvent such as methanol
- a base such as barium carbonate
- the preferred conditions employ an excess, (2-4 molar equivalents) of an orgnic sulfonyl chloride, preferably p-t ⁇ luene-sulfonyl chloride, which is reacted with the oxi e, as free base or acid salt) in a mixture of a lower ketone, such as methyl ethyl ketone or acetone, and water containing a large molar excess of sodium bicarbonate, at a temperature of 0-50°C, preferably at 0-30°C.
- a lower ketone such as methyl ethyl ketone or acetone
- step 3 or Scheme 1 Imlno ether, of formula 3, is reduced to a mixture of the laotam, of formula 4, and the azalide, of formula 5.
- a reducing agent such as an organo-metallic catalyst such as platinum oxide in an acid solvent such as glacial acetic acid under a minimum of 50 psi hydrogen pressure.
- a preferred method for the production of 5 incorporates the u ⁇ e of boron reduoing reagents such as sodium borohydride, generally used in excess, 2-10 equivalents, in protic solvents such as methanol or ethylene glycol, at a temperature range of -5-40 ⁇ C, preferably 0-20°C.
- boron reduoing reagents such as sodium borohydride, generally used in excess, 2-10 equivalents
- protic solvents such as methanol or ethylene glycol
- azalide, of formula 5 is converted to formula 6 via reductive methylatlon, using a methylating agent such as formaldehyde in the presence of a reducing agent, preferably formic acid.
- a methylating agent such as formaldehyde
- a reducing agent preferably formic acid.
- the preferred method requires at least one molar equivalent each of formaldehyde and formic acid in an inert solvent such as chloroform at 20-100°C, preferably at 30-60°C.
- step 1 of Scheme 2 the C-9 carbonyl, of formula J , is converted to the corresponding oxime at the C-9 position by reacting the macrolide with hydroxylamine or preferably, a hydroxylamine salt such as a hydrochloride.
- hydroxylamine or preferably, a hydroxylamine salt such as a hydrochloride.
- a protic solvent such as methanol
- a base such as barium carbonate
- step 3a of Scheme 2 oxime, of formula 2a, is rearranged to the corresponding imlno ether via a Beckman rearrangement as outlined In J. Chem. Soc. Perki ⁇ Trans. I, I9 ⁇ , 1181.
- the preferred conditions employ an excess, 2-4 molar equivalents) of an orgnic sulfonyl chloride, preferably p-toiuene-sulfonyl chloride, which is reacted with the oxime, as free base or acid salt) in a mixture of a lower ketone, such as methyl ethyl ketone or acetone, and water containing a large molar excess if sodium bicarbonate, at a temperature of 0-50 p C, preferably at 0-30°C.
- a lower ketone such as methyl ethyl ketone or acetone
- step 4a of Scheme 2 imino ether, of formula 3a, is reduced to the azalide, of formula 5a.
- a reducing agent such as an organo- meialllc catalyst such as for example, platinum oxide
- an acid solvent such as glacial acetic acid under a minimum of 50 psi hydrogen pressure.
- a preferred method for the production of incorporates the use of boron reducing reagents such as sodium borohydride, generally used in excess, 2-10 equivalents, in protic solvents such as methanol or ethylene glycol, at a temperature range of -5-40"C, preferably 0-20°C.
- azalide, of formula 5a is converted to formula 6a via reductive methylation, using a methylating agent such as formaldehyde, in the presence of a reducing agent, preferably formic acid.
- a methylating agent such as formaldehyde
- a reducing agent preferably formic acid.
- the preferred method requires at least one molar equivalent each of formaldehyde and formic acid in an inert solvent such as chloroform at 20- 100°C, preferably at SO- ⁇ o'O.
- the C-2' hydroxy group may be selectively protected by treating the compound of formula ) with one equivalent of acetic anhydride in dichlorometha ⁇ e in the absence of external base to provide the compound of formula 1_1 wherein R is acetyl.
- the acetyl protecting group may be removed by treating the compound of formula V ⁇ _ with methanol at 23 - 65 °C for about 10 to about 48 hours.
- the C-2' hydroxy may also be protected with other protecting groups such as the benzyloxycarbonyl (Cbz) group using methods familiar to those skilled in the art.
- the C-9a amino group may also require protection when Y is H before further synthetic modifications are performed.
- Suitable protecting groups for the amino moiety are Cbz and t-butyloxycarbonyl (Boc) groups.
- the macrolide may be treated with t-butyl dicarbonate in anhydrous tetrahydrofuran (THF) or benzyloxycarbonyl N-hydroxysuccinimide ester or benzylchloroformate to protect the amino group as its t-butyl or benzyl carbamate.
- THF tetrahydrofuran
- benzyloxycarbonyl N-hydroxysuccinimide ester or benzylchloroformate to protect the amino group as its t-butyl or benzyl carbamate.
- Both the C-9a and the C-2' bydroxy may be selectively protected with the Cbz group in one step by treating the compound of formula 1J3, where Y is a hydrogen, with benzylchloroformate in THF and water.
- the Boc group may be removed by acid treatment and the Cbz group may be removed by conventional catalytic hydrogenation.
- the C-9a amino moiety and the C-2' hydroxy group are protected and deprotected as would be deemed appropriate by one skilled in the art.
- step 2 of Scheme 3A the C-4" hydroxy group of the compound of formula 1_1 is oxidized to the corresponding ketone, formula _12, by methods familiar to those skilled in the art
- Compounds of formula 14 can be generated by treating compounds of formula 2 with R 19 MgX ⁇ or R 19 -Li and Mg(X 1 ) 2 .
- X 1 is a halide such as chloro or bromo, in a solvent such as THF, ethylene glycol, dimethyl ether (DME), diisopropyl ether, toluene, diethyl ether, or tetramethylethylenediamine (TMEDA), or a mixture of the foregoing solvents, preferably an ether solvent, at a temperature ranging from about -78°C to about room temperature (20- 25°C).
- a solvent such as THF, ethylene glycol, dimethyl ether (DME), diisopropyl ether, toluene, diethyl ether, or tetramethylethylenediamine (TMEDA), or a mixture of the foregoing solvents, preferably an ether solvent, at a temperature ranging from about -78°C to about room temperature (20- 25°C).
- Scheme 3B Illustrates the preparation of compounds of formula 14 using an epoxide intermediate.
- the compound of formula __3 may be generated by two methods.
- a base such as potassium tert-butoxide, sodium ethoxide, sodium hydride, 1,1,3,3-tetramethylguanidine, 1 ,8-diazabicyclo[5.4.0]undec-7-ene, 1,5- diazabicylo[4.3.0]n ⁇ n-5-ene, potassium ethoxide, or sodium methoxide, preferably a sodium- containing base such as sodium hydride, in a solvent such as THF, an ether solvent, dimethylformamide (DMF), or methyl solfoxide (DMSO), or a mixture of the foregoing solvents, at
- the compound of formula 13 is generated in which the following configuration of the epoxide moiety may predominate:
- the compound of formula 13 may be converted to a compound of formula 14 wherein R 1B is hydroxy and R 19 is a group that is attached to the C-4" carbon through a methyi ⁇ ne group, such as where R 19 is -CH 2 N ⁇ R a or -CH z S( ⁇ ) n R 8 wherein n, R 6 , and R ⁇ are as defined above.
- the compound of formula 13 may be treated with a compound of formula HNR ⁇ R 8 , wherein R 6 and R 8 are as defined above, in the absence or presence of a solvent such as water, methanol, or THF, or a mixture of the foregoing solvents, at a temperature ranging from about room temperature to about 100°C, preferably 60°C, optionally in the presence of a metal halide such as potassium iodide, pyridinium hydrochloride, or tetraalkylammonium haiide reagent such as benzene or toluene at a temperature ranging from about room temperature to about 120°C.
- a solvent such as water, methanol, or THF, or a mixture of the foregoing solvents
- step 1 of Scheme 4 the C-9 carbonyl, of formula
- hydroxylamine or preferably, a hydroxylamine salt such as a hydrochloride.
- at least one molar equivalent, usually an excess, 5-10 equivalents is usually employed in a weakly basic, tertiary amine (preferably pyridine) as a solvent; at a temperature range 20-80°C.
- a protic solvent such as methanol, can also be used in conjunction with a base, such as barium carbonate, to provide a weakly basic solvent.
- the oxime of formula 6 may be reduced to the corresponding imine by treating with a metal halide such at TiCl 3 in an acetate buffered solvent such as methanol or ethanol, methanol preferred.
- a metal halide such at TiCl 3
- an acetate buffered solvent such as methanol or ethanol, methanol preferred.
- Reduction is also possible with divalent vanadium (prepared by Zn/Hg/HCI reduction of vanadyl sulphate) in a protic organic solvent such as methanol or ethanol, methanol preferred.
- the imine of formula 17 may be reduced to the corresponding amine, of formula Iji, by various methods familiar to those skilled in the art.
- a preferred method incorporates the use of boron reducing reagents such as sodium borohydride, generally used in excess, 2-10 equivalents, in protic solvents such as methanol or ethylene glycol, at a temperature range of -5-40°C, preferably 0-20°C.
- the compounds of the present invention may have asymmetric carbon atoms and therefore exist in different enantiomeric and diastereomeric forms.
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods known to those skilled in the art, for example, by chromatography or fractional crystallization.
- Ena ⁇ tiomers may be separated by converting the enantiomeric mixtures into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., alcohol), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g., alcohol
- the compounds of the present invention that are basic in nature are capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to mammals, it is often desirable in practice to initially isolate the compound of the present invention from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert the latter back to the free base compound by treatment with an alkaline reagent and subsequently convert the latter free base to a pharmaceutically acceptable acid addition salt.
- the acid addition salts of the base compounds of this invention are readily prepared by treating the base compound with a substantially equivalent amount of the chosen mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent, such as methanol or ethanol. Upon careful evaporation of the solvent, the desired solid salt is readily obtained.
- the desired salt can also be precipitated from a solution of the free base in an organic solvent by adding to the solution an appropriate mineral or organic acid.
- Those compounds of the present invention that are acidic in nature are capable of forming base salts with various cations.
- base salts include the alkali metal or alkaline-earth metal salts and particularly the sodium, amine and potassium salts. These salts are all prepared by conventional techniques.
- the chemical bases which are used as reagents to prepare the pharmaceutically acceptable base salts of this invention are those which form non-toxic base salts with the acidic compounds of the present invention.
- Such non-toxic base salts include those derived from such pharmacologically acceptable cations as sodium, potassium, calcium, magnesium, various amine cations, etc.
- These salts can easily be prepared by treating the corresponding acidic compounds with an aqueous solution containing the desired pharmacologically acceptable bases with cations such as sodium, potassium, calcium, magnesium, various amine cations, etc., and then evaporating the resulting solution to dryness, preferably under reduced pressure.
- they may also be prepared by mixing lower alkanolic solutions of the acidic compounds and the desired alkali metal alkoxide together, and then evaporating the resulting solution to dryness In the same manner as before.
- stoichi ⁇ metric quantities of reagents are preferably employed in order to ensure completeness of reaction and maximum yields of the desired final product.
- the activity of the compounds of the present invention against bacterial and protozoa pathogens is demonstrated by the compound's ability to inhibit growtn of defined strains of human (Assay I) or animal (Assays II and 111) pathogens.
- Assay I Assay I, descnbed below, employs conventional methodology and interpretation criteria and is designed to provide direction for chemical modifications that may lead to compounds that circumvent defined mechanisms of macrolide resistance.
- Assay I a panel of bacterial strains is assembled to include a variety of target pathogenic species, including representatives of macrolide resistance mechanisms that have been characterized.
- Bacterial pathogens that comprise the screening panel are shown in the table below in many cases, both the macrolide-susceptible parent strain and the macrolide-resistant strain derived from it are available to provide a more accurate assessment of the compound's ability to circumvent the resistance mechanism.
- Strains that contain the gene with the designation of ormAAtrmBfermC are resistant to macrolides, lincosamides, and streptogramin B antibiotics due to modifications (methylation) of 23S rRNA molecules by an Erm methylase, thereby generally prevent the binding of all three structural classes.
- msrA encodes a component of an efflux system in staphylococci that prevents the entry of macrolides and streptogramins while m ⁇ fA/E encodes a transmembrane protein that appears to efflux only macrolides.
- Inactivation of macrolide antibiotics can occur and can be mediated by either a phosphorylation of the 2'-hydroxyl (mph) or by cleavage of the macrocyclic lactone (esterase)
- the strains may be characterized using conventional polymerase chain reaction (PCR) technology and/or by sequencing the resistance determinant The use of PCR technology in this application is described in J.
- Assay II is utilized to test for activity against Pasteurella multocida and Assay III is utilized to test for activity against Pasteurella haemolytica.
- Assay II This assay is based on the liquid dilution method in microtiter format. A single colony of
- P. mult ⁇ ida (strain 59A067) is inoculated into 5 ml of brain heart infusion (BHl) broth.
- the test compounds are prepared by solubilizing 1 mg of the compound in 125 ⁇ l of dimethylsulfoxide (DMSO). Dilutions of the test compound are prepared using uninoculated BHl broth. The concentrations of the test compound used range from 200 ⁇ g/ml to 0.098 ⁇ g/ml by two-fold serial dilutions.
- the P. multocida inoculated BHl is diluted with uninoculated BHl broth to make a 10 4 cell suspension per 200 ⁇ l.
- the BHl cell suspensions are mixed with respective serial dilutions of the test compound, and incubated at 37°C for 18 hours.
- the minimum inhibitory concentration (MIC) Is equal to the concentration of the compound exhibiting 100% inhibition of growth of P. multocida as determined by comparison with an uninoculated control.
- This assay Is based on the agar dilution method using a Steers Replicator. Two to five colonies isolated from an agar plate are inoculated into BHl broth and incubated overnight at 37°C with shaking (200 rpm). The next morning, 300 ⁇ l of the fully grown P. haemolytica preculture is inoculated into 3 ml of fresh BHl broth and is incubated at 37 D C with shaking (200 rpm). The appropriate amounts of the test compounds are dissolved in ethanol and a series of two-fold serial dilutions are prepared. Two ml of the respective serial dilution is mixed with 18 ml of molten BHl agar and solidified. When the inoculated P.
- haemolytica culture reaches 0.5 McFarland standard density, about 5 ⁇ l of the P. haemolytica culture is inoculated onto BHl agar plates containing the various concentrations of the test compound using a Steers Replicator and incubated for 18 hours at 37 C. initial concentrations of the test compound range from 100-200 ⁇ g/ml. The MIC is equal to the concentration of the test compound exhibiting 100% inhibition of growth of P. haemolytica as determined by comparison with an uninoculated control.
- the jn yj o activity of the compounds of formula (I) can be determined by conventional animal protection studies well known to those skilled in the art, usually carried out in mice. Mice are allotted to cages (10 per cage) upon their amval, and allowed to acclimate for a minimum of 48 hours before being used. Animals are inoculated with 0.5 ml of a 3 x 10 3 CFU/ml bacterial suspension (P. multocida strain 59A006) intrapentoneally. Each experiment has at least 3 non-medicated control groups including one infected with 0.1X challenge dose and two infected with 1X challenge dose; a 10X challenge data group may also be used.
- mice in a given study can be challenged within 30-90 minutes, especially if a repeating syringe (such as a Cornwall® syringe) is used to administer the challenge. Thirty minutes after challenging has begun, the first compound treatment is given. It may be necessary for a second person to begin compound dosing if all of the animals have not been challenged at the end of 30 minutes.
- the routes of administration are subcutaneous or oral doses. Subcutaneous doses ere administered into the loose skin in the back of the neck whereas oral doses are given by means of a feeding needle In both cases, a volume of 02 ml is used per mouse Compounds are administered 30 minutes, 4 hours, and 24 hours after challenge. A control compound of known efficacy administered by the same route is included in each test. Animals are observed daily, and the number of survivors in each group is recorded. The P. multocida model monitoring continues for 96 hours (four days) post challenge.
- the PDso is a calculated dose at which the compound tested protects 50% of a group of mice from mortality due to the bacterial infection which would be lethal in the absence of drug treatment.
- the compounds of formula 1, 11, S and 0 and the pharmaceutically acceptable salts thereof may be adminstered through oral, parenteral, topical, or rectal routes in the treatment of bacterial and protozoa infections.
- these compounds are most desirably administered in dosages ranging from about 0.2 mg per kg body weight per day (mg/kg/day) to about 200 mg/kg/day in single or divided doses (i.e., from 1 to 4 doses per day), although variations will necessanly occur depending upon the species, weight and condition of the subject being treated and the particular route of administration chosen.
- a dosage level that is in the range of about 4 mg/kg/day to about 50 mg/kg/day is most desirably employed, Vanations may nevertheless occur depending upon the species of mammal, fish or bird being treated and its individual response to said medicament, as well as on the type of pharmaceutical formulation chosen and the time period and interval at which such administration is earned out.
- dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effects, provided that such larger doses are first divided into several small doses for administration throughout the day.
- the active compounds may be administered alone or in combination with pharmaceutically acceptable carriers or diluents by the routes previously indicated, and such administration may be carried out in single or multiple doses.
- the active compounds may be administered in a wide variety of different dosage forms, I.e., they may be combined with various pharmaceutically acceptable inert carriers in the form of tablets, capsules. lozenges, troches, hard candies, powders, sprays, creams, salves, suppositories, jellies, gels, pastes, lotions, ointments, aqueous suspensions, injectable solutions, elixirs, syrups, and the like.
- Such carriers include solid diluents or fillers, sterile aqueous media and various non-toxic organic solvents, etc.
- oral pharmaceutical compositions can be suitably sweetened and/or flavored.
- the active compounds are present in such dosage forms at concentration levels ranging from about 5.0% to about 70% by weight.
- tablets containing various excipients such as microcrystalline cellulose, sodium citrate, calcium carbonate, dicalcium phosphate and glycine may be employed along with various disintegrants such as starch (and preferably com, potato or tapioca starch), alginic acid and certain complex silicatea, together with granulation binders like polyvinylpyrrolidone, sucrose, gelatin and acacia, Additionally, lubricating agents such as magnesium stearate, sodium lauryl sulfate and talc are often very useful for tabletting purposes.
- compositions of a similar type may also be employed as fillers in gelatin capsules; preferred materials In this connection also include lactose or milk sugar as well as high molecular weight polyethylene ⁇ lycols.
- preferred materials In this connection also include lactose or milk sugar as well as high molecular weight polyethylene ⁇ lycols.
- the active compound When aqueous suspensions and/or elixirs are desired for oral administration, the active compound may be combined with various sweetening or flavoring agents, coloring matter or dyes, and, if so desired, emulsifying and/or suspending agents as well, together with such diluents as water, ethanol, propylene glycol, glycerin and various like combinations thereof.
- solutions of an active compound in either sesame or peanut oil or in aqueous propylene glycol may be employed.
- the aqueous solutions should be suitably buffered (preferably pH greater than 8) if necessary and the liquid diluent first rendered isotonic.
- These aqueous solutions are suitable for intravenous injection purposes,
- the oily solutions are suitable for intraarticular. intramuscular and subcutaneous injection purposes.
- the preparation of all these solutions under sterile conditions is readily accomplished by standard pharmaceutical techniques will known to those skilled in the art.
- the active compounds of the present invention may be administered topically and this may be done by way of creams, jellies, gels, pastes, patches, ointments and the like, in accordance with standard pharmaceutical practice.
- the active compounds may be administered in the feed of the animals or orally as a drench composition.
- the active compounds may also be adminstered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from a variety of phosphollplds, such as cholesterol, stearylamine or phosphatidylcholines.
- the active compounds may also be coupled with soluble polymers as targetable drug carriers.
- soluble polymers can include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylam.de phenyl, polyhydroxyethylaspartamide-phenol. or polyethyleneoxide-potylysine substituted with palmitoylre ⁇ idue ⁇ .
- the active compounds may be coupled to a class of biodegradable polymers useful in achieving controlled release of a drug, for example, polylactic acid, polygtycolic acid, copolymers of polylactic and polyglycolic acid, polyepsilon caprolacto ⁇ e, polyhydroxy butyric acid, polyorthoesters. polyacetals, polydihydropyrans, polycyanoacrylates and cross-linked or amphipathic block copolymers of hydrogels.
- Example 1-1 400 mg of the corresponding macrolide of formula _1_, where R 1 is isopropyl and R 2 is H, was dissolved in 8 mL of anhydrous pyridine. Hydroxylamine hydrochloride (0.2B5 g, 7.5 equiv.) was added and the solution heated to 60" C and stirred Tor 24 hours. The reaction was worked up by decanting into 50 mL of a 1:1 mixture of methylene chloride and water. The pH was adjusted to 9 using 1N NaOH, extracted 3 x 25 mL methylene chloride, and dried over Na 2 S0 4 . Filtration and concentration of filtrate yielded a light yellow solid product. The product (0.403 g) was carried on to the next step without further purification. Preparation for Example 1-2 (Table 1)
- Example 1-3 Preparation for Example 1-3 (Table 1) 250 mg of the corresponding macrolide of formula 1_, where R 1 is see-butyl and R 2 is H, was dissolved in 5 mL of anhydrous pyridine. Hydroxylamine hydrochloride (0.175 g, 7.5 equiv.) was added and the solution heated to 60° C and stirred for 24 hours. The reaction was worked up by decanting into 50 mL of a 1:1 mixture of methylene chloride and water. The pH was adjusted to 9 using 1N NaOH, extracted 3 x 25 mL methylene chloride, and dried over NaaSO «, Filtration and concentration of filtrate yielded a light yellow solid product. The product (0.246 g) was carried on to the next step without further purification.
- Hydroxylamine hydrochloride 0.175 g, 7.5 equiv.
- Example 1-4 Preparation for Example 1-4 (Table 1) 250 mg of the corresponding macrolide of formula 1, where R 1 is cyclobutyl and R 2 is H, was dissolved in 5 mL of anhydrous pyridine. Hydroxylamine hydrochloride (0.175 g, 7.5 equiv.) was added and the solution heated to 60" C and stirred for 24 hours. The reaction was worked up by decanting into 50 mL of a 1:1 mixture of methylene chloride and water. The pH was adjusted to 9 using 1 N NaOH. extracted 3 x 25 mL methylene chloride, and dried over Na 2 S0 . Filtration and concentration of filtrate yielded a light yellow solid product. The product (0.249 g) was carried on to the next step without further purification.
- Hydroxylamine hydrochloride 0.175 g, 7.5 equiv.
- Example 1-6 Preparation for Example 1-6 (Table 1) 95 mg of the corresponding macrolide of formula 1_, where R 1 is methylthioethyl and R 2 is H, was dissolved in 1.9 mL of anhydrous pyridine. Hydroxylamine hydrochloride (0.065 g, 7.5 equiv.) was added and the solution heated to 60° C and stirred for 24 hours. The reaction was worked up by decanting into 50 mL of a 1:1 mixture of methylene chloride and water. The pH was adjusted to 9 using 1N NaOH, extracted 3 x 25 mL methylene chloride, and dried over Na 2 S0 4 . Filtration and concentration of filtrate yielded a light yellow solid product. The product (0.091 g) was carried on to the next step without further purification.
- Hydroxylamine hydrochloride 0.065 g, 7.5 equiv.
- Example 1-8 (Table 1) 4.9(3 g of the corresponding macrolide of formula 1_, where R 1 Is cyclobutyl and R 2 Is OH. was dissolved in 50.0 mL of anhydrous pyridine. Hydroxylamine hydrochloride (3.4 g. 7.5 equjv.) was added and the solution heated to 60° C and stirred for 24 hours. The reaction was worked up by decanting into 50 mL of a 1:1 mixture of methylene chloride and water. The pH was adjusted to 9 using 1N NaOH, extracted 3 x 25 mL methylene chloride, and dried over Na S j. Filtration and concentration of filtrate yielded a light yellow solid product. The product (4.24 g) was carried on to the next step without further purification.
- Hydroxylamine hydrochloride 3.4 g. 7.5 equjv.
- Example 2-7 Preparation for Example 2-7 (Table 2) 60 mg of the corresponding oxime of formula 2, where R 1 is cyclopropyl and R 2 is OH, was dissolved in 1 mL of acetone. A 0.1 M aqueous solution of Na 2 HC0 3 (0.027 g in 0.5 mL water) was added and the resulting mixture was cooled to 0° C. A solution of para- toluenesulfonyl chloride (0.031 g) in acetone (0.5mL). cooled to 0° C, was added and the mixture stirred overnight The reaction was worked up by decanting into 25 L of a 1:1 mixture of methylene chloride and water.
- Preparation 4 An amount of 11-250 mg of the corresponding imidate of formula 3 was dissolved in THF and ethylene glycol then cooled to 0-5° C. NaBH 4 (5-10 equiv.) was added and the reaction stirred tor 4 hours at 0-5 6 C and then warmed to room temperature. The reaction was worked up by decanting into 10 mL of a 1:1 mixture of methylene chloride and water. The aqueous was re-extracted 3 x 5 mL methylene chloride. The organic layers were combined and dried over Na 2 S0 4 . Filteration and concentration yielded a solid product
- Example 3-1 (Table 3) 150 mg of the corresponding imidate of formula 3, where R 1 is isopropyl and R 2 is H, was dissolved in 3.75 mL tetrahydrofuran and 7.5 mL ethylene glycol and then cooled to 0-5° C. Na ⁇ H 4 (0.039 g) was added and the reaction stirred for 6 hours at 0-5° C and then warmed to room temperature. The reaction was worked up by decanting into 10 mL of a 1:1 mixture of methylene chloride and water. The aqueous was re-extracted 3 x 5 mL methylene chloride. The organic layers were combined and dried over Na z S0 4 . Filteration and concentration yielded a solid product (0.1 9 g).
- Example 3-2 165 mg of the corresponding imidate of formula 3, where R 1 is cyclopropyl and R 2 is H, was dissolved In glacial acetic acid (20 mL) . Platinum oxide catalyst (0.026 g, 50 mole %) was added, the reaction flushed with nitrogen, placed under 50 psi hydrogen and shaken at room temperature for 24 hours. Additional platinum oxide catalyst (50 mole %) was added, the reaction flushed with nitrogen, placed under 50 psi hydrogen and shaken at room temperature for additional 24 hours. Reaction was worked up by filteration through CeliteTM.
- Example 3-4 (Table 3) 250 mg of the corresponding imidate of formula 3, where R 1 cyclobutyl and R 2 is H, was dissolved in 3.83 mL tetrahydrofuran and 5.0 mL ethylene glycol and then cooled to 0-5 p C. NaBH 4 (0.191 g) was added and the reaction stirred for 6 hours at 0-5° C and then warmed to room temperature. The reaction was worked up by decanting into 10 mL of a 1:1 mixture of methylene chloride and water. The aqueous was re-extracted 3 x 5 L methylene chloride. The organic layers were combined and dried over Na 2 S0 4 . Filteration and concentration yielded a solid product (0.201 g).
- T e nmr data for compounds included within those of formula 5 is shown in Tables 3A and 38 below.
- Preparation 6 The corresponding oxime of formula 2 is dissolved in ethanol. Lithium hydroxide monohydrate (2 equivalents) is added and the reaction mixture stirred ovemight at room temperature. The reaction is concentrated under vacuum and partitioned between brine and ethyl acetate, the pH of the reaction mixture is adjusted to 9-10, the reaction mixture is extracted with ethyl acetate, and dried over Na 2 S0 4 . A 4:1 ratio of Z:E isomers is produced. Isolation of isomers is accomplished by either silica chromatography or crystallization from nitromethane.
- Preparation 7 The corresponding oxime of formula 2a is dissolved in acetone. A 0.1 M aqueous solution of Na g HCO a (2 equiv.) is added and the resulting mixture is cooled to 0-5° C. A 0.1 M solution of para-toluenesulfonyl chloride in acetone is added and the mixture stirred overnight. The reaction is worked up by decanting into 25 mL of a 1:1 mixture of methylene chloride and water. The pH of the reaction mixture is adjusted to 9-10 using 1N NaOH, the reaction mixture is extracted with 3 x 20 mL methylene chloride, and dried over Na 2 S0 4 . Filtration and concentration of filtrate yields a solid product. The product is carried on to the next step without further purification. Table 7
- Preparation 9 The corresponding imidate of formula 3a is dissolved in 0.5 mL MeOH and cooled to 0-5° C. NaBH 4 (10 equiv.) is added and the reaction stirred for 4 hours at 0-5° C, warmed to room temperature, and stirred ovemight. The reaction is worked up by decanting into 10 mL of a 1:1 mixture of methylene chloride and water, the pH of the reaction mixture adjusted to ⁇ -g using i N NaOH, extracted 3 x 5 mL methylene chloride, and dried over Na-jSO,. Filtration and concentration yields a solid product Purification is accomplished by HPLC.
- Preparation 10 The corresponding azalide of formula 5 ⁇ is dissolved in chloroform. 37% formaldehyde (1.0 equiv.) and formic acid (1.0 equiv.) is added and the solution stirred at 45-50° C for 48-72 hours. The reaction mixture is then decanted into a 1:1 mixture of chloroform and water, the pH of the reaction mixture is adjusted to 9-10 using 1N NaOH, the reaction mixture is extracted with chloroform, and dried over Na 2 S0 4 . Filtration and concentration yields a solid product. Isolation of product is accomplished by either silica chromatography or HPLC.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
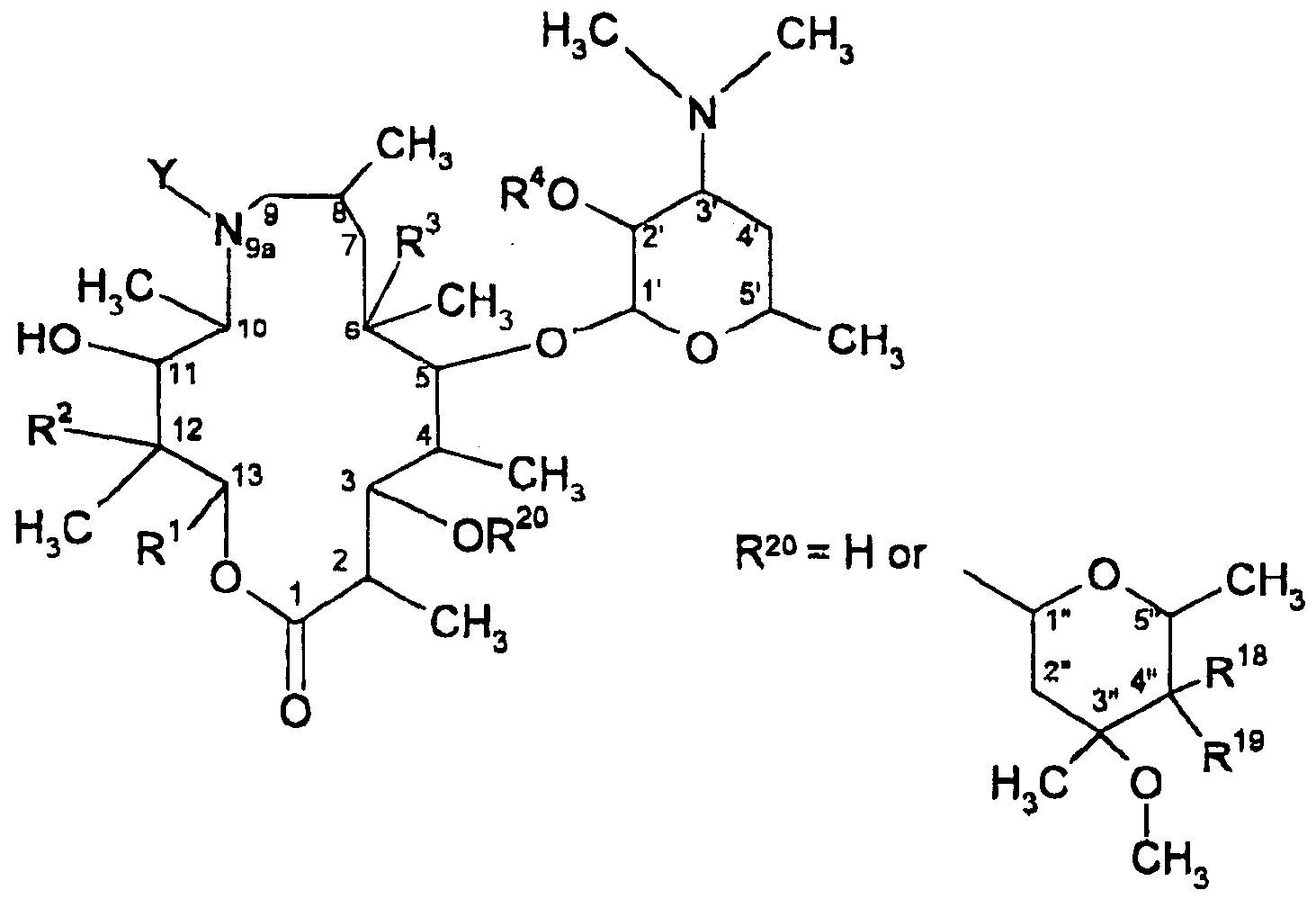


















































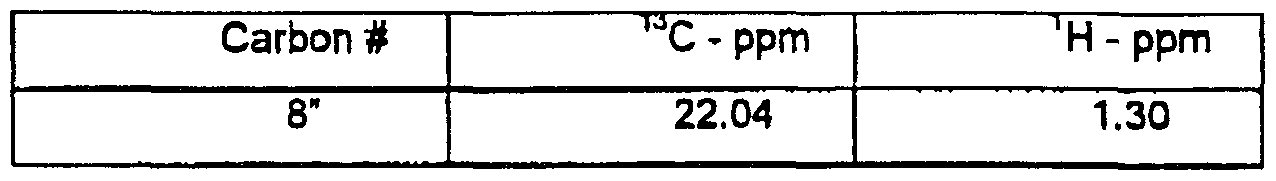















Claims





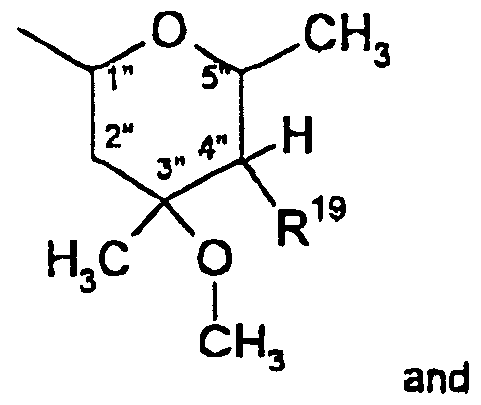















Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL98341642A PL341642A1 (en) | 1998-01-02 | 1998-12-21 | Novel macrolydes |
| BR9814601-7A BR9814601A (en) | 1998-01-02 | 1998-12-21 | "macrolides" |
| HU0100420A HUP0100420A3 (en) | 1998-01-02 | 1998-12-21 | Novel macrolides and process for preparing them |
| US09/554,988 US6472371B1 (en) | 1998-01-02 | 1998-12-21 | Macrolides |
| HR20000442A HRP20000442A2 (en) | 1998-01-02 | 1998-12-21 | Novel macrolides |
| CA002316919A CA2316919C (en) | 1998-01-02 | 1998-12-21 | Novel macrolides |
| JP2000527552A JP2002500230A (en) | 1998-01-02 | 1998-12-21 | New macrolide antibiotics |
| AU14447/99A AU756409B2 (en) | 1998-01-02 | 1998-12-21 | Novel macrolides |
| KR1020007007313A KR100352809B1 (en) | 1998-01-02 | 1998-12-21 | Novel macrolides |
| IL13702498A IL137024A0 (en) | 1998-01-02 | 1998-12-21 | Novel macrolides |
| EP98958384A EP1044207A1 (en) | 1998-01-02 | 1998-12-21 | Novel macrolides |
| SK978-2000A SK9782000A3 (en) | 1998-01-02 | 1998-12-21 | Macrolides, method for production thereof and pharmaceutical compositions on their base |
| NZ504986A NZ504986A (en) | 1998-01-02 | 1998-12-21 | Erythromycin and azalide derivatives and pharmaceutical compositions thereof, useful as antibacterial agents and antoprotozoa agents |
| EA200000594A EA200000594A1 (en) | 1998-01-02 | 1998-12-22 | NEW MACROLIDES |
| IS5528A IS5528A (en) | 1998-01-02 | 2000-06-09 | New macrolide |
| NO20003347A NO20003347L (en) | 1998-01-02 | 2000-06-27 | New macrolides |
| BG104644A BG104644A (en) | 1998-01-02 | 2000-07-28 | Novel macrolides |
| US10/209,682 US7015203B2 (en) | 1998-01-02 | 2002-07-30 | Macrolides |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US7034398P | 1998-01-02 | 1998-01-02 | |
| US60/070,343 | 1998-01-02 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09554988 A-371-Of-International | 1998-12-21 | ||
| US09/554,988 A-371-Of-International US6472371B1 (en) | 1998-01-02 | 1998-12-21 | Macrolides |
| US10/209,682 Division US7015203B2 (en) | 1998-01-02 | 2002-07-30 | Macrolides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1999035156A1 true WO1999035156A1 (en) | 1999-07-15 |
| WO1999035156B1 WO1999035156B1 (en) | 1999-10-21 |
Family
ID=22094723
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB1998/002099 Ceased WO1999035156A1 (en) | 1998-01-02 | 1998-12-21 | Novel macrolides |
Country Status (34)
| Country | Link |
|---|---|
| US (2) | US6472371B1 (en) |
| EP (1) | EP1044207A1 (en) |
| JP (1) | JP2002500230A (en) |
| KR (1) | KR100352809B1 (en) |
| CN (1) | CN1284081A (en) |
| AP (1) | AP9801420A0 (en) |
| AR (4) | AR014948A1 (en) |
| AU (1) | AU756409B2 (en) |
| BG (1) | BG104644A (en) |
| BR (1) | BR9814601A (en) |
| CA (1) | CA2316919C (en) |
| CO (1) | CO4970821A1 (en) |
| CZ (1) | CZ20002458A3 (en) |
| DZ (1) | DZ2698A1 (en) |
| EA (1) | EA200000594A1 (en) |
| GT (1) | GT199800206A (en) |
| HR (1) | HRP20000442A2 (en) |
| HU (1) | HUP0100420A3 (en) |
| IL (1) | IL137024A0 (en) |
| IS (1) | IS5528A (en) |
| MA (1) | MA24729A1 (en) |
| NO (1) | NO20003347L (en) |
| NZ (1) | NZ504986A (en) |
| OA (1) | OA11438A (en) |
| PA (1) | PA8465301A1 (en) |
| PE (1) | PE20000049A1 (en) |
| PL (1) | PL341642A1 (en) |
| SK (1) | SK9782000A3 (en) |
| TN (1) | TNSN98238A1 (en) |
| TW (1) | TW520374B (en) |
| UY (2) | UY25330A1 (en) |
| WO (1) | WO1999035156A1 (en) |
| YU (1) | YU41500A (en) |
| ZA (1) | ZA9811939B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0984019A1 (en) * | 1998-08-19 | 2000-03-08 | Pfizer Products Inc. | C11 carbamates of macrolide antibacterials |
| US6399582B1 (en) | 1999-04-16 | 2002-06-04 | Ortho-Mcneil Pharmaceutical, Inc. | Ketolide antibacterials |
| US6590083B1 (en) | 1999-04-16 | 2003-07-08 | Ortho-Mcneil Pharmaceutical, Inc. | Ketolide antibacterials |
| WO2003072589A1 (en) * | 2002-02-26 | 2003-09-04 | Meiji Seika Kaisha, Ltd. | Novel 15-membered cyclic azalide, novel 16-membered cyclic diazalide derivative, and process for producing these |
| US6794366B2 (en) | 1999-04-16 | 2004-09-21 | Kosan Biosciences, Inc. | Macrolide antiinfective agents |
| EP1437360A3 (en) * | 1998-08-19 | 2005-04-06 | Pfizer Products Inc. | C11 Carbamates of macrolide antibacterials |
| US7015203B2 (en) | 1998-01-02 | 2006-03-21 | Pfizer Inc. | Macrolides |
| EP1754700A2 (en) | 1999-01-27 | 2007-02-21 | Kosan Biosciences, Inc. | Synthesis of oligoketides |
| CN103965273A (en) * | 2013-08-23 | 2014-08-06 | 普莱柯生物工程股份有限公司 | Macrolide compound |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AP1060A (en) | 1998-01-02 | 2002-04-23 | Pfizer Prod Inc | Novel erythromycin derivatives. |
| EA200100983A1 (en) | 1999-05-24 | 2002-10-31 | Пфайзер Продактс Инк. | DERIVATIVES 13-METHYLERITROMYCIN |
| BRPI0515917A (en) | 2004-09-23 | 2008-08-12 | Schering Plough Ltd | control of parasites in animals by the use of novel trifluoromethanesulfonanilide oxime ether derivatives |
| EP1811841B1 (en) * | 2004-11-19 | 2009-10-28 | Schering-Plough Ltd. | Control of parasites in animals by the use of parasiticidal 2-phenyl-3-(1h-pyrrol-2-yl) acrylonitrile derivatives |
| UA85937C2 (en) | 2004-12-21 | 2009-03-10 | Пфайзер Продактс Інк. | Macrolide compounds |
| WO2006135648A2 (en) * | 2005-06-09 | 2006-12-21 | Schering-Plough Ltd. | Control of parasites in animals by n-[(phenyloxy)phenyl]-1,1,1-trifluoromethanesulfonamide and n-[(phenylsulfanyl)phenyl]-1,1,1-trifluoromethanesulfonamide derivatives |
| US8119667B2 (en) * | 2005-12-29 | 2012-02-21 | Schering-Plough Animal Health Corporation | Carbonates of fenicol antibiotics |
| US20070238700A1 (en) * | 2006-04-10 | 2007-10-11 | Winzenberg Kevin N | N-phenyl-1,1,1-trifluoromethanesulfonamide hydrazone derivative compounds and their usage in controlling parasites |
| WO2008076259A1 (en) * | 2006-12-13 | 2008-06-26 | Schering-Plough Ltd. | Water-soluble prodrugs of florfenicol and its analogs |
| CA2672795A1 (en) * | 2006-12-13 | 2008-06-26 | Schering-Plough Ltd. | Water-soluble prodrugs of chloramphenicol, thiamphenicol, and analogs thereof |
| TWI430995B (en) | 2007-06-26 | 2014-03-21 | Du Pont | Naphthalene isoxazoline invertebrate pest control agents |
| KR101769728B1 (en) | 2007-06-27 | 2017-08-18 | 이 아이 듀폰 디 네모아 앤드 캄파니 | Animal pest control method |
| WO2009064953A1 (en) * | 2007-11-15 | 2009-05-22 | Enanta Pharmaceuticals, Inc. | Use of bridged macrolides or tylosin derivatives in treating inflammatory bowel diseases |
| TWI468407B (en) | 2008-02-06 | 2015-01-11 | Du Pont | Mesoionic pesticides |
| WO2010014566A2 (en) * | 2008-07-30 | 2010-02-04 | Intervet International B.V. | Process for preparing oxazoline-protected aminodiol compounds useful as intermediates to florfenicol |
| UA110924C2 (en) | 2009-08-05 | 2016-03-10 | Е. І. Дю Пон Де Немур Енд Компані | Mesoionic pesticides |
| KR20120050467A (en) | 2009-08-05 | 2012-05-18 | 이 아이 듀폰 디 네모아 앤드 캄파니 | Mesoionic pesticides |
| US20120122680A1 (en) | 2009-08-05 | 2012-05-17 | E.I. Du Pont De Nemours And Company | Mesoionic pesticides |
| UA107804C2 (en) | 2009-08-05 | 2015-02-25 | Du Pont | MIXTURES OF MESOIONIC PESTICIDES |
| UA108881C2 (en) | 2010-05-27 | 2015-06-25 | CRYSTAL FORM 4- $ 5- $ 3-CHLORINE-5- (TRIFLUOROMETHYL) PHENYL] -4,5-DIHYDRO-5- (TRIFLUORMETHYL) -3-ISOXAZOLYL] -N- $ 2-OXO-2 - $ 2,2 -TRIFLUOROTHYL) AMINO] ETHYL] -1-NAPHTHALINE CARBOXAMIDE | |
| WO2012087630A1 (en) | 2010-12-20 | 2012-06-28 | E.I. Du Pont De Nemours And Company | Pyridine and pyrimidine compounds for controlling invertebrate |
| KR20140004187A (en) * | 2011-01-31 | 2014-01-10 | 메이지 세이카 파루마 가부시키가이샤 | Novel macrolide derivative |
| JP2014520151A (en) | 2011-06-20 | 2014-08-21 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | Heterocyclic compounds for treating helminth infections |
| CN102260306B (en) * | 2011-07-22 | 2012-07-18 | 山东鲁抗舍里乐药业有限公司 | Novel method for preparing tulathromycin |
| TW201326128A (en) | 2011-11-28 | 2013-07-01 | Du Pont | Sulfonamide anthelmintics |
| WO2013158422A1 (en) | 2012-04-17 | 2013-10-24 | E. I. Du Pont De Nemours And Company | Heterocyclic compounds for controlling invertebrate pests |
| WO2014099837A1 (en) | 2012-12-18 | 2014-06-26 | E. I. Du Pont De Nemours And Company | Sulfonamide anthelmintics |
| CN105669798B (en) * | 2013-08-23 | 2019-07-26 | 普莱柯生物工程股份有限公司 | A kind of macrolides compound |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0283055A2 (en) * | 1987-09-03 | 1988-09-21 | SOUR PLIVA farmaceutska, Kemijska prehrambena i kozmeticka industrija, n.sol.o. | 10-Dihydro-10-deoxo-11-azaerythronolide-A-compounds, methods and intermediates for the manufacture thereof and their use in pharmaceuticals and in the manufacture thereof |
| FR2691464A1 (en) * | 1992-05-21 | 1993-11-26 | Roussel Uclaf | New 1-oxa-6-aza-cyclopentadecane-13,15-di:one derivs. - useful as antibiotics for treating wide range of bacterial infections |
| EP0619320A1 (en) * | 1991-12-27 | 1994-10-12 | Taisho Pharmaceutical Co. Ltd | 5-o-desosaminylerythronolide derivative |
| WO1998056802A1 (en) * | 1997-06-11 | 1998-12-17 | Pfizer Products Inc. | 4'-substituted-9-deoxo-9a-aza-9a-homoerythromycin a derivatives |
| WO1998056801A1 (en) * | 1997-06-11 | 1998-12-17 | Pfizer Products Inc. | C-4''-substituted macrolide derivatives |
Family Cites Families (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3652537A (en) | 1969-11-21 | 1972-03-28 | Lilly Co Eli | Epierythromycylamine and epierythromycyl b amine |
| US3923784A (en) | 1973-09-10 | 1975-12-02 | Hoffmann La Roche | Erythromycin a derivatives |
| SI7910768A8 (en) | 1979-04-02 | 1996-06-30 | Pliva Pharm & Chem Works | Process for pripering 11-aza-4-0-cladinosyl-6-0-desosaminyl-15-ethyl- 7,13,14-trihydroxy-3,5,7,9,12,14-hexamethyl- oxacyclopentadecane-2-one and their derivatives |
| US4474768A (en) | 1982-07-19 | 1984-10-02 | Pfizer Inc. | N-Methyl 11-aza-10-deoxo-10-dihydro-erytromycin A, intermediates therefor |
| US4585759A (en) | 1985-01-22 | 1986-04-29 | Pfizer Inc. | Antibacterial derivatives of a neutral macrolide |
| US5141926A (en) | 1990-04-18 | 1992-08-25 | Abbott Laboratories | Erythromycin derivatives |
| US5824513A (en) | 1991-01-17 | 1998-10-20 | Abbott Laboratories | Recombinant DNA method for producing erythromycin analogs |
| WO1993013663A1 (en) | 1992-01-17 | 1993-07-22 | Abbott Laboratories | Method of directing biosynthesis of specific polyketides |
| US5985844A (en) | 1992-03-26 | 1999-11-16 | Merck & Co., Inc. | Homoerythromycin A derivatives modified at the 4"-and 8A-positions |
| CA2065218A1 (en) | 1991-04-11 | 1992-10-12 | Robert R. Wilkening | Process for the preparation of 9-deoxo-8a-aza-8a-homoerythromycin a and its 8a-alkyl derivatives |
| US5332807A (en) * | 1993-04-14 | 1994-07-26 | Merck & Co., Inc. | Process of producing 8A- and 9A-azalide antibiotics |
| US5441939A (en) * | 1994-03-04 | 1995-08-15 | Pfizer Inc. | 3"-desmethoxy derivatives of erythromycin and azithromycin |
| US6271255B1 (en) | 1996-07-05 | 2001-08-07 | Biotica Technology Limited | Erythromycins and process for their preparation |
| WO1998051696A1 (en) | 1997-05-09 | 1998-11-19 | Pfizer Products Inc. | Erythromycin derivatives |
| US6407074B1 (en) | 1997-06-11 | 2002-06-18 | Pfizer Inc | C-4″-substituted macrolide derivatives |
| US20020025937A1 (en) | 2000-03-20 | 2002-02-28 | Yong-Jin Wu | 9-oxime erythromycin derivatives |
| WO1999000124A1 (en) | 1997-06-26 | 1999-01-07 | Merck & Co., Inc. | 9a-azalides, compositions containing such compounds and methods of treatment |
| US6339063B1 (en) | 1997-09-10 | 2002-01-15 | Merck & Co., Inc. | 9a-azalides as veterinary antimicrobial agents |
| PA8461401A1 (en) | 1997-10-29 | 2000-05-24 | Pfizer Prod Inc | TRICYCLIC ERYTHROMYCINE DERIVATIVES |
| HN1998000159A (en) | 1997-10-29 | 1999-02-09 | Monsanto Co | DERIVATIVES OF 9- AMINO - 3 CETO ERITROMICINA |
| AP1060A (en) | 1998-01-02 | 2002-04-23 | Pfizer Prod Inc | Novel erythromycin derivatives. |
| AP9801420A0 (en) | 1998-01-02 | 1998-12-31 | Pfizer Prod Inc | Novel macrolides. |
| PT941998E (en) | 1998-03-03 | 2004-12-31 | Pfizer Prod Inc | MACROLIDAN ANTIBIOTICS OF TYPE 3,6-CETAL |
| AU3439899A (en) | 1998-06-03 | 1999-12-20 | Pfizer Products Inc. | Tricyclic 3-keto derivatives of 6-o-methylerythromycin |
| US6043227A (en) | 1998-08-19 | 2000-03-28 | Pfizer Inc. | C11 carbamates of macrolide antibacterials |
| US6100240A (en) | 1998-10-09 | 2000-08-08 | Pfizer Inc | Macrolide derivatives |
| KR20010083944A (en) | 1998-11-03 | 2001-09-03 | 실버스타인 아써 에이. | Novel macrolide antibiotics |
| UA70972C2 (en) | 1998-11-20 | 2004-11-15 | Пфайзер Продактс Інк. | 13-membered azalides and use thereof as antibiotics |
| EA200100983A1 (en) | 1999-05-24 | 2002-10-31 | Пфайзер Продактс Инк. | DERIVATIVES 13-METHYLERITROMYCIN |
| EP1101769A3 (en) | 1999-11-18 | 2001-10-24 | Pfizer Products Inc. | Nitrogen containing erythromycin derivatives |
| JP4311203B2 (en) | 2001-08-08 | 2009-08-12 | 大正製薬株式会社 | 11a-azalide compound and method for producing the same |
-
1998
- 1998-12-17 AP APAP/P/1998/001420A patent/AP9801420A0/en unknown
- 1998-12-21 IL IL13702498A patent/IL137024A0/en unknown
- 1998-12-21 SK SK978-2000A patent/SK9782000A3/en unknown
- 1998-12-21 CA CA002316919A patent/CA2316919C/en not_active Expired - Fee Related
- 1998-12-21 EP EP98958384A patent/EP1044207A1/en not_active Withdrawn
- 1998-12-21 YU YU41500A patent/YU41500A/en unknown
- 1998-12-21 AU AU14447/99A patent/AU756409B2/en not_active Ceased
- 1998-12-21 CN CN98813253A patent/CN1284081A/en active Pending
- 1998-12-21 NZ NZ504986A patent/NZ504986A/en unknown
- 1998-12-21 HU HU0100420A patent/HUP0100420A3/en unknown
- 1998-12-21 JP JP2000527552A patent/JP2002500230A/en active Pending
- 1998-12-21 PL PL98341642A patent/PL341642A1/en unknown
- 1998-12-21 CZ CZ20002458A patent/CZ20002458A3/en unknown
- 1998-12-21 KR KR1020007007313A patent/KR100352809B1/en not_active Expired - Fee Related
- 1998-12-21 WO PCT/IB1998/002099 patent/WO1999035156A1/en not_active Ceased
- 1998-12-21 BR BR9814601-7A patent/BR9814601A/en not_active IP Right Cessation
- 1998-12-21 HR HR20000442A patent/HRP20000442A2/en not_active Application Discontinuation
- 1998-12-21 US US09/554,988 patent/US6472371B1/en not_active Expired - Fee Related
- 1998-12-22 EA EA200000594A patent/EA200000594A1/en unknown
- 1998-12-23 PA PA19988465301A patent/PA8465301A1/en unknown
- 1998-12-23 TW TW087121542A patent/TW520374B/en not_active IP Right Cessation
- 1998-12-28 PE PE1998001287A patent/PE20000049A1/en not_active Application Discontinuation
- 1998-12-29 AR ARP980106717A patent/AR014948A1/en not_active Application Discontinuation
- 1998-12-29 GT GT199800206A patent/GT199800206A/en unknown
- 1998-12-30 UY UY25330A patent/UY25330A1/en not_active Application Discontinuation
- 1998-12-30 CO CO98077585A patent/CO4970821A1/en unknown
- 1998-12-30 DZ DZ980308A patent/DZ2698A1/en active
- 1998-12-30 TN TNTNSN98238A patent/TNSN98238A1/en unknown
- 1998-12-30 MA MA25407A patent/MA24729A1/en unknown
- 1998-12-30 ZA ZA9811939A patent/ZA9811939B/en unknown
-
1999
- 1999-06-18 UY UY25574A patent/UY25574A1/en not_active Application Discontinuation
- 1999-11-29 AR ARP990106063A patent/AR020851A2/en not_active Application Discontinuation
- 1999-11-29 AR ARP990106062A patent/AR019787A2/en not_active Application Discontinuation
- 1999-11-29 AR ARP990106064A patent/AR020551A2/en not_active Application Discontinuation
-
2000
- 2000-06-09 IS IS5528A patent/IS5528A/en unknown
- 2000-06-27 NO NO20003347A patent/NO20003347L/en unknown
- 2000-06-30 OA OA1200000192A patent/OA11438A/en unknown
- 2000-07-28 BG BG104644A patent/BG104644A/en unknown
-
2002
- 2002-07-30 US US10/209,682 patent/US7015203B2/en not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0283055A2 (en) * | 1987-09-03 | 1988-09-21 | SOUR PLIVA farmaceutska, Kemijska prehrambena i kozmeticka industrija, n.sol.o. | 10-Dihydro-10-deoxo-11-azaerythronolide-A-compounds, methods and intermediates for the manufacture thereof and their use in pharmaceuticals and in the manufacture thereof |
| EP0619320A1 (en) * | 1991-12-27 | 1994-10-12 | Taisho Pharmaceutical Co. Ltd | 5-o-desosaminylerythronolide derivative |
| FR2691464A1 (en) * | 1992-05-21 | 1993-11-26 | Roussel Uclaf | New 1-oxa-6-aza-cyclopentadecane-13,15-di:one derivs. - useful as antibiotics for treating wide range of bacterial infections |
| WO1998056802A1 (en) * | 1997-06-11 | 1998-12-17 | Pfizer Products Inc. | 4'-substituted-9-deoxo-9a-aza-9a-homoerythromycin a derivatives |
| WO1998056801A1 (en) * | 1997-06-11 | 1998-12-17 | Pfizer Products Inc. | C-4''-substituted macrolide derivatives |
Non-Patent Citations (2)
| Title |
|---|
| DJOKIC, SLOBODAN ET AL: "Erythromycin series. Part 11. Ring expansion of erythromycin A oxime by the Beckmann rearrangement", J. CHEM. SOC., PERKIN TRANS. 1 (1986), (11), 1881-90, XP002913052 * |
| DJOKIC, SLOBODAN ET AL: "Erythromycin series. Part 13. Synthesis and structure elucidation of 10-dihydro-10-deoxo-11-methyl-11-azaerythromycin A", J. CHEM. RES., SYNOP. (1988), (5), 152-3, XP002913051 * |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7015203B2 (en) | 1998-01-02 | 2006-03-21 | Pfizer Inc. | Macrolides |
| EP1437360A3 (en) * | 1998-08-19 | 2005-04-06 | Pfizer Products Inc. | C11 Carbamates of macrolide antibacterials |
| EP0984019A1 (en) * | 1998-08-19 | 2000-03-08 | Pfizer Products Inc. | C11 carbamates of macrolide antibacterials |
| EP1754700A2 (en) | 1999-01-27 | 2007-02-21 | Kosan Biosciences, Inc. | Synthesis of oligoketides |
| US6590083B1 (en) | 1999-04-16 | 2003-07-08 | Ortho-Mcneil Pharmaceutical, Inc. | Ketolide antibacterials |
| US6794366B2 (en) | 1999-04-16 | 2004-09-21 | Kosan Biosciences, Inc. | Macrolide antiinfective agents |
| US6458771B1 (en) | 1999-04-16 | 2002-10-01 | Ortho-Mcneil Pharmaceutical, Inc. | Ketolide antibacterials |
| US6399582B1 (en) | 1999-04-16 | 2002-06-04 | Ortho-Mcneil Pharmaceutical, Inc. | Ketolide antibacterials |
| WO2003072589A1 (en) * | 2002-02-26 | 2003-09-04 | Meiji Seika Kaisha, Ltd. | Novel 15-membered cyclic azalide, novel 16-membered cyclic diazalide derivative, and process for producing these |
| CN103965273A (en) * | 2013-08-23 | 2014-08-06 | 普莱柯生物工程股份有限公司 | Macrolide compound |
| WO2015024298A1 (en) * | 2013-08-23 | 2015-02-26 | 普莱柯生物工程股份有限公司 | Macrolide compound |
| CN103965273B (en) * | 2013-08-23 | 2016-05-25 | 普莱柯生物工程股份有限公司 | A macrolide compound |
| US9765105B2 (en) | 2013-08-23 | 2017-09-19 | Pulike Biological Engineering, Inc. | Macrolide compound |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6472371B1 (en) | Macrolides | |
| CA2292359C (en) | Novel azalides and methods of making same | |
| AU759019B2 (en) | Novel erythromycin derivatives | |
| US6777543B2 (en) | 13-methyl erythromycin derivatives | |
| IL189430A (en) | 13-membered azalides and their use as antibiotic agents | |
| WO1998056800A1 (en) | 9-oxime erythromycin derivatives | |
| EP0984019B1 (en) | C11 carbamates of macrolide antibacterials | |
| MXPA00006604A (en) | Novel macrolides | |
| EP1437360A2 (en) | C11 Carbamates of macrolide antibacterials | |
| MXPA00001110A (en) | Novel azalides and methods of making same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 137024 Country of ref document: IL Ref document number: P-415/00 Country of ref document: YU Ref document number: 98813253.2 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: B1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: B1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09554988 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 504986 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9782000 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14447/99 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998958384 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2316919 Country of ref document: CA Ref document number: 2316919 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007007313 Country of ref document: KR Ref document number: PV2000-2458 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20000442A Country of ref document: HR Ref document number: 200000594 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2000/006604 Country of ref document: MX |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998958384 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007007313 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-2458 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020007007313 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 14447/99 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998958384 Country of ref document: EP |