Dérivés d'ascididémine et leurs applications thérapeutiques
La présente invention concerne des compositions pharmaceutiques à base de composés polyaromatiques utiles notamment comme médicaments antitumoraux.
En 1999, les traitements cytotoxiques (chimiothérapie) utilisés pour réduire la taille des tumeurs cancéreuses, contenir le développement du processus tumoral voire, dans trop peu de cas encore, supprimer les amas de cellules cancéreuses et le risque de métastases, combinent des substances chimiques d'introduction récente avec d'autres qui sont utilisées depuis quelques dizaines d'années. Par exemple, au 5-fluorouracil (5-FU), reconnu depuis près de 40 ans comme l'un des traitements les plus actifs du cancer colo-rectal, peut être substitué l'un ou l'autre des inhibiteurs spécifiques de la topoisomérase I (irinotécan ou topotécan) lorsque la tumeur n'est plus sensible au 5-FU. Plus généralement, l'arsenal thérapeutique disponible pour traiter les tumeurs colo-rectales va également s'enrichir avec la mise à disposition de l'oxaliplatine, des nouveaux "donneurs" in situ de 5-FU ou des inhibiteurs sélectifs de la thymidylate synthétase. Cette coexistence ne se limite pas au traitement des cancers colo-rectaux puisque, également, la chimiothérapie des cancers du sein, de l'ovaire, du poumon fait maintenant largement appel à la famille des dérivés des taxanes (paclitaxel, docetaxel). Le besoin de traitements plus efficaces et mieux tolérés, améliorant ainsi la survie et la qualité de vie des malades est impérieux puisque, en prenant toujours l'exemple des tumeurs colo-rectales, il a été estimé (S.L. Parker, T. Tong, S. Bolden et al., CA Cancer J. Clin., 1997) que, rien qu'aux Etats-Unis plus de 131 000 nouveaux cas ont été diagnostiqués en 1997, dont 54 000 étaient responsables du décès des patients. C'est la connaissance de cette situation qui a incité les inventeurs à s'intéresser à une famille de composés polyaromatiques encore peu étudiés, identifiés chez des Ascidies de mers chaudes, pour développer une chimie médicinale originale destinée à sélectionner des composés synthétiques issus d'un travail de conception/modulation chimique et doués d'une activité cytotoxique significative au plan thérapeutique. Les mers et les océans qui couvrent plus de 70 % de la surface du globe, hébergent des plantes marines et des éponges dont l'étude pharmacognosique systématique progressive montre que ces espèces vivantes peuvent contenir des alcaloïdes complexes présentant des propriétés pharmacologiques intéressantes.
Par exemple, les éponges Cryptotheca crypta et Halichondria okadai font l'objet d'études approfondies depuis la découverte de la présence, dans leurs cellules, de cytarabine ou d'halichondrine B. Il en est de même pour la famille des tuniciers, depuis l'isolement de l'aplidine du tunicier >4p//d//jm albicans qui vit dans les îles Baléares (Espagne). Des alcaloïdes à structure tétrahydroisoquinolone ont été isolés de l'ascidie Ecteinascidia turbinata. Parmi ceux-ci, l'ecteinascidin- 743 fait l'objet de travaux pré-cliniques approfondis (E. Igbicka et al., NCI-EORTC symposium, 1998; Abst. 130 p.34), ainsi que d'essais cliniques destinés à définir son potentiel thérapeutique comme médicament anticancéreux (A. Bowman et al., NCI-EORTC symposium, 1998; Abst. 452 p.118 ; M.Villanova-Calero et al., NCI-EORTC symposium, 1998; Abst. 453 p.118 ; M.J.X. Hillebrand et a , NCI- EORTC symposium, 1998; Abst. 455 p.119; E. Citkovic et a , NCI-EORTC symposium, 1998; Abst. 456 p.119). De nouveaux dérivés d'acridines pentacycliques font également l'objet de travaux de pharmaco-chimie (D.J. Hagan et al., J. Chem. Soc, Perkin Transi, 1997; 1 : 2739-2746).
Autre alcaloïde naturel d'origine marine, l'ascididémine a été extraite du tunicier Didemnum sp. (J. Kobayashi et a , Tehahedron, lett. 1988 ; 29 : 1177- 80) et de l'ascidie Cystodytes dellechiajei (I. Bonnard et al, Anti-cancer Drug design 1995 ; 10 : 333-46). L'ascididémine possède des propriétés antiproliferatives mises en évidence sur le modèle de leucémie murine (lignées P388 ou L1210) et décrites par F. Schmitz et al. (J. Org. Chem. 1991 ; 56 : 804- 8), B. Lindsay et al. (Bioorg. Med. Chem. Lett. 1995 ; 5 : 739-42) et J. Kobayashi et al. (Tehahedron lett. 1988 ; 29 : 1177-80) et sur le modèle de leucémie humaine décrites par I. Bonnard et al. (Anti-cancer Drug design 1995 ; 10 : 333- 46). Plusieurs voies de synthèse de l'ascididémine ont été rapportées par différents auteurs : F. Bracher et al. (Heterocycles 1989 ; 29 : 2093-95), C.J. Moody et al. (Tetrahedron Lett. 1992 ; 48 : 3589-602) et G. Gellerman et al. (Synthesis 1994 ; 239-41 ).
On peut également citer la 2-bromoleptoclinidone (selon la dénomination de S.J. Bloor ef al. 1987) et isolée de l'ascidie Leptoclinides sp. par S.J. Bloor et al. (J. Ann. Chem. Soc. 1987 ; 109 : 6134-6) et synthétisée par F. Bracher ef al. (Heterocycles 1989 ; 29 : 2093-95) puis par M.E. Jung ét al. (Heterocycles 1994 ; 39 ; 2 : 767-778). La 2-bromoleptoclinidone présente une cytoxicité sur le modèle
cellulaire de leucémie avec une DE 50 de 0,4 μg/ml. Les propriétés cytotoxiques ont été confirmées, par F. Bracher (Pharmazie 1997 ; 52 : 57-60) aussi bien in vitro - sur soixante lignées cellulaires tumorales en culture - que in vivo sur les modèles de xénogreffes de lignées cellulaires tumorales humaines (tumeurs du colon SW-620 et HTC116, tumeur rénale A498 et mélanome LOX IM VI) implantées chez des souris.
D'autres composés dérivés de l'ascididémine tels que la 11-hydroxy ascididémine, la 11-méthoxy ascididémine, les 11-phényle et 11- nitrophényle ascididémines, les 1-nitro et 3-nitro ascididémines et la néocalliactine ont été décrits au plan chimique (selon la numérotation de S.J. Bloor ef al. 1987) par différentes équipes telles que celles de F.J. Schmitz (J. Org. Chem. 1991 ; 56 : 804-8) et de Y. Kitahara et al. (Heterocycles 1993 ; 36 : 943-46 ; Tetrahedron Lett. 1997 ; 53, 17029-38), G. Gellerman ef al. (Tetrahedron lett. 1993 ; 34 : 1827-30), S. Nakahara ef al (Heterocycles 1993; 36 : 1139-44), I. Spector ef al. (US Patent Number : 5,432,172, Jul. 11 , 1995).
La présente invention a pour objet une composition pharmaceutique comprenant une quantité efficace d'un composé choisi parmi les composés de formules générales I et la suivantes :
dans lesquelles :
- X est choisi parmi l'oxygène, le groupe =NH, le groupe = N-OH,
- Ri est choisi parmi l'hydrogène, les halogènes, le groupe nitro et les groupes -NRβRg dans lesquels Rδ et Rg sont choisis indépendamment l'un de l'autre parmi l'hydrogène et les groupes alkyle (C1-C4),
- R2 est choisi parmi l'hydrogène, les halogènes,
- R3 est choisi parmi l'hydrogène, les halogènes, les groupes alkyle (C1-C4), les groupes alcoxy (C-|-C6), un groupe guanidino, les groupes -NR10R1 1 dans lesquels R-10 et Ru sont choisis, indépendamment l'un de l'autre, parmi l'hydrogène, les groupes alkyle (C1-C4), phénylalkyle (C1-C4), et les groupes - (CH2)n-Y avec Y est choisi parmi les halogènes et les groupes CN, -CH(0-Et)2, alcoxy(Cι-C6), -O-(CH2)2-N(CH3)2, -N(CH3)2 et n = 1 à 3,
- R4 est choisi parmi l'hydrogène, les halogènes, les groupes nitro, et les groupes -NR-12R13 dans lesquels Rη2 et R13 sont choisis indépendamment l'un de l'autre parmi l'hydrogène et les groupes alkyle (C1-C4),
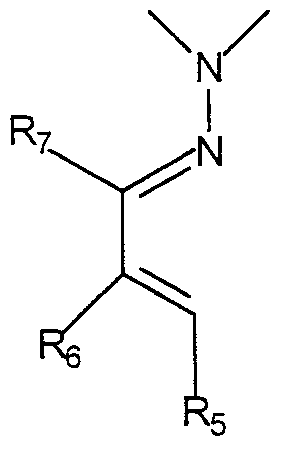
- R5, R6 et R7 sont choisis parmi : l'hydrogène, un atome d'halogène, les groupes alkyle en C1-C6, hydroxyle, alcoxy en C -C6, alcoxy (C-j-
C6)alkyle(Ci-C6), alkyl(Cι-C4) carbonyloxyalkyle(Cι-C4), -CHO, -COOH, -CN, - CO2R14. -CONHR14, -CONR14R15, les groupes -NHCOR14 et -NR14R15, dans lesquels R14 et R15 sont choisis indépendamment l'un de l'autre parmi l'hydrogène, les groupes alkyle (Cι-C6),-phényle-CO-CH3 et -CH2-CH2-
N(CH3)2, les groupes -phényle-CO-CH3 ou -phényle-CO-CH=CH-N(CH3)2, morpholino, nitro, SO3H, les groupes :
-CH2 - N -COOR16 , -CH2 - N- COOR16 , I I
CH2-COOR17 CH2-Ar
R16 et R17 étant choisis parmi les groupes alkyle en C-j-Cô et Ar étant un groupe aryle en C6-C*i4ι à l'exclusion des composés de formule I dans lesquels existe la combinaison: X = O, et, ou bien : R-j , R2, R3, R4, R5, RQ, R7 = H, ou bien : R-j , R3, R4, R5, RQ, RJ = H et R2 = Br,
et à l'exclusion du composé de formule la dans lequel existe la combinaison X = O et Ri , R2> R3- R4. R5- R6. R7 = H. et les sels d'addition de ces composés avec des acides pharmaceutiquement acceptables. La présente invention a plus particulièrement pour objet une composition pharmaceutique comprenant une quantité efficace d'un composé choisi parmi les composés de formule I dans laquelle :
- X est choisi parmi l'oxygène, le groupe =NH, le groupe = N-OH,
- Ri est choisi parmi l'hydrogène, les halogènes, le groupe nitro et les groupes -NR8R9 dans lesquels RQ et Rg sont choisis indépendamment l'un de l'autre parmi l'hydrogène et les groupes alkyle (C1-C4),
- R2 est choisi parmi l'hydrogène, les halogènes,
- R3 est choisi parmi l'hydrogène, les halogènes, les groupes alkyle (C1-C4), les groupes alcoxy (C1-C6), un groupe guanidino, les groupes -NR10R11 dans lesquels R10 et Ru sont choisis, indépendamment l'un de l'autre, parmi l'hydrogène, les groupes alkyle (C1-C4), phénylalkyle (C1-C4), -(CH2)2-N(CH3)2, et -(CH2)2-O-(CH2)2-N(CH3)2,
- R4 est choisi parmi l'hydrogène, les halogènes, les groupes nitro, et les groupes -NR12R 3 dans lesquels R 2 et R13 sont choisis indépendamment l'un de l'autre parmi l'hydrogène et les groupes alkyle (C1-C4),
- R5, R6 et R7 sont choisis parmi : l'hydrogène, un atome d'halogène, les groupes alkyle en C1-C6, hydroxyle, alcoxy en C1-C6, -CHO, - COOH, -CN, -CO2R14, -CONHR14, -CONR14R15, les groupes -NHCOR14 et - NRi4Ri5_ dans lesquels R14 et R15 sont choisis indépendamment l'un de l'autre parmi l'hydrogène, les groupes alkyle (C1-C6) et -CH2-CH2-N(CH3)2, les groupes -phényle-CO-CH3 ou -phényle-CO-CH=CH-N(CH3)2, morpholino, nitro, SO3H,
les groupes :
-CH2 - N - COOR16 , -CH2 - N- COOR16 , CH2- COOR17 CH2-Ar
R16 et R 7 étant choisis parmi les groupes alkyle en C1-C6 et Ar étant un groupe aryle en C6-C14, à l'exclusion des composés dans lesquels X = O et, ou bien : R→\ , R2, R3, R4, R5, RQ, R7 = H, ou bien : Ri , R3, R4, R5, RQ, RJ = H et R2 = Br, et les sels d'addition de ces composés avec des acides pharmaceutiquement acceptables.
La présente invention a plus particulièrement pour objet une composition pharmaceutique comprenant une quantité efficace d'un composé choisi parmi les composés de formule I dans laquelle :
- X représente l'oxygène,
- Ri est choisi parmi l'hydrogène et le groupe amino,
- R2 est choisi parmi l'hydrogène et les halogènes, - R3 est choisi parmi l'hydrogène, les halogènes, les groupes alkyle (C1-C4), les groupes alcoxy (C1 -C6), un groupe guanidino, les groupes -NR10R1 1 dans lesquels R10 et R 1 sont choisis, indépendamment l'un de l'autre, parmi l'hydrogène, les groupes méthyle, phénylalkyle (C1 -C4), -(CH2)2-N(CH3)2, -
(CH2)2-0-(CH2)2-N(CH3)2 ; - R4 est choisi parmi l'hydrogène, les halogènes et le groupe nitro et amino,
- R5, R6 , R7 représentent un hydrogène, à l'exclusion des composés dans lesquels R→\ , R2, R3, R4, R5, RQ, RJ = H, ou
Ri , R3, R4, R5, R6, R7 = H et R2 = Br, et leurs sels d'addition de ces composés avec des acides pharmaceutiquement acceptables.
Dans sa forme préférée, la présente invention a plus particulièrement pour objet une composition pharmaceutique comprenant une quantité efficace d'un composé choisi parmi les composés de formules I et la dans lesquelles :
- X représente l'oxygène, - Ri est choisi parmi l'hydrogène et le groupe amino,
- R2 est choisi parmi l'hydrogène et les halogènes,
- R3 est choisi parmi l'hydrogène, les halogènes, les groupes alkyle (C1-C4), les groupes alcoxy (C1 -C6), un groupe guanidino, les groupes -NR10R11 dans lesquels R10 et Ru sont choisis, indépendamment l'un de l'autre, parmi l'hydrogène, les groupes méthyle, phénylalkyle (C1 -C4) et les groupes -(CH2)n-Y avec Y est choisi parmi les halogènes et les groupes CN, -CH(O-Et)2, alcoxy(Cι- C6), -O-(CH2)2-N(CH3)2, -N(CH3)2 et n = 1 à 3,
- R4 est choisi parmi l'hydrogène, les halogènes et les groupes nitro et amino,
- R5 est choisi parmi un hydrogène, un halogène ou un groupe méthoxy, - R6, R7 sont choisis parmi l'hydrogène, les groupes alcoxy(Cι-C6), alcoxy
(Cι-C6)alkyle(Cι -C6) et -CH2OCOCH3, à l'exclusion des composés de formule I dans lesquels R , R2, R3, R4, R5, RQ, RJ = H, ou Ri , R3, R4, R5, Re, R7 = H et R2 = Br, et du composé de formule la dans lequel Ri , R2, R3, R4, R5, RQ, RJ = H. et les sels d'addition de ces composés avec des acides pharmaceutiquement acceptables.
La présente invention a également pour objet les composés de formule I telle que définie précédemment à l'exclusion des composés dans lesquels X=O, et, ou bien Ri , R2, R3, R4, R5, RQ, RJ = H, ou bien Ri , R3, R4, R5, RQ, RJ = H et R2 = Br, ou bien Ri , R2, R4, R5, R6, R7 = H et R3,= OCH3, ou bien Ri , R2, R3, R4, RQ, RJ = H et R5 = OH ou OCH3, ou bien Ri ,= NO2 et R2, R3, R4, RQ, RJ = H. et les sels d'addition de ces composés avec des acides pharmaceutiquement acceptables.
La présente invention a également pour objet les composés de formule la telle que définie précédemment à l'exclusion du composé dans lequel X=0 et R , R2,
R3, R , R5, R6, R7 = H, et les sels d'addition de ces composés avec des acides pharmaceutiquement acceptables.
Les "sels d'addition avec des acides pharmaceutiquement acceptables" désignent les sels qui donnent les propriétés biologiques des bases libres, sans avoir d'effet indésirable. Ces sels peuvent être notamment ceux formés avec des acides minéraux, tels que l'acide chlorhyd que, l'acide bromhydnque, l'acide sulfurique, l'acide nitrique, l'acide phosphorique; des sels métalliques acides, tels que l'orthophosphate disodique et le sulfate monopotassique, et des acides organiques.
De manière générale, les composés de formule I sont obtenus selon le schéma réactionnel général décrit par F. Bracher et al (Heterocycles 1989 ; 29 : 2093-95) pour l'ascididémine. Selon ce schéma, les composés sont préparés par amination oxydative d'une 5,8-quinone substituée avec une ortho- aminoacétophénone substituée, suivi de la cyclisation de la diaryl aminé obtenue (composés de formule II) en quinone tétracyclique intermédiaire (composés de formule III). L'énamine formée par réaction du composé de formule III avec le diéthyl acétal du diméthylformamide conduit au dérivé final par cyclisation :
Schéma I
Formule III
L'ascididémine (ou 9-H-quino[4,3,2-de][1 ,10]phénanthrolin-9-one) a été préparée selon le procédé décrit par F. Bracher et al. (Heterocycles 1989 ; 29 : 2093-95) et est référencée, dans le présent document, sous le numéro CRL 8274.
Certains composés peuvent être préparés directement à partir de l'ascididémine ou à partir d'un composé de formule I utilisé comme intermédiaire de synthèse.
C'est ainsi en particulier que les composés de formule I dans lesquels R3 est un groupe -NR10R11 , avec R10 et/ou R11 différents de l'hydrogène, peuvent
être obtenus à partir d'un composé de formule I dans laquelle R3 est un groupe - NH2,
De même, les composés de formule la peuvent être obtenus selon le schéma réactionnel général II. Selon ce schéma, les composés sont préparés par condensation d'un acide chlorobenzoïque substitué et de la diméthoxyaniline pour former les composés de formule lia. Après transformation de la fonction acide en méthylcétone, cyclisation puis oxydation, une quinone tricyclique intermédiaire (composé de formule llla) est obtenue. Une cycloaddition de Diels Aider avec un 1-azadiène conduit à la formation d'une quinone tétracyclique (composé de formule IVa). L'addition du diéthylacétal de diméthylformamide sur cette quinone conduit à un intermédiaire énamine qui se cyclise, en présence de chlorure d'ammonium, en composé final de formule la.
Schéma II
Formule lia
DMFDEA, NH4CI
Formule la
Certains composés peuvent être préparés directement à partir de l'isomère de l'ascididémine appelé 9-H-quino[4,3,2-de][1 ,7]phénanthrolin-9-one, ou à partir d'un composé de formule la utiiisé comme intermédiaire de synthèse.
Certains composés peuvent être préparés directement à partir de l'isomère de l'ascididémine appelé 9-H-quino[4,3,2-de][1 ,7]phénanthrolin-9-one, ou à partir d'un composé de formule la utilisé comme intermédiaire de synthèse. C'est ainsi en particulier que les composés de formule la dans lesquels R3 est un groupe -NR10R11 , avec R10 et/ou R11 différents de l'hydrogène, peuvent être obtenus à partir d'un composé de formule la dans laquelle R3 est un groupe -NH2.
A - Préparation des produits intermédiaires de formule II (Schéma I)
A-1 - Synthèse de la 6-(2-acétyl-4-méthyl-phénylamino)-quinoline-5,8-dione (intermédiaire Ai) (CRL 8322)
On additionne lentement une solution de quinoline-5,8-dione (0,215 g, 1 ,35 mmol) dans 12 ml d'éthanol, à une solution de chlorure de cérium (1g, 2,7 mmol) et de 5-méthyl-2 amino acétophénone (0,402 g, 2,7 mmol) dans 5 ml d'éthanol. Le milieu réactionnel (rouge) est laissé sous agitation à température ambiante une nuit. On hydrolyse par 30 ml d'une solution aqueuse d'acide acétique à 10 % et on extrait 4 fois au chloroforme. Les phases organiques sont séchées sur MgSθ4 puis évaporées. Le brut obtenu est purifié par flash-chromatographie sur colonne de silice (CH2Cl2/MeOH 95 : 5) pour donner 0,405 g du composé tricyclique attendu sous forme de poudre :
• Rrendement = 98 %.
• RMN 1 H (CDCI3) : 2,42 (s, 3H), 2,77 (s, 3H), 6,86 (s, 1 H), 7,38 (dd, 1 H, J = 8 et 1 .6 Hz), 7,52 (d, 1 H, J = 8 Hz), 7,61 (dd, 1 H, J = 5,2 et 7,6 Hz), 7,74 (d, 1 H, J = 1 .6 Hz), 8,46 (dd, 1 H, J = 7,6 et 5,2 Hz), 9,02 (dd, 1 H, J = 2 et 5,2 Hz), 1 1 , 18 (s, 1 H).
A-2 - Synthèse de la 6-(2-acétyl-4-chloro-phénylamino)-quinoline-5,8-dione (Intermédiaire A2)
Préparation selon le procédé décrit dans le chapitre A-1 : quinoline-5,8-dione (0,1 88 g, 1 ,18 mmol), chlorure de cérium (0,88 g, 2,36 mmol), 5-chloro-2-
aminoacétophénone (0,4 g, 3,14 mmol), éthanol (10 + 4 ml), acide acétique (25 ml). On obtient 0,3 g de poudre rouge :
• Rendement = 78 %
• RMN - (CDCI3) : 2, 65 (s, 3H), 6,84 (s, 1 H), 7,52 (dd, 1 H, J = 8.8 Hz), 7,57 (d, 1 H, J = 8,8 Hz), 7,63 (dd, 1 H, J = 8 et 4,4 Hz), 7,89 (d, 1 H, J = 2,4 Hz), 8,46
(dd, 1 H, J = 0,8 et 8 Hz), 9,02 (dd, 1 H, J = 2 et 5,2 Hz), 11 ,18 (s, 1 H).
• RMN 13c (CDCI3) : 28,5 ; 107,36 ; 121 ,86 ; 126,69 ; 126,85 ; 127,49 ; 128,39 ; 132,10 ; 134,06 ; 134,75 ; 138,36 ; 143,29 ; 148,32 ; 155,22 ; 181 ,28 ; 182,63 ; 200,39.
A-3 - Synthèse de la 6-(2-acétyl-4-benzylamino-phénylamino)-quinoline-5,8- dione (Intermédiaire A3)
Préparation selon le procédé décrit dans le chapitre A-1 : quinoline-5,8-dione
(0,250 g, 1 ,57 mmol), chlorure de cérium (0,77 g, 3,14 mmol), 5-benzylamino-2 aminoacétophénone (0,603 g, 3,14 mmol), éthanol (15 + 7 ml), acide acétique
(35 ml). On obtient 0,56 g de poudre rouge :
• Rendement = 91 %.
• RMN 1 H (CDCI3) : 2,54 (s, 3H), 4,38 (s, 2H), 6,70 (s, 1 H), 6,83 (dd, 1 H, J = 9.6 et 3,2 Hz), 7,08 (d, 1 H, J = 3.2 Hz), 7,30-7,37 (m, 5H), 7,43 (d, 1 H, J = 9,6 Hz), 7,58 (dd, 1 H, J = 7,6 et 4,8 Hz), 8,43 (dd, 1 H, J = 7,6 et 2 Hz), 9,03 (dd, 1 H, J = 2 et 4,8 Hz), 10,67 (s, 1 H).
A-4 - Synthèse de la 6-(2-acétyl-5-bromo-phénylamino)-quinoline- 5,8 dione (Intermédiaire A4) (CRL8268) Préparation selon le procédé décrit par F. Bracher, Liebigs Ann. Chem. 1990,
205-206.
A-5 - Synthèse de la 6-(2-acétyl-4-diméthylamino-phénylamino)-quinoline- 5,8-dione (Intermédiaire A5) Préparation selon le procédé décrit dans le chapitre A-1 : quinoline-5,8-dione
(0,36 g, 2,26 mmol), chlorure de cérium (1 ,67 g, 4,49 mmol), 5-diméthylamino-2
aminoacétophénone (0,8 g, 4,49 mmol), éthanol (20 + 10 ml), acide acétique (50 ml). On obtient 1 ,26 g de poudre rouge :
• Rendement = 84 %.
• RMN H (CDCI3) : 2,85 (s, 3H), 3,12 (s, 6H), 6,72 (s, 1 H) 6,90 (dd, 1 H, J = 2.8 et 9.2 Hz), 7,15 (d, 1 H, J = 2,8 Hz), 7,49 (d, 1H, J = 9,2 Hz), 7,58 (dd, 1H, J =
8Hz et 4,4 Hz), 8,43 (dd, 1 H, J = 1 ,6 et 8 Hz), 9,00 (dd, 1 H, J = 1.6 et 4,4 Hz), 10,69 (s, 1 H).
A-6 - Synthèse de la 6-(2-acétyl-4-méthoxy-phénylamino)-quinoline-5,8- dione (Intermédiaire Aβ)
Préparation selon le procédé décrit dans le chapitre A-1 : quinoline-5,8-dione (3,51 g, 22,08 mmol), chlorure de cérium (16,4 g, 44,03 mmol), 5-méthoxy-2- amino acétophénone (7,29 g, 44,18 mmol), éthanol (200 + 90 ml), acide acétique (500 ml). On obtient 4,25 g de poudre rouge : • Rendement = 60 %.
• RMN 1 H (CDCI3) : 2,65 (s, 3H), 3,87 (s, 3H), 6,76 (s, 1 H) 7,12 (dd, 1 H, J =
2,8 et 8,8 Hz), 7,42 (d, 1 H, J = 2,8 Hz), 7,55 (d, 1 H, J= 8,8 Hz), 7,61 (dd, 1 H, J =
7,6 et 4,4 Hz), 8,45 (dd, 1 H, J = 1 ,6 et 7,6 Hz), 9,01 (dd, 1 H, J = 1 ,6 et 4,4 Hz),
10,80 (s, 1 H).
A-7 - Synthèse de la 4,6-bis(2-acétylanilino)-quinoline-5,8-dione (Intermédiaire Aγ) Préparation selon le procédé décrit dans le chapitre A-1 : 4-chloro-quinoline-
5,8-dione (3,5 g, 18 mmol), chlorure de cérium (13,5 g, 36,24 mmol), 2- aminoacétophénone (4,4 ml, 36 mmol), éthanol (160 + 70 ml), acide acétique
(400 ml). On obtient 2,32 g de poudre rouge :
• Rendement = 30 %.
• RMN 1 H (CDCI3) : 2,69 (s, 3H), 2,72 (s, 3H), 6,85 (s, 1 H), 7,18 (ddd, 1 H, J =
7,6 et 7,6 et 0,8 Hz), 7,28 (m, 1 H), 7,30 (d, 1 H, J = 6,4 Hz), 7,54-7,59 (m, 3H), 7,63 (d, 1 H, J = 7,6 Hz), 7,91 (dd, 1 H, J = 1 ,6 et 8,4 Hz), 7,94 (dd, 1 H, J = 1 ,2 et 8,4 Hz), 8,47 (d, 1 H, J = 6,4 Hz), 11 ,35 (s, 1 H), 12,35 (s, 1 H).
A-8 - Synthèse de la 6-(2-acétyl-4-bromo-phénylamino)-4-méthoxy- quinoline-5,8-dione (Intermédiaire As)
Préparation selon le procédé décrit dans le chapitre A-1 : 4-méthoxyquinoline- 5,8-dione (1 ,57 g, 9,1 mmol), chlorure de cérium (3,1 g, 8,3 mmol), 5-bromo-2- amino acétophénone (Léonard, Boyd, J.Org. Chem. 1946;1 1 , 419-423) (1 ,95g, 9,1 mmol), éthanol (200 ml), acide acétique (180 ml). On obtient après purification par flash-chromatographie sur colonne de silice (CH2Cl2 MeOH 95 : 5) 1 ,22 g de poudre orange :
• Rendement = 37 %. • RMN 1 H (CDCI3) : 3,15 (s, 3H), 4,58 (s, 3H), 7,61 (d, 1 H, J = 6 Hz), 7,74 (s,
1 H), 7,99 (d, 1 H, J = 8,8 Hz), 8,14 (dd, 1 H, J = 8,8 et 2,4 Hz), 8,51 (d, 1 H, J = 2,8 Hz), 9,32 (d, 1 H, J = 6 Hz), 11 ,68 (s, 1 H).
A-9 - Synthèse de la 2-méthoxy-6-(2-acétyl-phénylamino)-quinoline-5,8- dione (Intermédiaire Ag)
A une suspension de 2-méthoxyquinoline-5,8-dione (0,54 g, 2,8 mmol) et de chlorure de cérium (1 ,16 g, 4,7 mmol) dans l'éthanol (100 ml), on ajoute une solution d'o-aminoacétophénone (0,41 g, 3,1 mmol) dans l'éthanol (6 ml). Le milieu réactionnel est agité, à température ambiante, pendant 40 h. Après concentration à l'évaporateur rotatif, le brut obtenu est purifié par filtration sur silice (CHCI3/heptane 98 : 2) pour donner le produit de condensation attendu sous forme de poudre rouge (0,35 g).
• Rendement = 38 %.
• Point de fusion = 258 °C. • 1H RMN (CDCI3) : 2,67 (s, 3H) ; 4,15 (s, 3H) ; 6,79 (s, 1 H) ; 6,98 (d, 1 H, J
= 8,8 Hz) ; 7, 18 (ddd, 1 H, J = 8, 1 , 8,4 et 1 ,5 Hz) ; 7,56 (dd, 1 H, J = 8,4 et 1 ,5 Hz) ; 7,61 (ddd, 1 H, J = 8,1 et 8,4 et 1 ,1 Hz) ; 7,94 (dd, 1 H, J = 8,1 et 1 ,5 Hz) ; 8,31 (d, 1 H, J = 8,8 Hz).
• 13C RMN (CDCI3) : 28,51 ; 54,73 ; 106,02 ; 1 15,22 ; 120,78 ; 122,50 ; 123,1 1 ; 125,70 ; 132,34 ; 134,24 ; 137,18 ; 140,05 ; 143,30 ; 148,21 ; 167,75 ;
180,88 ; 183,05 ; 201 ,41.
• IR (CHCi3) : 1668 ; 1644 cm"1.
A-10 - Synthèse de la 3-hydroxyméthyl-6-(2-acétylphénylamino)-quinoline- 5,8-dione (Intermédiaire A10)
a) - 3-hydroxyméthyl-5,8-diméthoxyquinoline
A une solution d'éthyl-5,8-diméthoxyquinoline-3-carboxylate (180 mg, 0,689 mmol) dans 60 ml de THF, une solution de LiA!H4 1 M/Et2θ (5 ml, 5 mmol) est ajoutée goutte-à-goutte et sous azote. Le mélange est agité à température ambiante pendant 15 heures puis versé dans 15 ml de NaOH 1 N et 40 ml d'eau. Après extraction au CH2CI2 (3 x 100 ml), puis séchage de la phase organique sur MgSθ4, l'extrait est concentré à l'évaporateur rotatif. Le brut obtenu est purifié par flash-chromatographie (CH2CI2 : MeOH 95 : 5) pour donner le produit attendu sous forme de poudre marron (72 mg) :
• Rendement = 48 %. • Point de fusion = 150 °C.
• 1H RMN (CDCI3) : 3,92 (s, 3H) ; 4,00 (s, 3H) ; 4,88 (s, 2H) ; 6,72 (d, 1 H, J = 8,4 Hz) ; 6,88 (d, 1 H, J = 8,4 Hz) ; 8,47 (d, 1 H, J = 2,2 Hz) ; 8,87 (d, 1 H, J = 2,2 Hz).
• 3C RMN (CDCI3) : 55,76 ; 56,00 ; 63,09 ; 103,95 ; 106,70 ; 121 ,25 ; 128,62 ; 133,35 ; 139,80 ; 148,61 ; 149,21 ; 149,41.
• IR (CDCI3) : 3607, 3417, 1622, 1605 cm-1.
b) - 3-hydroxyméthylquinoline-5,8-dione
Une solution de 3-hydroxyméthyl-5,8-diméthoxyquinoline (55 mg, 0,25 mmol) et de nitrate de cérium ammonium (550 mg, 1 mmol) dans un mélange
CH3CN/H2O (3 ml/ 1 ml) est agitée à température ambiante pendant 40 min.
Après addition de 5 ml d'H2θ et de 10 ml d'une solution saturée de NaHCθ3, le milieu est extrait au CH2CI2 (6 x 30 ml) et on sèche les phases organiques sur
MgSθ3. Après évaporation du solvant à l'évaporateur rotatif, la quinone attendue est obtenue sous forme de poudre marron (11 mg) ;
• Rendement = 22 %.
• Point de fusion = 150 °C.
• 1H RMN (CDCI3) : 4,95 (s, 2H) 7,06 (d, 1H, J = 10,2 Hz) ; 7,15 (d, 1 H, J = 10,2 Hz) ; 8,43 (s, 1 H) ; 9,03 (s, 1 H).
. 13C RMN (CDCI3) : 62,03 ; 128,86 ; 132,26 ; 138,00 ; 139,11 ; 141 ,33 ; 146,58 ; 153,10 ; 183,12 ; 184,54.
• IR (CHCI3) : 3413 ; 1680 ; 1596 cm-1.
c) - Synthèse de la 3-hydroxyméthyl-6-(2-acétyIphénylamino)-quinoline-5,8- dione (Intermédiaire Afo) A une suspension de 3-hydroxyméthylquinoline-5,8-dione (0,22 g, 1 ,16 mmol) et de chlorure de cérium (0,6 g, 2,43 mmoll) dans l'éthanol (40 ml), est ajoutée une solution de 2-aminoacétophénone (0,18 g, 1 ,33 mmol). Le milieu réactionnel est agité à température ambiante dans l'obscurité pendant 3 h. Après concentration à l'évaporateur rotatif, le brut obtenu est purifié par filtration sur silice (CH2Cl2 MeOH 98 : 2) pour donner le produit de condensation attendu sous forme de poudre violette (0,16 g) :
• Rendement = 42 %.
• Point de fusion = 258 °C.
• 1H RMN (DMSO-d6) : 2,67 (s, 3H) ; 4,73 (d, 2H, J = 5,5 Hz) ; 5,67 (t, 1 H, J = 5,5 Hz) ; 6,64 (s, 1 H) ; 7,30 (m, 1 H) ; 7,71 (m, 2H) ; 8,12 (d, 1 H, J = 8,0 Hz) ;
8,35 (d, 1 H, J = 2,0 Hz) ; 8,93 (d, 1 H, J = 2,0 Hz) ; 11 ,02 (s, 1 H).
• 13C RMN (CDCI3) : 28,81 ; 60,08 ; 106,52 ; 120,96 ; 123,44 ; 126,14 ; 127,23 ; 131 ,52 ; 132,69 ; 134,43 ; 138,91 ; 141 ,75 ; 143,55 ; 146,62 ; 152,81 ; 181 ,62 ; 181 ,84 ; 202,02. • IR (CHCI3) : 3440, 1690, 1661 , 1640 cm-1.
B - Préparation des produits intermédiaires de formule III (Schéma II)
B-1 - Synthèse de la 9,11 -diméthyl-1 ,6-diaza-naphtacène-5,12-dione (intermédiaire β-/)(CRL 8324;
A une solution de tricycle intermédiaire A (0,4 g, 1 ,3 mmol) dans 12 ml d'acide acétique, on ajoute lentement 1 ,9 ml d'acide sulfuhque en solution dans 9,6 ml d'acide acétique. Le milieu réactionnel est porté à reflux 30 minutes, puis versé, après refroidissement, dans un bêcher contenant de la glace pilée. On neutralise par NH4OH puis on extrait 4 fois au dichlorométhane. Les phases organiques sont séchées sur MgSθ4 puis évaporées. Le brut obtenu est purifié par flash-chromatographie sur colonne de silice (CH2Cl2/MeOH 95 : 5) pour donner 0,325 g de composé tétracycle attendu. • Rendement = 86 %. • RMN H (CDCI3) : 2,64 (s, 3H), 3,29 (s, 3H), 7,74 (dd, 1 H, J = 7,6 et 4,8 Hz),
7,75 (dd, 1 H, J = 8,4 et 1 ,6 Hz), 8,12 (dd, J = 1 ,6 Hz), 8,33 (d, 1 H, J = 8,4), 8,71 (dd, 1 H, J = 2 et 7,6 Hz), 9,13 (dd, 1 H, J = 2 et 4,8 Hz).
B-2 - Synthèse de la 9-chloro-11 -méthyl-1 ,6-diaza-naphtacène-5,12-dione (Intermédiaire B2)
Préparation selon le procédé décrit dans le chapitre B-1 : tricycle intermédiaire A2 (0,289 g, 0,88 mmol), acide sulfuhque (1 ,3 ml), acide acétique (8 + 6,5 ml). Après purification par flash-chromatographie (CH2θ2/MeOH 95 : 5), on obtient 0,26 g de tétracycle :
• Rendement : 95 %.
• RMN 1 H (CDCI3) : 3,25 (s, 3H), 7,76 (dd, 1 H, J = 8 et 4,8 Hz), 7,85 (dd, 1 H, J = 8,8 et 2 Hz), 8,33 (dd, 1 H, J = 2 Hz), 8,38 (d, 1 H, J = 8,8), 8,71 (dd, 1 H, J = 1.6 et 8 Hz), 9,15 (dd, 1 H, J = 1 ,6 et 4,8 Hz).
B-3 - Synthèse de la 9-benzylamino-11 -méthyl-1 , 6-diaza-naphtacène-5, 12- dione (Intermédiaire B3)
Préparation selon le procédé décrit dans le chapitre B-1 : intermédiaire A3 (4 g, 10 mmol), acide sulfuhque (15,1 ml), acide acétique (92 + 75 ml). Après traitement, on obtient 3,58 g de tétracycle.
• Rendement = 98 %.
. RMN 1 H (CDCI3) : 3,09 (s, 3H), 4,52 (d, 2H), 4,86 (t, 1 H), 7,06 (d, 1 H, J = 2.8 Hz), 7,29 (dd, 1 H, J = 9,2 et 2,8 Hz), 7,3-7,43 (m, 5H), 7,71 (dd, 1 H, J = 4,8 et 8 Hz), 8,20 (d, 1 H, J = 9,8 Hz), 8,69 (dd,1 H, J = 1 ,6 et 8 Hz), 9,09 (dd, 1 H, J = 1 ,6 et 4,8 Hz).
B-4 - Synthèse de la 8-bromo-11 -méthyl-1 ,6-diaza-naphtacène-5,12-dione (Intermédiaire B4)
Préparation selon le procédé décrit par F. Bracher, Liebigs Ann. Chem. 1990, 205-206.
B-5 - Synthèse de la 9-diméthylamino-11 -méthyl-1 ,6-diaza-naphtacène-5,12- dione (intermédiaire B5)
Préparation selon le procédé décrit dans le chapitre B-1 : tricycle intermédiaire A5 (0,76 g, 2,27 mmol), acide sulfurique (3,5 ml), acide acétique (20 + 18 ml). Après traitement on obtient 0,67 g de tétracycle.
• Rendement = 93 %.
. RMN 1 H (CDCI3) : 3,17 (s, 3H), 3,21 (s, 6H), 7,04 (d, 1 H, J = 3,2 Hz), 7,51 (dd, 1 H, J = 3,2 et 9,2 Hz), 7,71 (dd, 1 H, J = 8 et 4,4 Hz), 8,26 (d, 1 H, J = 9,2 Hz), 8,7 (dd, 1 H, J = 1 ,6 et 8 Hz) 9,09 (dd, 1 H, J = 1 ,6 et 4,4 Hz).
B-6 - Synthèse de la 9-méthoxy-11 -méthyl-1 ,6-diaza-naphtacène-5,12-dione (Intermédiaire Bβ)
Préparation selon le procédé décrit dans le chapitre B-1 : tricycle intermédiaire Q (4,25 g, 13,18 mmol), acide sulfurique (20 ml), acide acétique (110 + 100 ml). Le produit obtenu par flash-chromatographie (CH2Cl2/MeOH 100 : 3) est lavé à l'éther éthylique pour donner 2,9 g de tétracycle.
• Rendement = 72 %.
. RMN H (CDCI3) : 3,25 (s, 3H), 4,02 (s, 3H), 7,49 (d, 1 H, J = 3,3 Hz), 7,56
(dd, 1 H, J = 3,3 et 9,3 Hz), 7,74 (dd, 1 H, J = 8,3 et 4,3 Hz), 8,34 (d, 1 H, J = 9,3 Hz), 8,71 (dd, 1 H, J = 2,5 et 8,3 Hz) 9,12 (dd, 1 H, J = 2,5 et 4,3 Hz).
B-7 - Synthèse de la 4-(2-acétylanilino)-11 -méthyl-1 , 6-diaza-naphtacène- 5,12-dione(7nferméd7a/re Bγ) (CRL 8332)
Préparation selon le procédé décrit dans le chapitre B-1 : tricycle intermédiaire A7 (1 g, 2,35 mmol), acide sulfurique (3,5 ml), acide acétique (18 ml). Le produit obtenu par flash-chromatographie (CH2Cl2/MeOH 100 : 3) est lavé à l'éther éthylique pour donner 0,6 g de tétracycle sous forme de poudre orange.
• Rendement = 63 %.
• RMN 1 H (CDCI3) : 2,59 (s, 3H), 3,25 (s, 3H), 7,29 (ddd, 1 H, J = 7,2 et 7,2 et 1 ,2 Hz), 7,37 (d, 1 H, J = 6 Hz), 7,54 (ddd, 1 H, J = 6,8 et 6,8 et 1 ,6 Hz), 7,59 (d,
1 H, J = 6,8 Hz), 7,74 (dd, 1 H, J = 7,2 et 1 ,2 Hz), 7,76 (dd, 1 H, J = 6,8 et 1 ,6 Hz), 7,87-7,918 (m, 2H), 8,34 (d, 1 H, J = 8,4 Hz), 8,43 (d, 1 H, J = 8,4 Hz), 8,54 (d, 1 H, 6 Hz), 12,5 (s, 1 H).
B-8 - Synthèse de la 4-méthoxy-9-bromo-11 -méthyl-1, 6-diaza-naphtacène- 5,12-dione (Intermédiaire Bs)
Préparation selon le procédé décrit dans le chapitre B-1 : tricycle intermédiaire As (1 ,22 g, 3,04 mmol), acide sulfurique (4,5 ml), acide acétique (27 + 23 ml). Le produit obtenu par flash-chromatographie (CH2Cl2/MeOH 100 : 3) est lavé à l'éther éthylique pour donner 0,76 g de tétracycle sous forme de poudre jaune.
• Rendement = 65 %.
• RMN 1 H (CDCI3) : 3,21 (s, 3H), 4,10 (s, 3H), 7,18 (d, 1 H, J = 6 Hz), 7,96 (dd,
1 H, J = 8,8 et 2 Hz), 8,27 (d, 1 H, J = 8,8 Hz), 8.47 (d, 1 H, J = 2 Hz), 8,89 (d, 1 H, J = 6 Hz).
B-9 - Synthèse de 2-méthoxy-11 -méthyl-1 ,6-diaza-naphtacène-5,12-dione, (Intermédiaire Bg)
Une solution de 2-méthoxy-6-(2-acétyl-phénylamino)-quinoline-5,8-dione (0,34 g, 1 ,1 mmol) dans un mélange acide acétique/acide sulfurique (25 mol/2, 7 ml) est chauffée à 90 °C pendant 45 min. Après refroidissement le milieu réactionnel est versé dans un mélange eau/glace (200 ml) puis alcalinisé à pH 8
par du K2CO3 et extrait au CHCI3 (3 x 200 ml). Les phases organiques sont séchées sur MgS0 puis concentrées à l'évaporateur rotatif. Le brut obtenu est purifié par filtration sur silice (CHCI3) pour donner le tétracycle attendu sous forme de poudre beige (0,23 g) : • Rendement = 71 %.
• Point de fusion = 260 °C.
• H RMN (CDCI3) : 3,32 (s, 3H) ; 4,23 (s, 3H) ; 7,14 (d, 1H, J = 8,8 Hz) ; 7,79 (ddd, 1 H, J = 8,6, 8,4 et 1 ,2 Hz) ; 7,91 (ddd, 1 H, J = 8,4, 8,6 et 1 ,2 Hz) ; 8,38 (dd, 1 H, J = 8,6 et 1 ,2 Hz) ; 8,46 (dd, 1 H, J = 8,4 et 1 ,2 Hz) ; 8,58 (d, 1 H, J = 8,8 Hz).
• 13C RMN (CDCI3) : 16,63 ; 54,76 ; 117,29 ; 125,29 ; 125,50 ; 125,64 ; 129,62 ; 129,75 ; 132,37 ; 132,57 ; 138,12 ; 147,73 ; 148,63 ; 149,69 ; 152,28 ; 167,77 ; 181 ,10 ; 183,55.
• IR (CHCl3) : 1683 ; 1599 cm-1.
B-10- Synthèse 3-acétoxyméthyl-11 -méthyl-1 ,6-diazanaphtacène-5,12-dione (Intermédiaire B10)
Une solution de 3-hydroxyméthyl-6-(2-acétylphénylamino)-quinoline-5,8- dione (Intermédiaire A10), (0,248 g, 0,77 mmol) dans un mélange acide acétique/acide sulfurique (16 ml/1 ,3 ml) est chauffée à 90 °C pendant 2h30.
Après refroidissement le milieu réactionnel est versé dans un mélange eau/glace
(15 ml) puis alcalinisé à pH 9 au Na2Cθ3_ Le milieu est ensuite extrait au
CH2CI2 (3 x 150 ml). Les phases organiques sont séchées sur MgSO4 puis concentrées à l'évaporateur rotatif. Le brut obtenu est purifié par filtration sur silice (CH2CI2 / MeOH 98 : 2) pour donner le composé attendu sous forme de poudre marron (0,21 g).
• Rendement = 85%.
• Point de fusion = 210 °C.
• 1H RMN (CDCI3) : 2,18 (s, 3H) ;3,30 (s, 3H) ; 5,31 (s, 2H) ; 7,78 (ddd, 1 H, J = 1 ,1 , 6,8 et 8,1 Hz) ; 7,92 (ddd, 1 H, J = 1 ,1 , 6,8 et 8,1 Hz) ; 8,37 (dd, 1 H, J =
,1 et 1 ,1 Hz) ; 8,43 (dd, 1 H, J = 8,1 et 1 ,1 Hz) ; 8,66 (d, 1 H, J = 2,2 Hz) ; 9,09 (d, 1 H, J = 2,2 Hz).
. 13C RMN (CDCI3) : 16,06 ;20,11 ; 62,06 ; 124,81 ; 124,91 ; 129,06 ; 129,18 ; 129,29 ; 131 ,70 ; 132,18 ; 134,06 ; 136,09 ; 146,86 ; 147,97 ; 149,01 ; 152,19 ; 154,30 ; 169,72 ; 180,96 ; 182,34.
• IR (CHCI3) : 3420, 1746, 1692 cm-1.
C - Préparation des produits intermédiaires de formule Nia (Schéma II)
1) Synthèse de l'acide N-(2,5-diméthoxyphényl)anthranilique (composé 4)
Un mélange d'acide 2-chlorobenzoïque (9,2 g, 60 mmol), de diméthoxyaniline (10 g, 65 mmol), de cuivre (0,96 g), de Cu2O (0,96 g) et de K2CO3 (10,4 g) dans 120 ml de diglyme est porté à reflux pendant une nuit. Après évaporation du solvant, le milieu réactionnel est alcalinisé à la soude 1 N. De l'éther est ajouté puis le milieu filtré sur silice et la phase éthérée éliminée. La phase aqueuse est acidifiée à l'HCI concentré puis extraite à l'acétate d'éthyle. Après séchage sur MgSO4 et évaporation du solvant à l'évaporateur rotatif, le brut obtenu est purifié par filtration sur silice (CH2CI2) pour donner le produit de condensation attendu sous forme de poudre jaune (14,5 g). • Rendement = 89 %.
• Point de fusion = 138 °C.
• RMN 1 H RMN (CDCI3) : 3,77 (s, 3H) ; 3,85 (s, 3H) ; 6,57 (dd, 1 H, J = 8,8 et 2,9 Hz) ; 6,77 (ddd, 1 H, J = 1 ,9 et 7,5 Hz) ; 6,87 (d, 1 H, J =9,2 Hz) ; 7,04 (d, 1 H, J = 2,9 Hz) ; 7,3 à 7,4 (m, 2H) ; 9,35 (slarge, 1 H). • 13C RMN (CDCI3) : 55,76 ; 56,45 ; 107,30 ; 107,71 ; 1 12,00 ; 1 12,26 ; 1 14,70
; 1 17,53 ; 130,78 ; 132,60 ; 134,09 ; 145,98 ; 147,71 ; 153,75 ; 172,95.
• IR (CHCI3) : 3327 ; 1685 cm-1.
2) Synthèse de la 2-(2,5-diméthoxyphényiamino)acétophénone (composé 5)
A un mélange d'acide N-(2,5-diméthoxyphényl)anthranilique (2 g, 73 mmol) dans 14 ml de THF, sont ajoutés 16 ml de MeLi (1 ,4 M / Et2O) à 0 °C et sous N2 . Après remontée de la température, le milieu est porté à reflux pendant 2 h puis
100 ml d'eau sont ajoutés et le mélange extrait à l'éther (3 x 100 ml). Après séchage sur MgS0 , le solvant est évaporé à l'évaporateur rotatif pour donner le dérivé attendu sous forme de solide jaune (1 ,49 g).
• Rendement = 75 %. • Point de fusion = 79 °C.
• 1H RMN (CDCI3) : 2,64 (s, 3H) ; 3,76 (s, 3H) ; 3,84 (s, 3H) ; 6,55 (dd, 1 H, J = 8,8 et 2,9 Hz) ; 6,73 (dd, 1 H, J = 1 ,4 et 7,5 Hz) ; 6,85 (d, 1 H, J =8,8 Hz) ; 7,04 (d, 1 H, J = 2,9 Hz) ; 7,3 à 7,4 (m, 2H) ; 7,81 (dd, 1 H, J = 1 ,5 et 8,0 Hz) ; 10,5 (slarge, 1 H). • 13C RMN (CDCI3) : 48,15 ; 55,73 ; 56,36 ; 107,10 ; 107,72 ; 112,05 ; 114,80 ;
116,84 ; 120,09 ; 130,68 ; 132,42 ; 134,35 ; 145,98 ; 146,67 ; 153,62 ; 201 ,00.
• IR (CHCI3) : 3350 ; 1642 cm"1.
3) Synthèse de la 1 ,4-diméthoxy-9-méthylacridine (composé 6) Un mélange de 2-(2,5-diméthoxyphénylamino)acétophénone (1 ,3 g, 48 mmol) et d'acide polyphosphorique (13 g, 133 mmol) est chauffé à 100 °C pendant 1 h. Après addition de 50 ml d'eau, le mélange est neutralisé à la soude 4M puis extrait au CHCI3 (3 x 100 ml). Après séchage sur MgSO4 et évaporation du solvant, le brut obtenu est purifié par filtration sur silice (CH2CI2) pour donner quantitativement le dérivé thcyclique attendu sous forme d'un solide orange- marron.
• Point de fusion = 136 °C.
• 1H RMN (CDCI3) : 3,36 (s, 3H) ; 3,96 (s, 3H) ; 4,09 (s, 3H) ; 6,68 (d, 1 H, J = 8,0 Hz) ; 6,89 (d, 1 H, J = 8,4 Hz) ; 7,54 (m, 1 H) ; 7,73 (m, 1 H) ; 8,32 (d, 1 H, J = 8,4) ; 8,36 (d, 1 H, J = 8,8 Hz).
• 13C RMN (CDCI3) : 17,78 ; 55,66 ; 56,13 ; 102,43 ; 105,18 ; 120,25 ; 124,28 ; 125,62 ; 126,59 ; 129,44 ; 130,81 ; 142,45 ; 144,23 ; 147,21 ; 149,46 ; 151 ,45.
• IR (CHCl3) : 1685 ; 1661 cm"1.
3) Synthèse de la 9-méthylacridine-1,4-dione (Composé 7)
Une solution de 1 ,4-diméthoxy-9-méthylachdine (20 mg, 0,079 mmol) et de nitrate de cérium ammonium (196 mg, 0,357 mmol) dans un mélange
CH2CI2/H2O (0,5 ml / 0,25 ml) est agitée à 0 °C pendant 20 min. Après addition de 1 ,4 ml de H2O et de 0,4 ml d'une solution saturée de NaHCO3, le milieu est laissé sous agitation, puis extrait au CH CI2 (3 x 3ml). Les phases organiques sont séchées sur MgSO4. Après évaporation du solvant à l'évaporateur rotatif, la quinone attendue est obtenue sous forme de poudre marron (15 mg).
• Rendement = 90 %.
• Point de fusion > 260 °C.
• 1H RMN (CDCI3) : 3,22 (s, 3H) 7,09 (d, 1 H, J = 10,3 Hz) ; 7,18 (d, 1 H, J = 10,3 Hz) ; 7,78 (dd, 1 H, J = 8,5 et 8,5 Hz) ; 7,91 (dd, 1 H, 8,5 et 8,5 Hz) ; 8,32 (d, 1 H, J = 8,5) ; 8,43 (d, 1 H, J = 8,5 Hz).
• 13C RMN (CDCI3) 15,87 ; 124,40 ; 125,41 ; 126,30 ; 129,61 ; 132,32 ; 132,52 ; 137,88 ; 141 ,61 ; 147,05 ; 148,23 ; 151 ,23 ; 183,43 ; 186,69.
• IR (CHCl3) : 1701 ; 1661 cm"1.
D - Préparation des produits intermédiaires de formule IVa (Schéma II)
D-1 - Synthèse de la 6-méthyl-1 ,11-diaza-naphtacène-5,12-dione (Intermédiaire D→
Une solution de 9-méthylacridine-1 ,4-dione (200 mg, 0,896 mmol), d'acroleine-N,N-diméthylhydrazone (96 mg, 0,984 mmol) et d'anhydride acétique (1 ml) dans 20 ml de CH2CI2 est agitée sous N2, à température ambiante pendant 30 min. Après concentration du solvant, le milieu est purifié par filtration sur silice (CH2CI2) pour récupérer le produit d'addition non complètement aromatique. Une suspension de ce composé et de Pd/C 10 % (20 mg) dans 4 ml de toluène est chauffée à reflux pendant 30 min. Après concentration, le brut obtenu est purifié par flash-chromatographie sur silice (CH2CI2/MeOH 98 : 2) pour donner le tétracycle attendu sous forme de poudre beige (23 mg),
• Rendement = 13 %.
• 1H RMN (CDCI3) : 3,32 (s, 3H) 7,78-7,83 (m, 2H) ; 7,95 (ddd, 1 H, J = 8,4, 7,7 et 1 ,5 Hz) ; 8,39 (dd, 1 H, J = 8,8 et 1 ,5 Hz) ; 8,51 (dd, 1 H, J = 7,7 et 1 ,5 Hz) ;
8,68 (dd, 1 H, J = 8,1 et 1 ,9 Hz) ; 9,16 (dd, 1 H, J = 4,8 et 1 ,9 Hz).
• 13C RMN (CDCI3) 16,67 ; 124,59 ; 125,44 ; 128,39 ; 129,76 ; 129,89 ; 132,25 ; 132,54 ; 132,88 ; 135,93 ; 148,00 ; 148,59 ; 148,73 ; 152,48 ; 155,31 ; 180,81 ; 184,37.
• IR (CHCI3) : 1703 ; 1663 cm .
D-2 - Synthèse de la 3-méthoxy-6-méthyl-1 ,11 -diazanaphtacène-5,12- dione (Intermédiaire D2)
La 3-méthoxy-6-méthyl-1 ,11-diazanaphtacène-5,12-dione est préparée selon la procédure décrite dans D-1, à partir d'une solution de 9- méthylacridine-1 ,4-dione (composé 7) (200 mg, 0,896 mmol), de 2-méthoxy-
2-propénal-diméthylhydrazone (126 mg, 0,984 mmol) et d'anhydride acétique (1 ml) dans 20 ml de CH2CI2.
EXEMPLE 1 5-méthyl-9H-quino [4, 3, 2 -de] [1 ,10] phénanthrolin-9-one
(CRL 8323)
On porte à reflux pendant 1 heure une solution de tétracycle intermédiaire B (1 g, 3,47 mmol) et de diméthylformamide diéthyl acétal (2 ml, 10,41 mmol) dans 7 ml de DMF. Après évaporation à sec, on ajoute du chlorure d'ammomium (2,77 g, 52 mmol) et 50 ml d'éthanol. Le milieu réactionnel est à nouveau porté à reflux 30 minutes. Après évaporation du solvant, le brut est repris à l'eau et extrait 4 fois au dichlorométhane. Les phases organiques sont séchées sur MgSθ4 puis évaporées. Après recristallisation dans 125 ml de méthanol, on obtient 0,7 g du composé CRL 8323 attendu sous forme d'un solide jaune moutarde.
• Rendement = 67 %.
• Point de fusion = 200° C.
• RMN 1 H (CDCI3) : 2,69 (s, 3H), 7,65 (dd, 1 H, J = 8 et 4,8 Hz), 7,81 (dd, 1 H, J = 8 et 1 ,2 Hz), 8,44 (d, 1 H, J = 1 ,2 Hz), 8,49 (d, 1 H, J = 8 Hz), 8,50 (d, 1 H, J = 5,6 Hz), 8,78 (dd, 1 H, J = 2 et 8 Hz), 9,15 (dd, 1 H, J = 4,8 et 2 Hz), 9,24 (d, 1 H, J = 5,6 Hz).
• RMN 3c (CDCI3) : 22,06 ; 116,54 ; 117,87 ; 122,15 ; 123,12 ; 125,24 ;
128,74 ; 132,58 ; 133,47 ; 136,25 ; 137,19 ; 141 ,63 ; 143,88 ; 144,79 ; 149,16 ; 149,31 ; 152,09 ; 155,15 ; 181 ,53.
• SM (m/z) : 297 (17,6); 296 (34,3); 268 (25,4); 149 (50,3).
EXEMPLE 2
5-chloro-9H-quino [4,3,2-cfe] [1,10] phénantrolin-9-one (CRL 8301) Préparation selon le procédé décrit dans l'exemple 1 à partir du tétracycle intermédiaire B2 (0,25 g, 0,81 mmol) et le diméthylformamide diéthyl acétal (1 ,5 ml, 8,75 mmol) dans le DMF (4,5 ml). Chlorure d'ammomium (2,95 g, 55 mmol), éthanol (50 ml). Après purification par flash-chromatographie (CH2Cl2/MeOH 98 : 2), on obtient 60 mg du composé CRL 8301 attendu sous forme d'un solide jaune. • Rendement = 23 %.
• Point de fusion = 200° C.
• RMN H (CDCI3) : 7,68 (dd, 1 H, J = 8,4 et 4,8 Hz), 7,94 (dd, 1 H, J= 8,8 et 2
Hz), 8,46 (d, 1 H, J = 5,6 Hz), 8,55 (d, 1H, J = 8,8 Hz), 8,63 (d, 1 H, J = 2 Hz), 8,79 (dd, 1 H, J = 2 et 8,4 Hz), 9,18 (dd, 1 H, J = 4,8 et 2 Hz), 9,30 (d, 1 H, J = 5,6 Hz). • RMN 3c (CDCI3) : 117,07 ; 118,46 ; 122,98 ; 124,82 ; 126,12 ; 129,34 ;
133,02 ; 134,81 ; 137,00 ; 137,42 ; 137,79 ; 144,45 ; 146,35 ; 150,24 ; 150,45 ; 152,55 ; 156,02 ; 181 ,9.
• SM (m/z) : 319 (43); 317 (100); 291 (14,5); 290 (18); 289 (100).
EXEMPLE 3
5-(benzylamino)-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8241)
Préparation selon le procédé décrit dans l'exemple 1 à partir du tétracycle intermédiaire B3 (3,58 g, 9,45 mmol) et le diméthylformamide diéthyl acétal (5,7 ml, 33,26 mmol) dans du DMF (19 ml). Chlorure d'ammomium (2,95 g, 55 mmol), éthanol (50 ml). Après purification par flash-chromatographie (CH2Cl2/MeOH 96 :
4), on obtient 2 g du composé CRL 8241 attendu sous forme de poudre lie-devin.
• Rendement = 55 %.
• Point de fusion = 219° C.
• RMN 1 H (CDCI3) : 4,61 (d, 2H), 5,10 (t, 1 H), 7,31 (dd, 1 H, J = 8,8 Hz, J = 2.4 Hz), 7,452-7,327 (m, 5H), 7,55 (d, 1 H, J = 2,4 Hz), 7,63 (dd, 1 H, J = 4,4 Hz, J = 8,4 Hz), 8,29 (d, 1 H, J= 5,2 Hz), 8,36 (d, 1 H, J = 8,8 Hz), 8,79 (dd, 1 H, J = 1 ,2 Hz, J = 8,4 Hz), 9,13 (dd, 1 H, J = 4,4 et 1 ,2 Hz), 9,14 (d, 1 H, J = 5,2 Hz).
• SM (m/z) : 388 (7); 387 (100); 386 (85); 385 (25); 369 (99); 368 (44).
EXEMPLE 4
5-(diméthylamino)-9H-quino[4,3,2-cfe][1 ,10] phénanthrolin-9-one (CRL 8325)
Préparation selon le procédé décrit dans l'exemple 1 à partir du tétracycle intermédiaire B5 (0,25 g, 0,79 mmol) et le diméthylformamide diéthyl acétal (0,5 ml, 2,98 mmol) dans le DMF (5 ml). Chlorure d'ammomium (1 g, 18,7 mmol), éthanol (16 ml). Après purification par flash-chromatographie (CH2Cl2/MeOH 100 : 5), on obtient 170 mg du composé CRL 8325 attendu sous forme d'une poudre violette. • Rendement = 66 %.
• Point de fusion > 260°C.
• RMN H (CDCI3) : 3,25 (s, 6H), 7,45 (dd, 1 H, J = 9,2 Hz, J = 3Hz), 7,57 (d,
1 H, J = 3 Hz), 7,63 (dd, 1 H, J = 4,4 et 8Hz), 8,41 (d, 1 H, J = 9,2 Hz), 8,43 (d, 1 H, J = 5,6 Hz), 8,81 (dd, 1 H, J = 2 et 7,6 Hz), 9,13 (dd, 1 H, J = 4,4 et 2 Hz), 9,17 (d, 1 H, J = 5,6 Hz).
• RMN "I 3c (CDCI3) : 40,45 ; 100,84 ; 1 16,81 ; 1 18,69 ; 1 18,99 ; 125,19 ; 126,10 ; 129,46 ; 134,62 ; 136,03 ; 136,30 ; 139,00 ; 140,69 ; 148,16 ; 149,15 ; 151 ,53 ; 152,47 ; 154,83 ; 181 ,65.
• SM(m/z) : 326 (34,5); 325 (100); 324 (100); 254 (15,5); 253 (13,4).
EXEMPLE 5
5-méthoxy-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one
(CRL 8297)
Préparation selon le procédé décrit dans l'exemple 1 à partir du tétracycle intermédiaire BQ (2 g, 6,57 mmol) et le diméthylformamide diéthyl acétal (4 ml, 23,34 mmol) dans le DMF (14 ml). Chlorure d'ammomium (8 g, 149,5 mmol), éthanol (130 ml). Après purification par flash-chromatographie (CH2Cl2/MeOH 100 : 5), on obtient 170 mg du composé CRL 8297, attendu, sous forme d'un solide verdâtre.
• Rendement = 66 %.
• Point de fusion > 260 °C. • RMN H (CDCI3) : 4,10 (s, 3H), 7,62 (dd, 1 H, J = 9,2 Hz, J = 2,4Hz), 7,66
(dd, 1 H, J = 4,4 et 8 Hz), 7,96 (d, 1 H, J = 2,4 Hz), 8,48 (d, 1 H, J = 2,4 Hz), 8,54 (d, 1 H, J = 9,2 Hz), 8,80 (dd, 1 H, J = 2,4 et 8 Hz), 9,16 (dd, 1 H, J = 4,4 et 2,4 Hz), 9,25 (d, 1 H, J = 5,2 Hz).
• RMN 3c (CDCI3) : 30,93 ; 1 16,86 ; 1 18,41 ; 122,44 ; 125,56 ; 129,25 ; 134,96 ; 136,55 ; 137,13 ; 141 ,52 ; 143,67 , 149,1 1 ; 149,77 ; 152,37 ; 155,38 ;
161 ,71 ; 181 ,93 ; 207,00.
• SM (m/z) : 313 (26); 312 (100); 285 (2); 284 (15); 269 (15); 242 (32.5).
EXEMPLE 6 7-nitro-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one
(CRL 8289)
A 0 °C, on ajoute, par portions, de l'ascididémine (2 g, 7,06 mmol) à un mélange de 45 ml d'acide sulfurique et de 45 ml d'acide nitrique. Le milieu réactionnel est chauffé à 130°C pendant 2 heures puis versé après refroidissement dans un erlen contenant 400 g de glace. Après filtration, on obtient un précipité jaune que l'on rince plusieurs fois à l'éther. On le reprend ensuite dans un mélange CH2CI2/NH4OH/H2O 600 : 1 : 300. On récupère la phase organique et on extrait 3 fois la phase aqueuse au CH2CI2. Après séchage sur MgS04, les phases organiques sont évaporées pour donner 1 ,62 g du composé CRL 8289, attendu, sous forme d'un solide jaune.
• Rendement = 70 %.
• Point de fusion = 224° C.
• RMN H (CDCI3) : 7,69 (dd, 1 H, J = 4,4 et 8 Hz), 8,04 (dd, 1 H, J = 8 et 8 Hz), 8,28 (dd, 1 H, J = 8 Hz), 8,56 (d, 1 H, J = 5,2 Hz), 8,75 (dd, 1 H, J = 2 et 8 Hz), 8,89 (dd, 1 H, J = 1 ,2 et 8 Hz), 9,18 (dd, 1 H, J = 4,4 et 2 Hz), 9,37 (d, 1 H, J = 5,6 Hz).
. RMN 3C (CDCI3) : 79,20 ; 117,61 ; 118,39 ; 124,21 ; 124,89 ; 125,98 ;
127,54 ; 129,04 ; 130,14 ; 135,62 ; 1-36, 63 ; 148,17 ; 149,76 ; 149,94 ; 150,12 ; 151 ,66 ; 154,88 ; 180,56.
• SM (m/z) : 328 (18); 327 (100); 299 (22); 297 (9); 269 (10); 253 (24); 242 (1 1 ); 241 (33).
EXEMPLE 7
7-amino-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8344)
Une suspension de dérivé nitré CRL 8289 (0,4 g, 1 ,22 mmol) et de fer (0,37 g, 6,59 mmol) dans un mélange ACOH/H2O 10 : 10 est portée à reflux pendant 1 heure. On ajoute de l'EDTA (1 ,94 g, 6,59 mmol), puis on alcalinise le milieu réactionnel avec de la soude concentrée. On extrait 3 fois au CH2CI2. Après séchage sur MgSθ4 les phases organiques sont évaporées pour donner 0,32 g du composé CRL 8344 attendu sous forme d'un solide bleu. • Rendement = 88 %
• Point de fusion > 260 °C
• RMN 1 H (CDCI3) : 5,68 (s, 2H), 7,16 (d, 1 H, J= 7,8 Hz), 7,66 (dd, 1 H, J = 7,6 et 4,8 Hz), 7,69 (dd, 1 H, J = 7,8 et 7,8 Hz), 7,91 (d, 1 H, J = 7,8 Hz), 8,46 (d, 1 H, J = 5,2 Hz), 8,77 (dd, 1 H, J = 1 ,6 et 7,6 Hz), 9,17 (dd, 1 H, J = 1 ,6 et 4,8 Hz), 9,21 (d, 1 H, J = 5,2 Hz).
• RMN 3c (CDCI3) : 109,42 ; 112,71 ; 117,70 ; 118,43 ; 124,29 ; 125,64 ;
129,12 ; 132,63 ; 132,81 ; 135,53 ; 137,27 ; 141 ,68 ; 148,68 ; 148,89 ; 149,03 ; 151 ,96 ; 154,68 ; 180,71.
• SM (m/z) : 298 (34,7); 297 (100); 269 (11 ); 268 (8).
EXEMPLE 8
5-bromo-9H-quino [4,3,2-de] [1,10] phénantrolin-9-one (CRL 8248)
A une solution d'ascididémine (0,5 g, 1 ,77 mmol) dans 20 ml d'acide acétique, on ajoute goutte à goutte une solution de brome (0,2 ml, 3,88 mmol) dans 5 ml d'acide acétique. Le milieu réactionnel est porté à reflux (réfrigérant bouché) 24 heures. Après refroidissement, on neutralise par une solution saturée de NaHCθ3 et on extrait 4 fois au CH2CI2. Les phases organiques sont séchées sur MgSθ4 puis évaporées. Le brut obtenu est purifié par flash-chromatographie sur colonne de silice (CH2Cl2/MeOH 96 : 4) pour donner 0,548 g du composé CRL 8248 attendu sous forme d'un solide jaune.
• Rendement = 86 %
• Point de fusion = 208 °C
• RMN 1 H (CDCI3) : 7,68 (dd, 1 H, J = 4,4 et 8 Hz), 8,09 (dd, 1 H, J = 8,8 Hz, J = 2 Hz), 8,48 (d, 1 H, J = 8,8 Hz), 8,49 (d, 1 H, J = 6Hz), 8,79 (dd, 1 H, J = 2 et 8 Hz), 8,82 (d, 1 H, J = 2 Hz), 9,18 (dd, 1 H, J = 2 Hz, J = 4,4 Hz), 9,30 (d, 1 H, J = 6 Hz).
• RMN 13c (CDCI3) : 1 16,76 ; 1 17,04 ; 1 18,26 ; 124,76 ; 125,81 ; 125,93 ; 129,05 ; «134,52 ; 135,43 ; 136,72 ; 137,01 ; 144,41 ; 146,24 ; 149,93 ; 150, 12 ; 152,27 ; 155,67 ; 181 ,69. • SM (m/z) : 363 (99); 362 (83); 361 (100); 360 (27); 255 (9); 254 (51 ).
EXEMPLE 9
5-amino-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8347) A une solution d'ascididémine bromée : CRL 8248 (2,3 g, 6,33 mmol) dans
460 ml de DMF, on ajoute l'azidure de sodium (2,34 g, 36,1 mmol). Le milieu réactionnel est porté à reflux pendant 4 heures. Après refroidissement, on évapore à sec et on reprend le solide obtenu à l'eau. On extrait 4 fois au CH2CI2. Après séchage sur MgSO4 et évaporation du solvant, le brut est purifié par flash chromatographie sur colonne de silice (HCCl3/MeOH 90 : 10) pour donner 1 15 mg du composé CRL 8347 attendu sous forme d'une poudre noire.
• Rendement = 6 %
• point de fusion > 260 °C
• RMN 1 H (CDCI3) : 7,43 (dd, 1 H, J = 8,8 et 2,4 Hz), 7,74 (dd, 1 H, J = 4,8 et 8 Hz), 7,81 (d, 1 H, J = 2,4 Hz), 8,48 (d, 1 H, J = 6Hz), 8,50 (d, 1 H, J = 8,8 Hz), 8,90 (dd, 1 H, J = 2 et 8 Hz), 9,25 (dd, 1 H, J = 2 et 4,8 Hz), 9,29 (d, 1 H, J = 6 Hz). • RMN 3c (DMSO) : 102,26 ; 117,13 ; 118,54 ; 121 ,62 ; 123,20 ; 125,34 ;
126,11 ; 129,18 ; 133,80 ; 134,83 ; 135,47 ; 138,42 ; 147,65 ; 148,29 ; 151 ,63 ; 152,39 ; 154,32 ; 180,35.
• SM (m/z) : 298 (32); 297 (100); 269 (4); 268 (0,5).
EXEMPLE 10
10-méthoxy-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8368)
Préparation selon la procédure décrite par Y. Kitahara ét al., Heterocycles, 1993, 36, 943-946.
EXEMPLE 11
10-hydroxy-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8387)
Préparation selon la procédure décrite Y. Kitahara et al., Tetrahedron, 1997, 53, 17029-17038.
EXEMPLE 12 :
9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-imine (CRL 8290) 100 mg (0,353 mmol) d'ascididémine sont mis en solution dans une solution contenant 5 ml d'ammoniaque et 2 ml d'EtOH. Le milieu réactionnel est porté à reflux (réfrigérant bouché) pendant 72 heures. Après évaporation du solvant à l'évaporateur rotatif, le résidu est purifié par flash-chromatographie sur alumine (CH2Cl2/MeOH 99 : 1 ), pour donner 87 mg du composé CRL 8290. • Rendement =87 %
• Point de fusion > 260 °C
• RMN 1 H (CDCI3) : 7,61 (dd, 1 H, J = 5 et 8 Hz) ; 7,86 (dd, 1 H J = 8 et 8 Hz) ; 7,97 (dd, 1 H, J = 8 et 8 Hz) ; 8,40 (d, 1 H, J = 8 Hz) ; 8,43 (d, 1 H, J = 6 Hz) ; 8,64 (d, 1 H, J = 8 Hz) ; 9,04 (dd, 1 H, J = 8 et 2,5 Hz) ; 9,08 (dd, 1 H, J = 5 et 2,5 Hz) ; 9,22 (d, 1 H, J = 6 Hz), 12,48 (s, 1 H).
EXEMPLE 13 :
9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-oxime
(CRL 8292)
500 mg (1 ,77 mmol) d'ascididémine et 500 mg de NH2OH, 1/2 H2SO4 sont mis en solution dans 1 ml de pyridine et 10 ml d'EtOH. Le milieu réactionnel est porté à reflux pendant 48 heures. Après évaporation du solvant à l'évaporateur rotatif, 20 ml d'eau sont ajoutés, et le milieu est extrait au HCCI3 (3 fois 20 ml).
Les phases organiques sont séchées sur MgSθ4 puis évaporées à l'évaporateur rotatif. Le résidu est purifié par flash-chromatographie sur alumine (CH2Cl2/MeOH 99 : 1 ) pour donner 240 mg de l'oxime CRL 8292 sous forme de poudre jaune.
• Rendement = 46 %
• Point de fusion > 260 °C
• RMN H (CDCI3) : 7,68 (dd, 1 H, J = 4,4 et 8,4 Hz) ; 7,98 (ddd, 1 H, J = 7,6 et 7,6 et 1 ,6 Hz) ; 8,07 (ddd, 1 H, J = 7,6 et 7,6 et 1 ,6 Hz) ; 8,30 (dd, 1 H, J = 7,6 et 1 ,6 Hz) ; 8,56 (d, 1 H, J = 6 Hz) ; 8,75 (dd, 1 H, J = 7,6 et 1 ,6 Hz) ; 9,00 (dd, 1 H, J = 8,4 et 1 ,2 Hz) ; 9,12 (dd, 1 H, J = 4,4 et 1 ,2 Hz) ; 9,41 (d, 1 H , J = 6 Hz).
• RMN 13c (CDCI3) : 1 15,06 ; 1 16,14 ; 123,16 ; 123,29 ; 125,46 ; 128,33 ; 128,77 ; 129,54 ; 131 ,86 ; 132,16 ; 138,48 ; 140,94 ; 141 ,37 ; 145,82 ; 146,75 ; 151 ,27 ; 151 ,65 ; 151 ,80.
• SM (m/z) : 298 (64,5); 268 (100); 266 (21 ,9).
EXEMPLE 14 :
10-(2-acétylanilino)-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8333)
Préparation selon le procédé décrit dans l'exemple 1 à partir du tétracycle intermédiaire B7 (0,4 g, 0,98 mmol) et du diméthylformamide diéthyl acétal (0,6
ml, 3,43 mmol) dans le DMF (4 ml). Chlorure d'ammomium (1 ,2 g, 22,4 mmol), éthanol (20 ml). Après purification par flash-chromatographie (CH2Cl2/MeOH 100 : 5), on obtient 144 mg du composé CRL 8333 attendu sous forme d'un solide rouge-brun. • Rendement = 35 %.
• Point de fusion > 260 °C.
• RMN 1 H (CDCI3) : 2,88 (s, 3H), 3,12 (s, 3H), 5,54 (d, 1H), 7,13 (d, 1 H, J = 6 Hz), 7,30 (ddd, 1 H, J = 7,6 et 7,6 et 1 ,2 Hz), 7,45 (ddd, 1 H, J = 7,6 et 7,6 et 1 ,2 Hz), 7,51 (d, 1 H, J = 7,6 Hz), 7,65 (s large, 1 H), 7,69 (d, 1 H, J = 7,6 Hz), 7,88 (ddd, 1 H, J = 7,6 et 7,6 et 1 ,2 Hz), 7,96 (ddd, 1 H, J = 7,6 et 7,6 et 1 ,2 Hz), 8,48 (d, 1 H, J = 6 Hz), 8,51 (d, 1 H, J = 6 Hz), 8,57 (dd, 1 H, J = 7,6 et 1 ,2 Hz), 8,64 (dd, 1 H, J = 7,6 et 1 ,2 Hz), 9,23 (d, 1 H, J = 6 Hz).
• RMN 3c (CDCI3) : 37,22 ; 45,05 ; 109,94, 113,94 ; 116,56 ; 117,38 ; 122,92
; 123,34 ; 125,43 ; 125,90 ; 129,47 ; 130,27 ; 131 ,61 ; 132,94 ; 135,87 ; 137,17 ; 137,57 ; 145,87 ; 146,93 ; 149,81 ; 150,28 ; 153,30 ; 154,27 ; 154,54 ; 154,61 ; 183,73.
EXEMPLE 15 : Diiodure de 10-hydroxy-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8369)
500 mg (1 ,597 mmol) de composé de l'exemple 10 (CRL 8368) et 40 ml d'acide acétique dans 100ml d'acide iodhydrique (57 %) sont chauffés à 100 °C pendant 30 min. Après refroidissement, le milieu réactionnel est versé dans 500 ml d'eau additionnée de glace puis neutralisé par NaHCθ3 (solide). Après plusieurs extractions par un mélange de 5 % de MeOH dans HCCI3 (6 fois 500 ml), les phases organiques sont séchées sur MgSθ4 puis concentrées à l'évaporateur rotatif pour donner 0,36 g du composé CRL 8369 sous forme de poudre lie-de-vin. • Rendement = 41 %
• Point de fusion > 260 °C
. RMN 1 H (DMSO) 6,24 (d, 1 H, J = 7,6 Hz) ; 6,86 (td, 1 H, J = 8 et 4 Hz) ; 7,27 (d, 2H, J = 4 Hz) ; 7,57 (d, 1 H, J = 5,2 Hz) ; 7,89 (d, 1 H, J = 8 Hz) ; 7,93 (dd, 1 H, J = 7,6 et 7,6 Hz) ; 8,51 (d, 1 H, J = 5,2 Hz) ; 9,54 (s, 1 H) ; 12,62 (m large, 1 H) ; 14,42 (s, 1 H).
• RMN 13C RMN (DMSO) 107,81 ; 109,87 ; 114,24 ; 115,36 ; 116,31 ; 117,33 ; 120,11 ; 120,97 ; 124,14 ; 127,63 ; 132,18 ; 132,81 ; 134,89 ; 139,24 ; 139,35 ; 141 ,15 ; 148,72 ; 181 ,29.
EXEMPLE 16
10-chloro-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8373)
50 mg (0,09 mmol) de sel de l'exemple 15 (CRL 8369) en solution dans 4 ml de POCI3 sont portés à reflux pendant 2 heures. Après évaporation du POCI3 à l'évaporateur rotatif, le milieu réactionnel est neutralisé par une solution saturée de NaHCθ3. Après plusieurs extractions par un mélange 5 % MeOH dans HCCI3 (5 fois 20 ml), les phases organiques sont séchées sur MgSθ4 puis concentrées à l'évaporateur rotatif. Le résidu obtenu est purifié par flash-chromatographie sur colonne de silice (CH2CI2 / MeOH 95 : 5) pour donner 20 mg du composé CRL 8373attendu sous forme de poudre jaune.
• Rendement = 77 %.
• Point de fusion > 260 °C.
• RMN 1 H (CDCI3) : 7,67 (d, 1 H, J = 5,6 Hz) ; 7,95 (ddd, 1 H, J = 8 et 8 et 0,8 Hz) ; 8,03 (ddd, 1 H, J = 8 et 8 et 1 ,2 Hz) ; 8,57 (d, 1 H, J = 5,6 Hz) ; 8,61 (ddd, 1 H,
J = 8 et 1 ,2 Hz) ; 8,68 (ddd, 1 H, J = 8 et 0,8 Hz) ; 8,97 (d, 1 H, J = 5,6 Hz) ; 9,30 (d, 1 H, J = 5,6 Hz).
• RMN 3c RMN (CDCI3) 117,60 ; 117,84 ; 123,31 ; 123,60 ; 126,69 ; 129,10 ; 131 ,17; 132,38 ; 133,47 ; 138,21 ; 146,24 ; 146,51 ; 147,26 ; 149,40 ; 150,32 ; 154,30 ; 154,94 ; 180,47.
• SM (m/z) : 318 (9,6); 316 (70,2); 290 (29,6) ; 288 (100); 255 (23,4); 253 (26,8).
EXEMPLE 17
5-bromo-10-méthoxy-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8389) Préparation selon le procédé décrit dans l'exemple 1 à partir du tétracycle intermédiaire Bg (0,74 g, 1 ,93 mmol) et de diméthylformamide diéthyl acétal (1 ,3 ml, 7,24 mmol) dans 15 ml de DMF. Chlorure d'ammomium (1 ,96 g, 36,4 mmol), éthanol (200 ml). Après purification par flash-chromatographie (CH2Cl2/MeOH
95: 5), on obtient 210 mg du composé CRL 8389 attendu sous forme de poudre orange.
• Rendement = 42 %
• Point de fusion > 260° C
• RMN 1 H (CDCI3) : 4,14 (s, 3H), 7,14 (d, 1 H, J = 5,6 Hz), 8,05 (dd, 1 H, J = 2 et 8,8 Hz), 8,43 (d, 1 H, J = 6 Hz), 8,44 (d, 1 H, J = 8,8 Hz), 8,76 (d, 1 H, J = 2 Hz), 8,95 (d, 1 H, J = 6 Hz), 9,27 (d, 1 H, J = 5,6 Hz).
• RMN c (CDCI3) : 57,12 ; 109,52 ; 1 17,00 ; 117,76 ; 1 19,46 ; 121 ,58 ; 124,81 ; 125,52 ; 134,72 ; 135,49 ; 137,00 ; 144,85 ; 146,51 ; 147,24 ; 147,92 ; 150,43 ; 156,21 ; 167,98 ; 180,57.
• SM (m/z) : 393 (100); 392 (61.7); 391 (99,2); 390 (17,4) ; 362 (9,2) ; 333 ( 9,8) ; 254 (34,5).
EXEMPLE 18
5-amino-11 -méthoxy-9H-quino[4,3,2de][1 ,10] phénanthrolin-9-one (CRL 8389) Une solution de composé CRL 8389 (0,5 g, 1 ,3 mmol) et de NaN3 (0,5 g, 7,7 mmol) dans 20 ml de DMF est chauffée à 90 °C pendant 10 h. Après concentration, le résidu est repris par KOH 1 N (35 ml) , puis extrait au CH2Cl2/MeOH 95 : 5 (4 x 200 ml). Après séchage sur MgSO4 et concentration à l'évaporateur rotatif, le brut obtenu est purifié par flash-chromatographie sur silice (CH2Cl2/MeOH 80 : 20) pour donner du composé CRL 8389 attendu sous forme de poudre violette (65 mg).
• Rendement = 15 %.
• Point de fusion > 260 °C.
. RMN 1 H (DMSO-d6) : 4,07 (s, 3H) ; 6,62 (s, 2H) ; 7,36 (d, 1 H, J = 8,8 Hz) ; 7,41 (d, 1 H, J = 5,9 Hz) ; 7,74 (s, 1 H) ; 8,08 (d, 1 H, J = 8,8 Hz) ; 8,48 (d, 1 H, J = 5,2 Hz) ; 8,86 (d, 1 H, J = 5,9 Hz) ; 9,08 (d, 1 H, J = 5,2 Hz).
• IR (KBr) : 3420, 3196, 1636, 1616 cm-1.
EXEMPLE 19
Chlorhydrate de la 5-amino-9H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8406)
Une solution de 5-amino-9H-quino[4,3,2-de][1 ,10]phénanthrolin-9-one (1 g,
3,35 mmol) et d'HCI concentré (0,56 ml) dans 200 ml de methanol est agitée à température ambiante pendant 1 h. On ajoute 200 ml d'éther et après avoir laisser précipiter le sel, le milieu est filtré pour récupérer le composé CRL 8406 attendu sous forme de poudre noire (1 g).
• Rendement = 90 %).
• RMN 1 H (DMSO-d6) : 7,44 (dd, 1 H, J = 8,8 et 2,2 Hz) ; 7,81 (d, 1 H, J = 2,2 Hz) ; 7,93 (dd, 1 H, J = 5,6 et 5,9 Hz) ; 8,12 (d, 1 H, J = 8,8 Hz) ; 8,66 (d, 1 H, J = 5,6 Hz) ; 8,75 (d, 1 H, J = 5,9 Hz) ; 9,07 (d, 1 H, J = 5,9 Hz) ; 9,14 (d, 1 H, J = 5,9 Hz).
• IR (KBr) : 3404, 3287, 3170, 1691 , 1676, 1649 cm-1.
EXEMPLE 20
Chlorhydrate de la 5-(diméthylamino)-9H-quino[4,3,2-de][1,10] phénanthrolin-9-one (CRL 8407)
Une solution de 5-(diméthylamino)-9/-/-quino[4,3,2-de][1 ,10]phénanthrolin-9- one (1 g, 3,06 mmol) et d'HCI concentré (0,3 ml) dans 120 ml de CHCI3 est agitée à température ambiante pendant 45 min. Après addition de 350 ml d'éther, puis précipitation du sel, le milieu est filtré pour récupérer le produit attendu sous forme de poudre bleu marine (0,97 g).
• Rendement = 87 %.
• Point de fusion > 260 °C.
EXEMPLE 21
Chlorhydrate de la 5-(benzylamino)-9fY-quino[4,3,2-de][1,10] phénanthrolin-9-one (CRL 8416) Une solution de 5-(benzylamino)-9H-quino[4,3,2-de][1 ,10]phénanthrolin-9-one
ASC20 (0,94 g, 2,42 mmol) et d'HCI concentré (0,2 ml) dans 40 ml de CHCI3 est agitée à température ambiante pendant 30 min. On évapore le solvant et on ajoute 150 ml d'éther et après avoir laisser précipiter le sel, on filtre pour récupérer le du composé CRL 8416 attendu sous forme de poudre noire (0,98 g). • Rendement = 95 %
• Point de fusion > 260 °C.
EXEMPLE 22
5-(diméthylamino-2-éthyl)amino-9H-quino[4,3,2de][1 ,10] phénanthrolin-9- one (CRL 8419)
A un mélange du composé CRL 8347 (2,56 g, 8,59 mmol) et de diméthylformamide de diéthyl acétal (7,9 ml, 43,3 mmol), sont ajoutés 25 ml (166 mmol) d'acide trifluoroacétique à 0°C. Le milieu réactionnel est maintenu sous agitation pendant 5 minutes puis du cyanoborohydrure de sodium (8,2 g, 130 mmol) est ajouté par portions. Le milieu réactionnel est alors chauffé et maintenu à 95°C. Au bout de 18 heures, le mélange est alcalinisé, à pH 8, par une solution saturée de NaHCθ3 (environ 600 mi), puis extrait au CHCl3/MeOH 95: 5 (3 x 800 ml). Les phases organiques sont lavées à l'eau puis séchées sur MgSÛ4 . Après évaporation du solvant à l'évaporateur rotatif, le brut obtenu est purifié par filtration sur alumine (CHCI3 puis CHCl3/MeOH 95) pour donner 1 ,15g du composé CRL 8419 attendu sous forme de poudre noire.
• Rendement = 36 %.
• Point de fusion : se décompose avant de fondre.
• RMN 1 H (CDCI3) : 2,37 (s, 6H), 2,62 (t, 2H, J = 7,32 Hz), 3,70 (t, 2H, J = 7,32 Hz), 7,39 (dd, 1 H, J = 9,2 et 3 Hz), 7,62 (dd, 1 H, J = 8,0 et 4,5 Hz), 7,66 (d,
1 H, J = 3 Hz), 8,35 (d, 1 H, J = 9,2 Hz), 8,38 (d, 1 H, J = 5,7 Hz), 8,79 (dd, 1H, J = 8,0 et 1 ,8 Hz), 9,12 (dd, 1 H, J = 4,5 et 1 ,8 Hz), 9,15 (d, 1 H, J = 5,7 Hz.
• RMN 13c (CDCI3) : 45,97 ; 50,31 ; 56,40 ; 101 ,05 ; 116,81 ; 1 18,48 ; 118,89 ; 125,22 ; 126,30 ; 129,35 ; 134,87 ; 135,97 ; 136,32 ; 138,91 ; 140,55 ; 148,25 ; 148,98 ; 149,69 ; 152,23 ; 154,82 ; 181 ,37 .
• IR (CHCl3) : 1663 cm-1. • SM (m/z) : 369 (100); 354 (15); 236 (37).
EXEMPLE 23
Chlorhydrate de la 5-(diméthylamino-2-éthyl)amino-9H- quino[4,3,2de][1,10] phénanthrolin-9-one (CRL 8418) A 1 ,2g (3,25 mmol) de composé CRL 8419 en solution dans 60 ml de chloroforme, sont ajouté 265μl (3,25 mmol) d'acide chlorhydhque concentré. Le milieu réactionnel est maintenu sous agitation 2 heures à température ambiante. Le précipité formé est filtré puis lavé à l'éther. Le composé CRL 8418 (0.93g) est obtenu sous forme de poudre noire. • Rendement = 70 %.
• RMN 1 H (DMSO àQ) : 2,67 (s, 6H), 3,09 (m, 2H), 4,01 (m, 2H), 7,67 (dm, 1 H,
J = 9,2 Hz), 7,80 (dd, 1 H, J = 8,0 et 4,5 Hz), 7,94 (m, 1 H), 8,26 (d, 1 H, J = 9,2 Hz), 8,64 (d, 1 H, J = 5,7 Hz), 9,09 (m, 1 H), 9,12 (dd, 1 H, J = 4,5 et 1 ,8 Hz), 9,14 (d, 1 H, J = 5,7 Hz).
EXEMPLE 24
5-bis(2-chloroéthyl)amino-9H-quino[4,3,2de][1 ,10] phénanthrolin-9-one (CRL 8422)
A une solution de 5-amino-9H-quino[4,3,2-de][1 ,10]phénanthrolin-9-one (1 g, 3,95 mmol) et de chloroacetaldehyde (50 % aqueux, 2,6 ml ; 16,8 mmol) dans l'acide acétique (30 ml), sont ajoutées, par petites portions et à 0°C, 10 mmol de cyanoborohydrure de sodium NaBH3CN (0,63 g). Le milieu réactionnel est maintenu sous agitation à 0 °C pendant 5 minutes, puis à température ambiante pendant 30 minutes. Ensuite, le milieu est alcalinisé à l'aide d'une solution saturée de carbonate acide de sodium NaHC03, puis extrait par un mélange CHCl3/MeOH 95 : 5. Les phases organiques sont séchées sur sulfate de magnésium MgSθ4 et concentrées à l'évaporateur rotatif. Le brut obtenu est
purifié par filtration sur silice (CHCI3 puis CHCl3/MeOH 99 : 1 ) pour donner deux composés : CRL 8422 et CRL 8423 (décrit dans l'exemple 25).
La 5-bis(chloroéthyl)amino-9H-quino[4,3,2de][1 ,10] phénanthrolin-9-one (CRL 8422) a été obtenue sont forme de poudre rose (0,14 g) : • Rendement = 10%.
• Point de fusion = 220 °C.
• IR (KBr) :1666 ; 1650 cm"1 .
• RMN 1 H (CDCI3) : 3,83 (t, 4H, J = 7,0 Hz) ; 4,04 (t, 4H, J = 7,0 Hz) ; 7,47 (dd, 1 H, J = 9,5 et 2,9 Hz) ; 7,66 (dd, 1 H, J = 8,0 et 4,4 Hz) ; 7,70 (d, 1 H, J = 2,9 Hz) ; 8,42 (d, 1 H, J = 5,6 Hz) ; 8,50 (d, 1 H, J = 9,5 Hz) ; 8,81 (dd, 1 H, J = 8,0 et 1 ,8 Hz) ; 9,16 (dd, 1 H, J = 4,4 et 1 ,8 Hz) ; 9,23 (d, 1 H, J = 5,6 Hz).
. RMN c (CDCI3) : 40,16 ; 53,60 ; 101 ,70 ; 1 16,60 ; 1 18,37 ; 1 18,68 ; 125,39 ; 125,91 ; 129,25 ; 135,13 ; 136,12 ; 136,38 ; 139,42 ; 141 ,93 ; 148,24 ; 148,73 ; 149,34 ; 152,22 ; 155,08 ; 181 ,43.
EXEMPLE 25
5-(2-chioroéthyl)amino-9H-quino[4,3,2de][1 ,10] phénanthrolin-9-one (CRL 8423)
Selon le procédé décrit dans l'exemple 24, 0,22 g de composé CRL 8423 sont obtenus sous forme de poudre violette. Les caractéristiques du composé CRL 8423 sont les suivantes :
• Rendement = 18 %.
• Point de fusion = 196°C.
• IR (KBr) 3413 ; 3275 ; 1654 ; 1617cm"1. • RMN 1 H (CDCI3) : 3,81 (t, 2H, J = 5,5 Hz) ; 3,88 (t, 2H, J = 5,5 Hz) ; 5,01
(slarge, 1 H) ; 7,34 (dd, 1 H, J = 8,8 et 2,5 Hz) ; 7,60 (d, 1 H, J = 2,5 Hz) ; 7,65 (dd, 1 H, J = 7,5 et 4,4 Hz) ; 8,41 (d, 1 H, J = 5,8 Hz) ; 8,43 (d, 1 H, J = 8,8 Hz) ; 8,82 (dd, 1 H, J = 7,5 et 1 ,5 Hz) ; 9,15 (dd, 1 H, J = 4,4 et 1 ,5 Hz) ; 9,21 (dd, 1 H, J = 5,8 Hz).
• RMN 13c (CDCI3) : 42,83 ; 45,01 ; 100,76 ; 116,81 ; 118,78 ; 120,85 ;
125,38 ; 126,35 ; 129,35 ; 135,04 ; 136,04 ; 136,43 ; 140,22 ; 141 ,56 ; 148,49(2C); 149,41 ; 152,30 ; 155,07 ; 181 ,57.
EXEMPLE 26
12-méthoxy-9-r7-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8472)
A une suspension du 2-méthoxy-1 1 -méthyl-1 ,6-diazanaphtacène-5, 12- dione (Intermédiaire Bg) (0,23 g, 0,75 mmol) dans le DMF (7 ml), sont ajoutés 0,54 ml (3 mmol) de diméthylformamide diéthylacétal sous N2. Le milieu réactionnel est chauffé à 120 °C pendant 1 h. Après concentration sous vide, de l'éthanol (45 ml) et du NH CI (0,46 g) sont ajoutés puis le mélange est porté à reflux pendant 30 min. Après concentration à l'évaporateur rotatif, 30 ml d'eau sont ajoutés puis le milieu est extrait au CHCI3 (2 x 30 ml). Les phases organiques sont séchées sur MgS0 et concentrées. Le brut obtenu est purifié par flash-chromatographie sur silice (CHCI3) pour donner le composé CRL 8472 attendu sous forme de poudre marron (50 mg).
• Rendement = 21 %.
• Point de fusion > 260 °C. • 1H RMN (CDCI3) : 4,31 (s, 3H) ; 7,04 (d, 1 H, J = 8,5 Hz) ; 7,92 (ddd, 1 H,
J = 8,1 , 7,0 et 1 ,5 Hz) ; 8,00 (ddd, 1 H, J = 8,4, 7,0 et 1 ,5 Hz) ; 8,52 (d, 1 H, J = 5,5 Hz) ; 8,63 (dd, 1 H, J = 8,1 et 1 ,5 Hz) ; 8,66 (d, 1 H, J = 8,5 Hz) ; 8,67 (dd, 1 H, J = 8,4 et 1 ,5 Hz) ; 9,27 (d, 1 H, J = 5,5 Hz).
• 13C RMN (CDCI3) : 54,69 ; 1 14,43 ; 1 16,75 ; 118,15 ; 122,92 ; 123,41 ; 124,51 ; 130,62 ; 131 ,81 ; 133,15 ; 137,86 ; 139,17 ; 145,80 ; 146,28 ; 149,59 ;
149,99 ; 152,24 ; 167,75 ; 181 ,04.
• IR (CHCl3) : 1671 ; 1588 cm-1 .
• MS : m/z 313 (50) ; 312 (91 ) ; 284 (17) ; 283 (100) ; 254 (23) ; 193 (51 ).
EXEMPLE 27
4-bromo-5-amino-9-H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8478)
A une suspension de composé CRL 8347 (0,2 g, 0,67 mmol) dans l'acide acétique (8 ml), on ajoute du brome (35 μL, 0,67 mmol). Le milieu réactionnel est chauffé à 50 °C pendant 6 h. Après concentration, le milieu est alcalinisé à la soude 5N (20 ml) puis extrait avec un mélange 5 % MeOH/CHCl3 (400 ml). Après séchage sur MgSO4 et évaporation du solvant, le composé CRL 8478 est obtenu sous forme d'une poudre violette qui est recristallisée dans un mélange CHCI3/pentane 20 ml/15 ml (152 mg).
• Rendement = 61 %.
• Point de fusion > 260 °C. • 1H RMN (DMSO-d6) : 7,07 (slarge, 2H) ; 7,61 (d, 1 H, J = 8,8 Hz) ; 7,77
(dd, 1 H, J = 7,7 et 4,0 Hz) ; 8,18 (d, 1 H, J = 8,8 Hz) ; 8,61 (d, 1 H, J = 7,7 Hz) ; 9,10 (d, 1 H, J = 4,0 Hz) ; 9,14 (d, 1 H, J = 5,9 Hz) ; 9,91 (d, 1 H, J = 5,9 Hz).
• IR (CHCI3) : 3501 ; 3400 ; 1673 cm"1 .
• MS : m/z 378 (42) ; 377 (100) ; 376 (48) ; 375 (27).
EXEMPLE 28
11 -acétoxyméthyl-9-H-quino[4,3,2-de][1 ,10] phénanthrolin-9-one (CRL 8528)
A une suspension du 3-acétoxyméthyl-1 1 -méthyl-1 , 6-diazanaphtacène-
5,12-dione (Intermédiaire B10) (0,1 1 g, 0,31 mmol) dans le DMF (4 ml), on ajoute sous N2_ du diméthylformamide diéthylacétal (0,27 ml, 1 ,5 mmol). Le milieu réactionnel est chauffé à 120 °C pendant 1 h. Après concentration sous vide, de l'éthanol (25 ml) et du NH4CI (0,23 g) sont ajoutés puis le mélange est porté à reflux pendant 30 min. Après concentration à l'évaporateur rotatif, 30 ml d'eau sont ajoutés, puis le milieu est extrait au CHCI3 (2 x 30 ml). Les phases organiques sont séchées sur MgS04. Après évaporation du solvant à l'évaporateur rotatif, et purification par flash-chromatographie sur silice (CHCI3), 65 mg de composé CRL 8528 sont obtenus.
• Rendement = 60 %.
• Point de fusion = 206-210 °C. • 1H RMN (CDCI3) : 2,19 (s, 3H) ; 5,32 (s, 2H) ; 7,96 (ddd, 1 H, J = 1 , 1 ,8 et
8 Hz) ; 8,03 (ddd, 1 H, J = 1 , 1 ,8 et 8,4 Hz) ; 8,56 (d, 1 H, J = 5,5 Hz) ; 8,64 (dd,
1H, J = 1 ,1 et 8,4 Hz) ; 8,71 (dd, 1 H, J = 1 ,1 et 8,0 Hz) ; 8,77 (d, 1 H, J = 2,4 Hz) ; 9,14 (d, 1 H, J = 2,4 Hz) ; 9,28 (d, 1 H, J = 5,5 Hz).
• MS : m/z 355 (88) ; 313 (100) ; 296 (25) ; 267 (7).
EXEMPLE 29
9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8529)
A une suspension de 6-méthyl-1 ,11-diaza-naphtacène-5,12-dione (Intermédiaire D- ) (52 mg, 0,875 ml), le diméthylformamide diéthylacétal (0,15 ml, 0,875 mmoles) est ajouté sous N2. Le milieu réactionnel est chauffé à 120 °C pendant 30 min. Après concentration sous vide, de l'éthanol (60 ml) et du NH4CI (0,34 g) sont ajoutés et le mélange est porté à reflux pendant 30 min. Après concentration à l'évaporateur rotatif, 10 ml d'eau sont ajoutés puis le milieu extrait au CH2CI2 (2 x 10 ml). Les phases organiques sont séchées sur MgSO et concentrées. Le brut obtenu est purifié par flash-chromatographie sur silice (CH2CI2/MeOH 99 : 1) pour donner le composé CRL 8529 attendu sous forme de solide jaune (6 mg).
• Rendement = 11 %.
• 1H RMN (CDCI3) : 7,78 (dd, 1 H, J = 8,1 et 4,8 Hz) ; 7,97 (ddd, 1 H, J = 8,0, 7,4 et 1 ,2 Hz) ; 8,04 (ddd, 1 H, J = 8,0, 7,4 et 1 ,2 Hz) ; 8,51 (d, 1 H, J = 5,9 Hz) ; 8,69 (dd, 2H, J = 8,0 et 1 ,5 Hz) ; 9,08 (dd, 1 H, J = 4,8 et 1 ,9 Hz) ; 9,13 (d, 1 H, J = 5,9 Hz) ; 9,27 (d, 1 H, J = 1 ,9 et 8,1 Hz).
• 13C RMN (CDCI3) : 1 1 5, 15 ; 121 ,76 ; 122,28 ; 127,13 ; 127,16 ; 129,65 ; 130,82 ; 132,21 ; 132,60 ; 132,99 ; 136,88 ; 139,91 ; 144,85 ;148,00 ; 148,03 ; 151 ,75 ; 151 ,84 ; 179,94. • MS : m/z 283 (54) ; 255 (100) ; 228 (10).
EXEMPLE 30
5-bromo-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8839)
Le composé CRL 8839 est préparé selon la procédure décrite dans l'exemple 8 à partir de la 9-H-quino[4,3,2-de][1 ,7]phénanthrolin-9-one (CRL 8529) (0,5g, 1 ,77 mmol) ; 20 ml d'acide acétique ; solution de brome (0.2ml, 3,88 mmol/5ml d'acide acétique) ; reflux 24 heures.
EXEMPLE 31
5-amino-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8836)
Le composé CRL 8836 est préparé selon la procédure décrite dans l'exemple 9 à partir de la 5-bromo-9-/-/-quino[4,3,2-de][1 ,7]phénanthrolin-9-one (CRL 8839) (1 ,15g, 18 mmol) ; 250 ml de DMF ; azidure de sodium (1 ,2g, 1 ,85 mmol) ; reflux 4 heures.
EXEMPLE 32 5-(diméthylamino-2-éthyl)amino-9-W-quino[4,3,2-de][1 ,7] phénanthrolin-9- one (CRL 8840)
Le composé CRL 8840 est préparé selon la procédure décrite dans l'exemple
22 à partir de la 5-amino-9-H-quino[4,3,2-de][1 ,7]phénanthrolin-9-one (CRL 8836)
(1 ,28g, 4,3 mmol) ; diméthylformamide de diacétal (4 ml, 21 ,9 mmol) ; acide trifluoroacétique (12,5 ml, 83 mmol) ; cyanoborohydrure de sodium (4,1 g, 65 mmol) ; 90 °C pendant 8 heures.
EXEMPLE 33
5-bis(chloroéthylamino-2-éthyl)amino-9-H-quino[4,3,2- de][1,7]phénanthrolin-9-one (CRL 8841)
Le composé CRL8841 est préparé selon la procédure décrite dans l'exemple
24 à partir de la 5-amino-9-/-/-quino[4,3,2-de][1 ,7]phénanthrolin-9-one (CRL 8836)
(1g, 3,95 mmol) ; chloroacetaldehyde (2,6 ml, 16,8 mmol) ; acide acétique (30 ml) ; cyanoborohydrure de sodium (0,63 g, 10 mmol) ; 30 minutes à température ambiante.
EXEMPLE 34
5-(chloroéthylamino-2-éthyl)amino-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9- one (CRL 8842)
Le composé CRL 8842 est également obtenu lors de la procédure décrite dans l'exemple précédant à partir de la 5-amino-9-r7-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8836).
EXEMPLE 35
4-bromo-5-amino-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8843)
Le composé CRL 8843 est préparé selon la procédure décrite dans l'exemple 27 à partir de la 5-amino-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8836) (0,6g, 2,01 mmol) ; 24 ml d'acide acétique ; brome (35 μl, 0,67 mmol) ; 50°C pendant 6 heures.
EXEMPLE 36
7-nitro-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8838)
Le composé CRL 8838 est préparé selon la procédure décrite dans l'exemple 6 à partir de 9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8529) (1 g, 3,53 mmol) ; 23 ml d'acide sulfurique et 23 ml d'acide nitrique ; 130 °C pendant 2 heures.
EXEMPLE 37 7-amino-9-r -quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8837)
Le composé CRL 8837 est préparé selon la procédure décrite dans l'exemple 7 à partir de 7-nitro-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8838) (0,2g ; 0,61 mmol) ; fer (0,19g, 3,38 mmol), 10 ml de mélange d'acide acétique/eau (50/50).
EXEMPLE 38
12-méthoxy-9-H-quino[4,3,2-de][1 ,7] phénanthrolin-9-one (CRL 8844)
Le composé CRL 8844 est préparé selon la procédure décrite dans l'exemple 29 à partir de la 3-méthoxy-6-méthyl-1 ,11-diaza-naphtacène-5,12- dione (Intermédiaire D2) (0,76 g, 25 mmol) ; diacétal de diméthylformamide (2 ml,
11 ,67 mmol) ; 120 °C pendant 30 min ; NH4CI (4,5g) ; 500 ml d'éthanol ; reflux pendant 30 minutes.
Les résultats des essais pharmacologiques, présentés ci-après, mettent en évidence les propriétés cytotoxiques des composés de formules I et la , ainsi que les doses maximales tolérées. Ces données permettent d'apprécier l'intérêt thérapeutique des composés revendiqués.
1- Détermination de la dose maximale tolérée (DMT)
L'évaluation de la dose maximale tolérée a été réalisée chez des souris B6D2F1/Jico âgées de 4 à 6 semaines. Les composés ont été administrés par voie intrapéritonéale à des doses croissantes s'échelonnant de 2,5 à 160 mg/kg. La valeur de la DMT (exprimée en mg/kg) est déterminée à partir de l'observation du taux de survie des animaux sur une période de 14 jours après une administration unique du produit considéré. L' évolution pondérale des animaux est également suivie pendant cette période. Lorsque la valeur de la DMT est supérieure à 160 mg/kg, la valeur de la DMT est assimilée à 160 mg/kg par défaut.
Les résultats de l'estimation de la dose maximale tolérée (DMT) sont rassemblés dans le tableau I suivant :
TABLEAU I
La majorité des produits décrits dans la famille de l'ascididémine ou de son isomère ne présentent pas de toxicité directe (DMT > 160 mg/kg) et peuvent donc être utilisés in vivo à concentrations tissulaires élevées, donc à des posologies fortes.
2- Activité cytotoxique sur des lignées cellulaires tumorales en culture
L'influence des composés de formules I et la sur les cellules néoplasiques a été évaluée à l'aide du test colorimétrique MTT. Le principe du test MTT est basé sur la réduction mitochondriale par les cellules vivantes métaboliquement actives du produit MTT (3-(4,5-diméthylthiazol-2-yl)-2,5 diphényltétrazolium bromide) de couleur jaune en un produit de couleur bleue, le formazan. La quantité de formazan ainsi obtenue est directement proportionnelle à la quantité de cellules vivantes présentes dans le (ou les) puit(s) de culture. Cette quantité de formazan est mesurée par spectrophotométhe.
Les lignées cellulaires sont maintenues en culture monocouche à 37°C dans des boîtes de culture à bouchon fermé contenant du milieu de base MEM 25 MM HEPES (Minimum Essential Médium). Ce milieu, adapté à la croissance d'une gamme de cellules variées diploïdes ou primaires de mammifères, est ensuite additionné : - d'une quantité de 5% de SVF (Sérum de Veau Fœtal) décomplémenté à
56°C pendant 1 heure,
- de 0,6 mg/ml de L-glutamine,
- de 200 lU/ml de pénicilline,
- de 200 mg/ml de streptomycine,
- de 0,1 mg/ml de gentamicine.
Les 12 lignées cellulaires cancéreuses humaines utilisées ont été obtenues auprès de l'American Type Culture Collection (ATCC, Rockville, MD, USA). Ces 12 lignées cellulaires sont :
- U-373MG (code ATCC : HTB-17) et U-87MG (code ATCC : HTB-14) qui sont deux glioblastomes,
- SW1088 (code ATCC : HTB-12) qui est un astrocytome,
- A549 (code ATCC : CCL-185) et A-427 (code ATCC : HTB-53) qui sont deux cancers du poumon non-à-petites-cellules,
- HCT-15 (code ATCC : CCL-225) et LoVo (code ATCC : CCL-229) qui sont deux cancers colorectaux,
- T-47D (code ATCC : HTB-133) et MCF7 (code ATCC : HTB-22) qui sont deux cancers du sein, - J82 (code ATCC : HTB-1 ) et T24 (code ATCC : HTB-4) qui sont deux cancers de la vessie,
- PC-3 (code ATCC : CRL-1435) qui est un cancer de la prostate.
Au plan expérimental : 100 μl d'une suspension cellulaire contenant 20 000 à 50 000 (selon le type cellulaire utilisé) cellules/ml de milieu de culture sont ensemencés en plaques multi-puits de 96 puits à fond plat et sont mis à incuber à
37°C, sous atmosphère comprenant 5% de CO2 et 70% d'humidité. Au bout de
24 heures d'incubation, le milieu de culture est remplacé par 100 μl de milieu frais contenant soit les différents composés à tester à des concentrations variant de
-5 -10 10 à 10 M soit le solvant ayant servi à la mise en solution des produits à tester (condition contrôle). Après 72 heures d'incubation dans les conditions précédentes, le milieu de culture est remplacé par 100 μl d'une solution jaunâtre de MTT dissous à raison de 1 mg/ml dans du RPMI 1640. Les microplaques sont remises à incuber pendant 3 heures à 37°C puis centrifugées pendant 10 minutes
à 400 g. La solution jaunâtre de MTT est éliminée et les cristaux de formazan bleu formés au niveau cellulaire sont dissous dans 100 μl de DMSO. Les microplaques sont ensuite mises sous agitation pendant 5 minutes. L'intensité de la coloration bleue résultant donc de la transformation du produit MTT jaune en formazan bleu par les cellules encore vivantes au terme de l'expérience est quantifiée par spectrophotométrie à l'aide d'un appareil de type DYNATECH IMMUNOASSAY SYSTEM aux longueurs d'onde de 570 nm et 630 nm correspondant respectivement aux longueurs d'ondes d'absorbance maximale du formazan et au bruit de fond. Un logiciel intégré au spectrophotomètre calcule les valeurs moyennes de densité optique ainsi que les valeurs de déviation standard (Dév. Std.) et d'erreur standard sur la moyenne (ESM).
L'activité inhibitrice de la croissance cellulaire des composés de formules I et la sur les différentes lignées cellulaires tumorales a été comparée à celle du produit naturel, l'ascididémine (CRL 8274). L'ensemble des composés présentent une activité inhibitrice significative de la prolifération cellulaire des 12 lignées tumorales humaines : U-87MG, U-373MG, SW 1088, T24, J82, HCT-15, LoVo, MCF7, T-47D, A549, A-427 et PC-3 avec une concentration inhibitrice 50 % (Cl
50) qui est comprise entre 10-^M et 10"1θM, selon les composés et les lignées tumorales testés. A titre d'exemple, les valeurs des concentrations encadrant les CI50 obtenues sur les différentes lignées cellulaires sont données dans le tableau II :
TABLEAU II
Concentrations inhibitrices (M) encadrant la valeur de la concentration inhibitrice 50 % des composes de formule I, II (*) ou III (**) sur les lignées cellulaires
On donnera, dans le tableau III, les résultats des CI50 (en nM) moyennes (calculées à partir de l'activité cytotoxique sur les 12 lignées tumorales étudiées) et les ratios DMT/CI50 (ces ratios sont calculés en faisant le rapport des DMT et des CI50 exprimées en nombres sans dimension).
TABLEAU III
* : le ratio DMT/IC50 des différents composés a été estimé en prenant comme référence un ratio égal à 1 pour l'ascididémine.
Les composés décrits présentent, sur les modèles de lignées cellulaires tumorales des Cl 50 (nM) supérieures ou équivalentes à celle de l'ascididémine.
A l'exception du CRL 8289 (dont la dose maximale tolérée est équivalente à celle de l'ascididémine mais dont la CI50 est dix fois inférieure à celle de l'ascididémine), les doses maximales tolérées des composés décrits, considérées par défaut équivalente à 160 mg/kg, sont nettement supérieures à celle de l'ascididémine (20 mg/kg). Ces résultats suggèrent que cette nouvelle famille de composés ne présentent pas de toxicité directe. En conséquence, les ratios tolérance/activité cytotoxique des composés exemplifiés dans la présente invention, sont nettement supérieurs à celui de l'ascididémine naturelle. Ces composés peuvent donc être utilisés comme médicament anti-tumoraux, pour leurs propriétés cytotoxiques à des concentrations tissulaires plus élevées que celles induites par l'ascididémine naturelle. Ils sont donc caractérisés par une meilleure maniabilité thérapeutique. Le CRL 8289 dont la CI50 de 10 nM présente également une meilleure maniabilité thérapeutique que l'ascididémine.
Grâce à leurs propriétés cytotoxiques, les composés de formules I et la tels que décrits ou sous forme de sels ou solvates pharmaceutiques acceptables, peuvent être utilisés comme principes actifs de médicaments.
Les composés de formules I et la sont généralement administrés en unités de dosage établies soit par m2 de surface corporelle, soit par kg de poids. Les dites unités de dosage sont de préférence formulées dans des compositions pharmaceutiques dans lesquelles le principe actif est mélangé avec un (ou plusieurs) excipient(s) pharmaceutique(s).
Les composés de formules I et la peuvent être utilisés selon la pathologie cancéreuse du sujet à traiter à des doses comprises entre 0,05 et 350 mg/m2 de surface corporelle, de préférence à des doses de 0,5 à 50 mg/m /jour pour un traitement curatif dans sa phase aiguë en fonction du nombre de cycles de
traitement de chaque cure. Pour un traitement d'entretien, on utilisera avantageusement les composés de formules I et la à des doses de 0,05 à
25 mg/m2/jour, de préférence à des doses de 0,1 à 1 ,5 mg/m2/jour selon le nombre de cycles de traitement de la cure. Ils pourront être associés aux médicaments anti-tumoraux utilisés dans les protocoles validés de polychimiothérapie intensive.
Dans les compositions pharmaceutiques de la présente invention pour l'administration par voie orale, intraveineuse, les principes actifs peuvent être administrés sous formes unitaires d'administration, en mélange avec des supports pharmaceutiques classiques adaptés à la thérapeutique humaine. Les formes unitaires d'administration appropriées comprennent les formes par voie orale telles que les comprimés, éventuellement sécables, ou les gélules, les implants et les formes d'administration intraveineuse.
Pour une administration parentérale (perfusion intraveineuse à débit constant), on utilise des suspensions aqueuses stériles, des solutions salines isotoniques stériles ou des solutions stériles et injectables qui contiennent des agents de dispersion et ou des agents solubilisants pharmacologiquement compatibles, par exemple le propylèneglycol, le polyéthyièneglycol, ou une β cyclodextrine.
Ainsi, pour préparer une solution aqueuse injectable par voie intraveineuse et destinée à une perfusion réalisée sur 1 à 24 h, on peut utiliser un cosolvant : un alcool tel que l'éthanol, un glycol tel que le polyéthyièneglycol ou le propylèneglycol et un tensioactif hydrophile tel que le Tween 80.
Lorsque l'on prépare une composition solide sous forme de comprimés, on peut ajouter au principe actif, micronisé ou non, un agent mouillant tel que le laurylsulfate de sodium et on mélange le tout avec un véhicule pharmaceutique tel que la silice, la gélatine, l'amidon, le lactose, le stéarate de magnésium, le talc, la gomme arabique ou analogues. On peut enrober les comprimés de saccharose, de divers polymères ou d'autres matières appropriées ou encore les traiter de telle sorte qu'ils aient une activité prolongée ou retardée et qu'ils libèrent d'une façon continue une quantité prédéterminée de principe actif.
On obtient une préparation en gélules en mélangeant le principe actif avec un diluant tel qu'un glycol ou un ester de glycérol et en incorporant le mélange obtenu dans des gélules molles ou dures.
Le principe actif peut être formulé également sous forme de microcapsules ou microsphères, éventuellement avec un ou plusieurs supports ou additifs.
Le principe actif peut être également présenté sous forme de complexe avec une cyclodextrine, par exemple α-, β- ou γ-cyclodextrine, 2-hydroxypropyl-β- cyclodextrine ou méthyl-β-cyclodextrine.
Les composés de formules I seront utilisés dans le traitement de la plupart des tumeurs solides du fait de leurs activités cytotoxiques puissantes, en particulier pour traiter les tumeurs cérébrales, les cancers du poumon, les tumeurs de l'ovaire et du sein, les cancers de l'endomètre, les cancers colo-rectaux, les cancers de la prostate et les tumeurs testiculaires.