WO1999061411A1 - Processes for producing cyanophenyl derivatives - Google Patents
Processes for producing cyanophenyl derivatives Download PDFInfo
- Publication number
- WO1999061411A1 WO1999061411A1 PCT/JP1999/002857 JP9902857W WO9961411A1 WO 1999061411 A1 WO1999061411 A1 WO 1999061411A1 JP 9902857 W JP9902857 W JP 9902857W WO 9961411 A1 WO9961411 A1 WO 9961411A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- cyanobenzaldehyde
- reaction
- producing
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/30—Preparation of carboxylic acid nitriles by reactions not involving the formation of cyano groups
Definitions
- the present invention relates to a method for producing a cyanophenyl derivative. More specifically, the present invention relates to a method for producing a cyanobenzaldehyde compound, a cyanobenzoic acid octalide compound, and a cyanobenzoic acid compound using a cyanobenzylamine compound as a raw material.
- the cyanobenzaldehyde compound, cyanobenzoic acid halide compound and cyanobenzoic acid compound obtained by the present invention are important intermediates such as pharmaceuticals, pesticides, liquid crystals, and functional polymer monomers.
- p_cyanobenzaldehyde converts p_cyanobenzoic acid to p_cyanobenzoyl chloride with a chlorinating agent such as thionyl chloride, which is then reduced to Rosemmmd. (Rapoport et al., J. Am. Chem. Soc, 25, 1125 (1953)).
- the above method for synthesizing p-cyanobenzaldehyde has the following problems.
- a method for producing a cyanobenzoic acid halide compound a method in which a corresponding cyanobenzoic acid compound is reacted with an acid halide agent has been proposed.
- a method of reacting p-cyanobenzoic acid with an acid chloride to obtain p-cyanobenzoic acid chloride will be described.
- the method (6) using thionyl chloride has a problem in that the separation and detoxification of sulfur dioxide is costly because the sulfur dioxide that causes air pollution is by-produced.
- the method using oxalyl chloride of (7) also has a problem that it costs much to detoxify it because it produces by-produced carbon monoxide which causes air pollution.
- the method using phosphorus pentachloride in (8) produces by-products containing phosphorus, and these by-products containing phosphorus become eutrophic sources such as lakes and rivers and cause environmental pollution. It must be disposed of after processing.
- P-cyanobenzoic acid is synthesized by the Sandmeyer reaction in which (9) p-aminobenzoic acid is diazotized and then reacted with copper cyanide.
- (11) p-cyanobenzoic acid can be synthesized by the carbonylation of 4_chlorocyanobenzene using a palladium monophosphine catalyst in the presence of carbon monoxide (Japanese Patent Application Laid-Open No. 64-47).
- Prior arts related to the present invention include: (12) phase transfer in a two-layer system of water and an organic solvent, using p-tolunitrile as a ruthenium compound as an oxidation catalyst, sodium chlorite as an oxidizing agent; There is a method of oxidizing in the presence of a catalyst (Yod et al., J. Org. Chem., 51, 2880 (1986)). According to this document, p-cyanobenzoic acid is formed from tolunitrile via p-cyanobenzaldehyde.
- the Sandmeyer method (9) requires dangerous copper cyanide, and it is difficult to isolate and purify P-cyanobenzoic acid under acidic conditions under which hydrogen cyanide is released.
- (11) is not an economical method because expensive palladium and phosphine are used.
- the method using a ruthenium compound in (12) is not an economical method because it requires an expensive ruthenium compound as an essential component to the raw material at 1 mol% and a phase transfer catalyst at 5 mol%. .
- the method of (13) using the oxidation of p-cyanobenzaldehyde has a low yield in both the oxygen oxidation method using a cobalt catalyst and the method using sodium perborate.
- the cyanobenzoic acid compound has a problem that it is difficult to obtain a high-purity form by a conventionally known technique because it is complicated to synthesize and a raw material is not easily available. Problems to be solved by the invention
- an object of the present invention is to provide a method for industrially and advantageously producing a cyanobenzaldehyde compound in a small number of reaction steps using easily available raw materials.
- Another object of the present invention is to use a cyanobenzaldehyde compound obtained efficiently by the method of the present invention as a raw material, to reduce the burden on the environment, to easily detoxify by-products, to obtain an industrially high yield,
- An object of the present invention is to provide a method capable of producing a cyanobenzoic acid octalide compound with high purity.
- Still another object of the present invention is to provide a method for producing a cyanobenzoic acid compound in a high yield and a high purity by an industrially advantageous method using a cyanobenzaldehyde compound obtained efficiently by the method of the present invention as a raw material. Is to do. Disclosure of the invention
- Shianoben Jiruamin compounds may be easily synthesized from phthalonitrile compound such as a starting material, aminomethyl group without compromising Shiano group on the benzene ring by an oxidation reaction using an oxidizing agent of the reagent (- CH 2 NH 2 ) was efficiently converted to an aldehyde group (_CHO), and a cyanobenzaldehyde compound was obtained.
- the present inventors using a cyanobenzaldehyde compound as a starting material, convert an aldehyde group (—CH ⁇ ) into an acid halide group (one COY: Y is a chlorine atom or a bromine atom without damaging the cyano group on the benzene ring).
- COY aldehyde group
- Y is a chlorine atom or a bromine atom without damaging the cyano group on the benzene ring.
- the present inventors have found a method for producing a cyanobenzoic acid halide compound which has never been known before and can be converted into
- the present inventors have found that the cyanobenzaldehyde compound is used as a raw material to react the cyanobenzaldehyde compound and the hypohalous acid compound with water or water in an aprotic polar solvent, whereby the cyanobenzoic acid compound is highly purified. It was found that it can be obtained with high purity and purity.
- the present invention provides the following cyano benzaldehyde compounds, cyano benzoic acid halide compounds, and methods for producing cyano benzoic acid compounds.
- a method for producing a cyanobenzaldehyde compound comprising reacting a cyanobenzylamine compound with an oxidizing agent.
- —CHO and —X represent a substituent on a benzene ring, —CHO represents the m-position or p-position of one CN, X represents a chlorine atom or a fluorine atom, and n represents 0 or 1-4. Wherein, when n is 2 or more, X may be the same or different.
- the cyanobenzylamine compound represented by the general formula (I) is m— or p—cyanobenzylamine, and the compound represented by the general formula ( ⁇ ) corresponds to the corresponding m— or p—sia 15.
- a method for producing a cyanobenzoic acid halide compound characterized in that:
- a cyano anhydride characterized by converting an aldehyde group to an acid halide group without damaging the cyano group on the benzene ring of the cyanobenzaldehyde compound.
- a method for producing a benzoic acid octalide compound A cyano anhydride characterized by converting an aldehyde group to an acid halide group without damaging the cyano group on the benzene ring of the cyanobenzaldehyde compound.
- the cyanobenzaldehyde compound represented by the general formula ( ⁇ ) is m-cyanobenzaldehyde or p-cyanobenzaldehyde, and the cyanobenzoic acid chloride of the general formula (IV) corresponds to the m-cyanobenzoic acid chloride.
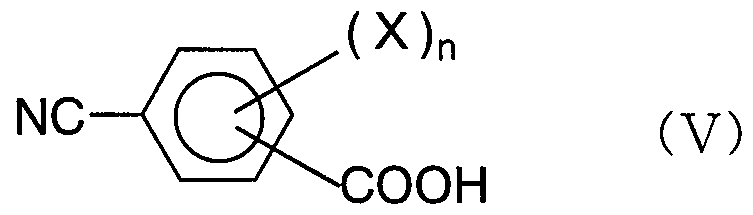
- the cyanobenzaldehyde compound represented by the general formula ( ⁇ ⁇ ) is m-cyanobenzaldehyde or p-cyanobenzaldehyde, and the cyanobenzoic acid compound represented by the general formula (V) corresponds to the m-cyanobenzaldehyde compound.
- Examples of the unsubstituted cyanobenzylamine compound used as a raw material in the method of the present invention include p-cyanobenzylamine and m_cyanobenzylamine, which are terephthalonitrile and isophthaloyl, respectively. It can be easily synthesized by reducing one nitrile group of two nitrile groups of nitrile (JP-A-49-85041)
- the substituent is not particularly limited, but may be any as long as it is inert in the reaction of the present invention.
- a halogen atom an alkyl group (Preferably C 1 to C 5), and an alkoxy group (preferably C 1 to C 5).
- a cyano benzylamine compound substituted with halogen will be described as a preferable example.
- a chlorinated cyanobenzylamine compound such as 4-cyano-2,3,5,6-tetrachloromouth benzylamine and 3-cyano-1,2,4,5,6-tetrachlorobenzylamine is terephthalic nitrile Or one of the two nitrile groups of a chlorinated terephthalonitrile compound such as tetrachloromouth terephthalonitrile obtained by chlorination of isophthalonitrile, or a chlorinated isoflononitrile compound such as tetrachloroisophthalonitrile It can be easily synthesized by reducing the group.
- Fluorinated cyanobenzylamine compounds such as 4-cyano_2,3,5,6-tetrafluorobenzylamine and 3-cyano 2,4,5,6-tetrafluorobenzylamine are tetrachlorotereph
- Chlorinated terephthalonitrile compounds such as nitronitrile or chlorinated isophthalonitriles such as tetrachloroisophthalonitrile Tetrafluorotereph obtained by the fluorination reaction of isophthalonitrile compounds It can be easily synthesized by reducing one nitrile group of two nitryl groups of a fluorinated isophthalonitrile compound such as phthalonitrile.
- a direct or indirect electrochemical oxidation reaction is not used as a synthesis reaction of the cyanobenzaldehyde compound, and the aminomethyl group of the cyanobenzylamine compound is used as an oxidizing agent for the reagent. Utilize the oxidation reaction.
- the oxidizing agent used here may be any organic or inorganic compound that directly or indirectly converts an aminomethyl group into an aldehyde group.
- Specific examples include (i) a method using a transition metal compound, (ii) a method using ammonia and formaldehyde or a condensate thereof, (iii) a method using persulfates, and (iv) a reaction with a halogenating agent. Later, it is reacted with a basic compound, Hydrolysis with a neutral aqueous solution.
- a transition metal oxide in a higher oxidation state such as chromium, manganese, iron, or ruthenium, it is possible to directly synthesize a cyanobenzaldehyde compound from a cyanobenzylamine compound.
- the dimer of cyanobenzylamine can be derived to a cyanobenzaldehyde compound. That is, a cyanobenzylamine compound can be obtained by hydrolyzing a cyanobenzylamine dimer, recovering the cyanobenzylamine compound and reusing it as a raw material.
- the compound obtained by the dehydrogenation of the cyanobenzylamine compound may be a compound other than the imine compound, such as an acetate compound or a hydroxylamine compound, which is in the same oxidation state as the aldehyde compound.
- These synthetic intermediates may be isolated and hydrolyzed, or when they cannot be isolated, they may be hydrolyzed as they are in the same reaction system.
- Transition metal compounds suitable for this reaction include copper compounds, palladium compounds, ruthenium compounds, cobalt compounds, chromium compounds, manganese compounds, iron compounds, tungsten compounds, molybdenum compounds, and the like.
- transition metal compounds may be used in a higher oxidation state alone in a stoichiometric amount or more with respect to the cyanobenzylamine compound, but other oxidizing agents such as oxygen (air) and peroxide coexist. This allows the reaction to proceed with a stoichiometric or less catalytic amount. In particular, a combination of a transition metal compound and air is preferred.
- oxidation reaction of the cyanobenzylamine compound according to the present invention (ii) ammonia and formaldehyde or its formaldehyde; A method using a condensate, (iii) a method using persulfates, (iv) a reaction with a halogenating agent, a reaction with a basic compound, and a method of hydrolyzing with an acidic aqueous solution are particularly preferable.
- a typical method for converting a cyanobenzylamine compound into a cyanobenzylaldehyde compound using an oxidizing agent will be described below.
- a cyanobenzylamine compound, ammonia and formaldehyde or a condensate thereof (such as hexamethylenetetramine) are charged into a reaction vessel under acidic conditions in the presence of water, and the reaction temperature is increased with stirring to a predetermined temperature. Until time It is performed by heating and stirring.
- the charging of the reaction and the performance of the reaction can be performed under atmospheric pressure.
- Glass and acid-resistant metal containers are suitable for the reactor.
- an oxidation-reduction reaction occurs between an imine compound (formula (2)) dehydrated and condensed from formaldehyde and ammonia and p_cyanobenzylamine to produce methylamine and p_cyanobenzylimine (formula ( 3))
- P-cyanobenzylimine is hydrolyzed with an acidic aqueous solvent to form P-cyanobenzaldehyde (Formula (4))
- Hexamethylenetetramine is known as a typical dehydration condensation compound of ammonia and formaldehyde (formula (6))
- Ammonia In addition to the ammonia molecules themselves, compounds that release ammonia under reaction conditions can be used.
- the formaldehyde not only the formaldehyde molecule itself, but also a compound that releases formaldehyde under the reaction conditions can be used.
- This reaction requires the presence of ammonia and formaldehyde, or a condensate of ammonia and formaldehyde, such as hexamethylenetetramamine.
- ammonia gaseous ammonia and aqueous ammonia as an aqueous solution can be used.
- organic and inorganic salts such as ammonium acetate and ammonium carbonate which can release ammonia under the reaction conditions can be used.
- formaldehyde gaseous formaldehyde, formalin as an aqueous solution, a dehydrated condensate such as paraformaldehyde capable of releasing formaldehyde under reaction conditions, or formaldehyde acetates such as formaldehyde dimethyl acetal, etc. are used. be able to.
- the amount of ammonia used in the present method is preferably 1 to 8 in a molar ratio to the cyanobenzylamine compound.
- the amount of formaldehyde used in the present method is preferably 1 to 12 in a molar ratio to the cyanobenzylamine compound.
- a condensate of 1 mol of formaldehyde is preferably 1 to 12 with respect to the cyanobenzylamine compound.
- the molar ratio is preferably 0.5 to 2, more preferably 0.7 to 1.3, based on the cyanobenzylamine compound. If the amount of hexamethylenetetramamine is too small relative to the amount of the cyanobenzylamine compound, it takes time to complete the reaction.If the amount is too large, the reaction produces by-products of organic and inorganic compounds. Separation operation is required.
- the water may be used in a solvent amount or in a trace amount.
- the water may be entirely added at the time of preparing the reaction, or may be added as the reaction proceeds.
- This reaction is performed under acidic conditions using an acid.
- the acids used are organic and inorganic protic acids.
- inorganic acid sulfuric acid, nitric acid, hydrochloric acid, phosphoric acid and the like are suitable.
- organic acid carboxylic acids such as acetic acid and butyric acid, and sulfonic acids such as tosylic acid can be used, and particularly, low-boiling organic sulfonic acids that can also serve as a solvent are preferable.
- Lewis acids such as aluminum chloride and stannic chloride which can react with a protic solvent and release a protonic acid can be used. The entire amount of the acid may be added during the preparation of the reaction, or may be added as the reaction proceeds.
- the reaction when the reaction is carried out with an acidic aqueous solution using only water as a medium, the reaction raw materials, intermediates, products, and the like may be precipitated, and therefore, an organic solvent can be used to prevent the precipitation of the raw materials and products.
- an organic solvent when an organic solvent is used, the reaction can be performed by appropriately adding an amount of water necessary for the reaction.
- organic solvent examples include hydrocarbons such as toluene and xylene, alcohols such as methanol and ethanol, carboxylic acids such as acetic acid and propionic acid, chloroform, 1,2—
- the reaction may be carried out as a homogeneous solution by adding acetic acid or ethanol, or may be carried out in a two-layer system using toluene-1,2-dichlorobenzene.
- the solvent may be selected according to the purification method.
- the amount of the solvent in this reaction is preferably 3 to 10 times, more preferably 4 to 6 times the total weight of the cyanobenzylamine compound and ammonia, formaldehyde or a condensate thereof (such as hexamethylenetetramine). Is desirable.
- the reaction temperature is preferably 50 ° (: to 150 ° C, more preferably It is desirable to select between 70 ° C and 130 ° C.
- reaction time of this reaction depends on the reaction temperature and the composition of the solvent, but is preferably 30 minutes to 10 hours.
- P- or m-cyanobenzaldehyde will be described by way of a typical example of a method for purifying a cyanobenzaldehyde compound obtained by this reaction.
- p- or m_cyanobenzaldehyde may be extracted with hot water without melting by heating.
- p- or m_cyanobenzaldehyde is co-distilled with water (the boiling point is 100 ° C or higher, so there is no azeotropic distillation). Or m—cyanobenzaldehyde is obtained.
- water is distilled off at a boiling point of 100.5 ° C to 105 ° C (normal pressure), and about 0.5 g to 3 g is obtained per 100 g of water.
- the cyanobenzaldehyde compound can be purified by distillation or recrystallization.
- This reaction is preferably carried out by charging a cyanobenzylamine compound, a persulfate and water in a reaction vessel, and heating and stirring at a predetermined reaction temperature for a predetermined time.
- the charging of the reaction and the execution of the reaction can be performed under atmospheric pressure.
- Glass and acid-resistant metal containers are suitable for the reactor.
- Cyanobenzylamine compounds undergo a dehydrogenation reaction with sodium persulfate to produce the corresponding imine. At this time, the imine undergoes dimerization due to liquidity (neutral to basic) or reacts with unreacted p_cyanobenzylamine to produce an imine dimer.
- sodium peroxysulfate is converted to sodium monohydrogen sulfate by the dehydrogenation of P-cyanobenzylamine, and the reaction system becomes acidic. Under acidic conditions, the dimer of the imine and the P-cyanobenzylamine compound hydrolyzes to produce P-cyanobenzaldehyde.
- the persulfate ammonium persulfate, sodium persulfate, persulfuric acid lime, or the like can be used.
- the acidity of the reaction system gradually increases with the consumption of persulfate. Since the persulfate gradually decomposes in an acidic condition, it is better to use the persulfate in a slightly excessive amount relative to the cyanobenzylamine compound in order to complete the reaction.
- the amount of the persulfate is preferably from 1 to 1.8, more preferably from 1.1 to 1.3, in a molar ratio to the cyanobenzylamine compound.
- a transition metal compound can be used as a catalyst.
- Transition metal compounds act on persulfate activation, and can react mildly at lower temperatures than under non-transition metal catalysts.
- a transition metal compound that causes a one-electron redox reaction such as a silver compound, a copper compound, an iron compound, a cerium compound, a manganese compound, and a titanium compound is used.
- monovalent silver compounds such as silver nitrate, monovalent copper compounds such as copper chloride, divalent iron compounds such as ferrous sulfate, trivalent cerium compounds such as cerium trichloride, and acetic acid (II)
- a transition metal ion compound having an appropriate oxidation number such as a divalent manganese compound such as manganese and a trivalent titanium compound such as titanium trichloride is used, there is no induction period of activation and the reaction time can be shortened.
- the transition metal compound is used in a molar ratio to the persulfate of 0.0001 to 0.01.
- reaction raw materials, intermediates, products, etc. may precipitate out of the solution, so use an organic solvent together to prevent the precipitation of the raw materials, products, etc. be able to.
- hydrocarbons such as toluene and xylene
- alcohols such as methanol and ethanol
- chloroform halogens
- 1,2-dichloroethane nitriles
- acetonitrile and propionitrile dioxane
- Organic solvents such as ethers such as dimethoxyethane can be used.
- the reaction may be performed by adding acetonitrile or ethanol to make a homogeneous solution.
- the reaction may be performed in a two-layer system using toluene or 1,2-dichloromethane.
- the reaction system solvent may be selected according to the solvent used in the purification method.
- the amount of the solvent used in this reaction is preferably 3 to 30 times, and more preferably 5 to 10 times the weight of the cyanobenzylamine compound.
- reaction temperature is too low, the reaction rate is low. If the reaction temperature is too high, the generated cyanobenzaldehyde compound is decomposed to lower the yield.
- a temperature between 40 ° C and 80 ° C is selected.
- the transition metal compound is added, it can be carried out at 0 ° C to 80 ° C, but preferably at 20 ° C to 70 ° C.
- the reaction time is influenced by the starting compound, the reaction temperature, the composition of the solvent, and the like, but is usually preferably 20 minutes to 10 hours.
- a cyanobenzylamine compound and a halogenating agent are charged into a reaction vessel, the temperature is raised to a reaction temperature with stirring, and the mixture is heated and stirred for a predetermined time to cause a reaction, and the cyanobenzylamine compound disappears. Thereafter, a basic compound is charged into a reaction vessel and subjected to a dehalogenation reaction, and then the solution is made acidic in the presence of water.
- the preparation of the reaction and the execution of the reaction are not particularly limited, they can be usually performed under atmospheric pressure. Glass and acid-resistant metal containers are suitable for the reactor.
- halogenating agent used in the present reaction means a compound capable of introducing a halogen atom into an amino group of a cyanobenzylamine compound.
- the “base” used in this reaction means a compound capable of capturing a proton acid by an acid-base reaction.
- a reaction from P_cyanobenzilamine to P-cyanobenzaldehyde will be described.
- P-cyanobenzylamine reacts with the halogenating agent (XY) to halogenate the amino group of the primary amine (formula (8), this reaction is hereinafter referred to as "halogenation").
- the base is preferably added after the reaction between p-cyanobenzylamine and the halogenating agent is completed.
- the process may be shortened by adding a base in the presence of a halogenating agent, but dehydrogenogenation occurs in the same reaction system to produce imine.
- This imine reacts with unreacted p-cyanobenzylamine to form a dimer of p-cyanobenzylamine as a by-product, resulting in a decrease in the yield of p-cyanobenzaldehyde.
- a volatile halogenating agent is used for halogenation, introduce a gas such as nitrogen after the reaction.
- the halogenating agent can be removed, the non-volatile halogenating agent may be removed by decomposition using an appropriate reducing agent or the like.
- the acid hydrolysis is preferably carried out after completion of the dehydrohalogenation to produce the aldehyde.
- N-dihalogen of a cyanobenzylamine compound can be obtained due to the presence of an excess of a halogenating agent in the halogenation of the formula (8) or a low selectivity of a monohalogen.
- the N-monohalogen can be directly hydrolyzed without using a reducing agent, but the N-monohalogen is said to be an oxime equivalent, and requires relatively severe conditions for hydrolysis. It is not preferable because the cyano group may be decomposed.
- the product halogenoimine may further undergo a dehydrohalogenation reaction to form nitrile. As described above, when N-dihalogenation is performed, a route for generating an extra reduction operation or a by-product is generated. Therefore, it is preferable that the halogenation be stopped with an N-monohalogen.
- the halogenating agent that can be used in this reaction is not particularly limited, and a halogenating agent generally used in organic synthesis can be used.
- halogen molecules such as chlorine, bromine and iodine
- mixed halogen molecules such as bromine chloride (BrCl) and iodine bromide (IBr)
- haloimides such as N-chlorosuccinimide, N-bromosuccinimide and N-bromoacetamide
- Hypohalogenous acids such as amides, loamides, hypochlorous acid, and hypobromite
- hypohalogenates such as calcium hypochloride (Ca (C10) 2 ) and t-butyl hypochloride
- hypohalogens Chloride and bromide such as acid esters, sulfuryl chloride and sulfuryl bromide are used.
- the amount of the halogenating agent to be used is preferably 1 mol equivalent to 1 mol equivalent of the cyanobenzylamine compound.
- the reaction temperature is about 20 to 120 ° C, preferably 40 to 80 ° C.
- the reaction time is preferably 0.5 to 8 hours.
- radical initiator examples include azobiss such as azobisisobutylylonitrile; disilcapoxides such as benzoyl peroxide; dialkyl peroxides such as di-t-butyl peroxide and dicumyl peroxide; Hydroperoxides such as tert-butyl hydrooxide and cumene hydrooxide, and alkyl esters such as tert-butyl peracetate and tert-butyl benzoate are mentioned.
- azobiss such as azobisisobutylylonitrile
- disilcapoxides such as benzoyl peroxide
- dialkyl peroxides such as di-t-butyl peroxide and dicumyl peroxide
- Hydroperoxides such as tert-butyl hydrooxide and cumene hydrooxide
- alkyl esters such as tert-butyl peracetate and tert-butyl benzoate are mentioned.
- the solvent used in the reaction of the compound obtained by the reaction of the cyanobenzylamine compound with the halogenating agent and the base is a solvent containing the cyanobenzylamine compound and the halogenating agent. May be the same as the reaction solvent described above, or a mixed solvent may be added by adding another solvent, or the solvent may be substituted.
- the reaction temperature is about 0 to 80 ° C, preferably 10 to 50 ° C. If the reaction temperature is lower than 0 ° C, the time required for the completion of the reaction is significantly increased, and if the reaction temperature is higher than 80 ° C, the reaction substrate is decomposed and the yield of the dehydrohalogenation reaction is lowered.
- the reaction time is preferably from 30 minutes to 10 hours.
- the reaction time depends on the amount of the base added and the reaction temperature.At least an equimolar amount of the base of the charged cyanobenzylamine compound is required, and when the solution is a two-layer system or a weak base is used. When the excess base is used, the rate of the dehydrohalogenation reaction can be improved. Also, when an acidic compound is generated by the halogenation reaction in the previous step, it is necessary to add an amount of a base capable of capturing excess acid as a salt.
- an organic or inorganic basic compound can be used as the base that can be used in the present invention.
- organic basic compound amines, nitrogen-containing heterocyclic compounds and the like can be used, and tertiary amines and nitrogen-containing heterocyclic compounds are preferable.
- amines, nitrogen-containing heterocyclic compounds and the like can be used, and tertiary amines and nitrogen-containing heterocyclic compounds are preferable.
- pyridine, triethylamine, N-methyl Morpholine and the like are preferred.
- Inorganic basic compounds include alkali and alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide and magnesium hydroxide, alkaline earth metal oxides such as magnesium oxide and calcium oxide, sodium peroxide, Alkali metal peroxides such as potassium peroxide, alkali metal superoxides such as sodium superoxide and superoxide, alkali metal carbonates such as sodium hydrogencarbonate, lithium hydrogencarbonate, sodium carbonate and potassium carbonate, sodium Alkoxides of alkali metals such as methoxide, sodium ethoxide and potassium tertiary butyloxide can be used.
- alkali and alkaline earth metal hydroxides such as sodium hydroxide, potassium hydroxide and magnesium hydroxide
- alkaline earth metal oxides such as magnesium oxide and calcium oxide
- sodium peroxide Alkali metal peroxides such as potassium peroxide
- alkali metal superoxides such as sodium superoxide and superoxide
- alkali metal carbonates such as sodium hydrogen
- the acid hydrolysis in the final step will be described.
- the acid hydrolysis reaction is preferably performed in an acidic solution containing water.
- the pH of the solution is between 1 and 6.
- the pH is 3-5.
- the amount of the acid is at least equimolar to the imine formed in the dehydrohalogenation reaction, and it is necessary to add an amount of the acid corresponding to the excess base in order to capture the excess base as a salt. It is.
- the reaction temperature is about 20 to 100 ° C, preferably 40 to 80 ° C.
- the reaction time is preferably 0.5 to 8 hours.
- the same solvent may be used as the reaction solvent for the halogenation step and the dehydrohalogenation reaction, or a mixed solvent may be added by adding another solvent, or the solvent may be substituted. If water is not used in the halogenation step and the dehydrohalogenation step, water must be at least equimolar to imine. Water may be added as a solvent as long as the reaction compound does not precipitate from the reaction system.
- Acids that can be used for acid hydrolysis in the present invention are organic and inorganic protic acids.
- organic acid carboxylic acids such as acetic acid, propionic acid, and trifluoroacetic acid, and sulfonic acids such as methanesulfonic acid and p-toluenesulfonic acid can be used.
- solvents that can be used in this reaction include, for example, ethers such as dioxane and tetrahydrofuran, alcohols such as methanol, ethanol, propanol and butanol, nitriles such as acetonitrile and propionitrile, and dichloromethane.
- Halogen compounds such as 1,2-dichloroethane and aprotic polar solvents such as dimethylformamide and dimethylsulfoxide.
- Water can also be used as a solvent.However, the reaction raw materials, intermediates, and products may be precipitated.Therefore, in order to prevent the precipitation of the raw materials and products, the water is mixed with the above organic solvent to carry out the reaction. Can be performed. In this case, water and an organic solvent may be mixed to form a homogeneous system, or an organic solvent compatible with water may be used. If present, the reaction may be performed in a two-layer system. The same solvent may be used in each of the steps of halogenation, dehalogenation, and acid hydrolysis, or the solvent may be mixed or substituted according to the solubility of each raw material, intermediate, or product.
- a cyanobenzaldehyde compound obtained by the above-mentioned method of the present invention is preferably used, but is not limited thereto.
- an unsubstituted cyanobenzaldehyde compound may be oxidized by a reduction reaction of one nitrile group of the corresponding benzenedinitrile (Japanese Patent Application Laid-Open No. 49-85041), such as a Sommdet reaction of cyanobenzylamine. It can be synthesized by a typical deamination reaction.
- Chlorinated cyanobenzaldehyde compounds such as aldehydes are obtained by chlorinating isophthalonitrile or terephthalonitrile to synthesize tetrachloroisophthalonitrile or tetrachloroterephthalonitrile, and then isolating tetrachloroisophthalonitrile or tetrachloroterephthalonitrile.
- the fluorinated cyanobenzaldehyde compound may be tetrafluoroisophthalonitrile or tetrafluoroteretrile obtained by a fluorination reaction of a chlorinated phthalonitrile compound such as tetrachloroisophthalone nitrile or tetrachloroterephthalonitrile.
- a chlorinated phthalonitrile compound such as tetrachloroisophthalone nitrile or tetrachloroterephthalonitrile.
- nitrile groups of fluorinated phthalonitrile compounds such as lonitrile, 3-shear obtained by the reduction reaction of one nitrile group It can be synthesized by oxidative deamination of 2,4,5,6-tetrafluorobenzylamine or 41-cyano_2, amine.
- a halogenating agent is used for acid octylation of an aldehyde group.
- halogenating agent is a general term for reagents that can introduce a halogen atom into an aldehyde group of a cyanobenzaldehyde compound and convert it into an acid halide group. This reaction is hereinafter referred to as acid halide conversion.
- halogen molecules such as chlorine and bromine
- mixed halogen molecules such as bromine chloride (BrCl)
- octaimides such as N-chlorosuccinimide, N-bromosuccinimide, and N-bromoacetamide.
- Octal amides calcium hypochloride (Ca (Cl ⁇ ⁇ ⁇ ) 2 ), t-butyl hypochlorite and other hypohalites, hypohalous esters, sulfuryl chloride, sulfuryl bromide and other chlorides and bromides
- a halogenating agent generally used in organic synthesis can be used.
- the halogenating agent is preferably used in an amount of 0.8 mol 1 equivalent to 3 mol 1 equivalent to 1 mol equivalent of the cyanobenzaldehyde compound.
- the reaction temperature is preferably about 50 to 150 ° C, and more preferably 40 to 100 ° C.
- the reaction time is preferably 0.5 to 8 hours.
- radical initiator examples include azobis such as azobisisobutyronitrile, disil peroxides such as benzoyl peroxide, and dialkyl peroxides such as di-tert-butyl peroxide and dicumyl peroxide.
- Hydroxides such as t-butyl hydroxide, t-butyl hydroperoxide and cumene hydroperoxide, and alkyl peresters such as t-butyl peracetate and t-butyl perbenzoate can be suitably used.
- the reaction can be carried out in a molten state by raising the temperature of the cyanobenzaldehyde compound to the melting point without using a solvent.
- a reaction product of a cyano benzoic acid octalide compound corresponding to the cyano benzaldehyde compound of the reaction raw material is added to the cyano benzaldehyde compound of the raw material, and the melting point is lowered to carry out the reaction at a lower temperature than that of the cyano benzaldehyde compound alone.
- the cyanobenzoic acid octalide compound to be added is preferably used in a molar ratio of 0.05 to 10 with respect to the cyanobenzaldehyde compound.
- a solvent can be used. Any solvent can be used as long as it does not decompose the halogenating agent and the cyanobenzoic acid halide compound and does not adversely affect the reaction.
- halogens such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, chloroform benzene, dichloromethane benzene, ethers such as 1,2-dimethoxyethane, dioxane, diglyme, and benzene
- aromatic hydrocarbons nitriles such as acetonitrile and propionitrile, and tertiary alcohols such as t-butanol.
- nitriles such as acetonitrile and propionitrile
- tertiary alcohols such as t-butanol.
- the amount of the solvent used is preferably 1 to 50 times the weight of the cyanobenzaldehyde compound.
- a method for producing a cyanobenzoic acid compound according to the present invention comprises the steps of: The reaction is carried out by charging a metal compound and a hypohalous acid compound together with water or water and an aprotic polar solvent as a solvent in a reaction vessel, and reacting the mixture under stirring at a predetermined temperature for a predetermined time.
- the raw materials can be charged and the reaction can be performed under normal pressure or under pressure. Preferably, it is performed under normal pressure. Glass, acid-resistant metal containers, etc. are used as the reactor.
- cyano benzaldehyde compounds As the starting material of the cyano benzaldehyde compound used in this reaction, those obtained by the method of the present invention described above are preferably used, but are not limited thereto, and are not limited to the above-mentioned (b) of the cyano benzoic acid halide compound.
- Unsubstituted cyano benzaldehyde compounds, chlorinated cyano benzaldehyde compounds, fluorinated cyano benzaldehyde compounds, and the like, which are synthesized by the method described in the section, are used.
- a hypohalous acid compound is used for the oxidation of the aldehyde group.
- the hypohalous compound can be used in a relatively wide pH range of acidic, neutral and basic.However, if the pH of the reaction solution is too low, the reaction of the hypohalogen compound can be performed. Involvement of hypohalous compounds until the completion of the reaction becomes significant, and the consumption of hypohalous compounds increases until the reaction is completed. If the pH is too high, cyanobenzal. Dehydration of the nitrile group of the aldehyde compound or the cyanobenzoic acid compound generated by the reaction is decomposed. The reactions are likely to occur simultaneously, and the purity of the cyanobenzoic acid compound will be low.
- the pH is preferably in the range of 5 to 10.
- hypohalous acid compound may be added all at once at the start of the reaction.However, since the reaction may occur rapidly and a side reaction may occur, it is usually added over 5 minutes to 10 hours. preferable.
- hypohalous compounds that can be used in the production of the cyanobenzoic acid compound of the present invention include hypohalous acids such as hypochlorous acid, hypobromite, and hypoiodic acid, sodium hypochlorite, and the like. Potassium hypochlorite, calcium hypochlorite And hypohalites such as barium hypochlorite, sodium hypobromite, potassium hypobromite, sodium hypoiodite, and hypoiodite potassium lime.
- the amount of the hypohalous acid compound used in the reaction of the present invention is preferably 1 to 5 in a molar ratio to the cyanobenzaldehyde compound.
- the cyanobenzaldehyde compound As the cyanobenzaldehyde compound is oxidized, the cyanobenzoic acid compound is generated and starts to precipitate.
- the pH of the reaction solution is 4 or less, a large amount of cyanobenzoic acid compound rapidly precipitates, which makes stirring difficult, and the unreacted cyanobenzaldehyde compound is incorporated into the precipitated cyanobenzoic acid compound to complete the reaction. There are problems such as difficulty.
- the base can be added to the reaction system, the cyanobenzoic acid compound can be dissolved as a salt in the reaction solution, and the reaction can proceed efficiently if the reaction is carried out in a homogeneous solution.
- the base may be initially added in a necessary amount all at once, or may be added continuously as the reaction proceeds so as not to precipitate the cyanobenzoic acid compound.
- Bases that can be used in the production of the cyanobenzoic acid compound of the present invention include alkali metals such as lithium hydroxide, sodium hydroxide, potassium hydroxide, magnesium hydroxide, and calcium hydroxide, and alkaline earth metals.
- Alkali metal bicarbonate such as metal hydroxide, sodium hydrogen carbonate, potassium bicarbonate, lithium carbonate, sodium carbonate, potassium carbonate, magnesium carbonate, calcium carbonate, etc.
- Metal oxides of alkaline earth metals such as carbonate, magnesium oxide and calcium oxide can be used.
- the amount of the base used in the above depends on the type and amount of the coexisting hypochlorous acid compound, but the total amount of the base contained in the hypochlorous acid compound and the base to be added to the reaction is equimolar with the cyanobenzaldehyde compound.
- the reaction system during the reaction. The pH should be such that the pH range can be maintained between 5 and 10.
- the reaction can be carried out in an aqueous solution. If the solubility of the cyanobenzaldehyde compound is low when water is used as the solvent, the reaction can be carried out efficiently by coexisting a non-protonic polar solvent.
- aprotic polar solvents examples include ethers such as dioxane and diglyme, amides such as dimethylformamide, iodine-containing systems such as dimethylsulfoxide and sulfolane, and nitriles such as acetonitrile.
- the amount of the aprotic polar solvent used in this reaction is at least 0.1 (part by weight) with respect to 1 (part by weight) of the cyanobenzaldehyde compound, and can be used in an amount that can be mixed with water. Preferably, it is 0.3 to 3 (parts by weight) with respect to 1 (parts by weight) of the cyanobenzaldehyde compound.
- the reaction temperature is preferably 10 to 80 ° C, more preferably 30 to 50 ° C.
- the reaction time of this reaction depends on the pH, the composition of the solvent and the like, but is preferably 10 minutes to 12 hours.
- Example 24 13.2 g of p-cyanobenzylamine and 200 g of acetic acid are mixed, and 160 g (14% by weight) of sodium hypochlorite aqueous solution is added dropwise with stirring at 50 ° C over 30 minutes, and further 1 hour The mixture was stirred at the same temperature. Analysis by high performance liquid chromatography showed that the yield of P-cyanobenzaldehyde was 43%.
- Example 25 13.2 g of p-cyanobenzylamine and 200 g of acetic acid are mixed, and 160 g (14% by weight) of sodium hypochlorite aqueous solution is added dropwise with stirring at 50 ° C over 30 minutes, and further 1 hour The mixture was stirred at the same temperature. Analysis by high performance liquid chromatography showed that the yield of P-cyanobenzaldehyde was 43%.
- Example 25 13.2 g of p-cyanobenzylamine and 200 g of acetic acid are mixed, and 160 g (14% by weight) of sodium hypo
- the toluene was concentrated under reduced pressure, and 48 kg of a toluene solution was recovered. After returning to normal pressure, 54 kg of water was added, and the mixture was concentrated under reduced pressure until the distillation temperature became constant (toluene was completely distilled off), and 21 kg of toluene and 6 kg of water were recovered.
- the mixture was cooled to room temperature with stirring (it became an aqueous solution in which p-cyanobenzaldehyde had precipitated.).
- a 13.5 wt% aqueous solution of sodium hypochlorite (125 kg) was heated to about 40 ° C ( ⁇ 5 ° C).
- a sulfuric acid aqueous solution containing 300 g of 98% by weight sulfuric acid and 165 g of water was added dropwise in 20 minutes. The mixture was further stirred at 100 ° C for 1 hour. After cooling the internal temperature to 50 ° C, 300 g of 98% by weight sulfuric acid was added. The reaction solution was concentrated to dryness under reduced pressure. The concentrated residue was separated with toluene water, washed with water, and the obtained toluene layer was concentrated to dryness under reduced pressure. 900 g of 1% by weight sulfuric acid was added to the concentrated residue, and the mixture was vigorously stirred at 100 ° C. for 3 hours and cooled to room temperature over 2 hours.
- a cyanobenzaldehyde compound can be easily produced with high yield and purity by oxidizing a cyanobenzylamine compound easily obtained from a phthalonitrile compound using an oxidizing agent.
- the load on the environment is significantly reduced as compared with the conventional method under the simple reaction conditions using a cyanobenzaldehyde compound, which can be easily obtained in a large amount and at low cost, as a starting material by the above method.
- a cyanobenzoic acid halide compound can be industrially produced with high purity and high yield.
- the cyanobenzaldehyde compound, cyanobenzoic acid halide compound and cyanobenzoic acid compound obtained by the method of the present invention are important intermediates such as pharmaceuticals, pesticides, liquid crystals, and functional polymer monomers.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description











Claims






Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP99922570A EP1083165A4 (en) | 1998-05-28 | 1999-05-28 | METHOD FOR PRODUCING CYANOPHENYL DERIVATIVES |
| AU39564/99A AU3956499A (en) | 1998-05-28 | 1999-05-28 | Processes for producing cyanophenyl derivatives |
Applications Claiming Priority (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US60/123,983 | 1998-03-11 | ||
| JP10/147746 | 1998-05-28 | ||
| JP14774698A JPH11343273A (ja) | 1998-05-28 | 1998-05-28 | シアノベンズアルデヒド化合物の製法 |
| US9130998P | 1998-06-30 | 1998-06-30 | |
| US60/091,309 | 1998-06-30 | ||
| JP10/296330 | 1998-10-19 | ||
| JP29633098A JP4402186B2 (ja) | 1998-10-19 | 1998-10-19 | シアノ安息香酸化合物の製造方法 |
| JP32078798A JP4482165B2 (ja) | 1998-11-11 | 1998-11-11 | シアノベンズアルデヒド化合物の製造法 |
| JP10/320787 | 1998-11-11 | ||
| JP35558298A JP4335339B2 (ja) | 1998-12-15 | 1998-12-15 | シアノベンズアルデヒドの製造方法 |
| JP10/355582 | 1998-12-15 | ||
| US12398399P | 1999-03-11 | 1999-03-11 | |
| JP11086058A JP2000281637A (ja) | 1999-03-29 | 1999-03-29 | シアノ安息香酸ハライドの製造方法 |
| JP11/86058 | 1999-03-29 | ||
| US13242799P | 1999-05-04 | 1999-05-04 | |
| US60/132,427 | 1999-05-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999061411A1 true WO1999061411A1 (en) | 1999-12-02 |
Family
ID=27572785
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1999/002857 Ceased WO1999061411A1 (en) | 1998-05-28 | 1999-05-28 | Processes for producing cyanophenyl derivatives |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1083165A4 (ja) |
| AU (1) | AU3956499A (ja) |
| WO (1) | WO1999061411A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0989115A3 (en) * | 1998-09-24 | 2001-04-04 | Showa Denko Kabushiki Kaisha | Process for producing cyanobenzoic acid derivatives |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0967302A (ja) * | 1995-08-29 | 1997-03-11 | Nippon Light Metal Co Ltd | ベンゼンカルボニルクロライド誘導体の製造方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5124494B2 (ja) * | 1972-12-20 | 1976-07-24 |
-
1999
- 1999-05-28 WO PCT/JP1999/002857 patent/WO1999061411A1/ja not_active Ceased
- 1999-05-28 EP EP99922570A patent/EP1083165A4/en not_active Ceased
- 1999-05-28 AU AU39564/99A patent/AU3956499A/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0967302A (ja) * | 1995-08-29 | 1997-03-11 | Nippon Light Metal Co Ltd | ベンゼンカルボニルクロライド誘導体の製造方法 |
Non-Patent Citations (6)
| Title |
|---|
| MCKILLOP A, KEMP D: "FURTHER FUNCTIONAL GROUP OXIDATIONS USING SODIUM PERBORATE", TETRAHEDRON, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 45, no. 11, 1 January 1989 (1989-01-01), AMSTERDAM, NL, pages 3299 - 3306, XP002919780, ISSN: 0040-4020, DOI: 10.1016/S0040-4020(01)81008-5 * |
| PUNNIYAMURTHY T, SWINDER JEET SINGH KALRA, JAVED IQBAL: "COBALT CATALYSED SYNTHESIS OF 1,2-DIONES FROM AROMATIC ALDEHYDES INTHE PRESENCE OF N-BUTANAL", TETRAHEDRON LETTERS, PERGAMON, GB, vol. 35, no. 18, 1 January 1994 (1994-01-01), GB, pages 2959/2960, XP002919779, ISSN: 0040-4039, DOI: 10.1016/S0040-4039(00)76671-8 * |
| SASSON Y, ZAPPI G D, NEUMANN R: "LIQUID-PHASE OXIDATION OF DEACTIVATED METHYLBENZENES BY AQUEOUS SODIUM HYPOCHLORITE CATALYZED BY RUTHENIUM SALTS UNDER PHASE-TRANSFER CATALYTIC CONDITIONS", THE JOURNAL OF ORGANIC CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 51, 1 January 1986 (1986-01-01), US, pages 2880 - 2883, XP002919778, ISSN: 0022-3263, DOI: 10.1021/jo00365a005 * |
| See also references of EP1083165A4 * |
| SEMMELHACK M F, SCHMID C R: "NITROXYL-MEDIATED ELECTROOXIDATION OF AMINES TO NITRILES AND CARBONYL COMPOUNDS", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, AMERICAN CHEMICAL SOCIETY, US, vol. 105, no. 22, 1 January 1983 (1983-01-01), US, pages 6732 - 6734, XP002919777, ISSN: 0002-7863, DOI: 10.1021/ja00360a042 * |
| SMITH J R L, MORTIMER D N: "OXIDATIVE N-DEALKYLATION OF N,N-DIMETHYLBENZYLAMINES BY METALLOPORPHYRIN-CATALYSED MODEL SYSTEMS FOR CYTOCHROME P450 MONO-OXYGENASES", JOURNAL OF THE CHEMICAL SOCIETY, CHEMICAL COMMUNICATIONS., CHEMICAL SOCIETY. LETCHWORTH., GB, 1 January 1985 (1985-01-01), GB, pages 64/65, XP002919776, ISSN: 0022-4936, DOI: 10.1039/c39850000064 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0989115A3 (en) * | 1998-09-24 | 2001-04-04 | Showa Denko Kabushiki Kaisha | Process for producing cyanobenzoic acid derivatives |
| EP1508567A3 (en) * | 1998-09-24 | 2005-03-02 | Showa Denko Kabushiki Kaisha | Process for producing cyanobenzoic acid derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| AU3956499A (en) | 1999-12-13 |
| EP1083165A1 (en) | 2001-03-14 |
| EP1083165A4 (en) | 2003-06-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6262292B1 (en) | Method for producing cyanophenyl derivatives | |
| KR19990077617A (ko) | 메타크릴산메틸의제조방법 | |
| JPS6212212B2 (ja) | ||
| CN100398500C (zh) | 一种生态友好的溴苯合成方法 | |
| WO1999061411A1 (en) | Processes for producing cyanophenyl derivatives | |
| IL131974A (en) | 3 cyano - 2, 4, 5 - trifluorobenzoyl fluoride and intermediates to form it | |
| EP0921115B1 (en) | Process for making aromatic nitriles | |
| KR100626400B1 (ko) | 시아노페닐 유도체 제조방법 | |
| KR100674024B1 (ko) | 시아노페닐 유도체 제조방법 | |
| EP1721892A1 (en) | Method for producing cyanophenyl derivatives | |
| US6353126B1 (en) | Process for the production of malononitrile | |
| JP3930647B2 (ja) | シアノ安息香酸の製造法 | |
| EP1721891A1 (en) | Method for producing cyanophenyl derivatives | |
| JP4402186B2 (ja) | シアノ安息香酸化合物の製造方法 | |
| EP1508567B1 (en) | Process for producing cyanobenzoic acid derivatives | |
| JP4275242B2 (ja) | シアノ基含有安息香酸化合物の製造方法 | |
| JP4482165B2 (ja) | シアノベンズアルデヒド化合物の製造法 | |
| JP2000281637A (ja) | シアノ安息香酸ハライドの製造方法 | |
| JP4335339B2 (ja) | シアノベンズアルデヒドの製造方法 | |
| JP5000031B2 (ja) | 芳香族−o−ジアルデヒド化合物の製造方法 | |
| JP2001114741A (ja) | 芳香族シアノ安息香酸化合物の製造方法 | |
| JP4275243B2 (ja) | シアノ基含有安息香酸化合物の製造方法 | |
| JPH0725817A (ja) | アルキルビフェニルカルボン酸の製造法 | |
| JPH08169868A (ja) | 4−シアノ−4’−ヒドロキシビフェニルの製造方法 | |
| JP2000143605A (ja) | シアノ安息香酸クロライド化合物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 99806714.8 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1999922570 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007013420 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IN/PCT/2000/871/CHE Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999922570 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007013420 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020007013420 Country of ref document: KR |