WO2000024711A1 - Synthesis of methylthiophenyl hydroxyketones - Google Patents
Synthesis of methylthiophenyl hydroxyketones Download PDFInfo
- Publication number
- WO2000024711A1 WO2000024711A1 PCT/US1999/025064 US9925064W WO0024711A1 WO 2000024711 A1 WO2000024711 A1 WO 2000024711A1 US 9925064 W US9925064 W US 9925064W WO 0024711 A1 WO0024711 A1 WO 0024711A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- process according
- formula
- compound
- group
- substantially non
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC*N1*CC*CCC1 Chemical compound CC*N1*CC*CCC1 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/14—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides
- C07C319/20—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides by reactions not involving the formation of sulfide groups
Definitions
- Non-steroidal, antiinflammatory drugs exert most of their antiinflammatory, analgesic and antipyretic activity and inhibit hormone-induced uterine contractions and certain types of cancer growth through inhibition of prostaglandin G/H synthase, also known as cyclooxygenase.
- cyclooxygenase-1 cyclooxygenase-1
- COX-2 cyclooxygenase-1
- COX-1 This enzyme is distinct from the COX-1 which has been cloned, sequenced and characterized from various sources including the sheep, the mouse and man.
- the second form of cyclooxygenase, COX-2 is rapidly and readily inducible by a number of agents including mitogens, endotoxin, hormones, cytokines and growth factors.
- prostaglandins have both physiological and pathological roles, we have concluded that the constitutive enzyme, COX-1, is responsible, in large part, for endogenous basal release of prostaglandins and hence is important in their physiological functions such as the maintenance of gastrointestinal integrity and renal blood flow.
- COX-2 the inducible form
- a selective inhibitor of COX-2 will have similar antiinflammatory, antipyretic and analgesic properties to a non-steroidal antiinflammatory drug, and in addition would inhibit hormone-induced uterine contractions and have potential anti-cancer effects, but will have a diminished ability to induce some of the mechanism-based side effects.
- such a compound should have a reduced potential for gastrointestinal toxicity, a reduced potential for renal side effects, a reduced effect on bleeding times and possibly a lessened ability to induce asthma attacks in aspirin-sensitive asthmatic subjects.
- such a compound will also inhibit prostanoid-induced smooth muscle contraction by preventing the synthesis of contractile prostanoids and hence may be of use in the treatment of dysmenorrhea, premature labour, asthma and eosinophil related disorders. It will also be of use in the treatment of Alzheimer's disease, for decreasing bone loss particularly in postmenopausal women (i.e. treatment of osteoporosis) and for the treatment of glaucoma.
- COX-2 inhibitors is given in an article by John Vane, Nature, Vol. 367, pp. 215-216, 1994, and in an article in Drug News and Perspectives. Vol. 7, pp. 501-512, 1994.
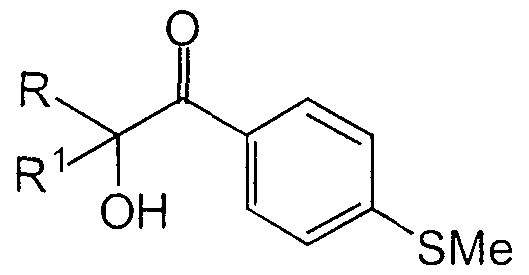
- This invention encompasses a novel process for synthesizing compounds represented by formula A:
- R and Rl are C ⁇ _6alkyl, comprising reacting a compound of formula B:
- X represents a 5 or 6-membered non-aromatic ring wherein X is selected from the group consisting of: C, N, O and S,
- the invention encompasses a process for synthesizing compounds represented by formula A:
- R and Rl are C ⁇ _6alkyl, comprising reacting a compound of formula B:
- X represents a 5 or 6-membered non-aromatic ring wherein X is selected from the group consisting of: C, N, O and S,
- the lithiating agent is selected from the group consisting of: n- butyllithium, hexyllithium and phenyllithium.
- the substantially non-reactive solvent is selected from the group consisting of: tetrahydrofuran, toluene, ethylene glycol dimethyl ether, t-butyl methyl ether and -
- Another embodiment of the invention encompasses a mixture of two or more of the aforesaid solvents.
- the reduced temperature ranges from about -78° C to about 0° C. In another preferred embodiment the reduced temperature is about -40° C.
- a preferred embodiment of the invention is that wherein the reaction is quenched with an aqueous acid.
- quenching acids include: sulfuric acid, hydrochloric acid, citric acid and acetic acid.
- Another embodiment of the invention is that wherein
- R is methyl and Rl is ethyl.
- the compound of formula A consists of two stereoisomers, one stereoisomer in enantiomeric excess with respect to the other.
- Another embodiment of the invention is that wherein the product yield of the compound of formula A is greater than about 90%.
- a preferred embodiment of the invention encompasses the process wherein the compound of formula B is produced by reacting a compound of formula D:
- R and Rl are Ci- ⁇ alkyl, with an activating agent in a substantially non-reactive solvent at reduced temperature and then with pyrrolidine at room temperature to produce a compound of formula B.
- an activating agent is carbonyldiimidazole.
- the substantially non-reactive solvent is selected from the group consisting of: tetrahydrofuran, toulene, isopropyl acetate, ethyl acetate, t- butlymethyl ether, ethylene glycol dimethyl ether and N,N- dimethylformamdide. Mixtures of two or more of the aforesaid solvents are also contemplated.
- the reduced temperature is in the range of about -25° C to about 10° C. More particularly the reduced temperature is about 0° C.
- a preferred embodiment is that wherein the product yield of the compound of formula B is greater than about 90%.
- R is methyl and Rl is ethyl.
- a subclass of this class encompasses a process wherein the compound of formula D consists of one stereoisomer that is in enantiomeric excess with respect to the other.
- a group of this subclass is a process wherein the compound of formula D is resolved by reacting the racemic mixture of the compound of formula D with a chiral amine resolving agent in a substantially non-reactive solvent.
- substantially non-reactive solvent include those selected from the group consisting of: acetone, ethyl acetate, hexane and isopropyl acetate. Additionally mixtures of two or more of the aforesaid solvents are included.
- a preferred embodiment is a process wherein the compound of formula D is resolved to an enantiomeric excess of about 98%.
- the product yield for the resolution is greater than about 65%.
- the compound of formula D is resolved to about 98% enatiomeric excess and the yield is about 60-70%.
- the invention is illustrated in connection with the following generic schemes A and B.
- R a represents C 3.6 alkyl
- b represents C 2-6 alkyl
- the racemic starting material is first separated into its diastereomers with a chiral amine resolving agent to provide the desired stereospecific hydroxy acid.
- a chiral amine resolving agent to provide the desired stereospecific hydroxy acid.
- the appropriate families of chiral amines may be used as described in T. Vries, et al., Angew Chem. Int. Ed. (1998) 37: 2349-2354.
- chiral amine resolving agents can be selected from the group consisting of:
- the resolved hydroxy acid is then activated using an appropriate activating agent, in a substantially non-reactive solvent, at reduced temperature, and then combined with a cyclic amine, providing compound B.
- the cyclic amine serves as a leaving group in the next step, when is displaced via a lithiation reaction, producing compound A.
- Compound A is oxidized to produce methyl sulfone D.
- a suitable oxidizing agent is Oxone®.
- Methyl sulfone D is then subjected to esteriflcation by reaction with a compound R OCH2CO2H, wherein Ra represents a C3-6 alkyl group.
- a suitable esteriflcation procedure involves the addition of the esterifying agent such as dicyclohexylcarbodiimide (DCC) to methyl sulfone D in the presence of an amine base, e.g., DABCO, in a solvent or solvent mixture at about 30 - 35 °C.
- DCC dicyclohexylcarbodiimide
- the ester is thereafter cyclized, and optionally deprotected, to provide compounds having COX-2 selective inhibitory activity.
- alkyl means linear, branched or cyclic structures and combinations thereof, containing one to twenty carbon atoms.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, s- and t-butyl, pentyl, hexyl, heptyl, octyl, nonyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, eicosyl, 3,7-diethyl-2,2-dimethyl- 4-propylnonyl, and the like.
- substantially non-reactive solvent includes tetrahydrofuran, toluene, acetone, ethyl acetate, hexane, isopropyl acetate, ethylene glycol dimethyl ether, t-butyl methyl ether and N,N-dimethylformamide.
- one stereoisomer that is in enantiomeric excess with respect to the other means that the mixture contains over 50% of one stereoisomer and under 50% of the other.
- This phrase also is meant to include an enantiomerically pure compound consisting essentially of 100% of a stereoisomer and ' essentially 0% of the corresponding enantiomer.
- room temperature means about 20° C.
- reduced temperature is meant to include any temperature less than room temperature.
- lithiumating agent includes n-butyllithium, hexyllithium and phenyllithium.
- activating agent means any compound that activates a particular site on any other compound for displacement by another group.
- An example is carbonyldiimidazole.
- chiral amine resolving agent is meant to include any amine compound that when reacted with a mixture of enantiomers yields a mixture of one stereoisomer that is in enantiomeric excess with respect to the other and where such excess is greater than any excess of the original mixture. Examples include (R)-(+)-l-(l-napthyl)ethylamine and (S)-(-)-l-(l- napthyl)ethylamine.
- Part B Salt break. Recovery of acid and amine
- reaction mixture was transferred via cannula into the quench, (2L THF rinse), with vigorous agitation.
- the layers were separated, and the aq. Layer was extracted with 30 L Toluene.
- the combined organics were washed with aq. NaHCO3 (5 wt%, 18
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pyrrole Compounds (AREA)
Abstract
Description


























Claims






Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000578282A JP2002528435A (en) | 1998-10-27 | 1999-10-25 | Synthesis of methylthiophenylhydroxyketone |
| AU11337/00A AU758349B2 (en) | 1998-10-27 | 1999-10-25 | Synthesis of methylthiophenyl hydroxyketones |
| CA002347768A CA2347768A1 (en) | 1998-10-27 | 1999-10-25 | Synthesis of methylthiophenyl hydroxyketones |
| EP99955168A EP1124798A4 (en) | 1998-10-27 | 1999-10-25 | SYNTHESIS OF METHYLTHIOPHENYL HYDROXYKETONES |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10583098P | 1998-10-27 | 1998-10-27 | |
| US60/105,830 | 1998-10-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000024711A1 true WO2000024711A1 (en) | 2000-05-04 |
Family
ID=22308018
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/025064 Ceased WO2000024711A1 (en) | 1998-10-27 | 1999-10-25 | Synthesis of methylthiophenyl hydroxyketones |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US6281391B1 (en) |
| EP (1) | EP1124798A4 (en) |
| JP (1) | JP2002528435A (en) |
| AU (1) | AU758349B2 (en) |
| CA (1) | CA2347768A1 (en) |
| WO (1) | WO2000024711A1 (en) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5663195A (en) * | 1994-10-19 | 1997-09-02 | Merck & Co., Inc. | Method of preventing bone loss |
| US5840746A (en) * | 1993-06-24 | 1998-11-24 | Merck Frosst Canada, Inc. | Use of inhibitors of cyclooxygenase in the treatment of neurodegenerative diseases |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0003002B1 (en) * | 1977-12-22 | 1984-06-13 | Ciba-Geigy Ag | Use of aromatic-aliphatic ketones as photoinitiators, photopolymerisable systems containing such ketones and aromatic-aliphatic ketones |
| US5474995A (en) * | 1993-06-24 | 1995-12-12 | Merck Frosst Canada, Inc. | Phenyl heterocycles as cox-2 inhibitors |
| DE69529690T2 (en) * | 1994-12-21 | 2003-11-13 | Merck Frosst Canada & Co., Halifax | DIARYL-2 (5H) -FUARANONE AS COX-2 INHIBITORS |
| US6020343A (en) * | 1995-10-13 | 2000-02-01 | Merck Frosst Canada, Inc. | (Methylsulfonyl)phenyl-2-(5H)-furanones as COX-2 inhibitors |
| UA57002C2 (en) * | 1995-10-13 | 2003-06-16 | Мерк Фросст Кенада Енд Ко./Мерк Фросст Кенада Енд Сі. | (methylsulfonyl)phenyl-2-(5n)-furanon derivative, a pharmaceutical composition and a method for treatment |
| CA2283100A1 (en) * | 1997-03-14 | 1998-09-24 | Merck Frosst Canada & Co. | (methylsulfonyl)phenyl-2-(5h)-furanones with oxygen link as cox-2 inhibitors |
-
1999
- 1999-10-25 AU AU11337/00A patent/AU758349B2/en not_active Ceased
- 1999-10-25 WO PCT/US1999/025064 patent/WO2000024711A1/en not_active Ceased
- 1999-10-25 CA CA002347768A patent/CA2347768A1/en not_active Abandoned
- 1999-10-25 JP JP2000578282A patent/JP2002528435A/en active Pending
- 1999-10-25 EP EP99955168A patent/EP1124798A4/en not_active Withdrawn
- 1999-10-27 US US09/427,413 patent/US6281391B1/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5840746A (en) * | 1993-06-24 | 1998-11-24 | Merck Frosst Canada, Inc. | Use of inhibitors of cyclooxygenase in the treatment of neurodegenerative diseases |
| US5663195A (en) * | 1994-10-19 | 1997-09-02 | Merck & Co., Inc. | Method of preventing bone loss |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1124798A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1124798A4 (en) | 2002-08-28 |
| AU758349B2 (en) | 2003-03-20 |
| EP1124798A1 (en) | 2001-08-22 |
| AU1133700A (en) | 2000-05-15 |
| CA2347768A1 (en) | 2000-05-04 |
| JP2002528435A (en) | 2002-09-03 |
| US6281391B1 (en) | 2001-08-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100549748B1 (en) | Asymmetric synthesis of pregabalin | |
| KR101045935B1 (en) | Method for preparing prostaglandin derivative | |
| WO2019062561A1 (en) | Synthesis methods of firocoxib and intermediate thereof | |
| AU758349B2 (en) | Synthesis of methylthiophenyl hydroxyketones | |
| JP2011157326A (en) | Maxacalcitol intermediate and process for producing the same | |
| CN115010692A (en) | Design, preparation and application of novel macrolides | |
| CN103508999A (en) | Maxacalcitol synthesizing intermediate and preparation method and application thereof | |
| CN111777538A (en) | Preparation method of bimatoprost | |
| JP2001233870A (en) | 3-(1-hydroxypentylidene)-5-nitro-3h-benzofuran-2-one, method for producing the same and use thereof | |
| Kamimura et al. | A simple preparation of syn-NH-amide aldols and amide-Baylis–Hillman adducts via a Michael–aldol tandem process | |
| CN114214651B (en) | A kind of synthesis method of α-carbonyl-α’-thiocyanatosulfoxide ylide under electrocatalysis | |
| CN114634407B (en) | Method for stereospecifically synthesizing 2-enal, 2-enone compound and deuterated compound thereof | |
| CN109970560B (en) | A kind of preparation method of tri-substituted 1,3 diolefin compounds | |
| JP2004339085A (en) | METHOD FOR PRODUCING alpha-SUBSTITUTED PHENYL-ALKANECARBOXYLIC ESTER DERIVATIVE | |
| CN116496136B (en) | Diaryl sulfone compound and preparation method thereof | |
| CN116789658B (en) | Preparation method of 1-chloro-4-(β-D-pyranoglucopyran-1-yl)-2-(4-tetrahydrofuran-3-yloxy-benzyl)-benzene | |
| Garcı́a et al. | Highly stereoselective hydrocyanation of optically pure α-sulfinylaldehydes | |
| CN115160198B (en) | Preparation method of thiourethane collector | |
| CN114989052B (en) | Guaiacol-austempering compound and preparation method thereof | |
| CN105343061A (en) | Application and preparation method of Balasubramide derivatives | |
| US6495713B2 (en) | Synthesis of ketosulfone esters | |
| JP2001509501A (en) | Method for producing 2-alkylthiobenzoic acid derivative | |
| CN114773247A (en) | Method for synthesizing asymmetric selenium sulfide by cross coupling of diselenide and sulfoxide | |
| JPH0723356B2 (en) | Process for producing 4,4-disulfonylbutanoic acid esters | |
| CN110256247B (en) | Synthesis method of 2- ((9-hydrofluoren-9-yl) methyl) malonate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref country code: AU Ref document number: 2000 11337 Kind code of ref document: A Format of ref document f/p: F |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2347768 Country of ref document: CA Ref country code: CA Ref document number: 2347768 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999955168 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 578282 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11337/00 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999955168 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 11337/00 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999955168 Country of ref document: EP |