WO2001049662A2 - Carbazole derivatives as inhibitors of spla2 - Google Patents
Carbazole derivatives as inhibitors of spla2 Download PDFInfo
- Publication number
- WO2001049662A2 WO2001049662A2 PCT/US2001/010850 US0110850W WO0149662A2 WO 2001049662 A2 WO2001049662 A2 WO 2001049662A2 US 0110850 W US0110850 W US 0110850W WO 0149662 A2 WO0149662 A2 WO 0149662A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- phenyl
- compound
- formula
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C*)(C=CC(C(O)=O)=C1*)C=C1NO Chemical compound CC(C*)(C=CC(C(O)=O)=C1*)C=C1NO 0.000 description 11
- SVDXUSHCQSAYDN-FQEVSTJZSA-N CC(C)C[C@@H](C(O)=O)NC(COc1cccc([n](Cc2ccccc2)c2ccc3)c1c2c3C(N)=O)=O Chemical compound CC(C)C[C@@H](C(O)=O)NC(COc1cccc([n](Cc2ccccc2)c2ccc3)c1c2c3C(N)=O)=O SVDXUSHCQSAYDN-FQEVSTJZSA-N 0.000 description 1
- WVVXSXLVTXBXHZ-UHFFFAOYSA-N NC(COc1cccc2c1c(c(C(N)=O)ccc1)c1[n]2Cc1ccccc1)=O Chemical compound NC(COc1cccc2c1c(c(C(N)=O)ccc1)c1[n]2Cc1ccccc1)=O WVVXSXLVTXBXHZ-UHFFFAOYSA-N 0.000 description 1
- KJKHKZWZYFLYPR-UHFFFAOYSA-N NC(c1c2c(c(OCC(NCC(O)=O)=O)ccc3)c3[n](Cc3ccccc3)c2ccc1)=O Chemical compound NC(c1c2c(c(OCC(NCC(O)=O)=O)ccc3)c3[n](Cc3ccccc3)c2ccc1)=O KJKHKZWZYFLYPR-UHFFFAOYSA-N 0.000 description 1
- KXEGYHWBRKNJDC-KRWDZBQOSA-O NC(c1c2c(c(OCC(N[C@@H](CC(O)=O)C(O[IH+])=O)=O)ccc3)c3[n](Cc3ccccc3)c2ccc1)=O Chemical compound NC(c1c2c(c(OCC(N[C@@H](CC(O)=O)C(O[IH+])=O)=O)ccc3)c3[n](Cc3ccccc3)c2ccc1)=O KXEGYHWBRKNJDC-KRWDZBQOSA-O 0.000 description 1
- LOEUKRWGLDWONT-UHFFFAOYSA-N NC(c1cccc([n](Cc2ccccc2)c2c(cc3)F)c1c2c3OCC(O)=O)=O Chemical compound NC(c1cccc([n](Cc2ccccc2)c2c(cc3)F)c1c2c3OCC(O)=O)=O LOEUKRWGLDWONT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- This invention relates to novel substituted tricyclic organic compounds useful for inhibiting sPLA2 mediated release of fatty acids for the treatment of inflammatory diseases.
- SPLA2 human non- pancreatic secretory phospholipase A2
- SP A2 is a rate limiting enzyme in the arachidonic acid cascade which hydrolyzes membrane phospholipids .
- fatty acids e.g., arachidonic acid.
- Such compounds would be of value in the general treatment of conditions induced and/or maintained by overproduction of sPLA2 such as septic shock, adult respiratory distress syndrome, pancreatitis, trauma-induced shock, bronchial asthma, allergic rhinitis, rheumatoid arthritis, etc.
- This invention provides tricyclic compounds as depicted in the general formula (I) below:
- Z is cyclohexenyl, or phenyl
- R ⁇ is selected from groups (a) , (b) and (c) where;
- (a) is - (C5-C20) alkyl, - (C5-C20) lkenyl, -( C5- C20) alkynyl , carbocyclic radicals, or heterocyclic radicals, or
- (b) is a member of (a) substituted with one or more independently selected non-interfering substituents;
- (c) is the group -(L) ⁇ R ; where, (L)-is a divalent linking group of 1 to 12 atoms selected from carbon, hydrogen, oxygen, nitrogen, and sulfur; wherein the combination of atoms in - (L) - are selected from the group consisting of (i) carbon and hydrogen only, (ii) one sulfur only, (iii) one oxygen only, (iv) one or two nitrogen and hydrogen only, (v) carbon, hydrogen, and one sulfur only, and (vi) an carbon, hydrogen, and oxygen only; and where R 80 is a group selected from (a) or (b) ; R 21 is a non-mterfe ⁇ ng substituent where f is 1-3;
- R 1 is -NHNH 2 , -NH 2 , or -C0NH 2 ;
- R is selected from the group consisting of -0(CH 2 ) t R where 5 ' 9 10 9 1_ ⁇
- R is -CONR R , where R and R is independently H, - (Ci- C 6 )alkyl, - (C ⁇ -C 6 ) alkyl substituted with -C0 2 H or a derivative thereof, or -CF 3 ; phenyl or phenyl substituted with - (C1-C ) alkyl; t is 1-5; or R is - (L ⁇ ) - (acylamino acid) wherein - (L ⁇ ) - is an acylamino acid linker having an acylamino- acid linker length of 1 to 7 ;
- R 3 ' i.s selected from non-mterferm. g substituent, carbocyclic radicals, carbocyclic radicals substituted with non-interfering substituents, heterocyclic radicals, and heterocyclic radicals substituted with non- interfering substituents; or a pharmaceutically acceptable racemate, solvate, tautomer, optical isomer, prodrug derivative or salt, thereof .
- the substituted tricyclic compounds of the present invention are effective in inhibiting human SPLA2 mediated release of fatty acids.
- This invention is also a pharmaceutical formulation comprising a compound of formula I in association with one or more pharmaceutically acceptable diluents, carriers and excipients .
- This invention is also a method of inhibiting SPLA2 comprising administering to a mammal in need of such treatment a therapeutically effective amount of a compound of formula I .
- a method of selectively inhibiting SPLA2 in a mammal in need of such treatment comprising administering to said mammal a therapeutically effective amount of a compound of formula I.
- This invention further provides a compound of formula I for use as a medicament in the treatment of inflammatory diseases such as septic shock, adult respiratory distress syndrome, pancreatitis, trauma- induced shock, bronchial asthma, allergic rhinitis, rheumatoid arthritis, cystic fibrosis, stroke, acute bronchitis, chronic bronchitis, acute bronchiolitis, chronic bronchiolitis, osteoarthritis, gout, spondylarthropathris, ankylosing spondylitis, Reiter's syndrome, psoriatic arthropathy, enterapathric spondylitis, Juvenile arthropathy or juvenile ankylosing spondylitis, Reactive arthropathy, infectious or post- infectious arthritis, gonoccocal arthritis, tuberculous arthritis, viral arthritis, fungal arthritis, syphilitic arthritis, Lyme disease, arthritis associated with "vasculitic syndromes”, polyarteritis nodosa, hypersensitivity vas
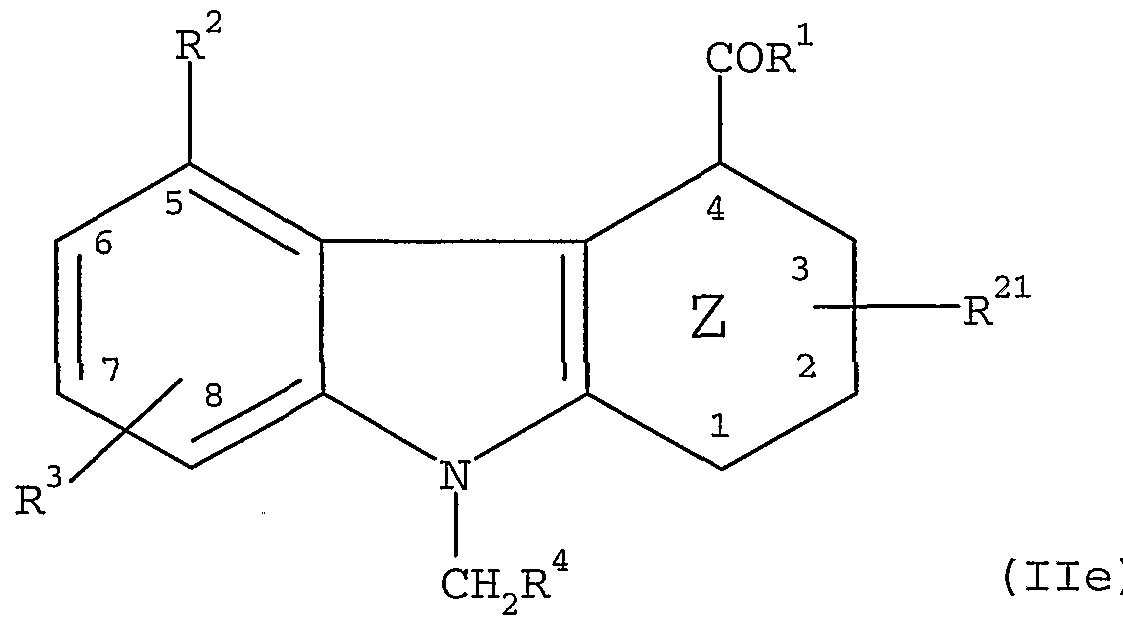
- This invention provides, in addition, a process for preparing compounds of formula II
- Z is cyclohexenyl, or phenyl
- R21 is a non-interfering substituent
- R 1 is -CONH 2 , -NHNH2 or -NH 2 ;
- R 2 ' is the group -0(CH2)t R5 ' where
- R 5 ' is -CONR 9 R 10 , where R 9 and R 10 are independently H, - (C ⁇ -Cg) alkyl or -CF 3 ; phenyl or phenyl substituted with
- R ⁇ ' is the group - (L ⁇ ) - (acylamino acid) group, wherein - (L ⁇ ) - is an acylamino acid linker having an acylamino acid linker length of 1 to 7;
- R 3 ' is H, -0(C ⁇ -C 4 ) alkyl, halo, - (C ⁇ Cg) alkyl, phenyl, -
- C 6 alkyl, halo, or -CF3; -CH 2 0Si (C1-C5) alkyl, furyl, thiophenyl, - (Ci-Cgjhydroxyalkyl, - (C ⁇ -Cg) alkoxy (C]_-
- R 8 is H, -CONH 2 , -NR 9 R 10 , -CN or phenyl where R 9 and R!0 are independently hydrogen, -CF3 , phenyl, - (C]_- C4)alkyl, - (C1-C4) alkylphenyl or -phenyl (C1-C4) alkyl and ⁇ n is 1 to 8; and R4 is H, - (C3-C14) alkyl, - (C3-C14) cycloalkyl, pyridyl, phenyl or phenyl substituted with from 1-5 substituents selected from the group consisting of - (C ⁇ -Cg) alkyl, halo, -CF 3 , -OCF3 , -(C ⁇ -C 4 )alkoxy,
- R 3 (a) is H, -0 (C ⁇ -C 4 ) alkyl, halo, - (C ⁇ -C 6 ) alkyl, phenyl, - (C 1 -C 4 ) alkylphenyl; phenyl substituted with - (Cv C 6 ) alkyl, halo or -CF 3 ; -CH 2 0Si (C ⁇ -C 6 ) alkyl, furyl, thiophenyl, - (C ⁇ -C 6 )hydroxyalkyl, - (Ci-C ⁇ ) alkoxy (C ⁇ C 6 ) alkyl, - (C ⁇ -C 6 ) alkoxy (C ⁇ -C 6 ) alkenyl; or -(CH 2 ) n R 8 where
- R° is H, -NR 9 R 10 , -CN or phenyl where R 9 and R 10 are independently hydrogen, -CF 3 , phenyl , - (C 1 -C 4 ) alkyl , - (Ci- C 4 ) alkylphenyl or -phenyl (C 1 -C 4 ) alkyl and n is 1 to 8 ; to form a compound of formula XII
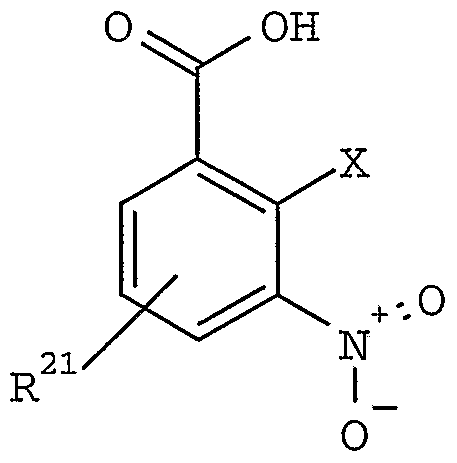
- step (I) coupling the free acid compound of step (I) with a C-terminal protected amino acid to from a compound of formula I .
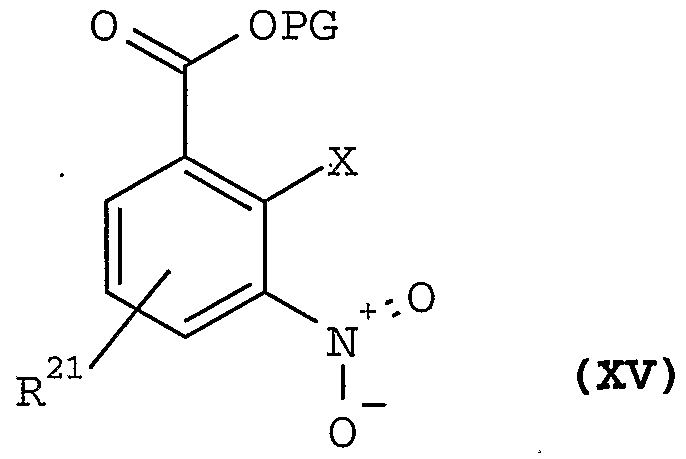
- This invention provides, in addition, a process for preparing compounds of formula II or a pharmaceutically acceptable racemate, solvate, tautomer, optical isomer, prodrug derivative or salt, thereof; which process comprises the steps of: a) esterifying a compound of formula XVI
- alkyl by itself or as part of another substituent means, unless otherwise defined, a straight or branched chain monovalent hydrocarbon radical such as methyl, ethyl, n-propyl, isopropyl, n-butyl, tertiary butyl, isobutyl, sec-butyl tert butyl, n-pentyl, isopentyl, neopentyl, heptyl, hexyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl and the like.
- alkyl includes -(C1-C2) alkyl, -(C1-C4) alkyl, - (C1-C6) alkyl, -(C5- C14) alkyl, and - (C1-C10) alkyl .
- alkenyl as used herein represents an olefinically unsaturated branched or linear group having at least one double bond.
- groups include radicals such as vinyl, allyl, 2-butenyl, 3- butenyl ,
- alkynyl denotes such radicals as ethynyl , propynyl , butynyl , pentynyl , hexynyl , heptynyl as well as di- and tri-ynes.
- halo means chloro, fluoro, bromo or iodo .
- - (C1-C4) alkoxy denotes a group such as methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, t-butoxy and like groups, attached to the remainder of the molecule by the oxygen atom.
- phenyl (C1-C4) alkyl refers to a straight or branched chain alkyl group having from one to four carbon atoms attached to a phenyl ring which chain is attached to the remainder of the molecule.
- Typical phenylalkyl groups include benzyl, phenylethyl, phenylpropyl , phenylisopropyl, and phenylbutyl .
- - (C1-C4) alkylthio defines a straight or branched alkyl chain having one to four carbon atoms attached to the remainder of the molecule by a sulfur atom.
- Typical - (C1-C4) alkylthio groups include methylthio, ethylthio, propylthio, butylthio and the like.
- cycloalkyl includes groups such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl, cyclododecyl, cyclotridecyl, cyclotetradecyl and the like.
- -(C3-C14) cycloalkyl includes groups such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl, cyclododecyl, cyclotridecyl, cyclotetradecyl and the like.
- C14) cycloalkyl includes and - (C3-C7) cycloalkyl .
- heterocyclic radical refers to radicals derived from onocyclic or polycyclic, saturated or unsaturated, substituted or unsubstituted heterocyclic nuclei having 5 to 14 ring atoms and containing from 1 to 3 hetero atoms selected from the group consisting of nitrogen, oxygen or sulfur.
- Typical heterocyclic radicals are pyridyl, thienyl, fluorenyl, pyrrolyl, furanyl, thiophenyl, pyrazolyl, imidazolyl, phenylimidazolyl, triazolyl, isoxazolyl, oxazolyl, thiazolyl, thiadiazolyl, indolyl, carbazolyl, norharmanyl, azaindolyl, benzofuranyl, dibenzofuranyl, thianaphtheneyl , dibenzothiophenyl, indazolyl, imidazo (1.2-A)pyridinyl, benzotriazolyl, anthranilyl, 1, 2-benzisoxazolyl, benzoxazolyl, benzothiazolyl, purinyl , pryidinyl , dipyridylyl , phenylpyridinyl , benzylpyridinyl
- carrier radical refers to radicals derived from a saturated or unsaturated, substituted or unsubstituted 5 to 14 membered organic nucleus whose ring forming atoms (other than hydrogen) are solely carbon atoms.
- Typical carbocyclic radicals are cycloalkyl, cycloalkenyl , phenyl, naphthyl, norbornanyl, bicycloheptadienyl, tolulyl, xylenyl, indenyl, stilbenyl, terphenylyl, diphenylethylenyl, phenylcyclohexeyl, acenaphthylenyl, and anthracenyl, biphenyl, bibenzylyl and related bibenzylyl homologues represented by the formula (bb) ,
- n is an integer from 1 to 8.
- non-interfering substituent refers to radicals suitable for substitution at positions 1, 2, 3,
- radical (s) suitable for substitution on the heterocyclic radical and carbocyclic radical as defined above are hydrogen, - (C1-C14) alkyl, - (C2-C6) alkenyl, - (C2-
- organic substituent refers to a monovalent radical consisting of carbon and hydrogen with or without oxygen, nitrogen, sulfur, halogen, or other elements.
- Illustrative organic substituents are c l ⁇ c 8 alkyl, aryl, C7-C14 aralkyl, C7-C14 alkaryl, C3- Cg cycloalkyl, alkoxyalkyl and these groups substituted with halogen, -CF3 , -OH, C]_-Cg alkyl, amino, carbonyl, and -CN.
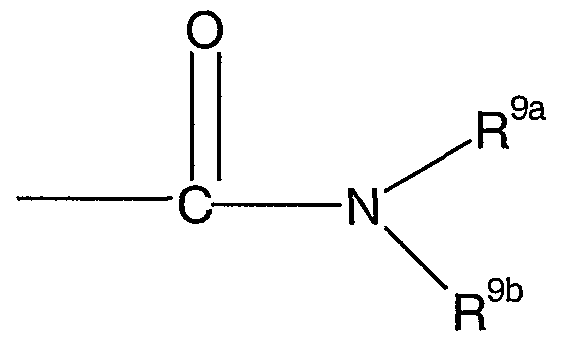
- acylamino acid is a group represented by the formula :
- R 9a is selected from the group consisting of H, (C1-C ) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl and aryl, -CF3 ; and wherein NR 9 -° is an amino acid residue of either a natural or unnatural amino acid with the nitrogen atom being part of the amino group of the amino acid.
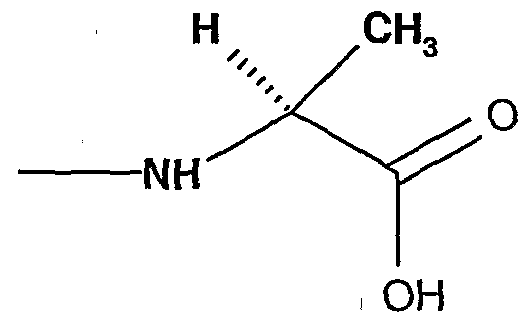
- a typical amino acid is selected from the group comprising isoleucine, valine, phenylalanine, aspartic acid, leucine, glycine, asparagine, cystein, glutamine, glutamic acid, histidine, lysine, methionine, serine, threonine, tryptophan, tyrosine and derivatives thereof.
- amino acid is 2-proline, -proline and derivatives thereof.
- amino acids are peptides, polypeptides and derivatives thereof.
- substituted group is an organic group substituted with one or more non-interfering substituents .
- C-terminal protected amino acid is an amino acid group wherein the carboxy terminal is protected by an acid protecting group known in the art to form groups such as the ester or amide, e.t.c, while the amino terminal is available for coupling with substrate carboxy groups by peptide coupling methodologies known to one of skill in the art.
- amino acid residue refers to the portion of the amino acid group coupled at the nitrogen atom of the amino terminus. It is the amino acid less a hydrogen atom from the amino terminus. It is further illustrated as used herein for the amino acid alanine attached at the nitrogen atom as shown below:
- acylamino acid linker refers to a divalent linking group symbolized as, -(Ifa)-, which has the function of joining the 5 - position of the tricyclic nucleus to an acylamino acid group in the general relationship:
- acylamino acid linker length refer to the number of atoms (excluding hydrogen) in the shortest chain of the linking group - (I/ ) - that connects the 5 - position of the tricyclic nucleus with the acylamino acid group.
- the presence of a carbocyclic ring in - (Lh) - counts as the number of atoms approximately equivalent to the calculated diameter of the carbocyclic ring.
- a benzene or cyclohexane ring in the acid linker counts as 2 atoms in calculating the length of - (Lh) - ⁇
- Illustrative acylamino acid linker groups are;
- groups (a) , (b) and (c) have acylamino acid linker lengths of 5, 7, and 2, respectively;
- acid linker means an organic group which when attached to a tricyclic nucleus, through suitable linking atoms (hereinafter defined as the “acid linker”), acts as a proton donor capable of hydrogen bonding.
- acidic group e.g., an organic group which when attached to a tricyclic nucleus, through suitable linking atoms (hereinafter defined as the “acid linker”), acts as a proton donor capable of hydrogen bonding.
- salts of the above tricyclic compounds are an additional aspect of the present invention.
- various salts may be formed which are more water soluble and physiologically suitable than the parent compound.
- Representative pharmaceutically acceptable salts include but are not limited to the alkali and alkaline earth salts such as lithium, sodium, potassium, calcium, magnesium, aluminum and the like. Salts are conveniently prepared from the free acid by treating the acid in solution with a base or by exposing the acid to an ion exchange resin.
- salts include the relatively non-toxic, inorganic and organic base addition salts of compounds of the present invention, for example, ammonium, quaternary ammonium, and amine cations, derived from nitrogenous bases of sufficient basicity to form salts with the compounds of this invention (see, for example, S. M. Berge, et al . , "Pharmaceutical Salts," J. Phar . Sci . , 66: 1-19 (1977)).
- Compounds of the invention may have chiral centers and exist in optically active forms. R- and S- isomers and racemic mixtures are contemplated by this invention.
- a particular stereoiso er may be prepared by known methods using stereospecific reactions with starting materials containing asymmetric centers already resolved or, alternatively, by subsequent resolution of mixtures of stereoisomers using known methods.
- Prodrugs are derivatives of the compounds of the invention which have chemically or metabolically cleavable groups and become by solvolysis or under physiological conditions the compounds of the invention which are pharmaceutically active in vivo .
- Prodrugs of the compounds of this invention have activity in both their acid and base derivative forms, but the acid derivative form often offers advantages of solubility, tissue compatibility, or delayed release in a mammalian organism (see, Bundgard, H. , Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985).
- Prodrugs include acid derivatives, such as, esters prepared by reaction of the parent acidic compound with a suitable alcohol, or amides prepared by reaction of the parent acid compound with a suitable amine.
- Simple aliphatic esters e.g., methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert-butyl
- aromatic esters derived from acidic groups pendent on the compounds of, this invention are preferred prodrugs.
- Other preferred esters include morpholinoethyloxy, diethylglycolamide and diethylaminocarbonylmethoxy.
- double ester type prodrugs such as (acyloxy) alkyl esters or ( (alkoxycarbonyl) oxy) alkyl esters .
- acid protecting group is used herein as it is frequently used in synthetic organic chemistry, to refer to a group which will prevent an acid group from participating in a reaction carried out on some other functional group in the molecule, but which can be removed when it is desired to do so.
- acid protecting group is discussed by T. W. Greene in chapter 5 of Protective Groups in Organic Synthesis, John Wiley and Sons, New York, 1981, incorporated herein by reference in its entirety.
- acid protecting groups include ester or amide derivatives of the acid group, such as, methyl, methoxymethyl , methyl-thiomethyl, tetrahydropyranyl , methoxyethoxymethyl, benzyloxymethyl , phenyl, aryl, ethyl, 2, 2 , 2-trichloroethyl, 2-methylthioethyl, t-butyl, cyclopentyl, triphenylmethyl , diphenylmethyl , benzyl, trimethylsilyl, N,N-dimethyl, pyrrolidinyl, piperidinyl, or o-nitroanilide.
- a preferred acid-protecting group is methyl .
- a preferred subclass of compounds of formula (I) are those wherein R21 is selected from the group hydrogen, halo, -(C1-C3) alkyl, - (C3-C4) cycloalkyl, -(C3-
- Another preferred subclass of compounds of formula (I) are those wherein for R ⁇ ' , - (L j - - is an alkyl chain of 1 or 2 carbon atoms .
- R ⁇ group R ⁇ O j_ s selected from the group consisting of cycloalkyl, cycloalkenyl, phenyl, naphthyl, norbornanyl, bicycloheptadienyl, tolulyl, xylenyl, indenyl, stilbenyl, terphenylyl, diphenylethylenyl, phenyl-cyclohexenyl, acenaphthylenyl, and anthracenyl, biphenyl, bibenzylyl and related bibenzylyl homologues represented by the formula (bb) ,
- n is a number from 1 to 8.
- R ⁇ is selected from the group consisting of
- R ⁇ 7 is a radical independently selected from halo, - (Ci-Cio) alkyl, - (Ci-Cio) alkoxy, -S- (Ci-Cio alkyl), and - (C ⁇ -C ⁇ o)haloalkyl, and w is a number from 0 to 5.
- R ⁇ ' is a substituent having an acylamino acid linker with an acylamino acid linker length of 2 or 3 and the acylamino acid linker group, -
- Q is selected from the group -(CH2)-, -0-, -NH-, and -S-, and R84 and R85 are each independently selected from hydrogen, - (C1-C10) alkyl, aryl, - (C1-C10) alkylaryl, -aryl (C1-C10) alkyl, carboxy, carbalkoxy, and halo.
- R84 and R85 are each independently selected from hydrogen, - (C1-C10) alkyl, aryl, - (C1-C10) alkylaryl, -aryl (C1-C10) alkyl, carboxy, carbalkoxy, and halo.
- Most preferred are compounds where the acylamino acid linker, -(Lft)-, for R2 ' is selected from the specific groups;
- R2 ' is a substituent having an acylamino acid linker with an acylamino acid linker length of 3 to 8 atoms and the acid linker group, - (Lh . ) for R2 ' is selected from;
- r is a number from 1 to 7 , s is 0 or 1, and Q is selected from the group -(CH2)-, -0-, -NH-, and -S-, and R84 and R85 are each independently selected from hydrogen, - (Ci-Cio) alkyl, aryl, - (C1-C10) alkylaryl, -aryl (C1-C10) alkyl, carboxy, carbalkoxy, and halo.
- Most preferred are compounds where the acylamino acid linker, -(Lh)- for R ⁇ ' is selected from the specific groups ;
- R84 and R85 are each independently selected from hydrogen, - (Ci-Cio) alkyl, aryl, - (Ci-Cio) alkaryl, - (C ⁇ C ⁇ o)aralkyl, carboxy, carbalkoxy, and halo.
- R3 ' is selected from hydrogen and non-interfering substituents, with the non-interfering substituents being selected from the group consisting of hydrogen, - (C1-C6) alkyl, - (C2-C6) alkenyl, - (C2- Cg) alkynyl,
- R ⁇ ' Another preferred group of substituents for R ⁇ ' include H, -0(C ⁇ -C4) alkyl, halo, - (C ⁇ -Cg) alkyl, phenyl, - (C ⁇ -
- R 9 and R ⁇ u are independently hydrogen, - (C1-C4) alkyl or -phenyl (C ⁇ _-C4) alkyl and n is 1 to 8;
- R3 ' include H, -0(C ] _-
- alkylphenyl phenyl substituted with - (C ⁇ -Cg) alkyl, halo or -CF3 ; -CH2 ⁇ Si (C ⁇ -Cg) alkyl, furyl, thiophenyl, -
- R 9 and R10 are independently - (C Q _- C4) alkyl or -phenyl (C1-C4) alkyl and n is 1 to 8.
- Preferred compounds of the invention are those having the general formula (Ila)
- R 1 is -CONH , -NHNH 2 , or -NH 2 ;
- R ⁇ is selected from the group consisting of -0(CH2) m ⁇ ' where
- R 5 ' is -CONR 9 R 10 , where R 9 and R 10 are independently
- - (Lh) - (acylamino acid) is a group wherein - (Lh) - is an acylamino acid linker having an acylamino acid linker length of 1 to 7 and m is 1-2;
- R 3 is H, -0(C ⁇ -C 4 ) alkyl, halo, - (C ⁇ -C 6 ) alkyl, phenyl, -(Ci- C 4 ) alkylphenyl ; phenyl substituted with - (Ci-C ⁇ ) alkyl, halo, or -CF 3 ; -CH 2 OSi ( L -C 6 ) alkyl, furyl, thiophenyl, - (C ⁇ -C 6 )hydroxyalkyl; or -(CH 2 ) n R 8 where R 8 is H, -C0NH 2 , - NR 9 R 10 , -CN or phenyl where R 9 and R 10 are independently - (C 1 -C 4 ) alkyl or -phenyl (C 1 -C 4 ) alkyl and n is 1 to 8 ; R 4 is H, - (C 1 -C 14 ) alkyl, - (C-C 14
- Z is cyclohexenyl, or phenyl; or a pharmaceutically acceptable racemate, solvate, tautomer, optical isomer, prodrug derivative or salt, thereof.
- Preferred substituents of compounds of formula I, II and la include the following:
- R 1 is -CONH2, -NH2, -NHNH2;
- R 1 is -CONH2
- R2 ' is -0(CH2)m ⁇
- R ⁇ is an acylamino acid group represented by the formula: wherein R a is selected from the group consisting of H, . (C ⁇ -Cg) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl and aryl, -CF3 ; and wherein R 3 is an "amino acid residue" of either a natural or unnatural amino acid with the nitrogen atom being part of the amino group of the amino acid.
- An amino acid residue is selected from the group comprising isoleucine, valine, phenylalanine, aspartic acid, leucine, glycine, asparagine, cystein, glutamine, glutamic acid, histidine, lysine, methionine, serine, threonine, tryptophan, 2-proline, d-proline, tyrosine and derivatives thereof .
- R 8 is H or phenyl;
- R 3 is H, or -0(C ⁇ -C4 alkyl);
- R 3 is -(CH2) n R 8 where R 8 is - (C1-C4) alkyl;
- R ⁇ is phenyl;
- (k) R ⁇ is phenyl substituted at the 2- and 6- position of the phenyl ring with - (C1-C4) alkyl, (C1-C4) alkoxy, halo or phenyl ;
- R ⁇ is phenyl substituted at the 2- or 6-position of the phenyl ring with - (C1-C4) alkyl, - (C1-C4) alkoxy, halo or phenyl ;
- R ⁇ is phenyl substituted at the 3- or 5-position of the phenyl ring with - (C1-C4) alkyl, - (C1-C4) alkoxy, halo or phenyl ;
- R 4 is - (Cg-Ci4) alkyl or - (Cg-Ci4) cycloalkyl;
- (o) Z is cyclohexenyl or phenyl
- R 5 is -C02H, -C02(Cl-C4 alkyl),
- R 5 is -CO2H, -CO2 (C1-C4 alkyl), phenyl, or phenyl substituted with -CO2H or
- R 3 is H, -0(C ⁇ -C 4 ) alkyl, halo, - (C ⁇ -C 6 ) alkyl, phenyl, - (C 1 -C 4 ) alkylphenyl; phenyl substituted with - (C ⁇ -C 6 ) alkyl, halo, or -CF 3 ; -CH 2 0Si (C ⁇ -C 6 ) alkyl, furyl, thiophenyl, -
- R 13 and R 14 are independently selected from a halogen, Ci to Cs alkyl, Cx to C 8 alkyloxy, Ci to C 8 alkylthio, aryl, heteroaryl, and Ci to Cs haloalkyl, is an oxygen atom or a sulfur atom, L 5 is a bond, -(CH 2 )v-,
- -C C-, -CC-, -0-, or -S-
- v is an integer from 0 to 2
- ⁇ is -CH 2 - or -(CH 2 ) -
- ⁇ is an oxygen atom or a sulfur atom
- b is an integer from 0 to 3
- d is an integer from 0 to 4
- f , p, and w are independently an integer from 0 to 5
- r is an integer from 0 to 7
- u is an integer from 0 to 4, or is (e) a member of (d) substituted with at least one substituent selected from the group consisting of Ci to C 6 alkyl, Ci to C 8 alkyloxy, Ci to Cs haloalkyloxy, Ci to Cs haloalkyl, aryl, and a halogen.
- Carbazole and tetrahydrocarbazole SPLA2 inhibitors and methods of making these compounds are set out in United States Patent Application SN 09/063066 filed April 21, 1998 (titled, "Substituted Carbazoles and 1,2,3,4- Tetrahydrocarbazoles") , the entire disclosure of which is incorporated herein by reference .
- the method of the invention includes treatment of a mammal with these compounds .
- the method of the invention is for treatment of a mammal, including a human, afflicted with inflammation, said method comprising administering to said human a therapeutically effective amount of carbazole or tetrahydrocarbazole represented by the following:
- a compound of the formula (Ie) represented by the following:
- A is phenyl or pyridyl wherein the nitrogen is at the 5-, 6-, 7- or 8-position; one of B or D is nitrogen and the other is- carbon; Z is cyclohexenyl, phenyl, pyridyl, wherein the nitrogen is at the 1-, 2-, or 3-position, or a 6-membered heterocyclic ring having one heteroatom selected from the group consisting of sulfur or oxygen at the 1-, 2- or 3- position, and nitrogen at the 1-, 2-, 3- or 4-position;
- R ⁇ is selected from groups (a) , (b) and (c) where;
- (a) is -(C 5 -C 2 o) lkyl, -(C 5 -C 20 ), - (C 5 -C 20 ) alkynyl, carbocyclic radicals, or heterocyclic radicals, or
- (b) is a member of (a) substituted with one or more independently selected non-interfering substituents;
- (c) is the group -(L)-R 8 ⁇ ; where, - (L) - is a divalent linking group of 1 to 12 atoms selected from carbon, hydrogen, oxygen, nitrogen, and sulfur; wherein the combination of atoms in -(L)- are selected from the group consisting of (i) carbon and hydrogen only, (ii) one sulfur only, (iii) one oxygen only, (iv) one or two nitrogen and hydrogen only, (v) carbon, hydrogen, and one sulfur only, and (vi) and carbon, hydrogen, and oxygen only; and where R 8 ⁇ is a group selected from (a) or (b) ; R21 is a non-interfering substituent; Rl' is -NHNH 2 , -NH or -C0NH ;
- R2 ' is the group -0(CH2)t R5 ' where
- R 3 ' is selected from non-interfering substituent, carbocyclic radicals, carbocyclic radicals substituted with non-interfering substituents, heterocyclic radicals, and heterocyclic radicals substituted with non- interfering substituents; or a pharmaceutically acceptable racemate, solvate, tautomer, optical isomer, prodrug derivative or salt thereof; provided that; when R 3 ' is H, R ⁇ is benzyl and m is 1 or
- R 2 ' cannot be -0(CH2) m H; and provided that when d is nitrogen, the heteroatom of z is selected from the group consisting of sulfur or oxygen at the 1-, 2- or 3-position and nitrogen at the 1-, 2-, 3- or 4-position.
- Z is cyclohexenyl, or phenyl
- R21 is a non-interfering substituent
- R 1 is -NHNH2, -CONH 2 OR -NH 2 ;
- R2 is the group -0(CH2)][R5 where
- R5 is - (acylamino acid) where the acylamino acid group is represented by the formula
- R a is selected from the group consisting of H, (C ⁇ -Cg) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl and aryl, -CF3 ; and wherein R 9 -k is an amino acid residue of either a natural or unnatural amino acid with the nitrogen atom being part of the amino group of the amino acid, and where is 1-3;
- R 3 is H, -0(C ⁇ -C4) alkyl, halo, - (C]_-Cg) alkyl, phenyl,
- R 4 is H, - (C5-C14) alkyl, - (C3-C14) cycloalkyl, pyridyl, phenyl or phenyl substituted with - (C]_-Cg) alkyl, halo, - CF3, -OCF3, -(C!-C ) alkoxy, -CN, - alkylthio, phenyl (CI-C4) alkyl, - (C -C4) alkylphenyl, phenyl, phenoxy or naphthyl; or a pharmaceutically acceptable racemate, solvate, tautomer, optical isomer, prodrug derivative or salt, thereof .
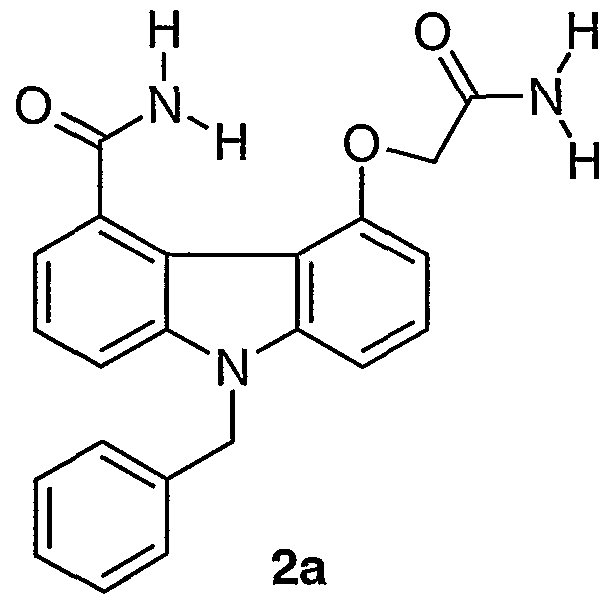
- Preferred specific compounds including all salts and prodrug derivatives thereof, for practicing the method of the invention are as follows:
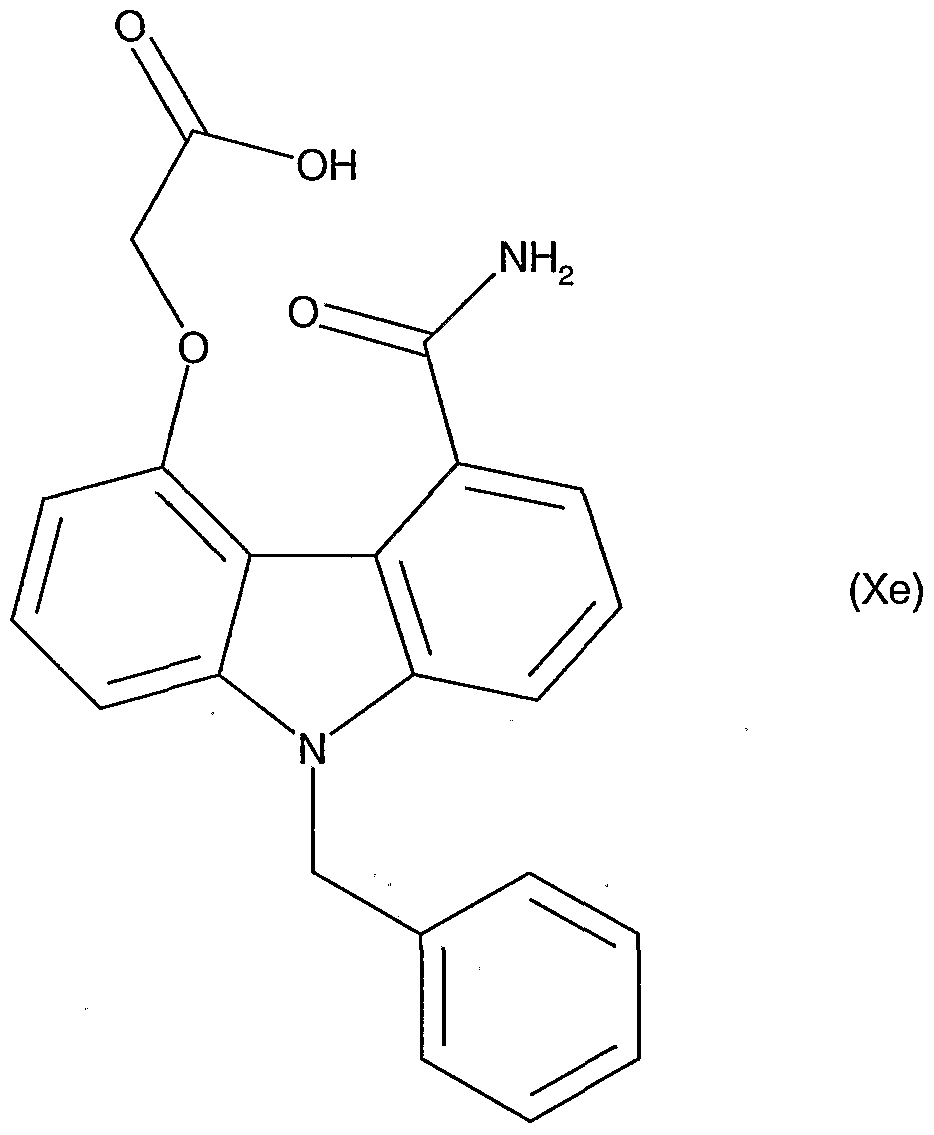
- Particularly preferred compounds useful as starting materials for preparation of compounds for the treatment of inflammation are represented by the formulae (Xe) and (Xle) below:
- Prodrugs are derivatives of SPLA2 inhibitors used in the method of the invention which have chemically or metabolically cleavable groups and become by solvolysis or under physiological conditions the compounds of the invention which are pharmaceutically active in vivo.
- Derivatives of the compounds of this invention have activity in both their acid and base derivative forms, but the acid derivative form often offers advantages of solubility, tissue compatibility, or delayed release in a mammalian organism (see, Bundgard, H. , Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985).
- Prodrugs include acid derivatives well known to practitioners of the art, such as, for example, esters prepared by reaction of the parent acidic compound with a suitable alcohol, or amides prepared by reaction of the parent acid compound with a suitable amine. Simple aliphatic or aromatic esters derived from acidic groups pendent on the compounds of this invention are preferred prodrugs. In some cases it is desirable to prepare double ester type prodrugs such as (acyloxy) alkyl esters or ( (alkoxycarbonyl) oxy) alkyl esters.
- Specific preferred prodrugs are ester prodrugs inclusive of methyl ester, ethyl ester, n-propyl ester, isopropyl ester, n-butyl ester, sec-butyl, tert-butyl ester, N,N- diethylglycolamido ester, and morpholino-N-ethyl ester.
- Methods of making ester prodrugs are disclosed in U.S. Patent No. 5,654,326. Additional methods of prodrug synthesis are disclosed in U.S. Provisional Patent Application Serial No.
- Carbazole and tetrahydrocarbazole SPLA2 inhibitor compounds useful for practicing the method of the invention may be made by the following general methods:
- R ⁇ ' is [tricyclic] -Lh ⁇ (acylamino acid) where the acylamino acid group is represented by the formula
- R 9a is selected from the group consisting of H, (C]_-Cg) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl and aryl, -CF3 ,- and wherein NR 9 -" is an amino acid residue of either a natural or unnatural amino acid with the nitrogen atom being part of the amino group of the amino acid, may be prepared by coupling the tetrahydrocarbazole or the carbazole of formula Ie, If, or Ig with an amino acid or protected amino acid such as the methyl ester as shown for example in Scheme 2
- the compounds of the present invention wherein R ⁇ ' is the group -Lh-acylamino acid group are prepared by base catalyzed amino acid coupling reactions .
- the reaction involves coupling of the free acid or acid derivative as shown in for example, Scheme 2, with a N- terminus amino acid (i.e, the C-terminal has been capped by derivatization to the ester for example) .
- the reaction may be catalyzed by a base. In general a base which minimizes the incidence of racemization, i.e, a hindered base is employed. Particularly preferred for this reaction is a tertiary base such as collidine.
- Various amino acid coupling agent may be utilized to afford the coupled product of formula I or derivative thereof . Coupling agents include but are not limited to HOBT (hydroxy benzotriazole) , dicyclohexyl carbodiimide
- BOP (dimethy1amino)phosphonium hexafluorophosphate
- PyBOP pyrrolidiniu BOP
- BOP pyrrolidiniu BOP
- an equivalent of the BOP reagent is added to a mixture (preferably 1:1) of the starting material and the N-terminus amino acid in a suitable solvent.
- a suitable solvent is inert to the substrates under the reaction condition.
- a preferred solvent is DMF .
- the coupling reaction may be performed at about 0-
- reaction is preferably performed at about room temperature.
- the reaction time is from about 10 minutes to about 48 hours.
- a preferred reaction time is from about 1 to 3 hours.
- the reaction product may be isolated after removing the ⁇ solvent and following chromatography. Other isolation protocols known to one of skill in the art are not precluded.
- the protected amino acid derivative compounds as shown in Scheme 2 may be further hydrolyzed to afford the free acid compounds as shown for example in scheme 3 (below),- the compounds and processes of which are further embodiments of the present invention.
- Protecting groups of the R ⁇ ' substituent, particularly when R5 ' is -Lh ⁇ may be removed by hydrolysis .
- Preferred is basic hydrolysis employing aqueous bases for example NaOH, OH, LiOH, Ca(OH)2' etc. Particularly preferred is lithium hydroxide.
- the hydrolysis reaction is performed in an aqueous solvent or a mixture of aqueous solvents . Preferred is a mixture of THF, methanol and water. The reaction is preferably performed at about room temperature or colder.
- the product may be isolated as the salt or as the free acid after acid hydrolysis (e.g., 5N HCl) .
- a further embodiment of the present invention and compounds thereof are prepared by subjecting the free amino acid product of step 3 to a subsequent coupling reaction as in Scheme 2 to form the di-peptide, tri- peptide and polypeptide.
- the process of Scheme 2 can be accomplished by utilizing a peptide (mono, di, tri or poly) to form the corresponding compounds of formula I .
- Procedure for generating the peptides and methods of coupling analogous to the above described are known torn one of skill in the art and are also provided in Amino Acid and Peptide Synthesis, John Jones, Oxford Chemistry Primers, Stephen G. Davis Editor, Oxford University Press Inc. New York, NY (1992).
- R 1 is -NE-2
- R 3 (a) is H, -0 (C1-C4) alkyl, halo, - (C ⁇
- R 8 is H, -NR 9 R 10 , -CN or phenyl where R 9 and R!0 are independently hydrogen, -CF3 , phenyl, - (C ⁇ C4) alkyl, - (C1-C4) alkylphenyl or -phenyl (C1-C4) lkyl and n is 1 to 8; R 2(a ) i s -0CH3 or -OH.
- An appropriately substituted nitrobenzene (1) can be reduced to the aniline (2) by treatment with a reducing agent, such as hydrogen in the presence of Pd/C, preferably at room temperature .
- a reducing agent such as hydrogen in the presence of Pd/C, preferably at room temperature .
- Compound (2) is N-alkylated at temperatures of from about 0 to 20 °C using an alkylating agent such as an appropriately substituted aldehyde and sodium cyanoborohydride to form (3).
- an appropriately substituted benzyl halide may be used for the first alkylation step.
- the resulting intermediate is further N-alkylated by treatment with 2-carbethoxy-6- bromocyclohexanone, preferably at temperatures of about 80 °C to yield (4) or by treatment with potassium hexamethyldisilazide and the bromoketoester .
- the product (4) is cyclized to the tetrahydrocarbazole (5) by refluxing with ZnCl2 in benzene for from about 1 to 2 days, preferably at 80 °C, (See Julia, M. ; Lenzi, J. Preparation d'acides tetrahydro-1, 2, 3, 4-carbazole-l ou -4. Bull . Soc . Chim. France, 1962, 2262-2263).
- Compound (5) is converted to the hydrazide (6) by treatment with hydrazine at temperatures of about 100 °C, or to the amide (7) by reacting with methylchloroaluminum amide in benzene (see Levin, J.I.; Turos, E.; Weinreb, S.M. An alternative procedure for the aluminum-mediated conversion of esters to amides. Syn . Comm. , 1982, 12, 989-993) .
- (7) may be produced by treatment of (6) with Raney nickel active catalyst.
- Compounds (6) and (7) may be dealkylated, preferably at
- R ⁇ (a) ⁇ s _OH which may then be further converted to compound (9), by realkylating with a base, such as sodium hydride, and an alkylating agent, such as Br(CH2) m R ⁇ , where R ⁇ is the carboxylate or phosphonic diester or nitrile as defined above. Conversion of R ⁇ to the carboxylic acid may be accomplished by treatment with an aqueous base.
- conversion to the tetrazole may be achieved by reacting with tri-butyl tin azide or conversion to the carboxamide may be achieved by reacting with basic hydrogen peroxide.
- conversion to the acid may be achieved by reacting with a dealkylating agent such as tri ethylsilyl bromide.
- the onoester may be accomplished by reacting the diester with an aqueous base.
- R2 and R 3 are both methoxy, selective demethylation can be achieved by treating with sodium ethanethiolate in dimethylformamide at 100 °C.
- R 3a is as defined in Scheme 1, above.
- the aniline (2) is N-alkylated with 2-carbethoxy-6- bro ocyclohexanone in dimethyl formamide in the presence of sodium bicarbonate for 8-24 hours at 50 °C .
- Preferred protecting groups include methyl, carbonate, and silyl groups, such as t-butyldimethylsilyl .
- the reaction product (4') is cyclized to (5') using the ZnCl2 i- n benzene conditions described in Scheme 1(a), above.
- N- alkylation of (5') to yield (5) is accomplished by treatment with sodium hydride and the appropriate alkyl halide in dimethylformamide at room temperature for 4-8 hours .
- carbazole (5) is hydrolyzed to the carboxylic acid (10) by treatment with an aqueous base, preferably at room temperature to about 100 °C.
- the intermediate is then converted to an acid chloride utilizing, for example, oxalyl chloride and dimethylformamide, and then further reacted with a lithium salt of (S) or (R) -4-alkyl-2-oxazolidine at a temperature of about -75 °C, to give (11a) and (lib) , which are separable by chromatography.
- the diastereomers are converted to the corresponding enantiomeric benzyl esters (12) by brief treatment at temperatures of about 0 °C to room temperature with lithium benzyl oxide.
- esters to amides may be accomplished by hydrogenation using, for example, hydrogen and palladium on carbon, as described above, to make the acid and then reacting with an acyl azide, such as diphenylphosphoryl azide followed by treatment with ammonia.
- an acyl azide such as diphenylphosphoryl azide followed by treatment with ammonia.
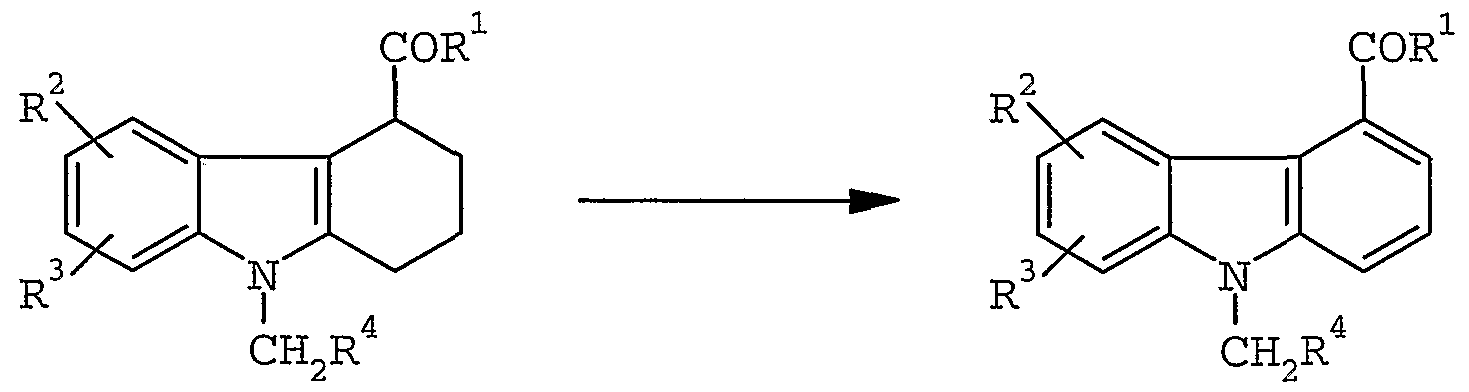
- a 1,2,3, 4-tetrahydrocarbazole-4-carboxamide or 4- carboxhydrazide (13) is dehydrogenated by refluxing in a solvent such as carbitol in the presence of Pd/C to produce the carbazole-4-carboxamide.
- a solvent such as carbitol
- Pd/C a solvent such as Pd/C
- carbazole-4-carboxamide a solvent such as carbitol
- treatment of (13) with DDQ in an appropriate solvent such as dioxane yields carbazole (14) .
- oxidation as described above may result in de-alkylation of the nitrogen.
- R 3 is substituted at the 8- position with methyl
- oxidation results in dealkylation of the nitrogen which may be realkylated by treatment with sodium hydride and the appropriate alkyl halide as described in Scheme 1(a) above to prepare the desired product (14) .
- the reactions are conducted at temperatures from about 0 to 100 °C. preferably at ambient temperature, and are substantially complete in about 1 to 48 hours depending on conditions .
- the aniline (25) and dione (15) are condensed under dehydrating conditions, for example, using the general procedure of Iida, et al . , (J of Org. Chem. 45, 2938 (1980)), with or without a non-interfering solvent, such as toluene, benzene, or methylene chloride, under dehydrating conditions at a temperature about 10 to 150°C.
- a non-interfering solvent such as toluene, benzene, or methylene chloride
- the water formed in the process can be removed by distillation, azeotropic removal via a Dean-Stark apparatus, or the addition of a drying agent, such as molecular sieves, magnesium sulfate, calcium carbonate, sodium sulfate, and the like.
- the process can be performed with or without a catalytic amount of an acid, such a p-toluenesulfonic acid or methanesulfonic acid.
- an acid such as a p-toluenesulfonic acid or methanesulfonic acid.
- suitable catalysts include hydrochloric acid, phenylsulfonic acid, calcium chloride, and acetic acid.
- solvents examples include tetrahydrofuran, ethyl acetate, methanol, ethanol, 1, 1, 2 , 2-tetrachloroethane, chlorobenzene, bromobenzene, xylenes, and carbon tetrachloride .
- the condensation of the instant process is preferably carried out neat, at a temperature about 100 to 150 °C with the resultant water removed by distillation via a stream of inert gas, such as, nitrogen or argon.
- inert gas such as, nitrogen or argon.
- the reaction is substantially complete in about 30 minutes to 24 hours.
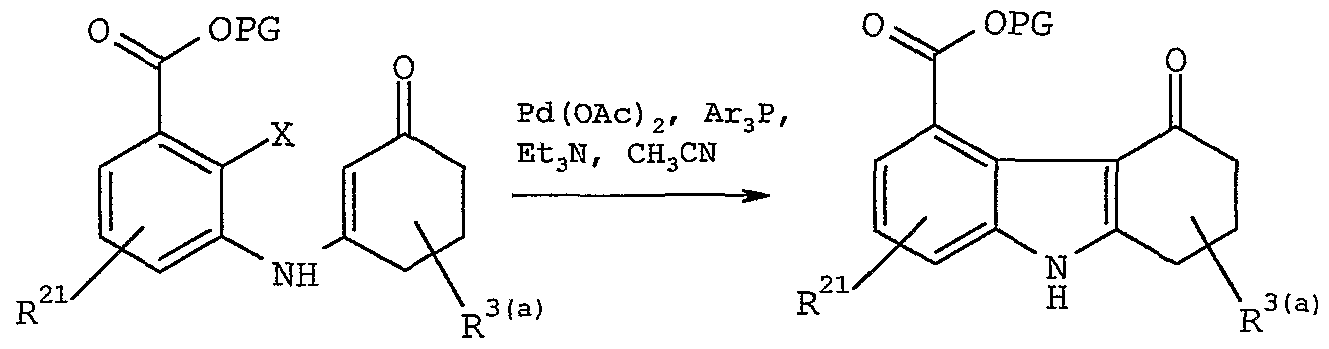
- Intermediate (26) may then be readily cyclized in the presence of a palladium catalyst, such as Pd(OAc)2 or Pd(PPb-3)4 and the like, a- phosphine, preferably a trialkyl- or triarylphosphine, such as triphenylphosphine, tri-o-tolylphosphine , or tricyclohexylphosphine, and the like, a base, such as, sodium bicarbonate, triethylamine, or diisopropylethylamme, in a non-interfering solvent, such as, acetonitrile, triethylamine, or toluene at a temperature about 25 to 200°C to form (19) .
- a palladium catalyst such as Pd(OAc)2 or Pd(PPb-3)4 and the like
- a- phosphine preferably a trialkyl- or triarylphosphine, such as triphenylpho
- solvents examples include tetrahydrofuran, benzene, dimethylsulfoxide, or dimethylformamide .
- Examples of other suitable palladium catalysts include Pd(PPh3)Cl 2 , Pd(OCOCF 3 ) 2 , [ (CH3C5H4) 3 P] 2 PdCl 2 ,
- phosphines examples include triisopropylphosphine, triethylphosphine, tricyclopentylphosphine, 1,2- bis (diphenylphosphino) ethane,
- Examples of other suitable bases include triisopropyl amine, 2 , 2 , 6, 6-tetramethylpiperidine, 1,5- diazabicyclo[2.2.2] octane (DABCO) , 1,8- diazabicyclo [5.4.0]undec-7-ene (DBU) , 1,5- diazabicyclo [4.3.0]non-5-ene, (DBN) sodium carbonate, potassium carbonate, and potassium bicarbonate.
- DABCO 1,8- diazabicyclo [5.4.0]undec-7-ene
- DBN 1,5- diazabicyclo [4.3.0]non-5-ene
- the cyclization of the instant process is preferably carried out with palladium (II) acetate as catalyst in the presence of either triphenylphosphine, tri-o- tolylphosphine, 1, 3-bis (diphenylphosphino) propane, or tricyclohexylphosphme in acetonitrile as solvent and triethylamine as base at a temperature about 50 to 150 °C.
- the reaction is substantially complete in about 1 hour to 14 days .
- a preferred process for cyclization consists of the reaction of intermediate (26) with a palladacycle catalyst such as trans-di ( ⁇ -acetato) -bis [o- (di-o-tolylphosphino) benzyl] dipalladium (II) in a solvent such as dimethylacetamide (DMAC) at 120-140 °C in the presence of a base such as sodium acetate.
- a palladacycle catalyst such as trans-di ( ⁇ -acetato) -bis [o- (di-o-tolylphosphino) benzyl] dipalladium (II) in a solvent such as dimethylacetamide (DMAC) at 120-140 °C in the presence of a base such as sodium acetate.
- Intermediate (19) may be alkylated with an alkylating agent XCH2R4, where X is halo in the presence of a base to form (20) .
- Suitable bases include potassium carbonate, sodium carbonate, lithium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, potassium hydroxide, sodium hydroxide, sodium hydride, potassium hydride, lithium hydride, and Triton B (N- benzyltrimethylammonium hydroxide) .
- the reaction may or may not be carried out in the presence of crown ether. Potassium carbonate and Triton B are preferred.
- the amount of alkylating agent is not critical, however, the reaction is best accomplished using an excess of alkyl halide relative to the starting material .
- a catalytic amount of an iodide such as sodium iodide or lithium iodide may or may not be added to the reaction mixture.
- the reaction is preferably carried out in an organic solvent, such as, acetone, dimethylformamide, dimethylsulfoxide, or acetonitrile.
- organic solvents include tetrahydrofuran, methyl ethyl ketone, and t-butyl methyl ether.
- the reaction is conducted at temperatures from about -10 to 100 °C. preferably at ambient temperature, and is substantially complete in about 1 to 48 hours depending on conditions.
- a phase transfer reagent such as tetrabutylammonium bromide or tetrabutylammonium chloride may be employed.
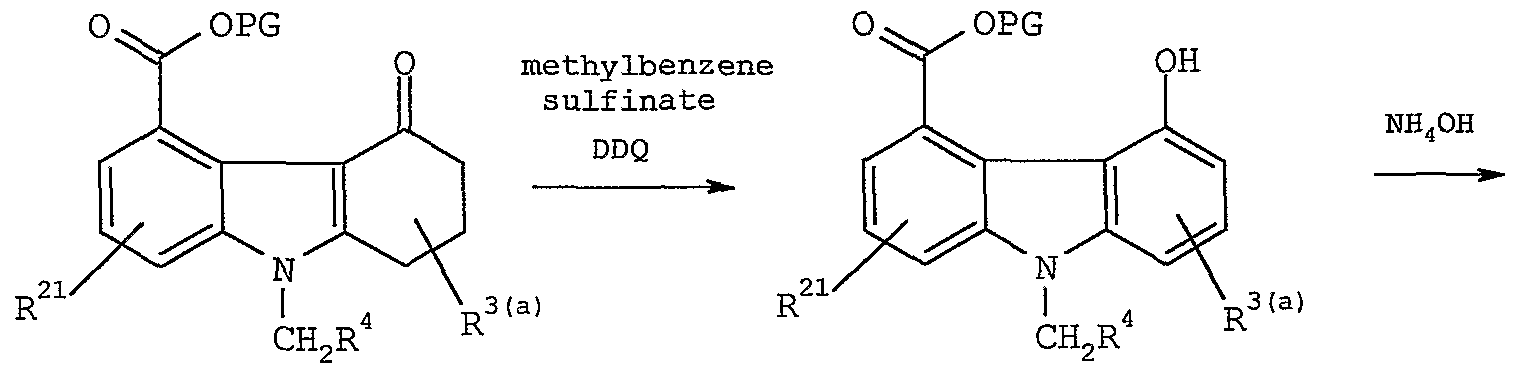
- Intermediate (20) may by dehydrogenated by oxidation with 2, 3-dichloro-5, 6-dicyano-l, 4-benzoquinone in a non- interfering solvent to form (21) .
- Suitable solvents include methylene chloride, chloroform, carbon tetrachloride, diethyl ether, methyl ethyl ketone, and t-butyl methyl ether. Toluene, benzene, dioxane, and tetrahydrofuran are preferred solvents.
- the reaction is carried out at a temperature about 0 to 120 °C . Temperatures from 50 to 120 °C are preferred. The reaction is substantially complete in about 1 to 48 hours depending on conditions.
- Intermediate (21) may be a inated with ammonia in the presence of a non-interfering solvent to form compound (22) .
- Ammonia may be in the form of ammonia gas or an ammonium salt, such as ammonium hydroxide, ammonium acetate, ammonium trifluoroacetate, ammonium chloride, and the like.
- Suitable solvents include ethanol, methanol, propanol, butanol, tetrahydrofuran, dioxane, and water. A mixture of concentrated aqueous ammonium hydroxide and tetrahydrofuran or methanol is preferred for the instant process.
- the reaction is carried out at a temperature about 20 to 100 °C . Temperatures from 50 to 60 °C are preferred.
- the reaction is substantially complete in about 1 to 48 hours depending on conditions.
- Alkylation of (22) is achieved by treatment with an alkylating agent of the formula XCH 2 R ⁇ where X is halo and R 70 is -C0 R 71 , -SO3R 71 , -P (O) (OR 71 ) 2 , or -
- Suitable bases include potassium carbonate, sodium carbonate, lithium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, potassium hydroxide, sodium hydroxide, sodium hydride, potassium hydride, lithium hydride, and Triton B (N-benzyltrimethylammonium hydroxide) .
- the reaction may or may not be carried out in the presence of a crown-ether.
- Cesium carbonate and Triton B are preferred bases.
- the amount of alkylating agent is not critical, however, the reaction is best accomplished using an excess of alkyl halide relative to the starting material.
- the reaction is preferably carried out in an organic solvent, such as, acetone, dimethylformamide, dimethylsulfoxide, or acetonitrile.
- organic solvents include tetrahydrofuran, methyl ethyl ketone, and t-butyl methyl ether.
- the reaction is conducted at temperatures from about -10 to 100 °C. preferably at ambient temperature, and is substantially complete in about 1 to 48 hours depending on conditions.
- a phase transfer reagent such as tetrabutylammonium bromide or tetrabutylammonium chloride may be employed.
- Intermediate (23) may be optionally hydrolyzed with a base or acid to form desired product (24) and optionally salified.
- Hydrolysis of (23) is achieved using a base such as sodium hydroxide, potassium hydroxide, lithium hydroxide, aqueous potassium carbonate, aqueous sodium carbonate, aqueous lithium carbonate, aqueous potassium bicarbonate, aqueous sodium bicarbonate, aqueous lithium bicarbonate, preferably sodium hydroxide and a lower alcohol solvent, such as, methanol, ethanol, isopropanol, and the like.
- a base such as sodium hydroxide, potassium hydroxide, lithium hydroxide, aqueous potassium carbonate, aqueous sodium carbonate, aqueous lithium carbonate, aqueous potassium bicarbonate, aqueous sodium bicarbonate, aqueous lithium bicarbonate, preferably sodium hydroxide and a lower alcohol solvent, such as, methanol, ethanol, isopropanol, and the like.
- a lower alcohol solvent such as, methanol, ethanol, isopropanol, and the like.
- suitable solvents include
- the acid protecting group may be removed by organic and inorganic acids, such as trifluoroacetic acid and hydrochloric acid with or without a non- interfering solvent.
- Suitable solvents include methylene chloride, tetrahydrofuran, dioxane, and acetone.
- the t- butyl esters are preferably removed by neat tri luoroacetic acid.
- the reaction is conducted at temperatures from about -10 to 100°C. preferably at ambient temperature, and is substantially complete in about 1 to 48 hours depending on conditions.
- a base preferably potassium carbonate or sodium carbonate
- a non-interfering solvent preferably dimethylformamide or dimethylsulfoxide.
- the preferred alkyl halide is methyl iodide.
- the reaction is conducted at temperatures from about 0 to 100°C. preferably at ambient temperature, and is substantially complete in about 1 to 48 hours depending on conditions.
- the starting material (16) may be prepared by condensation with an alcohol HOPG, where PG is an acid protecting group, in the presence of a dehydrating catalyst such as, dicyclohexylcarbodiimide (DCC) or carbonyl diimidazole.
- a dehydrating catalyst such as, dicyclohexylcarbodiimide (DCC) or carbonyl diimidazole.
- X is halo
- Compound (28) is converted to the carbazole product (29) by treatment with a trialkyl or triaryl phosphite or phosphine, such as, triethylphosphite or triphenyl phosphine, according to the general procedure of Cadogan, et al . (see J. Cadogan et al . , J. Chem. Soc . , 4831 (1965)
- Compound (29) is N-alkylated with an appropriately substituted alkyl or aryl halide XCH R in the presence of a base, such as sodium hydride or potassium carbonate, in a non-interfering solvent, such as toluene, dimethylformamide, or dimethylsulfoxide to afford carbazole (30) .
- a base such as sodium hydride or potassium carbonate
- a non-interfering solvent such as toluene, dimethylformamide, or dimethylsulfoxide
- Compound (30) is converted to the corresponding amide (22) by treatment with boron tribromide or sodium thioethoxide, followed by ammonia or an ammonium salt, such as ammonium acetate, in an inert solvent, such as water or alcohol, or with methylchloroaluminum amide in an inert solvent, such as toluene, at a temperature between 0 to 110 °C .
- an inert solvent such as water or alcohol
- methylchloroaluminum amide in an inert solvent, such as toluene
- Conversion to the desired prodrug may be accomplished by techniques known to the skilled artisan, such as for example, by treatment with a primary or secondary halide to make an ester prodrug.
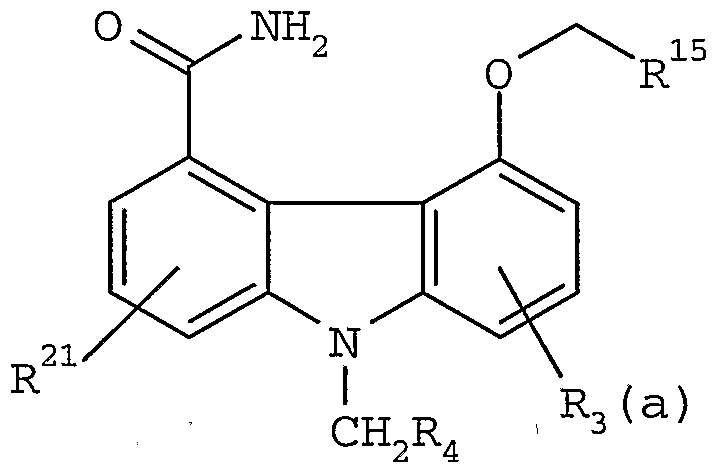
- Typical starting materials which may be converted to the corresponding compounds of formula I useful in the present invention, by the methods described supra, also include:
- the compounds described herein are believed to achieve their beneficial therapeutic action principally by direct inhibition of human SPLA2 , and not by acting as antagonists for arachidonic acid, nor other active agents below arachidonic acid in the arachidonic acid cascade, such as 5-lipoxygenases, cyclooxygenases, etc.
- the method of the invention for inhibiting SPLA2 mediated release of fatty acids comprises contacting sPLA with a therapeutically effective amount of the compound of Formula (I) or its salt.
- the compounds of the invention may be used in a method of treating a mammal (e.g., a human) to alleviate the pathological effects of septic shock, adult respiratory distress syndrome, pancreatitus, trauma, bronchial asthma, allergic rhinitis, and rheumatoid arthritis; wherein the method comprises administering to the mammal a compound of formula (I) in a therapeutically effective amount.
- a mammal e.g., a human
- a “therapeutically effective" amount is an amount sufficient to inhibit sPLA2 mediated release of fatty acid and to thereby inhibit or prevent the arachidonic acid cascade and its deleterious products.
- the therapeutic amount of compound of the invention needed to inhibit SP A2 may be readily determined by taking a sample of body fluid and assaying it for sPLA 2 content by conventional methods .
- the person or animal to be treated will be described as a "mammal", and it will be understood that the most preferred subject is a human.
- Use of the present compounds in non-human animals is also a contemplated aspect of the invention.
- the dosage ranges for other animals will necessarily be quite different from the doses administered to humans, and accordingly that the dosage ranges described will be recalculated.
- a small dog may be only l/10 th of a typical human's size, and it will therefore be necessary for a much smaller dose to be used.
- the determination of an effective amount for a certain non- human animal is carried out in the same manner described below in the case of humans, and veterinarians are well accustomed to such determinations .
- the compounds of this invention are useful for inhibiting SPLA2 mediated release of fatty acids such as arachidonic acid.
- inhibiting is meant the prevention or therapeutically significant reduction in release of sPLA2 initiated fatty acids by the compounds of the invention.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof .
- the compounds of the invention are most desirably administered at a dose that will generally afford effective results without causing any serious side effects and can be administered either as a single unit dose, or if desired, the dosage may be divided into convenient subunits administered at suitable times throughout the day.
- the specific dose of a compound administered according to this invention to obtain therapeutic or prophylactic effects will, of course, be determined by the particular circumstances surrounding the case, including, for example, the route of administration, the age, weight and response of the individual patient, the condition being treated and the severity of the patient's symptoms.
- Typical daily doses will contain a non-toxic dosage level of from about 0.01 mg/kg to about 50 mg/kg of body weight of an active compound of this invention.
- the pharmaceutical formulation is in unit dosage form.
- the unit dosage form can be a capsule or tablet itself, or the appropriate number of any of these.
- the quantity of active ingredient in a unit dose of composition may be varied or adjusted from about 0.1 to about 1000 milligrams or more according to the particular treatment involved. It may be appreciated that it may be necessary to make routine variations to the dosage depending on the age and condition of the patient.
- the dosage will also depend on the route of administration.
- a “chronic” condition means a deteriorating condition of slow progress and long continuance. As such, it is treated when it is diagnosed and continued throughout the course of the disease.
- An “acute” condition is an exacerbation of short course followed by a period of remission. In an acute event, compound is administered at the onset of symptoms and discontinued when the symptoms disappear.
- Pancreatitis, trauma-induced shock, bronchial asthma, allergic rhinitis and rheumatoid arthritis may occur as an acute event or a chronic event.
- the treatment of these conditions contemplates both acute and chronic forms.
- Septic shock and adult respiratory distress are acute conditions treated when diagnosed.
- the compound can be administered by a variety of routes including oral, aerosol, rectal, transdermal, subcutaneous, intravenous, intramuscular, and intranasal .
- compositions of the invention are prepared by combining (e.g., mixing) a therapeutically effective amount of the compounds of the invention together with a pharmaceutically acceptable carrier or diluent therefor.
- the present pharmaceutical formulations are prepared by known procedures using well known and readily available ingredients .
- the active ingredient will usually be admixed with a carrier, or diluted by a carrier, or enclosed within a carrier which may be in the form of a capsule, sachet, paper or other container.
- a carrier which may be in the form of a capsule, sachet, paper or other container.
- the carrier serves as a diluent, it may be a solid, semi- solid or liquid material which acts as a vehicle, or can be in the form of tablets, pills, powders, lozenges, elixirs, suspensions, emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium) , or ointment, containing, for example, up to 10% by weight of the active compound.
- the compounds of the present invention are preferably formulated prior to administration.
- any suitable carrier known in the art can be used.
- the carrier may be a solid, liquid, or mixture of a solid and a liquid.
- Solid form > formulations include powders, tablets and capsules.
- a solid carrier can be one or more substances which may also act as flavoring agents, lubricants, solubilisers, suspending agents, binders, tablet disintegrating agents and encapsulating material .
- Tablets for oral administration may contain suitable excipients such as calcium carbonate, sodium carbonate, lactose, calcium phosphate, together with disintegrating agents, such as maize, starch, or alginic acid, and/or binding agents, for example, gelatin or acacia, and lubricating agents such as magnesium stearate, stearic acid, or talc.
- suitable excipients such as calcium carbonate, sodium carbonate, lactose, calcium phosphate
- disintegrating agents such as maize, starch, or alginic acid
- binding agents for example, gelatin or acacia
- lubricating agents such as magnesium stearate, stearic acid, or talc.
- the carrier is a finely divided solid which is in admixture with the finely divided active ingredient.
- the active ingredient is mixed with a carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain from about 1 to about 99 weight percent of the active ingredient which is the novel compound of this invention.
- Suitable solid carriers are magnesium carbonate, magnesium stearate, talc, sugar lactose, pectin, dextrin, starch, gelatin, tragacanth, methyl cellulose, sodium carboxymethyl cellulose, low melting waxes, and cocoa butter.
- Sterile liquid form formulations include suspensions, emulsions, syrups and elixirs.
- the active ingredient can be dissolved or suspended in a pharmaceutically acceptable carrier, such as sterile water, sterile organic solvent or a mixture of both.
- a pharmaceutically acceptable carrier such as sterile water, sterile organic solvent or a mixture of both.
- the active ingredient can often be dissolved in a suitable organic solvent, for instance aqueous propylene glycol.
- suitable organic solvent for instance aqueous propylene glycol.
- Other compositions can be made by dispersing the finely divided active ingredient in aqueous starch or sodium carboxymethyl cellulose solution or in a suitable oil.
- Active ingredient refers to a compound according to Formula (I) , (II) or (III) or a pharmaceutically acceptable salt, solvate, or prodrug thereof.
- Hard gelatin capsules are prepared using the following ingredients:
- Formulation 2 A tablet is prepared using the ingredients below:
- the active ingredient is mixed with ethanol and the mixture added to a portion of the propellant 22, cooled to -30°C and transferred to a filling device. The required amount is then fed to a stainless steel container and ' diluted with the remainder of the propellant. The valve units are then fitted to the container .
- Formulation 4 Tablets each containing 60 mg of active ingredient, are made as follows:
- the active ingredient, starch and cellulose are passed through a No. 45 mesh U.S. sieve and mixed thoroughly.
- the aqueous solution containing polyvinylpyrrolidone is mixed with the resultant powder, and the mixture then is passed through a No . 14 mesh U.S. sieve.
- the granules so produced are dried at 50°C and passed through a No. 18 mesh U.S. sieve.
- the sodium carboxymethyl starch, magnesium stearate and talc, previously passed through a No. 60 mesh U.S. sieve, are then added to the granules which, after mixing, are compressed on a tablet machine to yield tablets each weighing 150 mg.
- Capsules each containing 80 mg of active ingredient, are made as follows:
- the active ingredient is passed through a No. 60 mesh U.S. sieve and suspended in the saturated fatty acid glycerides previously melted using the minimum heat necessary. The mixture is then poured into a suppository mold of nominal 2 g capacity and allowed to cool .
- Formulation 7 Suspensions, each containing 50 mg of active ingredient per 5 ml dose, are made as follows:
- the active ingredient is passed through a No. 45 mesh U.S. sieve and mixed with the sodium carboxymethyl cellulose and syrup to form a smooth paste.
- the benzoic acid solution, flavor and color are diluted with a portion of the water and added, with stirring. Sufficient water is then added to produce the required volume.
- Formulation 8 An intravenous formulation may be prepared as follows :
- the solution of the above ingredients generally is administered intravenously to a subject at a rate of 1 ml per minute.
- Bovine ⁇ Serum Albumin (fatty acid free) (1 g/L)
- TRITON X-100TM prepare at 6.249 mg/ml in reaction buffer to equal lOuM
- TRITON X-100TM is a polyoxy ethylene non- ionic detergent supplied by Pierce Chemical Company, 3747 N. Meridian Road, Rockford, Illinois 61101 .
- a measured volume of racemic dipheptanoyl thio PC supplied in chloroform at a concentration of 100 mg/ml is taken to dryness and redissolved in 10 millimolar TRITON X-100TM nonionic detergent aqueous solution.
- Reaction Buffer is added to the solution, then DTNB to give the Reaction Mixture .
- the reaction mixture thus obtained contains ImM diheptanoly thio-PC substrate, 0.29 mm Triton X-
- test compound or solvent blank
- % inhibition measured at 405 nanometers generated by enzyme reactions containing inhibitors relative to the uninhibited control reactions was determined. Each sample was titrated in triplicate and result values were averaged for plotting and calculation of IC50 values. IC50 were determined by plotting log concentration versus inhibition values in the range from 10-90% inhibition.
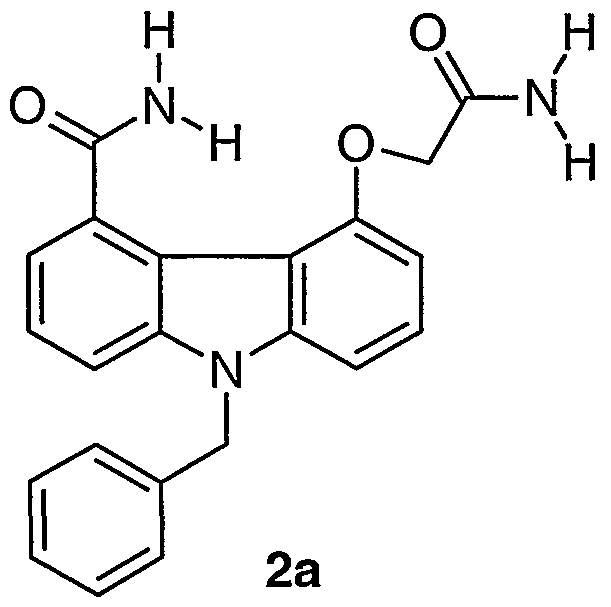
- reaction mixture was concentrated in vacuo to near dryness, then it was taken up in CH 2 CI 2 , chromatographed on a silica gel column (gradient 10-25% THF in CH 2 C1 2 ) and dried in an 80°C vacuum oven to give 0.155 g of 2b as an off-white solid in 96% yield.
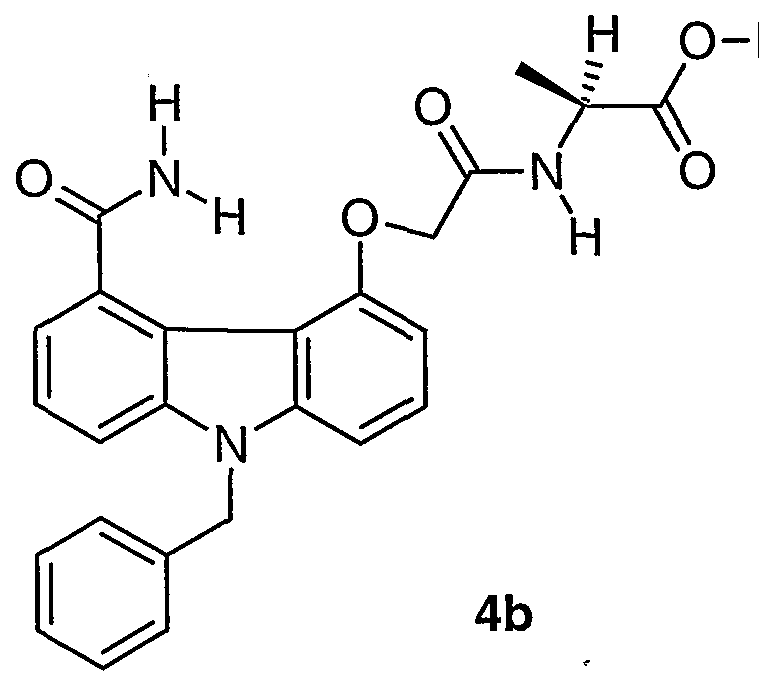
- reaction mixture was concentrated in vacua to near dryness, then it was taken up in CH 2 C1 2 and chromatographed on a silica gel column (gradient 20-40% THF in CH 2 C1 2 ) to give 0.168 g of 3a as a white solid in 71% yield.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description






































































































Claims















































Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU45996/01A AU4599601A (en) | 2000-01-07 | 2001-01-05 | Substituted tricyclics |
| EP01918984A EP1248769A2 (en) | 2000-01-07 | 2001-01-05 | Carbazole derivatives as inhibitors of spla2 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17502800P | 2000-01-07 | 2000-01-07 | |
| US60/175,028 | 2000-01-07 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/168,152 A-371-Of-International US20030096854A1 (en) | 2000-01-07 | 2001-01-05 | Substituted tricyclics |
| US10/830,380 Division US20040204473A1 (en) | 2000-01-07 | 2004-04-22 | Substituted tricyclics |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001049662A2 true WO2001049662A2 (en) | 2001-07-12 |
| WO2001049662A3 WO2001049662A3 (en) | 2002-06-27 |
Family
ID=22638535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/010850 Ceased WO2001049662A2 (en) | 2000-01-07 | 2001-01-05 | Carbazole derivatives as inhibitors of spla2 |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1248769A2 (en) |
| AU (1) | AU4599601A (en) |
| WO (1) | WO2001049662A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002079154A1 (en) * | 2001-03-28 | 2002-10-10 | Eli Lilly And Company | Substituted carbazoles as inhibitors of spla2 |
| WO2005037791A1 (en) * | 2003-10-15 | 2005-04-28 | Chiron Corporation | Compositions and methods for viral inhibition |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL180523B1 (en) * | 1994-04-01 | 2001-02-28 | Eli Lilly & Co | New compound, 1H-indole-3-glyoxylamide derivative and pharmaceutical agent PL PL PL PL PL PL PL PL PL PL |
| CA2310250A1 (en) * | 1997-11-14 | 1999-05-27 | August Masaru Watanabe | Treatment for alzheimer's disease |
-
2001
- 2001-01-05 WO PCT/US2001/010850 patent/WO2001049662A2/en not_active Ceased
- 2001-01-05 AU AU45996/01A patent/AU4599601A/en not_active Abandoned
- 2001-01-05 EP EP01918984A patent/EP1248769A2/en not_active Withdrawn
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002079154A1 (en) * | 2001-03-28 | 2002-10-10 | Eli Lilly And Company | Substituted carbazoles as inhibitors of spla2 |
| US6933313B2 (en) | 2001-03-28 | 2005-08-23 | Eli Lilly And Company | Substituted carbazoles as inhibitors of sPLA2 |
| WO2005037791A1 (en) * | 2003-10-15 | 2005-04-28 | Chiron Corporation | Compositions and methods for viral inhibition |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1248769A2 (en) | 2002-10-16 |
| AU4599601A (en) | 2001-07-16 |
| WO2001049662A3 (en) | 2002-06-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU753436B2 (en) | Substituted tricyclics | |
| US20030191175A1 (en) | Indole sPLA2 inhibitors | |
| CZ137199A3 (en) | Substituted tricyclic compounds | |
| WO2000007590A1 (en) | INDOLE sPLA2 INHIBITORS | |
| US6872743B2 (en) | sPLA2 inhibitors | |
| US20030096854A1 (en) | Substituted tricyclics | |
| JP2004518659A5 (en) | ||
| US6391908B1 (en) | Oxime amide indole type sPLA2 inhibitors | |
| US6448284B1 (en) | Substituted tricyclics | |
| US6933313B2 (en) | Substituted carbazoles as inhibitors of sPLA2 | |
| EP1202963B1 (en) | Spla2 inhibitors | |
| WO2001049662A2 (en) | Carbazole derivatives as inhibitors of spla2 | |
| US6706752B1 (en) | sPLA2 inhibitors | |
| EP1220839B1 (en) | Hydroxyfunctional amide 1h-indole derivatives active as spla2 inhibitors | |
| US6992100B2 (en) | sPLA2 inhibitors | |
| EP1349836B1 (en) | Tetracyclic derivatives as spla2 inhibitors | |
| EP1349832A2 (en) | Substituted benzoindoles as spla2 inhibitors | |
| HK1024912A (en) | Substituted carbazoles, process for their preparation and their use as spla2 inhibitors | |
| HK1024911A (en) | Substituted carbazoles, process for their preparation and their use as spla2 inhibitors | |
| HK1010546A (en) | Substituted tricyclics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10168152 Country of ref document: US |
|
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001918984 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001918984 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |