WO2000032640A1 - A process that produces polymers - Google Patents
A process that produces polymers Download PDFInfo
- Publication number
- WO2000032640A1 WO2000032640A1 PCT/US1999/027999 US9927999W WO0032640A1 WO 2000032640 A1 WO2000032640 A1 WO 2000032640A1 US 9927999 W US9927999 W US 9927999W WO 0032640 A1 WO0032640 A1 WO 0032640A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- support
- catalyst
- chromium
- process according
- polymerizing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/16—Copolymers of ethene with alpha-alkenes, e.g. EP rubbers
Definitions
- a PROCESS THAT PRODUCES POLYMERS FIELD OF THE INVENTION This invention is related to the field of processes that polymerize ethylene, or that polymerize ethylene and at least one other olefm, to produce a polymer.
- blow molding is useful for producing hollow plastic products.
- a principle advantage of blow molding is its ability to produce hollow shapes without having to join two or more separately molded parts.
- the inventors provide this invention so that such good quality polymers with high ESCR's are more readily obtainable, and readily useable in blow molding applications.
- a process comprises (or optionally “consists essentially of, or “consists of) polymerizing ethylene, or polymerizing ethylene and at least one other olefin, to produce a polymer, wherein said polymerizing is conducted in a polymerization zone, and wherein said polymerizing is conducted using a catalyst and a cocatalyst, and wherein said catalyst comprises chromium on a support, and wherein the amount of said chromium on said support is from about 0.5 to 5 weight percent, and wherein said support comprises silica, in major part, and wherein the amount of titanium in said support is greater than about 3.5 to about 10 weight percent based on the weight of said support, and wherein said support has a surface area from about 400 to about 800 square meters per gram, and wherein said support has a pore volume from about 1.8 to about 4 cubic centimeters per gram, and wherein said catalyst has been activated at a temperature in the range of about 315°C (about
- a polymer comprising the following properties: a density from about 0.94 to about 0.96, a high load melt index from about 5 to about 45 g/10 min., a shear ratio (high load melt index/melt index) from about 150 to about 400, a heterogeneity index from about 15 to about 55, an ESCR condition A greater than about 1000 hours, and ESCR Condition B greater than about 200 hours, a normalized die swell from about 0.8 to about 1.1, a weight swell from about 300 to about 500 percent, and an onset of melt fracture greater than about 2000 sec '1 .
- This polymerization can be carried out in any manner known in the art such as gas phase, solution or slurry polymerization conditions.
- a stirred reactor can be utilized for a batch process, or the reaction can be carried out continuously in a loop reactor.
- This polymerization is conducted in a polymerization zone. It is preferred to conduct this polymerization in a loop reactor. It is more preferred when said polymerization is conducted in a loop reactor under slurry polymerization conditions.
- the preferred diluent for slurry polymerization is isobutane.
- Loop reactors are known in the art, see, for example, U.S. patents 3,248,179; 4,424,341; 4,501,855; and 4,613,484; the entire disclosures of which are hereby incorporated by reference. Especially preferred processes are disclosed in U.S. Patents 4,589,957; 4,737,280; 5,597,892; and 5,575,979 the entire disclosures of which are also hereby incorporated by reference.
- a preferred polymerization technique is that which is referred to as a particle form, or slurry process, wherein the temperature is kept below the temperature at which polymer swells and fouls the reactor.
- Such polymerization techniques are well known in the art and are disclosed, for example, in Norwood, U.S. Pat. No. 3,248,179, the disclosure of which is hereby incorporated by reference.
- Two preferred polymerization methods for the slurry process are those employing a loop reactor of the type disclosed in Norwood and those utilizing a plurality of stirred reactors either in series, parallel or combinations thereof wherein the reaction conditions are different in the different reactors.
- the diluent before it enters the reactor, comprises isobutane. Additionally, before the diluent enters the reactor, the majority of said diluent is isobutane. It is preferred when the diluent contains 60-100, more preferably, 70-100, and most preferably 80-100 weight percent isobutane based on the weight of the diluent before it enters the reactor.
- the polymerization is conducted at a temperature from about 88°C (about 190°F) to about 110°C (about 230°F).
- said polymerizing is conducted at a temperature from about 90.5°C (about 195°F) to about 107.2°C (about 225°F) and it is even more preferred when said polymerizing is conducted at a temperature from 93.3°C (200°F) to 104.4°C (220°F).
- temperatures below about 88°C (about 190°F) the efficiency of the catalyst and the reactor is adversely affected.
- temperatures above about 110°C (about 230°F) the reactor could foul due to the swelling of the polymer.
- the pressure that the polymerization is conducted at is in the range of about 2756 kPa (about 400 psia) to about 5512 kPa (about 800 psia), preferably about 3445 kPa (about 500 psia) to 4823 kPa (about 700 psia).
- the catalyst used in this invention comprises chromium on a support, preferably in the form of chromium oxide on a support.
- the amount of chromium on said support is in the range of about 0.5 to about 5 weight percent, preferably about 1 to about 4 weight percent, and most preferably from 1.5 to 3 weight percent, where such weight percents are based on the weight of the support.
- the support comprises silica and titania. Additionally, such support has silica, as its major component by weight, and titania, as its minor component by weight. It is most preferred when said support consists essentially of silica and titania, with little, if any, impurities. It is even more preferred when the silica and titania are cogelled.
- the amount of titanium in the support is from about 3.5 to about 10 weight percent, preferably about 4 to about 8 percent, and most preferably from 4 to 6 weight percent, where said weight percents are based on the weight of the support.
- the amount of titanium is less than about 3.5 weight percent, the ESCR of the resin produced tends to be too low.
- the amount of titanium is greater than about 10 weight percent, the catalyst becomes thermally unstable and processability of the resin produced tends to be undesirable.
- the support should have a surface area from about 400 to about 800 square meters per gram. It is more preferred when the support has a surface area from about 425 to 700 square meters per gram, and it is most preferred when said support has a surface area from 450 to 650 square meters per gram. Surface areas below about 400 m 2 /g tend to have less activity, less ESCR, and too little die swell, while surface areas above about 800 m 2 /g produces polymers that have a die swell that is too high, an amount of long chain branching that is too low, and possibly, a melt index that is too low.
- the support should have a pore volume from about 1.8 to about 4 cubic centimeters per gram.
- the support has a pore volume from about 1.9 to about 3 cm 3 /g, and it is most preferred when said support has a pore volume from 2 to 2.7 cm 3 / gram. Pore volumes below about 1.8 cm 3 /g produce polymer with low ESCR, while pore volumes above about 4 cm 3 /g are difficult to handle in commercial operations.
- the catalyst should be activated in the presence of an oxidizing ambient (sometime referred to as "atmosphere") at a temperature greater than about 315°C (about 600°F) to about 593°C (about 1100°F). It is even more preferred when the temperature is from about 37PC (about 700°F) to less than 593°C (1100°F), and it is even more preferred when the temperature is from about 482°C (about 900°F) to about 588°C (about 1090°F), and it is most preferred when the temperature is from about 482°C (about 900°F) to 566°C (about 1050°F).
- an oxidizing ambient (sometime referred to as "atmosphere"
- the activity of the catalyst is reduced and the physical properties of the polymer are adversely affected.
- temperatures above about 593°C (about 1100°F) there is a loss of ESCR in the polymer.
- the preferred oxidizing ambient is air. This activation is carried out for a time period of about 1 minute to about 50 hours. This allows a portion of the chromium in a lower valance state to be converted to a hexavalent state.
- the ethylene used should be polymerization grade ethylene.
- the other olefins that can be used are alpha-olefins having from 4 to 12 carbon atoms. Currently, 1-butene, 1-hexene, and 1-octene are the most preferred olefins.
- the catalyst must be used in the presence of a cocatalyst that is an organoboron compound.
- Organoboron compounds, as used in this invention have the following general formula: B(X) 3 . In this formula (X) is a hydrocarbyl having from 1-20 carbon atoms. Currently, it is preferred when (X) is an alkyl having from 1 to 10 carbon atoms.
- (X) is selected from the group consisting of methyl, ethyl, propyl, butyl, and isobutyl.
- Examples of such compounds are as follows: trimethylboron; triethylboron; tripropylboron; tributylboron; and triisobutylboron.
- triethylboron is preferred.
- the amount of organoboron compound to use in this invention is from about 1 to about 15 parts per million by weight, based on the weight of the diluent before it enters the reactor. However, it is preferred when the amount is from about 1 to about 10, and it is most preferred when the amount is from 2 to 4 parts per million.
- the polymer produced needs to have the following properties in order to be a polymer that is good for blow molding applications.
- the density needs to be from about 0.94 to 0.96 grams per cubic centimeter. However, it is preferred when the density is from about 0.95 g/cm 3 to 0.96 g/cm 3 and it is more preferred when the density is from 0.953 g/cm 3 to 0.958 g/cm. This density is determined in accordance with ASTM D1505.
- the high load melt index needs to be from about 5 to about 45 grams per ten minutes. However, it is preferred when the high load melt index is from about 8 g/10 min to about 35 g/10 min. and it is even more preferred when the high load melt index is from 10 g/10 min. to 25 g/10 min. This high load melt index is determined in accordance with ASTM D 1238.
- the shear ratio (HLMI/MI) needs to be from about 150 to about 400. However, it is preferred when the shear ratio is from about 170 to about 350 and it is even more preferred when the shear ratio is from 180-320.
- the heterogeneity index (Mw/Mn) needs to be from about 15 to about 55. However, it is preferred when the heterogeneity index is from 20 to 50 and it is even more preferred when the heterogeneity index is from 25 to 45, and it is most preferred when the Heterogeneity index is from 30 to 40.
- the heterogeneity index was determined by gel permeation chromatography.
- the ESCR Condition A of the polymer is greater than 1000 hours.
- the ESCR Condition B of the polymer is greater than 200 hours, preferably greater than 300 hours. These ESCR's are measured according to ASTM D1693, Conditions A and B. Additionally, the polymer should have a bottle ESCR greater than 700 hours as measured in accordance with the examples below.
- the die swell is an indication of how much the molten polymer tends to flare out as it is extruded from the die.
- the normalized die swell should be between about 0.8 and about 1.1, preferably, about 0.9 and about 1.05, and most preferably, from 0.95 to 1.05. Normalized die swell outside this range leads to poor bottle molding. High die swell results in the parison extending beyond the mold, leading to, for example, "pinch-off or other problems. Low die swell can cause a failure to fill the mold.
- Weight swell is a measure of how much memory the polymer retains as it is extruded. A 300 weight percent swell indicates that the final bottle wall thickness is three times the die gap distance. If the polymer has a characteristically high weight swell, it requires a smaller die gap to produce the required wall thickness, and a smaller gap can restrict polymer flow, and thus machine output.
- the weight swell should be between about 300 and about 500 weight percent, preferably, about 325 and about 475 weight percent, and most preferably, from 350 to 450 weight percent.
- TESTS A "Quantachrome Autosorb-6 Nitrogen Pore Size Distribution Instrument” was used to determined the surface area and pore volume of the supports. This instrument was acquired from the Quantachrome Corporation, Syosset, N.Y.
- Polymer density was determined in grams per cubic centimeter (g/cc) on a compression molded sample, cooled at 15°C per hour, and conditioned for 40 hours at room temperature in accordance with ASTM D1505 and ASTM D1928, procedure C.
- High load melt index (HLMI, g/10 mins) was determined in accordance with ASTM D 1238 at 190°C. with a 21,600 gram weight.
- MI Melt index
- ESCR Environmental Stress Crack Resistance
- ASTM D1693, Conditions A and B The Heterogeneity index was determined using size exclusion chromatography (SEC) analyses that were preformed at 140°C. on a Water, model 150 GPC with a refractive index detector. A solution concentration of 0.25 weight percent in 1,2,4-triclorobenzene was found to give reasonable elution times.
- Polymer resins obtained by this invention are useful for blow molding applications.
- blow molding evaluations were conducted by blowing a 3.8 litre (one gallon) (105.0 + 0.5 gm) bottle on a Uniloy 2016 single head blow molding machine using a 6.4 cm (2.5 inch) diameter die, 20 degree diverging die, 32% accumulator position, 8.5 second blow time, 0.10 second blow delay, 0.75 second pre-blow delay and a 45 degree °F mold temperature.
- a reciprocating screw speed of 45 rpm was used, providing parison extrusion at shear rates greater than 10,000/sec through the die.
- Percent weight swell measures the amount the molten resin expands immediately as it exits the die. It is a measure of the "memory" of the polymer chains as they seek to relax and thus reform the polymer shape. Weight swell is an important parameter as it determines how tight the die gap must be adjusted to provide a constant bottle weight. If a resin has high weight swell, the die gap required will be tighter to make the proper part weight. In so doing, it will require higher stress to push the resin through the die than a lower weight swell resin. Weight swell is defined as the ratio of the final bottle wall thickness to the die gap. Another measurement of swell is die swell or diameter swell. This is the ratio of the parison diameter to the die diameter.
- a typical experiment would consist of extruding a polymer over a range of flow rates (screw RPM) using extruder, adapter, and die temperature settings of 170°C.
- screw RPM flow rate
- the pressure in the adapter, flow rate at various RPM were noted along with the RPM at which the onset of melt fracture occurred.
- Pressure drop versus flow rate data were also collected using the orifice die alone. Using standard calculations for flow through capillary dies, this data was then converted to true shear stress versus shear rate for each resin examined.
- the catalyst contained more than 1 weight percent chromium. In these cases extra chromium was added to the catalyst. This was accomplished by impregnating the catalyst to incipient wetness or somewhat less, with a methanol solution of chromium (III) nitrate containing 0.5 g Cr/100 mis.
- EXAMPLES 1-4 These polymers were prepared in a continuous, particle form process by contacting a catalyst system with monomers, which employed a liquid full 15.2 cm diameter pipe loop reactor having a volume of 23 gallons (87 liters), isobutane as the diluent, and occasionally some hydrogen to regulate the molecular weight of the product. The reactor was operated to have a residence time of 1.25 hours.
- the reactor temperature was varied over a range of 95 °C to 107°C, depending on the particular experiment, and the pressure was four Mpa (580 psi).
- the isobutane feed rate was about 46 liters per hour
- the ethylene feed rate was about 13.5 kg/hr (about 30 lbs/hr)
- the 1-hexene feed rate was varied to control the density of the polymer product.
- Polymer was removed from the reactor at the rate of about 11 kg/hr (about 25 lbs per hour) and recovered in a flash chamber.
- a Vulcan dryer was used to dry the polymer under nitrogen at about 60-80 degrees °C. Ethylene that had been dried over alumina was used as the monomer.
- a commercially available chromium catalyst system was purchased from the W.R. Grace Corporation. This chromium catalyst system was the 964
- Magnapore Catalyst It had a chromium content of about 1 weight percent based on the weight of the chromium catalyst system and about 5 weight percent titanium based on the weight of the total catalyst system.
- extra chromium was added to the 964 Magnapore catalyst. This was accomplished by impregnating the catalyst to incipient wetness or somewhat less, with a me hanol solution of chromium (III) nitrate containing 0.5 g Cr/100 mis.
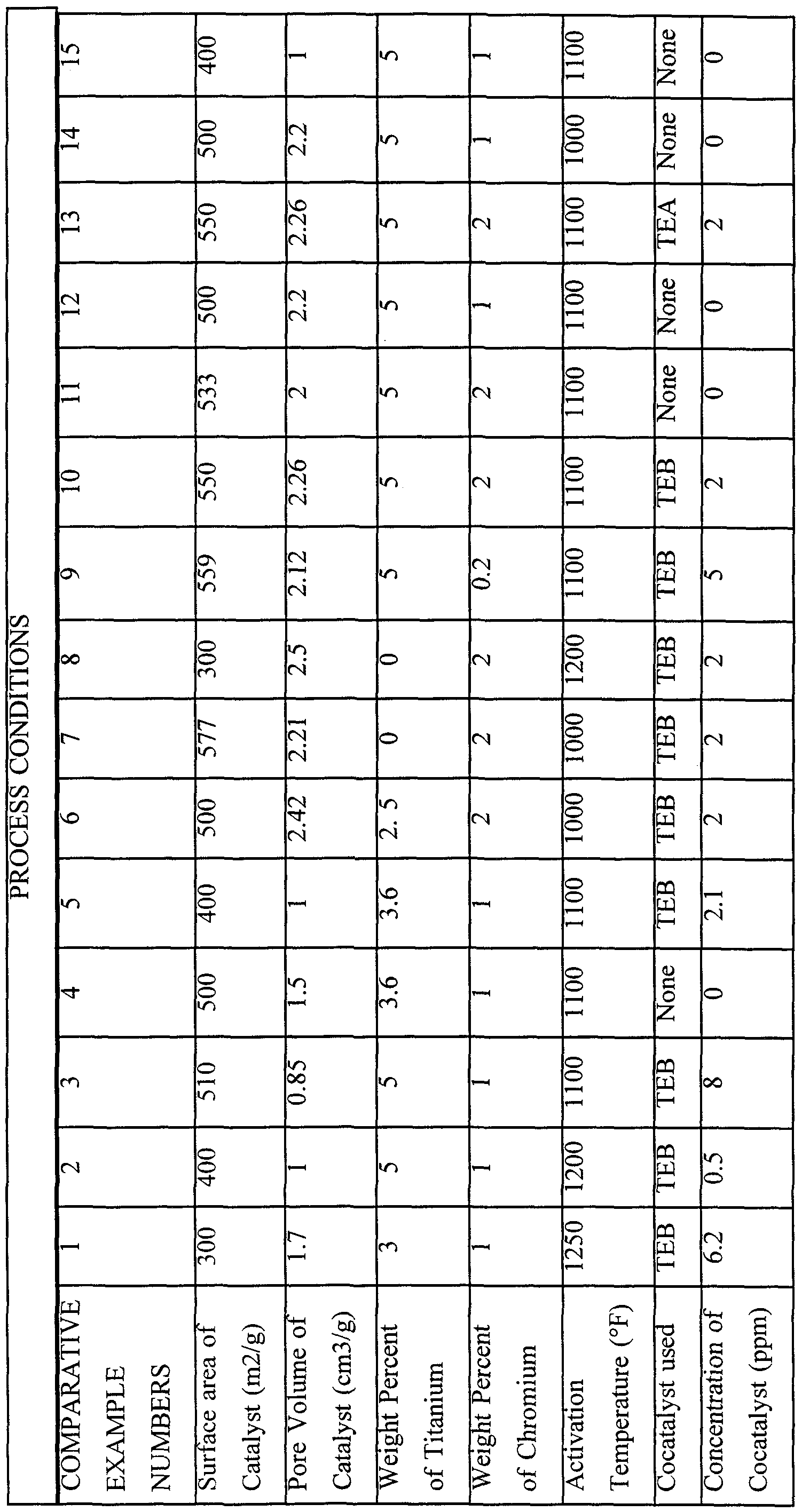
- COMPARATIVE EXAMPLES 1-15 These polymers were prepared in the same reactor and under the same process parameters as described above. Various catalysts and cocatalysts were used in these runs as indicated in the table and descriptions below.
- chromium catalyst system was purchased from the W.R. Grace Corporation. This chromium catalyst system was the 969 catalyst. Titanium was added by first drying the catalyst in dry nitrogen in a fluidized bed at 204-260°C (400-500°F), then lowering the temperature to 121-204°C (250°F-400°F) during which time titanium isopropoxide liquid was added over a period of about one hour. The titanium isopropoxide evaporated while transported by the nitrogen in a 3.17mm (1/8") stainless steel coil which introduced the vapor into the bottom of the bed. After all the titanium had been added, the nitrogen gas stream was replaced by dry air and the temperature was ramped up to the desired activation temperature in the usual fashion. The final catalyst composition was analyzed after activation.
- chromium catalyst system In comparative examples 2 and 15 a commercially available chromium catalyst system was purchased from the W.R. Grace Corporation. This chromium catalyst system was sold under the name of 965 Sylopore. This chromium catalyst was treated with titanium during activation as described earlier. In comparative example 3 a chromium catalyst system was obtained from the W.R. Grace Corporation by spray drying a silica-titania-chromia hydrogel. This chromium catalyst system was called the SD Tergel catalyst.
- chromium catalyst system was purchased from the W.R. Grace Corporation known as HA-30. This chromium catalyst was treated with titanium during activation as described earlier.
- chromium catalyst system was purchased from the W.R. Grace Corporation. This chromium catalyst system was sold under the name of 965 Sylopore.
- chromium catalyst system was purchased from the W.R. Grace Corporation. This chromium catalyst system was the 963 Magnapore catalyst. Extra chromium was added to the catalyst. This was accomplished by impregnating the catalyst to incipient wetness or somewhat less, with a methanol solution of chromium (III) nitrate containing 0.5 g Cr/100 mis.
- a chromium catalyst system was obtained from the W.R. Grace Corporation, designated HPVSA indicating its relatively high pore volume and surface area compared to standard 969MS grades. Extra chromium was added to the catalyst. This was accomplished by impregnating the catalyst to incipient wetness or somewhat less, with a methanol solution of chromium (III) nitrate containing 0.5 g Cr/100 mis.
- chromium catalyst system In comparative examples 10 to 14 a commercially available chromium catalyst system was purchased from the W.R. Grace Corporation. This chromium catalyst system was sold under the name 964 Magnapore. In examples 10, 11, and 13, extra chromium was added to the catalyst. This was accomplished by impregnating the catalyst to incipient wetness or somewhat less, with a methanol solution of chromium (III) nitrate containing 0.5 g Cr/100 mis
- a 964 Magnapore catalyst was produced except that no chromium was used in the process. Extra chromium was added to the catalyst. This was accomplished by impregnating the catalyst to incipient wetness or somewhat less, with a methanol solution of chromium (III) nitrate containing 0.5 g Cr/100 mis.
- Comparative Example 10 is considered a comparative run due to the values obtained for ESCR (B) and normalized die swell, these values are considered to be within the experimental error of these measurements.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
Abstract
Description


Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU20314/00A AU2031400A (en) | 1998-12-01 | 1999-11-29 | A process that produces polymers |
| ES99963987T ES2408596T3 (en) | 1998-12-01 | 1999-11-29 | A process that produces polymers |
| CA002349834A CA2349834C (en) | 1998-12-01 | 1999-11-29 | A process that produces polymers |
| EP99963987.5A EP1192188B1 (en) | 1998-12-01 | 1999-11-29 | A process that produces polymers |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/203,094 | 1998-12-01 | ||
| US09/203,094 US6201077B1 (en) | 1998-12-01 | 1998-12-01 | Process that produces polymers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000032640A1 true WO2000032640A1 (en) | 2000-06-08 |
Family
ID=22752478
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/027999 Ceased WO2000032640A1 (en) | 1998-12-01 | 1999-11-29 | A process that produces polymers |
Country Status (7)
| Country | Link |
|---|---|
| US (3) | US6201077B1 (en) |
| EP (1) | EP1192188B1 (en) |
| KR (1) | KR100625397B1 (en) |
| AU (1) | AU2031400A (en) |
| CA (1) | CA2349834C (en) |
| ES (1) | ES2408596T3 (en) |
| WO (1) | WO2000032640A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2142577A4 (en) * | 2007-04-30 | 2011-08-24 | Fina Technology | Polymerization process providing polyethylene of enhanced optical properties |
| EP2247382A4 (en) * | 2008-01-31 | 2012-05-09 | Fina Technology | Preparation of supported chromium catalyst and polymerization process |
| EP1907430B2 (en) † | 2005-07-27 | 2023-06-28 | Univation Technologies, LLC | Blow molding polyethylene resins |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050101983A1 (en) * | 1998-05-29 | 2005-05-12 | By-Pass,Inc. | Method and apparatus for forming apertures in blood vessels |
| US6201077B1 (en) * | 1998-12-01 | 2001-03-13 | Phillips Petroleum Company | Process that produces polymers |
| US6204346B1 (en) * | 1998-12-17 | 2001-03-20 | Phillips Petroleum Co. | Polymerization process |
| US6569960B2 (en) * | 1999-07-27 | 2003-05-27 | Phillips Petroleum Company | Process to produce polymers |
| CN1217733C (en) * | 1999-12-16 | 2005-09-07 | 菲利浦石油公司 | organometallic compound catalyst |
| US6830174B2 (en) | 2000-08-30 | 2004-12-14 | Cerebral Vascular Applications, Inc. | Medical instrument |
| US7632901B2 (en) * | 2002-06-06 | 2009-12-15 | Exxonmobil Chemical Patents Inc. | Catalyst system and process |
| US7223823B2 (en) * | 2002-06-06 | 2007-05-29 | Exxon Mobil Chemical Patents Inc. | Catalyst system and process |
| US7192901B2 (en) * | 2004-10-27 | 2007-03-20 | Exxonmobil Chemical Patents Inc. | Method of preparing a treated support |
| US7381778B2 (en) | 2002-06-06 | 2008-06-03 | Exxonmobil Chemical Patents Inc. | Method of preparing a treated support |
| US6753387B1 (en) | 2003-03-19 | 2004-06-22 | Chevron Phillips Chemical Company Lp | Temperature controlling system for olefin polymerization reactors |
| US7384885B2 (en) * | 2003-03-31 | 2008-06-10 | Exxonmobil Chemical Patents Inc. | Catalyst activation and resins therefrom |
| US20040192863A1 (en) * | 2003-03-31 | 2004-09-30 | Towles Thomas W. | Catalyst activation method and activated catalyst |
| AU2003284383A1 (en) * | 2003-10-31 | 2005-06-17 | Exxonmobil Chemical Patents Inc. | High molecular weight hdpe resins |
| US20050119426A1 (en) * | 2003-10-31 | 2005-06-02 | Roger Scott T. | High molecular weight HDPE resins |
| ES2450927T3 (en) * | 2004-04-22 | 2014-03-25 | Chevron Phillips Chemical Company Lp | Catalyst systems for producing polymers having wide molecular weight distributions and methods for preparing them |
| US6977235B2 (en) * | 2004-04-22 | 2005-12-20 | Chevron Phillips Chemical Company, Lp | Catalyst systems comprising a calcined chromium catalyst and a non-transition metal cyclopentadienyl cocatalyst |
| US7307133B2 (en) | 2004-04-22 | 2007-12-11 | Chevron Phillips Chemical Company Lp | Polymers having broad molecular weight distributions and methods of making the same |
| WO2005111098A1 (en) * | 2004-05-17 | 2005-11-24 | Sabic Polyethylenes B.V. | A process for the preparation of an ethylene copolymer |
| US7148298B2 (en) * | 2004-06-25 | 2006-12-12 | Chevron Phillips Chemical Company, L.P. | Polymerization catalysts for producing polymers with low levels of long chain branching |
| US20050288461A1 (en) * | 2004-06-25 | 2005-12-29 | Jensen Michael D | Polymerization catalysts for producing polymers with low levels of long chain branching |
| US7642330B2 (en) * | 2005-03-31 | 2010-01-05 | Exxonmobil Chemical Patents Inc. | Method of selecting polyolefins based on rheological properties |
| US7517939B2 (en) | 2006-02-02 | 2009-04-14 | Chevron Phillips Chemical Company, Lp | Polymerization catalysts for producing high molecular weight polymers with low levels of long chain branching |
| US7619047B2 (en) * | 2006-02-22 | 2009-11-17 | Chevron Phillips Chemical Company, Lp | Dual metallocene catalysts for polymerization of bimodal polymers |
| US7576163B2 (en) * | 2006-03-31 | 2009-08-18 | Chevron Phillips Chemical Company, Lp | Polymerization catalysts for producing polymers with low levels of long chain branching |
| IT1392101B1 (en) * | 2008-12-12 | 2012-02-09 | Zuccari Societa A Responsabilita Limitata In Breve Zuccari S R L | COMPOSITION INCLUDING ISOFLAVON |
| US8399580B2 (en) | 2010-08-11 | 2013-03-19 | Chevron Philips Chemical Company Lp | Additives to chromium catalyst mix tank |
| US8937139B2 (en) | 2012-10-25 | 2015-01-20 | Chevron Phillips Chemical Company Lp | Catalyst compositions and methods of making and using same |
| US8895679B2 (en) | 2012-10-25 | 2014-11-25 | Chevron Phillips Chemical Company Lp | Catalyst compositions and methods of making and using same |
| WO2014071119A1 (en) | 2012-11-01 | 2014-05-08 | Univation Technologies, Llc | Mixed compatible ziegler-natta/chromium catalysts for improved polymer products |
| US8877672B2 (en) | 2013-01-29 | 2014-11-04 | Chevron Phillips Chemical Company Lp | Catalyst compositions and methods of making and using same |
| US9034991B2 (en) | 2013-01-29 | 2015-05-19 | Chevron Phillips Chemical Company Lp | Polymer compositions and methods of making and using same |
| CN107405321A (en) | 2015-01-23 | 2017-11-28 | 坦普尔大学 | Application of short-chain fatty acids in cancer prevention |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5599887A (en) * | 1993-12-28 | 1997-02-04 | Phillips Petroleum Company | Chromium catalyst compositions and ethylene polymerization processes therewith |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3887494A (en) | 1970-11-12 | 1975-06-03 | Phillips Petroleum Co | Olefin polymerization catalyst |
| US4312967A (en) | 1980-02-06 | 1982-01-26 | Phillips Petroleum Co. | Polymerization catalyst and process |
| US5115053A (en) * | 1988-07-25 | 1992-05-19 | Phillips Petroleum Company | Twice-aged porous inorganic oxides, catalysts, and polymerization processes |
| US5115068A (en) | 1988-09-26 | 1992-05-19 | Phillips Petroleum Company | High strength linear, low density ethylene copolymer |
| CA2036929A1 (en) * | 1990-05-14 | 1991-11-15 | Donald R. Witt | Polymerization catalysts and processes |
| US5071927A (en) | 1990-10-09 | 1991-12-10 | Phillips Petroleum Company | High-temperature slurry polymerization of ethylene |
| US5274056A (en) | 1992-01-31 | 1993-12-28 | Phillips Petroleum Company | Linear, very low density polyethylene polymerization process and products thereof |
| EP0857736A1 (en) * | 1997-02-07 | 1998-08-12 | Fina Research S.A. | Production of polyethylene for blow moulding |
| AU5630198A (en) * | 1997-03-07 | 1998-09-10 | Phillips Petroleum Company | A process for polymerizing olefins |
| EP0882740A1 (en) * | 1997-06-06 | 1998-12-09 | Fina Research S.A. | Titanated chromium-based catalysts to produce polyethylene exhibiting better environmental stress crack resistance |
| US6201077B1 (en) * | 1998-12-01 | 2001-03-13 | Phillips Petroleum Company | Process that produces polymers |
-
1998
- 1998-12-01 US US09/203,094 patent/US6201077B1/en not_active Expired - Lifetime
-
1999
- 1999-11-29 EP EP99963987.5A patent/EP1192188B1/en not_active Expired - Lifetime
- 1999-11-29 CA CA002349834A patent/CA2349834C/en not_active Expired - Fee Related
- 1999-11-29 ES ES99963987T patent/ES2408596T3/en not_active Expired - Lifetime
- 1999-11-29 AU AU20314/00A patent/AU2031400A/en not_active Abandoned
- 1999-11-29 WO PCT/US1999/027999 patent/WO2000032640A1/en not_active Ceased
- 1999-11-29 KR KR1020017006763A patent/KR100625397B1/en not_active Expired - Fee Related
-
2001
- 2001-01-31 US US09/773,294 patent/US6642324B2/en not_active Expired - Lifetime
-
2003
- 2003-07-29 US US10/629,358 patent/US20040024160A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5599887A (en) * | 1993-12-28 | 1997-02-04 | Phillips Petroleum Company | Chromium catalyst compositions and ethylene polymerization processes therewith |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1907430B2 (en) † | 2005-07-27 | 2023-06-28 | Univation Technologies, LLC | Blow molding polyethylene resins |
| EP2142577A4 (en) * | 2007-04-30 | 2011-08-24 | Fina Technology | Polymerization process providing polyethylene of enhanced optical properties |
| EP2247382A4 (en) * | 2008-01-31 | 2012-05-09 | Fina Technology | Preparation of supported chromium catalyst and polymerization process |
Also Published As
| Publication number | Publication date |
|---|---|
| US6642324B2 (en) | 2003-11-04 |
| US20010004663A1 (en) | 2001-06-21 |
| US6201077B1 (en) | 2001-03-13 |
| KR100625397B1 (en) | 2006-09-19 |
| KR20010080634A (en) | 2001-08-22 |
| AU2031400A (en) | 2000-06-19 |
| CA2349834A1 (en) | 2000-06-08 |
| EP1192188B1 (en) | 2013-04-10 |
| CA2349834C (en) | 2005-08-30 |
| EP1192188A1 (en) | 2002-04-03 |
| US20040024160A1 (en) | 2004-02-05 |
| EP1192188A4 (en) | 2002-07-24 |
| ES2408596T3 (en) | 2013-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6201077B1 (en) | Process that produces polymers | |
| US6657023B2 (en) | Polymerization process | |
| EP1072618B1 (en) | A process to produce polymers | |
| CA2390617C (en) | Polymerization catalyst comprising chromium and silica-titania and process | |
| AU2011202295B2 (en) | Chromium based polymerization catalyst, the method to prepare it and polymers prepared therewith | |
| JP2009533511A (en) | Chromium-based catalyst | |
| CN1957003B (en) | Chromium-based polymerization catalysts, processes for their preparation and polymers prepared therewith | |
| EP1040131A1 (en) | Processes for producing copolymers of ethylene and at least one other olefin | |
| AU2012201734B2 (en) | Chromium based polymerization catalyst, the method to prepare it and polymers prepared therewith | |
| HK1099320B (en) | Chromium based polymerization catalyst, the method to prepare it and polymers prepared therewith |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2349834 Country of ref document: CA Ref country code: CA Ref document number: 2349834 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020017006763 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999963987 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020017006763 Country of ref document: KR |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999963987 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020017006763 Country of ref document: KR |