WO2000024712A1 - Vitamin d3 derivatives and remedies for inflammatory respiratory diseases containing the same - Google Patents
Vitamin d3 derivatives and remedies for inflammatory respiratory diseases containing the same Download PDFInfo
- Publication number
- WO2000024712A1 WO2000024712A1 PCT/JP1999/005826 JP9905826W WO0024712A1 WO 2000024712 A1 WO2000024712 A1 WO 2000024712A1 JP 9905826 W JP9905826 W JP 9905826W WO 0024712 A1 WO0024712 A1 WO 0024712A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- vitamin
- hydrogen atom
- pharmaceutically acceptable
- derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C401/00—Irradiation products of cholesterol or its derivatives; Vitamin D derivatives, 9,10-seco cyclopenta[a]phenanthrene or analogues obtained by chemical preparation without irradiation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/02—Nutrients, e.g. vitamins, minerals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
- A61P3/14—Drugs for disorders of the metabolism for electrolyte homeostasis for calcium homeostasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention is useful vitamin D 3 derivative or a pharmaceutically acceptable solvate thereof as medicament, therapeutic agent of using them and to pharmaceutical compositions containing them. More specifically, 1 alpha-hydroxycarboxylic vitamin D 3 derivative or a pharmaceutically acceptable solvate thereof with neutrophil infiltration inhibitory action and vitamin D 3 antagonism good, inflammatory respiratory to them valid Ingredient therapeutic agent for diseases based on disorders Ya vitamin D 3 over action, and to pharmaceutical compositions containing their these.
- Active vitamin D 3 derivative has calcium absorption promoting effect in the small intestine, the bone Hone ⁇ yield, has an action such as modulating bone formation, it is used as a therapeutic agent of various calcium metabolism abnormality in disease ing.
- immunomodulatory effects, cell growth inhibitory effects, and cell differentiation inducing effects have been found.
- therapeutic agents for malignant tumors JP-A-57-149224
- therapeutic agents for rheumatoid arthritis JP-A 56-26820
- an anti-allergic agent Japanese Patent Application Laid-Open No. 63-107928, British Patent No.
- Respiratory tract infection is a condition that occurs when a pathogen invades the airway infection prevention mechanism and is based on treatments that improve airway clearance, such as bronchodilators and expectorants.
- treatments that improve airway clearance such as bronchodilators and expectorants.
- antimicrobial treatment is strongly applied to causative bacteria.
- most underlying illnesses often worsen with repeated acute exacerbations.
- resistant bacteria such as MRSA has led to a review of current treatments that rely too much on antimicrobial agents.
- Chronic lower respiratory tract infection is a general term for bacterial infections such as chronic bronchitis, diffuse panbronchiolitis, and bronchiectasis (bronchial asthma with other infections, chronic emphysema, pulmonary tuberculosis, and sequelae of pulmonary tuberculosis). Etc. may be included). Although these have different disease names, they are all known to have common conditions such as large amounts of purulent sputum, exertional dyspnea, and hypoxemia.
- erythromycin acts on inflammatory cells, especially neutrophils, which accumulate in the respiratory tract rather than the bacteria themselves.
- neutrophils infiltrate tissues by various stimuli based on infection and release protease ⁇ active oxygen, which causes epithelial damage, cilia motility disorder, mucus hypersecretion and adversely affects respiratory physiology. It is believed that erythromycin acts on these processes. From this idea, a drug that suppresses neutrophil infiltration into lung tissue or its function can be said to be useful as a therapeutic agent for inflammatory dyspnea, for example, chronic lower respiratory tract infection.
- vitamin D if the control of vitamin D production becomes abnormal due to a disease or the like, and if the concentration in the living body increases and the physiological action is excessively expressed, various diseases caused by excessive vitamin D are caused.
- sarcoidosis it is known that neoplastic macrophage-like cells produce excessive amounts of vitamin D (J. Clin. Invest. Journal J. Clin. Invest. ), 64, 218-225, 1979), resulting in hypercalcemia.
- Glucocorticoids are mainly used for this treatment, but side effects have been observed with long-term administration of large amounts of glucocorticoids.
- vitamin D exerts a physiological action via the vitamin D receptor present in the cells, so that the action of the overexpressed vitamin D can be suppressed by specific action for the receptor-mediated action. Vitamin D 3 antagonist is considered to be effective.
- active vitamin D 3 regulates the production of parathyroid hormone (hereinafter also referred to as PTH) in the living body, and the production of active vitamin D 3 decreases the production of PTH. Therefore, it is thought that the use of a vitamin D 3 antagonist can correct the decrease in PTH production due to increased production of active vitamin D 3 and further promote the production of PTH. It is known that various diseases are caused by decreased PTH production, one of which is hypoparathyroidism. Although the administration of PTH seems to be ideal for this treatment, no oral PTH preparation has been developed so far. On the other hand, Vitamin D 3 antagonists since it is possible to orally administered is considered to be useful as an ideal therapeutic agent for hypoparathyroidism.
- PTH has an effect on the proliferation and differentiation of chondrocytes and the synthesis of cartilage matrix (Cellular and calcium), Vol. 16, pp. 112-122, 1994.
- Tissue International Calculated T issue International; 50, 61-66, 1992
- a growth promoting effect on osteoblasts Endocrinology, 118, 2445- 2449, 1986
- Collagen synthesis-promoting action Journal of Oblin Clinical Investigation (J. Clin. Invest.), 83, 60-65, 1989 Etc. reported Have been.
- vitamin D 3 antagonist can be administered orally, which can solve these problems, and is considered to be useful as an ideal therapeutic agent for cartilage metabolism disorders and bone metabolism disorders.
- Prior art relating to the compounds of the present invention includes the following.
- the compounds disclosed in the present invention do not include the above-mentioned compounds, and the gazette does not disclose whether or not the compounds described therein have a neutrophil infiltration inhibitory action or a vitamin D antagonistic action. There is no mention or suggestion.
- Compounds having an ⁇ -.hydroxyl- ⁇ -methyllactone structure are indicated, and the latter publication suggests indications as therapeutic agents for hypercalcemia, anticancer agents, osteoporosis, and the like.
- the compounds disclosed in the present invention do not include the above-mentioned compounds, and whether the compounds described in the above-mentioned documents have a neutrophil infiltration inhibitory action or a vitamin D 3 antagonist action is described. Does not mention or suggest anything.
- an object of the present invention is to provide a novel vitamin D : 3 ⁇ 4 derivative effective as a therapeutic agent for inflammatory respiratory diseases having a neutrophil infiltration inhibitory action.
- an object of the present invention is to provide a novel vitamin D 3 derivative which is effective as a therapeutic agent for a disease based on a vitamin D 3 excess action having a vitamin D 3 antagonistic action.
- Another object of the present invention is to provide a method for treating inflammatory respiratory diseases using them vitamin D 3 derivative as an active ingredient.
- Another object of the present invention is to provide a method for treating a disease based on vitamin D 3 over effects using these vitamin D 3 derivatives as active ingredients.
- object of the present invention is to provide a medical drug composition containing them vitamin D 3 derivative as an active ingredient.
- the above object of the present invention is achieved by the following general formula (1)
- R 0 1 and R. 2 are each independently a hydrogen atom, a trimethylsilyl group
- Toryechi represents a 'group, a t-butyldimethylsilyl group, an acetyl group, a methoxymethyl group, or a tetrahydro 4H-pyran-12-yl group.
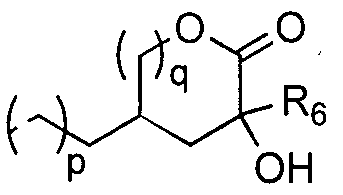
- Z represents one of the following equations (1-1), (1-2), (1-3), (1-4) and (1-5).
- n an integer of 0 to 2.
- n an integer of 0 to 2.
- X ′ represents an oxygen atom or NH.
- Ri and R 12 are identical or different, to display the hydrogen atom or a C ' ⁇ Ji alkyl group Tewi.
- K, L, and M are all hydrogen atoms; M is a hydrogen atom, K and L together represent a single bond, and represent a double bond together with the specified single bond; K is a hydrogen atom; L and M Represents together a single bond and represents a double bond together with the specified single bond.
- R 21, R 22, and are the same or different, a hydrogen atom; a C 2 -C 5; hydroxyl; carboxyl group; triflate Ruo Russia methyl; Arukiruokishi force Ruponiru group of C, -C 4; penta full O Roe methyl group Ashiruokishi group; C] Arukiruokishi group -C 4; or hydroxyl group, Ashiruokishi group of C 2 -C 5 or Ji! ⁇ .
- C which may be substituted with an alkyloxy group R 2 i and R 22 together with the carbon atom to which they are attached may represent a C 3 -C 6 cyclic alkyl group.
- Q is> C (- F) one R 31 or> N-R 3 represents a], wherein R 3, a hydrogen atom; water group; triflate Ruo Russia methyl; penta full O Roe methyl group; C 2 ⁇ Ji ⁇ Ji of Arukiruokishi group; Ashiruokishi group C 5 or a hydroxyl group, C 2 -C 5 in Ashiruokishi group, if Ku for c, which may be substituted with Arukiruokishi group to c 4 c, alkyl of to c 4 Represents a group.
- R 32 , R 33 , R : i 4 , and R 35 are the same or different and represent a hydrogen atom, a hydroxyl group, a C, -C 4 alkyl group, or a C 2 -C 5 acyloxy group.
- a and B are the same or different and each represent a hydrogen atom or a hydroxyl group, or together represent a single bond, and form a double bond with the specified single bond.
- X and Y together represent a carbonyl group together with the carbon atom to which they are attached Force, indicating that either or the other a hydrogen atom and the other is a hydroxyl group, one or the other is other person a hydrogen atom is Ashiruokishi group C 2 ⁇ C 5.
- R 41 and R 42 are the same or different and each represents a hydrogen atom; a hydroxyl group; a trifluoromethyl group; a pentafluoroethyl group; a C 2 to C 5 acyloxy group; Or a hydroxyl group, a C 2 -C 5 acyloxy group, or an alkyl group which may be substituted with a 4- ( 4 alkyloxy group), or together form C j
- Or represents an alkylidene group to c 5, or c 3 to c together with the carbon atoms to which they are attached
- R 43 and R 44 are the same or different, and each represents a hydrogen atom; a hydroxyl group; a trifluoromethyl group; a pentafluoroethyl group; an acyloxy group of c 2 to c 5 ; an alkyloxy group of cc ⁇ ; or a hydroxyl group, C 2 to C 5 of Ashiruokishi group or Ct ⁇ of or represents an alkyl group which may be C i to C 4 substituted with Arukiruokishi group, or both together a connexion C,!
- Or represents an alkylidene group to c 5, or c 3 to c together with the carbon atoms to which they are attached
- R 45 and R 46 are the same or different and each is a hydrogen atom; a hydroxyl group; a trifluoromethyl group; a pentafluoroethyl tomb; a C 2 to C 5 acyloxy group; an alkyloxy group of Ci C; or a hydroxyl group, C 2 to Represents a C i -C 4 alkyl group which may be substituted with a C 5 acyloxy group or a Ci ⁇ alkyloxy group.
- D and E both represent a hydrogen atom, D is a hydroxyl group and E represents a hydrogen atom, or both D and E together represent a single bond and represent a double bond together with the specified single bond; Or E represents a single bond together with R 41 to represent a single bond and a double bond, in which case D represents a hydrogen atom or a hydroxyl group, and R 42 represents a hydrogen atom; a hydroxyl group; a trifluoromethyl group A pentafluoroethyl group; a C 2 to C 5 acyloxy group; a di-to-dialkyloxy group; or a hydroxyl group, a C 2 to C 5 acyloxy group, or a CC alkyloxy group.
- R 51 represents —CONR 511 R 512 , one COR 513 , or one C (OH) R 514 R 515 , wherein R 511 and R 512 are the same or different and are a hydrogen atom, an alkyl group of Represents a C 3 to C 8 nitrogen-containing alkyl ring or a morpholino group together with a nitrogen atom bonded together; R 513 , R 514 , and R 515 are the same or different, and represent an alkyl group of ( ⁇ to ⁇ .
- R 52 represents a methyl group, an ethyl group, a trifluoromethyl group, or a pentafluoroethyl group.
- R 01 and R. 2 are the same as defined in the above formula (1), the configuration at the 20-position is (R) configuration, and R 7 represents a methyl group or a methylene group. If 7 represents a methylene group, the bond between R 7 and 26 of the Ru is achieved by vitamin D 3 derivative or a pharmaceutically acceptable solvate thereof represented by the representative.) a double bond.
- the compound structure when the compound structure contains an asymmetric carbon, its configuration may be any of the (S) configuration and the (R) configuration unless otherwise specified.
- the present invention includes a mixture of these various isomers in any ratio.
- the object of the present invention is to provide a method for treating inflammatory respiratory diseases, comprising as an active ingredient a therapeutically effective amount of the above-mentioned vitamin 30 derivative or a pharmaceutically acceptable solvate thereof. Achieved.
- the above object of the present invention contains a therapeutically effective amount of the vitamin D 3 derivative or a pharmaceutically acceptable solvate thereof as an active ingredient Vitamin D: i excessive work It is achieved by a method of treating a disease based on use.
- a pharmaceutical composition comprising a disc are the vitamin D 3 derivative or a pharmaceutically acceptable solvates and pharmaceutically acceptable.
- the object of the present invention contains a vitamin D 3 derivative or a pharmaceutically acceptable solvate thereof represented by the therapeutically effective amount of the following general formula (3), accomplished by inflammatory respiratory diseases agents Is done.
- the Ashiruokishi group C 2 -C 5 represents a linear, branched or cyclic aliphatic hydrocarbon carbonylation Ruokishi groups, 2 to 5 carbon atoms.
- specific examples include acetoxy, propionyloxy, butyryloxy, isobutyryloxy, valeryloxy, isovaleryloxy, vivaloyoxy, cyclopropylcarbonyloxy, cyclopropylacetoxy, cyclobutylcarbonyloxy, and the like. Group.
- the above-mentioned alkyloxy group means a linear, branched or cyclic aliphatic hydrocarbon oxy group having 1 to 4 carbon atoms.
- specific groups include methoxy, ethoxy, propoxy, butoxy, isopropoxy, isobutoxy, sec-butoxy, tert-butoxy, cyclopropylpyrmethyloxy and the like.
- the Oh Rui represents an aliphatic hydrocarbon Okishikarubo two Le group ring.
- the cyclic alkyl group represented by i represents a cyclic aliphatic hydrocarbon group having 3 carbon atoms and 6 carbon atoms.
- the nitrogen-containing alkyl ring of S 8 to C 8 represents an aliphatic hydrocarbon ring having 3 to 8 carbon atoms and including a nitrogen atom in the ring.
- specific groups include aziridine, azetidine, pyrrolidine, imidazolidine, virazolidine, piperidine, piperazine ring and the like.
- R 0 i and R. 2 each independently represents a hydrogen atom, a trimethylsilyl group, a triethylsilyl group, a t-butyldimethylsilyl group, an acetyl group, a methoxymethyl group, or a tetrahydro-4H-pyran-12-yl group.
- R 0 1 and R. Among these 2 is preferably a hydrogen atom, a trinotylsilyl group, a triethylsilyl group, or a t-butyldimethylsilyl group, and most preferably a hydrogen atom.
- Z represents any one of the following formulas (111), (1-2), (1-3), (1-4) and (1-5).
- m represents an integer of 0 to 2. Of these, 0 or 1 is preferred.
- n represents an integer of 0 to 2. Of these, 0 or 1 is preferred.
- X ′ represents an oxygen atom or NH. Of these, an oxygen atom is preferred.
- R 12 are the same or different, and represent a hydrogen atom or Represents an alkyl group. Among them, a hydrogen atom, a methyl group or an ethyl group is preferred.
- K, L and M are all hydrogen atoms; M is a hydrogen atom, and K and L are linked together to represent a single bond and a double bond together with the specified single bond; Is a hydrogen atom, and L and M together represent a single bond and represent a double bond with the specified single bond.
- M is a hydrogen atom
- K and L are joined together to represent a single bond.
- K is a hydrogen atom
- L and M together form a single bond to form a double bond with the specified single bond
- R 2 , R 22 and R 23 are the same or different and each represents a hydrogen atom; a hydroxyl group; a carboxyl group; a trifluoromethyl group; a pentafluoroethyl group; Arukiruoki sheet group CC;; Ashiruokishi group C 2 -C 5; alkoxycarbonyl group or a hydroxyl group, Ashiruokishi groups c 2 to c 5, or c, Ji may be substituted with Arukiruoki sheet group to c 4 ⁇ .
- R 21 and R 22 together with the carbon atom to which they are attached may represent a C 3 -C 6 cyclic alkyl group.
- R 21 and R 22 are the same or different, and a hydrogen atom, a hydroxyl group
- the alkyl group, or R 21 and R 22 together represent a C 3 -C 6 cyclic alkyl group together with the carbon atom to which they are attached, and more preferably a hydrogen atom, a hydroxyl group, a methyl group, an ethyl group
- R 23 is preferably a hydrogen atom or a hydroxyl group.
- R 2 examples have R 22, and R 23, (a) R 2 have R 22, R 23 are all hydrogen atoms) 1 ⁇ 21 Oyobi 1 22 is a methyl group, R 23 is a hydrogen atom, (0) combination of scale 21 and 1 22 is a methyl group and a hydroxyl group, R 23 is a hydrogen atom, (d) in Kumiawasegame methyl group and a hydroxyl group of R 21 and R 22, R 23 is a hydroxyl group, (e It is preferred that R 21 and R 22 together form a cyclopropyl group with the carbon atoms to which they are attached, and wherein R 23 is a hydrogen atom.
- Q represents> C (-1F) —R 31 or> N—R 31 , wherein R 3 j is a hydrogen atom; a hydroxyl group; a trifluoromethyl group; a pentafluoroethyl group A C 2 -C 5 alkoxy group; a C ⁇ alkyloxy group; or a hydroxyl group, a C 2 -C 5 alkoxy group, or Represents an alkyl group of C!
- R 31 is preferably a hydrogen atom; a hydroxyl group; or a hydroxyl group, a C 2 -C 5 acyloxy group, or a Ci C alkyl group which may be substituted by a CC alkyloxy group, and more preferably a hydrogen atom, a hydroxyl group. Or a methyl group is most preferred.
- R 32, R 33, R 34, and R 35 are identical or different, represent a hydrogen atom, a hydroxyl group, an alkyl group of C! C, Ashiruokishi group C 2 -C 5. Of these, a hydrogen atom or a C 4 to C 4 alkyl group is preferred, and a hydrogen atom is most preferred.
- a and B are the same or different and each represent a hydrogen atom or a hydroxyl group, or represent a single bond together and form a double bond with the specified single bond.
- a and B are both hydrogen atoms, A is a hydroxyl group and B is a hydrogen atom, or both A and B represent a single bond and form a double bond with the specified single bond Is preferred Les ,.
- ⁇ and ⁇ together represent a carbonyl group together with the carbon atom to which they are bonded, or one of them is a hydrogen atom and the other is a hydroxyl group. There indicating that the other is a Ashiruokishi group c 2 ⁇ c 5 a hydrogen atom.
- X and Y preferably represent a carbonyl group together with the carbon atom to which they are bonded.
- R 4 and R 42 are the same or different and each represents a hydrogen atom; a hydroxyl group; a trifluoromethyl group; a pentafluoroethyl group; a C 2 to C 5 acyloxy group; and a Ci to C 4 alkyloxy group. It represents a or a hydroxyl group, C 2 -C Ashiruokishi group 5 or Ci ⁇ C 4 Al Kiruokishi group optionally substituted c 1 even if to c 4 alkyl group; group. Further, either become both together represent an alkylidene group of Ci Cg, represents a cyclic alkyl group of C 3 to c 6 together with the carbon atoms to which they are attached. Among them, it is preferred that both represent a hydrogen atom or both represent a methylene group.
- R 43 and R 44 are the same or different and each represents a hydrogen atom; a hydroxyl group; a trifluoromethyl group; a pentafluoroethyl group; a C 2 to C 5 acyloxy group; and a C or C 4 alkyloxy group. group;. represents a or a hydroxyl group, C 2 -C 5 in Ashiruokishi group, or ( ⁇ ⁇ Ji Al Kiruokishi alkyl group - where Ji may be substituted with a group also, C i to C 5 become both together or it represents a alkylidene group, a cyclic alkyl group of C 3 to c 6 together with the carbon atoms to which they are attached. Above all, may both represent a hydrogen atom or both together a connexion methylene group.
- R 45 and R 46 are the same or different, and each represents a hydrogen atom; a hydroxyl group; a trifluorophenol group; a pentafluoroethyl group; a C 2 to C 5 acyloxy group; Or a hydroxyl group, a C 2 -C 5 alkoxy group, or a C 1 -C 4 alkyl group which may be substituted with a C 4 alkyl group, especially a hydrogen atom, a hydroxyl group, a methyl group, or an ethyl group.
- Groups are preferred, and hydrogen atoms are most preferred.
- D and E both represent a hydrogen atom
- D is a hydroxyl group and E represents a hydrogen atom
- both D and E represent a single bond, and are both double bonds with the specified single bond.
- the E may also represent a double bond with the single bond explicitly represent connexion single bond such together with R 41, in this case, D is a hydrogen atom or a hydroxyl group, R 42 represents a hydrogen atom, a hydroxyl group, triflic Oromechiru group; penta full O Roe butyl group; Arukiruokishi group c ⁇ c 4;; C 2 ⁇ C Ashiruokishi group 5 or hydroxyl, Ashiruokishi groups c 2 to c 5, if Ku Arukiruokishi's Ci ⁇ C 4 to display the alkyl group which may c] to c 4 optionally substituted by a group.
- D and E are both hydrogen atoms, D and E together represent a single bond and form a double bond with the specified single bond, D is a hydrogen atom and E is R 41 It is preferred that they together represent a single bond and form a double bond with the specified single bond.
- R 5 1 represents an CONR 51, R 51 2, one COR 51 3 or a C (OH) R 514 R 515 , wherein R 51, and R 512 are the same or different, R 513 , R 514 , and R 515 are the same as a hydrogen atom, a C-C 4 alkyl group, or a C 3 -C «nitrogen-containing alkyl ring or a morpholino group together with a nitrogen atom bonded together. Or differently, it represents a C 1 to C 4 alkyl group.
- R 51 is preferably one of CONR 51 , R 512 and one COR 513 .
- R 51 and R 512 are each a methyl group or an ethyl group, or when they are taken together to form an aziridine ring, a pyrrolidine ring, a piperidine ring or a morpholino ring together with the nitrogen atom to which they are bonded; But preferred.
- R 513 , R 5 , 4 and R 515 are preferably a methyl group or an ethyl group.
- R 52 represents a methyl group, an ethyl group, a trifluoromethyl group, or a pentafluoroethyl group. Of these, a methyl group is preferred. Vitamin D 3 derivatives of the present invention can be converted to a solvate of their pharmaceutically acceptable as needed.
- Such solvents include water, methanol, ethanol, propyl alcohol, isopropyl alcohol, butanol, t-butanol, acetonitrinole, acetone, methyl ethyl ketone, chlorophonolem, ethyl acetate, getyl ether ethanol, t-butyl methyl ether, benzene , Toluene, DMF, DMSO and the like.
- water, methanol, ethanol, propyl alcohol, isopropyl alcohol, acetonitrile, acetone, methyl ethyl ketone, and ethyl acetate are preferred.
- vitamin D 3 derivatives of the present invention represented by the aforementioned formula (1)
- Table 1 one 1-1, 1-2- 1, 1 one 3-1, 1-3-2, 1 One four one one, one four-two, one one five-one.
- the configuration when an asymmetric carbon is contained in the compound structure, unless otherwise specified, the configuration includes both the (S) configuration and the (R) configuration. L and M, A and B, D and E, or E and R 4 ! When they form a double bond together, the configuration of the double bond includes both the (E) configuration and the (Z) configuration.
- the configuration of the double bond includes both the (E) configuration and the (Z) configuration.
- Table 1-1-1-1-1 describes a shaped portion subscript in the table in normal size.
- H ⁇ is is * ". U Me, Et ⁇ » ⁇ FS *
- H NH 0 0 H, Me K hydrogen atom, L, two- ⁇
- Ri 1 1 carbonyl group OH H, H H,
- Mouth 1 1 OH radical, OH H, ⁇ H,
- R42 0H H, H H, H
- ⁇ R42 0H H, OH H, H
- H un c — n ⁇ ⁇ , ⁇ ⁇ methylene H, H 4UD H, H ⁇ - ⁇ F— u ⁇ , ⁇ ⁇ methylene H, H 4U / H, H one H, HH, H 0 H, H uu— t— H, HH, H uy H, H-H, HH, H
- ⁇ mouth R42 H H, H H, H
- R 0 and R 2 are hydrogen atoms and R 7 R and R are compounds wherein is a methylene group.
- Compounds in which 2 is a hydrogen atom and R 7 is a methyl group can be mentioned.
- the configuration when the compound structure contains an asymmetric carbon, the configuration includes both (S) configuration and (R) configuration unless otherwise specified.
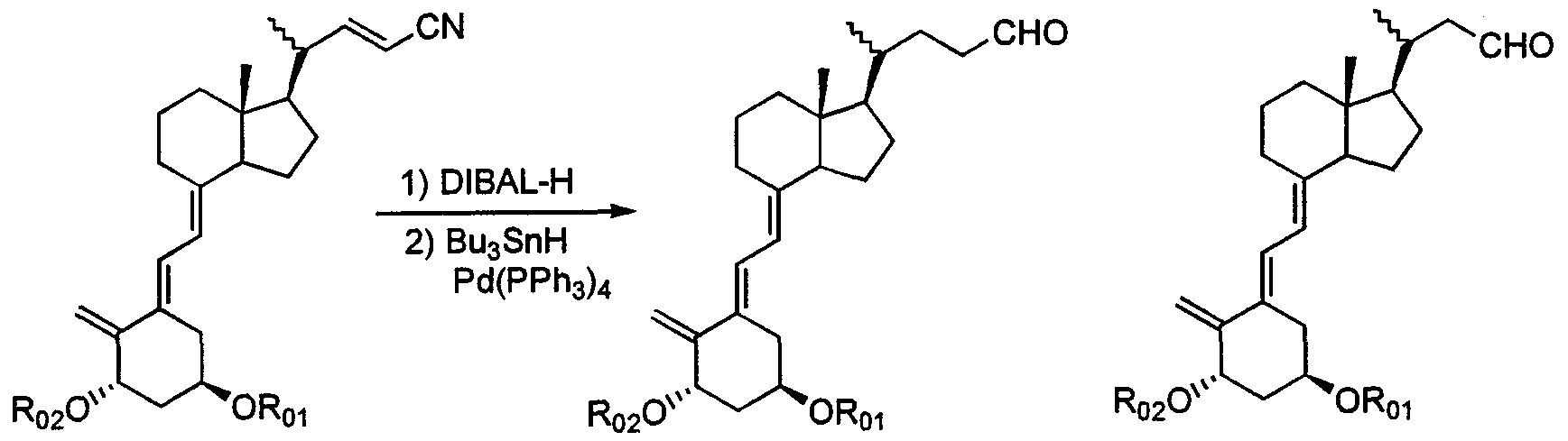
- Production of vitamin D 3 derivative represented by the above formula (1) may, for example, Trost (T rost) et al, (Journal 'O Bed' American 'Chemical' Society one (J. Am. C h em .
- Z, R. and R. 2 are the same as defined in the above formula (1), and Y represents a bromine atom or an iodine atom.
- the palladium catalyst used in the coupling reaction for example, a mixture of a zero-valent or divalent organic palladium compound and a trisubstituted phosphorus compound (molar ratio, 1: 1 to 1:10) is used.
- a palladium compound include tetrakis (triphenylphosphine) palladium, tris (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) palladium chromate form, and palladium acetate.
- the trisubstituted phosphorus compound include, for example, triphenylphosphine, triptylphosphine, and the like.
- a palladium catalyst combining both, a combination of tris (dibenzylideneacetone) palladium and trifenylphosphine, a combination of tris (dibenzylideneacetone) palladium chlorophonolem and trifenylphosphine (molar ratio, 1: 1 to 1:10) Is preferred. Also from 1 to 1 0 0 mol% relative to the compound represented by an organic palladium compound is the formula (4), preferably 5 to 3 0 mol 0 /. Used in the range. Further, the trisubstituted phosphorus compound is used in an amount of 1 to 10 equivalents based on the organic palladium compound to produce active palladium.
- the compound represented by the above formula (4) and the enyne compound represented by the above formula (5) undergo an equimolar reaction stoichiometrically. It is usually desirable to use a small excess of the readily available one.
- Examples of the organic solvent used in the coupling reaction include hydrocarbon solvents such as hexane and toluene; ether solvents such as tetrahydrofuran and dioxane; water-soluble solvents such as N, N-dimethylformamide and acetonitrile; It is preferable to use after degassing sufficiently.
- the reaction temperature generally ranges from room temperature to the boiling point of the solvent.
- the reaction time varies depending on the reaction solvent and the reaction temperature used.
- the compound represented by the above formula (4) or the enyne compound represented by the above formula (5) is analyzed using an analytical means such as thin layer chromatography. It is desirable to do this until one of them disappears.
- the reaction is preferably performed in the presence of a base such as, for example, triethylamine or diisopropylethylamine.
- a base such as, for example, triethylamine or diisopropylethylamine.
- the amount of the base for this is determined by the compound represented by the above formula (4). The amount is preferably 1 equivalent or more based on the product, and it can be used also as a solvent if necessary.
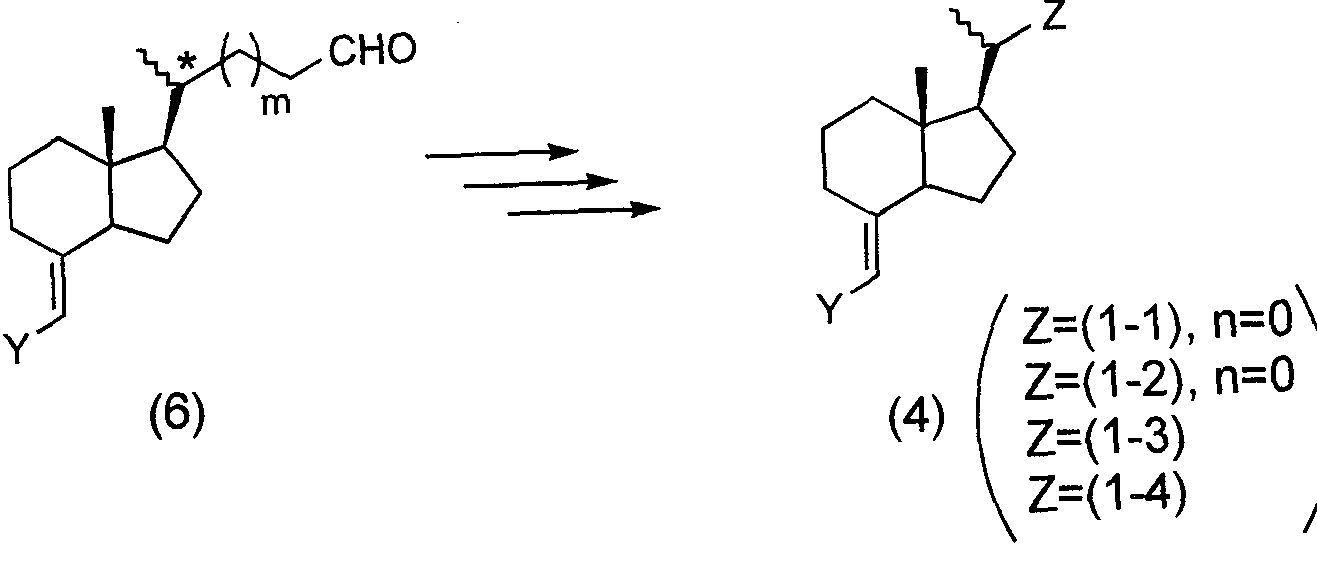
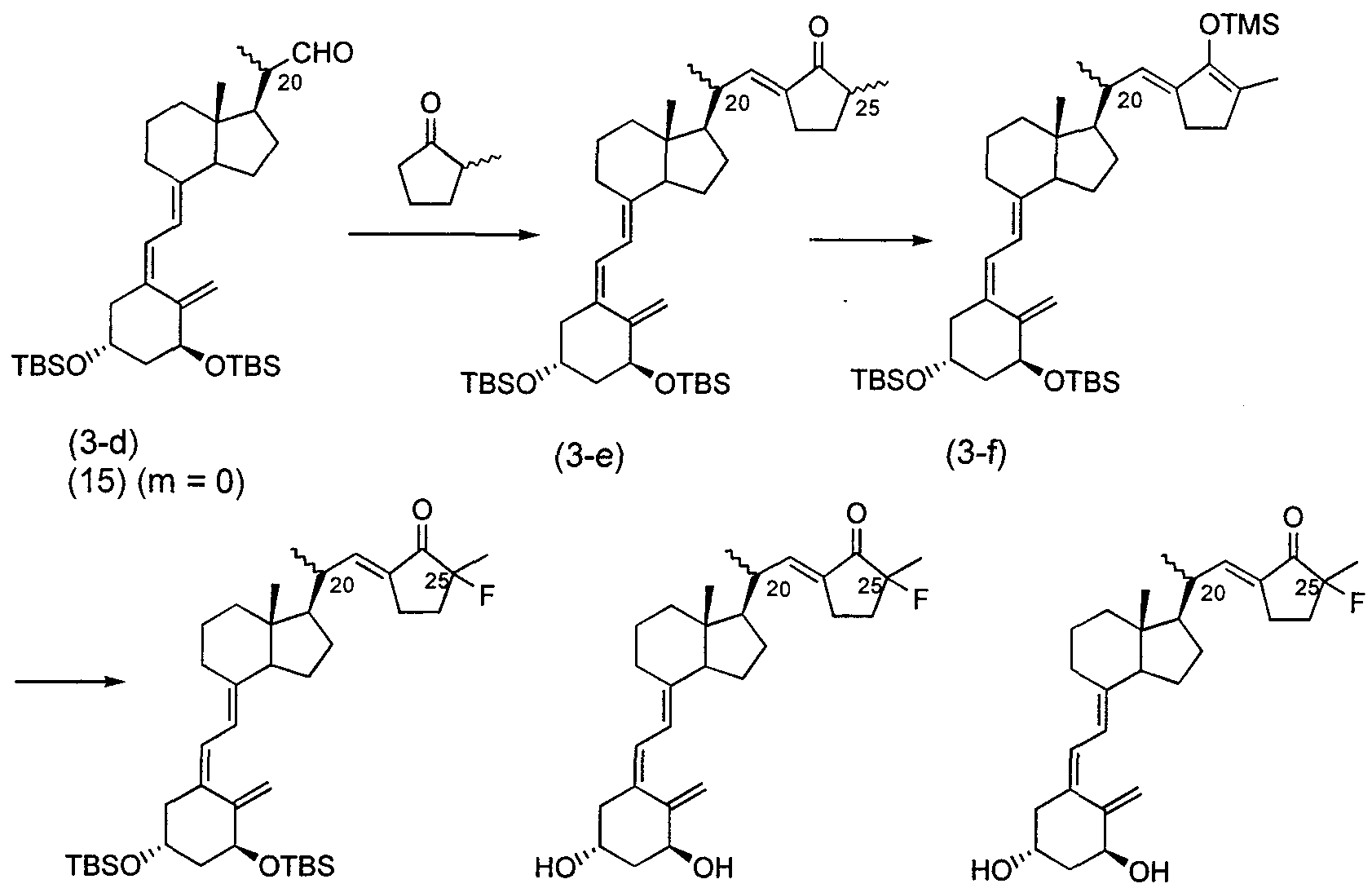
- the aldehyde compound (6) used here in which the * configuration of carbon is (R) and m is 0, 1, and 2, is shown in Schemes 3, 4, and 5 below, for example. It can be obtained by combining known methods as described above.
- m and Z are the same as defined in the above formula (1), Y represents a bromine atom or an iodine atom, and n is an integer of 1 or 2.
- PG represents a hydroxyl-protecting group.
- the compound (7) used herein, in which m is 1, 2 and 3, is, for example, an aldehyde compound obtained from the above-mentioned intermediate of Schemes 3 to 6 ( 10) and can be obtained by combining known methods as shown in Scheme 15 below.
- the compound (12) in which the asymmetric center marked with an asterisk (*) has the (S) configuration can be obtained, for example, by treating the intermediate (13) in Scheme 26 with a base and reacting the resulting epimer in the same manner as in Scheme 26. Can be obtained. Further, the production of the vitamin D 3 derivative represented by the above formula (1) is carried out by, for example, deriving from a compound (15) obtained by photoisomerizing a compound represented by (14) as shown in Scheme 27 below. Or by photoisomerizing the compound (16) derived from (14). The induction of the compound (14) to the force (16) can be performed in the same manner as the induction of the compound (15) to the compound (1) described below. Scheme 27
- Inorganic base catalysts such as potassium carbonate, lithium hydroxide, sodium hydroxide, potassium hydroxide, calcium hydroxide, and sodium hydride; 1,8-diazabicyclo [5.4.0] pendene (DBU)
- organic base catalysts such as lithium diisopropylamide, lithium hexamethyldisilamide, and sodium hexamethyldisilamide.
- base catalysts are used in an amount of 0.1 to 10 equivalents, preferably 0.5 to 3 equivalents, based on the starting aldehyde. If necessary, an additive for promoting the reaction may be added to the reaction system. Is also good.
- the aldehyde represented by the above formula (15) and the compound represented by the above formula (17) undergo a stoichiometric equimolar reaction, but the reaction is surely completed. Therefore, it is desirable to use one of them, which is easily available, in small excess.
- Organic solvents used in the aldol reaction include alcohol solvents such as methanol and ethanol; halogen solvents such as methylene chloride, chloroform, and carbon tetrachloride; hydrocarbon solvents such as hexane and toluene; tetrahydrofuran; Ether solvents such as dioxane; water-soluble solvents such as N, N-dimethylformamide and acetonitrile; and mixed solvents thereof.
- the solvent can be selected in consideration of the solubility and reactivity of the compound.
- the reaction temperature is generally in the range of 178 ° C to the boiling point of the solvent. The reaction time depends on the base catalyst used, the reaction solvent and the reaction temperature.
- the compound represented by the above formula (17) or the compound represented by the above formula (15) is analyzed using an analytical means such as thin-layer chromatography. It is advisable to do so until any of the aldehydes disappear.
- Examples of the dehydrating agent used in the dehydration reaction include acids such as potassium hydrogen sulfate, oxalic acid, p-toluenesulfonic acid, iodine, and anhydrous copper sulfate; halogenating agents such as thioyrucu mouth light and phosphoric acid chloride; and methanesulfuric acid Examples thereof include a sulfonating agent such as a mouth lid and the like, and it is used in an amount of 1 to 10 equivalents, preferably 1 to 5 equivalents, based on a raw material.
- sodium cesium borohydride, diisobutylaluminum dimethyl hydride (DIB AH), 9-borabic port [3.3.1] nonane (9-BBN), lithium n-butylborohydride, K One-select triode, tri-i-primary minimum, etc. can be used.
- fluorinating agent used for the fluorination reaction examples include N-fluoropyridinium triflate, N-fluoropyridinium trifluoromethanesulfonate, and N-fluoro 2,4,6-trimethylpyridini Dimethyl triflate, N-fluoro-3,5 dichloromouth pyridinium triflate, N-fluoro-2,6-dichloropyridinium triflate, N-fluoro-4,6-dimethylpyridinium-12-sulfonate, N— 4-N-funoleo 1-6- (Snorrephonate), N-Funole 1-6- (Trifnorelomethyl) pyridinium 2-Snolephonate, N-Fluoro-4,6-bis (trifluoromethyl) pyridinium 2 _
- the above reduction reaction can be performed by reduction with a hydride reagent such as sodium borohydride, lithium aluminum hydride, or the like.
- a hydride reagent such as sodium borohydride, lithium aluminum hydride, or the like.
- halogenation reaction for example, a combination of carbon tetrabromide or carbon tetrachloride and triflate phosphine can be used.
- metal reagent for the metal-halogen exchange reaction examples include butyllithium, metal magnesium, and samarium iodide, and are used in an amount of 1 to 5 equivalents based on the raw material.
- the metal-metal exchange reaction is carried out, if necessary, subsequent to the metal-halogen exchange reaction.
- copper iodide, copper bromide or an appropriate complex thereof, copper cyanide, or the like is used. be able to.
- an additive can be used.
- Silylating agents such as triflate and chlorotrimethylsilane; coordinating compounds such as hexaphosphoric acid triamide (HMPA) and triphenylphosphine; and combinations thereof are mentioned.
- HMPA hexaphosphoric acid triamide
- triphenylphosphine and combinations thereof are mentioned.
- HMP A combinations are preferred.
- metal-to-metal exchange reaction to metal-to-metal exchange reaction in Scheme 30 above is preferably performed continuously in the same system without any post-treatment.
- N-methylanilinium Oroacetate and paraformaldehyde can be used.
- the methyleneation divided into two steps can be performed by adding an imidium salt and then deaminating.
- the base include potassium tert-butoxide, sodium hydride, potassium hydride, sodium ethoxide, lithium diisopropylamide, lithium hexamethyldisilylamide, and the like.
- examples thereof include N, N-dimethyl (methylene) ammonium iodide and N, N-dimethyl (methylene) ammonium trifluoracetate.
- the imidium salt may be generated in the system from a secondary amine and formaldehyde or an equivalent thereof.
- the deamination is carried out by heating or induction into an ammonium salt.
- the enol obtained by coupling the compound (15) with an unsaturated enone as shown in Scheme 32 below This can be done from ether (19).
- compounds (1 5) to (1) (Z (l-4)); the combination of D and E both represent a hydrogen atom, D represents a hydroxyl group and E represents a hydrogen atom, And E together form a single bond and represent a double bond with the specified single bond; the combination of R 4 , and R 42 is (hydrogen atom and hydroxyl group), (hydrogen atom and C 2 -C 5 Ashiruokishi group), (hydroxyl group, Ashiruokishi group of C 2 -C 5 or Ji,] ⁇ .
- Arukiruokishi may be substituted with group C, alkyl group and a hydroxyl group in -C 4), or (a hydroxyl group, C 2 ⁇ Ashiruokishi group C 5 or induction and C, which may be substituted with Arukiruokishi group -C 4 C, to what is Ashiruokishi group) of the alkyl groups and C 2 -C 5 of -C 4, is for example the following As in Scheme 33, the aldehyde (15) and ketone ⁇ -carbonyl group is protected by acetal.
- the aldol reaction can be carried out with chloroalkanone (20), followed by a dehydration reaction, a reduction reaction or a reaction with an organometallic reagent, an acetal deprotection or the like, if necessary.
- organometallic reagent used in the reaction with the organometallic reagent examples include an organolithium compound, a Grignar_d reagent, an organic cellum reagent, and the like.
- the aldehyde represented by the above formula (14) can be produced, for example, as in the following Schemes 34 and 35.
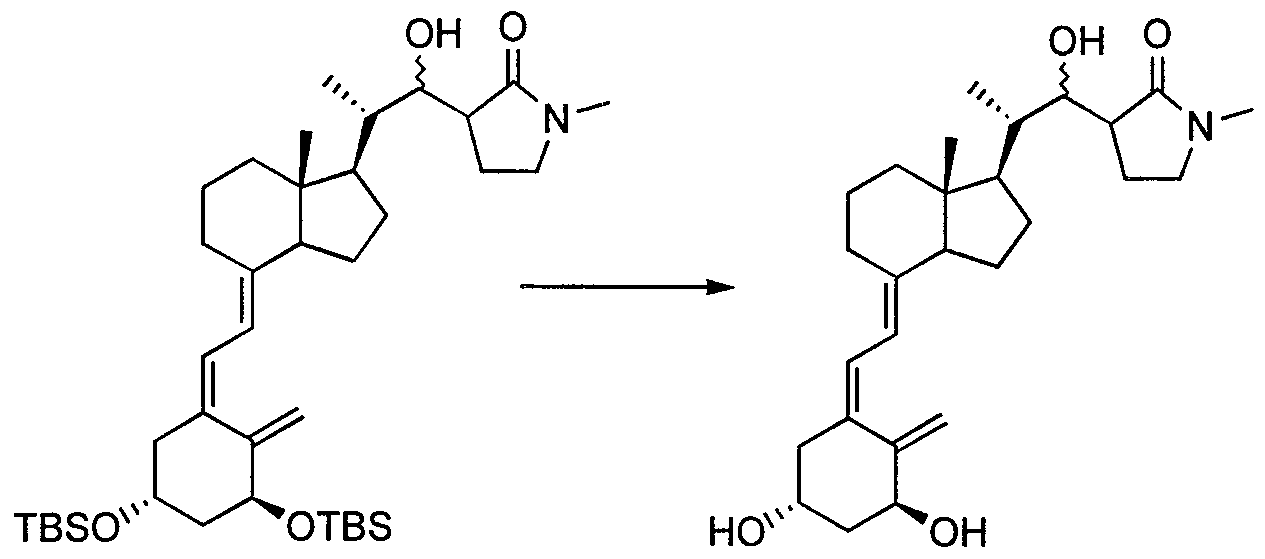
- the deprotection reaction can be performed as follows.
- R 01 and R 02 are acetyl groups
- ordinary alkali hydrolysis, potassium cyanide, ammonia-methanol, or the like can be used.
- R 01 and R 02 are a methoxymethyl group or a tetrahydro-14H-pyran-2-yl group
- acid conditions such as hydrochloric acid, acetic acid, trifluoroacetic acid and the like, pyridinium p-toluenesulfonate (PPTS), Li BF 4 or the like can be used.
- PPTS pyridinium p-toluenesulfonate
- R 01 and R 02 are a trimethylsilyl group, a triethylsilyl group, or a t-butyldimethylsilyl group
- a known method for example, Cavalyl (Caver 1 y), tetrahedron (Te trahedron), Vol. 20, No. 4609) — 4619, 1987
- a deprotecting agent for example, tetrabutylammonium fluoride, pyridinum p-toluenesulfonate, hydrogen fluoride and the like can be used.
- organic solvent used in the reaction examples include halogen-based solvents such as methylene chloride, chloroform, and carbon tetrachloride; hydrocarbon-based solvents such as hexane and toluene; ether-based solvents such as tetrahydrofuran and dioxane; N-dimethylformamide, acetoni A water-soluble solvent such as tolyl; or a mixed solvent thereof, etc., can be used, and can be selected in consideration of the solubility and reactivity of the compound.
- the reaction temperature is generally in the range of 120 ° C to the boiling point of the solvent.
- the reaction time varies depending on the dehydrating agent, deprotecting agent, reaction solvent and reaction temperature used, and it is usually desirable to use an analytical means such as thin layer chromatography or the like until the starting materials disappear.
- R 0 1 and R 0 2 is a trimethylsilyl group, preparative Riechirushiriru group, when a t one-heptyl dimethylsilyl group, a combination of tetrafurfuryl O Lobo, single Toarukari metal salt and a mineral acid It can also be performed with a reagent consisting of Lithium tetrafluoroborate, sodium tetrafluoroborate, and potassium tetrafluoroborate are used as the alkali metal salts of tetrafluoroborate, and hydrochloric acid, sulfuric acid, etc. are used as the mineral acids. Can be.
- the vitamin D 3 derivative obtained as described above can be converted to the above-mentioned pharmaceutically acceptable solvate, if necessary.
- the present invention is a therapeutic agent for inflammatory respiratory diseases, comprising a therapeutically effective amount of the vitamin D 3 derivative represented by the above formula (1) or (3) or a pharmaceutically acceptable solvate thereof. And a method for treating the disease group using the same.
- Inflammatory respiratory diseases targeted by the therapeutic agent or method of the present invention include, for example, acute upper respiratory tract infection, chronic sinusitis, allergic rhinitis, chronic lower respiratory tract infection, emphysema, pneumonia, bronchial asthma
- inflammatory respiratory diseases selected from the group consisting of pulmonary tuberculosis sequelae, acute respiratory distress syndrome, cystic fibrosis, and pulmonary fibrosis can be mentioned as preferred.
- the inflammatory respiratory diseases of the present invention include, for example, common cold, acute pharyngitis, acute rhinitis, acute sinusitis, acute tonsillitis, acute laryngitis, acute epiglottitis, and acute bronchitis
- acute upper respiratory tract infections selected from the group consisting of, for example, chronic bronchitis, diffuse panbronchiolitis, and one or more chronic lower bronchitis selected from the group consisting of bronchiectasis Respiratory tract infections may be mentioned as preferred.
- the present invention also provides a therapeutically effective amount of the vitamin D 3 derivative represented by the above formula (1) or It is a therapeutic agent for a disease selected from the group consisting of malignant tumor, rheumatoid arthritis, osteoporosis, diabetes mellitus, hypertension, alopecia, acne, psoriasis, and dermatitis, containing its pharmaceutically acceptable solvate. And a method for treating the disease group using the same.
- the present invention provides a therapeutically effective amount of a vitamin D 3 derivative represented by the above formula (1), which comprises a compound having a vitamin D 3 antagonist effect, and comprising hypercalcemia based on vitamin D 3 excess.
- a therapeutic agent for a disease selected from the group consisting of hypoparathyroidism and cartilage metabolic disorder, and a method for treating the disease group using the same.
- hypoparathyroidism examples include idiopathic or postoperative hypoparathyroidism due to decreased PTH production.
- cartilage metabolic disorder include diseases in which cartilage components are degraded due to degradation or destruction of synthesizing ability of collagen and proteodalican in chondrocytes or matrix, and cartilage is reduced.
- the disease examples include osteoarthritis, rheumatoid arthritis, rheumatic fever and the like.
- the therapeutic agent for various diseases of the present invention can be administered orally, parenterally such as intravenously, subcutaneously, intramuscularly, transdermally, nasally, rectally, or by inhalation.
- Dosage forms for oral administration include tablets, pills, powders, granules, solutions, suspensions, syrups, and capsules.
- excipients such as ratatose, starch, calcium carbonate, crystalline cellulose, or caicic acid
- binders such as carboxymethylcellulose, methylcellulose, calcium phosphate, or polyvinylpyrrolidone
- Disintegrants such as sodium acid, sodium bicarbonate, sodium sodium radium sulfate and stearic acid monoglyceride
- Wetting agents such as glycerin
- Absorbents such as kaolin and colloidal silica
- Lubricants such as talc and granular phosphoric acid Formulation is made using the agent.
- Pills, powders, and granules are also formulated in the usual manner using additives as described above.
- Liquid preparations such as liquid preparations, suspensions and syrups are also formulated in accordance with ordinary methods.
- Carriers include, for example, dalycerol esters such as tricaprylin, triacetin, and fatty acid poppy oil fatty acid esters; water; alcohols such as ethanol; liquid bases such as liquid paraffin, coconut oil, soybean oil, sesame oil, and corn oil. Is used.
- Capsules are made by filling powders, granules, liquid preparations, and the like, into capsules such as gelatin. '
- Intravenous, subcutaneous and intramuscular dosage forms include injections in the form of sterile aqueous or non-aqueous solutions.
- aqueous solution for example, physiological saline is used.
- Non-aqueous solutions include, for example, propylene glycol, polyethylene glycol or olive Vegetable oils such as oil, injectable organic esters such as ethyl oleate, and oleic poppy oil fatty acid esters are used.
- These preparations are added, if necessary, with isotonic agents, preservatives, wetting agents, and emulsifiers. Dispersants, stabilizers, etc. are also added. Filtration through bacteria-retaining filters, blending of bactericides, irradiation, etc. Sterility can be achieved by appropriate treatment.
- a sterile solid preparation can be manufactured and dissolved in sterile water or a sterile solvent for injection immediately before use.
- the compound of the present invention can also be used after forming an inclusion compound with, for example, methylated cyclodextrin or ⁇ - or ⁇ -cyclodextrin. Also, an injection in the form of liposome may be used.
- Examples of the dosage form of the drug for transdermal administration include ointments, creams, lotions, liquids and the like.
- Bases for ointments include, for example, fatty oils such as castor oil, olive oil, sesame oil, and safflower oil; lanolin; white, yellow or hydrophilic petrolatum; wax; oleyl alcohol, isostearyl alcohol, octyldodecanol, hexyl Higher alcohols such as decanol; dalicols such as glycerin, diglycerin, ethylene glycol, propylene glycol, sorbitol, and 1,3-butanediol.
- ethanol, dimethyl sulfoxide, polyethylene glycol, or the like may be used as a solubilizing agent for the compound of the present invention.
- a preservative such as paraoxybenzoate, sodium benzoate, salicylic acid, sorbic acid, and boric acid; and an antioxidant such as butylhydroxyanisole and dibutylhydroxytoluene may be used.
- an absorption enhancer such as diisopropyl adipate, getyl sebacate, ethyl caproate, and ethyl laurate may be added.
- the compound of the present invention can be used after forming an inclusion compound with ⁇ -cyclodextrin or methylated cyclodextrin.
- An ointment can be manufactured by a usual method.
- the cream the form of an oil-in-water cream is preferred for stabilizing the compound of the present invention.
- the base the above-mentioned fatty oils, higher alcohols, glycols and the like are used, and emulsifiers such as diethylene glycol, propylene glycol, sorbitan monofatty acid ester, polysorbate 80 and sodium lauryl sulfate are used. . If necessary, the above-mentioned preservatives, antioxidants, etc. may be added.
- the compound of the present invention can be used as an inclusion compound of cyclodextrin and methylated cyclodextrin. Creams can be manufactured by usual methods.
- Examples of the lotion include a suspension type, an emulsion type and a solution type lotion.
- Suspension lotions use a suspending agent such as sodium alginate, tragacanth, sodium carboxymethyl cellulose, and add antioxidants and preservatives as needed. can get.
- the emulsifying lotion can be obtained by a usual method using an emulsifying agent such as sorbitan monofatty acid ester, polysorbate 80, sodium sodium sulfate and the like.
- an emulsifying agent such as sorbitan monofatty acid ester, polysorbate 80, sodium sodium sulfate and the like.
- the solvent include those obtained by dissolving the compound of the present invention in an alcohol solution such as ethanol, and adding an antioxidant, a preservative and the like as necessary.
- dosage forms such as pasta, cataplasm, aerosol and the like can be mentioned.
- These preparations can be manufactured by a usual method.
- Formulations for nasal administration are provided as liquid or powdered compositions.
- a base of the liquid agent water, saline, phosphate buffer, acetate buffer and the like are used, and may further contain a surfactant, an antioxidant, a stabilizer, a preservative, and a viscosity-imparting agent.
- a base of the powdered agent a water-absorbing base is preferable.
- polyacrylates such as sodium polyacrylate, potassium polyacrylate, and ammonium polyacrylate, and water-soluble polymethacrylate
- methinoresenorelose Cellulose lower alkyl ethers such as hydroxypropyl cellulose, hydroxypropyl cellulose, carboxymethyl cellulose sodium, polyethylene glycol, polybutylpyrrolidone, amylose, pullulan, etc.
- Cellulose cellulose such as cross-linked sodium carboxymethylcellulose, starch such as hydroxypropyne starch, carboxymethyl starch, cross-linked starch, amylose, amylopectin, pectin, gelatin, casein, casein
- proteins such as thorium, gums such as gum arabic, gum tragacanth, and darco mannan, polyvinyl polypyrrolidone, cross-linked polyacrylic acid and its salts, cross-linked polybutyl alcohol, and the like.
- an antioxidant, a coloring agent, a preservative, a preservative, a preservative, and the like may be added to the powder.
- liquid preparations and powder preparations can be administered using, for example, a spray device.
- a spray device for rectal administration, usual suppositories such as gelatin soft capsules are used.
- spray, nebulizer one using a dosing device of the atomizer first class, powdered or vitamin D 3 derivatives of the active ingredient of the present invention alone or in combination with suitable biocompatible excipient It can be administered to a disease site as a liquid composition. Alternatively, it can be administered to the diseased site by suspending or dissolving it in an aerosol propellant such as CFC alternative, and filling it into pMDI (quantitative atomizer). It can also be administered to the diseased site by dissolving it in an aqueous ethanol solution and filling it in an appropriate sprayer.
- pMDI quantitative atomizer
- the therapeutically effective amount of the active ingredient of the present invention varies depending on the administration route, patient age, sex, and the degree of the disease, but is usually about 0.001 to 100 ⁇ g / day, more preferably 0 to 100 ⁇ g / day.
- the dose is usually about 1 to 50 / g / day, and the administration frequency is usually 1 to 3 times / day. It is preferable to prepare a preparation so as to satisfy such conditions.
- the therapeutic agent for various diseases of the present invention can be used in combination with an existing drug.
- the usefulness of the vitamin D 3 derivative represented by the above formula (1) of the present invention for inflammatory respiratory diseases is demonstrated by the lipopolysaccharide commonly used as an inflammatory lung disease model, as specifically shown in Examples described later. (LPS) induced pulmonary inflammation in experiments with hamsters. That is, it was found that the compound of the present invention significantly suppressed LPS-induced lung inflammation by intratracheal administration and oral administration.
- the usefulness of the vitamin D 3 derivative represented by the above formula (1) of the present invention for a disease based on an excessive action of vitamin D 3 is determined by using HL-60 cells, as specifically shown in Examples described later.
- the effect of induction of differentiation was shown as an index. That is, the compound of the present invention specifically inhibits the differentiation of HL-60 cells induced by active vitamin D 3 (1 ⁇ , 25-dihydroxyvitamin D 3 ). it became clear that act as vitamin D 3 antagonists.
- Compound No. 51 07 is 1/11/1.
- the vitamin D 3 derivative represented by the above formula (1) has realized the separation of the anti-inflammatory action expression concentration and the vitamin D 3 antagonistic action from the blood calcium concentration increasing action expression concentration, and Is not considered to be expressed.
- a therapeutic agent containing the vitamin D 3 derivative represented by the above formula (1) as an active ingredient is useful for inflammatory respiratory diseases, ie, diseases caused by excessive action of vitamin D 3. .
- vitamin D 3 has been reported to have various effects on cell metabolism, examples thereof, for example, stimulation of cell maturation and differentiation (Tanaka et al., (Biochemical Journal ( J.), Vol. 204, pp. 7 13—7 19, 1982; Amento et al., Journal of 'Clini Force, Nore Investigation (J. C.) lin. Inves t.), Vol. 73, pp. 7311-739, 1998; Coleston et al., End Clinology-(En dcrinology), Vol. 108, pp. 1083--1086, 1 98 1; Abetet 1 et al., Proceedings of the National Aka. Sci., Proc. Natl. Acad.
- the vitamin D 3 derivative represented by the above formula (1) also has cell differentiation inducing ability as described above.
- the vitamin D 3 derivative represented by the above formula (1) can be used for, for example, malignant tumors, inflammatory diseases such as psoriasis, rheumatoid arthritis, dermatitis and autoimmune diseases, infectious diseases (particularly bacterial and viral diseases). And fungi) have therapeutic potential in various areas, such as in adjuvants and other therapeutic modalities involving mononuclear phagocytes.
- Some vitamin D 3 derivative represented by the above formula (1) of the present invention 1 shed, 25-binding ability of the dihydric mud carboxymethyl vitamin 0 3 receptor 1 alpha, 25-dihydrazide mud carboxymethyl vitamin D 3
- the compounds of the present invention can be expected to be useful as therapeutic agents for osteoporosis Motodzure was for having bone metabolism maintain operation of the active vitamin D 3.
- Diastereomer 2 ((R-20) or (R-21))
- Triphenyl phosphine 1 3.7 mg (52 ⁇ mo 1), Tris (dibenzylideneacetone) dipalladium (0) — 9 mg (8.6 / mo 1) of the form adduct of chloroform are dissolved in 1.0 ml of dry toluene, and the mixture is stirred at room temperature under a nitrogen atmosphere for 15 minutes. did.
- No.1104a (more polar)
- No.1104b (less polar)
- Example 1-1 (4) As in Example 1-1 (4), 209 mg (0.529 mmo 1) of (1-1i) was used as a raw material, and (3S), (5R) —3,5-bis Use (3S), (5R) -3,5-bis (t-butyldimethylsilyloxy) -11-octene-1-7- ⁇ f instead of (trimethylsilyloxy) -1-one Then, a coupled product and No. 1104a were obtained. Coupling product: 3 13 mg, yield 87%.
- triphenylphosphine (8 mg), tris (dibenzylideneacetone) dipalladium (0) -chloroform adduct (6 mg) were mixed with anhydrous toluene (0.5 ml) and diisopropylethylamine (0.5 ml). It was dissolved in a mixed solvent and stirred at room temperature for 15 minutes.
- Example 2-1 3.5 g (9.8 mmo 1) of (R-23) obtained in Reference Example 4 was converted to (2—g) low-polar substance by 1.8 g (yield 36%) and 1.24 g (yield 24%) of (2-g) highly polar compound were obtained. These are stereoisomers based on the asymmetric point to which the hydroxyl group is bonded.
- Example 2-1 In the same manner as in (2) and (3), (2-i) was obtained using (2-h) obtained above. In this two-step reaction, only the isomers whose asymmetric point to which the MOMCH 2 group was bonded had the configuration described above proceeded. After the second-step reaction, the target product was purified by silica gel column chromatography to obtain optically pure (2-i). (3) Example 2-1 (4) Similarly to (7), No. 22009 c was obtained from (2-i) obtained above.
- Example 2-1 (2) ′ In the same manner as (7), No. 2110b was obtained by using the (2—g) highly polar substance obtained in Example 22 (1).
- No.2101a (more polar)
- No.2101b (less polar)
- the cooled p This titanium tetrachloride 0. 64 g (3. 41 mmo 1 ) was added to the Flip, further
- Example 3-1 In the same manner as (3), using (3-k) 6 Omg (0.094 mmo 1) as a raw material, 17.5 mg of No. 3401 was obtained. Yield 47%.
- Example 3-1 13.1 mg of No. 3513a was obtained from (3-1) 24 mg (0.036 mmol) of a low-polarity substance in the same manner as (3). Yield 83%. This sample appears to be a mixture of stereoisomers based on the 22 or 23 position asymmetric point.
- Example 3-1 In the same manner as (3), using (3-1) a highly polar substance of 9.9 mg (0.015 mmo 1) as a raw material, convert N 0.35 13 b to 7. 5 mg was obtained. Yield 1 15%. This sample appears to be a mixture of stereoisomers based on the 22 or 23 position asymmetric point.
- No.4102a (more polar) No.4107a (more polar) No.4102b (less polar) No.4107b (less polar)
- No. 41 07a and No. 41 07b are stereoisomers based on asymmetric points on the cyclopentanone ring.
- the solvent was concentrated to obtain a crude form of a silyl enol form of bromomethylene.
- Example 4-3 Similarly to (2), 1.03 g of a mixture of (4-1d) and (4-e) obtained in Example 4-3 (1) and cyclopentenone 0. From 506ml (4
- This sample is a mixture of stereoisomers based on the 20-position asymmetric point.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Rheumatology (AREA)
- Nutrition Science (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description






















































































Claims





Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/830,167 US6531460B1 (en) | 1998-10-23 | 1999-10-22 | Vitamin D, derivatives and remedies for inflammatory respiratory diseases containing the same |
| CA002348235A CA2348235A1 (en) | 1998-10-23 | 1999-10-22 | Vitamin d3 derivatives and treating agent for inflammatory respiratory disease using same |
| EP99949355A EP1123921A4 (en) | 1998-10-23 | 1999-10-22 | VITAMIN D 3 DERIVATIVES? AND MEDICINES FOR INFLAMMATORY RESPIRATORY DISEASES CONTAINING THEM |
| AU62281/99A AU758792B2 (en) | 1998-10-23 | 1999-10-22 | Vitamin D3 derivatives and remedies for inflammatory respiratory diseases containing the same |
| US10/035,217 US6960573B2 (en) | 1998-10-23 | 2002-01-04 | Vitamin D3 derivative and treating agent for inflammatory respiratory disease using same |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP10/302321 | 1998-10-23 | ||
| JP30232198 | 1998-10-23 | ||
| JP36282798 | 1998-12-21 | ||
| JP10/362827 | 1998-12-21 | ||
| JP10/365209 | 1998-12-22 | ||
| JP10/365207 | 1998-12-22 | ||
| JP10/365208 | 1998-12-22 | ||
| JP36520898 | 1998-12-22 | ||
| JP36520998 | 1998-12-22 | ||
| JP36520798 | 1998-12-22 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09830167 A-371-Of-International | 1999-10-22 | ||
| US10/035,217 Division US6960573B2 (en) | 1998-10-23 | 2002-01-04 | Vitamin D3 derivative and treating agent for inflammatory respiratory disease using same |
| US10/035,219 Division US6548489B2 (en) | 1998-10-23 | 2002-01-04 | Vitamin D3 derivative and treating agent for inflammatory respiratory disease using same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000024712A1 true WO2000024712A1 (en) | 2000-05-04 |
Family
ID=27530947
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1999/005826 Ceased WO2000024712A1 (en) | 1998-10-23 | 1999-10-22 | Vitamin d3 derivatives and remedies for inflammatory respiratory diseases containing the same |
Country Status (6)
| Country | Link |
|---|---|
| US (5) | US6531460B1 (ja) |
| EP (1) | EP1123921A4 (ja) |
| KR (1) | KR100676106B1 (ja) |
| AU (1) | AU758792B2 (ja) |
| CA (1) | CA2348235A1 (ja) |
| WO (1) | WO2000024712A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002015894A3 (en) * | 2000-08-23 | 2002-08-29 | Teijin Ltd | Use of vitamin d derivatives as bone resorption inhibitors |
| WO2003070716A1 (en) * | 2002-02-20 | 2003-08-28 | Teijin Limited | Vitamin d3 derivatives and remedies using the same |
| EP1321141A4 (en) * | 2000-08-30 | 2004-02-18 | Teijin Ltd | INHIBITORS OF PARATHORMONE PRODUCTION CONTAINING VITAMIN D3 DERIVATIVES |
| WO2004067525A1 (ja) | 2003-01-30 | 2004-08-12 | Teijin Pharma Limited | ビタミンd3ラクトン誘導体 |
| WO2005042482A1 (ja) * | 2003-11-04 | 2005-05-12 | Teijin Pharma Limited | ビタミンd3ラクタム誘導体 |
| WO2006019169A1 (ja) * | 2004-08-17 | 2006-02-23 | Teijin Pharma Limited | 3-エピビタミンd3誘導体およびそれを用いる治療剤 |
| US7524980B2 (en) | 2003-11-04 | 2009-04-28 | Teijin Pharma Limited | Vitamin D3 lactam derivative |
| WO2010053165A1 (ja) | 2008-11-04 | 2010-05-14 | 帝人ファーマ株式会社 | ビタミンd3ラクタム誘導体 |
| JP2013501790A (ja) * | 2009-08-14 | 2013-01-17 | バーグ バイオシステムズ,エルエルシー | 脱毛症を治療するためのビタミンd3およびその類似体 |
| CN116075309A (zh) * | 2020-09-15 | 2023-05-05 | 帝人制药株式会社 | 侧链具有环状胺的维生素d衍生物 |
Families Citing this family (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5763429A (en) * | 1993-09-10 | 1998-06-09 | Bone Care International, Inc. | Method of treating prostatic diseases using active vitamin D analogues |
| US6242434B1 (en) * | 1997-08-08 | 2001-06-05 | Bone Care International, Inc. | 24-hydroxyvitamin D, analogs and uses thereof |
| US20020128240A1 (en) * | 1996-12-30 | 2002-09-12 | Bone Care International, Inc. | Treatment of hyperproliferative diseases using active vitamin D analogues |
| US6566353B2 (en) * | 1996-12-30 | 2003-05-20 | Bone Care International, Inc. | Method of treating malignancy associated hypercalcemia using active vitamin D analogues |
| US20030129194A1 (en) * | 1997-02-13 | 2003-07-10 | Bone Care International, Inc. | Targeted therapeutic delivery of vitamin D compounds |
| US6087350A (en) * | 1997-08-29 | 2000-07-11 | University Of Pittsburgh Of The Commonwealth System Of Higher Education | Use of pretreatment chemicals to enhance efficacy of cytotoxic agents |
| WO1999049870A1 (en) * | 1998-03-27 | 1999-10-07 | Oregon Health Sciences University | Vitamin d and its analogs in the treatment of tumors and other hyperproliferative disorders |
| EP1301479A2 (en) * | 2000-07-18 | 2003-04-16 | Bone Care International, Inc. | STABILIZED 1$g(a)-HYDROXY VITAMIN D |
| US20040019024A1 (en) * | 2000-08-23 | 2004-01-29 | Seiichi Ishizuka | Use of vitamin d derivatives as bone resorption inhibitors |
| BR0214679A (pt) * | 2001-12-03 | 2004-12-14 | Novacea Inc | Composições farmacêuticas compreendendo compostos a base de vitamina d ativa |
| US7163933B2 (en) | 2002-02-22 | 2007-01-16 | Teijin Limited | Treating agent for Paget's disease of bone |
| EP1542996A4 (en) * | 2002-09-20 | 2009-11-18 | Merck & Co Inc | OCTAHYDRO-2H-NAPHTOc1,2-F INDOL-4-CARBONSUUREAMIDE DERIVATIVES AS SELECTIVE MODULATORS OF THE GLUCOCORTICOID RECEPTOR |
| AR041696A1 (es) * | 2002-10-23 | 2005-05-26 | Leo Pharm Prod Ltd | Compuestos analogos de vitamina d, composiciones que comprenden dichos compuestos analogos y su uso |
| US20050026877A1 (en) * | 2002-12-03 | 2005-02-03 | Novacea, Inc. | Pharmaceutical compositions comprising active vitamin D compounds |
| US20060189586A1 (en) * | 2003-06-11 | 2006-08-24 | Cleland Jeffrey L | Pharmaceutical compositions comprising active vitamin D compounds |
| US20050020546A1 (en) * | 2003-06-11 | 2005-01-27 | Novacea, Inc. | Pharmaceutical compositions comprising active vitamin D compounds |
| MY139521A (en) | 2004-03-18 | 2009-10-30 | Leo Pharma As | Stereoselective synthesis of vitamin d analogues |
| CA2555260C (en) | 2004-03-18 | 2011-12-06 | Leo Pharma A/S | Stereoselective synthesis of vitamin d analogues |
| US20060003950A1 (en) * | 2004-06-30 | 2006-01-05 | Bone Care International, Inc. | Method of treating prostatic diseases using a combination of vitamin D analogues and other agents |
| US7094775B2 (en) * | 2004-06-30 | 2006-08-22 | Bone Care International, Llc | Method of treating breast cancer using a combination of vitamin D analogues and other agents |
| AU2006230296B2 (en) * | 2005-03-29 | 2011-07-07 | Wisconsin Alumni Research Foundation | 2-methylene-19-nor-(23s)-25-dehydro-1alpha-hydroxyvitamin D3-26,23-lactone and 2-methylene-19-nor-(23R)-25-dehydro-1alpha-hydroxyvitamin D3-26,23-lactone |
| KR20080028860A (ko) * | 2005-04-22 | 2008-04-02 | 노바세아, 인크. | 활성 비타민 d 화합물 또는 그의 유사체에 의한 화학 요법또는 방사선 치료와 관련된 폐 장애의 치료, 예방 및 개선 |
| EP2004597B1 (en) * | 2006-04-10 | 2011-01-26 | Wisconsin Alumni Research Foundation | 1alpha-hydroxy-2-(3'-hydroxypropylidene)-19-nor-vitamin d compounds with a 1,1-dimethylpropyl side chain |
| US7569719B1 (en) * | 2006-10-25 | 2009-08-04 | Loctite (R&D) Limited | Method of preparing electron deficient olefins |
| US9119777B2 (en) | 2008-05-30 | 2015-09-01 | Microdose Therapeutx, Inc. | Methods and compositions for administration of oxybutynin |
| US8415390B2 (en) | 2008-05-30 | 2013-04-09 | Microdose Therapeutx, Inc. | Methods and compositions for administration of oxybutynin |
| US7973119B1 (en) | 2007-10-24 | 2011-07-05 | Loctite (R&D) Limited | Adhesive systems using imines and salts thereof and precursurs to electron deficient olefins |
| ES2662644T3 (es) * | 2007-10-24 | 2018-04-09 | Henkel IP & Holding GmbH | Reactivos de metileno |
| US8053589B1 (en) * | 2007-10-24 | 2011-11-08 | Henkel Ireland Limited | Imines and methods of preparing electron deficient olefins using such novel imines |
| US8686105B2 (en) | 2007-10-24 | 2014-04-01 | Henkel IP & Holding GmbH | Adhesive systems using imines and salts thereof, precursors to electron deficient olefins and coreactants therefor |
| WO2009053484A2 (en) | 2007-10-24 | 2009-04-30 | Loctite (R & D) Limited | Electron deficient olefins and curable compositions prepared therefrom |
| US8399698B1 (en) | 2008-10-24 | 2013-03-19 | Henkel Ireland Limited | Substituted activated methylene reagents and methods of using such reagents to form electron deficient olefins |
| US10196471B1 (en) | 2008-10-24 | 2019-02-05 | Henkel IP & Holding GmbH | Curable composition having an electron deficient olefin |
| WO2011002756A2 (en) * | 2009-07-01 | 2011-01-06 | Vitamin Derivatives, Inc. | Vitamin d compounds and methods for preparing same |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0619305A1 (en) * | 1993-02-05 | 1994-10-12 | Teijin Limited | Lactone compound and process of producing thereof |
| JPH07173133A (ja) * | 1993-11-02 | 1995-07-11 | Chugai Pharmaceut Co Ltd | 1β−ヒドロキシビタミン▲D3▼誘導体 |
| WO1998058909A1 (en) * | 1997-06-25 | 1998-12-30 | Teijin Limited | Vitamin d3 derivatives and remedies for inflammatory respiratory diseases prepared from the same |
| JPH1149747A (ja) * | 1997-08-04 | 1999-02-23 | Teikoku Hormone Mfg Co Ltd | 活性型ビタミンd誘導体 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5087619A (en) * | 1988-01-20 | 1992-02-11 | Hoffman-La Roche Inc. | Vitamin D3 analogs |
| US4851401A (en) * | 1988-07-14 | 1989-07-25 | Wisconsin Alumni Research Foundation | Novel cyclopentano-vitamin D analogs |
| EP0614455B1 (en) * | 1991-11-07 | 1996-02-21 | Research Institute For Medicine And Chemistry Inc. | Vitamin d amide derivatives |
| GB9203535D0 (en) | 1992-02-19 | 1992-04-08 | Leo Pharm Prod Ltd | Novel treatment iii |
| IL107185A (en) | 1992-10-06 | 1998-02-22 | Schering Ag | Vitamin d, 25-carboxylic acid derivatives and pharmaceutical compositions containing the same |
| GB9309422D0 (en) * | 1993-05-07 | 1993-06-23 | Res Inst Medicine Chem | Chemical compounds |
| US5354872A (en) | 1993-05-28 | 1994-10-11 | Alcon Laboratories, Inc. | Process for preparation of calcitriol lactone and related intermediates |
| DE69513367T2 (de) | 1994-06-07 | 2000-06-08 | Teijin Ltd., Osaka | Vitamin d3 derivat und verfahren zu dessen herstellung |
| JPH0892098A (ja) * | 1994-09-27 | 1996-04-09 | Teijin Ltd | 肺結核治療剤 |
| JPH1087495A (ja) * | 1996-09-17 | 1998-04-07 | Teijin Ltd | 活性型ビタミンd含有予防剤あるいは治療剤 |
| JP2003342047A (ja) * | 2002-05-23 | 2003-12-03 | Jfe Steel Kk | 溶融スラグの造粒方法および造粒装置 |
-
1999
- 1999-10-22 EP EP99949355A patent/EP1123921A4/en not_active Withdrawn
- 1999-10-22 WO PCT/JP1999/005826 patent/WO2000024712A1/ja not_active Ceased
- 1999-10-22 KR KR1020017004678A patent/KR100676106B1/ko not_active Expired - Fee Related
- 1999-10-22 US US09/830,167 patent/US6531460B1/en not_active Expired - Fee Related
- 1999-10-22 AU AU62281/99A patent/AU758792B2/en not_active Ceased
- 1999-10-22 CA CA002348235A patent/CA2348235A1/en not_active Abandoned
-
2002
- 2002-01-04 US US10/035,251 patent/US6867313B2/en not_active Expired - Fee Related
- 2002-01-04 US US10/035,219 patent/US6548489B2/en not_active Expired - Fee Related
- 2002-01-04 US US10/035,211 patent/US6689766B2/en not_active Expired - Fee Related
- 2002-01-04 US US10/035,217 patent/US6960573B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0619305A1 (en) * | 1993-02-05 | 1994-10-12 | Teijin Limited | Lactone compound and process of producing thereof |
| JPH07173133A (ja) * | 1993-11-02 | 1995-07-11 | Chugai Pharmaceut Co Ltd | 1β−ヒドロキシビタミン▲D3▼誘導体 |
| WO1998058909A1 (en) * | 1997-06-25 | 1998-12-30 | Teijin Limited | Vitamin d3 derivatives and remedies for inflammatory respiratory diseases prepared from the same |
| JPH1149747A (ja) * | 1997-08-04 | 1999-02-23 | Teikoku Hormone Mfg Co Ltd | 活性型ビタミンd誘導体 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1123921A4 * |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002015894A3 (en) * | 2000-08-23 | 2002-08-29 | Teijin Ltd | Use of vitamin d derivatives as bone resorption inhibitors |
| AU2001284657B2 (en) * | 2000-08-23 | 2006-01-19 | Board Of Regents, The University Of Texas System | Use of vitamin d derivatives as bone resorption inhibitors |
| EP1321141A4 (en) * | 2000-08-30 | 2004-02-18 | Teijin Ltd | INHIBITORS OF PARATHORMONE PRODUCTION CONTAINING VITAMIN D3 DERIVATIVES |
| WO2003070716A1 (en) * | 2002-02-20 | 2003-08-28 | Teijin Limited | Vitamin d3 derivatives and remedies using the same |
| AU2004207719B9 (en) * | 2003-01-30 | 2009-10-01 | Teijin Limited | Vitamin D3 lactone derivative |
| WO2004067525A1 (ja) | 2003-01-30 | 2004-08-12 | Teijin Pharma Limited | ビタミンd3ラクトン誘導体 |
| US9073885B2 (en) | 2003-01-30 | 2015-07-07 | Teijin Pharma Limited | Vitamin D3 lactone derivatives |
| CN1771239B (zh) * | 2003-01-30 | 2010-06-09 | 帝人制药株式会社 | 维生素d3内酯衍生物 |
| AU2004207719B2 (en) * | 2003-01-30 | 2009-08-06 | Teijin Limited | Vitamin D3 lactone derivative |
| WO2005042482A1 (ja) * | 2003-11-04 | 2005-05-12 | Teijin Pharma Limited | ビタミンd3ラクタム誘導体 |
| US7524980B2 (en) | 2003-11-04 | 2009-04-28 | Teijin Pharma Limited | Vitamin D3 lactam derivative |
| WO2006019169A1 (ja) * | 2004-08-17 | 2006-02-23 | Teijin Pharma Limited | 3-エピビタミンd3誘導体およびそれを用いる治療剤 |
| WO2010053165A1 (ja) | 2008-11-04 | 2010-05-14 | 帝人ファーマ株式会社 | ビタミンd3ラクタム誘導体 |
| JP2013501790A (ja) * | 2009-08-14 | 2013-01-17 | バーグ バイオシステムズ,エルエルシー | 脱毛症を治療するためのビタミンd3およびその類似体 |
| JP2017008064A (ja) * | 2009-08-14 | 2017-01-12 | バーグ エルエルシー | 脱毛症を治療するためのビタミンd3およびその類似体 |
| CN116075309A (zh) * | 2020-09-15 | 2023-05-05 | 帝人制药株式会社 | 侧链具有环状胺的维生素d衍生物 |
| CN116075309B (zh) * | 2020-09-15 | 2025-07-18 | 帝人制药株式会社 | 侧链具有环状胺的维生素d衍生物 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU6228199A (en) | 2000-05-15 |
| US20020091109A1 (en) | 2002-07-11 |
| EP1123921A4 (en) | 2003-08-20 |
| US20020103173A1 (en) | 2002-08-01 |
| CA2348235A1 (en) | 2000-05-04 |
| EP1123921A1 (en) | 2001-08-16 |
| US6548489B2 (en) | 2003-04-15 |
| US20020099039A1 (en) | 2002-07-25 |
| AU758792B2 (en) | 2003-03-27 |
| US6531460B1 (en) | 2003-03-11 |
| US6867313B2 (en) | 2005-03-15 |
| KR100676106B1 (ko) | 2007-02-28 |
| US6689766B2 (en) | 2004-02-10 |
| US20020132799A1 (en) | 2002-09-19 |
| US6960573B2 (en) | 2005-11-01 |
| KR20010085919A (ko) | 2001-09-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2000024712A1 (en) | Vitamin d3 derivatives and remedies for inflammatory respiratory diseases containing the same | |
| JP2711161B2 (ja) | 新規ビタミンd類似体 | |
| EP0460032B1 (en) | Novel vitamin d analogues | |
| CN1662245A (zh) | (20S)-1α-羟基-2α-甲基和2β-甲基-19-去甲-维生素D3及它们的用途 | |
| CN104211626A (zh) | 新型维生素d受体活化剂及制造方法 | |
| KR100593238B1 (ko) | 비타민 d3 유도체 및 이로부터 제조된 염증성 호흡기 질환 치료제 | |
| HUT62251A (en) | Process for producing cytotoxic bicyclo/7.3.1/tridec-4-ene-2,6-diine derivatives and pharmaceutical compositions comprising such compounds | |
| WO2001096293A1 (en) | Vitamin d derivatives | |
| WO2009129361A2 (en) | Macrocyclic compounds and methods of making and using thereof | |
| WO2001079166A1 (en) | Vitamin d derivatives | |
| CN100534981C (zh) | 2-亚丙基-19-去甲维生素d化合物 | |
| US4421690A (en) | Process for the preparation of 24,24-difluoro-1α,25-dihydroxy vitamin D3 and intermediates obtained therefrom | |
| JP4512490B2 (ja) | ビタミンd類似体、該類似体を含んで成る組成物およびその使用 | |
| JP2003506435A (ja) | 新規ビタミンd類縁体 | |
| JPWO2000024712A1 (ja) | ビタミンd3誘導体およびそれを用いる炎症性呼吸器疾患治療剤 | |
| JPWO2003070716A1 (ja) | ビタミンd3誘導体およびそれを用いる治療剤 | |
| EP0278732A1 (en) | Fluorine-containing vitamin D2 derivatives | |
| JP2004175763A (ja) | 6,7−置換−19−ノル−ビタミンd3誘導体 | |
| HK1086817B (en) | 2- propylidene-19-nor-vitamin d compounds | |
| WO2004067525A1 (ja) | ビタミンd3ラクトン誘導体 | |
| JPH11246520A (ja) | 20−エピ−22−エチル−23,24−デヒドロ−24,24−ジホモビタミンd誘導体及びその合成中間体 | |
| JPWO2000038692A1 (ja) | ビタミンd3誘導体を用いる嚢胞性線維症治療剤 | |
| TW200533643A (en) | 2-propylidene-19-nor-vitamin d compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2000 578283 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 62281/99 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999949355 Country of ref document: EP Ref document number: 1020017004678 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2348235 Country of ref document: CA Kind code of ref document: A Ref document number: 2348235 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09830167 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999949355 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020017004678 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 62281/99 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020017004678 Country of ref document: KR |