WO2001017956A1 - Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity - Google Patents
Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity Download PDFInfo
- Publication number
- WO2001017956A1 WO2001017956A1 PCT/GB2000/003314 GB0003314W WO0117956A1 WO 2001017956 A1 WO2001017956 A1 WO 2001017956A1 GB 0003314 W GB0003314 W GB 0003314W WO 0117956 A1 WO0117956 A1 WO 0117956A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- hydroxy
- methyl
- alkyl
- chloro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1cc(N)c(*)c(*)c1SC#N Chemical compound *c1cc(N)c(*)c(*)c1SC#N 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/12—Antidiuretics, e.g. drugs for diabetes insipidus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/32—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C317/34—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having sulfone or sulfoxide groups and amino groups bound to carbon atoms of six-membered aromatic rings being part of the same non-condensed ring or of a condensed ring system containing that ring
- C07C317/38—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having sulfone or sulfoxide groups and amino groups bound to carbon atoms of six-membered aromatic rings being part of the same non-condensed ring or of a condensed ring system containing that ring with the nitrogen atom of at least one amino group being part of any of the groups, X being a hetero atom, Y being any atom, e.g. N-acylaminosulfones
- C07C317/42—Y being a hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
- C07C317/48—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton the carbon skeleton being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C317/50—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton the carbon skeleton being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups at least one of the nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/64—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton
- C07C323/65—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton containing sulfur atoms of sulfone or sulfoxide groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/12—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms
- C07D295/135—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms with the ring nitrogen atoms and the substituent nitrogen atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
Definitions
- the present invention relates to compounds which elevate pyruvate dehydrogenase (PDH) activity, processes for their preparation, pharmaceutical compositions containing them as active ingredient, methods for the treatment of disease states associated with reduced PDH activity, to their use as medicaments and to their use in the manufacture of medicaments for use in the elevation of PDH activity in warm-blooded animals such as humans, in particular the treatment of diabetes mellitus, peripheral vascular disease and myocardial ischaemia in warm-blooded animals such as humans, more particularly to their use in the manufacture of medicaments for use in the treatment of diabetes mellitus in warm-blooded animals such as humans.
- PDH pyruvate dehydrogenase
- adenosine triphosphate provides the energy for synthesis of complex molecules and, in muscle, for contraction. ATP is generated from the breakdown of energy-rich substrates such as glucose or long chain free fatty acids. In oxidative tissues such as muscle the majority of the ATP is generated from acetyl CoA which enters the citric acid cycle, thus the supply of acetyl CoA is a critical determinant of ATP production in oxidative tissues. Acetyl CoA is produced either by ⁇ -oxidation of fatty acids or as a result of glucose metabolism by the glycolytic pathway.
- the key regulatory enzyme in controlling the rate of acetyl CoA formation from glucose is PDH which catalyses the oxidation of pyruvate to acetyl CoA and carbon dioxide with concomitant reduction of nicotinamide adenine dinucleotide (NAD) to NADH.
- PDH nicotinamide adenine dinucleotide
- NIDDM non-insulin dependent
- IDDM insulin-dependent diabetes mellitus
- oxidation of lipids is increased with a concomitant reduction in utilisation of glucose, which contributes to the hyperglycaemia.
- Reduced glucose utilisation in both IDDM and NIDDM is associated with a reduction in PDH activity.
- PDH activity may be that an increase in pyruvate concentration results in increased availability of lactate as a substrate for hepatic gluconeogenesis. It is reasonable to expect that increasing the activity of PDH could increase the rate of glucose oxidation and hence overall glucose utilisation, in addition to reducing hepatic glucose output.
- Oxidation of glucose is capable of yielding more molecules of ATP per mole of oxygen than is oxidation of fatty acids.
- energy demand may exceed energy supply, such as myocardial ischaemia, intermittent claudication, cerebral ischaemia and reperfusion, (Zaidan et al., 1998; J. Neurochem. 70: 233-241)
- shifting the balance of 5 substrate utilisation in favour of glucose metabolism by elevating PDH activity may be expected to improve the ability to maintain ATP levels and hence function.
- An agent which is capable of elevating PDH activity may also be expected to be of benefit in treating conditions where an excess of circulating lactic acid is manifest such as in certain cases of sepsis.
- PDH is an intramitochondrial multienzyme complex consisting of multiple copies of several subunits including three enzyme activities El, E2 and E3, required for the completion of the conversion of pyruvate to acetyl CoA (Patel and Roche 1990; FASEB J., 4: 3224-3233).
- El catalyses the non-reversible removal of CO 2 from pyruvate; E2 forms acetyl CoA and E3 0 reduces NAD to NADH.
- Two additional enzyme activities are associated with the complex: a specific kinase which is capable of phosphorylating El at three serine residues and a loosely-associated specific phosphatase which reverses the phosphorylation.
- Phosphorylation of a single one of the three serine residues renders the El inactive.
- the proportion of the PDH in its active (dephosphorylated) state is determined by a balance between the activity of the kinase 5 and phosphatase.
- the activity of the kinase may be regulated in vivo by the relative concentrations of metabolic substrates such as NAD/NADH, CoA/acetylCoA and adenine diphosphate (ADP)/ATP as well as by the availability of pyruvate itself.
- European Patent Publication Nos. 617010 and 524781 describes compounds which are capable of relaxing bladder smooth muscle and which may be used in the treatment of urge 0 incontinence. We have found that the compounds of the present invention are very good at elevating PDH activity, a property nowhere disclosed in EP 0617010 and EP 524781.
- the present invention is based on the surprising discovery that certain compounds elevate PDH activity, a property of value in the treatment of disease states associated with disorders of glucose utilisation such as diabetes mellitus, obesity, (Curto et al., 1997; Int. J. Obes. 21: 1137-1142), and lactic acidaemia. Additionally the compounds may be expected to have utility in diseases where supply of energy-rich substrates to tissues is limiting such as peripheral vascular disease, (including intermittent claudication), cardiac failure and certain cardiac myopathies, muscle weakness, hyperlipidaemias and atherosclerosis (Stacpoole et al., 1978; N. Engl. J. Med. 298: 526-530). A compound that activates PDH may also be useful in treating Alzheimer's disease (AD) (J Neural Transm (1998) 105, 855-870).
- AD Alzheimer's disease
- R 1 is chloro, fluoro, bromo, methyl or methoxy
- R 2 is selected from one of the following three groups: i) halo, nitro, hydroxy, amino or cyano; ii) -X ! -R 5 wherein X 1 is a direct bond, -O-, -S-, -SO-, -SO 2 -, -NR 6 -, -CO-, -CONR 6 -, -NR 6 CO-, -NR 6 SO 2 - or NR 6 CONR 7 -; wherein R 6 and R 7 are independently hydrogen or C 1-4 alkyl optionally substituted with one or more A; and R 5 is selected from C 1-6 alkyl optionally substituted with one or more A, C 3-7 cycloalkyl optionally substituted with one or more A, C 3- cycloalkylC 1-6 alkyl optionally substituted with one or more A, C 2-6 alkenyl optionally substituted with one or more A, C 2 .
- heteroaryl ring is a carbon linked 6-membered ring containing 1-2 nitrogen atoms or a carbon linked 5-membered ring containing 1-3 heteroatoms selected independently from O, N and S; and wherein if said 5-membered heteroaryl ring contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G; iii) a nitrogen-linked 4-8 membered heterocyclic group optionally substituted on a ring carbon by one or more D and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted
- R 3 is C ⁇ - alkyl optionally substituted with one or more A, C 3 . 7 cycloalkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D, a carbon-linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms optionally substituted on a ring carbon by one or more D, or a carbon linked 5-membered heteroaryl ring containing 1-3 heteroatoms selected independently from O, N and S optionally substituted on a ring carbon by one or more D and wherein if said 5-membered heteroaryl ring contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G;
- A is selected from hydroxy, amino, halo, carboxy, N-(C 1-4 alkyl)amino, N,N-di-(C 1-4 alkyl)amino, carbamoyl and C 1-6 alkoxy;
- D is selected from: i) -X a -R c wherein X a is a direct bond, -O-, -S-, -SO-, -SO 2 -, -CO-, - ⁇ R d SO 2 -, -NR d CO-, -NR d CONR e -, -NR d - or -CONR d -; wherein R d and R e are independently hydrogen or C 1-4 alkyl optionally substituted with one or more hydroxy or C 1-4 alkoxy; and R c is selected from hydrogen or C ⁇ aU yl optionally substituted with one or more hydroxy or C 1-4 alkoxy; ii) a 4-8 membered Het which is optionally substituted on a ring carbon with one or more groups selected from hydroxy, halo, C ⁇ -4 alkoxy, C 1-4 alkyl or cyano and wherein if said 4-8 membered Het contains an -NH- moiety
- R 4 is hydrogen or fluoro; or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof.
- alkyl includes both straight and branched chain alkyl groups but references to individual alkyl groups such as “propyl” are specific for the straight chain version only.
- C 1-6 alkyl includes C ⁇ -4 alkyl, C 2-4 alkyl, propyl, isopropyl and t-butyl.
- references to individual alkyl groups such as 'propyl' are specific for the straight chained version only and references to individual branched chain alkyl groups such as 'isopropyl' are specific for the branched chain version only.
- halo refers to fluoro, chloro, bromo and iodo.
- Suitable values for "a carbon-linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms” include pyridyl, pyrimidyl, pyrazinyl and pyridazinyl.
- a carbon-linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms is pyridyl.
- a carbon-linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms is pyridazinyl.
- Suitable values for "a carbon-linked 5-membered heteroaryl ring containing 1-3 heteroatoms" include pyrrolyl, furyl, thienyl, pyrazolyl, oxazolyl, oxadiazolyl, imidazolyl and triazolyl.
- a “nitrogen-linked 4-8 membered heterocyclic group” is a saturated, partially saturated or unsaturated, monocyclic ring containing 4-8 atoms of which at least one is a nitrogen atom with optionally 1-3 further heteroatoms selected independently from O, N and S wherein a -CH 2 - group can optionally be replaced by a -C(O)- and a ring nitrogen and/or sulphur atom may be optionally oxidised to form the N-oxide and or the S-oxides. It will be appreciated that in forming this nitrogen link, the nitrogen atom is not quaternised, i.e. a neutral compound is formed.
- Suitable values for "nitrogen-linked 4-8 membered heterocyclic group” include morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl, homopiperazinyl, pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolidinyl and triazolyl.
- nitrogen-linked 4-8 membered heterocyclic group include azetidinyl, morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl, homopiperazinyl, pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolidinyl and triazolyl.
- a "nitrogen-linked 4-8 membered heterocyclic group” is morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl or homopiperazinyl. More preferably a "nitrogen-linked 4-8 membered heterocyclic group” is azetidinyl, morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl or homopiperazinyl.
- nitrogen-linked 4-8 membered heterocyclic group include azetidinyl, morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, 1-oxothiomorpholino, 1,1-dioxothiomorpholino, pyrrolinyl, homopiperazinyl, pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolidinyl and triazolyl.
- a "nitrogen-linked 4-8 membered heterocyclic group” is morpholino, 1-oxothiomorpholino, 1,1-dioxothiomorpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl or homopiperazinyl. More preferably a "nitrogen-linked 4-8 membered heterocyclic group” is azetidinyl, morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, 1-oxothiomorpholino, 1,1-dioxothiomorpholino, pyrrolinyl or homopiperazinyl.
- R f as a "nitrogen-linked 4-8 membered heterocyclic group” is azetidinyl, morpholino or pyrrolidinyl.
- R 2 is a "nitrogen-linked 4-8 membered heterocyclic group” it is thiomorpholino.
- R 2 is thiomorpholino, piperazinyl, 1-oxothiomorpholino, 1,1-dioxothiomorpholino or morpholino.
- a “nitrogen-linked 4-6 membered heterocyclic group” is a saturated, partially saturated or unsaturated, monocyclic ring containing 4-6 atoms of which at least one is a nitrogen atom with optionally 1-3 further heteroatoms selected independently from O, N and S wherein a -CH 2 - group can optionally be replaced by a -C(O)- and a ring nitrogen and/or sulphur atom may be optionally oxidised to form the N-oxide and or the S-oxides. It will be appreciated that in forming this nitrogen link, the nitrogen atom is not quaternised, i.e. a neutral compound is formed.
- Suitable values for a "nitrogen-linked 4-6 membered heterocyclic group” include azetidinyl, morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl, pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolidinyl and triazolyl.
- a "nitrogen-linked 4-6 membered heterocyclic group” is azetidinyl, morpholino or pyrrolidinyl.
- a “nitrogen-linked 5 or 6 membered heterocyclic group” is a saturated, partially saturated or unsaturated, monocyclic ring containing 5 or 6 atoms of which at least one is a nitrogen atom with optionally 1-3 further heteroatoms selected independently from O, ⁇ and S wherein a -CH 2 - group can optionally be replaced by a -C(O)- and a ring nitrogen and/or sulphur atom may be optionally oxidised to form the N-oxide and or the S-oxides. It will be appreciated that in forming this nitrogen link, the nitrogen atom is not quaternised, i.e. a neutral compound is formed.
- Suitable values for a "nitrogen-linked 5 or 6 membered heterocyclic group” include morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl, pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, pyrazolidinyl and triazolyl.
- a "nitrogen-linked 5 or 6 membered heterocyclic group” is morpholino, piperidyl, piperazinyl, pyrrolidinyl, thiomorpholino or pyrrolinyl.
- a “nitrogen-linked 6 membered heterocyclic group” is a saturated, partially saturated or unsaturated, monocyclic ring containing 6 atoms of which at least one is nitrogen atom with optionally 1-3 further heteroatoms selected independently from O, N and S wherein a -CH 2 - group can optionally be replaced by a -C(O)- and a ring nitrogen and/or sulphur atom may be optionally oxidised to form the N-oxide and or the S-oxides. It will be appreciated that in forming this nitrogen link, the nitrogen atom is not quaternised, i.e. a neutral compound is formed.
- Suitable values for a "nitrogen-linked 6 membered heterocyclic group” include morpholino, piperidyl, piperazinyl, thiomorpholino.
- a "4-8 membered Het” is a saturated, partially saturated or unsaturated monocyclic ring containing 4-8 atoms including 1-4 heteroatoms selected independently from O, ⁇ and S, which may be carbon or nitrogen linked, wherein a -CH 2 - group can optionally be replaced by a -C(O)- and a ring nitrogen and/or sulphur atom may be optionally oxidised to form the N-oxide and or the S-oxides.
- Suitable values for "4-8 membered Het" are morpholino, piperidyl, pyridyl, pyranyl, pyrrolyl, isothiazolyl, thienyl, thiadiazolyl, piperazinyl, thiazolidinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl, homopiperazinyl, tetrahydropyranyl, imidazolyl, pyrimidyl, pyrazinyl, pyridazinyl, isoxazolyl, 4-pyridone, 2-pyrrolidone, 4-thiazolidone, pyridine-N-oxide and quinoline-N-oxide.
- a “5 or 6 membered Het” is a saturated, partially saturated or unsaturated monocyclic ring containing 4-8 atoms including 1-4 heteroatoms selected independently from O, ⁇ and S, which may be carbon or nitrogen linked, wherein a -CH 2 - group can optionally be replaced by a -C(O)- and a ring nitrogen and/or sulphur atom may be optionally oxidised to form the N-oxide and or the S-oxides.
- Suitable values for "5 or 6 membered Het" are morpholino, piperidyl, pyridyl, pyranyl, pyrrolyl, isothiazolyl, thienyl, thiadiazolyl, piperazinyl, thiazolidinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl, tetrahydropyranyl, imidazolyl, pyrimidyl, pyrazinyl, pyridazinyl, isoxazolyl, 4-pyridone, 2-pyrrolidone, 4-thiazolidone, pyridine-N-oxide and quinoline-N-oxide.
- Examples of "d- ⁇ alkoxycarbonyl” include methoxycarbonyl, ethoxycarbonyl, n- and t-butoxycarbonyl.
- Examples of “C 1-6 alkoxy” include C ⁇ . alkoxy, methoxy, ethoxy and propoxy.
- Examples of “C ⁇ -4 alkylsulphinyl” include methylsulphinyl and ethylsulphinyl.
- Examples of “C 1-6 alkylsulphonyl” include C 1-4 alkylsulphonyl, mesyl and ethylsulphonyl.
- Examples of “ - ⁇ alkanoyl” include C 1-4 alkanoyl, propionyl and acetyl.
- Examples of “C 3-7 cycloalkyl” are cyclopropyl and cyclohexyl.
- Examples of “C 2-6 alkenyl” are vinyl, allyl and 1-propenyl.
- Examples of “C 2-6 alkynyl” are ethynyl, 1-propynyl and 2-propynyl.
- Examples of “N-(C] -6 alkyl)carbamoyl” are methylaminocarbonyl and ethylaminocarbonyl.
- Examples of “N-(C 1-6 alkyl) carbamoyl” are dimethylaminocarbonyl and methylethylaminocarbonyl.
- N-(C 1-4 alkyl)amino examples include methylamino and ethylamino.
- N-(C 1-4 alkyl) 2 amino include di-N-methylamino, di-(N-ethyl)amino and N-ethyl-N-methylamino.
- phenylC 1-6 alkyl examples include phenylC 1-4 alkyl, benzyl and phenethyl.
- C 3- cycloalkylC 1-6 alkyl examples include cyclopropylmethyl, cyclopentylethyl and 2-cyclohexylpropyl.
- (heteroaryl ring)C 1-6 alkyl examples include pyridylmethyl, pyrazinylethyl and imidazolylpropyl.
- n is 1 or 2; R is chloro, fluoro, bromo, methyl or methoxy; R 2 is selected from one of the following three groups: i) halo, nitro, hydroxy, amino or cyano; ii) -X'-R 5 wherein X 1 is a direct bond, -O-, -S-, -SO-, -SO 2 -, - ⁇ R 6 -, -CO-, -CO ⁇ R 6 -, - ⁇ R 6 CO-, -NR 6 SO 2 - or NR 6 CONR 7 -; wherein R 6 and R 7 are independently hydrogen or C 1-4 alkyl optionally substituted with one or more A; and R 5 is selected from C ⁇ -6 alkyl optionally substituted with one or more A, C 3-7 cycloalkyl optionally substituted with one or more A, C 2 .
- R is C ⁇ -6 alkyl optionally substituted with one or more A, C 3-7 cycloalkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D, a carbon-linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms optionally substituted on a ring carbon by one or more D, or a carbon linked 5-membered heteroaryl ring containing 1-3 heteroatoms selected independently from O, N and S optionally substituted on a ring carbon by one or more D and wherein if said 5-membered heteroaryl ring contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G; A is selected from hydroxy, amino, halo, carboxy and C 1-6 alkoxy; D is selected from: i) -X a -R c wherein X a is a direct bond, -O-, -S-, -SO-, -SO 2 -, -CO-,
- G is selected from d- ⁇ alkyl optionally substituted with one or more A, C 1-6 alkanoyl optionally substituted with one or more A, d ealkylsulphonyl optionally substituted with one or more A, d- ⁇ alkoxycarbonyl optionally substituted with one or more A, carbamoyl, N-(C 1-6 alkyl)carbamoyl optionally substituted with one or more A, N-(C 1-6 alkyl) 2 carbamoyl optionally substituted with one or more A and benzoyl optionally substituted with one or more A; and R 4 is hydrogen or fluoro; or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof. Accordingly to an additional aspect of the present invention there is provided a compound of formula (I) (as depicted above) wherein: n is 1 or 2;
- R 1 is chloro, fluoro, bromo, methyl or methoxy
- R 2 is selected from one of the following three groups: i) halo, nitro, hydroxy, amino or cyano; ii) -X'-R 5 wherein X 1 is a direct bond, -O-, -S-, -SO-, -SO 2 -, -NR 6 -, -CO-, -CONR 6 -, -NR 6 CO-, -NR 6 SO 2 - or NR 6 CONR 7 -; wherein R 6 and R 7 are independently hydrogen or C 1- alkyl optionally substituted with one or more A; and R 5 is selected from C 1-6 alkyl optionally substituted with one or more A, C 3-7 cycloalkyl optionally substituted with one or more A, C 2-6 alkenyl optionally substituted with one or more A, C 2-6 alkynyl optionally substituted with one or more A, phenyl optionally substituted with one
- R 3 is C ⁇ -6 alkyl optionally substituted with one or more A, C 3-7 cycloalkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D, a carbon-linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms optionally substituted on a ring carbon by one or more D, or a carbon linked 5-membered heteroaryl ring containing 1-3 heteroatoms selected independently from O, N and S optionally substituted on a ring carbon by one or more D and wherein if said 5-membered heteroaryl ring contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G;
- A is selected from hydroxy, amino, halo, carboxy, N-(C 1-4 alkyl)amino, N,N-di-(C ⁇ -4 alkyl)amino and C 1-6 alkoxy;
- D is selected from: i) -X a -R c wherein X a is a direct bond, -O-, -S-, -SO-, -SO 2 -, -CO-, - ⁇ R d SO 2 -, -NR d CO-, -NR d CONR e -, -NR d - or -CONR d -; wherein R d and R e are independently hydrogen or C 1-4 alkyl optionally substituted with one or more hydroxy or C 1-4 alkoxy; and R c is selected from hydrogen or C 1-6 alkyl optionally substituted with one or more hydroxy or C 1-4 alkoxy; ii) a 4-8 membered Het which is optionally substituted on
- X c is -C(O)- or -SO 2 - and R f is a nitrogen-linked 4-8 membered heterocyclic group optionally substituted on a ring carbon by one or more groups selected from hydroxy, halo, C 1-4 alkoxy, C ⁇ -4 alkyl or cyano and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G; G is selected from C 1-6 alkyl optionally substituted with one or more A, C ⁇ -6 alkanoyl optionally substituted with one or more A, C 1-6 alkylsulphonyl optionally substituted with one or more A, C 1-6 alkoxycarbonyl optionally substituted with one or more A, carbamoyl, N-(C 1-6 alkyl)carbamoyl optionally substituted with
- R 4 is hydrogen or fluoro; or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof.
- R 1 , R 2 , R 3 and R 4 are as follows. Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
- n is 1. In another aspect of the invention preferably n is 2.
- R 1 is chloro, fluoro or bromo. More preferably R 1 is chloro or fluoro. Particularly R 1 is chloro.
- R 1 is methyl, chloro, fluoro or bromo. In another aspect of the invention, more preferably R is methyl, chloro or fluoro. In another aspect of the invention, particularly R 1 is methyl or chloro.
- R 2 is selected from group i):
- group i) is halo or hydroxy.
- group i) is halo.
- Particularly group i) is chloro or fluoro.
- group i) is chloro.
- R is selected from group i):
- group i) is nitro, halo, amino or hydroxy.
- group i) is nitro, amino or halo. Particularly group i) is nitro, bromo, iodo, amino, chloro or fluoro.
- R 2 is selected from group ii):
- X 1 is -S-, -SO-, -SO 2 -, -NR 6 - or -NR 6 CO-; preferably R 6 is hydrogen; and preferably R 5 is selected from C 1-6 alkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or a carbon linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms optionally substituted on a ring carbon by one or more D.
- X 1 is -S-, -SO-, -SO 2 - or -NR 6 CO-; more preferably R 6 is hydrogen; and more preferably R 5 is selected from C ⁇ -4 alkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or a carbon linked pyridyl optionally substituted on a ring carbon by one or more D.
- X 1 is -S- or -NR 6 CO-; particularly R 6 is hydrogen; and particularly R 5 is selected from methyl optionally substituted with one or more A, ethyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or a carbon linked pyridyl optionally substituted on a ring carbon by one or more D.
- R 2 is selected from group ii):
- X 1 is -S-, -SO-, -SO 2 -, -NR 6 - or -NR 6 CO-; preferably R 6 is hydrogen; and preferably R 5 is selected from d- ⁇ alkyl optionally substituted with one or more A or phenyl optionally substituted with one or more D.
- X 1 is -S-, -SO-, -SO 2 - or -NR 6 CO-; more preferably R 6 is hydrogen; and more preferably R 5 is selected from C 1-4 alkyl optionally substituted with one or more A or phenyl optionally substituted with one or more D.
- X 1 is : S- or -NR 6 CO-; particularly R 6 is hydrogen; and particularly R 5 is selected from methyl optionally substituted with one or more A, ethyl optionally substituted with one or more A or phenyl optionally substituted with one or more
- X 1 is -O-, -S-, -SO-, -SO 2 -, -NR 6 - or -NR 6 CO-; preferably R 6 is hydrogen; and preferably R 5 is selected from C 1-6 alkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or phenylC 1-6 alkyl optionally substituted with one or more D.
- X 1 is -O-, -S-, -SO-, -SO 2 - or -NR 6 CO-; more preferably R 6 is hydrogen; and more preferably R 5 is selected from C ⁇ -4 alkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or phenylC 1-4 alkyl optionally substituted with one or more D.
- R 2 is selected from group iii):
- group iii) is a nitrogen-linked 5 or 6 membered heterocyclic group optionally substituted on a ring carbon by one or more D and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G.
- group iii) is a nitrogen-linked 6 membered heterocyclic group optionally substituted on a ring carbon by one or more D and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G.
- group iii) is morpholino optionally substituted on a ring carbon by one or more D, piperidin-1-yl optionally substituted on a ring carbon by one or more D or piperazin-1-yl optionally substituted on a ring carbon by one or more D and optionally substituted on the -NH- moiety by a group selected from G.
- R 2 is selected from group iii):
- Particularly group iii) is morpholino optionally substituted on a ring carbon by one or more D, thiomorpholino optionally substituted on a ring carbon by one or more D, piperidin-1-yl optionally substituted on a ring carbon by one or more D or piperazin-1-yl optionally substituted on a ring carbon by one or more D and optionally substituted on the -NH- moiety by a group selected from G.
- group iii) is thiomorpholino.
- R 2 is selected from group iii): Particularly group iii) is morpholino, thiomorpholino, 1-oxothiomorpholino, 1,1-dioxothiomorpholino or piperazin-1-yl optionally substituted on the -NH- moiety by a group selected from G.
- A is hydroxy, amino, halo, carboxy and methoxy. More preferably A is hydroxy.
- A is hydroxy, amino, dimethylamino, halo, carboxy and methoxy.
- A is hydroxy, methoxy and dimethylamino.
- A is hydroxy, amino, dimethylamino, halo, carboxy, methoxy and carbamoyl.
- X a in group i) is -S-, -SO-, -SO 2 -, -NR d -, -NR d CONR e - or -CONR d -; preferably R d is hydrogen or C 1-4 alkyl optionally substituted with one or more hydroxy; and preferably R c is selected from hydrogen or C 1-6 alkyl optionally substituted with one or more hydroxy.
- X a in group i) is -S-, -SO-, -SO 2 -, -NR d - or -CONR d -; more preferably R d is hydrogen, methyl or ethyl optionally substituted with hydroxy; and more preferably R c is selected from hydrogen or C 1- alkyl optionally substituted with one or more hydroxy.
- X a in group i) is -SO-, -SO 2 -, -NR d - or -CONR d -; more preferably R d is hydrogen, methyl or ethyl optionally substituted with hydroxy; and more preferably R c is selected from hydrogen, methyl or ethyl optionally substituted with hydroxy.
- D is selected from group i):
- X a in group i) is -SO-, -SO 2 -, -NR d - or -CONR d -; more preferably R d is hydrogen, methyl or ethyl optionally substituted with hydroxy; and more preferably R c is selected from hydrogen, methyl, ethyl or butyl optionally substituted with hydroxy.
- D is selected from group ii):
- group ii) is a 5 or 6 membered Het which is optionally substituted on a ring carbon with one or more groups selected from hydroxy, halo, C 1-4 alkoxy, C ⁇ -4 alkyl or cyano and wherein if said 5 or 6 membered Het contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G.
- group ii) is a 5 or 6 membered Het which is optionally substituted on a ring carbon with one or more groups selected from hydroxy, halo, methyl, ethyl, methoxy, ethoxy or cyano and wherein if said 5 or 6 membered Het contains an -NH- moiety that nitrogen may be optionally substituted with a group selected from G.
- group ii) is morpholino, morpholinyl, piperidinyl or piperazinyl optionally substituted on the -NH- moiety by a group selected from G.
- D is selected from group iii):
- group iii) is -X a -C 2-4 alkyl-X b -R c wherein X a and R c are as defined hereinbefore and X is preferably -SO- or -SO 2 -.
- D is selected from group iv):
- group iv) is cyano, fluoro or chloro. More preferably group iv) is fluoro or chloro.
- group iv) is fluoro
- X c is -C(O)- and R f is a nitrogen-linked 4-6 membered heterocyclic group optionally substituted by hydroxy.
- X c is -C(O)- and R f is azetidinyl, morpholino or pyrrolidinyl (optionally substituted by hydroxy).
- X c is -C(O)- and R f is azetidinyl, morpholino or 3-hydroxypyrrolidinyl.
- G is C 1- alkanoyl optionally substituted with one or more A or C 1-6 alkyl optionally substituted by one or more A.
- G is C 1-4 alkanoyl optionally substituted with one or more A or C 1-4 alkyl optionally substituted by one or more A.
- G is acetyl or C 2-4 alkyl substituted by one or more A.
- G is acetyl.
- R 2 is chloro, fluoro, methylthio, acetylamino, hydroxy, C 1 . 4 alkylsulphinyl
- R is chloro, fluoro, methylthio, acetylamino, hydroxy, methylsulphinyl, ethylsulphinyl (optionally substituted with hydroxy), mesyl, ethylsulphonyl (optionally substituted with hydroxy), phenylsulphonyl [optionally substituted with halo, amino, N,N-dimethylcarbamoyl, N,N-diethylcarbamoyl (optionally substituted with hydroxy), N-methyl-N-ethylcarbamoyl (optionally substituted with hydroxy), N-methylcarbamoyl, N-ethylcarbamoyl (optionally substituted with hydroxy), methylamino, ethylamino (optionally substituted with hydroxy), N,N-dimethylamino, N,N-diethylamino (optionally substituted with hydroxy), N-methyl-N-ethylamino
- R is chloro, fluoro, methylthio, acetylamino or hydroxy.
- R 2 is chloro, fluoro, bromo, iodo, nitro, amino, methylthio, acetylamino, hydroxy, C 1-4 alkylsulphanyl (optionally substituted with hydroxy), C 1-4 alkylsulphinyl (optionally substituted with hydroxy), C ⁇ -4 alkylsulphonyl (optionally substituted with hydroxy), N-(C ⁇ -4 alkyl)amino (optionally substituted with hydroxy, methoxy or dimethylamino), phenylsulphonyl [optionally substituted with halo, amino, N-(C ⁇ -4 alkyl) 2 carbamoyl (optionally substituted with hydroxy), N-(C ⁇ -4 alkyl)carbamoyl (optionally substituted with hydroxy), N-(C ⁇ -4 alkyl)a
- R 2 is chloro, fluoro, bromo, iodo, nitro, hydroxy, amino, methylthio, acetylamino, C 1-4 alkylsulphanyl (optionally substituted with hydroxy), C 1-4 alkylsulphinyl, C 1- alkylsulphonyl, N-(C 1-4 alkyl)amino (optionally substituted with hydroxy, methoxy or dimethylamino), thiomorpholino, phenylsulphanyl (optionally substituted with N-(C 1-4 alkyl) carbamoyl) or phenylsulphinyl (optionally substituted with N-(C 1-4 alkyl) 2 carbamoyl).
- R 2 is chloro, fluoro, bromo, iodo, nitro, amino, methylthio, acetylamino, hydroxy, methylsulphanyl, ethylsulphanyl (optionally substituted with hydroxy), methylsulphinyl, ethylsulphinyl (optionally substituted with hydroxy), mesyl, ethylsulphonyl (optionally substituted with hydroxy), methylamino, ethylamino (optionally substituted with hydroxy, methoxy or dimethylamino), phenylsulphonyl [optionally substituted with halo, amino, N,N-dimethylcarbamoyl, N,N-diethylcarbamoyl (optionally substituted with hydroxy), N-methyl-N-ethylcarbamoyl (optionally substituted with hydroxy), N-methylcarbamoyl, N-e
- N-methyl-N-ethylamino (optionally substituted with hydroxy), methylsulphinyl, ethylsulphinyl (optionally substituted with hydroxy), methylamino, ethylamino (optionally substituted with hydroxy), mesyl, ethylsulphonyl (optionally substituted with hydroxy), 4-acetylpiperazin-l-yl, 4-methylpiperazin- 1 -yl, 4-(2-hydroxyethyl)piperazin- 1 -yl, 4-(3-hydroxypropyl)piperazin- 1 -yl or 4-(2-hydroxypropyl)piperazin-l-yl], methylamino, ethylamino (optionally substituted with hydroxy), morpholino, 4-acetylpiperazin-l-yl, 4-methylpiperazin-l-yl, 4-(2-hydroxyethyl)piperazin- 1 -yl, 4-(3-hydroxypropyl
- R is fluoro, chloro, bromo, iodo, nitro, amino, hydroxy, methylthio, ethylsulphinyl, mesyl, 2-hydroxyethylamino, 2-methoxyethylamino, 2-dimethylaminoethylamino, 2,3-dihydroxypropylamino, 2-hydroxyethylsulphanyl, acetylamino, 4-N,N-dimethylcarbamoylphenylsulphanyl, 4-N,N-dimethylcarbamoylphenylsulphinyl or thiomorpholino.
- R 2 is chloro, fluoro, bromo, iodo, nitro, amino, methoxy, acetylamino, hydroxy, C ⁇ -4 alkylsulphanyl (optionally substituted with hydroxy), C 1-4 alkylsulphinyl, C 1-4 alkylsulphonyl, N-(C 1-4 alkyl)amino (optionally substituted with hydroxy, methoxy, dimethylamino or carbamoyl), morpholino, 4-acetylpiperazin-l-yl, thiomorpholino, 1-oxothiomorpholino, 1,1-dioxothiomorpholino, benzylamino, phenoxy, phenylsulphanyl (optionally substituted with N-(C 1-4 alkyl) 2 carbamoyl) or phenylsulphinyl (optionally substituted with N-(C 1-4 alkyl) 2 carbamoyl
- R is chloro, fluoro, bromo, iodo, nitro, amino, methoxy, acetylamino, hydroxy, methylthio, 2-hydroxyethylthio, methylsulphinyl, mesyl, 2-hydroxyethylamino, 2-methoxyethylamino, carbamoylmethylamino, 2-dimethylaminoethylamino, 2,3-dihydroxypropylamino, morpholino, 4-acetylpiperazin-l-yl, thiomorpholino, 1-oxothiomorpholino, 1,1-dioxothiomorpholino, benzylamino, phenoxy,
- R 2 is methylthio, morpholino
- R 2 is amino, 2-hydroxyethylamino or 2-methoxyethylamino.
- R 2 is fluoro or chloro.
- R 3 is C 1-6 alkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or a carbon-linked 6-membered heteroaryl ring containing 1-2 nitrogen atoms optionally substituted on a ring carbon by one or more D. More preferably R 3 is C 1-4 alkyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or a carbon-linked pyridyl optionally substituted on a ring carbon by one or more D.
- R 3 is methyl optionally substituted with one or more A, ethyl optionally substituted with one or more A, phenyl optionally substituted with one or more D or a carbon-linked pyridyl optionally substituted on a ring carbon by one or more D.
- R 3 is methyl, ethyl optionally substituted with A, phenyl optionally substituted with one or more D or a carbon-linked pyridyl optionally substituted on a ring carbon by one or more D.
- R 3 is C 1- alkyl optionally substituted with one or more hydroxy, phenyl [optionally substituted with halo, amino, N-(C ⁇ -4 alkyl) 2 carbamoyl (optionally substituted with hydroxy), N-(C 1-4 alkyl)carbamoyl (optionally substituted with hydroxy), N-(C ⁇ -4 alkyl)amino (optionally substituted with hydroxy), N-(C 1-4 alkyl) amino (optionally substituted with hydroxy), C 1-4 alkylsulphinyl (optionally substituted with hydroxy), C 1- alkylsulphonyl (optionally substituted with hydroxy), 4-acetylpiperazin-l-yl, 4-methylpiperazin-l-yl, 4-(2-hydroxyethyl)- piperazin-1-yl, 4-(3-hydroxypropyl)piperazin-l-yl or 4-(2-hydroxypropyl)piperazin-l-yl
- R 3 is methyl, ethyl optionally substituted with hydroxy, phenyl [optionally substituted with halo, amino, N,N-dimethylcarbamoyl, N,N-diethylcarbamoyl (optionally substituted with hydroxy), N-methyl-N-ethylcarbamoyl (optionally substituted with hydroxy), N-methylcarbamoyl, N-ethylcarbamoyl (optionally substituted with hydroxy), methylamino, ethylamino (optionally substituted with hydroxy), N,N-dimethylamino, N,N-diethylamino (optionally substituted with hydroxy), N-methyl-N-ethylamino (optionally substituted with hydroxy), methylsulphinyl, ethylsulphinyl (optionally substituted with hydroxy), mesyl, ethylsulphonyl (optionally substituted with hydroxy), 4-
- R 3 is ethyl or 4-fluorophenyl. Therefore, in another aspect of the invention preferably R 3 is C 1-4 alkyl (optionally substituted with one or more hydroxy), phenyl [optionally substituted with halo, amino, N-(C 1-4 alkyl) 2 carbamoyl (optionally substituted with hydroxy), N-(C 1-4 alkyl)carbamoyl (optionally substituted with hydroxy), N-(C 1-4 alkyl)amino (optionally substituted with hydroxy), N-(C ⁇ -4 alkyl) 2 amino (optionally substituted with hydroxy), C 1-4 alkylsulphinyl (optionally substituted with hydroxy), C 1-4 alkylsulphonyl (optionally substituted with hydroxy), 4-acetylpiperazin-l-yl, 4-methylpiperazin-l-yl, 4-(2-hydroxyethyl)-piperazin-l-yl, 4-(3-hydroxypropyl)pipe
- R 3 is C 1-4 alkyl (optionally substituted with one or more hydroxy), phenyl [optionally substituted with halo, N-(C ⁇ - alkyl) carbamoyl, N-(C 1-4 alkyl)carbamoyl, N-(C ⁇ -4 alkyl)amino (optionally substituted with hydroxy), C 1-4 alkylsulphonyl, azetidinylcarbonyl, morpholinocarbonyl or pyrrolidinylcarbonyl (optionally substituted with hydroxy)], or carbon-linked pyridyl [optionally substituted with amino].
- R 3 is methyl, ethyl (optionally substituted with hydroxy), butyl (optionally substituted with hydroxy), phenyl [optionally substituted with halo, amino, N,N-dimethylcarbamoyl, N,N-diethylcarbamoyl (optionally substituted with hydroxy), N-methyl-N-ethylcarbamoyl (optionally substituted with hydroxy), N-methylcarbamoyl, N-ethylcarbamoyl (optionally substituted with hydroxy), methylamino, ethylamino (optionally substituted with hydroxy), N,N-dimethylamino, N,N-diethylamino (optionally substituted with hydroxy), N-methyl-N-ethylamino (optionally substituted with hydroxy), methylsulphinyl, ethylsulphinyl (optionally substituted with hydroxy), mesyl, ethylsulphin
- N,N-diethylamino (optionally substituted with hydroxy), N-methyl-N-ethylamino (optionally substituted with hydroxy), methylsulphinyl, ethylsulphinyl (optionally substituted with hydroxy), mesyl, ethylsulphonyl (optionally substituted with hydroxy), 4-acetylpiperazin-l-yl, 4-methylpiperazin- 1 -yl, 4-(2-hydroxyethyl)piperazin- 1 -yl, 4-(3-hydroxypropyl)piperazin- 1 -yl or 4-(2-hydroxypropyl)piperazin- 1 -yl] .
- R 3 is methyl, ethyl, 2-hydroxyethyl, 2-hydroxybutyl, 4-fluorophenyl, 4-mesylphenyl, 4-(2-hydroxyethylamino)phenyl, 4-(N-methylcarbamoyl)phenyl, 4-(N-ethylcarbamoyl)phenyl, 4-(N,N-dimethylcarbamoyl)phenyl, 4-(N-methyl-N-ethylcarbamoyl)phenyl, 4-(azetidinylcarbonyl)phenyl, 4-(morpholinocarbonyl)phenyl, 4-(3-hydroxypyrrolidinylcarbonyl)phenyl or 6-aminopyrid-2-yl.
- R 3 is methyl, ethyl (optionally substituted with hydroxy), isopropyl, butyl (optionally substituted with hydroxy), phenyl [optionally substituted with halo, N,N-dimethylcarbamoyl, N-methyl-N-ethylcarbamoyl, N-methylcarbamoyl, N-ethylcarbamoyl, ethylamino (optionally substituted with hydroxy), mesyl, azetidinylcarbonyl, morpholinocarbonyl or pyrrolidinylcarbonyl (optionally substituted with hydroxy)] or carbon-linked pyridyl [optionally substituted with amino].
- R 3 is methyl, ethyl, 2-hydroxyethyl, isopropyl, 2-hydroxybutyl, 4-fluorophenyl, 4-(2-hydroxyethylamino)phenyl, 4-mesylphenyl, 4-(N,N-dimethylcarbamoyl)phenyl, 4-(N-ethylcarbamoyl)phenyl, 4-(N-methyl-N-ethylcarbamoyl)phenyl, 4-(N-methylcarbamoyl)phenyl, 4-(azetidinylcarbonyl)phenyl, 4-(morpholinocarbonyl)phenyl, 4-(3-hydroxypyrrolidinylcarbonyl)phenyl or 2-aminopyrid-6-yl.
- R 3 is methyl, ethyl or isopropyl I
- R is 4-(N-methylcarbamoyl)phenyl or 4-(N,N-dimethylcarbamoyl)phenyl
- R 3 is 4-(N,N-dimethylcarbamoyl)phenyl.
- R 4 is hydrogen
- R 4 is fluoro.
- the R-configuration is generally the preferred stereochemistry.
- n is 1 or 2;
- R 1 is methyl, chloro or fluoro;
- R is chloro, fluoro, bromo, iodo, nitro, amino, methoxy, acetylamino, hydroxy, d. 4 alkylsulphanyl (optionally substituted with hydroxy), C 1-4 alkylsulphinyl, C ⁇ -4 alkylsulphonyl, N-(C 1- alkyl)amino (optionally substituted with hydroxy, methoxy, dimethylamino or carbamoyl), morpholino, 4-acetylpiperazin-l-yl, thiomorpholino, 1-oxothiomorpholino, 1,1-dioxothiomorpholino, benzylamino, phenoxy, phenylsulphanyl (optionally substituted with N-(C 1-4 alkyl) 2 carbamoyl) or phenylsulphinyl (optionally substituted with N-(C 1-4 alkyl) 2 carbamoyl);
- R 3 is methyl, ethyl (optionally substituted with hydroxy), isopropyl, butyl (optionally substituted with hydroxy), phenyl [optionally substituted with halo, N.N-dimethylcarbamoyl, N-methyl-N-ethylcarbamoyl, N-methylcarbamoyl, N-ethylcarbamoyl, ethylamino (optionally substituted with hydroxy), mesyl, azetidinylcarbonyl, morpholinocarbonyl or pyrrolidinylcarbonyl (optionally substituted with hydroxy)] or carbon-linked pyridyl [optionally substituted with amino]; and
- R 4 is hydrogen; or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof.
- R 1 is chloro;
- R 2 is methylthio, morpholino, 4-acetylpiperazin-l-yl, 1-oxothiomorpholino or
- R 3 is methyl, ethyl or isopropyl
- R 4 is hydrogen
- a preferred compound of the invention is any one of the Examples or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof. More preferred compounds of the invention are Examples 7, 8, 22, 23, 24, 28, 48, 64,
- more preferred compounds of the invention are Examples 32, 35 and 61 or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof. In a further aspect of the invention more preferred compounds of the invention are
- Preferred aspects of the invention are those which relate to the compound or a pharmaceutically acceptable salt thereof.
- a compound of the formula (I) or a salt thereof may exhibit the phenomenon of tautomerism and that the formulae drawings within this specification can represent only one of the possible tautomeric forms.
- the invention encompasses any tautomeric form which elevates PDH activity and is not to be limited merely to any one tautomeric form utilized within the formulae drawings.
- the formulae drawings within this specification can represent only one of the possible tautomeric forms and it is to be understood that the specification encompasses all possible tautomeric forms of the compounds drawn not just those forms which it has been possible to show graphically herein.
- the present invention encompasses any racemic, optically-active, enantiomerically pure, mixture of diastereoisomers, polymorphic or stereoisomeric form, or mixtures thereof, which form possesses properties useful in the elevation of PDH activity, it being well known in the art how to prepare optically-active forms (for example, by resolution of the racemic form by recrystallization techniques, by synthesis from optically-active starting materials, by chiral synthesis, by enzymatic resolution, (for example WO 9738124), by biotransformation, or by chromatographic separation using a chiral stationary phase) and how to determine efficacy for the elevation of PDH activity by the standard tests described hereinafter.
- optically-active forms for example, by resolution of the racemic form by recrystallization techniques, by synthesis from optically-active starting materials, by chiral synthesis, by enzymatic resolution, (for example WO 9738124), by biotransformation, or by chromatographic separation using a chiral stationary phase
- a compound of the formula (I), or salt thereof, and other compounds of the invention may be prepared by any process known to be applicable to the preparation of chemically-related compounds. Such processes include, for example, those illustrated in European Patent Applications, Publication Nos. 0524781, 0617010, 0625516, and in GB 2278054, WO 9323358 and WO 9738124.
- Another aspect of the present invention provides a process for preparing a compound of formula (I) or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof, which process (in which variable groups are as defined for formula (I) unless otherwise stated) comprises of:
- Suitable values for Pg are a benzyl group, a silyl group (for example a trialkylsilyl group or an alkyldiphenylsilyl group) or an acetyl protecting group.
- Suitable values for X include halo (for example chloro or bromo), anhydrides, aryloxys (for example 4-nitrophenoxy or pentafluorophenoxy) or imidazol-1-yl.
- Suitable reagents for deprotecting an alcohol of formula (II) are: 1) when Pg is benzyl:
- Pg is acetyl: i) mild aqueous base for example lithium hydroxide; or ii) ammonia or an amine such as dimethylamine.
- the reaction can be conducted in a suitable solvent such as EtOH, MeOH, acetonitrile, or DMSO and may conveniently be performed at a temperature in the range of -40 to 100°C.
- a suitable solvent such as EtOH, MeOH, acetonitrile, or DMSO
- Scheme 1 E is a carboxy protecting group. Suitable values for E include C 1-6 alkyl, such as methyl and ethyl.
- Suitable oxidising agents include potassium permanganate, OXONE, sodium periodate, tert-butyl hydroperoxide (as solution in toluene), peracids (such as for example 3-chloroperoxybenzoic acid), hydrogen peroxide, TPAP (tetrapropylammonium perruthenate) or oxygen.
- the reaction may be conducted in a suitable solvent such as ether, DCM, MeOH, EtOH, water, acetic acid, or mixtures of two or more of these solvents.
- the reaction may conveniently be performed at a temperature in the range of -40 to 100°C.
- X is a leaving group, suitable values for X include halo, mesyl and tosyl. dine,
- X is a leaving group, suitable values for X include halo, mesyl and tosyl.
- Compounds of formula (Ilia) and (Illd) are commercially available compounds, or they are known in the literature, or they are prepared by standard processes known in the art. Process c)
- the reaction can be conducted in the presence of a suitable coupling reagent.
- Standard peptide coupling reagents known in the art can be employed as suitable coupling reagents, for example conditions such as those described above for the coupling of (lid) and (IV), or for example dicyclohexyl-carbodiimide, optionally in the presence of a catalyst such as dimethylaminopyridine or 4-pyrrolidinopyridine, optionally in the presence of a base for example triethylamine, pyridine, or 2,6-di- ⁇ /fcy/-pyridines (such as 2,6-lutidine or 2,6-di-tert-butylpyridine) or 2,6-diphenylpyridine.
- Suitable solvents include DMA, DCM, benzene, THF, and DMF.
- the coupling reaction may conveniently be performed at a temperature in the range of -40 to 40°C.
- Compounds of formula (IV) are commercially available compounds, or they are known in the literature, or they are prepared by standard processes known in the art, for example they may be prepared by oxidising compounds of formula (Illf) (with the aniline protected with a suitable protecting group) under standard oxidation conditions, for example with hydrogen peroxide or meta-chloroperoxy benzoic acid (followed by de-protection), or they may be prepared according to the following scheme:
- EP 0524781 also for preparation of the (S)-(-) acid, except that (lS,2R)-norephedrine may be used in place of (S)-(-)-l-phenylethylamine.
- the chiral acid may also be prepared by using the enzymatic resolution method as described in Tetrahedron Asymmetry, 1999, 10, 679. Process d)
- This coupling may be achieved optionally in the presence of a base for example triethylamine, pyridine, 2,6-di- ⁇ /&y/-pyridines (such as 2,6-lutidine or 2,6-di-tert-butylpyridine) or 2,6-diphenylpyridine.
- a base for example triethylamine, pyridine, 2,6-di- ⁇ /&y/-pyridines (such as 2,6-lutidine or 2,6-di-tert-butylpyridine) or 2,6-diphenylpyridine.
- Suitable solvents include DMA, DCM, benzene, THF, and DMF.
- the coupling reaction may conveniently be performed at a temperature in the range of -40 to 40°C.
- the necessary starting materials for the procedures such as that described above may be made by procedures which are selected from standard organic chemical techniques, techniques which are analogous to the synthesis of known, structurally similar compounds, or techniques which are analogous to the above described procedure or the procedures described in the examples.
- aromatic substituents in the compounds of the present invention may be introduced by standard aromatic substitution reactions or generated by conventional functional group modifications or interconversions either prior to or immediately following the processes mentioned above, and as such are included in the process aspect of the invention.
- Such reactions and modifications include, for example, introduction of a substituent by means of an aromatic substitution reaction, reduction of substituents, alkylation of substituents and oxidation of substituents.
- the reagents and reaction conditions for such procedures are well known in the chemical art.
- aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acylhalide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halogeno group.
- an acyl group using, for example, an acylhalide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions
- Lewis acid such as aluminium trichloride
- modifications include the reduction of a nitro group to an amino group by, for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl using, for example, hydrogen peroxide in acetic acid with heating or 3-chloroperbenzoic acid.
- functional group interconversions are for example conversion of an aniline into a halophenyl by, for example, diazotization in the presence of cupurous halides.
- a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl.
- the deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an acyl group such as a t-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid such as, for example hydrochloric, sulphuric or phosphoric acid or TFA and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation in the presence of a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate).
- a suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example dimethylaminopropylamine, or with hydrazine.
- a suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl.
- the deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation in the presence of a catalyst such as palladium-on-carbon.
- a suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as TFA, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a base such as sodium hydroxide
- a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as TFA, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- the protecting groups may be removed at any convenient stage in the synthesis using conventional techniques well known in the chemical art.
- compositions of formula (I) are sufficiently basic or acidic to form stable acid or basic salts
- administration of the compound as a salt may be appropriate, and pharmaceutically acceptable salts may be made by conventional methods such as those described following.
- suitable pharmaceutically acceptable salts are organic acid addition salts formed with acids which form a physiologically acceptable anion, for example, tosylate, methanesulphonate, acetate, tartrate, citrate, succinate, benzoate, ascorbate, ⁇ -ketoglutarate, and ⁇ -glycerophosphate.
- Suitable inorganic salts may also be formed such as sulphate, nitrate, and hydrochloride.
- salts may be obtained using standard procedures well known in the art, for example by reacting a sufficiently basic compound of formula (I) (or its ester) with a suitable acid affording a physiologically acceptable anion. It is also possible with most compounds of the invention to make a corresponding alkali metal (e.g. sodium, potassium, or lithium) or alkaline earth metal (e.g. calcium) salt by treating a compound of formula (I) (and in some cases the ester) with one equivalent of an alkali metal or alkaline earth metal hydroxide or alkoxide (e.g. the ethoxide or methoxide) in aqueous medium followed by conventional purification techniques.
- a corresponding alkali metal e.g. sodium, potassium, or lithium
- alkaline earth metal e.g. calcium
- the compounds of the formula (I) may be administered in the form of a prodrug which is broken down in the human or animal body to give a compound of the formula (I).
- prodrugs include in vivo hydrolysable esters of a compound of the formula (I).
- An in vivo hydrolysable ester of a compound of the formula (I) containing carboxy or hydroxy group is, for example, a pharmaceutically acceptable ester which is hydrolysed in the human or animal body to produce the parent acid or alcohol.
- Suitable in vivo hydrolysable esters for a compound of the formula (I) containing a carboxy group include C 1-6 alkoxy methyl esters for example methoxymethyl, C ⁇ -6 alkanoyloxymethyl esters for example pivaloyloxymethyl, phthalidyl esters, C 3-8 cycloalkoxycarbonyloxyd.
- alkyl esters for example 1-cyclohexylcarbonyloxy ethyl
- l,3-dioxolen-2-onylmethyl esters for example 5-methyl-l,3-dioxolen-2-onylmethyl
- C 1-6 alkoxycarbonyloxy ethyl esters for example 1-methoxycarbonyloxyethyl and may be formed at any carboxy group in the compounds of this invention.
- Suitable in vivo hydrolysable esters of a compound of the formula (I) containing a hydroxy group includes inorganic esters such as phosphate esters and ⁇ -acyloxyalkyl ethers.
- inorganic esters such as phosphate esters and ⁇ -acyloxyalkyl ethers.
- ⁇ -acyloxyalkyl ethers include acetoxymethoxy and 2,2-dimethylpropionyloxymethoxy.
- hydrolysable ester forming groups for hydroxy include alkanoyl, benzoyl, phenylacetyl and substituted benzoyl and phenylacetyl, alkoxycarbonyl (to give alkyl carbonate esters), dialkylcarbamoyl and N-(dialkylaminoethyl)-N-alkylcarbamoyl (to give carbamates), dialkylaminoacetyl and carboxyacetyl.
- substituents for benzoyl include morpholino and piperazino linked from a ring nitrogen atom via a methylene group to the 3- or 4- position of the benzoyl ring.
- In vivo cleavable prodrugs of compounds of formula (I) also include in vivo hydrolysable amides of compounds of the formula (I) containing a carboxy group, for example, a N-C 1-6 alkyl or N-di-C 1-6 alkyl amide such as N-methyl, N-ethyl, N-propyl, N-dimethyl, N-ethyl-N-methyl or N-diethyl amide.
- a N-C 1-6 alkyl or N-di-C 1-6 alkyl amide such as N-methyl, N-ethyl, N-propyl, N-dimethyl, N-ethyl-N-methyl or N-diethyl amide.
- cD ⁇ A encoding PDH kinase may be obtained by Polymerase Chain Reaction (PCR) and subsequent cloning. This may be expressed in a suitable expression system to obtain polypeptide with
- rat PDHkinasell obtained by expression of recombinant protein in Escherichia coli (E. Coli), was found to display PDH kinase activity.
- rPDHKH Genbank accession number U 10357
- a 1.3kb fragment encoding the protein was isolated by PCR from rat liver cD ⁇ A and cloned into a vector (for example pQE32 - Quiagen Ltd.).
- the recombinant construct was transformed into E. coli (for example
- PDH kinases for use in assays, may be cloned and expressed in a similar manner.
- E. coli strain M15pRep4 cells were transformed with the pQE32 vector containing rPDHKII cD ⁇ A. This vector incorporates a 6-His tag onto the protein at its ⁇ -terminus. E. coli were grown to an optical density of 0.6 (600 nM) and
- 25 protein expression was induced by the addition of 10 ⁇ M isopropylthio- ⁇ -galactosidase.
- Cells were grown for 18 hours at 18°C and harvested by centrifugation.
- the resuspended cell paste was lysed by homogenisation and insoluble material removed by centrifugation at 24000xg for 1 hour.
- the 6-His tagged protein was removed from the supernatant using a nickel chelating nitrilotriacetic acid resin ( ⁇ i- ⁇ TA: Quiagen Ltd.) matrix (Quiagen) which was washed with
- PDH kinase activity was then initiated by the addition of 5 ⁇ M ATP, 2 mM magnesium chloride and 0.04 U/ml PDH (porcine heart PDH Sigma P7032) in a total volume of 50 ⁇ l and plates incubated at ambient temperature for a further 45 minutes.
- the residual activity of the PDH was then determined by the addition of substrates (2.5mM coenzyme A, 2.5mM thiamine pyrophosphate (cocarboxylase), 2.5mM sodium pyruvate, 6mM NAD in a total volume of 80 ⁇ l and the plates incubated for 90 minutes at ambient temperature.
- substrates 2.5mM coenzyme A, 2.5mM thiamine pyrophosphate (cocarboxylase), 2.5mM sodium pyruvate, 6mM NAD
- NADH reduced NAD
- the ED 50 for a test compound was determined in the usual way using results from 12 concentrations of the compound,
- (b) In vitro elevation of PDH activity in isolated primary cells This assay determines the ability of compounds to stimulate pyruvate oxidation in primary rat hepatocytes.
- Hepatocytes were isolated by the two-step collagenase digestion procedure described by Seglen (Methods Cell Biol. (1976) 13, 29-33) and plated out in 6- well culture plates (Falcon Primaria) at 600000 viable cells per well in Dulbecco's Modified Eagles Medium (DMEM, Gibco BRL) containing 10% foetal calf serum (FCS), 10% penicillin/streptomycin (Gibco BRL) and 10% non-essential amino acids (NEAA, Gibco BRL).
- DMEM Dulbecco's Modified Eagles Medium
- FCS foetal calf serum
- NEAA non-essential amino acids
- MEM Minimum Essential Medium
- HEPES phosphate buffered saline
- 1ml HEPES -buffered Krebs solution 25mM HEPES, 0.15M sodium chloride, 25 mM sodium hydrogen carbonate, 5mM potassium chloride, 2mM calcium chloride, lmM magnesium sulphate, 1 mM potassium dihydrogen phosphate
- Control wells contained 0.1% DMSO only and a maximum response was determined using a 10 ⁇ M treatment of a known active compound.
- the capacity of compounds to increase the activity of PDH in relevant tissues of rats may be measured using the test described hereinafter.
- an increase in the proportion of PDH in its active, nonphosphorylated form may be detected in muscle, heart, liver and adipose tissue after a single administration of an active compound. This may be expected to lead to a decrease in blood glucose after repeated administration of the compound.
- DCA a compound known to activate PDH by inhibition of PDH kinase (Whitehouse, Cooper and Randle (1974) Biochem. J. 141, 761-774)
- 150 mg/kg intraperitoneally, increased the proportion of PDH in its active form (Vary et al. (1988) Circ. Shock 24, 3-18) and after repeated administration resulted in a significant decrease in plasma glucose (Evans and Stacpoole (1982) Biochem. Pharmacol.31, 1295-1300).
- Groups of rats are treated with a single dose or multiple doses of the compound of interest by oral gavage in an appropriate vehicle.
- a control group of rats is treated with vehicle only.
- animals are terminally anaesthetised, tissues are removed and frozen in liquid nitrogen.
- muscle samples are disrupted under liquid nitrogen prior to homogenisation by one thirty-second burst in a Polytron homogenizer in 4 volumes of a buffer containing 40 mM potassium phosphate pH 7.0, 5 mM EDTA, 2mM DTT, 1% Triton X-100, lOmM sodium pyruvate, lO ⁇ M phenylmethylsulphonyl chloride (PMSF) and 2 ⁇ g/ml each of 5 leupeptin, pepstain A and aprotinin. Extracts are centrifuged before assay. A portion of the extract is treated with PDH phosphatase prepared from pig hearts by the method of Siess and Wieland (Eur. J.
- the activity of the untreated sample is compared with the activity of the dephosphorylated extract
- PDH activity is assayed by the method of Stansbie et al., (Biochem. J. (1976) 154, 225).
- 50 ⁇ l Extract is incubated with 0.75 mM NAD, 0.2 mM CoA, 1.5 mM thiamine pyrophosphate (TPP) and 1.5mM sodium pyruvate in the presence of 20 ⁇ g/ml p-(p-amino-phenylazo) benzene sulphonic acid (AABS) and 50 mU/ml arylamine transferase (AAT) in a buffer containing 100 mM tris(hydroxymethyl)aminomethane, 0.5 mM EDTA,
- AAT is prepared from pigeon livers by the method of Tabor et al. (J. Biol. Chem. (1953) 204, 127). The rate of acetyl CoA formation is determined by the rate of reduction of AABS which is indicated by a decrease in optical density at 460 nm.
- Liver samples are prepared by an essentially similar method, except that sodium 0 pyruvate is excluded from the extraction buffer and added to the phosphatase incubation to a final concentration of 5mM.
- Treatment of an animal with an active compound results in an increase in the activity of PDH complex in tissues. This is indicated by an increase in the amount of active PDH (determined by the activity of untreated extract as a percentage of the total PDH activity in the 5 same extract after treatment with phosphatase).
- a pharmaceutical composition which comprises a compound of the formula (I) as defined hereinbefore or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof, in association with a pharmaceutically acceptable excipient or carrier.
- compositions may be in a form suitable for oral administration, for example as a tablet or capsule, for parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion) for example as a sterile solution, suspension or emulsion, for topical administration for example as an ointment or cream or for rectal administration for example as a suppository.
- parenteral injection including intravenous, subcutaneous, intramuscular, intravascular or infusion
- sterile solution suspension or emulsion
- topical administration for example as an ointment or cream or for rectal administration for example as a suppository.
- the above compositions may be prepared in a conventional manner using conventional excipients.
- compositions of the present invention are advantageously presented in unit dosage form.
- the compound will normally be administered to a warm-blooded animal at a unit dose within the range 5-5000 mg per square metre body area of the animal, i.e. approximately 0.1-100 mg/kg.
- a unit dose in the range, for example, 1-100 mg/kg, preferably 1-50 mg/kg is envisaged and this normally provides a therapeutically-effective dose.
- a unit dose form such as a tablet or capsule will usually contain, for example 1-250 mg of active ingredient.
- a compound of the formula (I) or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof as defined hereinbefore for use in a method of treatment of the human or animal body by therapy.
- a further feature of the present invention is a compound of formula (I) and pharmaceutically acceptable salts or in vivo hydrolysable esters thereof for use as a medicament.
- this is a compound of formula (I), or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof, for use as a medicament for producing an elevation of PDH activity in a warm-blooded animal such as a human being.
- this is a compound of formula (I), or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof, for use as a medicament for treating diabetes mellitus in a warm-blooded animal such as a human being.
- this is a compound of formula (I), or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof, for use as a medicament for treating diabetes mellitus, peripheral vascular disease and myocardial ischaemia in a warm-blooded animal such as a human being.
- a compound of the formula (I), or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof in the manufacture of a medicament for use in the production of an elevation of PDH activity in a warm-blooded animal such as a human being.
- a compound of the formula (I), or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof in the manufacture of a medicament for use in the treatment of diabetes mellitus in a warm-blooded animal such as a human being.
- a compound of the formula (I), or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof in the manufacture of a medicament for use in the treatment of diabetes mellitus, peripheral vascular disease and myocardial ischaemia in a warm-blooded animal such as a human being.
- a method for producing an elevation of PDH activity in a warm-blooded animal, such as a human being, in need of such treatment which comprises administering to said animal an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof as defined hereinbefore.
- a method of treating diabetes mellitus in a warm-blooded animal, such as a human being, in need of such treatment which comprises administering to said animal an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof as defined hereinbefore.
- a method of treating diabetes mellitus, peripheral vascular disease and myocardial ischaemia in a warm-blooded animal, such as a human being in need of such treatment which comprises administering to said animal an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof as defined hereinbefore.
- the size of the dose required for the therapeutic or prophylactic treatment of a particular disease state will necessarily be varied depending on the host treated, the route of administration and the severity of the illness being treated.
- a daily dose in the range of 1-50 mg/kg is employed.
- the daily dose will necessarily be varied depending upon the host treated, the particular route of administration, and the severity of the illness being treated. Accordingly the optimum dosage may be determined by the practitioner who is treating any particular patient.
- the elevation of PDH activity described herein may be applied as a sole therapy or may involve, in addition to the subject of the present invention, one or more other substances and/or treatments. Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate administration of the individual components of the treatment.
- diabetes mellitus chemotherapy may include the following main categories of treatment: i) insulin; ii) insulin secretagogue agents designed to stimulate insulin secretion (for example glibenclamide, tolbutamide, other sulphonylureas); iii) oral hypoglycaemic agents such as metformin, thiazolidinediones; iv) agents designed to reduce the abso ⁇ tion of glucose from the intestine (for example acarbose); v) agents designed to treat complications of prolonged hyperglycaemia; vi) other agents used to treat lactic acidaemia; vii) inhibitors of fatty acid oxidation; viii) lipid lowering agents; ix) agents used to treat coronary heart disease and peripheral vascular disease such as aspirin, pentoxifylline, cilostazol; and/or x) thiamine.
- insulin secretagogue agents designed to stimulate insulin secretion for example glibenclamide, tolbutamide, other s
- the compounds defined in the present invention are of interest for their ability to elevate the activity of PDH.
- Such compounds of the invention may therefore be useful in a range of disease states including diabetes mellitus, peripheral vascular disease, (including intermittent claudication), cardiac failure and certain cardiac myopathies, myocardial ischaemia, cerebral ischaemia and reperfusion, muscle weakness, hyperlipidaemias, Alzheimer's disease and/or atherosclerosis.
- Such compounds of the invention may be useful in a range of disease states including peripheral vascular disease, (including intermittent claudication), cardiac failure and certain cardiac myopathies, myocardial ischaemia, cerebral ischaemia and reperfusion, muscle weakness, hyperlipidaemias, Alzheimer's disease and/or atherosclerosis in particular peripheral vascular disease and myocardial ischaemia.
- the compounds of formula (I) and their pharmaceutically acceptable salts are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of elevators of PDH activity in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutic agents.
- temperatures are given in degrees Celsius (°C); operations were carried out at room or ambient temperature, that is, at a temperature in the range of 18-25°C and under an atmosphere of an inert gas such as argon;
- chromatography means flash chromatography on silica gel; thin layer chromatography (TLC) was carried out on silica gel plates; where a silica Mega Bond Elut column is referred to, this means a column containing 10 g or 20 g or 50 g of silica of 40 micron particle size, the silica being contained in a 60 ml disposable syringe and supported by a porous disc, obtained from Varian, Harbor City, California, USA under the name "Mega Bond Elut SI"; "Mega Bond Elut” is a trademark; where a Biotage cartridge is referred to this means a cartridge containing KP-SILTM silica, 60A, particle size 32-63mM, supplied by Biotage, a division of Dyax Co ⁇ ., 1500 Avon Street Extended, Charlottesville, VA 22902, USA; (iv) where a Chem Elut column is referred to, this means a "Hydromatrix" extraction cartridge for adso ⁇ tion
- NMR data is in the form of delta values for major diagnostic protons, given in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard, determined at 300 MHz using perdeuterio dimethyl sulphoxide (DMSO- ⁇ 6 ) as solvent unless otherwise indicated, other solvents (where indicated in the text) include deuterated chloroform - CDCI 3 ; coupling constants (J) are given in Hz; Ar designates an aromatic proton when such an assignment is made;
- (xii) solvent ratios are given in volume : volume (v/v) terms; (xiii) mass spectra (MS) were run with an electron energy of 70 electron volts in the chemical ionisation (CI) mode using a direct exposure probe; where indicated ionisation was effected by electron impact (El), fast atom bombardment (FAB) or electrospray (ESP); values for m/z are given; generally, only ions which indicate the parent mass are reported and unless otherwise stated the value quoted is (M-H) ⁇ ; (xiv) Oxone is a Trademark of E.I. du Pont de Nemours & Co., Inc., and refers to potassium peroxymonosulphate; (xv) The following abbreviations are used: ether diethyl ether;
- Example 30 The title compound was also isolated from the mixture obtained in Example 30 as a yellow foam.
- Oxalyl chloride (0.75 ml) was added to a stirred suspension of (R)-(+)-2-hydroxy- 2-methyl-3,3,3-trifluoropropanoic acid (Method 25) (1.37 g) in DCM (10 ml) containing DMF (1 drop). The mixture was stirred at ambient temperature for 18 hours and was then added to a solution of 4-ethylsulphanyl-2,3-dichloroaniline (Method 7) (1.92 g) and 2,6-diphenylpyridine (2.0 g) in DCM (50 ml).
- Triisopropylsilane thiol (2.0 ml) was added to a stirred suspension of sodium hydride
- reaction mixture was quenched with water (100 ml), and DCM was added (100 ml) and the mixture was filtered through diatomaceous earth. The aqueous layer was separated and washed with DCM (3 x 50 ml). The organic extracts were combined and dried. The volatile material was removed by evaporation, and the product was purified by chromatography using a Mega Bond Elut column (20 g silica) eluting with 0-5% MeOH / DCM to yield the title compound as a white solid. (0.53 g) 80%.
- Oxalyl chloride (0.7 ml) was added to a stirred suspension of (R)-(+)-2-hydroxy-2-methyl-3,3,3-trifluoropropanoic acid (Method 25) (1.26 g) in DCM (40 ml) containing DMF (3 drops). The mixture was stirred at ambient temperature for 4 hours and 2,6-di-t-butylpyridine (2.25 ml) and 4-iodo-3-fluoro-2-methylaniline (Method 48) (1. 368 g) were added. The resulting mixture was stirred at ambient temperature for 72 hours.
- Di-Phosphorous pentoxide (923 mg) was added to a solution of 4-mercaptobenzoic acid (2000 mg) in DMF (10 ml). The mixture was stirred for sixteen hours at 150°C under argon. The mixture was allowed to cool then EtOAc (150 ml) was added. The solution was washed with water (100 ml), brine (50 ml) and dried. The volatile material was removed by evaporation and the residue was purified by chromatography on silica gel, eluted with 2.5% MeOH in DCM to give two samples.
- Iodine monochloride (0.28 ml) was added to a solution of the mixture (1919 mg) in glacial acetic acid (10 ml). The mixture was stirred for four hours at 70°C. After the mixture was allowed to cool to ambient temperature, saturated sodium sulphite solution (50 ml) added. The solution was extracted with EtOAc (100 ml), volatile material was removed by evaporation and the residue was partitioned between EtOAc (100 ml) and saturated sodium bicarbonate solution (50 ml). The organic phase was separated, washed with brine (50 ml) and dried.
- 1,3-Bis(trimethylsilyl)urea (5.73 g) was added to a solution of (R)-(+)-2-hydroxy-2-methyl-3,3,3-trifluoropropanoic acid (Method 25; 4.424 g) in DCM (75 ml) and the mixture was stirred for 16 hours at ambient temperature. A solid was filtered off and washed with DCM (20 ml). The organic solutions were combined, cooled in an ice bath,
- Example 85 The following illustrate representative pharmaceutical dosage forms containing the compound of formula (I), or a pharmaceutically acceptable salt or in vivo hydrolysable ester thereof (hereafter compound X), for therapeutic or prophylactic use in humans:-
- the above formulations may be obtained by conventional procedures well known in the pharmaceutical art.
- the tablets (a)-(c) may be enteric coated by conventional means, for example to provide a coating of cellulose acetate phthalate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Hospice & Palliative Care (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pyrrole Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
Description







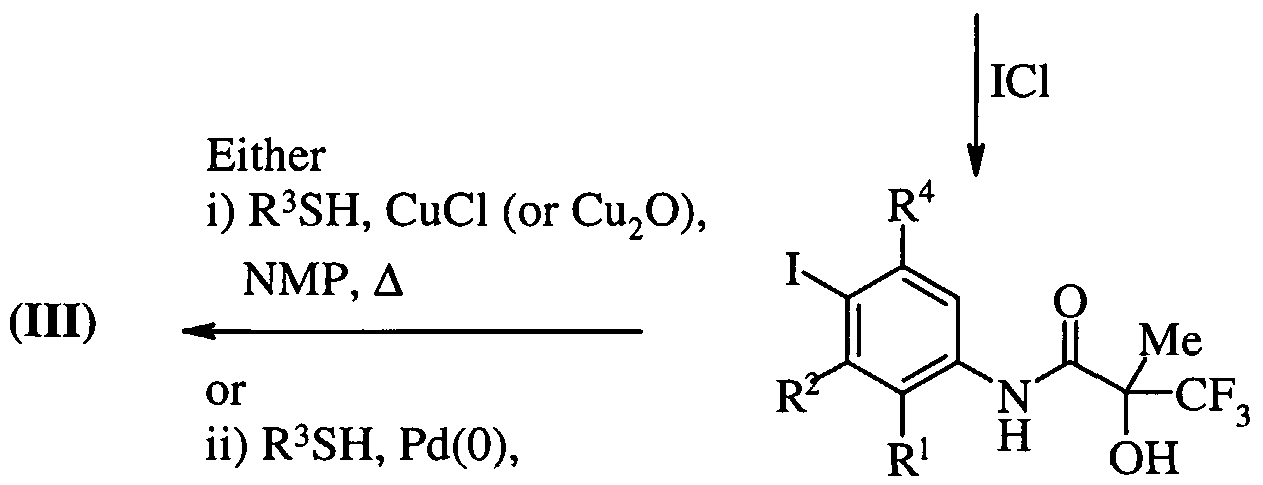
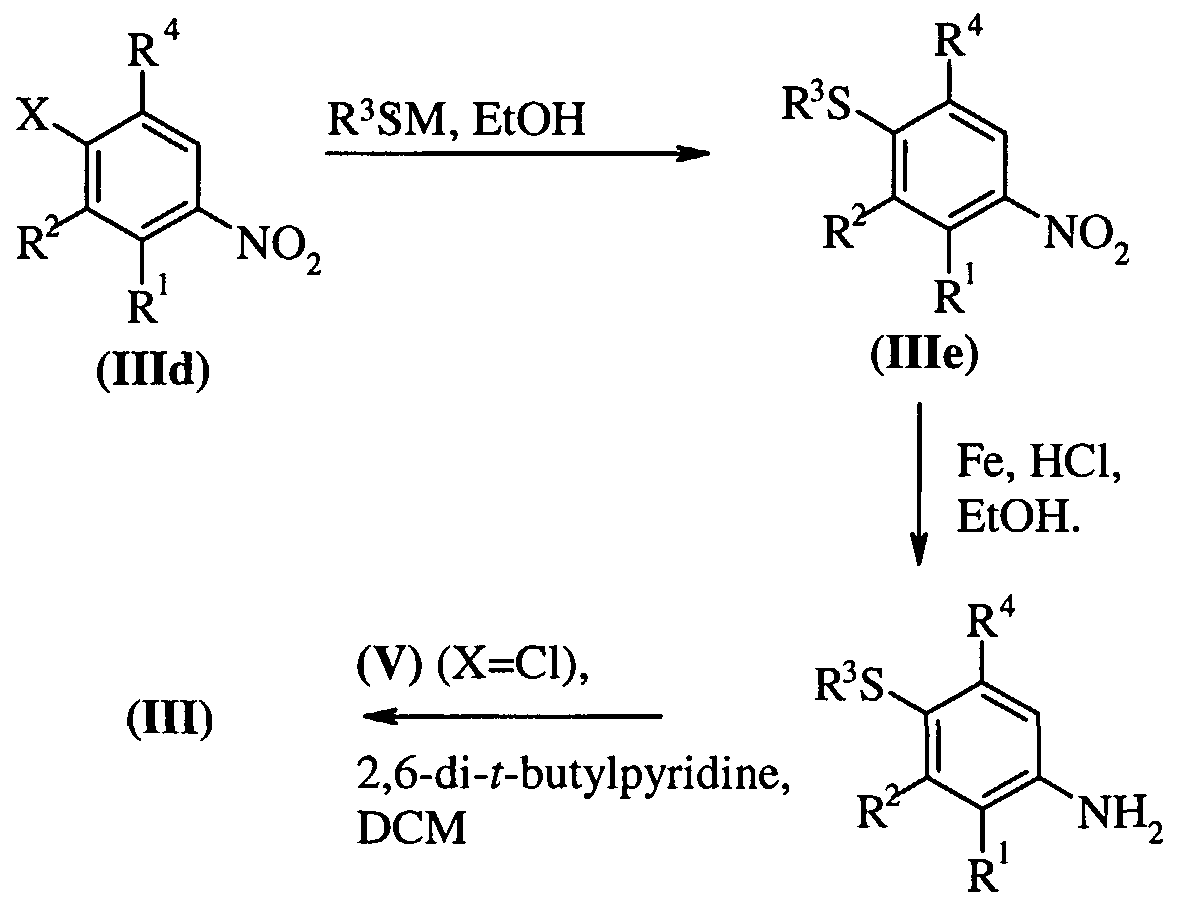








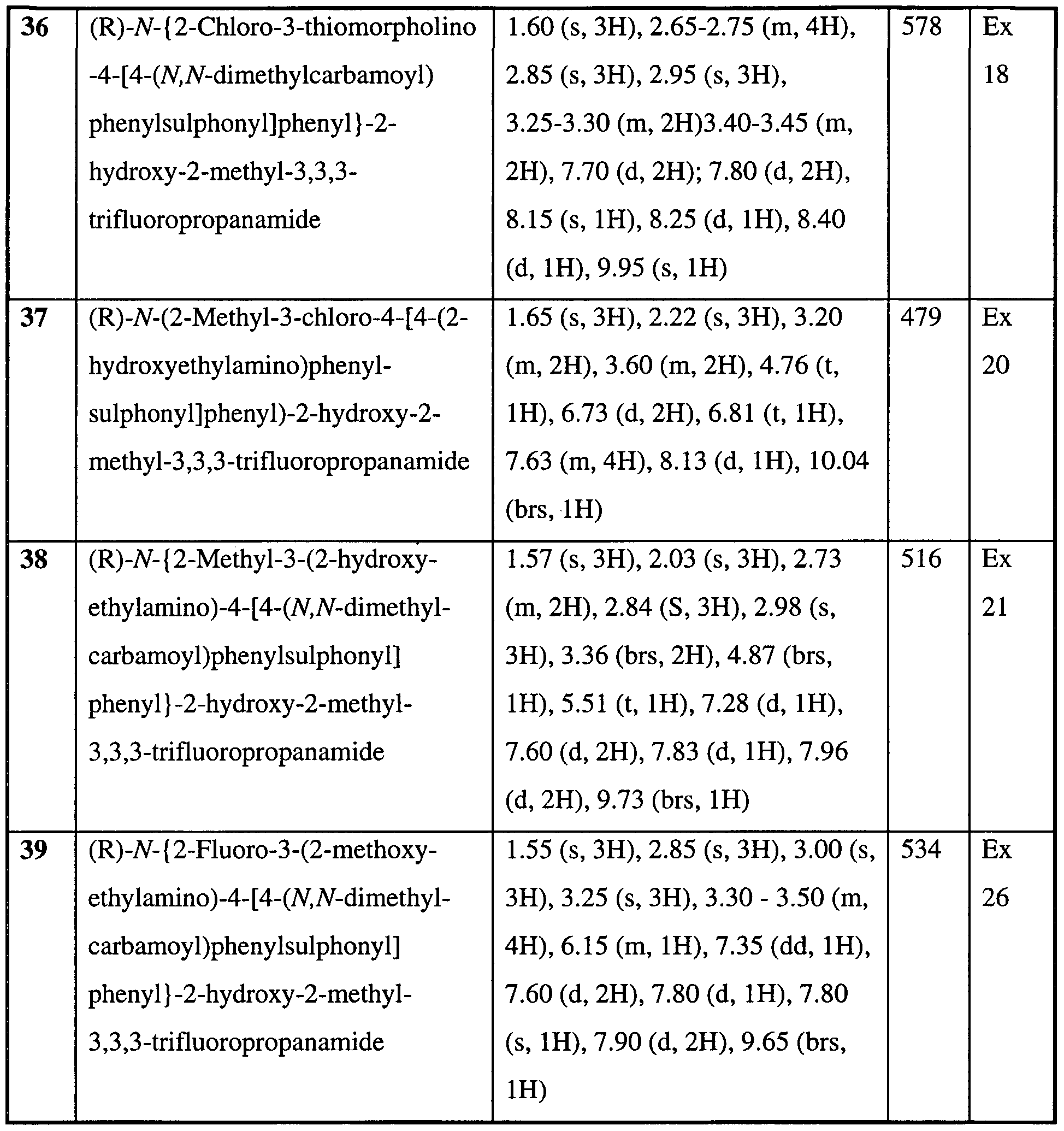



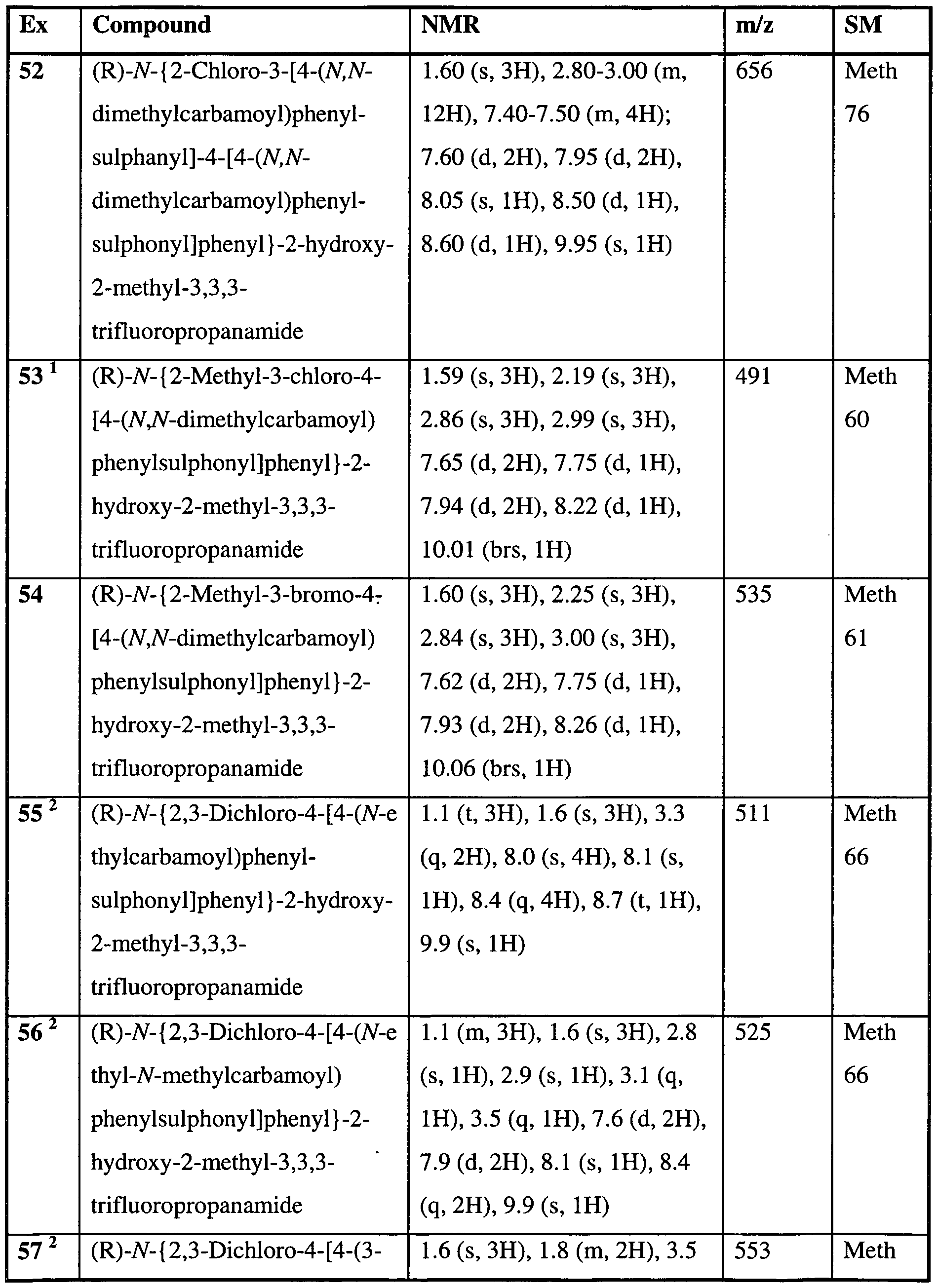







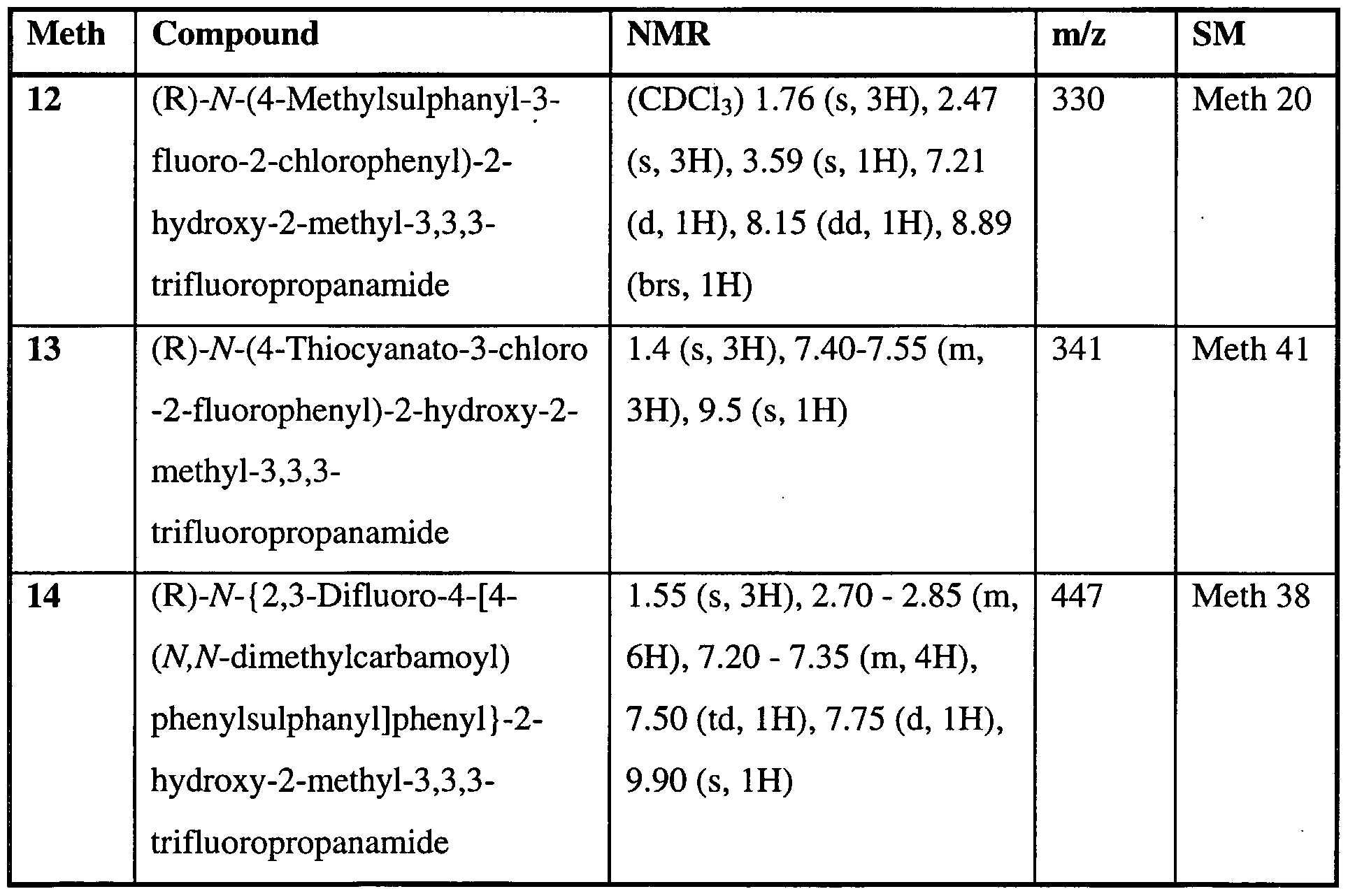





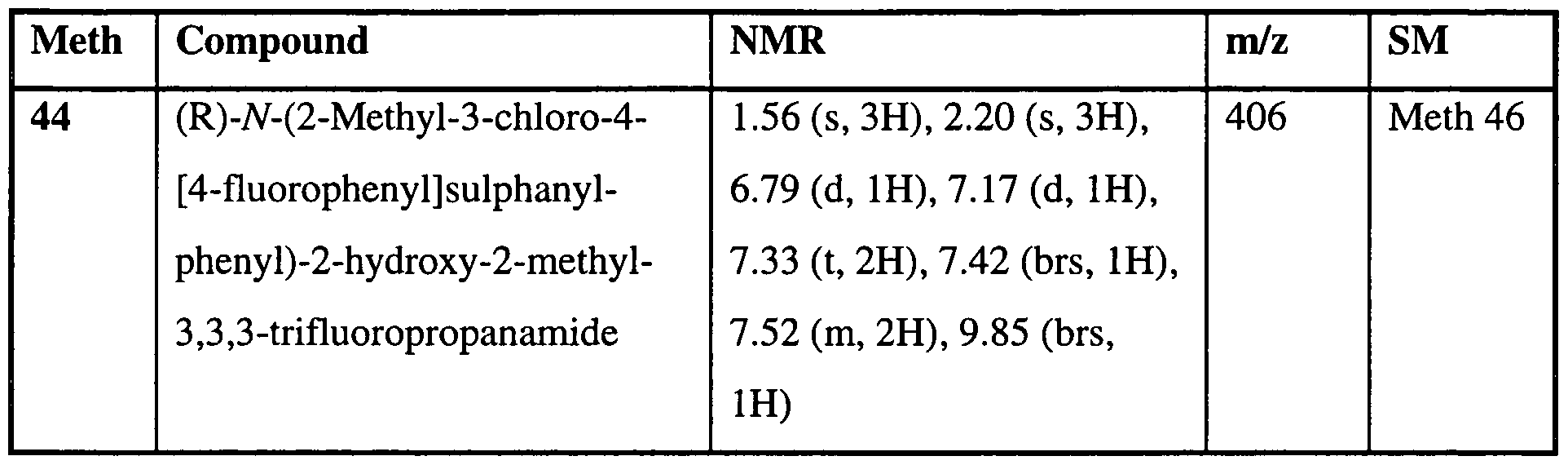






Claims



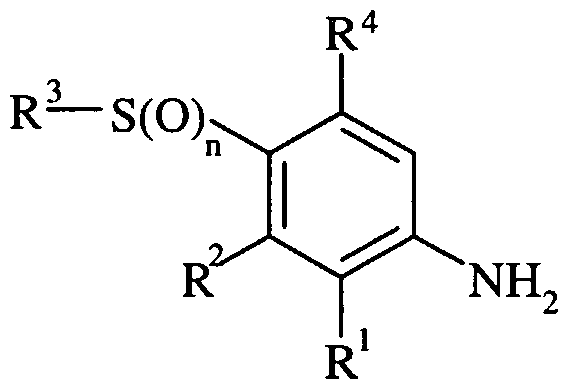

Priority Applications (21)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SI200030393T SI1214296T1 (en) | 1999-09-04 | 2000-08-30 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| HU0202725A HUP0202725A3 (en) | 1999-09-04 | 2000-08-30 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity, process for their preparation and pharmaceutical compositions containing them |
| UA2002042680A UA73319C2 (en) | 1999-09-04 | 2000-08-30 | Substituted derivatives of n-phenyl-2-hydroxy-2-methyl-3,3,3-trifluorpropanamide increasing pyruvatehydrogenase activity |
| AT00956672T ATE262507T1 (en) | 1999-09-04 | 2000-08-30 | SUBSTITUTED N-PHENYL-2-HYDROXY-2-METHYL-3,3,3-TRIFLUOROPANAMIDE DERIVATIVE TO INCREASE PYRUVATE DEHYDROGENASE ACTIVITY |
| DK00956672T DK1214296T3 (en) | 1999-09-04 | 2000-08-30 | Substituted N-phenyl-2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives that increase pyruvate dehydrogenase activity |
| DE60009319T DE60009319T2 (en) | 1999-09-04 | 2000-08-30 | SUBSTITUTED N-PHENYL-2-HYDROXY-2-METHYL-3,3,3-TRIFLUORPROPANAMID DERIVATIVES FOR INCREASING PYRUVATE DEHYDROGENASE ACTIVITY |
| BR0013694-8A BR0013694A (en) | 1999-09-04 | 2000-08-30 | Compound, process for preparing a compound, pharmaceutical composition, and use of a compound |
| US10/069,994 US6689909B1 (en) | 1999-09-04 | 2000-08-30 | Substituted N-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| JP2001521703A JP2003508515A (en) | 1999-09-04 | 2000-08-30 | Substituted N-phenyl-2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives that enhance pyruvate dehydrogenase activity |
| IL14811100A IL148111A0 (en) | 1999-09-04 | 2000-08-30 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| MXPA02002319A MXPA02002319A (en) | 1999-09-04 | 2000-08-30 | Substituted n phenyl 2 hydroxy 2 methyl 3,3, 3 trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity. |
| AU68543/00A AU760061B2 (en) | 1999-09-04 | 2000-08-30 | Substituted N-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| EEP200200113A EE05080B1 (en) | 1999-09-04 | 2000-08-30 | Substituted N-Phenyl-2-H-Droxy-2-Methyl-3,3,3-Trifluoropropanamide Derivatives Enhancing Pyruvate Dehydrogenase Activity |
| NZ517147A NZ517147A (en) | 1999-09-04 | 2000-08-30 | Substituted N-phenyl 2-hydroxy-2-methyl-3,3,3- trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| EP00956672A EP1214296B1 (en) | 1999-09-04 | 2000-08-30 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| SK300-2002A SK286774B6 (en) | 1999-09-04 | 2000-08-30 | Substituted N-phenyl-2-hydroxy-2-methyl-3,3,3- trifluoropropanamide derivatives, process for their preparation and pharmaceutical composition comprising the same |
| HK02107004.7A HK1045496B (en) | 1999-09-04 | 2000-08-30 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| CA002381581A CA2381581A1 (en) | 1999-09-04 | 2000-08-30 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| IL148111A IL148111A (en) | 1999-09-04 | 2002-02-11 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3- trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| IS6285A IS2069B (en) | 1999-09-04 | 2002-02-26 | Substituted N-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropamide derivatives that enhance pyruvate hydrogen sulfide activity |
| NO20021040A NO20021040L (en) | 1999-09-04 | 2002-03-01 | Substituted N-phenyl-2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which teach puruvate dehydrogenase activity |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9920814.2 | 1999-09-04 | ||
| GBGB9920814.2A GB9920814D0 (en) | 1999-09-04 | 1999-09-04 | Chemical compounds |
| GB0006641.5 | 2000-03-21 | ||
| GB0006641A GB0006641D0 (en) | 2000-03-21 | 2000-03-21 | Chemical compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001017956A1 true WO2001017956A1 (en) | 2001-03-15 |
Family
ID=26243905
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2000/003314 Ceased WO2001017956A1 (en) | 1999-09-04 | 2000-08-30 | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
Country Status (32)
| Country | Link |
|---|---|
| US (1) | US6689909B1 (en) |
| EP (1) | EP1214296B1 (en) |
| JP (1) | JP2003508515A (en) |
| KR (1) | KR100692232B1 (en) |
| CN (1) | CN1235877C (en) |
| AR (1) | AR035163A1 (en) |
| AT (1) | ATE262507T1 (en) |
| AU (1) | AU760061B2 (en) |
| BG (1) | BG65099B1 (en) |
| BR (1) | BR0013694A (en) |
| CA (1) | CA2381581A1 (en) |
| CO (1) | CO5190690A1 (en) |
| CZ (1) | CZ2002773A3 (en) |
| DE (1) | DE60009319T2 (en) |
| DK (1) | DK1214296T3 (en) |
| EE (1) | EE05080B1 (en) |
| ES (1) | ES2216940T3 (en) |
| HK (1) | HK1045496B (en) |
| HU (1) | HUP0202725A3 (en) |
| IL (2) | IL148111A0 (en) |
| IS (1) | IS2069B (en) |
| MX (1) | MXPA02002319A (en) |
| MY (1) | MY127036A (en) |
| NO (1) | NO20021040L (en) |
| NZ (1) | NZ517147A (en) |
| PL (1) | PL353887A1 (en) |
| PT (1) | PT1214296E (en) |
| RU (1) | RU2255085C2 (en) |
| SK (1) | SK286774B6 (en) |
| TR (1) | TR200200550T2 (en) |
| TW (1) | TW577867B (en) |
| WO (1) | WO2001017956A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001087850A1 (en) * | 2000-05-19 | 2001-11-22 | Astrazeneca Ab | Pyridazine derivative elevating pdhactivity |
| WO2003017995A1 (en) * | 2001-08-23 | 2003-03-06 | Astrazeneca Ab | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| US6878712B1 (en) | 1999-09-04 | 2005-04-12 | Astrazeneca Ab | Amides as inhibitors for pyruvate dehydrogenase |
| WO2012151448A1 (en) * | 2011-05-03 | 2012-11-08 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in therapy |
| US9108921B2 (en) | 2013-03-15 | 2015-08-18 | Agios Pharmaceuticals, Inc | Therapeutic compounds and compositions |
| US9458132B2 (en) | 2012-11-08 | 2016-10-04 | Agios Pharmaceuticals, Inc | Therapeutic compounds and compositions and their use as PKM2 modulators |
| US11364240B2 (en) | 2017-08-15 | 2022-06-21 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in treating blood disorders |
| WO2023032940A1 (en) * | 2021-09-01 | 2023-03-09 | 日本たばこ産業株式会社 | Nitrogen-containing tricyclic compound and pharmaceutical use thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100562516C (en) * | 2001-12-27 | 2009-11-25 | 第一制药株式会社 | Inhibitors of beta-amyloid production and secretion |
| CN109096186B (en) * | 2018-09-29 | 2020-08-04 | 中南大学 | Synthesis method of 2-arylsulfonyl quinoline derivative |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0524781A1 (en) * | 1991-07-25 | 1993-01-27 | Zeneca Limited | Therapeutic amides |
| EP0617010A1 (en) * | 1993-03-15 | 1994-09-28 | Zeneca Limited | Therapeutic compounds |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0002309B1 (en) | 1977-10-12 | 1982-12-01 | Imperial Chemical Industries Plc | Acylanilides, process for their manufacture and pharmaceutical and veterinary compositions containing them |
| US4191775A (en) | 1977-12-15 | 1980-03-04 | Imperial Chemical Industries Limited | Amide derivatives |
| NZ197008A (en) | 1980-05-22 | 1984-10-19 | Ici Ltd | Acylanilide derivatives and pharmaceutical compositions |
| AU8961382A (en) | 1981-11-06 | 1983-05-12 | Imperial Chemical Industries Plc | Amide derivatives |
| US4537618A (en) | 1982-05-26 | 1985-08-27 | Ciba Geigy Corporation | N-phenylsulfonyl-N'-pyrimidinylureas |
| EP0100172B1 (en) | 1982-07-23 | 1987-08-12 | Imperial Chemical Industries Plc | Amide derivatives |
| GB8617653D0 (en) | 1986-07-18 | 1986-08-28 | Ici Plc | Amide derivatives |
| GB8617652D0 (en) | 1986-07-18 | 1986-08-28 | Ici Plc | Acylanilide derivatives |
| GB9116069D0 (en) * | 1991-07-25 | 1991-09-11 | Ici Plc | Therapeutic amides |
| EP0539329A1 (en) | 1991-10-25 | 1993-04-28 | Ciba-Geigy Ag | Acetenyl compounds useful as leukotrien antagonists |
| SE9103397D0 (en) | 1991-11-18 | 1991-11-18 | Kabi Pharmacia Ab | NEW SUBSTITUTED SALICYL ACIDS |
| IL105558A (en) | 1992-05-18 | 1998-04-05 | Zeneca Ltd | Tertiary carbinols having potassium channel activity, process for their preparation and pharmaceutical compositions containing them |
| GB9309716D0 (en) | 1993-05-12 | 1993-06-23 | Zeneca Ltd | Heterocyclic derivatives |
| GB9310069D0 (en) | 1993-05-17 | 1993-06-30 | Zeneca Ltd | Heterocyclic compounds |
| GB9310095D0 (en) | 1993-05-17 | 1993-06-30 | Zeneca Ltd | Therapeutic compounds |
| GB2278054A (en) * | 1993-05-18 | 1994-11-23 | Zeneca Ltd | Compounds for the treatment of urinary incontinence |
| GB9505082D0 (en) | 1995-03-14 | 1995-05-03 | Zeneca Ltd | Medicament |
| GB9607458D0 (en) | 1996-04-10 | 1996-06-12 | Zeneca Ltd | Enzymatic process for stereoselective preparation of therapeutic amides |
| GB9804648D0 (en) * | 1998-03-06 | 1998-04-29 | Zeneca Ltd | Chemical compounds |
| GB9805520D0 (en) | 1998-03-17 | 1998-05-13 | Zeneca Ltd | Chemical compounds |
| GB9811427D0 (en) | 1998-05-29 | 1998-07-22 | Zeneca Ltd | Chemical compounds |
-
2000
- 2000-08-30 MX MXPA02002319A patent/MXPA02002319A/en active IP Right Grant
- 2000-08-30 MY MYPI20004037 patent/MY127036A/en unknown
- 2000-08-30 TR TR2002/00550T patent/TR200200550T2/en unknown
- 2000-08-30 JP JP2001521703A patent/JP2003508515A/en active Pending
- 2000-08-30 HK HK02107004.7A patent/HK1045496B/en not_active IP Right Cessation
- 2000-08-30 CA CA002381581A patent/CA2381581A1/en not_active Abandoned
- 2000-08-30 PL PL00353887A patent/PL353887A1/en not_active Application Discontinuation
- 2000-08-30 ES ES00956672T patent/ES2216940T3/en not_active Expired - Lifetime
- 2000-08-30 WO PCT/GB2000/003314 patent/WO2001017956A1/en not_active Ceased
- 2000-08-30 DE DE60009319T patent/DE60009319T2/en not_active Expired - Fee Related
- 2000-08-30 NZ NZ517147A patent/NZ517147A/en unknown
- 2000-08-30 CN CNB008152446A patent/CN1235877C/en not_active Expired - Fee Related
- 2000-08-30 DK DK00956672T patent/DK1214296T3/en active
- 2000-08-30 PT PT00956672T patent/PT1214296E/en unknown
- 2000-08-30 SK SK300-2002A patent/SK286774B6/en not_active IP Right Cessation
- 2000-08-30 IL IL14811100A patent/IL148111A0/en active IP Right Grant
- 2000-08-30 HU HU0202725A patent/HUP0202725A3/en unknown
- 2000-08-30 CZ CZ2002773A patent/CZ2002773A3/en unknown
- 2000-08-30 US US10/069,994 patent/US6689909B1/en not_active Expired - Fee Related
- 2000-08-30 EE EEP200200113A patent/EE05080B1/en not_active IP Right Cessation
- 2000-08-30 AT AT00956672T patent/ATE262507T1/en not_active IP Right Cessation
- 2000-08-30 KR KR1020027002841A patent/KR100692232B1/en not_active Expired - Fee Related
- 2000-08-30 BR BR0013694-8A patent/BR0013694A/en not_active Application Discontinuation
- 2000-08-30 RU RU2002108508/04A patent/RU2255085C2/en not_active IP Right Cessation
- 2000-08-30 EP EP00956672A patent/EP1214296B1/en not_active Expired - Lifetime
- 2000-08-30 AU AU68543/00A patent/AU760061B2/en not_active Ceased
- 2000-09-01 CO CO00065992A patent/CO5190690A1/en not_active Application Discontinuation
- 2000-09-04 AR ARP000104623A patent/AR035163A1/en unknown
- 2000-09-04 TW TW089118053A patent/TW577867B/en not_active IP Right Cessation
-
2002
- 2002-02-11 IL IL148111A patent/IL148111A/en not_active IP Right Cessation
- 2002-02-26 IS IS6285A patent/IS2069B/en unknown
- 2002-03-01 NO NO20021040A patent/NO20021040L/en not_active Application Discontinuation
- 2002-03-14 BG BG106520A patent/BG65099B1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0524781A1 (en) * | 1991-07-25 | 1993-01-27 | Zeneca Limited | Therapeutic amides |
| EP0617010A1 (en) * | 1993-03-15 | 1994-09-28 | Zeneca Limited | Therapeutic compounds |
Non-Patent Citations (1)
| Title |
|---|
| C.J. OHNMACHT, ET AL.: "N-Aryl-3,3,3-trifluoro-2-hydroxy-2-methylpropanamides: KATP potassium channel openers. Modifications on the western region", JOURNAL OF MEDICINAL CHEMISTRY, vol. 39, no. 23, 8 November 1996 (1996-11-08), Ameircan Chemical Society, Washington, DC, US, pages 4592 - 4601, XP002110510, ISSN: 0022-2623 * |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6878712B1 (en) | 1999-09-04 | 2005-04-12 | Astrazeneca Ab | Amides as inhibitors for pyruvate dehydrogenase |
| WO2001087850A1 (en) * | 2000-05-19 | 2001-11-22 | Astrazeneca Ab | Pyridazine derivative elevating pdhactivity |
| WO2003017995A1 (en) * | 2001-08-23 | 2003-03-06 | Astrazeneca Ab | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| RU2301805C2 (en) * | 2001-08-23 | 2007-06-27 | Астразенека Аб | Derivatives of substituted n-phenyl-2-hydroxy-2-methyl-3,3,3-trifluoropropaneamide, method for their preparing (variants), their using (variants) and pharmaceutical composition (variants) |
| AU2002321520B2 (en) * | 2001-08-23 | 2008-06-12 | Astrazeneca Ab | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| US9388164B2 (en) | 2011-05-03 | 2016-07-12 | Agios Pharmaceuticals, Inc | Methods of using pyruvate kinase activators |
| WO2012151448A1 (en) * | 2011-05-03 | 2012-11-08 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in therapy |
| US9458132B2 (en) | 2012-11-08 | 2016-10-04 | Agios Pharmaceuticals, Inc | Therapeutic compounds and compositions and their use as PKM2 modulators |
| US9108921B2 (en) | 2013-03-15 | 2015-08-18 | Agios Pharmaceuticals, Inc | Therapeutic compounds and compositions |
| US9365545B2 (en) | 2013-03-15 | 2016-06-14 | Agios Pharmaceuticals, Inc | Therapeutic compounds and compositions |
| US11364240B2 (en) | 2017-08-15 | 2022-06-21 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in treating blood disorders |
| US11464775B2 (en) | 2017-08-15 | 2022-10-11 | Agios Pharmaceuticals, Inc. | Pyruvate kinase modulators and use thereof |
| US11590132B2 (en) | 2017-08-15 | 2023-02-28 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in treating blood disorders |
| US11872225B2 (en) | 2017-08-15 | 2024-01-16 | Agios Pharmaceuticals, Inc. | Pyruvate kinase modulators and use thereof |
| US11957680B2 (en) | 2017-08-15 | 2024-04-16 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in treating blood disorders |
| US12133853B2 (en) | 2017-08-15 | 2024-11-05 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use in treating blood disorders |
| WO2023032940A1 (en) * | 2021-09-01 | 2023-03-09 | 日本たばこ産業株式会社 | Nitrogen-containing tricyclic compound and pharmaceutical use thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU744579B2 (en) | Benzenesulfonamide-derivatives and their use as medicaments | |
| EP1214287B1 (en) | Amides as inhibitors for pyruvate dehydrogenase | |
| AU740909B2 (en) | Use of compounds for the elevation of pyruvate dehydrogenase activity | |
| US6689909B1 (en) | Substituted N-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity | |
| EP1214295B1 (en) | Hydroxyacetamidobenzenesulphonamide derivatives | |
| WO1999062873A1 (en) | Benzenesulphonamide derivatives as pyruvate dehydrogenase activators | |
| ZA200201437B (en) | Substituted N-phenyl 2-hydroxy 2-methyl-3,3,3-trifluoropropanamide derivates which elevate pyruvate dehydrogenase activity. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2000956672 Country of ref document: EP Ref document number: 2381581 Country of ref document: CA Ref document number: 68543/00 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 148111 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 517147 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/01437 Country of ref document: ZA Ref document number: 200201437 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3002002 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2002-773 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/002319 Country of ref document: MX Ref document number: 2002/00550 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027002841 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2001 521703 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2000 106520 Country of ref document: BG Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2002 2002108508 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 008152446 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-773 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10069994 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2000956672 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027002841 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 68543/00 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 517147 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 517147 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2000956672 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 224/MUMNP/2005 Country of ref document: IN |